
Submitted:
01 February 2024
Posted:
02 February 2024
You are already at the latest version
Abstract
Traumatic brain injury (TBI) is associated with alcohol abuse and higher ethanol sensitivity later in life. Currently, it is poorly understood how ethanol sensitivity changes with time after TBI and whether there are sex-dependent differences in the relationship between TBI and ethanol sensitivity. This study uses the fruit fly Drosophila melanogaster to investigate how TBI affects alcohol sensitivity and whether the effects are sex-specific. Our results indicate that flies have a significantly higher sensitivity to the intoxicating levels of ethanol during the acute phase post-TBI, regardless of sex. The increased ethanol sensitivity decreases as time progresses; however, females take longer than males to recover from the heightened ethanol sensitivity. Dietary restriction does not improve the negative effects of alcohol post-TBI. We found that tau mutant flies exhibit similar ethanol sensitivity to TBI flies. However, TBI increased the ethanol sensitivity of dtauKO mutants, suggesting that TBI and dTau loss-of-function have additive effects on ethanol sensitivty.
Keywords:
TBI
; Alcohol
; Drosophila
; Tau
1. Introduction
Traumatic brain injury injuries (TBI) are one of the leading causes of neurological disabilities and death worldwide [1,2,3]. TBI-associated disabilities may cause a broad range of short- and long-term neurological, physiological, motor, and behavioral impairments [4,5,6]. TBI is prevalent among young adults, even more so for those involved in recreational and competitive sports [7,8,9]. TBI is also common amongst US veterans that have served in Afghanistan and Iraq [10,11].
TBI outcomes are divided into primary and secondary damage. Primary damage occurs during the initial mechanical impact to the brain resulting in brain tissue damage, axonal damage, edema or blood-brain barrier impairment, and intestinal barrier dysfunction [12]. Secondary injuries include cellular and molecular changes that occur over time due to the primary injuries. Secondary damage to the brain is initiated by damaged cells that release various intracellular proteins in the extracellular space leading to the activation of pro- and anti-inflammatory cytokines, chemokines, and reactive oxygen species (ROS) [13]. Together with these immune responses, the brain injury also leads to activation of the intrinsic apoptotic pathway through the activation of sFas and Caspase 3 [14,15].
Previous work in human and mouse models have shown that TBIs are linked to an increased risk of alcohol self-administration and alcohol use disorders (AUDs) [7,16,17,18]. In addition, TBI effects on alcohol use/misuse have been found prominently in young adults engaged in sports or military veterans [7,16,17]. Furthermore, some studies indicate that TBI is associated with an increase in the frequency of binge drinking [19,20] and higher sensitivity to the sedative effects of ethanol post-TBI [16,18].
Research on the effects of alcohol-related behaviors after TBI often considers the acute phase post-brain injury; however, the time needed for the brain to recover following brain injury is currently understudied. Furthermore, most of these studies do not account for sex differences, focusing more on males [4,7,16,18]. Men account for the majority of TBIs in the US [21,22], and binge drinking is almost twice as common in males than in females [23]. However, there are significant sex differences in the responses to TBIs in patients and animal models [24,25,26,27]. There are also substantial sex differences in the escalation of self-administration and brain damage associated with excessive alcohol consumption [28,29,30,31,32]. Hence, the time course of ethanol responses following TBI and sex differences should provide valuable information as to the biological changes that account for differences in AUD susceptibility following TBI.
Drosophila melanogaster is an important model organism for studying ethanol-related behaviors and TBI due to the behavioral and molecular mechanisms that are similar to those of mammals [33,34,35,36]. In this study, we used D. melanogaster to evaluate the effect that time post-TBI and sex have on ethanol sensitivity. Adult flies were exposed to a single high dose of 70% ethanol at different time intervals after TBI, and the sedation time of the flies was measured. Fly mortality rates were determined 24 hours after ethanol exposure. In addition, we investigated if dietary restriction affects ethanol sensitivity post-TBI [35].
At the molecular level, one of the major hallmarks of TBI is the accumulation of aberrant tau protein in the form of tau hyperphosphorylation or aggregates generating neuro-fibrillary tangles (NFTs) [37,38,39,40]. Tau is expressed predominantly in neurons, although it may also be expressed in glia [41]. In neurons, Tau is either bound to the inner side of the plasma membrane or to microtubules, where it stabilizes the microtubules or assists in motor-driven axonal transport [42,43]. In addition, Tau can also bind dendritic f-actin to help stabilize the cytoskeletal elements in spines [44]. Tau pathology is observed in all forms of TBI, ranging from mild to severe, and strongly correlates to TBI severity [45].
Drosophila Tau (dTau) is 44% identical and 66% similar to human Tau, and is predominantly expressed in the larval imaginal discs and adult retina and brain [46]. dTau is a true vertebrate ortholog, and its loss alters brain cytoskeletal dynamics and affects neuronal plasticity, causing retinal degeneration and impaired habituation [47,48]. To elucidate the importance of dTau in ethanol sensitivity post TBI we used the dtauKO mutants and flies expressing dtau RNAi, and assessed their role in alcohol sensitivity, with and without TBI.
2. Results
2.1. TBI Paradigm
It has previously been shown that the number of head strikes a fly receives with the HIT device increases the mortality rate [34]. We also found that increasing the number of strikes increases the mortality rate 24 hours after TBI (MI24) in both males and females (Figure 1). The flies exhibit a mild mortality rate after 2 and 4 - strikes, with a 4-13% mortality rate and a moderate to severe mortality rate as the number of strikes increased (6-10 hits). Furthermore, our results indicate that the effect of TBI on mortality is found in both sexes, inflicting a similar mortality rate on both males and females (Figure 1). These data are consistent with prior findings [34]. Hence, our assay is reproducible and consistent. Our data further suggest that 4 strikes produce a relatively mild TBI, with 90-95% of the flies surviving and having apparent normal mobility 10-15 minutes after the strikes. We, therefore, used four strikes separated by five-minute intervals as our standard protocol for all subsequent experiments.
2.2. TBI Alters Ethanol Sensitivity
An increasing body of evidence indicates that TBIs are a risk factor for AUDs [49]. To determine if TBIs change the behavioral responsiveness to ethanol in Drosophila, we initially tested the sedation sensitivity of males and females exposed to a high ethanol concentration (70%). Flies were placed in 70% ethanol, and the time required for 50% of the flies to sedate (ST50) was recorded. Ethanol sensitivity was measured 2, 24, and 48 hours after TBI (Figure 2). Our results indicate that males were very sensitive to the effects of ethanol during the acute TBI period (2 hrs later). However, ethanol sensitivity improved as the time passed (24 hrs later) and was restored to normal after 48 hrs. Surprisingly we found that control flies were also slightly more sensitive to ethanol 2 hours later compared to 24 and 48 hours later, indicating that handling plays an important role in ethanol sensitivity. Nevertheless, alcohol sensitivity in flies with TBI was significantly lower than in the control during the acute TBI period and was restored to normal levels after 48 hours (Figure 2a).
Prior studies of the role of sex on human brain injury are contradictory, with some studies finding no sex differences [34], while other studies indicate that females are more likely to die from TBI than males [50,51]. Sexual dimorphism in alcohol-related behaviors is also found in many organisms, including Drosophila [30,52]. Thus, we also tested the effect of ethanol sensitivity post-TBI in females (Figure 2b). Our results indicate that during the acute phase post-TBI, both males and females are more sensitive to the sedative effects of ethanol (p<0.01 and p< 0.0001, respectively). Surprisingly, while during the acute phase post-TBI, there is no sex difference in ethanol sensitivity, females remain sensitive to the effects of ethanol for a longer period compared to males (Figure 2c). These results demonstrate that TBI strongly affects ethanol sensitivity during the acute phase post-TBI. However, as time progresses, ethanol sensitivity recovery is faster in males than females.
2.3. Ethanol Exposure Post-TBI Increases the Mortality Rate
Next, we examined whether the high dose (70%) of ethanol given during the ethanol sensitivity assay affects the mortality rate. Flies exposed to alcohol post-TBI were transferred to a vial with food and water access and allowed to rest. The mortality rate (MI24) was recorded 24 hours later. The mortality rate of male flies exposed to ethanol 2 hours post-TBI was 37.9 ± 7.1%,, which is significantly higher than control TBI-treated flies that were not exposed to ethanol (Figure 3). When flies were exposed to ethanol 24 and 48 hours after TBI, the mortality rate was not significantly different from control, unexposed flies, demonstrating a time-limited effect of TBI on ethanol-induced mortality (Figure 3).
2.4. Post-TBI Diet Does Not Affect Ethanol Sensitivity and Mortality
Previous work in rodents and Drosophila indicates that diet may play an influential role during the acute phase of recovery from traumatic brain injury [35,53,54,55]. In Drosophila, a water-only (starvation) diet for up to 24 hours post-TBI significantly improves the survivorship of flies compared to a food diet [35]. Hence, we examined whether diet post-TBI impacts the sensitivity to the sedative effects of ethanol. Male and female flies were placed on food or a starvation diet following injury for 24 hours (or until an ethanol sensitivity assay was performed; Figure 4). This dietary manipulation immediately after the TBI does not affect the sedative effects of ethanol during the acute phase (2 hr) post-TBI (Figure 4a,b). Diet post-TBI also does not influence ethanol sensitivity 24 hours after the TBI (Figure 4a). Interestingly, as seen before (Figure 3), females are slower to recover normal sensitivity from the sedation effects of ethanol 24 hours post-TBI, and diet does not alter this sensitivity (Figure 4b). Our results indicate that while diet is important for attenuating some TBI-associated symptoms, it does not play a significant role in acute ethanol sensitivity post-TBI.
Since an intoxicating ethanol exposure post-TBI affects mortality, especially during the acute phase post-TBI, we further examined if diet post-TBI plays a role in ethanol-induced mortality (Figure 4c). Mortality rates were measured 24 hours after acute ethanol exposure following brain injury. Consistent with the ethanol sedation sensitivity data, diet did not improve mortality rates (MI24). Our results suggest that while a starvation diet can strongly influence post-TBI recovery [35], it does not affect ethanol sensitivity post-TBI in Drosophila.
2.5. Loss of dTau Exacerbates Acute Ethanol Sensitivity
In vertebrates, brain injury leads to axonal injury and accelerated tau pathology [37]. To determine whether dtau affects responses to ethanol sensitivity following TBI in Drosophila , we examined the ethanol sensitivity of tau mutants with and without TBI (Figure 5). We used a knock-out (tauKO) mutant lacking exons 2–6, including the tubulin-binding repeats; this deletion results in a null mutation lacking the 50kDa and 75kDa dTau isoforms [47]. The tauKO mutants exposed to acute ethanol sensitivity exhibit increased sensitivity to acute ethanol compared to wild-type Canton-S (CS) flies (Figure 5), regardless of sex (tauKO male vs. CS male p< 0.0001 and tauKO female vs. CS female p<0.0001).
In this experiment, we also subjected male and female wild type CS and dtau mutants to TBI, and measured changes in acute ethanol sensitivity 2 hours post-TBI. Heterozygous dtauKO males flies showed modest ethanol sensitivity compared to the control flies, dtauKO homozygous flies showed a significant decrease in ethanol sensitivity (p<0.0089). Homozygous dtauKO mutants, regardless of sex, show levels of ethanol sensitivity similar to CS-TBI (p>0.05). Consequentially, TBI increased the ethanol sensitivity of the dtauKO homozygotes, indicating that the chronic effect of TBI is at least partially independent of dtau.
To further validate our observation that tau expression is important for ethanol-related behaviors, we measured rapid ethanol tolerance using the loss of righting reflex (LORR) [56]. This LORR assay was used on dtauKO mutants and UAS-dtau RNAi transgenes driven exclusively in the nervous system by nSyb-Gal4 (Figure 6). Since our previous experiments suggested that the phenotype of dtauKO is not sex-specific, we used only males for these experiments. The results of the LORR assay are consistent with those obtained by the previous ethanol sensitivity assay, showing that dtauKO mutants exhibited an increased ethanol sedation sensitivity as indicated by lower LORR (Figure 6a). Therefore, using two different assays we found that homozygous dtauKO null mutant males are sensitive to both ethanol sensitivity and rapid tolerance. Interestestingly, the dtauKO mutants also displayed a reduced rapidethanol tolerance, suggesting dTau function may also be important for the neuroplasticity underlying tolerance formation (Figure 6a).
Next, we wanted to validate the effect of dtau using the targeted expression of dtau RNAi in the nervous system using the pan-neuronal driver nSyb-Gal4 (Figure 6b). Similar to the dtauKO mutants, flies with reduced dtau activity due to the expression of dtau RNAi in neurons exhibited increased ethanol sedation sensitivity. Hence, the dtau gene is critical in mediating the effects of ethanol sensitivity and rapid tolerance.
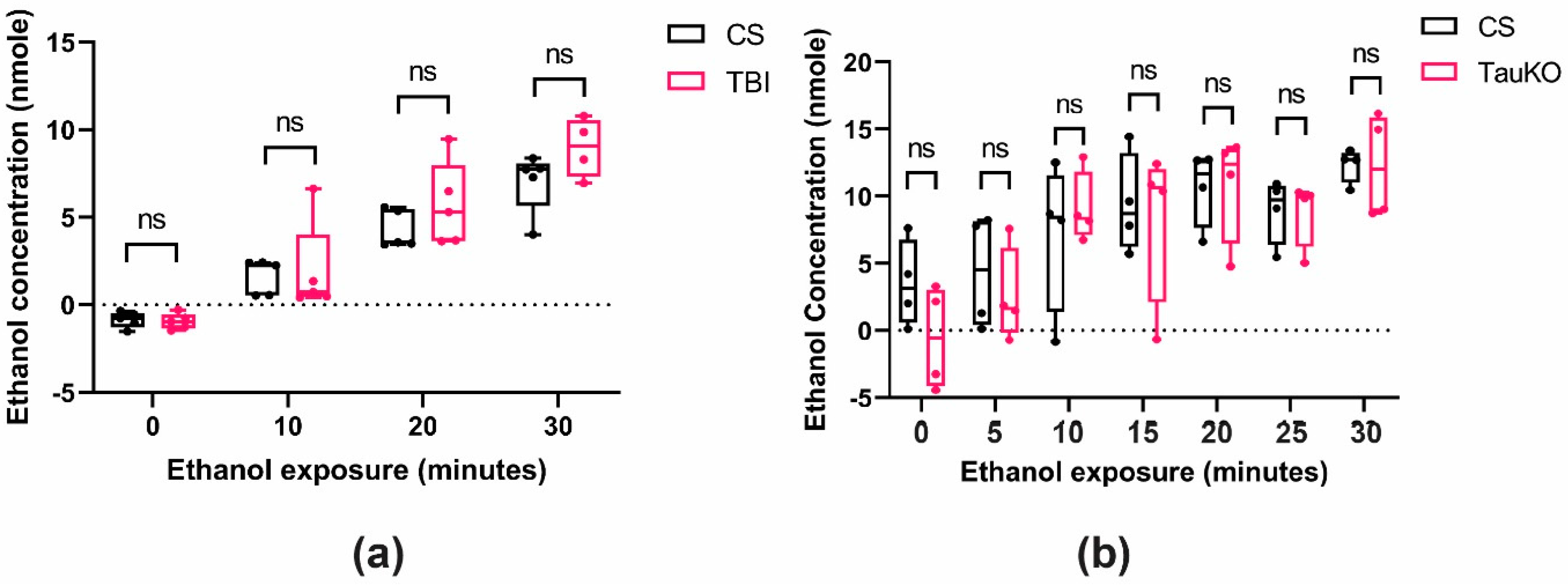
To observe if the increased ethanol sensitivity found after TBI or in the dtau loss-of-function mutant flies was caused by altered ethanol pharmacokinetics, we measured the internal alcohol absorption of wild-type Canton-S flies in the presence or absence of TBI, and in the dtauKO mutants. The tissue alcohol levels in flies exposed to TBI were not significantly different from the treatment controls (Figure 7a). Moreover, the ethanol absorption in the dtauKO mutants was also not significantly different from Canton-S (Figure 7b). These data support the hypothesis that TBI and dtau loss-of-function alter ethanol sedation sensitivity through functional changes in neuronal activity and not through differences in alcohol tissue concentrations.
3. Discussion
Herein, we have shown that TBI in Drosophila increases naïve ethanol sedation sensitivity. This increase in sedation sensitivity lasts for at least 24 hours post-TBI, with some effects still seen in females 48 hours after TBI treatments. The effect of TBI on ethanol sensitivity is seen in both males and females and is unaffected by starvation following TBI. Moreover, ethanol exposure 2 hours after TBI significantly increases mortality compared to TBI alone. However, this effect was gone by 24 hours post-TBI, suggesting acute TBI further increases ethanol intoxication sensitivity. We have also seen an increased ethanol sedation sensitivity in Oregon R and Canton-S genetic background strains. Hence, the ability of TBIs to increase the sensitivity to ethanol is robust and largely reversible over time.
We have further shown that loss-of-function dtau mutants have an increased ethanol sedation sensitivity phenotype, mimicking the effect of TBI. The pan-neuronal knockdown of dtau with RNAi also leads to an increase in ethanol sensitivity. This result verifies that reducing dtau function increases ethanol sensitivity and shows that this requirement for dTau is neuronal. Nevertheless, the effect of TBI on ethanol sedation sensitivity is additive with the dtauKO mutation. This additivity strongly suggests that the underlying causes of increased ethanol sedation sensitivity in TBI and dtauKO mutants are different.
3.1. TBI and Ethanol Sensitivity
TBIs in Drosophila are responsible for several types of injury, several of which may be at least partially responsible for an increase in ethanol sedation sensitivity and ethanol-induced mortality [57,58], including, but probably not limited to, disruption of the blood-brain barrier [34,59], mitochondrial dysfunction [60,61], and acute neuroinflammation [34,35,36,62]. The disruption of the Drosophila blood-brain barrier function can lead to changes in naïve ethanol sensitivity and tolerance formation [63,64,65]. An insertional mutation of the moody GPCRs, which are preferentially expressed in the subperineural glia, results in a decreased barrier function and increased resistance to ethanol sedation [64]. The disruption of blood-brain barrier function by TBI may also be expected to decrease ethanol sensitivity rather than increase sensitivity, as we have seen, assuming the acute response after injury is the same as the chronic disruption seen in moody mutants. However, subperineural glia knockdown of the scarface serine protease results in increased ethanol sedation sensitivity without affecting the paracellular permeability of the blood-brain barrier [65], suggesting there may be multiple ways for perturbing blood-brain barrier function that effect ethanol sensitivity. Further work is required to determine if moody or scarface pathways functionally interact with acute TBI to modify naïve ethanol sensitivity.
TBIs may also affect ethanol sensitivity through changes in mitochondrial function, although the effects of mitochondrial function on naïve alcohol sensitivity remain relatively unexplored [66]. In Drosophila, proteins required for the transport of mitochondria down axons are also required in Dopaminergic neurons to form a positive association between the hedonistic effects of ethanol and an odor [67]. This requirement is specific to the rewarding properties of ethanol since sugar reward was unaffected. In rodents, a single intoxicating dose of alcohol impacts mitochondrial function [68]. In C. elegans, intoxicating doses of ethanol lead to mitochondrial fragmentation [69]. In Zebrafish, acute presentation of ethanol stimulates mitochondrial respiration and O2 consumption [70]. Hence, across species, there appear to be exposure-induced differences in how mitochondria respond to alcohol, with changes in transport and function. Acute TBI-induced changes in mitochondrial function may sensitize neurons to ethanol inhibition, leading to increased sedation sensitivity.
TBI in Drosophila may also increase ethanol sedation sensitivity by activating neuroinflammation pathways [71,72]. TBIs activate the two main branches of the innate immune system: the Immune Deficiency Pathway (IMD) and the Toll pathway [34,35,36,57]. Sedating ethanol concentrations increases the transcription of genes within the Toll pathway [73,74]. Sedating doses of ethanol also activate the Toll pathway [72]. Genetically activating the Toll pathway increases ethanol resistance, while inhibiting the pathway increases ethanol sensitivity [72]. Since TBIs also activate Toll signaling, this injury may naively be expected to drive an increased resistance rather than increased sensitivity, as we have found. This assumption, however, does not account for differences in where Toll-signaling is employed and the outputs prioritized in the injured tissue. More information is needed on the specific effects of TBI on the Toll pathway before we understand how TBI-induced neuroinflammation affects ethanol sedation sensitivity. It is not currently known if the TBI-activated IMD pathway also affects ethanol sensitivity.
The Janus kinase/Signal transducer and activator of transcription (JAK/STAT) and c-Jun N-terminal kinase (JNK) pathways also potentially link TBI-induced immune responses and changes in alcohol responsiveness. In Drosophila, TBI induces both JAK/STAT and JNK pathways [62,75]. These two pathways are also activated by axonal injury and interact to regulate axon regeneration [76,77]. Interestingly, the knockdown of STAT92E, the only STAT ortholog in Drosophila, led to significantly higher ethanol-induced locomotor behavior in flies previously exposed to ethanol [78]. It is not currently known if this increased sensitivity to the activating properties of ethanol translates to an increased sensitivity to the sedating effects of this drug [78]. It remains possible that the loss-of-STAT92E activity would impair post-TBI axon recovery, leading to an increased sensitivity to ethanol sedation.
TBIs in Drosophila lead to wide-raging tissue damage and activation of stress and injury responses [57]. In addition to the pathways listed above, there are likely other effects of these head injuries that may also acutely influence the ethanol sensitivity we have found after the HIT protocol (e.g., calcium dysregulation [79]). It will be interesting to see how activating or inhibiting each of these known responses to TBI will affect acute ethanol sedation sensitivity and mortality. Since these pathways exhibit differences in when they are activated and required for recovery from TBIs, it will also be meaningful to see how these individual pathways alter the time course of TBI-induced hypersensitivity to ethanol.
3.2. Tau and Ethanol Sensitivity
Analogous to its vertebrate ortholog [80,81], dtau stabilizes microtubules, promotes actin polymerization and cytoskeletal dynamics, and is a negative regulator of translation; dtau has physiological roles in dendrites, nuclei, and axons [47]. In humans and vertebrate animal models, mild or severe TBIs are associated with tau pathology post-injury [37,82]. Most effects of tau on axonal degeneration are believed to be due to toxic gain-of-function posttranslational modifications of tau and not loss-of-function [41,83,84]. The exact role of dtau in Drosophila TBI-induced disorders is not currently known. Similar to mouse tau knock-out mutants, dtauKO flies have a normal survival rate and fertility [85,86]. Moreover, the dtauKO mutants maintain sexually dimorphic responses to TBI, suggesting that dtau does not play a role in these sex-dependent differences in TBI outcomes in Drosophila [87].
Several genes required for learning and memory in Drosophila also have ethanol sensitivity defects, pointing to shared synaptic mechanisms [88,89,90]. Loss-of-function dtauKO mutants have defects in habituation to electric foot shocks and demonstrate an increased ability to form protein-synthesis-dependent long-term memories (PSD-LTM) [47]. The reduction of tau expression through the expression of RNAi transgenes in the α’/β’ mushroom body neurons was sufficient to generate both habituation of PSD-LTM phenotypes [47]. Future work will establish if dTau’s requirement for normal ethanol sensitivity will be similarly localized the α’/β’ mushroom body neurons.
4. Materials and Methods
4.1. Fly Stocks and Maintenance
Oregon R flies were used as the control strain in the experiments shown in Figure 1, Figure 2, Figure 3 and Figure 4. Assays were performed on 4- to 7-day-old males or females that were not selected as virgins. The dtauKO (RRID:BDSC_64782) flies were a gift from Dr. E.M. Skoulakis (Institute for Fundamental Biomedical Research, Vari, Greece). Flies were reared on instant Drosophila medium (Carolina Biologicals, Burlington, NC) at 70% relative humidity.
Canton-S (CS) wildtype flies were used for the experiments shown in Figure 5, Figure 6 and Figure 7. These flies were raised on Cornmeal food at 25C in a 12:12 hour light dark cycle as previously described [91]. The dtauKO mutation and the nSyb-Gal4 driver (RRID:BDSC_51635) were backcrossed into the resident Cantonised-w1118 control isogenic background for six generations for this experiment. The UAS-dtauRNAi (RRID:BDSC_40875 ; P{y[+t7.7] v[+t1.8]=TRiP.HMS02042}attP40; gift from Dr. E.M. Skoulakis) was initially outcrossed for 6 generations into a Cantonized- y1 background, before replacing the the X chromsome with a wild-type Canton-S X chromosome. These flies were raised on a Standard Cornmeal media [92].
4.2. Traumatic Brain Injury (TBI) Paradigm
For these experiments, mature flies 4-7 days old were used. Flies were separated by sex, and 10-12 flies were placed into a Drosophila food vial. A TBI apparatus (HIT device) was constructed by connecting a tension spring to a board as previously described [34]. A 15ml conical tube was placed in a fixed position inside the spring to stabilize the fly vial to the concussion chamber spring. The narrow vials with flies could fit inside the conical tube, creating a stable and reproducible impact between the fly vial and the concussion chamber (Figure 6). The string was deflected at 90˚ angle and released on a pad. The velocity of the impact was ∼3.0 m/s (6.7 miles/h).
To reduce the impact of the hit, half a plug was placed on the bottom of the vial. The flies were confined to the bottom 1cm of the vial and let to rest for 5 minutes. The vial was then connected to the free end of the spring. The spring was deflected at 90 angle and released on the pad. There was a 5 minute recovery period between each strike. Following the completion of the injury, all fly cohorts were put into vials, which were placed on their sides to allow for a full recovery. Control flies were placed under the same conditions for the duration of the concussions; however, they were not subjected to any strike. Each experiment was reproduced at least 8 times (n= 80-130 flies). While the mortality rate was <10%, dead flies were excluded from further experiments.
4.3. Ethanol Sensitivity Assay and Mortality Rate
All the flies were separated into males and females. The ethanol sensitivity assay was as previously described [93] with slight modifications. In short, the treatment groups and the control were placed in an exposure chamber (vial) by placing half of a plug at the bottom of the chamber. The chambers had the same air space, allowing an equal ethanol concentration to volume ratio. 8-12 flies were placed into the ethanol exposure chamber and let to rest for 4 minutes. Then 1000 µL of 70% pure ethanol was added to a plug. Flies were subjected to the vial plug with ethanol, and the rate of ethanol sedation in each group was recorded every 4 minutes for a maximum of 60 minutes. ST50 was calculated as the median sedation time determined when half of the flies first became stationary (they were intoxicated and, therefore, lost balance and fell). Mortality rate 24 hrs (MI24) after ethanol exposure was also measured and recorded.
In Figure 6, the dtauKO and Tau RNAi mutants and transgenic lines were examined using the Loss-of-Righting Reflex (LORR) ethanol sensitivity assay)[56]. In this assay, 50% ethanol vapor is generated through a constant airflow of about 500 ml per minute and distributed to different vials containing 30 flies each. The time taken for 50% of flies to get sedated is measured by observing the loss of the righting reflex (LoRR)[56,94]. Naïve flies are exposed to 50%/50% ethanol/water vapor for ethanol sensitivity, and time for 50% LoRR is recorded. For rapid tolerance, 90% of naïve flies are sedated with 50%/50% ethanol/water vapor and incubated at room temperature for 4 hrs for recovery and further tested with a second exposure of 50% ethanol for the time taken for 50% LoRR of flies to be sedated is measured. Rapid tolerance is calculated by subtracting 50% LoRR of 1st exposure from 2nd exposure [95]. Alcohol absorption was measured as previously [91] using a colorimetric kit (Abcam, Cambridge, UK; ab65343).
4.4. Diet Studies
Flies were separated into males and females. Each sex was placed in food or starvation (water only) diet for a maximum of 24 hrs (or until ethanol sensitivity assay was performed). Following the treatment, flies were placed in an ethanol exposure chamber and ethanol sensitivity was measured. Control flies were placed into different vials similar to TBI-exposed flies, to control for the effect of handling and stress, but were not subjected to any strikes. Flies were later placed in food or water only diet similar to the TBI flies until ethanol sensitivity assay was performed.
4.5. Statistical Analyses
Statistical analyses were performed using GraphPad Prism, version 8.4.2 and version 9, and by SigmaPlot, version 15. Figures were made using GraphPad Prism, version 8.4.2. Data failing Shapiro-Wilk normality tests were Log10 transformed before ANOVA [96]. Statistical tests are specified in the figure legends. Experimental and control flies were always tested simultaneously. Following initial ANOVA, the Tukey multiple comparison test was performed. All data are expressed as means ± SE. The Kruskul-Wallis test was used to evaluate differences in the formation of rapid tolerance between Canton-S and dtauKO mutants [97].
References
- Zeiler, F.A.; Iturria-Medina, Y.; Thelin, E.P.; Gomez, A.; Shankar, J.J.; Ko, J.H.; Figley, C.R.; Wright, G.E.B.; Anderson, C.M. Integrative Neuroinformatics for Precision Prognostication and Personalized Therapeutics in Moderate and Severe Traumatic Brain Injury. Front. Neurol. 2021, 12, 729184. [Google Scholar] [CrossRef] [PubMed]
- Bigler, E.D. Neurobiology and neuropathology underlie the neuropsychological deficits associated with traumatic brain injury. Archives of Clinical Neuropsychology 2003, 18, 595–621. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hardy, J.; Zetterberg, H. The Neuropathology and Neurobiology of Traumatic Brain Injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Baratz, R.; Rubovitch, V.; Frenk, H.; Pick, C.G. The Influence of Alcohol on Behavioral Recovery after mTBI in Mice. J. Neurotrauma 2010, 27, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Masel, B.E.; DeWitt, D.S. Traumatic Brain Injury: A Disease Process, Not an Event. J. Neurotrauma 2010, 27, 1529–1540. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.M.; Donders, J. Correlates of verbal learning and memory after pediatric traumatic brain injury. Appl. Neuropsychol. Child 2017, 7, 298–305. [Google Scholar] [CrossRef]
- Alcock, B.; Gallant, C.; Good, D. The relationship between concussion and alcohol consumption among university athletes. Addict. Behav. Rep. 2018, 7, 58–64. [Google Scholar] [CrossRef]
- McCrory, P.; Meeuwisse, W.; Dvorak, J.; Aubry, M.; Bailes, J.; Broglio, S.; Cantu, R.C.; Cassidy, D.; Echemendia, R.J.; Castellani, R.J. Consensus statement on concussion in sport—the 5th international conference on concussion in sport held in Berlin, October 2016. British journal of sports medicine 2017, 51, 838–847. [Google Scholar] [CrossRef]
- Zuckerman, S.L.; Kerr, Z.Y.; Yengo-Kahn, A.; Wasserman, E.; Covassin, T.; Solomon, G.S. Epidemiology of sports-related concussion in NCAA athletes from 2009-2010 to 2013-2014: incidence, recurrence, and mechanisms. The American journal of sports medicine 2015, 43, 2654–2662. [Google Scholar] [CrossRef]
- Morissette, S.B.; Woodward, M.; Kimbrel, N.A.; Meyer, E.C.; Kruse, M.I.; Dolan, S.; Gulliver, S.B. Deployment-related TBI, persistent postconcussive symptoms, PTSD, and depression in OEF/OIF veterans. Rehabilitation psychology 2011, 56, 340. [Google Scholar] [CrossRef] [PubMed]
- Warden, D. Military TBI During the Iraq and Afghanistan Wars. J. Head Trauma Rehabilitation 2006, 21, 398–402. [Google Scholar] [CrossRef]
- Katzenberger, R.J.; Chtarbanova, S.; Rimkus, S.A.; Fischer, J.A.; Kaur, G.; Seppala, J.M.; Swanson, L.C.; Zajac, J.E.; Ganetzky, B.; Wassarman, D.A. Death following traumatic brain injury in Drosophila is associated with intestinal barrier dysfunction. eLife 2015, 4, e04790. [Google Scholar] [CrossRef]
- Gadani, S.P.; Walsh, J.T.; Lukens, J.R.; Kipnis, J. Dealing with Danger in the CNS: The Response of the Immune System to Injury. Neuron 2015, 87, 47–62. [Google Scholar] [CrossRef]
- Clark, R.S.B.; Kochanek, P.M.; Watkins, S.C.; Chen, M.; Dixon, C.E.; Seidberg, N.A.; Melick, J.; Loeffert, J.E.; Nathaniel, P.D.; Jin, K.L.; et al. Caspase-3 Mediated Neuronal Death After Traumatic Brain Injury in Rats. J. Neurochem. 2000, 74, 740–753. [Google Scholar] [CrossRef]
- Uzan, M.; Erman, H.; Tanriverdi, T.; Sanus, G.Z.; Kafadar, A.; Uzun, H. Evaluation of apoptosis in cerebrospinal fluid of patients with severe head injury. Acta Neurochir. 2006, 148, 1157–1164. [Google Scholar] [CrossRef]
- Schindler, A.G.; Baskin, B.; Juarez, B.; Lee, S.J.; Hendrickson, R.; Pagulayan, K.; Zweifel, L.S.; Raskind, M.A.; Phillips, P.E.; Peskind, E.R.; et al. Repetitive blast mild traumatic brain injury increases ethanol sensitivity in male mice and risky drinking behavior in male combat veterans. Alcohol. Clin. Exp. Res. 2021, 45, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.W.; Foster, E.; Comper, P.; Langer, L.; Hutchison, M.G.; Chandra, T.; Bayley, M. Cannabis, alcohol and cigarette use during the acute post-concussion period. Brain Inj. 2020, 34, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Lowing, J.L.; Susick, L.L.; Caruso, J.P.; Provenzano, A.M.; Raghupathi, R.; Conti, A.C. Experimental Traumatic Brain Injury Alters Ethanol Consumption and Sensitivity. J. Neurotrauma 2014, 31, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Ramchand, R.; Miles, J.; Schell, T.; Jaycox, L.; Marshall, G.N.; Tanielian, T. Prevalence and Correlates of Drinking Behaviors Among Previously Deployed Military and Matched Civilian Populations. Mil. Psychol. 2011, 23, 6–21. [Google Scholar] [CrossRef]
- Adams, R.S.; Larson, M.J.; Corrigan, J.D.; Horgan, C.M.; Williams, T.V. Frequent Binge Drinking After Combat-Acquired Traumatic Brain Injury Among Active Duty Military Personnel With a Past Year Combat Deployment. J. Head Trauma Rehabilitation 2012, 27, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Faul, M.; Coronado, V. Epidemiology of traumatic brain injury. Handbook of clinical neurology 2015, 127, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Coronado, V.G.; McGuire, L.C.; Sarmiento, K.; Bell, J.; Lionbarger, M.R.; Jones, C.D.; Geller, A.I.; Khoury, N.; Xu, L. Trends in Traumatic Brain Injury in the U.S. and the public health response: 1995–2009. J. Saf. Res. 2012, 43, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Bohm, M.K.; Liu, Y.; Esser, M.B.; Mesnick, J.B.; Lu, H.; Pan, Y.; Greenlund, K.J. Binge drinking among adults, by select characteristics and state—United States, 2018. American journal of transplantation 2021, 21, 4084–4091. [Google Scholar] [CrossRef]
- Gupte, R.P.; Brooks, W.M.; Vukas, R.R.; Pierce, J.D.; Harris, J.L. Sex Differences in Traumatic Brain Injury: What We Know and What We Should Know. J. Neurotrauma 2019, 36, 3063–3091. [Google Scholar] [CrossRef]
- Roof, R.L.; Duvdevani, R.; Stein, D.G. Gender influences outcome of brain injury: progesterone plays a protective role. Brain Res. 1993, 607, 333–336. [Google Scholar] [CrossRef]
- Farin, A.; Deutsch, R.; Biegon, A.; Marshall, L.F. Sex-related differences in patients with severe head injury: greater susceptibility to brain swelling in female patients 50 years of age and younger. J. Neurosurg. 2003, 98, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.K.; Bayir, H.; Ren, D.; Puccio, A.; Zafonte, R.D.; Kochanek, P.M. Relationships between Cerebrospinal Fluid Markers of Excitotoxicity, Ischemia, and Oxidative Damage after Severe TBI: The Impact of Gender, Age, and Hypothermia. J. Neurotrauma 2004, 21, 125–136. [Google Scholar] [CrossRef]
- Becker, J.B.; Koob, G.F. Sex Differences in Animal Models: Focus on Addiction. Pharmacol. Rev. 2016, 68, 242–263. [Google Scholar] [CrossRef] [PubMed]
- Radke, A.K.; Sneddon, E.A.; Frasier, R.M.; Hopf, F.W. Recent Perspectives on Sex Differences in Compulsion-Like and Binge Alcohol Drinking. Int. J. Mol. Sci. 2021, 22, 3788. [Google Scholar] [CrossRef]
- Ceylan-Isik, A.F.; McBride, S.M.; Ren, J. Sex difference in alcoholism: who is at a greater risk for development of alcoholic complication? Life sciences 2010, 87, 133–138. [Google Scholar] [CrossRef]
- Lenz, B.; Müller, C.P.; Stoessel, C.; Sperling, W.; Biermann, T.; Hillemacher, T.; Bleich, S.; Kornhuber, J. Sex hormone activity in alcohol addiction: Integrating organizational and activational effects. Prog. Neurobiol. 2012, 96, 136–163. [Google Scholar] [CrossRef] [PubMed]
- Erol, A.; Karpyak, V.M. Sex and gender-related differences in alcohol use and its consequences: Contemporary knowledge and future research considerations. Drug Alcohol Depend. 2015, 156, 1–13. [Google Scholar] [CrossRef]
- Kaun, K.R.; Devineni, A.V.; Heberlein, U. Drosophila melanogaster as a model to study drug addiction. Hum. Genet. 2012, 131, 959–975. [Google Scholar] [CrossRef]
- Katzenberger, R.J.; Loewen, C.A.; Wassarman, D.R.; Petersen, A.J.; Ganetzky, B.; Wassarman, D.A. A Drosophila Model of Closed Head Traumatic Brain Injury. Proc. Natl. Acad. Sci. USA 2013, 110, E4152–E4159. [Google Scholar] [CrossRef]
- Katzenberger, R.J.; Ganetzky, B.; A Wassarman, D. Age and Diet Affect Genetically Separable Secondary Injuries that Cause Acute Mortality Following Traumatic Brain Injury in Drosophila. G3 Genes|Genomes|Genetics 2016, 6, 4151–4166. [Google Scholar] [CrossRef]
- Barekat, A.; Gonzalez, A.; Mauntz, R.E.; Kotzebue, R.W.; Molina, B.; El-Mecharrafie, N.; Conner, C.J.; Garza, S.; Melkani, G.C.; Joiner, W.J. Using Drosophila as an integrated model to study mild repetitive traumatic brain injury. Sci. Rep. 2016, 6, 25252–25252. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic Brain Injury Induces Tau Aggregation and Spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, A.; Takeuchi, H.; Tanaka, F. Tau Pathology in Chronic Traumatic Encephalopathy and Alzheimer’s Disease: Similarities and Differences. Front. Neurol. 2019, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.-S.; Kubilus, C.A.; Stern, R.A. Chronic Traumatic Encephalopathy in Athletes: Progressive Tauopathy After Repetitive Head Injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Perry, G. Tau biology, tauopathy, traumatic brain injury, and diagnostic challenges. Journal of Alzheimer’s Disease 2019, 67, 447–467. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Pérez, M.; Hernández, F. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Trinczek, B.; Ebneth, A.; Mandelkow, E.M.; Mandelkow, E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 1999, 112, 2355–2367. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L.F. Differential Regulation of Dynein and Kinesin Motor Proteins by Tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef]
- Moraga, D.M.; Nuñez, P.; Garrido, J.; Maccioni, R.B. A τ Fragment Containing a Repetitive Sequence Induces Bundling of Actin Filaments. J. Neurochem. 1993, 61, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Widespread Tau and Amyloid-Beta Pathology Many Years After a Single Traumatic Brain Injury in Humans. Brain Pathol. 2011, 22, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Heidary, G.; E Fortini, M. Identification and characterization of the Drosophila tau homolog. Mech. Dev. 2001, 108, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Papanikolopoulou, K.; Roussou, I.G.; Gouzi, J.Y.; Samiotaki, M.; Panayotou, G.; Turin, L.; Skoulakis, E.M. Drosophila tau negatively regulates translation and olfactory long-term memory, but facilitates footshock habituation and cytoskeletal homeostasis. Journal of Neuroscience 2019, 39, 8315–8329. [Google Scholar] [CrossRef]
- Bolkan, B.J.; Kretzschmar, D. Loss of tau results in defects in photoreceptor development and progressive neuronal degeneration in Drosophila. Dev. Neurobiol. 2014, 74, 1210–1225. [Google Scholar] [CrossRef] [PubMed]
- Weil, Z.M.; Corrigan, J.D.; Karelina, K. Alcohol use disorder and traumatic brain injury. Alcohol research: current reviews 2018, 39, 171. [Google Scholar]
- Biegon, A. Considering Biological Sex in Traumatic Brain Injury. Front. Neurol. 2021, 12, 576366. [Google Scholar] [CrossRef]
- Gilthorpe, M.; Wilson, R.; Moles, D.; Bedi, R. Variations in admissions to hospital for head injury and assault to the head Part 1: age and gender. Br. J. Oral Maxillofac. Surg. 1999, 37, 294–300. [Google Scholar] [CrossRef]
- Devineni, A.V.; Heberlein, U. Acute ethanol responses in Drosophila are sexually dimorphic. Proc. Natl. Acad. Sci. 2012, 109, 21087–21092. [Google Scholar] [CrossRef]
- McDougall, A.; Bayley, M.; Munce, S.E. The ketogenic diet as a treatment for traumatic brain injury: a scoping review. Brain Inj. 2018, 32, 416–422. [Google Scholar] [CrossRef]
- Greco, T.; Glenn, T.C.; A Hovda, D.; Prins, M.L. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cereb. Blood Flow Metab. 2016, 36, 1603–1613. [Google Scholar] [CrossRef]
- Delventhal, R.; Wooder, E.R.; Basturk, M.; Sattar, M.; Lai, J.; Bolton, D.; Muthukumar, G.; Ulgherait, M.; Shirasu-Hiza, M.M. Dietary restriction ameliorates TBI-induced phenotypes in Drosophila melanogaster. Sci. Rep. 2022, 12, 1–16. [Google Scholar] [CrossRef]
- van der Linde, K.; Fumagalli, E.; Roman, G.; Lyons, L.C. The FlyBar: Administering Alcohol to Flies. Journal of visualized experiments: JoVE 2014. [CrossRef]
- Buhlman, L.M.; Krishna, G.; Jones, T.B.; Thomas, T.C. Drosophila as a model to explore secondary injury cascades after traumatic brain injury. Biomed. Pharm. 2021, 142, 112079. [Google Scholar] [CrossRef]
- Shah, E.J.; Gurdziel, K.; Ruden, D.M. Mammalian Models of Traumatic Brain Injury and a Place for Drosophila in TBI Research. Front. Neurosci. 2019, 13, 409. [Google Scholar] [CrossRef]
- Poznanski, P.; Lesniak, A.; Korostynski, M.; Sacharczuk, M. Ethanol consumption following mild traumatic brain injury is related to blood-brain barrier permeability. Addict. Biol. 2020, 25, e12683. [Google Scholar] [CrossRef]
- Saikumar, J.; Byrns, C.N.; Hemphill, M.; Meaney, D.F.; Bonini, N.M. Dynamic neural and glial responses of a head-specific model for traumatic brain injury inDrosophila. Proc. Natl. Acad. Sci. 2020, 117, 17269–17277. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Gurdziel, K.; Liu, J.; Qu, W.; Nuga, O.O.; Burl, R.B.; Hüttemann, M.; Pique-Regi, R.; Ruden, D.M. Smooth, an hnRNP-L Homolog, Might Decrease Mitochondrial Metabolism by Post-Transcriptional Regulation of Isocitrate Dehydrogenase (Idh) and Other Metabolic Genes in the Sub-Acute Phase of Traumatic Brain Injury. Front. Genet. 2017, 8, 175–175. [Google Scholar] [CrossRef]
- Shah, E.J.; Gurdziel, K.; Ruden, D.M. Drosophila Exhibit Divergent Sex-Based Responses in Transcription and Motor Function After Traumatic Brain Injury. Front. Neurol. 2020, 11, 511. [Google Scholar] [CrossRef]
- Parkhurst, S.J.; Adhikari, P.; Navarrete, J.S.; Legendre, A.; Manansala, M.; Wolf, F.W. Perineurial Barrier Glia Physically Respond to Alcohol in an Akap200-Dependent Manner to Promote Tolerance. Cell Rep. 2018, 22, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Bainton, R.J.; Tsai, L.T.-Y.; Schwabe, T.; DeSalvo, M.; Gaul, U.; Heberlein, U. moody Encodes Two GPCRs that Regulate Cocaine Behaviors and Blood-Brain Barrier Permeability in Drosophila. Cell 2005, 123, 145–156. [Google Scholar] [CrossRef]
- Contreras, E.G.; Glavic, Á.; Brand, A.H.; Sierralta, J.A. The serine protease homolog, Scarface, is sensitive to nutrient availability and modulates the development of the Drosophila blood–brain barrier. Journal of Neuroscience 2021, 41, 6430–6448. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.; Kaun, K.R. Alcohol, neuronal plasticity, and mitochondrial trafficking. Proceedings of the National Academy of Sciences 2022, 119, e2208744119. [Google Scholar] [CrossRef]
- Knabbe, J.; Protzmann, J.; Schneider, N.; Berger, M.; Dannehl, D.; Wei, S.; Strahle, C.; Tegtmeier, M.; Jaiswal, A.; Zheng, H.; et al. Single-dose ethanol intoxication causes acute and lasting neuronal changes in the brain. Proc. Natl. Acad. Sci. 2022, 119, e2122477119. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Carvajal, F.J.; Mira, R.G.; Arce, C.; Lerma-Cabrera, J.M.; Orellana, J.A.; Cerpa, W.; Quintanilla, R.A. Adolescent binge alcohol exposure affects the brain function through mitochondrial impairment. Molecular neurobiology 2018, 55, 4473–4491. [Google Scholar] [CrossRef]
- Oh, K.H.; Sheoran, S.; Richmond, J.E.; Kim, H. Alcohol induces mitochondrial fragmentation and stress responses to maintain normal muscle function in Caenorhabditis elegans. FASEB J. 2020, 34, 8204–8216. [Google Scholar] [CrossRef]
- Müller, T.E.; Nunes, M.E.; Rodrigues, N.R.; Fontana, B.D.; Hartmann, D.D.; Franco, J.L.; Rosemberg, D.B. Neurochemical mechanisms underlying acute and chronic ethanol-mediated responses in zebrafish: The role of mitochondrial bioenergetics. Neurochem. Int. 2019, 131, 104584. [Google Scholar] [CrossRef]
- Petruccelli, E.; Kaun, K.R. Insights from intoxicated Drosophila. Alcohol 2019, 74, 21–27. [Google Scholar] [CrossRef]
- Troutwine, B.R.; Ghezzi, A.; Pietrzykowski, A.Z.; Atkinson, N.S. Alcohol resistance in Drosophila is modulated by the Toll innate immune pathway. Genes, Brain Behav. 2016, 15, 382–394. [Google Scholar] [CrossRef]
- Kong, E.C.; Allouche, L.; Chapot, P.A.; Vranizan, K.; Moore, M.S.; Heberlein, U.; Wolf, F.W. Ethanol-Regulated Genes That Contribute to Ethanol Sensitivity and Rapid Tolerance in Drosophila. Alcohol. Clin. Exp. Res. 2010, 34, 302–316. [Google Scholar] [CrossRef]
- Morozova, T.V.; Anholt, R.R.; Mackay, T.F. Phenotypic and transcriptional response to selection for alcohol sensitivity in Drosophila melanogaster. Genome Biol. 2007, 8, R231–R231. [Google Scholar] [CrossRef]
- van Alphen, B.; Stewart, S.; Iwanaszko, M.; Xu, F.; Li, K.; Rozenfeld, S.; Ramakrishnan, A.; Itoh, T.Q.; Sisobhan, S.; Qin, Z.; et al. Glial immune-related pathways mediate effects of closed head traumatic brain injury on behavior and lethality in Drosophila. PLOS Biol. 2022, 20, e3001456. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, D.; Leyssen, M.; Koch, M.; Yan, J.; Srahna, M.; Sheeba, V.; Fogle, K.J.; Holmes, T.C.; Hassan, B.A. Axonal Injury and Regeneration in the Adult Brain of Drosophila. J. Neurosci. 2008, 28, 6010–6021. [Google Scholar] [CrossRef]
- Soares, L.; Parisi, M.; Bonini, N.M. Axon Injury and Regeneration in the Adult Drosophila. Sci. Rep. 2014, 4, srep06199. [Google Scholar] [CrossRef]
- Wilson, A.; Periandri, E.M.; Sievers, M.; Petruccelli, E. Drosophila Stat92E Signaling Following Pre-exposure to Ethanol. Neurosci. Insights 2023, 18, 26331055221146755. [Google Scholar] [CrossRef]
- Weber, J.T. Altered Calcium Signaling Following Traumatic Brain Injury. Front. Pharmacol. 2012, 3, 60. [Google Scholar] [CrossRef]
- He, H.J.; Wang, X.S.; Pan, R.; Wang, D.L.; Liu, M.N.; He, R.Q. The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol. 2009, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sealey, M.A.; Vourkou, E.; Cowan, C.M.; Bossing, T.; Quraishe, S.; Grammenoudi, S.; Skoulakis, E.M.; Mudher, A. Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol. Dis. 2017, 105, 74–83. [Google Scholar] [CrossRef]
- Walker, A.; Chapin, B.; Abisambra, J.; DeKosky, S.T. Association between single moderate to severe traumatic brain injury and long-term tauopathy in humans and preclinical animal models: a systematic narrative review of the literature. Acta Neuropathol. Commun. 2022, 10, 1–20. [Google Scholar] [CrossRef]
- Medina, M.; Hernández, F.; Avila, J. New Features about Tau Function and Dysfunction. Biomolecules 2016, 6, 21. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nature reviews neuroscience 2016, 17, 22–35. [Google Scholar] [CrossRef]
- Burnouf, S.; Grönke, S.; Augustin, H.; Dols, J.; Gorsky, M.K.; Werner, J.; Kerr, F.; Alic, N.; Martinez, P.; Partridge, L. Deletion of endogenous Tau proteins is not detrimental in Drosophila. Sci. Rep. 2016, 6, 23102. [Google Scholar] [CrossRef]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef]
- Shah, E.J.; Gurdziel, K.; Ruden, D.M. Sex-Differences in Traumatic Brain Injury in the Absence of Tau in Drosophila. Genes 2021, 12, 917. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.S.; DeZazzo, J.; Luk, A.Y.; Tully, T.; Singh, C.M.; Heberlein, U. Ethanol Intoxication in Drosophila: Genetic and Pharmacological Evidence for Regulation by the cAMP Signaling Pathway. Cell 1998, 93, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.H.; Kong, E.C.; Dubnau, J.; Tully, T.; Moore, M.S.; Heberlein, U. Ethanol Sensitivity and Tolerance in Long-Term Memory Mutants of Drosophila melanogaster. Alcohol. Clin. Exp. Res. 2008, 32, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Endo, K.; Wu, K.; Rodan, A.R.; Heberlein, U.; Davis, R.L. Drosophila fasciclinII Is Required for the Formation of Odor Memories and for Normal Sensitivity to Alcohol. Cell 2001, 105, 757–768. [Google Scholar] [CrossRef]
- Xu, S.; Pany, S.; Benny, K.; Tarique, K.; Al-Hatem, O.; Gajewski, K.; Leasure, J.L.; Das, J.; Roman, G. Ethanol Regulates Presynaptic Activity and Sedation through Presynaptic Unc13 Proteins inDrosophila. eneuro 2018, 5. [Google Scholar] [CrossRef]
- Myers, J.L.; Porter, M.; Narwold, N.; Bhat, K.; Dauwalder, B.; Roman, G. Mutants of the white ABCG Transporter in Drosophila melanogaster Have Deficient Olfactory Learning and Cholesterol Homeostasis. Int. J. Mol. Sci. 2021, 22, 12967. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.; Kollah, A.P.; Lewellyn, L.; Chan, R.F.; Grotewiel, M. An inexpensive, scalable behavioral assay for measuring ethanol sedation sensitivity and rapid tolerance in Drosophila. JoVE (Journal of Visualized Experiments) 2015, e52676. [CrossRef]
- van der Linde, K.; Fumagalli, E.; Roman, G.; Lyons, L.C. The FlyBar: Administering Alcohol to Flies. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.Y.; Wachi, Y.; Engdorf, E.; Fumagalli, E.; Wang, Y.; Myers, J.; Massey, S.; Greiss, A.; Xu, S.; Roman, G. Normal Ethanol Sensitivity and Rapid Tolerance Require the G Protein Receptor Kinase 2 in Ellipsoid Body Neurons in Drosophila. Alcohol. Clin. Exp. Res. 2020, 44, 1686–1699. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J Am Stat Assoc 1952, 47, 583–621. [Google Scholar] [CrossRef]
Figure 1.
Development of the TBI model. Flies were subjected to different numbers of strikes, and the mortality rate 24 hours post-concussion was recorded.
Figure 1.
Development of the TBI model. Flies were subjected to different numbers of strikes, and the mortality rate 24 hours post-concussion was recorded.
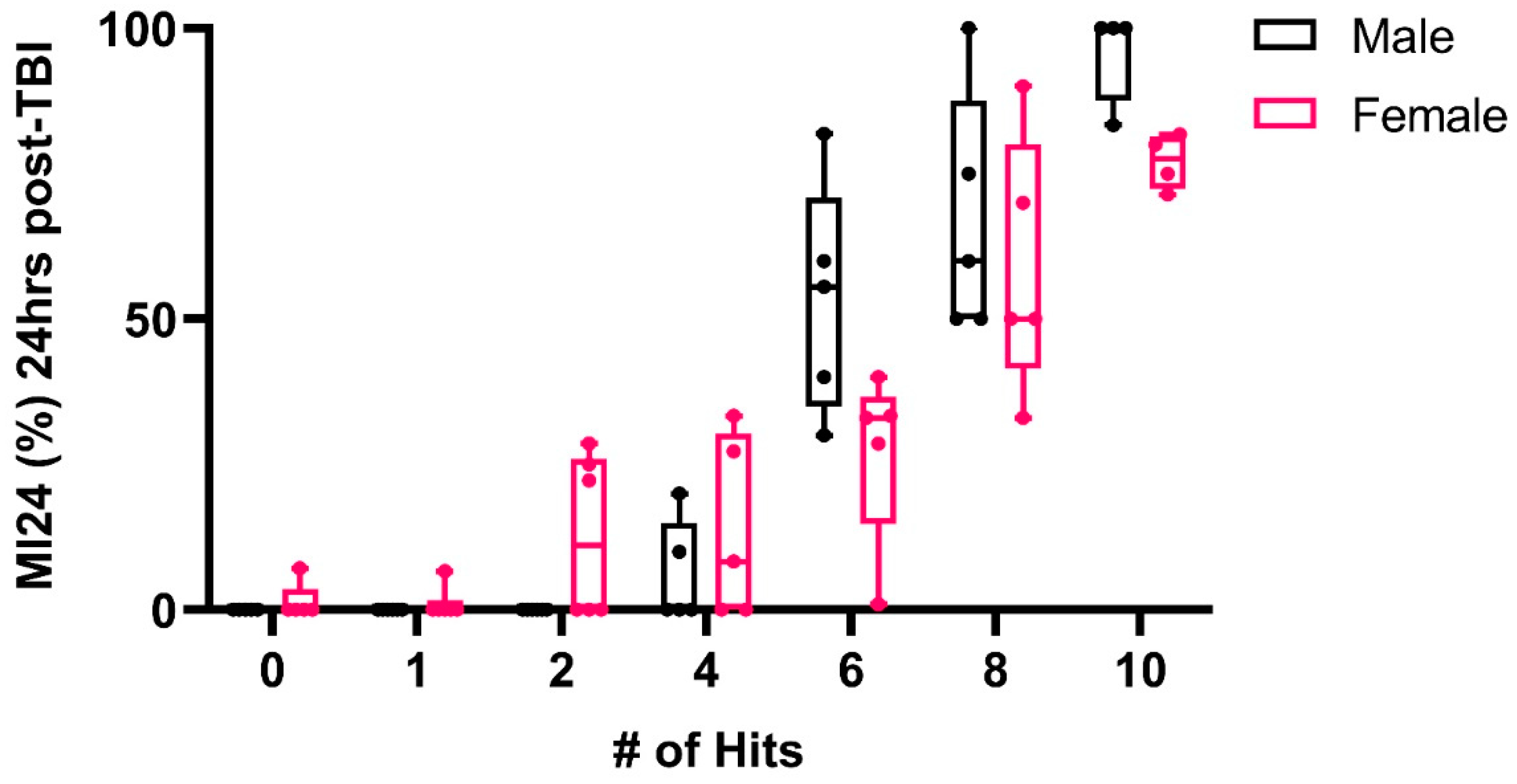
Figure 2.
Male and female flies were subjected to TBI, and ethanol sensitivity was measured at 2 hrs, 24 hrs, and 48 hrs post-TBI. (a) Male flies were exposed to ethanol post-TBI and then compared to control flies exposed to ethanol alone (2-way ANOVA: F(2,49)=12.34; **p<0.01, *p<0.05, ns; n=5-12). (b) Female flies exposed to ethanol post-TBI, compared to control flies exposed to ethanol alone (2-way ANOVA: F(2,37)=14.27; ***p<0.001, *p<0.05; n=5-9). (c) The effect of ethanol, administered at different times post-TBI, is shown. Males were compared to females at each timepoint by two-way ANOVA, followed by Tukey’s multiple comparison test (F (2,47)=26.94; p<0.0001; n>6).
Figure 2.
Male and female flies were subjected to TBI, and ethanol sensitivity was measured at 2 hrs, 24 hrs, and 48 hrs post-TBI. (a) Male flies were exposed to ethanol post-TBI and then compared to control flies exposed to ethanol alone (2-way ANOVA: F(2,49)=12.34; **p<0.01, *p<0.05, ns; n=5-12). (b) Female flies exposed to ethanol post-TBI, compared to control flies exposed to ethanol alone (2-way ANOVA: F(2,37)=14.27; ***p<0.001, *p<0.05; n=5-9). (c) The effect of ethanol, administered at different times post-TBI, is shown. Males were compared to females at each timepoint by two-way ANOVA, followed by Tukey’s multiple comparison test (F (2,47)=26.94; p<0.0001; n>6).
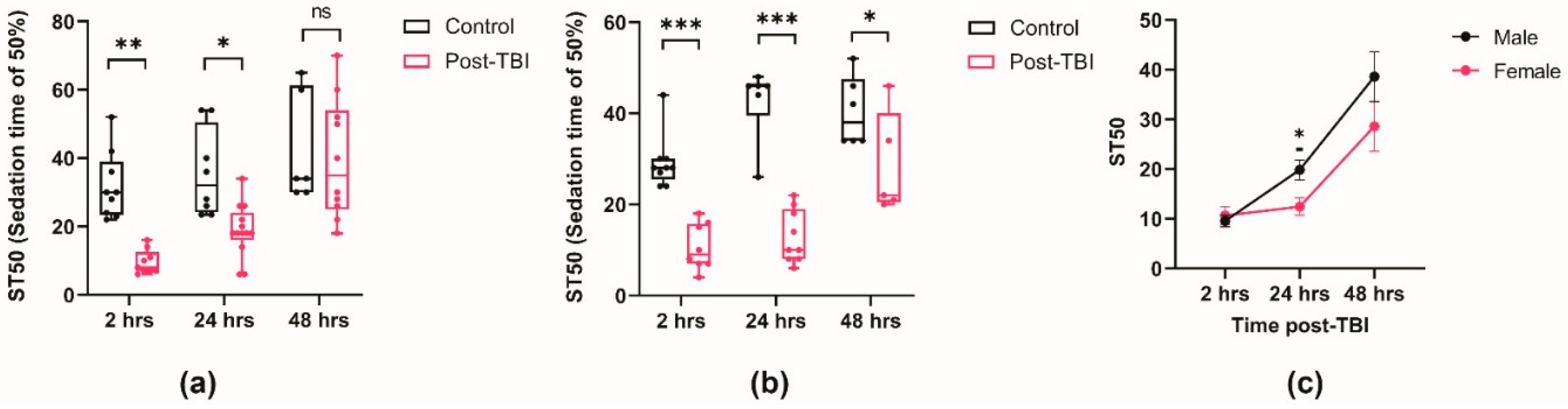
Figure 3.
TBI increases ethanol-induced mortality. % mortality post ethanol exposure in TBI-induced in male flies **p=0.002, n>8, Kruskal-Wallis.
Figure 3.
TBI increases ethanol-induced mortality. % mortality post ethanol exposure in TBI-induced in male flies **p=0.002, n>8, Kruskal-Wallis.
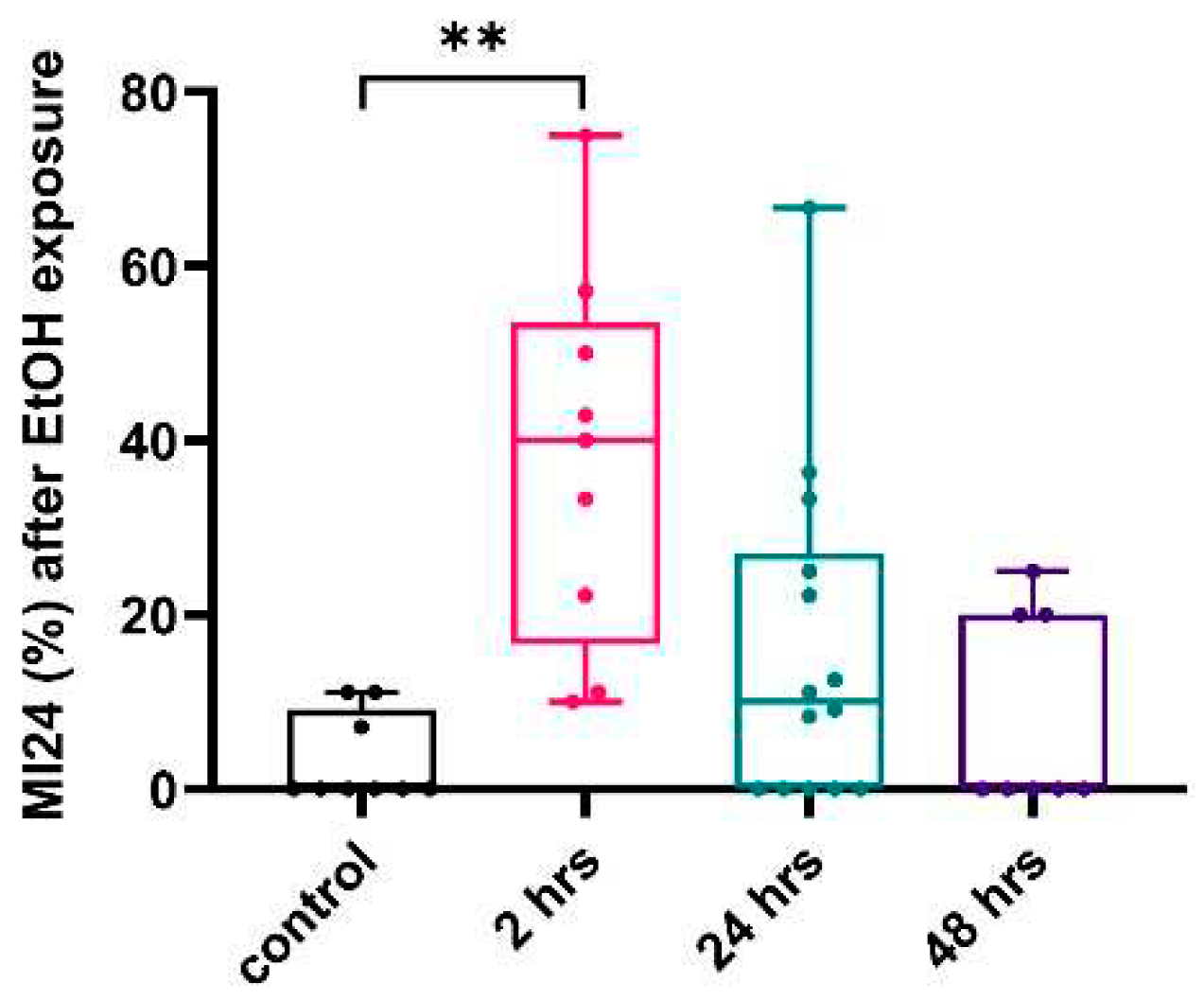
Figure 4.
Starvation after TBI does not affect ethanol sedation sensitivity. (a) Ethanol sensitivity was measured in control male flies without TBI, 2 hours post-TBI or 24 hours post-TBI. Kruskul-Wallis two-way ANOVA on ranks, factor; food, p=0.67, time post-TBI *p<0.011, **p<0.002, n>8. (b) Ethanol sensitivity was measured in control female flies without TBI, 2 hours post-TBI or 24 hours post-TBI. Kruskul-Wallis two-way ANOVA on ranks, factor; food, p=0.741, time post-TBI ***p<0.001, n>8. (c) The mortality 24 hours after ethanol exposure was measured for male and female flies that received a TBI at the indicated times with the indicated post-TBI diet. ( 3-way ANOVA followed by Tukey multiple comparisons test: Comparison Factor: Time post-TBI, **p<0.014, *** p<0.001; Diet and Sex, p>0.05, ns=not significantly different, n>7.).
Figure 4.
Starvation after TBI does not affect ethanol sedation sensitivity. (a) Ethanol sensitivity was measured in control male flies without TBI, 2 hours post-TBI or 24 hours post-TBI. Kruskul-Wallis two-way ANOVA on ranks, factor; food, p=0.67, time post-TBI *p<0.011, **p<0.002, n>8. (b) Ethanol sensitivity was measured in control female flies without TBI, 2 hours post-TBI or 24 hours post-TBI. Kruskul-Wallis two-way ANOVA on ranks, factor; food, p=0.741, time post-TBI ***p<0.001, n>8. (c) The mortality 24 hours after ethanol exposure was measured for male and female flies that received a TBI at the indicated times with the indicated post-TBI diet. ( 3-way ANOVA followed by Tukey multiple comparisons test: Comparison Factor: Time post-TBI, **p<0.014, *** p<0.001; Diet and Sex, p>0.05, ns=not significantly different, n>7.).
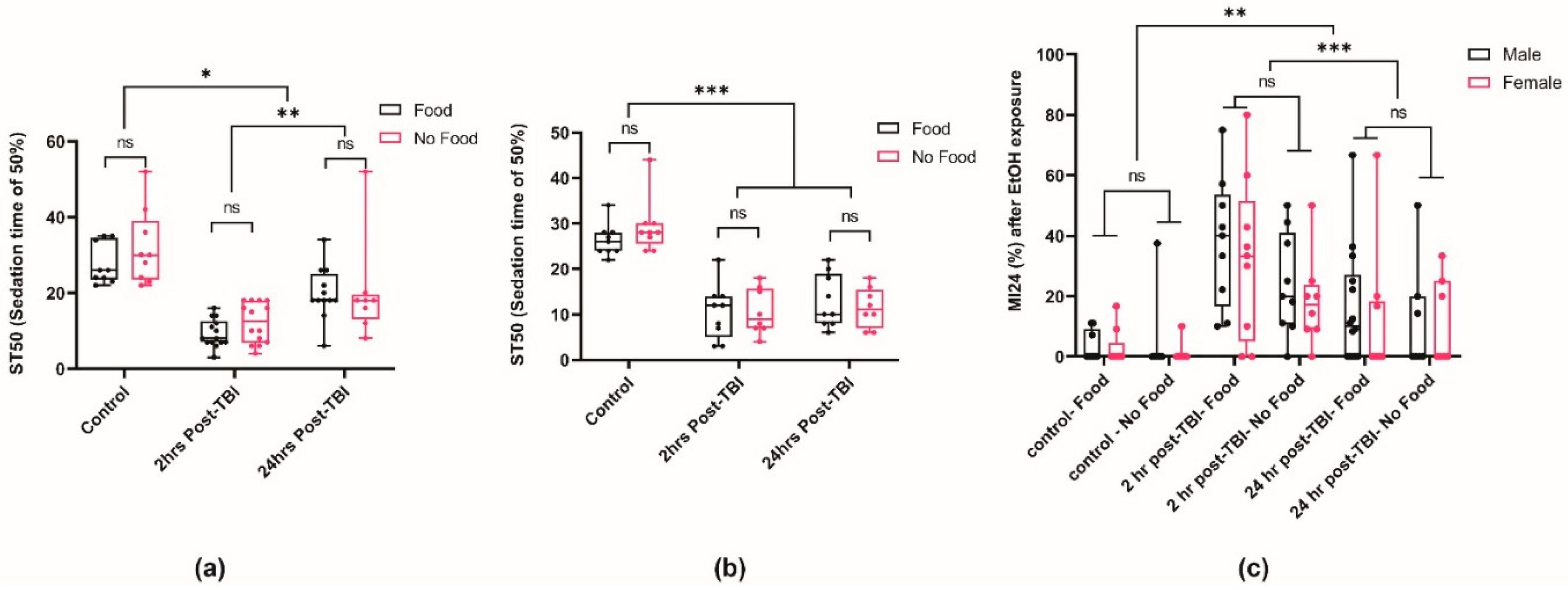
Figure 5.
TBI and dtau loss-of-function produces an additive effect in ethanol sedation sensitivity. dtauKO mutants and heterozygous were subjected to TBI and tested for ethanol sedation sensitivity with Canton-S (CS) as the wild-type control. (Three-way ANOVA: Tukey’s multiple comparison test, n>11 *p<0.0450, p**<0.0089, ***p<0.0003).
Figure 5.
TBI and dtau loss-of-function produces an additive effect in ethanol sedation sensitivity. dtauKO mutants and heterozygous were subjected to TBI and tested for ethanol sedation sensitivity with Canton-S (CS) as the wild-type control. (Three-way ANOVA: Tukey’s multiple comparison test, n>11 *p<0.0450, p**<0.0089, ***p<0.0003).
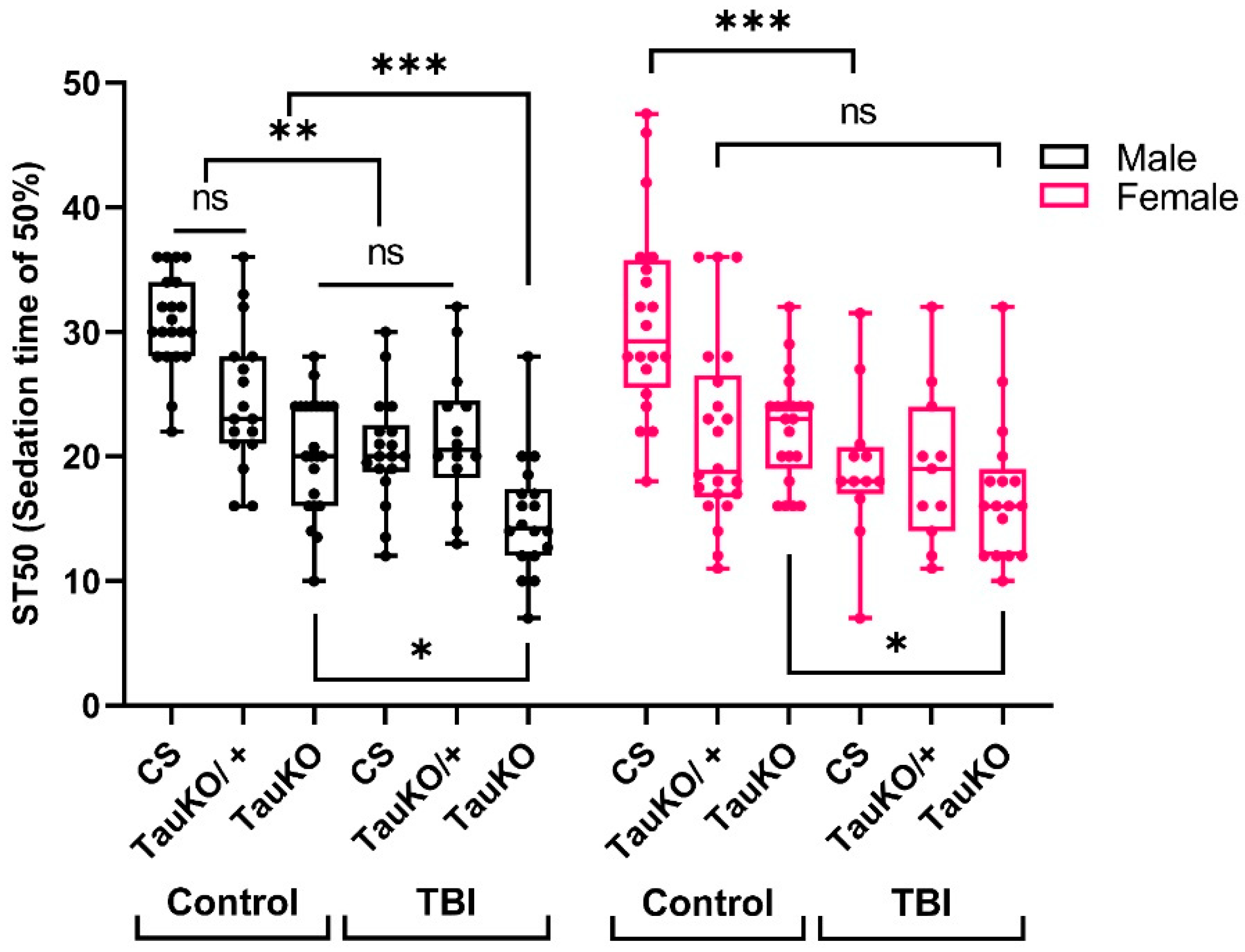
Figure 6.
Loss-of-function dtau homozygous mutants exhibit increased ethanol sedation sensitivity and altered rapid tolerance. (a) Males that were either heterozygous or homozygous for the dtauKO mutation were examined for Loss-of-Righting in the FlyBar assay [56]. Wildtype-Canton-S (CS) was used as a positive control. (Two-way ANOVA, Tukey test for 1st and 2nd exposure; Comparison for factor: treatment, p<0.001; genotype within 1st and 2nd exposure, p<0.001, n=13). Kruskal-Wallis One-Way ANOVA on Ranks for rapid tolerance, *p<0.027, **p<0.01, n=13). (b) Flies expressing dtau RNAi transgene in the brain (through nSyb-Gal4) were subjected to ethanol sedation sensitivity (One-way ANOVA, Tukey Test ; **p<0.012; *** p<0.001, n=13).
Figure 6.
Loss-of-function dtau homozygous mutants exhibit increased ethanol sedation sensitivity and altered rapid tolerance. (a) Males that were either heterozygous or homozygous for the dtauKO mutation were examined for Loss-of-Righting in the FlyBar assay [56]. Wildtype-Canton-S (CS) was used as a positive control. (Two-way ANOVA, Tukey test for 1st and 2nd exposure; Comparison for factor: treatment, p<0.001; genotype within 1st and 2nd exposure, p<0.001, n=13). Kruskal-Wallis One-Way ANOVA on Ranks for rapid tolerance, *p<0.027, **p<0.01, n=13). (b) Flies expressing dtau RNAi transgene in the brain (through nSyb-Gal4) were subjected to ethanol sedation sensitivity (One-way ANOVA, Tukey Test ; **p<0.012; *** p<0.001, n=13).
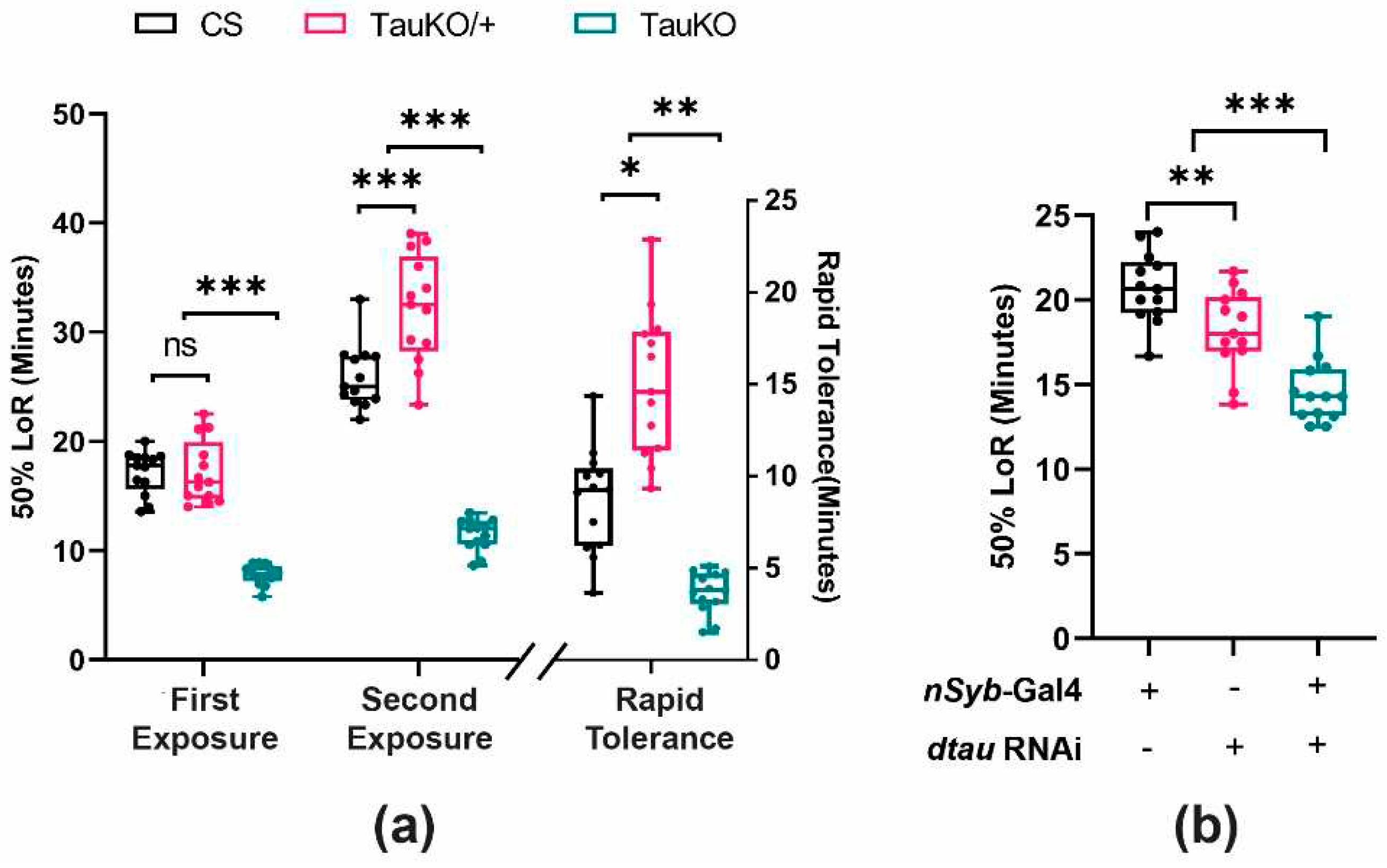
Figure 7.
Neither acute TBI nor dtau loss-of-function alters ethanol absorption in Drosophila. (a) To test for ethanol absorption, wild-type flies were tested in the presence and absence of TBI. Flies were subjected to ethanol at 10-minute intervals (Two-way ANOVA, Tukey test, Comparisons for factor: treatment; p=0.099, time; p<0.005, n=4-5). (b) Wild-type CS and dtauKO flies were tested for ethanol absorption at 5-minute intervals (n=4, Two-way ANOVA, Tukey test, comparison factor: genotype; p=0.466, time; p<0.001) ns = not significantly different .
Figure 7.
Neither acute TBI nor dtau loss-of-function alters ethanol absorption in Drosophila. (a) To test for ethanol absorption, wild-type flies were tested in the presence and absence of TBI. Flies were subjected to ethanol at 10-minute intervals (Two-way ANOVA, Tukey test, Comparisons for factor: treatment; p=0.099, time; p<0.005, n=4-5). (b) Wild-type CS and dtauKO flies were tested for ethanol absorption at 5-minute intervals (n=4, Two-way ANOVA, Tukey test, comparison factor: genotype; p=0.466, time; p<0.001) ns = not significantly different .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



