
Submitted:
02 February 2024
Posted:
08 February 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Alzheimer’s Disease (AD), a progressive and debilitating condition, is reported to be the most common type of dementia, with at least 55 million people believed to be currently affected. Many causation hypotheses of AD exist, yet the intriguing link between viral infection and its possible contribution to the known etiology of AD has become an attractive focal point of research for the field and a challenging study task. In this review, we will explore the historical perspective and milestones that led the field to investigate the viral connection to AD. Specifically, several viruses such as Herpes Simplex Virus 1 (HSV-1), Zika virus (ZIKV), and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), along with several others mentioned, include the various viruses presently considered within the field. We delve into the strong evidence implicating these viruses in the development of AD. We will also extend beyond these mere associations by carefully analyzing the potential mechanisms by which viruses may contribute to AD pathology. This includes but is not limited to direct neuronal infections, dysregulation of immune responses, and the impact on protein processing. Controversies and challenges of the viral-AD relationship emerge as we tease out these potential mechanisms considered. Looking forward, we emphasize the future directions the field should take to tackle the remaining unanswered questions and the glaring research gaps that persist. Overall, this review aims to provide a comprehensive survey of the past, present, and future of the potential link between viral infections and their association with AD development.
Keywords:
Alzheimer’s Disease
; AD pathology
; Viral-AD Hypothesis
; Zika
; Herpesvirus
; SARS-CoV-2
; Influenza
1. Introduction
Alzheimer’s Disease (AD) has challenged researchers for decades to attempt to uncover its elusive origins, multifaceted pathogenesis, and complicated causation. As we continue this quest for answers to ease the lives of aging people all over the globe, a compelling question arose and continues to resurface: could viral infections play a crucial role in the complex development of AD? As first described by Alois Alzheimer over a century ago, AD remains a pervasive health challenge, affecting millions of people worldwide (1). AD is characterized by progressive, irreversible cognitive decline, memory loss, and the presence of classical neuropathology, often including amyloid-beta (Ab) plaques and neurofibrillary tangles (2), neuropil threads, and dystrophic neurites, which are companies by astrogliosis and microglial cell activation (3). AD has a and contemporary research findings that specifically link virus infections to their potential to shape the progress of AD pathology and evaluate viral contributions to AD as not merely a speculative endeavor ore and consider the evolution of knowledge of the influence of viral infections on the development of AD. We will consider historical and contemporary research findings that specifically link virus infections to their potential to shape the progress of AD pathology and evaluate viral contributions to AD as not merely a speculative endeavor contemporary research findings that specifically link virus infections to their potential in shaping the progress of AD pathology and evaluate viral contributions to AD as not merely a speculative endeavor, but as a scientific inquiry supported by emerging evidence.
2. Overview of AD Pathology
To provide a solid foundation for exploring viral links to the development of AD, a comprehensive understanding of the pathological features of the disease is imperative. AD manifests as a progressive deterioration of cognitive functions, primarily encompassing the mutual characteristics of memory loss, impaired reasoning, and eventual debilitating disruptions to basic, everyday life (4). Two primary cardinal lesions often associate with AD are amyloid-beta (Ab) plaques and neurofibrillary tangles (NFTs) containing hyperphosphorylated Tau, although the presence of Aβ and Tau cannot completely conclude AD, as there are cognitively unimpaired individuals who can have biomarker evidence of both Aβ and Tau pathology but will often not develop clinical manifestations in their lifetime (5). Other lesions include cerebral amyloid angiopathy, glial responses, and neuronal and synaptic loss (3). Ab plaques are composed of aggregated Ab protein fragments that accumulate between nerve cells within the brain (6). These extracellular proteinaceous deposits are implicated in disrupting neuronal communication and triggering inflammatory responses, which accelerate the progression of the disease (7,8). NFTs result from the abnormal aggregation of tau protein within neurons due to hyperphosphorylation of tau. Neuronal function is thereby impaired in these patients affected with AD because their neurons are structurally unstable because of these intracellular tangles (9,10). While genetics and aging are acknowledged and accepted contributors to the development of AD, emerging evidence suggests that infectious agents, specifically viruses, could influence or modulate the balance of molecular events leading to AD pathology. There are studies that have highlighted the complexity in triggering or exacerbating AD including but not limited to genetic susceptibility, environmental influences, and the potential role of infections. Specifically, some of these studies highlight genetic susceptibility through well-described mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1), presenilin 2 (PSEN2), and apolipoprotein E (APOE) genes, which account for only 30-50% of the heritability of AD (11). Other studies have investigated the link of environmental risk factors or exposures such as traumatic brain injury (TBI), blood pressure, smoking, education, socioeconomic status, air pollution or geographical location, diet, and physical activity congruently with those previously mentioned gene interactions across individual age and duration of specific exposures (12,13). Further studies have contributed to exploring the potential role of viral infections in AD either through direct methods of infection and thereby modulation of neuronal function leading to neuronal damage, chronic neuroinflammation due to direct infection, processing of Ab, tau protein dysregulation, and finally immune system dysregulation, or indirectly, through virus-induced inflammation and subsequently encephalitis. Considering the dynamic nature of AD pathology is not only crucial for elucidating its origins but is also vital for the development of robust targeted therapeutic interventions.
3. Historical Perspectives on Viral Links to AD
Early observations hinted at a potential connection that viral infections contribute to AD development. Surveying these historical perspectives reveals the gradual evolution of thoughts that led to recent considerations and investigations of this viral hypothesis in AD. During the mid-20th century, there are records by clinicians and researchers reporting peculiar associations between infectious events and cognitive decline, generally (14–19). Specifically, the persuasive work by Oskar Fischer in 1907 established AD pathology, and it was not until decades later, specifically during the 1980’s, that infectious agents came under scrutiny for their potential influence on neurodegeneration. Attention slowly began to shift towards specific viral agents and their probable role in AD. The discovery of HSV DNA in postmortem AD brains really ignited the interest in the viral hypothesis of AD contributing causation (20,21). Subsequently, the presence of other herpesviruses, specifically Human Herpesviruses 6,7 (HHV-6,-7), Epstein-Barr virus (EBV), and Cytomegalovirus (CMV), were surveyed in AD brains, which provided additional support to an already convoluted hypothesis (22). As research progressed, questions regarding the nature of the association between viral infection and AD continued to emerge and grew more difficult to approach. For example, the concept of latent viral infections, particularly with individuals living with herpesviruses, gained prominence. Additionally, studies visited the possibility of viral reactivation within the aging brain, thereby highlighting the dynamic interplay between viruses and the neurodegenerative processes understood in AD. These approaches to this viral hypothesis, coupled with advancements in molecular and cellular biology, has boosted researchers to probe deeper into the possible precise mechanisms underlying the viral hypothesis to AD. In the following sections, we will cover several important viruses and their potential contribution to AD.
4. Selected Viral Examples Implicated in AD and Their Respective Role Elaborated
In this review, we use HSV-1, which has a long history of research on its contribution to AD, and two emerging viruses, ZIKV and SARS-CoV-2, as examples to discuss the viral-AD hypothesis in detail as shown in Figure 1 below.
4.1. Herpes Simplex Virus 1 (HSV-1)
The original and most famous example of viral involvement in AD is with Herpes Simplex Virus 1 (HSV-1). HSV-1 is a ubiquitous neurotropic double-stranded DNA viral pathogen that primarily infects epithelial cells of the oral and nasal mucosal regions. HSV-1 is a ubiquitous virus that affects more than 80% of people over 65 across the globe (23).
4.1.1. Supportive Evidence
HSV-1 was first considered a part of the microbe hypothesis in AD, which surfaced back in 1982 through the observation of individuals surviving a condition known as Herpes Simplex Encephalitis (HSE) and showed clinical signs reminiscent of AD in the forms of memory loss and cognitive impairment. To supplement this finding, brain regions affected in HSE (limbic system, frontal and temporal cortices) were the same regions involved in AD (24). Since beginning with Dr. McLauhlan’s 1980 work with confirmation of HSV-1 presence in AD brains via in situ hybridization and Dr. Ball’s 1982 study linking HSV-1, HSE, and AD, there have been several research groups that have conducted investigations providing substantiative support of HSV-1 involvement in AD pathogenesis (25). For example, The association between HSV-1 infection and AD shows mainly in people that harbor the APOE4 allele and an antiviral agent acyclovir (ACV) was shown to amend AD-related tauopathy because this antiviral halts viral replication (26,27). In addition, recurrent HSV-1 infection in a recently developed mice model induces hallmarks of neurodegeneration and cognitive deficits in mice (28). Moreover, HSV-1 infected human-induced neural stem cells (hiNSCs) led to resembled changes observed in AD brains including amyloid plaque-like formation (PLFs), gliosis, neuroinflammation, and decreased functionality (29).
4.1.2. Potential Mechanisms
HSV-1 has the propensity to enter sensory neurons near these mucosal regions through lytic replication and axonal transport, eventually reaching the trigeminal ganglion, where latent infection is established. Under stress from the host, usually in the form of a weakening immune system (which can be caused by diverse factors), HSV-1 can undergo periodic reactivation cycles where the virus will travel back to the site of the primary infection that occurred through those same sensory neurons, usually causing clinical signs of lesions known as cold sores or oral blisters, but also can reach the brain by traveling from the bipolar trigeminal ganglion to the trigeminal nuclei in the brainstem and cause acute neurological disorders such as encephalitis, specifically HSE (30). HSV-1 also can travel to neurons which project to the thalamus and reach the sensory cortex of the brain, causing HSE as well (30). Entering the central nervous system (CNS) through the bloodstream is another possible neurological invasion mechanism HSV-1 (31). Once entering CNS, HSV-1 likely invades through receptors for HSV-1 (herparin sulfate proteoglycans), and intracellular damage, cell death, and neurodegeneration often result from direct neurological HSV-1 invasion through receptor-mediated entry (32). Several receptors for HSV-1 are selectively enriched in the hippocampus of adult human brains, providing a plausible explanation to why this brain area is more afflicted in HSE patients (32). Other than this possible mechanism linking HSV-1 to the development of AD via the virus directly damaging neural tissue (29), triggering an inflammatory response (33), or interacting with genetic and environmental factors to increase the risk of developing AD (34) are also two other potential mechanisms of HSV-1.
4.1.3. Contrary Data
However, there are some instances not in support due to equivocal data. For example, two studies using small sample populations, < 35 patients reported no association between AD and levels of anti-HSV immunoglobulin G (IgG) (35,36). The data analysis to investigate the association between AD and the abundance of herpesvirus present in human brain samples needs to be carefully carried out. Recently a study supporting the existence of such a relationship in Neuron was challenged (37). The related claims pointed out several analysis errors, including mismatched gradients on color bars which consequently cause the interpretation of p-values to look indistinguishable and a lack of statistical robustness, subsequently establishing the study as statistically misleading (38). Recently, HSV-1 has also been shown not to induce Aβ pathology in a mice model of late onset AD (39).
4.1.4. Possible Future Directions to Conclude Role of HSV-1 in AD
Currently, some studies suggest a potential association between HSV-1 and AD, but the nature of this association remains unclear. There are several recent reviews that have listed and discussed many studies on the potential importance of HSV-1 in AD development (40–43). Herein, we take from some well-known or understood examples, which we hope to continue to build from through the perspective of the virus and the infection process and will pose possible experiments to connect HSV-1 to AD development distinctly. The original work identifying the presence of HSV-1 in the brain of AD patients was confirmed by molecular detections, specifically through PCR and in situ hybridization (44). In supporting the latter, in situ hybridization is used to identify the presence of virus proteins that could be present as the final products of viral particles. The former, PCR techniques could detect viral DNA/RNA from inactive or degraded particles and are harmless to the host. Therefore, the association of HSV-1 infection with AD development by comparing the expression of viral DNA/RNA between the control group and AD groups is difficult to conclude by PCR experiments alone. Despite the high prevalence of herpesvirus infections, our immune system most times can control it to be asymptomatic, while the individuals with immune deficiency are linked to higher risk of suffering HSE (45). Therefore, comparable HSV immunoglobulin between control and AD groups also cannot exclude the role of HSV-1 in AD without considering comorbidity. In fact, AD itself seems to be a significant risk factor for HSV-1 infection, as APOE4 carriers, the frequency of HSV-1 reactivation is supported by IgM positive or elevated levels of IgG, while no significant association was found in APOE4-negative subjects (46). Herein, we will explore what has been found and what could be further carried out to test the hypothesis on the contribution of HSV-1 infection to the development of AD.
One important question to ask is whether the productive infection or frequency of reactivation of HSV-1 is associated with AD onset or development. To address this question, comprehensive studies are needed. Since viral transcription, DNA replication, capsid assembly and DNA encapsulations of HSV-1 occur exclusively in the nucleus (47), perhaps reexamining viral genome copies of nucleus extracts of commonly AD affected areas in the brain such as the hippocampus, the frontal lobe, and temporal lobe could be done to investigate whether more genome copies, subsequently more competent for reactivation, is in samples from AD-affected brains. Whether AD risk factors, including age, genetics, lifestyle, and environmental influence, affect HSV-1’s impact on AD also should be explored by subgroup and correlation analysis. Some viral proteins such as VP16 is shown to be important in lytic transcription (48), therefore, quantifying viral proteins such as VP16, and compare their abundance in healthy and AD groups could be helpful to determine the role of HSV-1 in AD. Statistics wise, a large sample size and more sophistic analyses are appreciated for the correlation studies.
The animal models are often instructive and insightful to comprehensively test the role of viral infections in AD onset and development. The capability of the animal models for memory function assessment is also a surplus. Recently, it has been shown that recurrent HSV-1 by heat stress induces hallmarks of neurodegeneration and cognitive deficits in mice (28). Researchers may use the model to further study whether anti-HSV-1 treatment blocks the disease consequence related to AD, including Ab accumulation, tau hyperphosphorylation, neuroinflammation (astrogliosis and inflammatory cytokines/chemokines secretion), and/or cognitive deficits through measurable behavior testing. Whether HSV-1 mutants deficient in neuronal cell binding through glycoprotein B (49,50), HSV-1-encoded kinases that can phosphorylate tau to a hyperphosphorylated state (51), or nuclear egress of HSV-1 (52), lead to attenuated AD symptoms could also be carried out. Other disease parameters to be visited are granulovacuolar degeneration (53), atrophy of the gyri in frontal and temporal cortices (54), atrophy in posterior cortical areas (55) by functional imaging studies. These could also be carried out in animal models infected with HSV-1 or mutants. Taken together, HSV-1 as an etiological agent to AD development, like other viruses we will discuss, the viral hypothesis of AD remains a controversial and challenging field that needs further intense work mainly into the potential mechanisms and consistent associations with specific viral species.
4.2. Zika Virus (ZIKV)
Zika virus (ZIKV), a neurotropic and neuroinvasive non-segmented, positive-sense single-stranded RNA arbovirus of the Flaviviridae family, has a total of three global outbreaks over the last century with the most famous epidemic taking place between 2015 and 2016 in the Americas and Europe (56). ZIKV is mostly transmitted through the bite of an infected Aedes mosquito, but has also been horizontally transmitted through sexual activity, hospitalization, blood transfusion or organ transplantation and vertically transmitted from infected mother to fetus (57). Usually, ZIKV is a self-limiting disease because most people with ZIKV do not have any symptoms, like most arboviruses. However, about twenty percent of people will experience ZIKV-induced inflammation including fever, rash, joint pain, and its neurological impact such as reports where ZIKV has led to Guillain-Barré Syndrome (GBS) and microcephaly or ‘Congenital Zika Syndrome’ (CZS) (58–63). Therefore, a link between AD pathology and ZIKV infection could exist, mainly because of these two etiological factors, inflammation and neurological tropism with impact contributes to AD onset and development.
4.2.1. Supportive Evidence
One of the supportive pieces is the neurotropism of ZIKA. In neural progenitor cells (NPCs), neurotropism for ZIKV is canonically through the binding of the AUAG motif in the Xrn1-resistant RNA2 (xrRNA2) of ZIKV to human protein Musashi-1 (MSI1). There is also a non-canonical entry in NPCs through the interaction of viral RNA structure AGAA tetraloop with human MSI1 (64). Within human neuronal stem cells (NSCs), ZIKV also exhibits neurotropism due to the high expression a cell surface receptor the AXL protein, a receptor tyrosine kinase, implicated in viral cell entry (65). Because multipotent neural progenitor cells are targeted by ZIKV, embryogenesis is impeded due to the induction of apoptosis in these cells, which results in microcephaly, through in vivo neonate models of ZIKV infected mice (66). Other than NPCs and NSCs, ZIKV has preference for infecting neuroepithelial stem (NES) cells and resulting in cell death, proliferation reduction, and a decrease of neuronal cell-layer volume (67). ZIKV infection also causes structural disorganization and architectural impairment, which contributes to the observed deteriorating neurological defects of ZIKV (68,69).
Several groups have begun evaluating the possible connection between ZIKV and AD. A study by Drs. Kim and Kang explored the hypothesis that the persistent endoplasmic reticulum (ER) stress that ZIKV infection induces triggers the antiviral overactivation of the PERK-eIF2a pathway, and thereby results in synaptic failure, neuronal impairment, and cellular death. PERK-eIF2a pathway also activates unfolded protein response (UPR) resulting in increased presence of Ab and p-Tau through the upregulation of BACE1 and GSKa/b (70). Activated caspase 3, which is a cysteine protease-activated in apoptosis, was suggested to be a factor in the functional decline of those affected by AD and the infection of human mesenchymal stem cells with ZIKV leads to enhanced expression of caspase 3. Thereby, implicating that caspase 3 is involved in neuronal cell death and plaque formation in AD brains (71,72). Of the primary research published over the recent decade since the last epidemic of ZIKV, it has been shown that using an FDA-approved drug, known as memantine, which eases symptoms of AD, blocks N-Methyl-d-Aspartate receptors (NMDARs) through their overactivation and could disrupt the neuronal damage observed due to ZIKV infection. Their research not only provides a unique use of NMDAR blockers but also confirms again that there exists a link between ZIKV and neurodegeneration, specifically in the form of AD (73).
4.2.2. Contrary Evidence
However, of the reports of Ab peptide acting as an anti-microbial (74–76), the group of Drs. Zhang and Zheng explored this possible connection of ZIKV with AD and showed that APP gets stabilized by interacting with ZIKV and also inhibits ZIKV replication in both human neuronal progenitor stem cells or neuronal stem cells in neonatal mouse brains. They found that the ZIKV-APP interaction prevents APP being cleaved by BACE-1 by blocking BACE1-binding site for APP, resulting in a protective effect reducing the availability of ZIKV to other cells. This data thereby implicates that APP is an antagonist for ZIKV via receptor mimicry (77).
4.2.3. Potential Mechanisms & Possible Future Directions to Conclude Role of ZIKV in AD
ZIKV, compared with HSV-1, is a relatively newly considered virus in AD, therefore, there is not a clinical relevance study, yet (78). Although most ZIKV infections are characterized by subclinical or mild influenza-like illness, severe manifestations especially those involved in neurological system such as Guillain-Barre syndrome also exist (78). Therefore, following up those patients and investigating the long impact of ZIKV on neurodegenerative diseases including AD will be instructive. Also, not all babies born to infected mothers have microcephaly (79), therefore, following that population of babies to investigate the long impact of ZIKV on neurological development is also essential.
Despite one group suggesting that APP is an antagonist for ZIKV infection, the conclusion was made by enhanced ZIKV viral RNAs in medium of APP-null cells. The conclusion will be more solid if both intracellular and extracellular infectious particles were quantified, as it is possible that APP entraps viral RNAs to facilitate generation of infectious particles in cells, leading reduced animal survival, while in APP-null cells, viral RNAs are easier to be disseminated and then less infectious particles. Currently, animal models of ZIKV have demonstrated strong viral neurotropism enhanced by passive immunity with antibodies against other arboviruses. Different knockout models, such as Ifnar1-/- (80) and Stat2-/- (81) also have their effectiveness in recapitulating specific aspects of ZIKV pathogenesis and disease independently (82). Perhaps researchers should consider memory decline through observing behavior and reporting AD-like features including changes in Ab, p-Tau, and/or neuroimage after sham infection or infection with ZIKV using mouse models. ZIKV mutants which induce less neurological inflammatory responses could also be used to compare their impact on the memory function with wild type virus.
Other important experimental models for ZIKV-AD hypothesis would be human induced pluripotent stem cells (hiPSC)-derived three-dimensional (3D) cultures like brain organoids and spherical self-organized aggregates or hiPSC-derived neurological/immunoregulatory cells (83). These models have been widely used for drug reproposing. Could repurposing established drugs lead to studies that investigate further the combination of other neuroprotective drugs used in AD to slow the impact that ZIKV could have on the development of AD? Or possibly contributing to drug development of an antiviral for ZIKV that also has preventative impacts on developing AD-like signaling if previous infections of ZIKV have occurred? Some other experiments that could be done to build upon these imperative studies are desperately needed to expand the field. For example, there are specific mutations that exist between the French Polynesian strain and the Brazilian strain of ZIKV in three of the nonstructural (NS) proteins, 3 in NS1 (immune evasion), 1 in NS4B (evasion of type I IFN signaling), and 1 in NS5 (mask viral RNAs from viral RNA synthesis and replication) (84–88). Designing mutant construct and infection experiments in human neuronal progenitor or stem cell models and comparing their impact on the AD-like pathology or development with wild type viruses should be completed. Brain organoid models could continue to be useful tools in studying how ZIKV is implicated in provoking AD pathologies. Experiments designed to repeat critical events after viral infection with ZIKV such as cytokine storm stimulation or blood-brain barrier (BBB) leakage with advanced organoids are needed to demystify the role of immune cells and blood vessels in potential mechanisms of neurodegeneration, mainly neuroinflammation. The development of complex brain organoid models could also be used to study the role of cell death and its contribution to cell populations involved, including but not limited to neurons, immune cells of the brain, and endothelial cells of blood vessels associated with the BBB. Perhaps that the neuroinflammation is not just due to the presence of ZIKV causing an infection, but also due to the cell death that the virus also causes, which could accelerate cognitive decline. Taken together the previous evidence shown, ZIKV likely plays a role in AD onset and/or development but needs more studies to conclude.
4.3. SARS-CoV-2 (COVID-19) or Long-COVID
Recently, the entire world has been at the mercy of coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). SARS-CoV-2 is a positive-sense single-stranded RNA virus belonging to the family Coronaviridae (89). Still today, there are thousands of new cases and deaths reported daily. While it is not still much of an urgent burden as it was just a couple of years ago, SARS-CoV-2 has made lasting impacts through experiencing long-term effects or sequela from their acute or active infections, known as Long COVID or Post-COVID Conditions (PCC) (90). Sadly, Long COVID can include a wide range of ongoing health problems, which can last in an individual for weeks, months, or even years. Anyone who has been infected with COVID-19 can experience this condition, which is variable between individuals but there are some major common neurological symptomologies: headaches, difficulty thinking or concentrating (‘brain fog’), sleep problems, lightheadedness, change in smell or taste, and depression or anxiety. A lot of these same symptoms are reported in AD. AD was found to be one of the most common COVID-19 comorbidities, and the virus infection contributed to an increase in mortality in these individuals affected. Of the two main pathways that have been proposed by how viruses are involved in the development of AD pathology, direct (microbes infect the brain and promote the accumulation of Ab and hyperphosphorylated tau) and indirect (inflammatory effects of an infection with microbes). it seems that SARS-CoV-2 could be implicated in both. There exists strong evidence that is suggestive of SARS-CoV-2 ability to exhibit neurotropic properties, and thereby, allowing the virus to invade the CNS. Like the other sections of this review, we will take you through some of the viral lifecycle of SARS-CoV-2, with particular attention to the entry of the virus, and mention how the virus could be contributing to the development of AD, but also of any effective biomarkers that could lead to the implication of the viral causation of AD development. A review by Dr. Luisa Agnello’s group summaries some of the most common critical biomarkers that may overlap between the two conditions (91).
4.3.1. Supporting Evidence
The SARS-CoV-2 virus is transmitted via aerosolization of droplets riddled with the virus that are breathed in. In the upper respiratory tract, angiotensin-converting enzyme 2 (ACE2)-mediated entry plays a significant role for SARS-CoV-2 invasion (92). Because of the case reports and meta-analysis that included data relating COVID-19 to not only the development of AD, but to other devastating neurodegenerative conditions such as Parkinson’s Disease (PD), amyotrophic lateral sclerosis (ALS), several other dementias, and multiple sclerosis (MS), there seems to exist a link between SARS-CoV-2 infection and neurodegenerative disease impact, especially AD (93). Mechanistically wise, the BBB, consisting of endothelial cells, also express ACE2 receptors, which can mediate the possibility of SARS-CoV-2 invasion into the CNS (94). It has also been shown that viral infection from vascular endothelial cells through the BBB to the glial cells occurs, and then through infected neurons by transsynaptic transfer (95). Usually hematogenous spread through infected leukocytes, which operate as ‘trojan horses’ in this case, has also been suggested to carry the virus while migrating to the brain (96). The CNS invasion of virus through the olfactory nerve to the olfactory bulb by retrograde axonal transport has also be described (95). Within the brain, it has been reported that ACE2 can affect Ab42 synthesis (82) and SARS-CoV-2 infection is also able to change the expression of ACE2 (97), suggesting a possibility of SARS-CoV-2 as an additional AD regulatory factor by controlling ACE2 expression and subsequently affecting neurotoxic forms of Ab.
After entering the CNS, several neurobiological outcomes caused by crosstalk between COVID-19 and AD have been also suggested by several groups. During SARS-CoV-2 infection, there are potential mechanisms that may be involved in the development of AD and its corresponding sequelae, which comprise Ab deposition, genetic factors like the pathway of the APOEe4, neuroinflammation (signatures such as cytokines of IL-6, IL-1, and Gal-3), and microglial activation. Of some common biomarkers that can be considered for neuronal injury during COVID-19 and AD include p-tau, neurofilament light chain protein (NFL), and glial fibrillary acidic protein (GFAp) microvascular injury (98–103). These are all reported to be increased in both COVID-19 patients and AD (91,104). Overall, SARS-CoV-2 can ignite AD-like signaling and, subsequently, post-COVID-19 neurological syndrome after CNS invasion (105–107).
The link between SARS-CoV-2 infection and AD is also supported by observed impact of AD on SARS-COV-2 infection. Patients with AD seem more vulnerable to SARS-CoV-2 infection, partially due to AD-induced direct and indirect pathological alterations, in addition to other AD-associated adverse impacts, including age, a lack of capabilities to follow recommendations on public health precautions, and nutritional factors (108–110). Using a SARS-CoV-2 pseudovirus infection model, Dr. Shie’s group recently reported that the interaction of Ab42 and SARS-CoV-2 strengthened the binding of the viral spike protein to ACE2, leading to more prominent viral entry and subsequently enhanced inflammatory cytokine IL-6. These same outcomes were not observed with Ab40 (111). All these reports support the link between SARS-CoV-2 infection and AD.
4.3.2. Contrary Evidence
Because we still do not know much about how COVID-19 affects the body long-term, it is overall difficult to determine whether there is unsupportive or inconsistent evidence or whether the current evidence in support has absolute validity. However, based on an interaction study of the coronavirus S-protein binding alpha-secretase (α-secretase), which functions as an integrin and metalloproteinase-9 (ADAM-9), it was suggested that S-α-secretase interaction produces a protective affect against AD through the adhesion of the virus to the cellular membrane and prevents the cleavage of the APP within the Ab domain, thus preventing Ab generation (112).
4.3.3. Potential Mechanisms & Possible Future Directions to Conclude Role of SARS-CoV-2 or Long-COVID in AD
A recent retrospective study utilized the electronic health records of at least 6 million American adults over the age of 65 that were infected or had a history of an infection with SARS-CoV-2. From this study, it was determined through bidirectional relationships that these adults surveyed would have a significantly higher risk of developing AD (113,114). While this study does address the growing concerns of viruses affecting individuals long-term, especially because of the signs and symptomology associated with coronavirus and brain function reported, these studies do not consider the prior health of the patients affected with COVID-19 and eliminate the possibility that these patients could already be demonstrating signs of AD prior to COVID-19 infection. Therefore, follow-up studies on the relationship between AD development and COVID-19 severity/symptomatic length of adult patients, with all comorbidities considered, will need to be carried out. Subgroup studies will also help to determine the impact of age, sex, race, and preexisting health conditions on SARS-CoV-2’s effect on AD, if determined.
Recently, a mouse model studying the long COVID has been developed (115). This model demonstrated that a mouse adapted SARS-CoV-2 induces neuropathological outcomes several weeks after infection at similar rates of observed clinical prevalence of "Long COVID". Establishing the viability of this model is a key step towards the rapid development of novel therapeutic strategies to ameliorate neuroinflammation and restore brain function in those suffering from the persistent cognitive dysfunction of "Long-COVID". Using this model as a base, we could study the potential mechanisms that may be involved in the development of AD and its corresponding sequelae, which comprise Ab deposition, the expression of p-tau, NFL and GFAp, microvascular injury, genetic factors like the pathway of the APOEe4, neuroinflammation (signatures such as cytokines of IL-6, IL-1, and Gal-3), and microglial activation (98–103). Particularly, it could be noted that the experiments that could be developed to further strengthen the knowledge of neuroinflammation during COVID-19 and its implication to being a contributing factor of AD would be to study these cytokines and their effects in mouse models, such as with Tg2576 mice, which is a widely used mouse model to study AD through the overexpression of human APP695 along with a Swedish mutation (KM670/671NL) under a hamster prion promoter that results in an elevation of Ab levels and amyloid plaques (116), and APPNL-F and APPNL-G-F knock-in mice, both of which express the Swedish and Iberian mutations and accumulate Ab and recapitulate amyloid plaques, synaptic loss, neuroinflammation through astrocytosis and microgliosis) (117,118). Infecting those mice with SARS-CoV-2 to see if there is an acceleration and expression/production of AD-related biomarkers previously discussed or mentioned above could be a feasible and instructive study.
The mechanisms obtained from mouse models can be further validated using primary or iPSC-derived neurological cells or iPSC-derived brain organoid. For example, once microglia-mediated inflammation is shown to play an essential role in AD onset and development in response to SARS-CoV-2 infection in mice, SARS-COV-2-infected microglia could be used to study whether they can be polarized to a pro-inflammatory phenotype, leading to subsequent changes in neurodegenerative signaling. We could also compare the response of microglial cells to treatment with Ab and infection. From there inflammatory cytokines and chemokine secretion can be measured through expression and production.
5. Summary of Potential Mechanisms on Viral Contribution to AD
There are several other viruses including human herpes virus-6 (HHV-6), -7 (HHV-7) (34,119–123), Epstein-Barr virus (EBV) (124–130), cytomegalovirus (CMV) (26,131–134), varicella zoster virus (VZV) (135–138), human immunodeficiency virus (139–153), hepatitis C virus (HCV) (154), influenza A virus (IAV) (155–159), and measles (160–163), which have been also implicated for their roles in AD onset and development (see references listed). As discussed briefly throughout the review, there are several recognized potential mechanisms proposed for the viral-AD hypothesis, which encompass direct neuronal infection, chronic neuroinflammation, altered Ab processing, tau protein dysregulation, immune system dysregulation, blood-brain barrier disruption, and genetic and epigenetic interactions that may contribute to the development or exacerbation of AD as shown in Figure 2, which we will briefly elaborate on next.
5.1. Direct Neuronal Infection
Certain neurotropic viruses, such as various herpesviruses, establish latent infections within neuronal tissues. Their reactivation may result in direct neuronal infection, potentially contributing to AD pathogenesis due to the virus now replicating and synthesizing viral proteins and thereby causing whole virions to be produced and causing acute infection. Because the virus can remain in the body and act as a lifelong reservoir despite its latency could suggest an involvement of these types of neurotropic viruses in AD, leading to synaptic dysfunction of neurons and subsequent neuronal death (164).
5.2. Chronic Neuroinflammation
Microglia, the resident immune cells within the brain can be activated by viral infections and an essential factor for AD pathogenesis. This activation can lead to the release of pro-inflammatory cytokines like IL-1β, TNF-a, and IL-6, contributing to chronic neuroinflammation observed in AD (165). Consequently, dysregulation of inflammatory signaling pathways has also been proposed because of viral infections. Viruses can frequently disrupt typical immune responses and perpetuate these pro-inflammatory environments conducive to AD development (166–169).
5.3. Impacts on Ab Processing
Processing of APP may be influenced by viral infections, specifically by HSV-1. Altered APP processing could result in the accumulation of Ab plaques, an etiological feature of AD pathology (170). Interactions between specific viral components and Ab have been proposed. This could be direct interaction between viruses and Ab, which promote aggregation and contributes to the seeding and spreading of plaque pathology in the brain (171).
5.4. Tau Protein Dysregulation
Chronic viral infections may influence the phosphorylation of tau protein. Viral-induced alterations in tau phosphorylation could contribute to NFT formation (172). Just like with Ab, tau can also interact, aggregate, and spread pathological tau due to chronic viral infections (173).
5.5. Immune System Dysregulation
Viral infections can dysregulate immune responses in the brain. Chronic viral infection compromises normal immune response, potentially contributing to the persistence of AD pathology (174). Chronic viral infections may alter the function of immune cells responsible for clearing misfolded proteins. Impaired clearance mechanisms could exacerbate the accumulation of Ab and tau in AD (175). Viral infections could possess a potential role in disrupting the blood-brain barrier (BBB). This disruption may allow the entry of peripheral immune cells and pathogens into the brain, contributing to neuroinflammation and AD pathology (176).
5.6. Genetic and Epigenetic Interactions
Genetic and epigenetic factors may influence susceptibility to viral infections and modulate the host response (e.g., immunity), thereby shaping the overall impact on AD development. For example, meta-analyses indicate associations between AD, viral infections with HSV-1, especially in individuals with the APOE4 allele (177). With the development of RNA-seq and proteomics technologies, more common biomarkers for viral infections and neurogenerative diseases are identified. These emerging biomarkers of interest include lipid biomarker 7-ketocholesterol. This pro-oxidant and pro-inflammatory molecule appear to significantly contribute to the development of AD and is produced during COVID-19 infections alike (178). Non-coding RNAs (ncRNAs) are also another emerging regulatory family serving as study targets for viral-AD hypothesis. For example, microRNA-146a-5p, thoroughly reviewed by Pogue and Lukiw, may be a special biomarker that can be used for both viral infection and inflammatory neurodegeneration and could be influenced under viral infections of the brain such as by SARS-CoV-2 and subsequently leads to miRNA-regulated gene or protein changes for AD development (179). Recently, tRNA-derived RNA fragments (tRFs) have been shown to be essential regulators of many diseases including viral infectious diseases and neurodegenerative diseases (180–185). Commonly impacted tRFs by both neurotropic viral infection and AD are also identified. Whether there is tRF-mediated crosstalk between viral infection and AD development is also an interesting research topic for the research community. Iron metabolism could be an additional angle to test viral-AD hypothesis. A retrospective study exploring the higher serum level of myoglobin as a predictor of the worse prognosis of COVID-19 infections was carried out recently (186). In AD, there is an imbalance in iron homeostasis through excessive iron contributing to Ab deposition and the formation of NFTs (187). Evaluation of how iron metabolism during COVID-19 infections affects the development of AD has not been considered. The two conditions and their impact on iron metabolism and the outcome of disease has been studied separately but taken together to look out common pathogenesis pathway and myoglobin as a biomarker could serve as a novel perspective.
6. Challenges and Future Directions
Like any developing and evolving field, there will be challenges and controversies, and the viral-AD hypothesis is no different. Some of these key issues include causation challenges. Specifically, establishing causation is difficult, especially without longitudinal studies and intervention trials, which could supplement the extensive use of observational studies. The need for methodological variability across studies poses a significant challenge. Differences in study design, sample sizes, overly complicated diagnostic criteria, and lack of innovative detection methods for viral presence contribute to discrepancies in findings. Therefore, standardization is crucial to enhance reproducibility and reliability across viral-AD linked studies. Animal models, while effective, also have limitations in recapitulating the complexity of human AD. These inconsistencies between animals and human responses to viral infections will continue to pose challenges in reasoning findings. We must continue to develop more accurate animal models to advance this standard of translational research. AD often coexists with various comorbidities, such as type 2 diabetes or even another viral infection such as HIV (188). The combination of comorbidities is limitless, especially in human based studied, thereby confounding the interpretation of viral-AD associations. These comorbidities must be identified and considered, as well as disregarded, and these findings compared. Future research should adopt a holistic approach by integrating, for example, genetic and environmental factors to AD development. This could lead to refinement of risk prediction models and enhance clinical approaches to the viral-AD hypothesis. Lastly, the sophisticated nature of the viral-AD hypothesis urgently needs collaborative efforts across disciplines. Multidisciplinary approaches that involve virologists, neuroscientists, geneticists, and immunologists could unionize their diverse expertise and transform this field through achieving a more comprehensive understanding of the complexity of viral infections and AD.
7. Conclusions
The viral-AD hypothesis explored here provides consistent associations between viral pathogens and AD pathology that cannot be ignored. While these key findings point to intriguing associations and potential mechanisms, the field continues to struggle with methodological challenges. There is still a great deal of research that needs to be done to establish a direct link between AD and viral infection. The journey ahead demands a commitment to rigorous research, innovative methods, and collaborative approaches. We must explore this viral hypothesis from all angles, answering a critical consideration of whether AD may merely predispose people to viral infections or vice versa. In our pursuit to understand, treat, and hopefully cure AD, as researchers we must remain open, collaborative, and transformative to where the science will lead us.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. Conceptualization, M.D.R.B. and X.B.; , X.X.; writing—original draft preparation, M.D.R.B., and X.B.; writing—review and editing, M.D.R.B, W.W., J.D., M.P., X.F., and X.B.; visualization, X.B.; supervision, X.B; project administration, X.B.; funding acquisition, X.B., X.F. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This work was supported by grants from the US National Institute of Health (NIH) R21 AI66543 and R21AG069226 to XB, R61 AG075725 and TARCC Investigator-Initiated Research Award 952272 to XB and XF, R21 AG066060 to XF.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Yang HD, Kim DH, Lee SB, Young LD. History of Alzheimer’s Disease. Dement Neurocogn Disord. 2016 Dec;15(4):115–21.
- DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Molecular Neurodegeneration. 2019 Aug 2;14(1):32.
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011 Sep;1(1):a006189.
- Breijyeh Z, Karaman R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules. 2020 Dec 8;25(24):5789.
- Dubois B, Villain N, Frisoni GB, Rabinovici GD, Sabbagh M, Cappa S, et al. Clinical diagnosis of Alzheimer’s disease: recommendations of the International Working Group. Lancet Neurol. 2021 Jun;20(6):484–96.
- O’Brien RJ, Wong PC. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu Rev Neurosci. 2011;34:185–204.
- Chen GF, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin. 2017 Sep;38(9):1205–35.
- Murphy MP, LeVine H. Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis. 2010;19(1):311–23.
- Iqbal K, Alonso A del C, Chen S, Chohan MO, El-Akkad E, Gong CX, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005 Jan 3;1739(2–3):198–210.
- Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer Disease and Related Tauopathies. Curr Alzheimer Res. 2010 Dec;7(8):656–64.
- Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012 Oct 1;2(10):a006296.
- Knobel P, Litke R, Mobbs CV. Biological age and environmental risk factors for dementia and stroke: Molecular mechanisms. Front Aging Neurosci. 2022;14:1042488.
- Yuan S, Wu W, Ma W, Huang X, Huang T, Peng Mi, et al. Body mass index, genetic susceptibility, and Alzheimer’s disease: a longitudinal study based on 475,813 participants from the UK Biobank. J Transl Med. 2022 Sep 9;20(1):417.
- Katan M, Moon YP, Paik MC, Sacco RL, Wright CB, Elkind MSV. Infectious burden and cognitive function. Neurology. 2013 Mar 26;80(13):1209–15.
- Letenneur L, Pérès K, Fleury H, Garrigue I, Barberger-Gateau P, Helmer C, et al. Seropositivity to Herpes Simplex Virus Antibodies and Risk of Alzheimer’s Disease: A Population-Based Cohort Study. PLoS One. 2008 Nov 4;3(11):e3637.
- Kountouras J, Tsolaki M, Gavalas E, Boziki M, Zavos C, Karatzoglou P, et al. Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology. 2006 Mar 28;66(6):938–40.
- Aiello AE, Haan M, Blythe L, Moore K, Gonzalez JM, Jagust W. The influence of latent viral infection on rate of cognitive decline over 4 years. J Am Geriatr Soc. 2006 Jul;54(7):1046–54.
- Hernandez-Ruiz V, Letenneur L, Fülöp T, Helmer C, Roubaud-Baudron C, Avila-Funes JA, et al. Infectious diseases and cognition: do we have to worry? Neurol Sci. 2022 Nov;43(11):6215–24.
- Wennberg AM, Maher BS, Rabinowitz JA, Holingue C, Felder WR, Wells JL, et al. Association of common infections with cognitive performance in the Baltimore Epidemiologic Catchment Area study follow-up. Alzheimers Dement. 2023 Nov;19(11):4841–51.
- Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet. 1997 Jan 25;349(9047):241–4.
- Sochocka M, Zwolińska K, Leszek J. The Infectious Etiology of Alzheimer’s Disease. Curr Neuropharmacol. 2017;15(7):996–1009.
- Balin BJ, Hudson AP. Herpes viruses and Alzheimer’s disease: new evidence in the debate. Lancet Neurol. 2018 Oct;17(10):839–41.
- Piacentini R, De Chiara G, Li Puma DD, Ripoli C, Marcocci ME, Garaci E, et al. HSV-1 and Alzheimer’s disease: more than a hypothesis. Front Pharmacol. 2014 May 7;5:97.
- Ball MJ. Limbic predilection in Alzheimer dementia: is reactivated herpesvirus involved? Can J Neurol Sci. 1982 Aug;9(3):303–6.
- Middleton PJ, Petric M, Kozak M, Rewcastle NB, McLachlan DR. Herpes-simplex viral genome and senile and presenile dementias of Alzheimer and Pick. Lancet. 1980 May 10;1(8176):1038.
- Lövheim H, Olsson J, Weidung B, Johansson A, Eriksson S, Hallmans G, et al. Interaction between Cytomegalovirus and Herpes Simplex Virus Type 1 Associated with the Risk of Alzheimer’s Disease Development. J Alzheimers Dis. 2018;61(3):939–45.
- Wozniak MA, Frost AL, Preston CM, Itzhaki RF. Antivirals reduce the formation of key Alzheimer’s disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLoS One. 2011;6(10):e25152.
- De Chiara G, Piacentini R, Fabiani M, Mastrodonato A, Marcocci ME, Limongi D, et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019 Mar;15(3):e1007617.
- Cairns DM, Rouleau N, Parker RN, Walsh KG, Gehrke L, Kaplan DL. A 3D human brain–like tissue model of herpes-induced Alzheimer’s disease. Science Advances. 2020 May 6;6(19):eaay8828.
- Marcocci ME, Napoletani G, Protto V, Kolesova O, Piacentini R, Li Puma DD, et al. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends in Microbiology. 2020 Oct 1;28(10):808–20.
- Bello-Morales R, Andreu S, López-Guerrero JA. The Role of Herpes Simplex Virus Type 1 Infection in Demyelination of the Central Nervous System. Int J Mol Sci. 2020 Jul 16;21(14):5026.
- Lathe R, Haas JG. Distribution of cellular HSV-1 receptor expression in human brain. J Neurovirol. 2017 Jun 1;23(3):376–84.
- Xiao S, Chan P, Wang T, Hong Z, Wang S, Kuang W, et al. A 36-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate Alzheimer’s dementia. Alzheimers Res Ther. 2021 Mar 17;13(1):62.
- Itzhaki RF. Overwhelming Evidence for a Major Role for Herpes Simplex Virus Type 1 (HSV1) in Alzheimer’s Disease (AD); Underwhelming Evidence against. Vaccines (Basel). 2021 Jun 21;9(6):679.
- RENVOIZE EB, AWAD LO, HAMBLING MH. A SERO-EPIDEMIOLOGICAL STUDY OF CONVENTIONAL INFECTIOUS AGENTS IN ALZHEIMER’S DISEASE. Age and Ageing. 1987 Sep 1;16(5):311–4.
- Ounanian A, Seigneurin JM, Guilbert B, Avrameas S, Renverez JC. Antibodies to viral antigens, xenoantigens, and autoantigens in alzheimer’s disease. Journal of Clinical Laboratory Analysis. 1990;4(5):367–75.
- Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron. 2018 Jul 11;99(1):64-82.e7.
- Jeong HH, Liu Z. Are HHV-6A and HHV-7 Really More Abundant in Alzheimer’s Disease? Neuron. 2019 Dec 18;104(6):1034–5.
- Bocharova OV, Fisher A, Pandit NP, Molesworth K, Mychko O, Scott AJ, et al. Aβ plaques do not protect against HSV-1 infection in a mouse model of familial Alzheimer’s disease, and HSV-1 does not induce Aβ pathology in a model of late onset Alzheimer’s disease. Brain Pathol. 2023 Jan;33(1):e13116.
- Brady TF, Konkle T, Alvarez GA. A review of visual memory capacity: Beyond individual items and toward structured representations. J Vis. 2011 May 26;11(5):4.
- Feng S, Liu Y, Zhou Y, Shu Z, Cheng Z, Brenner C, et al. Mechanistic insights into the role of herpes simplex virus 1 in Alzheimer’s disease. Front Aging Neurosci. 2023;15:1245904.
- Cohen M, Austin E, Bradu S, Jagdeo J. The Association Between Herpes Simplex Virus and Alzheimer’s Disease: A Systematic Review. J Drugs Dermatol. 2023 Oct 1;22(10):1046–52.
- Itzhaki RF, Golde TE, Heneka MT, Readhead B. Do infections have a role in the pathogenesis of Alzheimer disease? Nat Rev Neurol. 2020 Apr;16(4):193–7.
- Walker DG, O’Kusky JR, McGeer PL. In situ hybridization analysis for herpes simplex virus nucleic acids in Alzheimer disease. Alzheimer Dis Assoc Disord. 1989;3(3):123–31.
- Zhang SY. Herpes simplex virus encephalitis of childhood: inborn errors of central nervous system cell-intrinsic immunity. Hum Genet. 2020 Jun;139(6–7):911–8.
- Linard M, Letenneur L, Garrigue I, Doize A, Dartigues JF, Helmer C. Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimer’s & Dementia. 2020;16(1):200–8.
- Heming JD, Conway JF, Homa FL. Herpesvirus Capsid Assembly and DNA Packaging. Adv Anat Embryol Cell Biol. 2017;223:119–42.
- Hafezi W, Lorentzen EU, Eing BR, Müller M, King NJC, Klupp B, et al. Entry of Herpes Simplex Virus Type 1 (HSV-1) into the Distal Axons of Trigeminal Neurons Favors the Onset of Nonproductive, Silent Infection. PLOS Pathogens. 2012 May 10;8(5):e1002679.
- Fan Q, Longnecker R, Connolly SA. Herpes Simplex Virus Glycoprotein B Mutations Define Structural Sites in Domain I, the Membrane Proximal Region, and the Cytodomain That Regulate Entry. J Virol. 95(22):e01050-21.
- Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J, et al. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer’s disease. PLoS One. 2012;7(12):e52056.
- Benetti L, Roizman B. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci U S A. 2004 Jun 22;101(25):9411–6.
- Maruzuru Y, Shindo K, Liu Z, Oyama M, Kozuka-Hata H, Arii J, et al. Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J Virol. 2014 Jul;88(13):7445–54.
- Köhler C. Granulovacuolar degeneration: a neurodegenerative change that accompanies tau pathology. Acta Neuropathol. 2016 Sep;132(3):339–59.
- Perl DP. Neuropathology of Alzheimer’s disease. Mt Sinai J Med. 2010;77(1):32–42.
- Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer’s disease. Brain. 2010 May;133(Pt 5):1352–67.
- Musso D, Gubler DJ. Zika Virus. Clin Microbiol Rev. 2016 Jul;29(3):487–524.
- Agumadu VC, Ramphul K. Zika Virus: A Review of Literature. Cureus. 2018 Jul 22;10(7):e3025.
- Cao-Lormeau VM, Blake A, Mons S, Lastère S, Roche C, Vanhomwegen J, et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. The Lancet. 2016 Apr 9;387(10027):1531–9.
- Beattie J, Parajuli S, Sanger M, Lee G, Pleninger P, Crowley G, et al. Zika Virus–Associated Guillain-Barré Syndrome in a Returning US Traveler. Infect Dis Clin Pract (Baltim Md). 2018 Nov;26(6):e80–4.
- Rivera-Correa J, de Siqueira IC, Mota S, do Rosário MS, Pereira de Jesus PA, Alcantara LCJ, et al. Anti-ganglioside antibodies in patients with Zika virus infection-associated Guillain-Barré Syndrome in Brazil. PLoS Negl Trop Dis. 2019 Sep;13(9):e0007695.
- Wen Z, Song H, Ming G li. How does Zika virus cause microcephaly? Genes Dev. 2017 May 1;31(9):849–61.
- CDC [Internet]. 2014 [cited 2024 Jan 21]. Zika Virus. Available from: https://www.cdc.gov/zika/index.html.
- CDC [Internet]. 2014 [cited 2024 Jan 21]. Zika and Pregnancy. Available from: https://www.cdc.gov/zika/pregnancy/index.html.
- Chen X, Wang Y, Xu Z, Cheng ML, Ma QQ, Li RT, et al. Zika virus RNA structure controls its unique neurotropism by bipartite binding to Musashi-1. Nat Commun. 2023 Feb 28;14(1):1134.
- Ramos da Silva S, Gao SJ. Zika virus: An update on epidemiology, pathology, molecular biology, and animal model. J Med Virol. 2016 Aug;88(8):1291–6.
- Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, et al. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell. 2016 Jul 7;19(1):120–6.
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, et al. Brain Region-specific Organoids using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016 May 19;165(5):1238–54.
- Onorati M, Li Z, Liu F, Sousa AMM, Nakagawa N, Li M, et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016 Sep 6;16(10):2576–92.
- Baggiani M, Dell’Anno MT, Pistello M, Conti L, Onorati M. Human Neural Stem Cell Systems to Explore Pathogen-Related Neurodevelopmental and Neurodegenerative Disorders. Cells. 2020 Aug 12;9(8):1893.
- Lee SE, Choi H, Shin N, Kong D, Kim NG, Kim HY, et al. Zika virus infection accelerates Alzheimer’s disease phenotypes in brain organoids. Cell Death Discov. 2022 Apr 2;8:153.
- Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW. Activated caspase-3 expression in Alzheimer’s and aged control brain: correlation with Alzheimer pathology. Brain Res. 2001 Apr 20;898(2):350–7.
- Louneva N, Cohen JW, Han LY, Talbot K, Wilson RS, Bennett DA, et al. Caspase-3 Is Enriched in Postsynaptic Densities and Increased in Alzheimer’s Disease. Am J Pathol. 2008 Nov;173(5):1488–95.
- Costa VV, Del Sarto JL, Rocha RF, Silva FR, Doria JG, Olmo IG, et al. N-Methyl-d-Aspartate (NMDA) Receptor Blockade Prevents Neuronal Death Induced by Zika Virus Infection. mBio. 2017 Apr 25;8(2):e00350-17.
- Kumar DKV, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016 May 25;8(340):340ra72.
- Chen D, Liu X, Chen Y, Lin H. Amyloid peptides with antimicrobial and/or microbial agglutination activity. Appl Microbiol Biotechnol. 2022 Dec 1;106(23):7711–20.
- Spitzer P, Condic M, Herrmann M, Oberstein TJ, Scharin-Mehlmann M, Gilbert DF, et al. Amyloidogenic amyloid-β-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci Rep. 2016 Sep 14;6:32228.
- Lingel A, Lin H, Gavriel Y, Weaver E, Polepole P, Lopez V, et al. Amyloid precursor protein is a restriction factor that protects against Zika virus infection in mammalian brains. J Biol Chem. 2020 Dec 11;295(50):17114–27.
- Plourde AR, Bloch EM. A Literature Review of Zika Virus. Emerg Infect Dis. 2016 Jul;22(7):1185–92.
- Santos GRBD, Aragão FBA, Lobão WJ de M, Lima FR, Andrade LMRL de, Furtado QR, et al. Relationship between microcephaly and Zika virus during pregnancy: a review. Rev Assoc Med Bras (1992). 2018 Jul;64(7):635–42.
- Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, et al. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe. 2016 May 11;19(5):720–30.
- Tripathi S, Balasubramaniam VRMT, Brown JA, Mena I, Grant A, Bardina SV, et al. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog. 2017 Mar;13(3):e1006258.
- Ziff OJ, Ashton NJ, Mehta PR, Brown R, Athauda D, Heaney J, et al. Amyloid processing in COVID-19-associated neurological syndromes. J Neurochem. 2022 Apr;161(2):146–57.
- Acharya P, Choi NY, Shrestha S, Jeong S, Lee MY. Brain organoids: A revolutionary tool for modeling neurological disorders and development of therapeutics. Biotechnol Bioeng. 2024 Feb;121(2):489–506.
- Xia H, Luo H, Shan C, Muruato AE, Nunes BTD, Medeiros DBA, et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat Commun. 2018 Jan 29;9(1):414.
- Hertzog J, Dias Junior AG, Rigby RE, Donald CL, Mayer A, Sezgin E, et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur J Immunol. 2018 Jul;48(7):1120–36.
- Peng NYG, Amarilla AA, Hugo LE, Modhiran N, Sng JDJ, Slonchak A, et al. The distinguishing NS5-M114V mutation in American Zika virus isolates has negligible impacts on virus replication and transmission potential. PLoS Negl Trop Dis. 2022 May;16(5):e0010426.
- Muñoz-Jordán JL, Laurent-Rolle M, Ashour J, Martínez-Sobrido L, Ashok M, Lipkin WI, et al. Inhibition of Alpha/Beta Interferon Signaling by the NS4B Protein of Flaviviruses. J Virol. 2005 Jul;79(13):8004–13.
- Fanunza E, Grandi N, Quartu M, Carletti F, Ermellino L, Milia J, et al. INMI1 Zika Virus NS4B Antagonizes the Interferon Signaling by Suppressing STAT1 Phosphorylation. Viruses. 2021 Dec 6;13(12):2448.
- Pal M, Berhanu G, Desalegn C, Kandi V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus. 12(3):e7423.
- DC. Centers for Disease Control and Prevention. 2023 [cited 2023 Dec 2]. Post-COVID Conditions. Available from: https://www.cdc.gov/coronavirus/2019-ncov/long-term-effects/index.html.
- Ciaccio M, Lo Sasso B, Scazzone C, Gambino CM, Ciaccio AM, Bivona G, et al. COVID-19 and Alzheimer’s Disease. Brain Sci. 2021 Feb 27;11(3):305.
- Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586–90.
- Zhang W, Xiao D, Mao Q, Xia H. Role of neuroinflammation in neurodegeneration development. Sig Transduct Target Ther. 2023 Jul 12;8(1):1–32.
- Reynolds JessicaL, Mahajan SD. SARS-COV2 Alters Blood Brain Barrier Integrity Contributing to Neuro-Inflammation. J Neuroimmune Pharmacol. 2021;16(1):4–6.
- Zubair AS, McAlpine LS, Gardin T, Farhadian S, Kuruvilla DE, Spudich S. Neuropathogenesis and Neurologic Manifestations of the Coronaviruses in the Age of Coronavirus Disease 2019 A Review. JAMA Neurol. 2020 Aug 1;77(8):1018–27.
- Whitmore HAB, Kim LA. Understanding the Role of Blood Vessels in the Neurologic Manifestations of Coronavirus Disease 2019 (COVID-19). Am J Pathol. 2021 Nov;191(11):1946–54.
- Mehrabadi ME, Hemmati R, Tashakor A, Homaei A, Yousefzadeh M, Hemati K, et al. Induced dysregulation of ACE2 by SARS-CoV-2 plays a key role in COVID-19 severity. Biomed Pharmacother. 2021 May;137:111363.
- Virhammar J, Nääs A, Fällmar D, Cunningham JL, Klang A, Ashton NJ, et al. Biomarkers for central nervous system injury in cerebrospinal fluid are elevated in COVID-19 and associated with neurological symptoms and disease severity. Eur J Neurol. 2021 Oct;28(10):3324–31.
- Zhou Y, Xu J, Hou Y, Leverenz JB, Kallianpur A, Mehra R, et al. Network medicine links SARS-CoV-2/COVID-19 infection to brain microvascular injury and neuroinflammation in dementia-like cognitive impairment. bioRxiv. 2021 Mar 22;2021.03.15.435423.
- Pilotto A, Masciocchi S, Volonghi I, De Giuli V, Caprioli F, Mariotto S, et al. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Encephalitis Is a Cytokine Release Syndrome: Evidences From Cerebrospinal Fluid Analyses. Clin Infect Dis. 2021 Nov 2;73(9):e3019–26.
- Toniolo S, Scarioni M, Di Lorenzo F, Hort J, Georges J, Tomic S, et al. Dementia and COVID-19, a Bidirectional Liaison: Risk Factors, Biomarkers, and Optimal Health Care. J Alzheimers Dis. 2021;82(3):883–98.
- Goyal A, Kushwah PS, Agrawal N, Pathak S. APOE4: A Culprit for the Vulnerability of COVID-19 in Alzheimer’s Patients. Curr Neurovasc Res. 2023;20(1):162–9.
- Tsagkaris C, Bilal M, Aktar I, Aboufandi Y, Tas A, Aborode AT, et al. Cytokine storm and neuropathological alterations in patients with neurological manifestations of COVID-19. Curr Alzheimer Res. 2022 Sep 8.
- Guasp M, Muñoz-Sánchez G, Martínez-Hernández E, Santana D, Carbayo Á, Naranjo L, et al. CSF Biomarkers in COVID-19 Associated Encephalopathy and Encephalitis Predict Long-Term Outcome. Frontiers in Immunology [Internet]. 2022 [cited 2023 Dec 22];13. Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2022.866153.
- Reiken S, Sittenfeld L, Dridi H, Liu Y, Liu X, Marks AR. Alzheimer’s-like signaling in brains of COVID-19 patients. Alzheimers Dement. 2022 May;18(5):955–65.
- Krasemann S, Haferkamp U, Pfefferle S, Woo MS, Heinrich F, Schweizer M, et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Reports. 2022 Jan 20;17(2):307–20.
- Gordon MN, Heneka MT, Le Page LM, Limberger C, Morgan D, Tenner AJ, et al. Impact of COVID-19 on the Onset and Progression of Alzheimer’s Disease and Related Dementias: A Roadmap for Future Research. Alzheimers Dement. 2022 May;18(5):1038–46.
- Pan AP, Meeks J, Potter T, Masdeu JC, Seshadri S, Smith ML, et al. SARS-CoV-2 Susceptibility and COVID-19 Mortality Among Older Adults With Cognitive Impairment: Cross-Sectional Analysis From Hospital Records in a Diverse US Metropolitan Area. Front Neurol. 2021 Jul 22;12:692662.
- Brown EE, Kumar S, Rajji TK, Pollock BG, Mulsant BH. Anticipating and Mitigating the Impact of the COVID-19 Pandemic on Alzheimer’s Disease and Related Dementias. Am J Geriatr Psychiatry. 2020 Jul;28(7):712–21.
- Hardan L, Filtchev D, Kassem R, Bourgi R, Lukomska-Szymanska M, Tarhini H, et al. COVID-19 and Alzheimer’s Disease: A Literature Review. Medicina (Kaunas). 2021 Oct 25;57(11):1159.
- Hsu JTA, Tien CF, Yu GY, Shen S, Lee YH, Hsu PC, et al. The Effects of Aβ1-42 Binding to the SARS-CoV-2 Spike Protein S1 Subunit and Angiotensin-Converting Enzyme 2. Int J Mol Sci. 2021 Jul 30;22(15):8226.
- Lichtenthaler SF. Alpha-secretase cleavage of the amyloid precursor protein: proteolysis regulated by signaling pathways and protein trafficking. Curr Alzheimer Res. 2012 Feb;9(2):165–77.
- Cohen K, Ren S, Heath K, Dasmariñas MC, Jubilo KG, Guo Y, et al. Risk of persistent and new clinical sequelae among adults aged 65 years and older during the post-acute phase of SARS-CoV-2 infection: retrospective cohort study. BMJ. 2022 Feb 9;376:e068414.
- Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. 2023;21(3):133–46.
- Gressett TE, Leist SR, Ismael S, Talkington G, Dinnon KH, Baric RS, et al. Mouse Adapted SARS-CoV-2 Model Induces “Long-COVID” Neuropathology in BALB/c Mice. bioRxiv. 2023 Mar 20;2023.03.18.533204.
- Tg2576 | ALZFORUM [Internet]. [cited 2024 Jan 21]. Available from: https://www.alzforum.org/research-models/tg2576.
- APP NL-G-F Knock-in | ALZFORUM [Internet]. [cited 2024 Jan 21]. Available from: https://www.alzforum.org/research-models/app-nl-g-f-knock.
- Nilsson P, Saito T, Saido TC. New Mouse Model of Alzheimer’s. ACS Chem Neurosci. 2014 May 22;5(7):499–502.
- Allnutt MA, Johnson K, Bennett DA, Connor SM, Troncoso JC, Pletnikova O, et al. Human Herpesvirus 6 Detection in Alzheimer’s Disease Cases and Controls across Multiple Cohorts. Neuron. 2020 Mar 18;105(6):1027-1035.e2.
- Agostini S, Mancuso R, Baglio F, Cabinio M, Hernis A, Guerini FR, et al. Lack of evidence for a role of HHV-6 in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis. 2016;49(1):229–35.
- Martín-García J, González-Scarano F. Glial Responses to Virus Infection. Encyclopedia of Neuroscience. 2009;861–9.
- Pathogen infection in Alzheimer’s disease: pathophysiology and therapeutic strategies. Ageing and Neurodegenerative Diseases. 2023 Jan 31;3(1):null-null.
- Lin WR, Wozniak MA, Cooper RJ, Wilcock GK, Itzhaki RF. Herpesviruses in brain and Alzheimer’s disease. J Pathol. 2002 Jul;197(3):395–402.
- Seo J, Park M. Molecular crosstalk between cancer and neurodegenerative diseases. Cell Mol Life Sci. 2020 Jul;77(14):2659–80.
- Yang Y, Gao F. Clinical characteristics of primary and reactivated Epstein-Barr virus infection in children. J Med Virol. 2020 Dec;92(12):3709–16.
- Wang W, Bu B, Xie M, Zhang M, Yu Z, Tao D. Neural cell cycle dysregulation and central nervous system diseases. Prog Neurobiol. 2009 Sep;89(1):1–17.
- Bonda DJ, Bajić VP, Spremo-Potparevic B, Casadesus G, Zhu X, Smith MA, et al. Review: Cell cycle aberrations and neurodegeneration. Neuropathol Appl Neurobiol. 2010 Apr;36(2):157–63.
- Humans IWG on the E of CR to. Epstein-Barr virus. In: Epstein-Barr Virus and Kaposi’s Sarcoma Herpesvirus/Human Herpesvirus 8 [Internet]. International Agency for Research on Cancer; 1997 [cited 2023 Dec 3]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK385500/.
- Biström M, Jons D, Engdahl E, Gustafsson R, Huang J, Brenner N, et al. Epstein-Barr virus infection after adolescence and human herpesvirus 6A as risk factors for multiple sclerosis. Eur J Neurol. 2021 Feb;28(2):579–86.
- Tiwari D, Mittal N, Jha HC. Unraveling the links between neurodegeneration and Epstein-Barr virus-mediated cell cycle dysregulation. Curr Res Neurobiol. 2022;3:100046.
- Caruso C, Buffa S, Candore G, Colonna-Romano G, Dunn-Walters D, Kipling D, et al. Mechanisms of immunosenescence. Immun Ageing. 2009 Jul 22;6:10.
- Liu Z, Liang Q, Ren Y, Guo C, Ge X, Wang L, et al. Immunosenescence: molecular mechanisms and diseases. Signal Transduct Target Ther. 2023 May 13;8(1):200.
- Lurain NS, Hanson BA, Martinson J, Leurgans SE, Landay AL, Bennett DA, et al. Virological and immunological characteristics of human cytomegalovirus infection associated with Alzheimer disease. J Infect Dis. 2013 Aug 15;208(4):564–72.
- Mody PH, Marvin KN, Hynds DL, Hanson LK. Cytomegalovirus infection induces Alzheimer’s disease-associated alterations in tau. J Neurovirol. 2023 Aug;29(4):400–15.
- Cairns DM, Itzhaki RF, Kaplan DL. Potential Involvement of Varicella Zoster Virus in Alzheimer’s Disease via Reactivation of Quiescent Herpes Simplex Virus Type 1. J Alzheimers Dis. 2022;88(3):1189–200.
- Bruno F, Abondio P, Bruno R, Ceraudo L, Paparazzo E, Citrigno L, et al. Alzheimer’s disease as a viral disease: Revisiting the infectious hypothesis. Ageing Res Rev. 2023 Nov;91:102068.
- Zotova E, Nicoll JA, Kalaria R, Holmes C, Boche D. Inflammation in Alzheimer’s disease: relevance to pathogenesis and therapy. Alzheimers Res Ther. 2010 Jan 22;2(1):1.
- Elhalag RH, Motawea KR, Talat NE, Rouzan SS, Reyad SM, Elsayed SM, et al. Herpes Zoster virus infection and the risk of developing dementia: A systematic review and meta-analysis. Medicine (Baltimore). 2023 Oct 27;102(43):e34503.
- Chai Q, Jovasevic V, Malikov V, Sabo Y, Morham S, Walsh D, et al. HIV-1 counteracts an innate restriction by amyloid precursor protein resulting in neurodegeneration. Nat Commun. 2017 Nov 15;8:1522.
- Zeinolabediny Y, Caccuri F, Colombo L, Morelli F, Romeo M, Rossi A, et al. HIV-1 matrix protein p17 misfolding forms toxic amyloidogenic assemblies that induce neurocognitive disorders. Sci Rep. 2017 Sep 4;7:10313.
- Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder — pathogenesis and prospects for treatment. Nat Rev Neurol. 2016 Apr;12(4):234–48.
- Kovalevich J, Langford D. Neuronal toxicity in HIV CNS disease. Future Virol. 2012 Jul 1;7(7):687–98.
- Canet G, Dias C, Gabelle A, Simonin Y, Gosselet F, Marchi N, et al. HIV Neuroinfection and Alzheimer’s Disease: Similarities and Potential Links? Front Cell Neurosci. 2018 Sep 11;12:307.
- Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D’Emilia DM, et al. Caspase Cascades in Human Immunodeficiency Virus-Associated Neurodegeneration. J Neurosci. 2002 May 15;22(10):4015–24.
- Garza RDL, Rodrigo H, Fernandez F, Roy U. The Increase of HIV-1 Infection, Neurocognitive Impairment, and Type 2 Diabetes in The Rio Grande Valley. Curr HIV Res. 2019;17(6):377–87.
- Bachis A, Biggio F, Major EO, Mocchetti I. M- and T-tropic HIVs Promote Apoptosis in Rat Neurons. J Neuroimmune Pharmacol. 2009 Mar;4(1):150–60.
- Ferrarese C, Aliprandi A, Tremolizzo L, Stanzani L, De Micheli A, Dolara A, et al. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology. 2001 Aug 28;57(4):671–5.
- Sami Saribas A, Cicalese S, Ahooyi TM, Khalili K, Amini S, Sariyer IK. HIV-1 Nef is released in extracellular vesicles derived from astrocytes: evidence for Nef-mediated neurotoxicity. Cell Death Dis. 2017 Jan;8(1):e2542.
- Wang Y, Santerre M, Tempera I, Martin K, Mukerjee R, Sawaya BE. HIV-1 Vpr disrupts mitochondria axonal transport and accelerates neuronal aging. Neuropharmacology. 2017 May 1;117:364–75.
- Chen X, Hui L, Geiger NH, Haughey NJ, Geiger JD. Endolysosome involvement in HIV-1 Tat-induced neuronal amyloid beta production. Neurobiol Aging. 2013 Oct;34(10):2370–8.
- Ortega M, Ances BM. Role of HIV in Amyloid Metabolism. J Neuroimmune Pharmacol. 2014 Sep;9(4):483–91.
- Cho YE, Lee MH, Song BJ. Neuronal Cell Death and Degeneration through Increased Nitroxidative Stress and Tau Phosphorylation in HIV-1 Transgenic Rats. PLoS One. 2017 Jan 20;12(1):e0169945.
- Hategan A, Bianchet MA, Steiner J, Karnaukhova E, Masliah E, Fields A, et al. HIV Tat protein and amyloid-β peptide form multifibrillar structures that cause neurotoxicity. Nat Struct Mol Biol. 2017 Apr;24(4):379–86.
- Wong MY, Lewis M, Doherty JJ, Shi Y, Cashikar AG, Amelianchik A, et al. 25-Hydroxycholesterol amplifies microglial IL-1β production in an apoE isoform-dependent manner. J Neuroinflammation. 2020 Jun 17;17(1):192.
- Sadasivan S, Zanin M, O’Brien K, Schultz-Cherry S, Smeyne RJ. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS One. 2015;10(4):e0124047.
- White MR, Kandel R, Hsieh IN, De Luna X, Hartshorn KL. Critical role of C-terminal residues of the Alzheimer’s associated β-amyloid protein in mediating antiviral activity and modulating viral and bacterial interactions with neutrophils. PLoS One. 2018;13(3):e0194001.
- Amyloid beta protein forms ion channels: implications for Alzheimer’s disease pathophysiology - PubMed [Internet]. [cited 2023 Dec 3]. Available from: https://pubmed.ncbi.nlm.nih.gov/11689468/.
- Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J Mol Biol. 2001 Oct 5;312(5):1103–19.
- Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D’Ursi AM, Temussi PA, et al. Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur J Biochem. 2002 Nov;269(22):5642–8.
- McQuaid S, Allen IV, McMahon J, Kirk J. Association of measles virus with neurofibrillary tangles in subacute sclerosing panencephalitis: a combined in situ hybridization and immunocytochemical investigation. Neuropathol Appl Neurobiol. 1994 Apr;20(2):103–10.
- CDC. Centers for Disease Control and Prevention. 2022 [cited 2023 Dec 3]. Measles Complications. Available from: https://www.cdc.gov/measles/symptoms/complications.html.
- Miyahara H, Akagi A, Riku Y, Sone J, Otsuka Y, Sakai M, et al. Independent distribution between tauopathy secondary to subacute sclerotic panencephalitis and measles virus: An immunohistochemical analysis in autopsy cases including cases treated with aggressive antiviral therapies. Brain Pathol. 2022 Apr 4;32(6):e13069.
- Qi C, Hasegawa M, Takao M, Sakai M, Sasaki M, Mizutani M, et al. Identical tau filaments in subacute sclerosing panencephalitis and chronic traumatic encephalopathy. Acta Neuropathol Commun. 2023 May 5;11:74.
- De Chiara G, Marcocci ME, Sgarbanti R, Civitelli L, Ripoli C, Piacentini R, et al. Infectious agents and neurodegeneration. Mol Neurobiol. 2012 Dec;46(3):614–38.
- Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, et al. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation. 2012 Jul 23;9:179.
- Sait A, Angeli C, Doig AJ, Day PJR. Viral Involvement in Alzheimer’s Disease. ACS Chem Neurosci. 2021 Mar 9;12(7):1049–60.
- Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018 Jan 23;9(6):7204–18.
- Talwar P, Gupta R, Kushwaha S, Agarwal R, Saso L, Kukreti S, et al. Viral Induced Oxidative and Inflammatory Response in Alzheimer’s Disease Pathogenesis with Identification of Potential Drug Candidates: A Systematic Review using Systems Biology Approach. Curr Neuropharmacol. 2019 Apr;17(4):352–65.
- Xie J, Van Hoecke L, Vandenbroucke RE. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front Immunol. 2022 Jan 6;12:796867.
- Wozniak M, Mee A, Itzhaki R. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. The Journal of Pathology. 2009 Jan;217(1):131–8.
- Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron. 2018 Jul 11;99(1):56-63.e3.
- Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, et al. Microbes and Alzheimer’s Disease. J Alzheimers Dis. 2016;51(4):979–84.
- Šimić G, Babić Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milošević N, et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules. 2016 Jan 6;6(1):6.
- Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2010;21(3):927–38.
- Rubio-Perez JM, Morillas-Ruiz JM. A Review: Inflammatory Process in Alzheimer’s Disease, Role of Cytokines. ScientificWorldJournal. 2012 Apr 1;2012:756357.
- Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J Exp Med. 2017 Nov 6;214(11):3151–69.
- Carter CJ. Genetic, Transcriptome, Proteomic, and Epidemiological Evidence for Blood-Brain Barrier Disruption and Polymicrobial Brain Invasion as Determinant Factors in Alzheimer’s Disease. J Alzheimers Dis Rep. 2017 Sep 28;1(1):125–57.
- Ghzaiel I, Sassi K, Zarrouk A, Nury T, Ksila M, Leoni V, et al. 7-Ketocholesterol: Effects on viral infections and hypothetical contribution in COVID-19. J Steroid Biochem Mol Biol. 2021 Sep;212:105939.
- Pogue AI, Lukiw WJ. microRNA-146a-5p, Neurotropic Viral Infection and Prion Disease (PrD). Int J Mol Sci. 2021 Aug 25;22(17):9198.
- Deng J, Ptashkin RN, Chen Y, Cheng Z, Liu G, Phan T, et al. Respiratory Syncytial Virus Utilizes a tRNA Fragment to Suppress Antiviral Responses Through a Novel Targeting Mechanism. Mol Ther. 2015 Oct;23(10):1622–9.
- Wang Q, Lee I, Ren J, Ajay SS, Lee YS, Bao X. Identification and functional characterization of tRNA-derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol Ther. 2013 Feb;21(2):368–79.
- Wu W, Lee I, Spratt H, Fang X, Bao X. tRNA-Derived Fragments in Alzheimer’s Disease: Implications for New Disease Biomarkers and Neuropathological Mechanisms. J Alzheimers Dis. 2021;79(2):793–806.
- Wu W, Choi EJ, Wang B, Zhang K, Adam A, Huang G, et al. Changes of Small Non-coding RNAs by Severe Acute Respiratory Syndrome Coronavirus 2 Infection. Frontiers in Molecular Biosciences [Internet]. 2022 [cited 2024 Jan 4];9. Available from: https://www.frontiersin.org/articles/10.3389/fmolb.2022.821137.
- Zhou J, Liu S, Chen Y, Fu Y, Silver AJ, Hill MS, et al. Identification of two novel functional tRNA-derived fragments induced in response to respiratory syncytial virus infection. J Gen Virol. 2017 Jul;98(7):1600–10.
- Wu W, Choi EJ, Lee I, Lee YS, Bao X. Non-Coding RNAs and Their Role in Respiratory Syncytial Virus (RSV) and Human Metapneumovirus (hMPV) Infections. Viruses. 2020 Mar 21;12(3):345.
- Rotondo C, Corrado A, Colia R, Maruotti N, Sciacca S, Lops L, et al. Possible role of higher serum level of myoglobin as predictor of worse prognosis in Sars-Cov 2 hospitalized patients. A monocentric retrospective study. Postgrad Med. 2021 Aug;133(6):688–93.
- Liu JL, Fan YG, Yang ZS, Wang ZY, Guo C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front Neurosci. 2018 Sep 10;12:632.
- Avitan I, Halperin Y, Saha T, Bloch N, Atrahimovich D, Polis B, et al. Towards a Consensus on Alzheimer’s Disease Comorbidity? J Clin Med. 2021 Sep 24;10(19):4360.
Figure 1.
Virus Examples Implicated in AD. The specific viruses HSV-1, ZIKV, and SARS-CoV-2 are discussed here. Careful examination of the possible biomarkers to consider, along with cells or regions of the brain affected are highlighted. Figure created in BioRender.com.
Figure 1.
Virus Examples Implicated in AD. The specific viruses HSV-1, ZIKV, and SARS-CoV-2 are discussed here. Careful examination of the possible biomarkers to consider, along with cells or regions of the brain affected are highlighted. Figure created in BioRender.com.
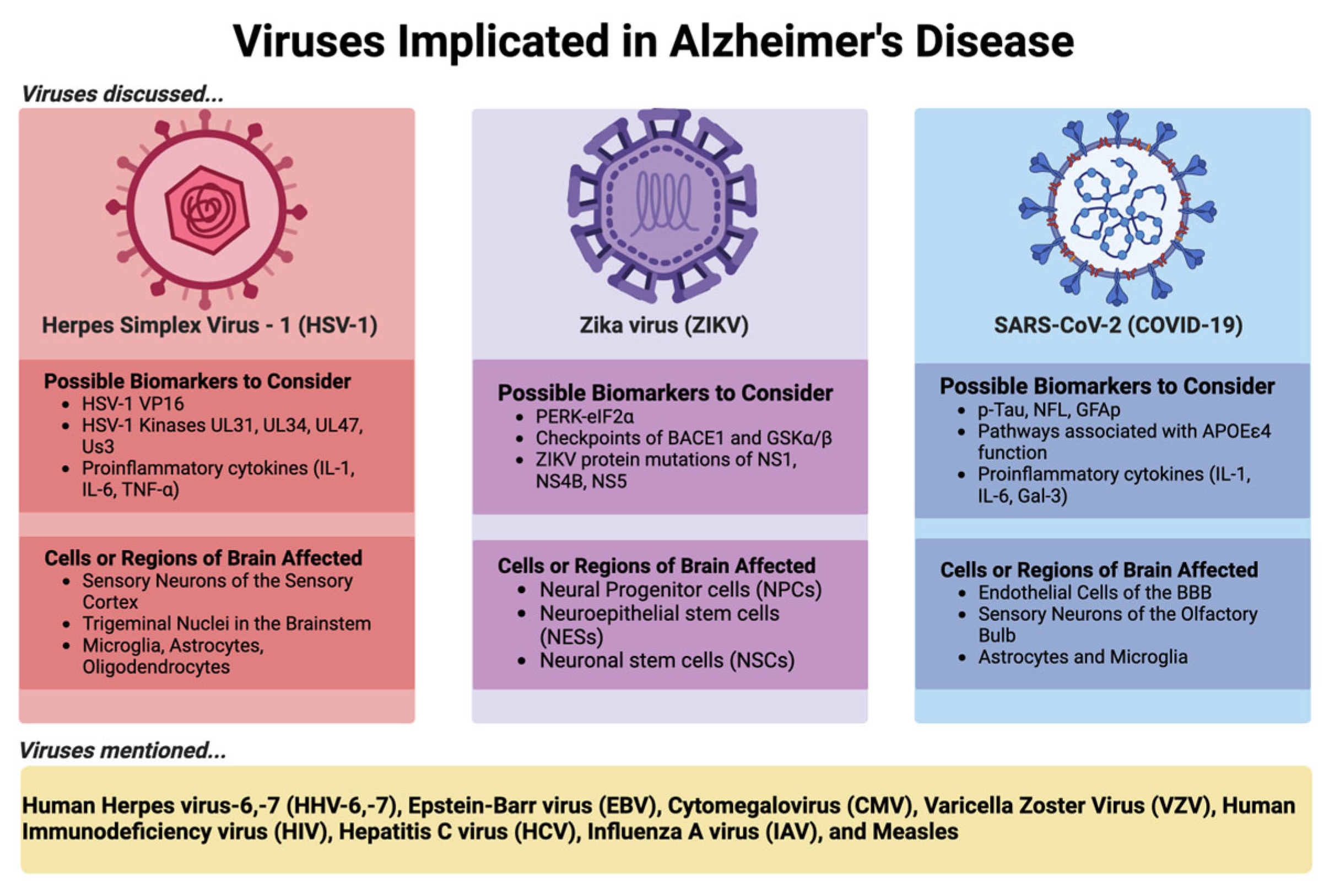
Figure 2.
A Summary of the Potential Mechanisms on Viral Contribution to AD. As elaborated on in the main text, the six potential mechanisms of the viral-AD hypothesis are illustrated. Figure created in BioRender.com.
Figure 2.
A Summary of the Potential Mechanisms on Viral Contribution to AD. As elaborated on in the main text, the six potential mechanisms of the viral-AD hypothesis are illustrated. Figure created in BioRender.com.
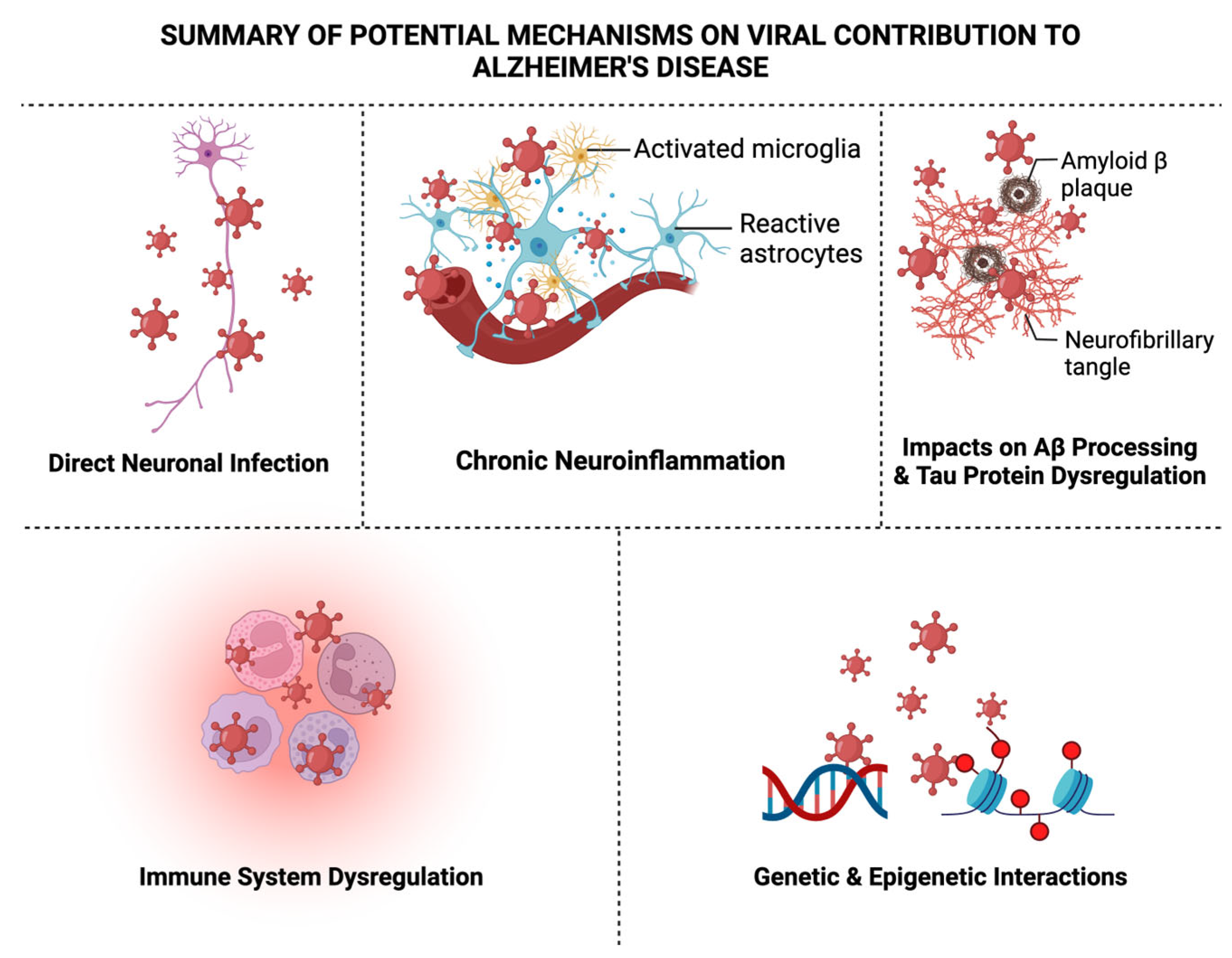
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



