
Submitted:
02 February 2024
Posted:
05 February 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
To better understand how immunity develops against SARS-CoV-2 during the pandemic period of 2020 to 2022, we analyzed the immune response of a small group of university staff and students who were either infected or vaccinated. We investigated the levels of receptor-binding domain (RBD)-specific and nucleocapsid (N)-specific IgG and IgA antibodies in serum and saliva samples taken early (around 10 days after infection or vaccination) and later (around 1 month later), as well as N-specific T-cell responses. One patient who had been infected in 2020 developed serum RBD and N-specific IgG antibodies, but declined 8 months later, then mRNA vaccination in 2021 produced a higher level of anti-RBD IgG than natural infection. In the vaccination of naïve individuals, vaccines induced anti-RBD IgG, but it declined after 6 months. A third vaccination boosted the IgG level again, albeit to a lower level than after the second. In 2022, when the Omicron variant became dominant, familial transmission occurred even among vaccinated people. In infected individuals, the levels of serum anti-RBD IgG antibodies increased later, while anti-N IgG peaked earlier. The N-specific activated T cells expressing IFN- or CD107a were detected early. In saliva, SARS-CoV-2-specific IgG but not IgA was detected. Interestingly, two individuals showed a temporary peak in RBD- and N-specific IgA antibodies in their saliva on the second day after infection. Thus, SARS-CoV-2 infection triggers typical immune responses in humans against acute viral infections, but mucosal immune responses, which are temporarily induced early during infection, may be more important for protection than current intramuscular vaccines.
Keywords:
SARS-CoV-2
; COVID-19
; infection and vaccination
; serum and saliva
; RBD and N-specific
; IgG and IgA
; T-cell responses
Introduction
Since the outbreak of the first novel coronavirus caused severe acute respiratory syndrome (SARS-CoV) in Guandong, China, in November 2002 [1], another novel coronavirus emerged in Wuhan, China, in December 2019 [2,3] and rapidly caused the global pandemic. The virus, officially designated as SARS-CoV-2, is an enveloped single-stranded RNA virus belonging to a b-coronavirus family [4]. The SARS-CoV-2 infection occurred directly in the lung tissue through an angiotensin-converting enzyme (ACE)-II as a primary receptor [5], thus rapidly developing severe pneumonia. The disease caused by SARS-CoV-2 is called COVID-19.
The SARS-CoV-2 accumulated mutations continuously during human-to-human transmission and in chronic infections [6]. The WHO worked with the reported genetic mutation of the virus and assigned simple labels for key variants as variants of interest (VOIs) and variants of concern (VOC) in May 2021 (https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/). From the original virus defined as B.1, Alpha (B.1.1 lineage) and Beta (B.1.35 lineage) variants were diverged, followed by the Delta (B.1.617 lineage) variant in October 2020 in India. At the end of 2021, the Omicron (B.1.1.529 lineage) variant emerged in South Africa and became a significant variant worldwide. Although Omicron continues to expand as various sub-lineages, they have changed to preferably infect the upper respiratory tract (versus lower respiratory tract), as compared to pre-Omicron VOCs (https://www.who.int/news/item/16-03-2023-statement-on-the-update-of-who-s-working-definitions-and-tracking-system-for-sars-cov-2-variants-of-concern-and-variants-of-interest) resulting in the attenuated phenotype. Currently, we need to be concerned about the virus spreading mainly among individuals with comorbidity and older people.
The rapid development of novel vaccine technology has helped us achieve herd immunity against SARS-CoV-2 infection in the general population. The COVID-19 vaccine, which started in late 2020, substantially altered the course of the pandemic [7]. In Japan, mRNA-based vaccines such as BNT162b2 (Pfizer/BioNTech) and mRNA-1273 (Moderna), as well as a defective adenovirus-based vaccine called ChAdOx1-S (Oxford), were introduced in The government first provided the vaccination program to medical workers and eligibility for free vaccines has since been extended to all age groups to achieve herd immunity in Japan (https://www.niid.go.jp/niid/ja/diseases/ka/corona-virus/2019-ncov/2484-idsc/10569-covid19-53.html#). However, decay of vaccine-induced neutralizing antibody response and the increase of SARS-CoV-2 re-infection were of great concern [8]. In this context, based on the experimental coronavirus infection study [9], the re-infection of human common-cold coronaviruses has been known to occur. The COVID-19 Forecasting Team recently showed that past-infection-induced protection against re-infection from pre-omicron variants was very high [10]. However, the protection was substantially lower and shorter for the Omicron BA.1 variant [10]. Of note, the natural infection appears to link to a lower incidence of SARS-CoV-2 infection than mRNA primary series vaccination [11]. Despite nationwide vaccinations, we have encountered eight epidemic peaks in Japan at the end of 2023 (https://www.niid.go.jp/niid/ja/basic-science/epidemi/12252-epi-2023-02.html). Thus, these findings indicate that re-infection frequently occurs after vaccinations and even after natural infections, probably associated with continuous virus mutations. While the COVID-19 vaccine cannot prevent infection, it can reduce the severity of the disease [7,8,10,12].
During the COVID-19 pandemic in early 2020, the government restricted all social activity in Japan: offices and schools were closed, and people could not go outside their homes. This strategy was successful for an initial epidemic wave for some time in Japan, but the second wave soon started in the summer of When vaccination began in 2021, we monitored the immune status of university students to understand the vaccine's effectiveness. The students in our university were relatively free from COVID-19 during several waves of epidemics in 2020 and 2021, probably by avoiding close contact among students and staff. On the other hand, when an omicron strain became dominant, we had a substantial number of students infected with SARS-CoV-2, mostly from family members. However, significant gaps in our understanding remain regarding the differential dynamics of both IgG and IgA antibodies in blood and saliva following SARS-CoV-2 infection and vaccination, highlighting a critical area of research yet to be thoroughly explored.
To gain a deeper understanding of how immunity develops against SARS-CoV-2 during the pandemic period of 2020 to 2022, we analyzed humoral and cellular immune responses in vaccinated and infected individuals, including close contact individuals who lived with infected family members and remained PCR or antigen-test-negative. We previously showed that the saliva IgA was induced early after influenza virus infection and could help evaluate mucosal immune responses [13]. Therefore, we also analyzed saliva IgA antibody responses in SARS-CoV-2 infection, including close contact with the infected.
Materials and Methods
2.Subject and sample collection
In 2020, one family member of a staff working in a hospital was infected with SARS-CoV-2, and samples were collected periodically. In 2021, university staff members recruited volunteers, including their families and students, before vaccination. We collected blood and saliva samples before (pre) and two weeks after (1st, second, and third post-vaccination) from twenty-two vaccinated donors (mostly primed from June to August 2021), as shown in Table We obtained some peripheral blood mononuclear cells (PBMCs) as described previously [13].
Since February 2022, SARS-CoV-2 infection has spread among young students who received vaccination in 2021, the information of which is shown in Table The samples from infected donors (17 donors) were collected early (10~14 days) and late (~1 month) post-infection. We collected the samples from individuals who were in close contact with infected families (PCR+) but test-negative, asymptomatic (10 donors) at two similar time points (early and late). Their information is shown in Table
Additionally, a member of one of the authors’ family had a mild fever and sore throat, which was followed by her sister, who developed a high fever. Their infection was confirmed through an antigen test. In this specific case, we were able to collect saliva samples in the early days after the infection.
For saliva sample collection, SalivaBio (Salimetris, Carlsbad, CA, USA) was used and centrifuged at 1,710g for 15 min, and the supernatants were transferred to new tubes as previously described [13]. All these samples were collected with written informed consent and kept in a −80˚C freezer until use. In a previous study, we stocked blood and saliva samples from volunteers in our division until before the influenza season 2019 [13]. We utilized some of them as pre-COVID-19 samples. The study followed the Helsinki Declaration and was approved by the ethical committee of the Tokyo University of Technology (No. E18HS-023).
2.Antigens
The nucleocapsid (N) is relatively conserved among human -coronaviruses in terms of amino acid sequences and structures [14]. We used frozen stock of SARS-CoV-1 N protein, which was produced in 2007 as described before using the pET-SUMO system [15], because the N protein of SARS-CoV-2 was shown to have approximately 90% homology with SARS-CoV-1 [16]. Wen et al. recently showed that COVID-19 patient-derived monoclonal antibodies were cross-reactive to the N protein of SARS-CoV-1 but little to other human b-coronaviruses [17]. The Receptor binding domain (RBD) derived from SARS-CoV-2 Wuhan-1 spike antigen was obtained commercially (Sino Biological, cat. 40592-VNAH). We used these antigens for the ELISA test.
For the in vitro stimulation of PBMC, a codon-humanized SARS-CoV-2 N DNA with Strep-tagII peptide coding sequence (WSHPQFEK) at 3' end was synthesized and cloned into an expression plasmid produced by GenScript Japan Co.Ltd. (Tokyo, Japan). The Expi293TM Expression System Kit (Thermo Fisher Scientific, Waltham, MA, USA) made recombinant N protein under the company's instruction. Both cell supernatant and lysates were combined, and we purified SARS-CoV-2 N using Strep-Tactin® Sepharose® (IBA Lifesciences GmbH, Goettingen, Germany) using the protocol provided by the company.
2.Standard antibodies
We obtained the RBD-specific IgG S309 (PMID: 32422645) by transfecting Expi293 cells with plasmids expressing S309 IgG heavy chain and light chain using the 293fectin transfection reagent (Thermo Fisher Scientific). On day 5, after transfection, the cell supernatant was clarified by centrifugation and filtered. The S309 antibody was purified from the supernatant by HiTrap rProtein A FF Column (Cytiva, Tokyo, Japan) using the AKTA go (Cytiva). We also utilized these heavy and light chain genes to produce RBD-specific IgA expression plasmid as described previously [13]. We had the recombinant anti-RBD-IgA antibody by transfecting this plasmid as the IgG antibody described above. The culture supernatant was purified using Peptide M/agarose (InvivoGen Inc., San Diego, CA, USA). These RBD-specific IgG and IgA antibodies were served as a standard to measure RBD-specific antibodies. IgG and IgA antibodies were purified from the patient's serum in Figure 1 at one-month post-infection and used to assess the level of N-specific IgG and IgA antibodies as standard.
2.ELISA
The amount of RBD or N-specific IgA and IgG in each sample was measured using the ELISA system described before [13]. In brief, a Nunc 96-well microtiter plate (Thermo Fisher Scientific) was coated with RBD-Spike or N protein at 1 μg/mL in PBS and kept at 4˚C overnight. The plate was washed and blocked with PBS/0.5% BSA at RT for 1.5 hrs, and the samples were added to the plate. Then, the plate was incubated for 1 hr with a biotinylated anti-human IgG or IgA antibody (Southern Bio-Tech, Birmingham, UK), followed by HRP–streptavidin (1:2000 dilution with PBST, BioLegend) for 30 min. Finally, the TMB substrate (Sigma-Aldrich, St. Louis, MO, USA) was added, and the color development was measured at OD450 using a microplate reader iMARKTM (BioRad, Hercules, CA, USA).
The amount of total IgA in saliva was measured as described previously [13]. As a standard, purified myeloma IgA1 protein (Sigma-Aldrich) was used. Note that the amount of total IgA was determined for saliva samples, and antigen-specific IgA titer was normalized with total IgA as previously described [13].
2.In vitro stimulation and flow cytometry
Frozen PBMCs were thawed, and 1 million cells were distributed to wells of 96-flat-bottom culture plate (Corning Inc., Corning, NY, USA) in 0.1 ml of RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 1/100 volume of Glutamax and Penicillin/Streptomycin (Thermo Fisher Scientific). The medium only or the medium containing 10 µg/ml of purified SARS-N antigen protein was added to make a final volume of 0.2 ml/well. A well containing SEB (Streptococcus enterotoxin B: Sigma-Aldrich) antigen at 1 µg/ml was prepared for each donor sample as a positive control. The plate was placed in a CO2 incubator (5% CO2) at 37ºC overnight. Sixteen hours later, brefeldin A (BFA) and monensin (both from Sigma-Aldrich) were added at a final concentration of 5 µg/mL and two µM, respectively. Simultaneously, 1 µl of PE-conjugated anti-CD107a antibody (BioLegend, San Diego, CA, USA) was added to each well, and the plate was incubated further for 5 hrs.
Cells were collected into 5 ml polystyrene round tubes (Falcon®352008, Corning), washed once with PBS, reacted with LIVE/DEAD™ Fixable Aqua Dead Cell Stain (Thermo Fisher Scientific) for 15 min at room temperature (RT), and then, incubated for 20 min on ice with the following antibody mixture: FITC-conjugated anti-human CD69, PerCP-Cy5.5-conjugated anti-human CD8, PE-Cy7-conjugated anti-human CD45RA, APC-Cy7-conjugated anti-human CD27, BV421-conjugated anti-human CD3 and FcReceptor blocker. For intracellular staining, cells were washed twice with PBS and fixed by 100 µl/sample of fixation buffer supplied in eBioscience™ Intracellular Fixation & Permeabilization kit (Thermo Fisher Scientific) for 20 min on ice. These cells were washed twice with 500 µl/sample of permeabilization buffer in the kit and incubated with APC-conjugated anti-human IFN-γ. APC-anti-mouse IgG1 was used as an isotype control. After 20 min incubation on ice, cells were washed with PBS/2%FBS/0.05%NaN3 (SB) and resuspended in SB for flow cytometer analysis. All these mouse monoclonal antibodies were purchased from BioLegend.
Finally, stained cells were acquired using FACSVerse™ Cell Analyzer (Becton Dickinson, and Co.(BD), Franklin Lakes, NJ, USA) and re-analyzed by a Flowjo ver 10.8.1 (BD). After removing doublets, live and CD3-positive T cells were gated for analysis. Then, the CD8+ and CD8- T cells were separately analyzed.
2.Statistical analysis
The amount of SARS-CoV-2 specific IgA and IgG antibodies was calculated based on standard IgG and IgA antibody titers using Microplate Manager 6 (BioRad). Statistical analysis was performed using GraphPad Prism version 10.1.The differences between groups were analyzed using the Non-parametric T-test of the Wilcoxon matched-pairs signed rank test. A P-value <0.05* or <0.01** was considered statistically significant.
Results
3.Immune responses to SARS-CoV-2 infection at early phase of pandemic, 2020
Compared to European countries, our university had minor infections in However, we had an opportunity to analyze the serum samples of one medical doctor infected with SARS-CoV-2 in the hospital in August He had a slight fever fever for only half a day, while other symptoms were mild. Serum samples were collected consecutively at day 14, followed by 1, 4, and 8 months post-infection. The serum taken in October 2019 for the influenza vaccine study was used as a pre-infection serum. We measured anti-RBD and anti-N IgG antibodies using an in-house ELISA system. As shown in Figure 1a, both the anti-RBD and anti-N IgG levels increased after infection and peaked one-month post-infection (p.i.). The levels of these IgG gradually declined but remained high at eight months p.i. At eight months p.i., he received the first COVID-19 mRNA vaccine, followed by a booster three weeks later. As expected, the level of anti-RBD IgG was augmented at nine months p.i. and slightly increased by the booster vaccination at ten months p.i. On the other hand, the level of anti-N decreased to the almost basal level at nine months p.i.
We conducted an analysis of T-cell responses to SARS-CoV-2 infection. We stimulated PBMCs were stimulated with a purified recombinant SARS-CoV-2 N protein in vitro. After 18 hrs of stimulation, we studied IFN-γ and CD107a production in activated (CD69+) T cells by flow cytometry. The representative results of flow cytometry, taken one month after infection are shown in Figure 1b. The data at four-time points (pre, 1, 4, and 8 months) are depicted in Figure 1c. We observed that the percentage of N-specific CD8+ T cells producing IFN-γ+ and CD107a+ cells was high at 1-month p.i. Although the rates of IFN-γ+ and CD107a+ cells are high even without antigen, the net increase of percentages is still at a peak at one month after infection. The results suggest that cytotoxic T cells are active at an early stage of infection. Based on these results, we suggest that cytotoxic T cells become active at an early stage of the disease. Therefore, it may be necessary to analyze T-cell responses as early as possible.
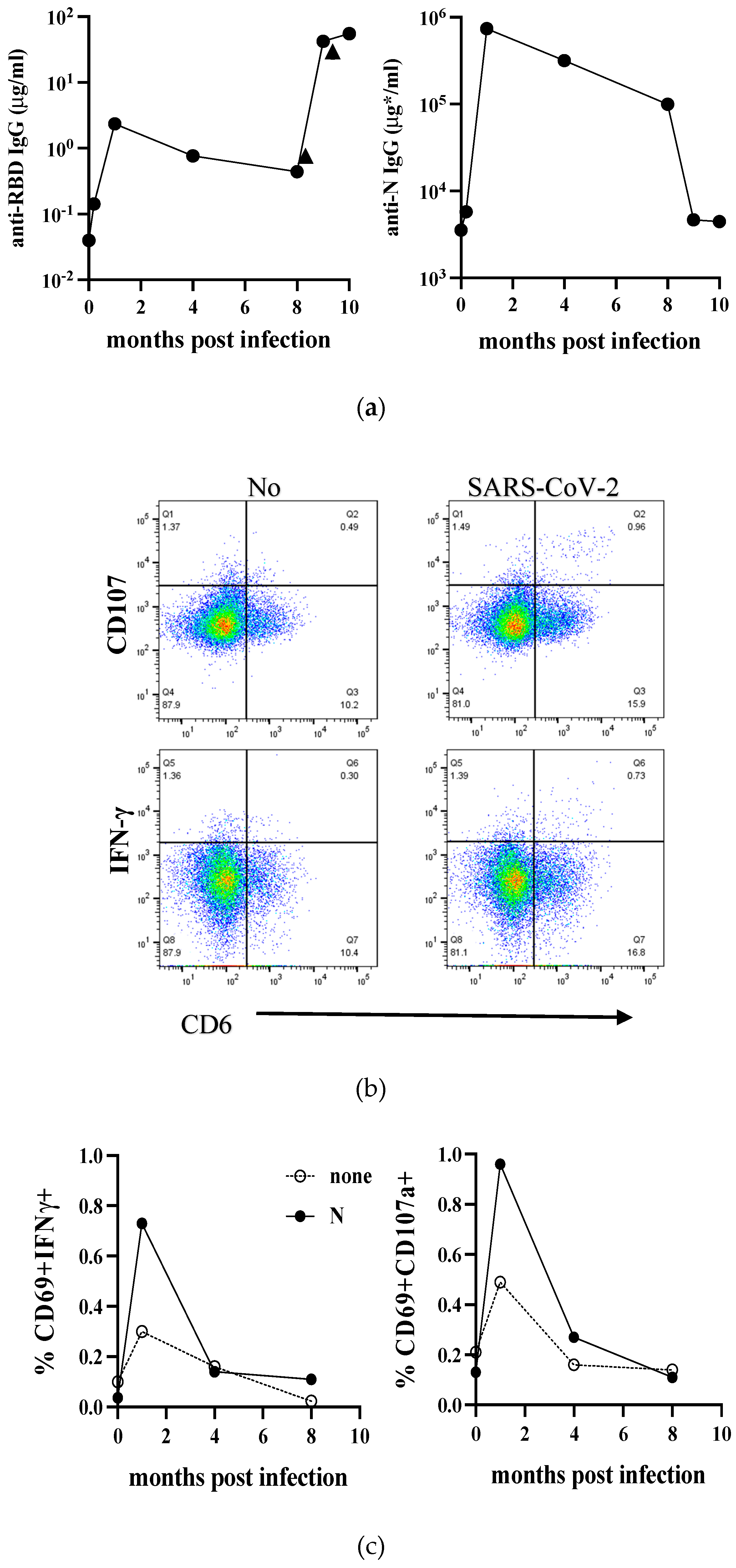
Figure 1.
Time course of Immune responses to SARS-CoV-2 in infected individuals during the early phase of the pandemic. (a) The levels of anti-RBD and anti-N IgG before and after SARS-CoV-2 infection are depicted in the left and right panels, respectively. The vertical and horizontal axes represent the amount of IgG and the time since infection. In the anti-N IgG panel, the amount of IgG was determined based on the amount of IgG purified from this individual’s serum at peak level (μg*). Arrowheads indicate two COVID-19 mRNA vaccinations. (b) PBMCs were either unstimulated (none) or stimulated with N protein (N) overnight and analyzed by flow cytometry. Live CD3+CD8+ T cell population was gated, and the frequencies of CD69+IFN-γ+ (left) or CD69+CD107a+ cells were depicted. The representative flow cytometer results one month after infection were shown. (c) Frequencies of CD69+IFN-γ+ (left) or CD69+CD107a+ cells at all time points were plotted.
Figure 1.
Time course of Immune responses to SARS-CoV-2 in infected individuals during the early phase of the pandemic. (a) The levels of anti-RBD and anti-N IgG before and after SARS-CoV-2 infection are depicted in the left and right panels, respectively. The vertical and horizontal axes represent the amount of IgG and the time since infection. In the anti-N IgG panel, the amount of IgG was determined based on the amount of IgG purified from this individual’s serum at peak level (μg*). Arrowheads indicate two COVID-19 mRNA vaccinations. (b) PBMCs were either unstimulated (none) or stimulated with N protein (N) overnight and analyzed by flow cytometry. Live CD3+CD8+ T cell population was gated, and the frequencies of CD69+IFN-γ+ (left) or CD69+CD107a+ cells were depicted. The representative flow cytometer results one month after infection were shown. (c) Frequencies of CD69+IFN-γ+ (left) or CD69+CD107a+ cells at all time points were plotted.
3.The vaccine-induced anti-SARS-CoV-2 RBD-specific antibodies in 2021
When the country-wide vaccination started in early 2021, we called for volunteers who will receive COVID-19 vaccines. The serum samples at five-time points, before immunization (pre), ten days after priming (V1), 10-14 days after the second booster (V2), six months after the second (6M post-V2), and 10-14 days after 3rd immunization (V3), were obtained from 22 staff or students in our medical technology department. The sample information is shown in Table The anti-RBD IgG and IgA levels are summarized in Fig.2a and 2b, respectively. After the first priming (V1), anti-RBD IgG slightly increased, and the level was augmented after the second booster (V2). At six months post-V2 vaccination, the level of anti-RBD IgG decreased to a basal level. Although the IgG level re-increased after the third vaccination (V3), the level was significantly lower than that of V2 (P<0.05). As reported by Keshavarz et al. [18], the difference in the induced levels of anti-RBD IgG between vaccine BNT162b2 (Pfizer) and mRNA-1273 (Moderna) was not observed after the second vaccination (V2).
This trend was also observed in the level of anti-RBD IgA. However, because the level of anti-RBD IgA was 100-fold lower than that of anti-RBD IgG, the difference between V2 and V3 was insignificant. Note that these samples for the vaccination study were collected in 2021 before the delta lineage virus became a significant lineage in Japan (https://www.niid.go.jp/niid/ja/basic-science/epidemi/12252-epi-2023-02.html) and only a few students were infected in our university.
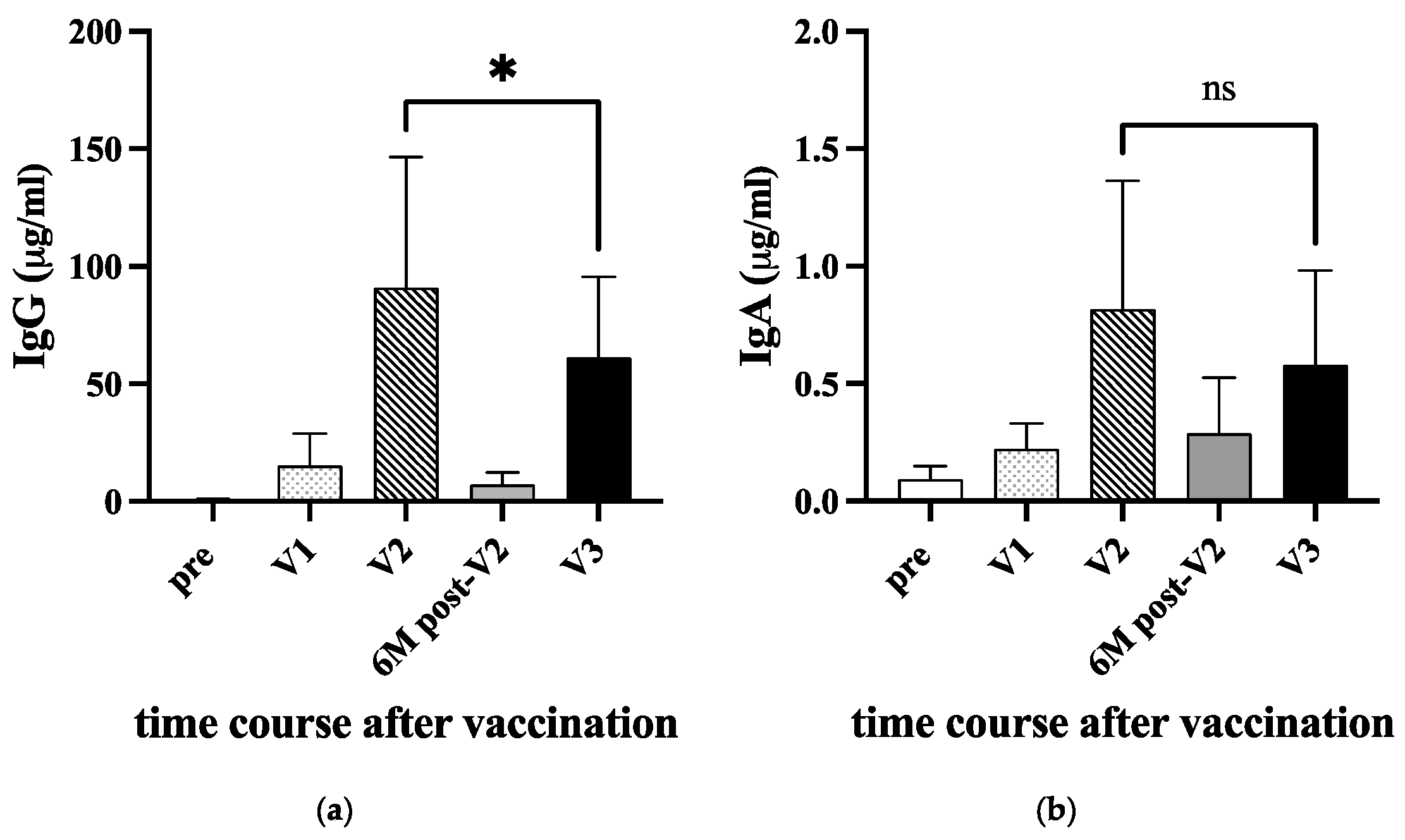
Figure 2.
Anti-RBD IgG and IgA antibodies in sera after vaccinations. Twenty-two donors were immunized either with BNT162b2 or mRNA-1273 COVID-19 vaccine in 2021 (see Table 1). The sera were collected before immunization (pre), ten days after priming (V1), 10-14 days after the second booster (V2), six months after the second (6M post-V2), and 10-14 days after 3rd immunization (V3). The amount of anti-RBD IgG (a) and IgA (b) were measured by ELISA. A P-value (* <0.05) and standard deviation (bars) are shown. ns: not significant.
Figure 2.
Anti-RBD IgG and IgA antibodies in sera after vaccinations. Twenty-two donors were immunized either with BNT162b2 or mRNA-1273 COVID-19 vaccine in 2021 (see Table 1). The sera were collected before immunization (pre), ten days after priming (V1), 10-14 days after the second booster (V2), six months after the second (6M post-V2), and 10-14 days after 3rd immunization (V3). The amount of anti-RBD IgG (a) and IgA (b) were measured by ELISA. A P-value (* <0.05) and standard deviation (bars) are shown. ns: not significant.
3.Serum Antibody responses to SARS-CoV-2 in infected and close contact individuals
By accumulating mutations of SARS-CoV-2, a highly transmissible Omicron VOC emerged in November 2021, became dominant in 2022, and permitted escape from infection- or vaccine-induced immunity [19]. On the other hand, attenuated replication and pathogenicity of omicron have been noted [20]. Since early 2022, we had many COVID-19 cases among students. We collected serum and saliva samples from 17 of them. They were all vaccinated in 2021 at least twice. Case C22-3 was unique because he was initially infected in the middle of 2021, before the vaccination started, and re-infected in April 2022 after immunization. The information on their infection/vaccination status is summarized in Table We took samples early, within 14 days after infection (day 0 was defined based on the fever followed by PCR positivity), and at late, 1-month p.i. We also used serum samples stocked before the end of 2019 as pre-COVID-19 samples. In-house ELISA measured the levels of anti-RBD and anti-N IgG in the serum.
Serum anti-RBD and anti-N IgG were upregulated after infection. Anti-RBD IgG increased early, and the level was further increased later (Figure 3a, left). Because donors are all vaccinated, a higher level of anti-RBD IgG early may not necessarily reflect the infection. However, the further increase of IgG levels later indicated that the anti-RBD IgG response was augmented by infection. In contrast, serum anti-N IgG increased only early by infection, and a further increase was not observed later (Figure 3a, right).
While Omicron was more transmissible than Delta, it exhibited reduced disease severity in the period it co-existed with Delta [6]. This results in many asymptomatic infections, and new infections often occur at home among family members. We collected ten samples of individuals with family members who had PCR-positive COVID-19 cases but remained uninfected. These close contacts were asymptomatic and coronavirus test negative. The information of these donors is shown in Table 3, and the levels of serum anti-RBD and anti-N IgG are depicted in Figure 3b. A significant variation in the level of anti-RBD IgG was observed (Figure 3a, left), and only a late increase of anti-RBD IgG was statistically significant (Figure 3b, left). However, anti-N IgG did not increase (Figure 3b, right). Because they were all vaccinated in 2021, we assumed their anti-RBD antibody response was boosted due to close contact with the infected. Asymptomatic contact individuals may be exposed to a limited amount of the virus transiently, but their immune systems are competent enough to eliminate it.
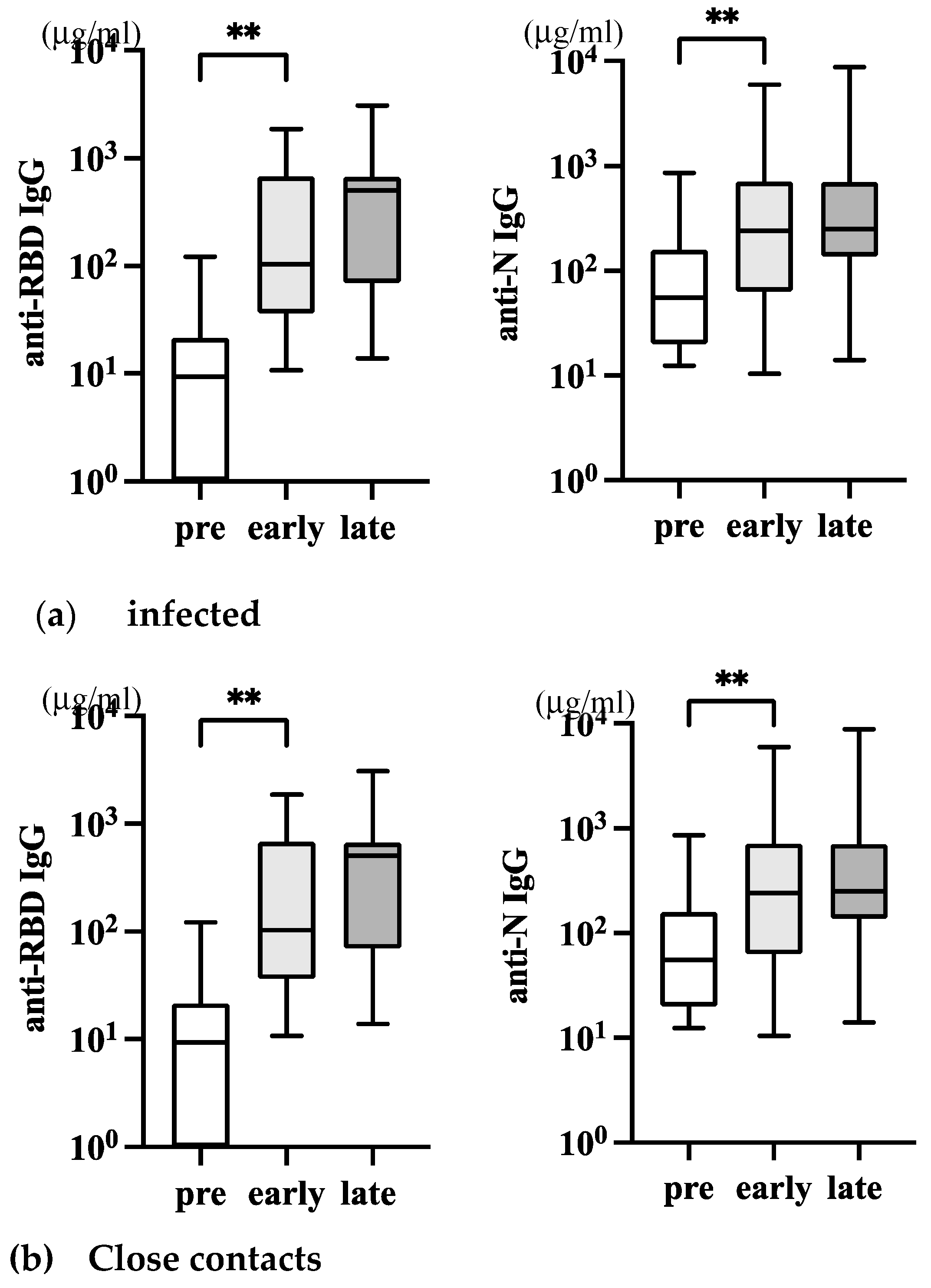
Figure 3.
Serum anti-SARS-CoV-2 IgG antibodies in infected and close contacts. (a) The level of serum IgG in infected individuals (n=17, Table 2) was depicted. The left panel is anti-RBD IgG, and the right panel is anti-N IgG. (b) The level of serum IgG in asymptomatic close contacts with infected individuals (n=10, Table 3) was depicted. The P-values (* <0.05 and **<0.01), mean (horizontal), and standard deviation (vertical) bars are depicted.
Figure 3.
Serum anti-SARS-CoV-2 IgG antibodies in infected and close contacts. (a) The level of serum IgG in infected individuals (n=17, Table 2) was depicted. The left panel is anti-RBD IgG, and the right panel is anti-N IgG. (b) The level of serum IgG in asymptomatic close contacts with infected individuals (n=10, Table 3) was depicted. The P-values (* <0.05 and **<0.01), mean (horizontal), and standard deviation (vertical) bars are depicted.

Table 2.
Characteristics of Infected individuals.
| ID | Infection in 2022 | Vaccination (times) |
Month after last vaccination | T-cell analysis (PBMCs) |
|---|---|---|---|---|
| CV26 | Feb | 2 | 5 | late |
| CV27 | Mar | 2 | 6 | late |
| CV28 | Mar | 2 | 5 | late |
| C22-1 | Apr | 2 | 6 | early |
| C22-2 | Apr | 2 | 6.5 | NA |
| C22-3 | Apr (2nd)* | 2 | 4 | late |
| C22-8 | Jul | 2 | 6 | NA |
| C22-9 | Jul | 3 | 1 | early |
| C22-10 | Jul | 3 | 2 | early |
| C22-11 | Jul | 3 | 2.5 | early |
| C22-13 | Jul | 3 | 3.5 | NA |
| C22-14 | Aug | 3 | 1.5 | early |
| C22-15 | Aug | 3 | 5 | NA |
| C22-16 | Aug | 3 | 3 | NA |
| C22-21 | Sep | 3 | 4.5 | early |
| C22-22 | Aug | 3 | 4 | early |
| C22-23 | Aug | 3 | 3 | NA |
æ 1st infection occurred in July NA denotes data not available, early refers to approximately 14 days after infection, and late refers to approximately 3 months after infection.
Table 3.
Characteristics of asymptomatic close contacts.
| ID | PCR test | Contacts in 2022 | Vaccination (times) |
Month after last vaccination |
|---|---|---|---|---|
| CV29 | negative | Apr | 2 | 6.5 |
| C22-5 | negative | May | 2 | 7 |
| C22-6 | negative | May | 2 | 7 |
| C22-7 | negative | May | 2 | 9 |
| C22-12 | negative | Jul | 3 | 3.5 |
| C22-17 | ND: Ag test | Aug | 3 | 2.5 |
| C22-18 | ND: Ag test | Aug | 3 | 3 |
| C22-19 | ND: Ag test | Aug | 3 | 3 |
| C22-20 | ND: Ag test | Aug | 2 | 10 |
| C22-24 | ND: Ag test | Aug | 3 | 5 |
ND denotes no PCR test carried out. Instead, the Ag test was negative.
3.T-cell responses against SARS-CoV-2 in infected individuals
The T-cell responses against Spike and Nucleocapsid develop after SARS-CoV-2 infection or vaccination and are believed to be necessary for the protection [21,22,23]. We obtained PBMCs from 7 infected donors at around ten days after infection (early) and from 4 infected donors over three months after infection (late) (Table 2). We recruited four additional donors (donors in Table 1) who were infected at the end of 2022, and their blood was taken 10 to 14 days after infection. Because they are all vaccinated, we used a N antigen for in vitro stimulation. PBMCs were cultivated in the absence (no Ag) or in the presence of SARS-CoV-2 N antigen (SARS-N) overnight, then frequencies of activated (CD69+) and IFN-γ or CD107a positive CD4+ or CD8 T+ cells were analyzed by flow cytometry.
As shown in Figure 4a, the SARS-CoV-2 N specific IFN-γ responses in CD8+ (left panel) and CD4+ (right panel) T cells were variably detected, though the background level (no Ag) was high in some donors. The positivity of 11 samples collected at early infection was significantly increased in the presence of SARS-CoV-2 N antigen; p=0.0186 in CD8+, but not in CD4+ T cells (p=0.0830). Although the statistical evaluation of 4 samples collected much later after infection is not feasible, N-specific IFN-γ+ memory T cells will likely become marginal in both CD8+ and CD4+ T cells.
As shown in Figure 4b, the frequency of CD107a+ T cells also significantly increased at early infection; p=0.0273 in CD8+ and p=0.0068 in CD4+ T cells. As regards the four donors at the late phase of infection, N-specific CD107a+ CD4+ T cells with cytotoxic granules may have a trend to be maintained longer than N-specific CD107a+ CD8+ T cells (Figure 4b, compare right to left panel).
Taken together, SARS-CoV-2-infected individuals can develop substantial SARS-N-specific CD8+ and CD4+ T cells early after infection, but the longevity of SARS-N-specific memory T cells needs to be studied further.
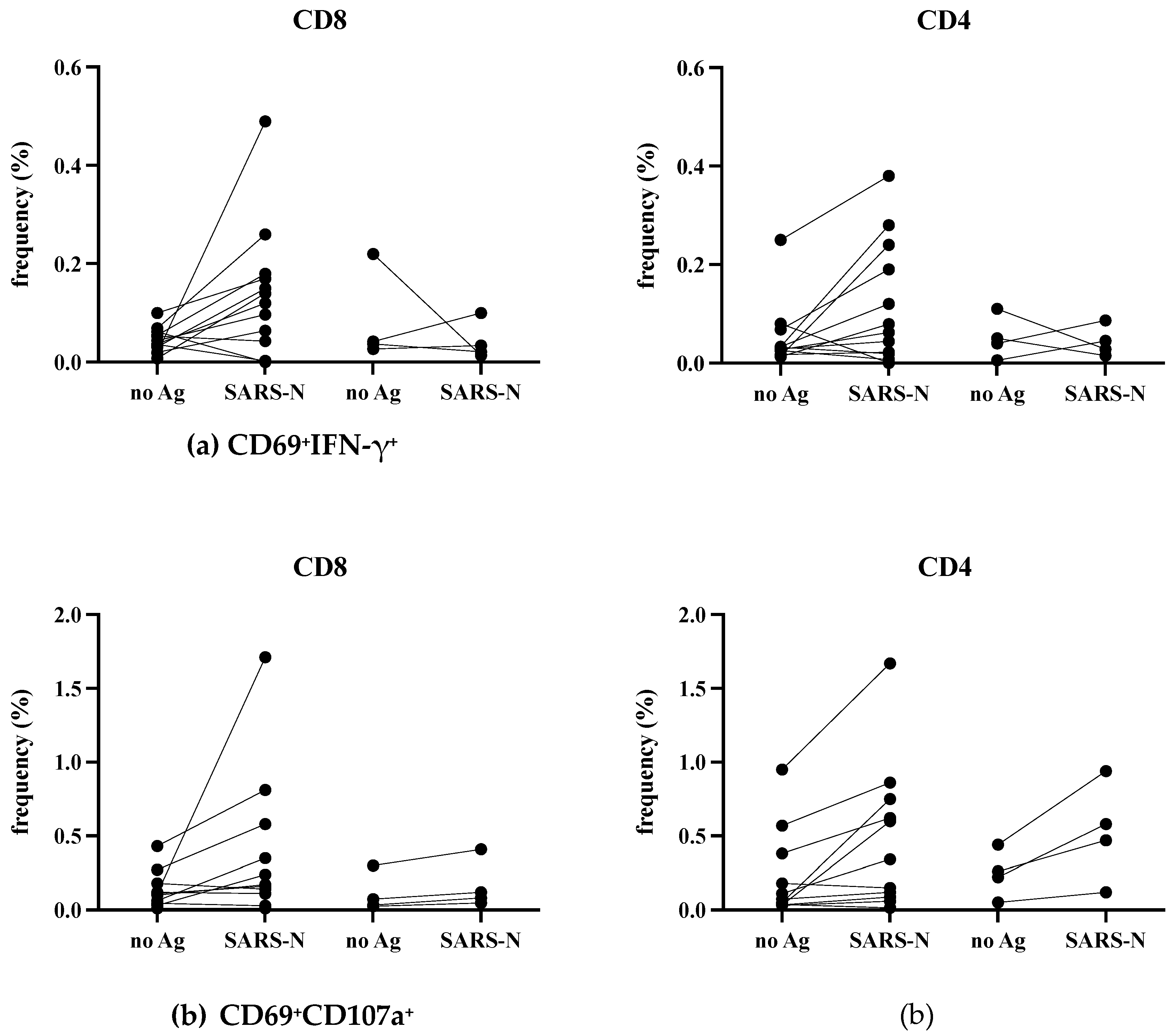
Figure 4.
N-specific T cell responses in infection. The PBMCs were either unstimulated or stimulated with N protein overnight and analyzed by flow cytometry. The frequencies of IFN-γ+ (a) and CD107a+ (b) in CD3+CD8+ (left panel) and CD3+non-CD8+ T cells (right panel) with activated phenotype (CD69+) are shown. early: 10~14 days after infection, late: more than three months after infection. The donors are indicated in Table 2.
Figure 4.
N-specific T cell responses in infection. The PBMCs were either unstimulated or stimulated with N protein overnight and analyzed by flow cytometry. The frequencies of IFN-γ+ (a) and CD107a+ (b) in CD3+CD8+ (left panel) and CD3+non-CD8+ T cells (right panel) with activated phenotype (CD69+) are shown. early: 10~14 days after infection, late: more than three months after infection. The donors are indicated in Table 2.
3.Saliva antibody responses in infected and close contact individuals
Saliva IgA represents upper mucosal immune responses. In influenza infection, virion-specific IgA antibody in saliva was mainly detected early at day 7~10 p.i. In comparison, in saliva, systemically induced IgG increased later at 3 to 4 weeks [13]. We collected saliva samples from infected and close contacts to understand the mucosal immune response in SARS-CoV-2 infection. However, salivary glands are known to be a reservoir for SARS-CoV-2 [24]. The saliva is a valuable specimen comparable to a nasopharyngeal swab sample for detecting SARS-CoV-2 in COVID-19 [25,26]. Therefore, it was impossible for ordinary individuals to collect saliva samples soon after infection for antibody analysis because of its infectivity. The initial university regulation for COVID-19 did not allow students to return to school earlier than two weeks. Thus, saliva samples were collected simultaneously with serum samples at more than 14 days p.i. as before, followed by more than one-month p.i. as late. As shown in Figure 5, no significant level of RBD nor N-specific IgA antibodies were detected in both infected (Figure 5a) and close contacts (Figure 5b).
Concerning saliva IgG antibody, a significant level of anti-RBD IgG was detectable compared to pre-COVID-19 in both infected and close contacts, but no further increase was noted later (Figure 5c, left). Interestingly, the increase of saliva anti-RBD IgG was more evident in close contacts (Figure 5d, left) than in infected (Figure 5c, left). The results suggest that the anti-RBD IgG in saliva is derived from systemically induced IgG by vaccination, which may, to some extent, contribute to the protection in these close contacts. Saliva anti-N IgG was detected in neither of these groups (Figure 5c and d, right panels).
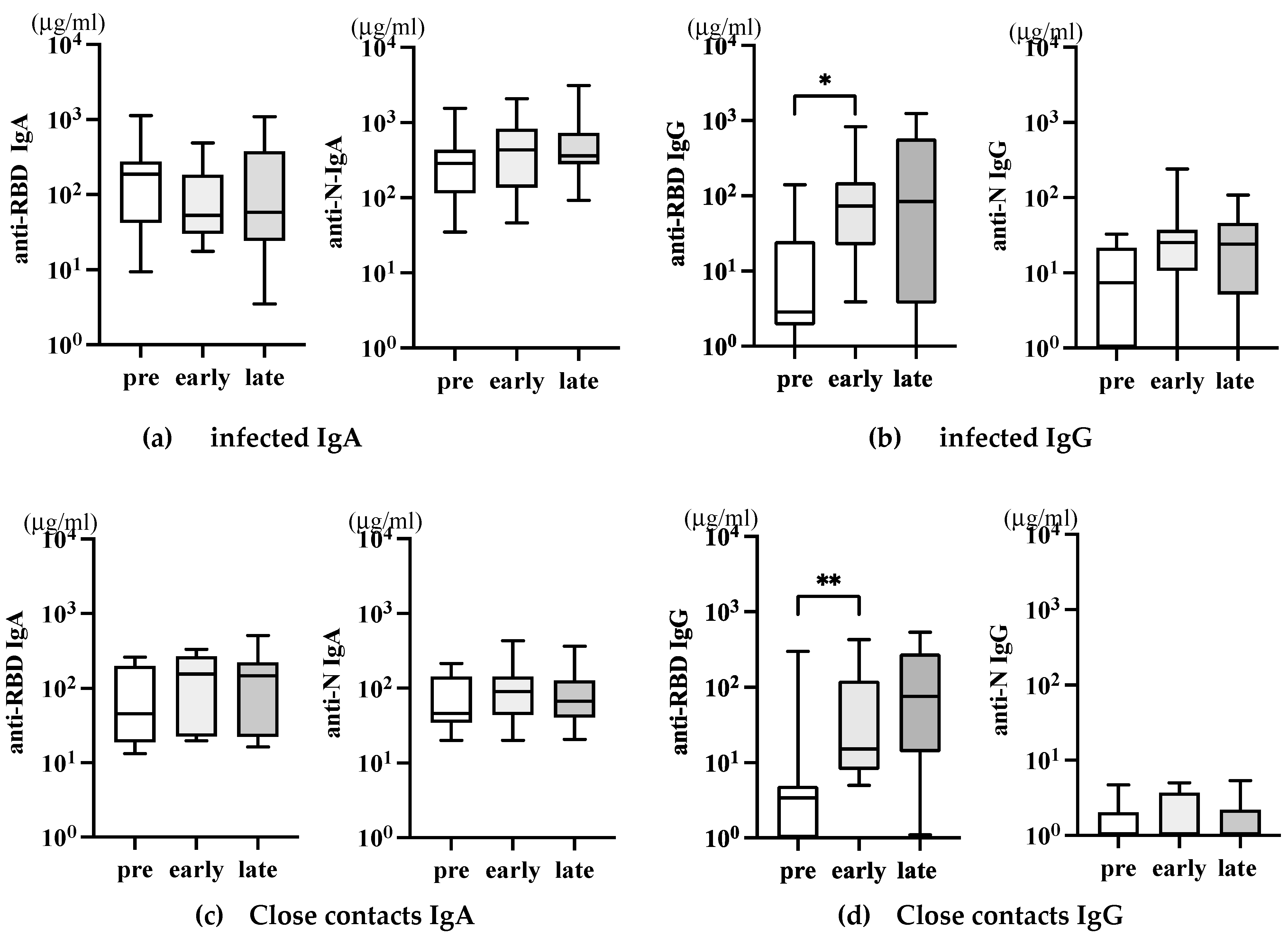
Figure 5.
Saliva anti-SARS-CoV-2 IgA antibodies after infection and close contact individuals. The amounts of anti-RBD (a~d, left panels) and anti-N (a~d, right panels), IgA (a and c), and IgG (b and d) in the saliva of the same individuals described in Figure 3 were analyzed. The P-values (* <0.05 and **<0.01), mean (horizontal), and standard deviation (vertical) bars are depicted.
Figure 5.
Saliva anti-SARS-CoV-2 IgA antibodies after infection and close contact individuals. The amounts of anti-RBD (a~d, left panels) and anti-N (a~d, right panels), IgA (a and c), and IgG (b and d) in the saliva of the same individuals described in Figure 3 were analyzed. The P-values (* <0.05 and **<0.01), mean (horizontal), and standard deviation (vertical) bars are depicted.
3.Early kinetics of saliva antibody responses in infected individuals
Thus, our collected saliva samples may not be suitable for analyzing early mucosal antibody responses in SARS-CoV-2 infection. The advantage of saliva is that saliva sampling has self-collection capabilities. When the Omicron raged in Japan, one of the authors was infected with SARS-CoV-2, and she developed a mild fever and sore throat (a). The next day, her younger sister developed a high fever (b). They were confirmed to be SARS-CoV-2 infected. Their saliva samples were collected consecutively every two days, starting from the day with fever (as day 0), 2, 5, 7, 9, and later. These saliva samples were heat-inactivated and analyzed for anti-RBD and anti-N antibodies by ELISA. The result is shown in Figure In the first case, both anti-RBD and anti-N IgA antibodies peaked on day two, and the second peak of anti-RBD IgA was observed on day 9 (Figure 6a). Also, in the second case, anti-RBD as well as anti-N IgA transiently increased on day The second peak of anti-N IgA was observed on day However, at a lower level than the first case (Figure 6b). In the second case, anti-N IgG increased on day 9, and anti-RBD IgG became high on day 14, whereas in the first case, both anti-RBD and anti-N IgG were only slightly increased one month later. Thus, mucosal IgA response was induced very early after infection, and IgG response appeared to follow much later after infection. The result suggests that we can obtain more definitive information only if self-collection of saliva on day two post-infection is feasible in all infected and close contacts.
Discussion
The outbreak of SARS-CoV-2 pandemic compelled us to change our lifestyle, but it proved to be a beneficial opportunity to study the immunological process in humans who have never encountered an emerging virus before. In this study, we chronologically analyzed immune responses to SARS-CoV-2 infection, by vaccination, and to post-vaccination infections.
The study of a single first case of COVID-19 infection in our surroundings has revealed some interesting findings. We observed a typical antibody response against acute virus infection that peaked at one month, gradually declining. Anti-N IgG returned to near the basal level at nine months. Feng et al. reported that RBD- and full-length spike-IgG decreased during the first six months but remained stable up to 1 year after hospital discharge, then declined [23]. In our case, the earlier decay of anti-N IgG could be due to the disease severity, which is very mild without any pulmonary damage. By vaccination at eight months after infection, the level of anti-RBD IgG became 1-log higher than the peak achieved by infection. Thus, vaccination can significantly enhance the antibody response elicited by SARS-CoV-2 infection. The long-term follow-up study by Koerber et al. showed that virus-neutralizing antibody titers rapidly declined in convalescents after asymptomatic or mild SARS-CoV-2 infection over nine months. Still, vaccination enhanced both antibody and cellular responses, especially against SARS-CoV-2 spike [22]. Unfortunately, because of the lack of RBD protein for in vitro stimulation, we followed only N-specific T-cell responses starting from one month after infection. We detected activated (CD69+) CD107a+CD8+ T cells only transiently at one month, not IFN-γ+ CD8+ T cells. While our research detected weak T-cell memory responses one month after infection, recent technology [21,22,23,27] can help improve our assay sensitivity and detect such responses earlier. These findings highlight the importance of vaccination as a critical strategy in the fight against COVID-19, especially for individuals who have previously been infected with the virus. These findings emphasize the importance of immunization as a vital strategy in the battle against COVID-19, especially for individuals who have previously been infected with the virus.
The level of RBD-IgG is important for protection against COVID-19, as it positively correlates with serum neutralizing capacity [28]. To assess the effectiveness of mRNA vaccines coding for the Spike gene, we analyzed the level of anti-RBD IgG and IgA in uninfected individuals' serum. Following the second booster, both IgG and IgA levels in vaccinees significantly increased. However, the IgG level gradually decreased over time, as reported earlier [22]. With the emergence of the delta variant, a third booster vaccination has been introduced [29,30,31]. In our cohort, the level of IgG increased again after the third vaccination but did not increase beyond the level seen after the second vaccination, which was also noted in a study by Kesharvarz et al. [18]. It is important to note that vaccine effectiveness increases significantly after the third vaccination [29,30,31,32]. Therefore, the difference in IgG levels between the second and third doses may have little impact. This data could be influenced by factors such as the age distribution of vaccine recipients, the timing of sampling, and the method of measurement.
The COVID-19 vaccines have shown efficacy in clinical trials and effectiveness studies. However, the level and duration of antibody response may not be high enough. A study conducted by Wei et al. indicated that the protection against infection would last for 5-8 months after two BNT162b2 doses without prior infection, compared to 1-2 years in unvaccinated individuals after natural infection [33]. Additionally, almost all symptoms were reported less frequently in infected and vaccinated individuals than in infected unvaccinated individuals. The vaccinated were also more likely to be completely asymptomatic [34]. Carazo et al. showed in their study of healthcare workers that those who had two doses of mRNA vaccine and previous Omicron infection were better protected from re-infection compared to a third vaccine dose without infection, indicating the limited benefit from additional vaccine doses for people with hybrid immunity [35]. In other words, current vaccination may not provide better immunity than natural infection.
It has been observed that cellular immune responses, specifically the spike-specific CD8+ T-cell responses that were produced by the original vaccines, are still very effective in responding to the Omicron variant [36]. This highlights the significance of T-cell responses in providing protection. During our study, we detected low but significant N-specific CD4+ and CD8+ T-cell responses two weeks after infection, but these responses were not detected 3-5 months later. Due to the small number of samples and limitations in our methodology, we are unable to determine the longevity of T-cell responses. However, T-cells triggered by vaccination or infection against SARS can help in reducing the severity of the disease, as previously reported [36].
Reinfection of SARS-CoV-2 is becoming more common even after vaccination, particularly with the emergence of the Omicron variant. This is because the vaccine-elicited neutralizing antibodies have been greatly reduced [37]. Additionally, the Omicron variant has reduced virus pathogenicity [20], making reinfection or asymptomatic infection more likely than before [6]. In our study, all infected individuals were previously vaccinated with original mRNA vaccines in 2021, and their disease was mild. It was unknown what the vaccine-induced anti-RBD IgG level was just before infection. However, the increased level of serum anti-N IgG shows the SARS-CoV-2 infection. Anti-N IgG increased early but was not augmented later, suggesting the transient nature of virus invasion. Close contacts of infected individuals did not show an increase in anti-N IgG, but their anti-RBD IgG levels were higher than pre-pandemic levels, likely due to previous mRNA vaccination. An interesting finding in the study was that close contacts who did not have detectable infection or antibodies gained T-cell immunity against SARS-CoV-2 [38]. This was determined by stimulating PBMCs in vitro with peptide pools for ten days. The authors suggested that exposure to the virus led to T-cell immunity even without a successful infection. However, more research is needed to determine if these T-cells can provide protective immunity. Alternatively, early mucosal IgA responses may have contributed to the inhibition of spreading infection in these close contacts.
We were unable to find any SARS-CoV-2-specific IgA antibodies in the saliva samples. The levels of anti-RBD IgG found in the saliva could result from the systemic IgG produced after vaccination [39]. In a previous study of influenza infection, we detected the presence of anti-flu IgA antibodies in saliva after 7 to 10 days of infection [13]. Therefore, at more than two weeks after the SARS-CoV-2 infection, the saliva sample collection appeared unsuitable for detecting early IgA response. In fact, Shan et al. reported that N protein presents early (day 1~7 p.i.) in blood and saliva in SARS-CoV-2 infection [40].
We had a valuable opportunity to obtain early saliva samples from two individuals at home. By analyzing SARS-specific antibodies in consecutive saliva samples starting from day 0, we found that SARS-CoV-2-specific IgA commonly peaked at day two, followed by a second peak around day nine p.i. These results suggest that the initial IgA-dominant mucosal response is weak and transient, whereas secondary memory responses involving systemic IgG are active after day nine p.i. Thus, if the self-collection of saliva samples is feasible, saliva will be more beneficial for detecting SARS-CoV-2 infection at mucosal sites. This may help us to understand how close contacts exposed to the virus are transiently infected but blocked the infection to a limited mucosal area by mucosal IgA or local tissue-resident T-cells, essentially conferring the protection as suggested by Wang et al. [38]. Using bronchoalveolar lavage samples, Mitsui et al. demonstrated that donors with a history of both infection and vaccination have more airway mucosal SARS-CoV-2 antibodies and memory B cells than those only vaccinated, suggesting that peripheral vaccination alone fails to induce durable lung mucosal immunity against SARS-CoV-2 [41]. Thus, as in the case of influenza vaccine [42], mucosal priming must be considered.
During the SARS-CoV-2 pandemic, we gained a lot of knowledge about how the human body develops immunity against acute virus infections. This information is crucial for us to prepare for any future emergence of unknown viruses. Nowadays, vaccines can be developed quickly once the genetic structure of a virus is determined. However, current vaccines are not efficient in blocking upper respiratory mucosal infections and they don't provide long-lasting protection. Therefore, we need to put in more efforts to develop highly effective and long-lasting vaccines. We also need better ways to predict the emergence of new pathogens.
Author Contributions
Conceptualization, YTY; software, WI, YK, and YTY.; validation, ASM and TY.; formal analysis, WI, YK, and SO.; investigation, WI and YK.; resources, WI, YK and TN.; data curation, YTY.; writing—original draft preparation, YTY.; writing—review and editing, WI, ASM, TN and TY.; visualization, YTY.; project administration, YTY, ASM and TY.; funding acquisition, ASM. All authors have read and agreed to the published version of the manuscript.
Funding
This research essentially received no funding, but one of the co-authors, ASM, is partly funded for collaboration by Competitive Research Funds for the Tokyo University of Technology.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Ethics Committee of Tokyo University of Technology (No. E21HS-004, May 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The raw data (excel sheets) of this article used for figures will be made available by the authors on request to the corresponding author.
Acknowledgments
We would like to express our gratitude to the staff and students of the Department of Medical Technology, School of Health Sciences, Tokyo University of Technology for their valuable contributions in collecting and analyzing samples in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ksiazek, T G.; Erdman, D.; Goldsmith, C.S., et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 2003; 348: 1953-1966. [CrossRef]
- Wu, F.; Zhao, S.; Yu, B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020; 579: 265-269. [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W, et al. A Novel Coronavirus from Patients with Pneumonia in China, N Engl J Med 2020; 382: 727-733. [CrossRef]
- Masters, P.; Perlman, S. Coronaviridae. In Fields virology, Knipe DM, PM, H., Eds. Lippincott Williams & Silkins: 2013; Vol. 1, pp. 825-858.
- Zhou, P.; Yang, X L.; Wang, X G., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020; 579: 270-273. [CrossRef]
- Carabelli, A M.; Peacock, T P.; Thorne, L G, et al. SARS-CoV-2 variant biology: immune escape, transmission and fitness. Nat Rev Microbiol 2023; 21: 162-177. [CrossRef]
- Watson, O J.; Barnsley, G.; Toor, J., et al. Global impact of the first year of COVID-19 vaccination: a mathematical modelling study. Lancet Infect Dis 2022; 22: 1293-1302. [CrossRef]
- Cromer, D.; Juno, J A.; Khoury, D., et al. Prospects for durable immune control of SARS-CoV-2 and prevention of reinfection. Nat Rev Immunol 2021; 21: 395-404. [CrossRef]
- Callow, K A.; Parry, H F.; Sergeant, M., et al. The time course of the immune response to experimental coronavirus infection of man. Epidemiol Infect 1990; 105: 435-446. [CrossRef]
- Team, C-F. Past SARS-CoV-2 infection protection against re-infection: a systematic review and meta-analysis. Lancet 2023; 401: 833-842. [CrossRef]
- Chemaitelly, H.; Ayoub, H H.; AlMukdad, S., et al. Protection from previous natural infection compared with mRNA vaccination against SARS-CoV-2 infection and severe COVID-19 in Qatar: a retrospective cohort study. Lancet Microbe 2022; 3: e944-e955. [CrossRef]
- Perry, J.; Osman, S.; Wright, J, et al. Does a humoral correlate of protection exist for SARS-CoV-2? A systematic review. PLoS One 2022; 17: e0266852. [CrossRef]
- Tsunetsugu-Yokota, Y.; Ito, S.; Adachi, Y, et al. Saliva as a useful tool for evaluating upper mucosal antibody response to influenza. PLoS One 2022; 17: e0263419. [CrossRef]
- Fung, T.S.; Liu, D X. Similarities and Dissimilarities of COVID-19 and Other Coronavirus Diseases. Annu Rev Microbiol 2021; 75: 19-47. [CrossRef]
- Tsunetsugu-Yokota, Y.; Ato, M.; Takahashi, Y., et al. Formalin-treated UV-inactivated SARS coronavirus vaccine retains its immunogenicity and promotes Th2-type immune responses. Jpn J Infect Dis 2007; 60: 106-112. [CrossRef]
- Oliveira, S C; de Magalhaes, M T Q.; Homan, E J. Immunoinformatic Analysis of SARS-CoV-2 Nucleocapsid Protein and Identification of COVID-19 Vaccine Targets. Front Immunol 2020; 11: 587615. [CrossRef]
- Wen, Y.; Guo, W.; Min, Y, et al. Patient-derived monoclonal antibodies to SARS-CoV-2 nucleocapsid protein N-terminal and C-terminal domains cross-react with their counterparts of SARS-CoV, but not other human betacoronaviruses. Front Immunol 2023; 14: 1093709. [CrossRef]
- Keshavarz, B.; Richards, N E.; Workman, L.J., et al. Trajectory of IgG to SARS-CoV-2 After Vaccination With BNT162b2 or mRNA-1273 in an Employee Cohort and Comparison With Natural Infection. Front Immunol 2022; 13: 850987. [CrossRef]
- Flemming, A. Omicron, the great escape artist. Nat Rev Immunol 2022; 22: 75. [CrossRef]
- Shuai, H.; Chan, J F.; Hu, B, et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022; 603: 693-699. [CrossRef]
- Rydyznski Moderbacher, C.; Ramirez, S I.; Dan, J.M., et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020; 183: 996-1012 e1019. [CrossRef]
- Koerber, N.; Priller, A.; Yazici, S., et al. Dynamics of spike-and nucleocapsid specific immunity during long-term follow-up and vaccination of SARS-CoV-2 convalescents. Nat Commun 2022; 13: 153. [CrossRef]
- Feng, C.; Shi, J.; Fan, Q, et al. Protective humoral and cellular immune responses to SARS-CoV-2 persist up to 1 year after recovery. Nat Commun 2021; 12: 4984. [CrossRef]
- Matuck, B F.; Dolhnikoff, M.; Duarte-Neto, A.N., et al. Salivary glands are a target for SARS-CoV-2: a source for saliva contamination. J Pathol 2021; 254: 239-243. [CrossRef]
- Williams, E.; Bond, K.; Zhang, B, et al. Saliva as a Noninvasive Specimen for Detection of SARS-CoV-J Clin Microbiol 2020; 58. [CrossRef]
- Yee, R.; Truong, T T.; Pannaraj, P.S., et al. Saliva Is a Promising Alternative Specimen for the Detection of SARS-CoV-2 in Children and Adults. J Clin Microbiol 2021; 59. [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I., et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020; 181: 1489-1501 e1415. [CrossRef]
- Garcia-Beltran, W F.; Lam, E C.; Astudillo, M G, et al. COVID-19-neutralizing antibodies predict disease severity and survival. Cell 2021; 184: 476-488 e411. [CrossRef]
- Bar-On, Y M.; Goldberg, Y.; Mandel, M., et al. Protection of BNT162b2 Vaccine Booster against Covid-19 in Israel. N Engl J Med 2021; 385: 1393-1400. [CrossRef]
- Barda, N.; Dagan, N.; Cohen, C, et al. Effectiveness of a third dose of the BNT162b2 mRNA COVID-19 vaccine for preventing severe outcomes in Israel: an observational study. Lancet 2021; 398: 2093-2100. [CrossRef]
- Pouwels, K B.; Pritchard, E.; Matthews, P.C., et al. Effect of Delta variant on viral burden and vaccine effectiveness against new SARS-CoV-2 infections in the UK. Nat Med 2021; 27: 2127-2135. [CrossRef]
- Xia, H.; Zou, J.; Kurhade, C, et al. Neutralization and durability of 2 or 3 doses of the BNT162b2 vaccine against Omicron SARS-CoV-Cell Host Microbe 2022; 30: 485-488 e483. [CrossRef]
- Wei, J.; Pouwels, K B.; Stoesser, N., et al. Antibody responses and correlates of protection in the general population after two doses of the ChAdOx1 or BNT162b2 vaccines. Nat Med 2022; 28: 1072-1082. [CrossRef]
- Antonelli, M.; Penfold, R S.; Merino, J., et al. Risk factors and disease profile of post-vaccination SARS-CoV-2 infection in UK users of the COVID Symptom Study app: a prospective, community-based, nested, case-control study. Lancet Infect Dis 2022; 22: 43-55. [CrossRef]
- Carazo, S.; Skowronski, D M.; Brisson, M., et al. Protection against omicron (B.1.1.529) BA.2 reinfection conferred by primary omicron BA.1 or pre-omicron SARS-CoV-2 infection among health-care workers with and without mRNA vaccination: a test-negative case-control study. Lancet Infect Dis 2023; 23: 45-55. [CrossRef]
- Liu, J.; Chandrashekar, A.; Sellers, D., et al. Vaccines elicit highly conserved cellular immunity to SARS-CoV-2 Omicron. Nature 2022; 603: 493-496. [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S., et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2022; 602: 654-656. [CrossRef]
- Wang, Z.; Yang, X.; Zhong, J, et al. Exposure to SARS-CoV-2 generates T-cell memory in the absence of a detectable viral infection. Nat Commun 2021; 12: 1724. [CrossRef]
- Hettegger, P.; Huber, J.; Passecker, K., et al. High similarity of IgG antibody profiles in blood and saliva opens opportunities for saliva based serology. PLoS One 2019; 14: e0218456. [CrossRef]
- Shan, D.; Johnson, J M.; Fernandes, S.C., et al. N-protein presents early in blood, dried blood and saliva during asymptomatic and symptomatic SARS-CoV-2 infection. Nat Commun 2021; 12: 1931. [CrossRef]
- Mitsi, E.; Diniz, M O.; Reine, J., et al. Respiratory mucosal immune memory to SARS-CoV-2 after infection and vaccination. Nat Commun 2023; 14: 6815. [CrossRef]
- Belyakov, I.M.; Ahlers, J D. What role does the route of immunization play in the generation of protective immunity against mucosal pathogens? J Immunol 2009; 183: 6883-6892. [CrossRef]
Figure 6.
Early kinetics of saliva antibody responses after infection. The saliva samples from two infected family members were collected consecutively starting on day 0 of the SARS-COV-2 infection. The levels of antibodies are depicted. The closed circle is anti-RBD IgA, the open circle is anti-RBD IgG, the closed triangles are anti-N IgA, and the open triangles are anti-N IgG.
Figure 6.
Early kinetics of saliva antibody responses after infection. The saliva samples from two infected family members were collected consecutively starting on day 0 of the SARS-COV-2 infection. The levels of antibodies are depicted. The closed circle is anti-RBD IgA, the open circle is anti-RBD IgG, the closed triangles are anti-N IgA, and the open triangles are anti-N IgG.
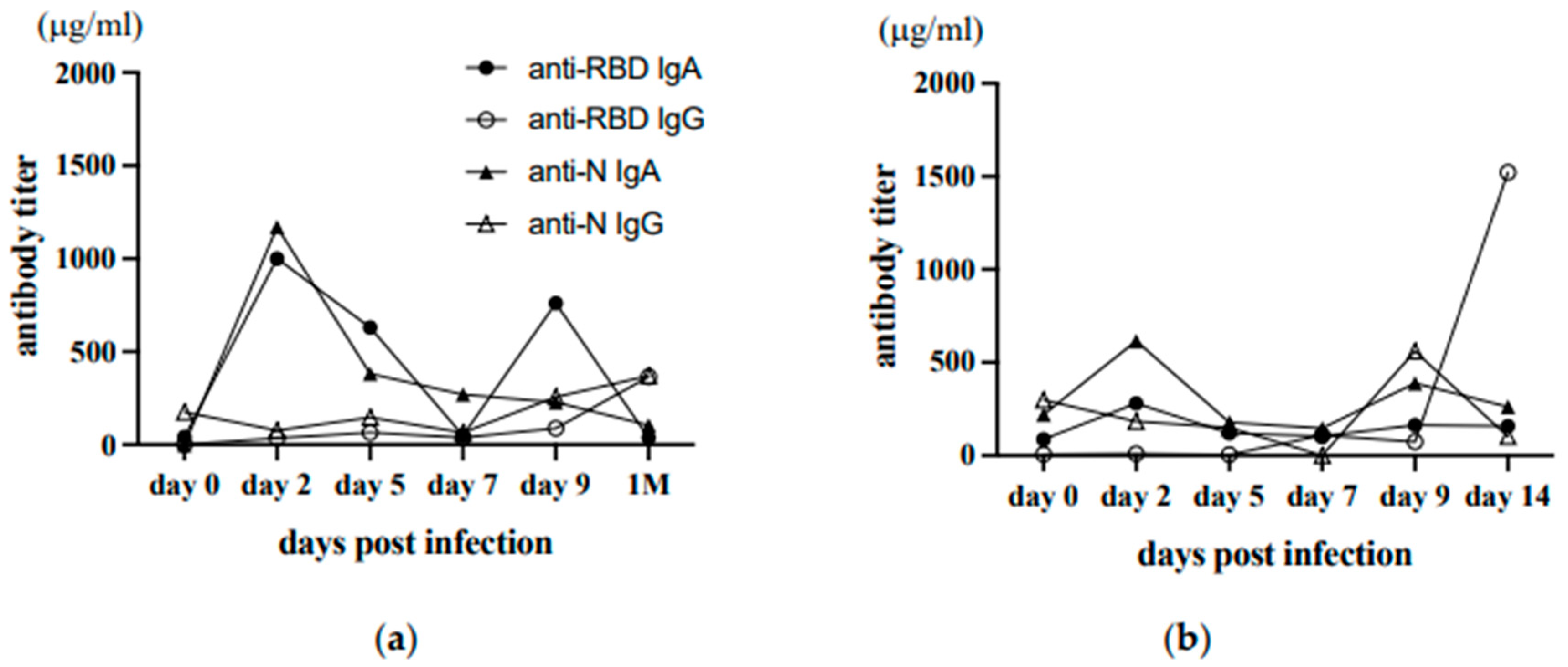
Table 1.
Information of donors who received COVID-19 vaccines in 2021.
| Total number | 22 | |
| age | 20~681 | |
| sex | male | 11 |
| female | 11 | |
| vaccination 2 doses |
BNT162b2 | 7 |
| mRNA-1273 | 15 | |
| 3 doses | 15 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



