Submitted:
02 February 2024
Posted:
05 February 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection can cause potentially life-threatening coronavirus disease (COVID-19). COVID-19 is a multisystem disease and is associated with significant respiratory distress, systemic hyper inflammation, vasculitis and multi-organ failures. SARS-CoV-2 causes deterioration of numerous systems with increasing evidence implying that COVID-19 affects endothelium and vascular function. The endothelium is important for preserving vascular tone and homeostasis. The overactivation and dysfunction of endothelial cells are significant outcomes of severity in patients with COVID-19. The Angiopoietin 1/Tie 2 pathway plays an important role in endothelium quiescence and vessel stability. The disruption of Angiopoietins/Tie balance affects vessel contact barrier and leads to vessel leakage, and this in turn causes endothelial dysfunction. Although vascular instability through SARS-CoV-2 is associated with endothelial dysfunction, it is still not understood if the virus affects Angiopoietin/Tie axis directly or via other mechanisms such as cytokine storm and/or immune response associated with the infection. This review provides an overview of the impact SARS-CoV-2 has on endothelial function and more specifically the Angiopoietin/Tie pathway.
Keywords:
SARS-CoV-2
; Endothelial cell
; Angiopoietin
; Tie1/2
; ACE2
; inflammatory cytokines
2. Endothelium
The circulatory system contains numerous blood vessels including arteries, veins, capillaries and lymph vessels which transport blood, haemocytes, oxygen, nutrients, hormones, vitamins, minerals and lymphatic fluids, which are important to support survival of the human body [25]. Endothelial cells are a thin monolayer that mainly line the interior surface of vasculature, and are commonly shielded by pericytes that promote vascular stability. These cells have various roles and are predominately surrounded by smooth muscle and connective tissue. The main function of endothelial cells is regulating haemostasis, coagulation, thrombosis, permeability, inflammatory processes, remodelling of new vessels, blood vessel tone, vasorelaxation and vasoconstriction, blood flow and pressure, and forming the barrier between vessels and tissues [26,27,28]. The endothelium has a capacity to respond to physical damage and chemical molecules. Stability of the endothelium depends on regulation of the vascular contractile activity of vascular smooth muscle cells, vasculitis and cellular attachment to other cells [29]. Physiological activity of certain endothelium signalling mediators including nitric oxide, prostacyclins and angiopoietins are important for all features of blood vessel stability and integrity. Therefore, the metabolic function of endothelial cells is crucial for the constant adaptation of blood vessel tone, which in turn regulates blood pressure. Additionally, endothelial cells have a key role in decreasing the formation of blood clots and blood thickness equilibrium in the blood stream, including the normal control of white blood cells transport from blood vessel to tissues [30].
2.1. Angiopoietin family
Angiopoietins belong to a family of vascular growth factor glycoproteins which control the formation of new blood vessels, promote angiogenesis and foetal/adult vascular development. The four isoforms, Angiopoietin (Angpt) 1, 2, 3, 4, are structurally comparable and are ligands of the Tunica Interna endothelial cell tyrosine kinase (Tie) receptor, predominantly expressed in endothelial cells [31]. Angpt 1 and 2 are key isoforms of this glycoprotein family and have been thoroughly studied, whereas Angpt 3 and 4 are less well-studied. Although Angpt 3 is expressed in mouse and humans, it is not biologically active in human endothelial cells. The specific and precise equilibrium of Angpt 1 and 2 is important to control vascular stability [32]. The alteration of these protein level ratios is connected with multiple diseases such as cancer, sepsis and coronary artery diseases [33].
2.2. The function of Angiopoietin 1
Angpt 1 has a molecular weight of 70 kDa and consists of 498 residues [34]. Angpt 1 is suggested to be the significant angiogenic growth factor and constitutive paracrine agonist of tyrosine kinase receptor (Tie2), and supports blood vessel stability and integrity. Witzenbichler et al (1998) and Jones et al (1999) reported that binding of Angpt 1 to Tie 2 in vascular endothelial cells leads to phosphorylation of several tyrosine residues on the carboxy terminus of the receptor, initiating various intracellular secondary messengers that contribute to endothelial function including vessel sprouting and cell viability [35]. The absence of Angpt 1 was shown to lead to defects in blood vessel growth and foetal blood vessel development in Angpt 1 knock out mice [36]. The upregulation of Angpt 1 in a genetically modified mouse model increased angiogenesis and decreased vascular permeability [37]. Angpt 1 does not bind to the Tie 1 receptor, however indirectly activates it through transphosphorylation of active Tie 2 receptor [38]. Angpt 1-induced PI3K/Akt leads to the addition of phosphate groups and inhibition of the forkhead transcription factor Foxo1 in vascular endothelial cells [39]. This transcription factor is involved in endothelial cell death and controls the synthesis of various mediators such as Angpt 2. Angpt 1 phosphorylation of the Tie 2 receptor also facilitates adhesion with pericytes and smooth muscle cells, which in turn leads to blood vessel protection and stability [40].
2.3. The function of Angiopoietin 2
Angpt 1 and 2 have 60% homology in their amino acid sequence. Like Angpt 1, Angpt 2 is capable of binding to Tie 2 receptor. Angpt 2 is stored in Weibel-Palade bodies (WPBs) in endothelial cells. The release of Angpt 2 is highly controlled and exerts an autocrine action [41]. Angpt 2 has the opposite effect from Angpt 1; it facilitates vascular permeability, obstructs endothelial barrier activity, and impairs vascular integrity [42]. Although Angpt 2 obstructs the vascular protective activity of Angpt 1 after it binds to Tie 2 receptor, in some cases this antagonist ligand can stimulate Tie 2 signalling through an unknown mechanism. In the presence of vascular endothelial growth factor (VEGF), Angpt 2 promotes angiogenesis, blood vessel sprouting and destabilisation [43]. Also, Angpt 2 promotes pericyte detachment by decreasing the cooperation between endothelial cell and pericytes, vascular smooth muscle cells and extracellular matrix [44]. Also, some studies reported that upregulation of Angpt 2 in vitro activates vascular endothelial cell movement, during the reorganisation stage of angiogenesis and increased cell viability via Tie 2 interaction and PI3K/Akt stimulation [45]. In physiological conditions, Angpt 2 is generally released at regions of blood vessel structural changes such as gonads, womb and placenta. The release is rapidly activated by various stimuli including thrombin, histamine, hypoxia, and VEGF. Angpt 2 also stimulates vascular endothelial cells to release inflammatory cytokines including TNF-α [46]. Angpt 2 interacts with white blood cells including monocytes, macrophages, and neutrophils throughout the inflammation process [47].
2.4. Tie receptors
Tie 1 and Tie 2 are members of a specific receptor tyrosine kinase family (RTKs) which are expressed predominantly on endothelium and hematopoietic cells. The Tie 2 receptor interacts with Angpt 1, 2, 4, whereas Tie 1 is thought to be incapable to bind to Angpts and alternative ligands of Tie 1 are not yet discovered [48]. Tie receptors contains three epidermal growth factor (EGF) domains amidst the two immunoglobulin (Ig)-like domains and three fibronectin type 3 domains. The Ig-like domains and EGF-like domains are important and sufficient to bind Angpt 1 and Angpt 2 [49]. Tie receptors play a role in the structural change and maturation of blood vessels during embryogenesis [50]. The Tie 1 orphan receptor is important for blood vessel formation, integrity and sprouting in the embryonic stage. Tie 1 synthesis decreases in adulthood but levels remain the same in endothelial cells of renal, cardiovascular and respiratory systems [51]. Tie 2 is the main signalling receptor of Angpt pathway and receptor clustering is important for stimulation of this transmembrane receptor [52]. Tie 1 is capable of forming heterodimer complexes with Tie 2 which decreases the activation of Angpt 1 and supports the activity of Angpt 2, whereas Tie2/Tie2 homodimers activate Angpt 1 signalling and vascular integrity. The process of the activation of Tie 2 receptor is still not well-understood, apart from the binding of carboxyl terminus domain of Angpt to the ligand-binding domain of this receptor [53].
2.5. Angiopoietin 1/Tie 2 signalling
After binding of the five subunits of multimeric Angpt 1 to the Tie 2 receptor at cell-to-cell junctional complex, the Tie 2 transmembrane receptor is phosphorylated and starts the signalling pathway resulting in regulating multiple processes including blood vessels stability and endothelial cell survival as illustrated in Figure 1 [53,54]. Angpt 1 activates PI3K by recruitment of regulatory p85 subunit in Tie 2, and that results in stimulation of Akt and nitric oxide synthase 3 (eNOS) to inhibit endothelial apoptosis. The stimulation of Akt by Angpt 1/Tie 2 signalling pathway leads to phosphorylation and obstruction of Forkhead transcription factor (FKHR) gene to support cell survival and vessel stability [55,56,57,58,59,60]. Angpt 1/Tie pathway is also involved in cortical actin cytoskeleton stability by regulating GTPase and decreasing the inflammatory molecules through inhibition of NFκB protein [61,62,63,64,65,66].
As discussed above, Tie 1/Tie 2 heterodimer receptors can affect Angpt 1/Tie 2 signalling. Tumour necrosis factor-α (TNF-α) is a crucial inflammatory cytokine that is able to stimulate Tie 1 ectodomain cleavage in endothelial cells. TNF-α was also shown to elevate Tie 2 expression leading to change in Tie 1 to Tie 2 ratio. Similar observations have been found with VEGF, which is capable of both cleaving ectodomains of Tie 1 and Tie 2, leading to regulation of Angpt 1 signalling [67,68,69,70,71]. Tie 1 decreases the capacity of Angpt 1 bind to Tie 2, and therefore Tie 2 activation by its ectodomain. Cleavage of the extracellular domain of Tie 1 enhances the Angpt 1 binding to Tie 2 and thus increases Angpt 1/Tie 2 activation [72]. The change in Angpt 1 and Angpt 2 levels also regulate Tie 2 signalling. A decrease of Angpt 1 and increase of Angpt 2 promotes vessel destabilisation under various disease states, and has been associated with acute lung injury [73]. Some studies reported that the release of Angpt 1 changes after ischemic stroke and increased level of this protein decreases the complication of the stroke [74]. During atherosclerosis, Angpt 2 elevation promotes vessel inflammatory processes by stimulating NFκB and traffics white blood cells through the vessel to the affected regions [75]. Also, inflammation caused by infection leads to decrease in Angpt 1 and suppression of the Angpt 1/Tie 2 pathway, leading to vascular instability and increased permeability. Several studies have mentioned that Angpt could be utilised as a molecular marker or treatment method to avoid severity of lung inflammation and sepsis [76,77]. Some studies suggest that elevated Angpt 2 in various tumours stimulate neovascularisation in cancer mice models. Angpt 1/Tie 2 pathway therapies have been connected with recovery and survival rate in specific types of tumours such as cervical and ovarian cancers [78,79,80].
3. The impact of SARS-CoV-2 on endothelial cells
Post-mortem studies in COVID-19 patients have reported procoagulant state and microvascular damage in deceased patients, demonstrating that vascular pathology caused by endothelial dysfunction is a crucial factor for the pathogenesis of SARS-CoV-2 [81,82,83]. Endothelial dysfunction during SARS-CoV-2 infection results in pro-inflammation, vascular permeability, and leads to vascular destabilisation through mechanisms shown in Figure 2 [84]. As mentioned above, the ACE2 receptor is one of the most important receptors that binds to the spike protein of SARS-CoV-2, and is expressed in various cells including vascular endothelial cells. SARS-CoV-2 has been connected with substantial immune activation that directly influences endothelial cells via cytokine storm [85]. Lilian et al. (2021) suggested that SARS-CoV-2 can infect host endothelial cells if high viral loads are used, and this leads to activation of cell death. Infection of endothelial cells has been suggested to be less efficient compared to epithelial cells due to lower expression of the ACE2 receptor and protease in endothelial cells [100].
Endothelial tissues cooperate with epithelial tissues in the respiratory tract airway barrier to control tissue stability and defence against infectious microorganisms. Epithelial cells communicate with endothelial cells during pathogenic infections to initiate immune cell activation. These two cells create a single layer bound by solid junctions that control vascular permeability, whereas adherent junctions control cell-to-cell and cell-to-matrix cooperation [86]. The endothelium in the lung contains macro- and micro-vessel endothelial cells with the latter belonging to the epithelial and endothelial contact junction [87]. The increase in inflammatory cytokines causes local endothelial dysfunction and supports blood clotting and platelet activation [88]. The damage of endothelial cells caused by intense immune reaction could affect the structure of blood vessel and airways junction, and cause vessel leakage, lung inflammation, white blood cell extravasation, and oxygen shortage [89]. The lung epithelial cells stimulate NOD-like receptor protein 3 (NLPR3) and inflammatory cytokines such as TNF-α, IL-6 during COVID-19 in order to start intense immune reactions and these processes result in apoptosis, cell injury, increased vessel permeability and lung inflammation [90]. In addition, unregulated synthesis of neutrophil extracellular traps (NETs) and reactive oxygen species (ROS) impair endothelial cells, activate platelet and blood clotting, and aid acute respiratory distress syndrome and tissue injury. Nitric oxide and prostacyclin are vasodilators that help with vascular integrity. Levels of these hormones are reduced in SARS-CoV-2 and lead to endothelial dysfunction [91].
Intracellular adhesion molecules bind to fibrinogen and support fibrin aggregation to endothelial cells, which is involved in abnormal function of endothelial cells, blood clotting, and vessel narrowing [92]. The hyperactivation of endothelial cells and dysfunction is a significant hallmark of severity in patients with SARS-CoV-2. It is not clear if SARS-CoV-2 can directly and productively infect endothelial cells despite endothelial cells expressing the ACE2 receptor, which is necessary for viral entry [93,94,95,96]. Enrichment of SARS-CoV-2 RNA was found in lung endothelial cells in deceased COVID-19 patients [97], and infection was observed in in vitro cultured endothelial cells [98,99]. On the other hand, infection of primary endothelial cells was not observed [100]. Despite this, it has been suggested that infection of neighbouring cells such as epithelial and vascular pericyte cell cause the elevation of inflammatory response and cytokine storm and leads to endothelial dysfunction [100].
Some studies have speculated that SARS-CoV-2-infected epithelial cells detach at the contact barriers and that permits the virus to transmit from the base surfaces of epithelial cells to endothelial cells in the lung. It has also been suggested that virus might infect endothelial cells through blood stream via the apical surface [101]. Also, others have suggested that ACE2 is released at high levels in small vessel pericytes and pericyte injury caused by SARS-CoV-2, and might play a role in endothelial dysregulation. The loss or detachment of SARS-CoV-2-infected and injured pericytes cause endothelial dysfunction through declining endothelial barrier activity and endothelial activation [93].
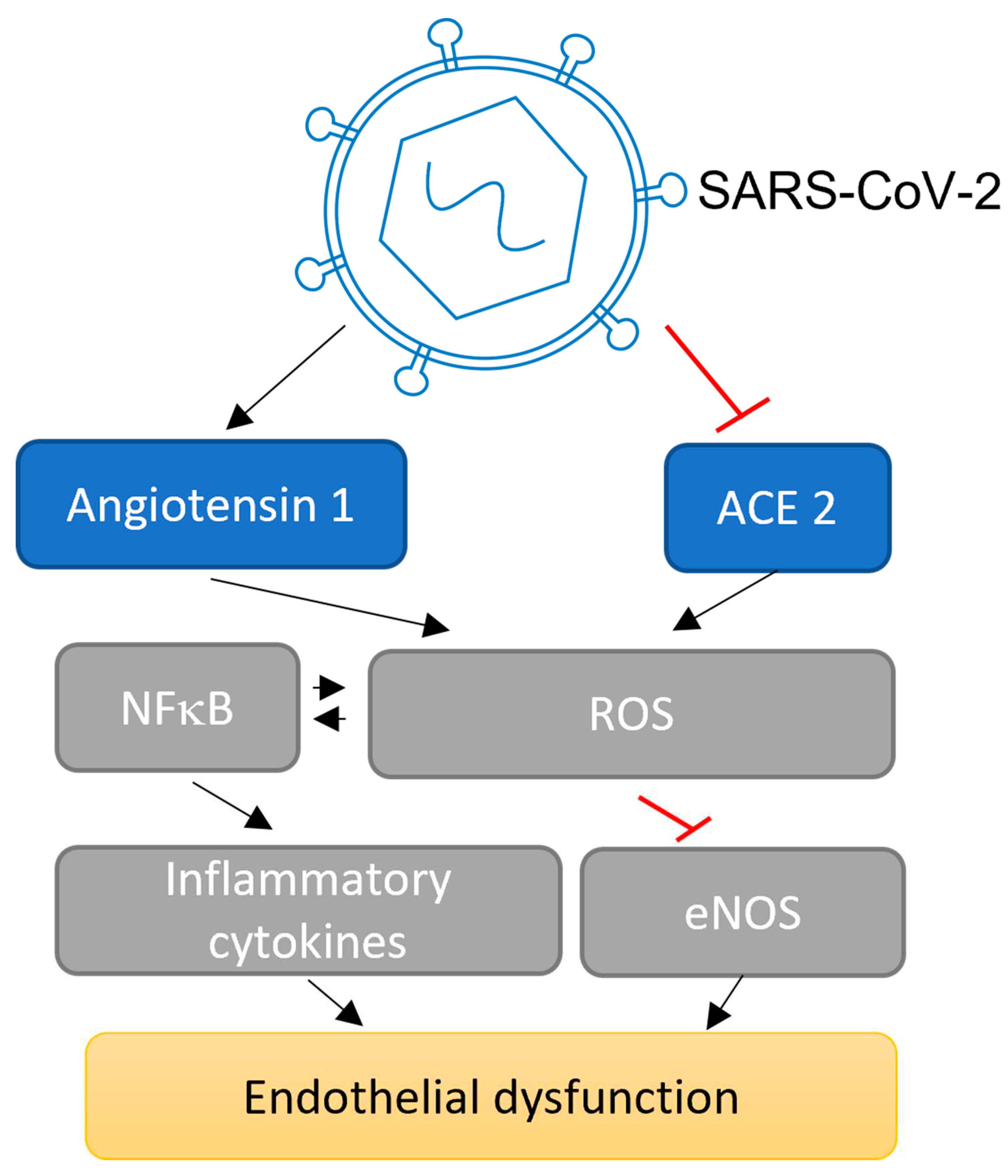
It has also been reported that the SARS-CoV-2 spike protein stimulated the damage of the vessel contact barrier in cerebral small vessel endothelial cells [101]. Endothelial cells sense and respond to damage associated molecular signals from surrounding virus-affected epithelial cells and endothelial cells. Furthermore, vascular endothelial cells regulated inflammatory process is one of the important parts in the pathological mechanism of COVID-19 [52]. During the infection, the inflammatory process activates endothelial cells to synthesise tissue factors that result in blood clotting, hyperpermeability of the small vessels, lung damage and increased level of cytokines such as TNF-α and IL-6 associated with amplified coagulation factor 1 or fibrinogen [102]. The extracellular activation of ACE2 is decreased and renin angiotensin system (RAS) is stimulated after SARS-CoV-2 entry into the host cell. ACE2 is important for supporting vessel relaxation molecules including angiotensins 1-7 to prevent the activation of reactive oxygen species (ROS) through inhibition of angiotensin II type 1 receptor (AT1R), and regulates the equilibrium of the RAS [103]. Downregulation of nitric oxide and elevation in vessel narrowing molecules such as angiotensin 2 are important aspects of endothelial dysregulation during COVID-19. The reduction of nitric oxide by upregulation of NFκB leads to reduction of endothelial nitric oxide synthase (eNOS), and synthesis of ROS which are involved in disruption of endothelial function (Figure 2) [104]. During SARS-CoV-2 infection and cytokine storm, increase in angiotensin type 2 receptor and reduction in angiotensin 1-7 is involved in decreasing vessel dilation, supporting white blood cell and thrombocytes adhesion, and therefore activates a pro-thrombotic, pro-coagulative state and inhibits the fibrinolysis process [105]. Moreover, angiotensin type 2 receptor upregulates nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase 2), and increased expression of angiotensin type 2 receptor leads to elevation of oxidative stress and destabilisation of vascular activity [106]. People infected with SARS-CoV-2 displayed elevated amounts of fibrinogen, fibrin dissolving fragments such as D-dimer, and blood clotting protein including factor 8, and decreased amounts of endothelial plasminogen activator inhibitor 1 [107].
3.1. Potential impact of SARS-CoV-2 on Angpt/Tie signalling
Angpt 2 plays an important role in elevated vascular inflammation and leakage through inactivating Angpt 1 and Tie signalling. Elevated levels of Angpt 2 have been detected in the blood serum of patients with vascular leakage, lung inflammation, acute respiratory distress syndrome and linked with complications like sepsis [108]. Lu et al. (2022) reported that SARS-CoV-2 infection declines the vascular endothelial contact barrier structure due to damage of vascular endothelial cadherin barrier junctions and an increase in inflammatory mediators including IL-6, IL-8 and Angpt 2 in vitro. Also, these cytokines were increased in SARS-CoV-2 infections compared to other coronaviruses such as NL63 [109]. The stimulation of phosphorylated mixed lineage kinase domain-like protein (pMLKL), which supports necrotic apoptosis occurred in small vessels with increased Angpt 2, connecting this to vessel damage in patients with COVID-19 [110]. Some studies suggested that thrombosis, vessel damage, and vascular endothelial cells apoptosis were observed in the tissue of deceased COVID-19 patients using pulmonary imaging mass cytometry. This result supports the speculation that COVID-19 is a vascular disorder [111]. SARS-CoV-2 uses cellular machinery of the infected cells, similar to other viruses, in order to produce the proteins which are necessary for the transcription and assembly of the pathogen, resulting in atypical hyperstimulation of signalling mechanism of PI3/Akt/mTOR pathway [112]. Hypoxia caused by SARS-CoV-2 infection is also involved in angiogenesis. Lack of oxygen connected with continuing inflammation leads to hypoxia inducible factor 1 alpha (HIF-1α) stabilisation, which interacts with molecular markers including VEGF, Angpt and Tie 2 receptor. Also, angiopoietin like 4 proteins (Angpt 4) is released in response to oxygen starvation and malnutrition by a HIF1-α related mechanism. Angpt 4 is involved in several activities including controlling vessel damage, permeability, wound healing, vessel function and cholesterol homeostasis [113]. Some studies reported that increased level of Angpt 4 was related to acute respiratory distress syndrome and severity of SARS-CoV-2 infection [114]. SARS-CoV-2 infection activates HIF-1α, and upregulation of this factor also involved in the pathophysiological mechanism of this pathogen including a decrease in ACE2 receptor [115].
White blood cell endothelial adhesion molecules such as soluble E-selectin are activated by inflammatory cytokines. Elevated levels of soluble E-selectin and Angpt 2 have been reported in patients with SARS-CoV-2 infections [116]. Thrombocytes, except pericytes, produce large numbers of Angpt 1 protien. Thrombocytopenia caused by apoptosis of thrombocytes, and build-up of thrombus aggregation leads to decline of serum Angpt 1 in patients with severe COVID-19 [117]. It has been speculated that after the first stage of SARS-CoV-2 infection, induced expression of inflammatory cytokines and local hypoxia due to upregulated Angpt 2 results in development of lung vessel injuries. Serum Angpt 2/Angpt 1 ratio is substantially elevated in patients with severe symptoms or those who are critically ill than patients with less severe symptoms [118].
4. Conclusion
Endothelial dysfunction plays a crucial role in the pathophysiology of SARS-CoV-2. The effect of SARS-CoV-2 on Tie receptor expression and Angpt 1 levels is not well-studied, however some studies indicate that infection induces changes in the levels of Angpts and Tie 2, which potentially may destabilise the vasculature, promote thrombosis, coagulation, inflammation and permeability during severe COVID-19. It is also possible that SARS-CoV-2 may indirectly affect Angpt/Tie 2 signalling through inflammatory cytokine storm and stimulation of a pro-thrombotic state. Inflammatory cytokines such as TNF-α and IL-6 upregulate Angpt 2 by inhibition of nitric oxides and NFκB activation, and thus suppress Angpt 1/Tie 2 signalling. Direct endothelial virus infection or infection of neighbouring cells may cause disruption of Angpt 1/Tie 2 and endothelial dysfunction during the immune activation against infection. As previously mentioned, SARS-CoV-2 is associated with long term vascular pathology and endothelial injury. Therefore, based on the evidence more research is required in order to understand molecular impact SARS-CoV-2 has on endothelial dysfunction and Angiopoietin/Tie axis to improve treatment and care.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., Zhao, X., Huang, B., Shi, W., Lu, R., Niu, P., Zhan, F., Ma, X., Wang, D., Xu, W., Wu, G., Gao, G.F., Tan, W. and China Novel Coronavirus Investigating and Research Team. A Novel Coronavirus from Patients with Pneumonia in China. The New England journal of medicine, 2020, 382(8), 727-733. [CrossRef]
- Banerjee, A., Kulcsar, K., Misra, V., Frieman, M. and Mossman, K. Bats and Coronaviruses. Viruses. 2019, 11(1), 41.
- Philip V’kovski, Annika Kratzel, Silvio Steiner, Hanspeter Stalder & Volker Thiel. Coronavirus biology and replication: implications for SARS-CoV-2. Nature review microbiology. 2020, 19, 155-170.
- Li, Q., Guan, X., Wu, P., Wang, X., Zhou, L., Tong, Y., Ren, R., Leung, K.S.M., Lau, E.H.Y., Wong, J.Y., Xing, X., Xiang, N., Wu, Y., Li, C., Chen, Q., Li, D., Liu, T., Zhao, J., Liu, M., Tu, W., Chen, C., Jin, L., Yang, R., Wang, Q., Zhou, S., Wang, R., Liu, H., Luo, Y., Liu, Y., Shao, G., Li, H., Tao, Z., Yang, Y., Deng, Z., Liu, B., Ma, Z., Zhang, Y., Shi, G., Lam, T.T.Y., Wu, J.T., Gao, G.F., Cowling, B.J., Yang, B., Leung, G.M. and Feng, Z. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. The New England journal of medicine. 2020, 382(13), 1199-1207. [CrossRef]
- Gaunt, E.R., Hardie, A., Claas, E.C., Simmonds, P. and Templeton, K.E. Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method. Journal of clinical microbiology. 2010. 48(8), 2940-2947. [CrossRef]
- Naskalska, A., Dabrowska, A., Szczepanski, A., Milewska, A., Jasik, K.P. and Pyrc, K. Membrane Protein of Human Coronavirus NL63 Is Responsible for Interaction with the Adhesion Receptor. Journal of virology. 2019, 93(19), e00355-19. [CrossRef]
- Corman, V.M., Muth, D., Niemeyer, D. and Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Advances in Virus Research. 2018, 100, 163-188. [CrossRef]
- Lalchhandama K. The chronicles of coronaviruses: the bronchitis, the hepatitis and the common cold. Science vision. 2020, 20 (1), 43-53. [CrossRef]
- Gierer, S., Bertram, S., Kaup, F., Wrensch, F., Heurich, A., Kramer-Kuhl, A., Welsch, K., Winkler, M., Meyer, B., Drosten, C., Dittmer, U., von Hahn, T., Simmons, G., Hofmann, H. and Pohlmann, S. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. Journal of virology. 2013, 87(10), 5502-5511. [CrossRef]
- Neuman, B.W., Kiss, G., Kunding, A.H., Bhella, D., Baksh, M.F., Connelly, S., Droese, B., Klaus, J.P., Makino, S., Sawicki, S.G., Siddell, S.G., Stamou, D.G., Wilson, I.A., Kuhn, P. and Buchmeier, M.J. A structural analysis of M protein in coronavirus assembly and morphology. Journal of structural biology. 2011, 174(1), 11-22. [CrossRef]
- Mart M. Lamers and Bart L. Haagmans. SARS-CoV-2 pathogenesis. Nature Reviews Microbiology. 2022, 20, 270–284.
- Markus Hoffmann , Hannah Kleine-Weber , Simon Schroeder , Nadine Krüger , Tanja Herrler , Sandra Erichsen , Tobias S Schiergens , Georg Herrler , Nai-Huei Wu, Andreas Nitsche , Marcel A Müller, Christian Drosten , Stefan Pöhlmann. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020, 181(2), 271-280. [CrossRef]
- Ou, X., Liu, Y., Lei, X., Li, P., Mi, D., Ren, L., Guo, L., Guo, R., Chen, T., Hu, J., Xiang, Z., Mu, Z., Chen, X., Chen, J., Hu, K., Jin, Q., Wang, J. and Qian, Z. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nature communications. 2020, 11(1), 1620-9.
- Kawase, M., Shirato, K., van der Hoek, L., Taguchi, F. and Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. Journal of virology. 2012, 86(12), 6537-6545. [CrossRef]
- Letko, M., Marzi, A. and Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nature microbiology. 2020, 5(4), 562-569. [CrossRef]
- W. Wang, L. Ye, L. Ye, B. Li, B. Gao, Y. Zeng, L. Kong, X. Fang, H. Zheng, Z. Wu, Y. She. Up-regulation of IL-6 and TNF-α induced by SARS-coronavirus spike protein in murine macrophages via NF-κB pathway. Virus Res. 2007. 128 (1–2), pp. 18, 10.1016/j. [CrossRef]
- Xin Yin, Laura Riva, Yuan Pu, Laura Martin-Sancho, Jun Kanamune, Yuki Yamamoto, Kouji Sakai, Shimpei Gotoh, Lisa Miorin, Paul D. De Jesus, Chih-Cheng Yang, Kristina M. Herbert, Sunnie Yoh, Judd F. Hultquist, Adolfo García-Sastre, and Sumit K. Chanda. MDA5 Governs the Innate Immune Response to SARS-CoV-2 in Lung Epithelial Cells. Cell Reports. 2021, 34(2), 108628.
- Lokugamage, K.G., Hage, A., de Vries, M., Valero-Jimenez, A.M., Schindewolf, C., Dittmann, M., Rajsbaum, R. and Menachery, V.D. Type I Interferon Susceptibility Distinguishes SARS-CoV-2 from SARS-CoV. Journal of virology. 2020, 94(23), e01410-20.
- Ann-Kathrin Reuschl , Lucy G Thorne, Matthew V X Whelan , Roberta Ragazzini , Wilhelm Furnon , Vanessa M Cowton , Giuditta De Lorenzo , Dejan Mesner , Jane L E Turner , Giulia Dowgier , Nathasha Bogoda , Paola Bonfanti, Massimo Palmarini , Arvind H Patel, Clare Jolly , Greg J Towers. Evolution of enhanced innate immune suppression by SARS-CoV-2 Omicron subvariants.Nature Microbiology. 2024. [CrossRef]
- Kim, Y. and Shin, E. Type I and III interferon responses in SARS-CoV-2 infection. Experimental & molecular medicine. 2021, 53(5), 750-760.
- Donghyuk Shin, Rukmini Mukherjee, Diana Grewe, Denisa Bojkova, Kheewoong Baek, Anshu Bhattacharya, Laura Schulz, Marek Widera, Ahmad Reza Mehdipour, Georg Tascher, Paul P. Geurink, Alexander Wilhelm, Gerbrand J. van der Heden van Noort, Huib Ovaa, Stefan Müller, Klaus-Peter Knobeloch, Krishnaraj Rajalingam, Brenda A. Schulman, Jindrich Cinatl, Gerhard Hummer, Sandra Ciesek & Ivan Dikic. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020. 587, 657-662.
- Shang, J., Ye, G., Shi, K., Wan, Y., Luo, C., Aihara, H., Geng, Q., Auerbach, A. and Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020, 581(7807), 221-224.
- Morse, J.S., Lalonde, T., Xu, S. and Liu, W. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. ChemRxiv: the preprint server for chemistry. 2020, 2,21 (5), 730-738. [CrossRef]
- Roberts, K.A., Colley, L., Agbaedeng, T.A., Ellison-Hughes, G.M. and Ross, M.D. Vascular Manifestations of COVID-19 - Thromboembolism and Microvascular Dysfunction. Frontiers in cardiovascular medicine. 2020, 7, 598400. [CrossRef]
- Pugsley, M.K. and Tabrizchi, R. The vascular system. An overview of structure and function. Journal of pharmacological and toxicological methods. 2000, 44(2), 333-340.
- Aird, W.C. 'Endothelium and haemostasis', Hamostaseologie, 2015, 35(1), 11-16. [CrossRef]
- G. Jia, A. R. Aroor, C. Jia, J. R. Sowers,Biochim. Biophys. Acta - Mol.Basis Dis. 2019,1865, 1802.
- Lo Gullo,C.O. Aragona,M.Scuruchi,A.G.Versace,A.Saitta,E.Imbalzano, S. Loddo, G. M. Campo, G. Mandraffino,Vascul. Phar-macol.2018,108, 8.
- Deanfield, J.E., Halcox, J.P. and Rabelink, T.J. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007. 115(10), 1285-1295.
- Pearson, J.D. 'Normal endothelial cell function'. Lupus. 2000, 9(3), 183-188. [CrossRef]
- Buhimschi, C.S., Bhandari, V., Dulay, A.T., Thung, S., Razeq, S.S.A., Rosenberg, V., Han, C.S., Ali, U.A., Zambrano, E., Zhao, G., Funai, E.F. and Buhimschi, I.A. Amniotic fluid angiopoietin-1, angiopoietin-2, and soluble receptor tunica interna endothelial cell kinase-2 levels and regulation in normal pregnancy and intraamniotic inflammation-induced preterm birth. The Journal of clinical endocrinology and metabolism. (2010). 95(7), 3428-3436. [CrossRef]
- Daly, C., Eichten, A., Castanaro, C., Pasnikowski, E., Adler, A., Lalani, A.S., Papadopoulos, N., Kyle, A.H., Minchinton, A.I., Yancopoulos, G.D. and Thurston, G. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer research. 2013, 73(1), 108-118. [CrossRef]
- Bilimoria, J. and Singh, H. The Angiopoietin ligands and Tie receptors: potential diagnostic biomarkers of vascular disease, Journal of receptor and signal transduction research. 2019, 39(3), 187-193. [CrossRef]
- Maisonpierre, P.C., Suri, C., Jones, P.F., Bartunkova, S., Wiegand, S.J., Radziejewski, C., Compton, D., McClain, J., Aldrich, T.H., Papadopoulos, N., Daly, T.J., Davis, S., Sato, T.N. and Yancopoulos, G.D. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science (New York, N.Y.). 1997, 277(5322), 55-60. [CrossRef]
- Witzenbichler, B., Maisonpierre, P.C., Jones, P., Yancopoulos, G.D. and Isner, J.M. Chemotactic properties of angiopoietin-1 and -2, ligands for the endothelial-specific receptor tyrosine kinase Tie2. The Journal of biological chemistry. 1998, 273(29), 18514-18521. [CrossRef]
- Suri, C., Jones, P.F., Patan, S., Bartunkova, S., Maisonpierre, P.C., Davis, S., Sato, T.N. and Yancopoulos, G.D. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell, 1996, 87(7), 1171-1180. [CrossRef]
- Chae, J.K., Kim, I., Lim, S.T., Chung, M.J., Kim, W.H., Kim, H.G., Ko, J.K. and Koh, G.Y. Coadministration of angiopoietin-1 and vascular endothelial growth factor enhances collateral vascularization. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000, 20(12), 2573-2578. [CrossRef]
- Saharinen, P., Eklund, L., Miettinen, J., Wirkkala, R., Anisimov, A., Winderlich, M., Nottebaum, A., Vestweber, D., Deutsch, U., Koh, G.Y., Olsen, B.R. and Alitalo, K. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nature cell biology. 2008, 10(5), 527-537. [CrossRef]
- Papapetropoulos, A., Fulton, D., Mahboubi, K., Kalb, R.G., O'Connor, D.S., Li, F., Altieri, D.C. and Sessa, W.C. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. The Journal of biological chemistry. 2000, 275(13), 9102-9105. [CrossRef]
- Cai, J., Kehoe, O., Smith, G.M., Hykin, P. and Boulton, M.E. The angiopoietin/Tie-2 system regulates pericyte survival and recruitment in diabetic retinopathy. Investigative ophthalmology & visual science. 2000, 49(5), 2163-2171. [CrossRef]
- Stratmann, A., Risau, W. and Plate, K.H. Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. The American journal of pathology. 1998. 153(5), 1459-1466. [CrossRef]
- Holash, J., Maisonpierre, P.C., Compton, D., Boland, P., Alexander, C.R., Zagzag, D., Yancopoulos, G.D. and Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science (New York, N.Y.). 1999, 284(5422), 1994-1998. [CrossRef]
- Lobov, I.B., Brooks, P.C. and Lang, R.A. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2002, 99(17), 11205-11210. [CrossRef]
- Dumont, D.J., Gradwohl, G., Fong, G.H., Puri, M.C., Gertsenstein, M., Auerbach, A. and Breitman, M.L. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes & development. 1994. 8(16), 1897-1909. [CrossRef]
- Felcht, M., Luck, R., Schering, A., Seidel, P., Srivastava, K., Hu, J., Bartol, A., Kienast, Y., Vettel, C., Loos, E.K., Kutschera, S., Bartels, S., Appak, S., Besemfelder, E., Terhardt, D., Chavakis, E., Wieland, T., Klein, C., Thomas, M., Uemura, A., Goerdt, S. and Augustin, H.G. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. The Journal of clinical investigation. 2012, 122(6), 1991-2005. [CrossRef]
- Fiedler, U., Scharpfenecker, M., Koidl, S., Hegen, A., Grunow, V., Schmidt, J.M., Kriz, W., Thurston, G. and Augustin, H.G. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood. 2004, 103(11), 4150-4156. [CrossRef]
- Scholz, A., Plate, K.H. and Reiss, Y. Angiopoietin-2: a multifaceted cytokine that functions in both angiogenesis and inflammation. Annals of the New York Academy of Sciences. 2015, 1347, 45-51. [CrossRef]
- Kontos, C.D., Cha, E.H., York, J.D. and Peters, K.G. The endothelial receptor tyrosine kinase Tie1 activates phosphatidylinositol 3-kinase and Akt to inhibit apoptosis. Molecular and cellular biology. 2002, 22(6), 1704-1713. [CrossRef]
- Fiedler, U., Krissl, T., Koidl, S., Weiss, C., Koblizek, T., Deutsch, U., Martiny-Baron, G., Marme, D. and Augustin, H.G. Angiopoietin-1 and angiopoietin-2 share the same binding domains in the Tie-2 receptor involving the first Ig-like loop and the epidermal growth factor-like repeats. The Journal of biological chemistry. 2003, 278(3), 1721-1727. [CrossRef]
- Koblizek, T.I., Weiss, C., Yancopoulos, G.D., Deutsch, U. and Risau, W. Angiopoietin-1 induces sprouting angiogenesis in vitro. Current biology: CB. 1998, 8(9), 529-532. [CrossRef]
- Korhonen, E.A., Lampinen, A., Giri, H., Anisimov, A., Kim, M., Allen, B., Fang, S., D'Amico, G., Sipila, T.J., Lohela, M., Strandin, T., Vaheri, A., Yla-Herttuala, S., Koh, G.Y., McDonald, D.M., Alitalo, K. and Saharinen, P. Tie1 controls angiopoietin function in vascular remodeling and inflammation. The Journal of clinical investigation. 2016, 126(9), 3495-3510. [CrossRef]
- Procopio, W.N., Pelavin, P.I., Lee, W.M. and Yeilding, N.M. Angiopoietin-1 and -2 coiled coil domains mediate distinct homo-oligomerization patterns, but fibrinogen-like domains mediate ligand activity. The Journal of biological chemistry. 1999, 274(42), 30196-30201. [CrossRef]
- Leppanen, V., Saharinen, P. and Alitalo, K. Structural basis of Tie2 activation and Tie2/Tie1 heterodimerization. Proceedings of the National Academy of Sciences of the United States of America. 2017, 114(17), 4376-4381.
- Jeansson M, Gawlik A, Anderson G, et al. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest. 2011, 1 21, 2278–2289. [CrossRef]
- Kwak HJ, So J, Lee SJ, et al. Angiopoietin-1 is an apoptosis survival factor for endothelial cells. FEBS Lett. 1999, 448, 249–253. [CrossRef]
- Kwak HJ, Lee SJ, Lee Y, et al. Angiopoietin-1 inhibits irradiation and mannitol-induced apoptosis in endothelial cells. Circulation. 2000, 101, 2317. [CrossRef]
- Kim I, Kim HG, So J, et al. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Circ Res. 2000, 8 6, 24–29. [CrossRef]
- Papapetropoulos A, Fulton D, Mahboubi K, et al. Angiopoietin-1 inhibits endothelial cell apoptosis via the akt/survivin pathway. J Biol Chem. 2000, 275, 9102–9105. [CrossRef]
- Kontos CD, Stauffer TP, Yang W, et al. Tyrosine 1101 of Tie2 is the major site of association of p85 and is required for activation of phosphatidylinositol 3-kinase and Akt. Mol Cell Biol. 1998, 18, 4131–4140. [CrossRef]
- Jones N, Master Z, Jones J, et al. Identification of tek/Tie2 binding partners. Binding to a multifunctional docking site mediates cell survival and migration. J Biol Chem. 1999, 274, 30896–30905.
- Jones N, Chen SH, Sturk C, et al. A unique autophosphorylation site on Tie2/tek mediates dok-R phosphotyrosine binding domain binding and function. Mol Cell Biol. 2003, 23, 2658–2668.
- Harfouche R, Gratton J, Yancopoulos GD, et al. Angiopoietin-1 activates both anti- and proapoptotic mitogen-activated protein kinases. FASEB J. 2003, 17, 1523–1525.
- Kichina JV, Goc A, Al-Husein B, et al. PAK1 as a therapeutic target. Expert Opin Ther Tar. 2010, 14, pp 703–725. [CrossRef]
- Hughes DP, Dunmore BJ, et al. ABIN-2 protects endothelial cells from death and has a role in the antiapoptotic effect of Angiopoietin-1. Blood. 2003, 102, 4407–4409. [CrossRef]
- Tadros A, Hughes DP, Dunmore BJ, et al. ABIN-2 protects endothelial cells from death and has a role in the antiapoptotic effect of Angiopoietin-1. Blood. 2003, 102, 4407–4409. [CrossRef]
- Van Huffel S, Delaei F, Heyninck K, et al. Identification of a novel A20-binding inhibitor of nuclear factor-kappaB activation termed ABIN-2. J Biol Chem. 2001, 276, 30216–30223.
- Korhonen EA, Lampinen A, Giri H, et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. JCI. 2016, 126, 3495–3510. [CrossRef]
- Inoue M, Itoh H, Ueda M, et al. Vascular endothelial growth factor (VEGF) expression in human coronary atherosclerotic lesions: possible pathophysiological significance of VEGF in progression of atherosclerosis. Circulation. 1998, 98, 2108–2016.
- Singh H, Hansen TM, Patel N, et al. The molecular balance between receptor tyrosine kinases Tie1 and Tie2 is dynamically controlled by VEGF and TNFa and regulates angiopoietin signalling. PLoS ONE. 2012, 7, 29319.
- Marron MB, Singh H, Tahir TA, et al. Regulated proteolytic processing of Tie1 modulates ligand responsiveness of the receptortyrosine kinase Tie2. J Biol Chem. 2007, 282, 30509–30517. [CrossRef]
- Findley CM, Cudmore MJ, Ahmed A, et al. VEGF induces Tie2 shedding via a phosphoinositide 3-kinase/Akt-dependent pathway to modulate Tie2 signaling. Arterioscl Throm Vas Biol. 2007, 27, pp 2619–2626. [CrossRef]
- Seegar TC, Eller B, Tzvetkova-Robev D, et al. Tie1-Tie2 interactions mediate functional differences between angiopoietin ligands. Mol Cell. 2010, 37, 643–655. [CrossRef]
- Schlosser K, Taha M, Deng Y, et al. High circulating Angiopoietin 2 levels exacerbate pulmonary inflammation but not vascular leak or mortality in endotoxin-induced lung injury in mice. Thorax. 2018, 73, 248–261. [CrossRef]
- Zhang ZG, Zhang L, Croll SD, et al. Angiopoietin-1 reduces cerebral blood vessel leakage and ischemic lesion volume after focal cerebral embolic ischemia in mice. Neuroscience. 2002, 113, 683–687. [CrossRef]
- Zernecke A, Weber C. Inflammatory mediators in atherosclerotic vascular disease. Basic Res Cardiol. 2005, 100, 93–101. [CrossRef]
- Davis JS, Yeo TW, Piera KA, et al. Angiopoietin-2 is increased in sepsis and inversely associated with nitric oxide-dependent microvascular reactivity. Crit Care. 2010, 14, R89. [CrossRef]
- Ghosh CC, Thamm K, Berghelli AV, et al. Drug repurposing screen identifies Foxo1-dependent Angiopoietin-2 regulation in sepsis. Crit Care Med. 2015, 43, 230–240. [CrossRef]
- Leow CC, Coffman K, Inigo I, et al. MEDI3617, a human anti angiopoietin 2 monoclonal antibody, inhibits angiogenesis and tumor growth in human tumor xenograft models. Int J Oncol. 2012, 40, 1321–1330. [CrossRef]
- White RR, Shan S, Rusconi CP, et al. Inhibition of rat corneal angiogenesis by a nuclease-resistant RNA aptamer specific for Angiopoietin-2. PNAS. 2003, 100, 5028–5033. [CrossRef]
- Yang, P. et al. (2017) The ratio of serum Angiopoietin-1 to Angiopoietin-2 in patients with cervical cancer is a valuable diagnostic and prognostic biomarker. Peer J. 2017, 5, 3387. [CrossRef]
- Ackermann M, Verleden SE, Kuehnel M et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020, 383, 120–128.
- Goshua G, Pine AB, Meizlish ML et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [CrossRef]
- Smadja DM, Mentzer SJ, Fontenay M et al. COVID-19 is a systemic vascular hemopathy: insight for mechanistic and clinical aspects. Angiogenesis. 2021, 24, 755–788. [CrossRef]
- Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol. 2020, 20, 389–391.
- Varga, Z., Flammer, A.J., Steiger, P., Haberecker, M., Andermatt, R., Zinkernagel, A.S., Mehra, M.R., Schuepbach, R.A., Ruschitzka, F. and Moch, H. 'Endothelial cell infection and endotheliitis in COVID-19', Lancet (London, England). 2020, 395(10234), 1417-1418. [CrossRef]
- Garcia-Ponce, A., Chanez Paredes, S., Castro Ochoa, K.F. and Schnoor, M. Regulation of endothelial and epithelial barrier functions by peptide hormones of the adrenomedullin family. Tissue barriers. 2016, 4(4), e1228439. [CrossRef]
- Millar, F.R., Summers, C., Griffiths, M.J., Toshner, M.R. and Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: insights and therapeutic opportunities. Thorax. 2016. 71(5), 462-473. [CrossRef]
- Noris, M., Benigni, A. and Remuzzi, G. The case of complement activation in COVID-19 multiorgan impact. Kidney international. 2020, 98(2), 314-322. [CrossRef]
- Pelaia, C., Tinello, C., Vatrella, A., De Sarro, G. and Pelaia, G. Lung under attack by COVID-19-induced cytokine storm: pathogenic mechanisms and therapeutic implications. Therapeutic advances in respiratory disease. 2020, 14, 1-9. [CrossRef]
- Freeman, T.L. and Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Frontiers in immunology. 2020, 11, 1518. [CrossRef]
- Bernard, I., Limonta, D., Mahal, L.K. and Hobman, T.C. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses. 2020, 13(1), 29.
- Grobler, C., Maphumulo, S.C., Grobbelaar, L.M., Bredenkamp, J.C., Laubscher, G.J., Lourens, P.J., Steenkamp, J., Kell, D.B. and Pretorius, E. Covid-19: The Rollercoaster of Fibrin (Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. International journal of molecular sciences. 2020, 21(14), 5168. [CrossRef]
- Nicin L, Abplanalp WT, Mellentin H et al. Cell type specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur Heart J. 2020, 41, 1804–1806.
- Sluimer JC, Gasc JM, Hamming I et al. Angiotensin converting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J Pathol. 2008, 215, 273–279. [CrossRef]
- Lu R, Zhao X, Li J et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020, 395, 565–574. [CrossRef]
- Hikmet F, Mear L, Edvinsson A, Micke P, Uhlen M, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol Syst Biol. 2020, 16, 9610. [CrossRef]
- Toni M. Delorey, Carly G. K. Ziegler, Graham Heimberg, Rachelly Normand, Yiming Yang, Åsa Segerstolpe, Domenic Abbondanza, Stephen J. Fleming, Ayshwarya Subramanian, Daniel T. Montoro, Karthik A. Jagadeesh, Kushal K. Dey, Pritha Sen, Michal Slyper, Yered H. Pita-Juárez, Devan Phillips, Jana Biermann, Zohar Bloom-Ackermann, Nikolaos Barkas, Andrea Ganna, James Gomez, Johannes C. Melms, Igor Katsyv, Erica Normandin, Aviv Regev. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. 2021. 595, 107-113.
- Peng Wang, Ronghua Luo, Min Zhang, Yaqing Wang, Tianzhang Song, Tingting Tao, Zhongyu Li, Lin Jin, Hongyi Zheng, Wenwen Chen, Mengqian Zhao, Yongtang Zheng, Jianhua Qin. A cross-talk between epithelium and endothelium mediates human alveolar-capillary injury during SARS-CoV-2 infection. Cell death and disease. 2020. 11(12), 1042.
- Vanessa Monteil , Hyesoo Kwon, Patricia Prado, Astrid Hagelkrüys, Reiner A Wimmer, Martin Stahl, Alexandra Leopoldi, Elena Garreta, Carmen Hurtado Del Pozo, Felipe Prosper, Juan Pablo Romero, Gerald Wirnsberger, Haibo Zhang, Arthur S Slutsky, Ryan Conder, Nuria Montserrat, Ali Mirazimi, Josef M Penninger. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell. 2020. 14;181 (4), 905-913.
- Schimmel, L., Chew, K.Y., Stocks, C.J., Yordanov, T.E., Essebier, P., Kulasinghe, A., Monkman, J., Dos Santos Miggiolaro, A.F.R., Cooper, C., de Noronha, L., Schroder, K., Lagendijk, A.K., Labzin, L.I., Short, K.R. and Gordon, E.J. Endothelial cells are not productively infected by SARS-CoV-2. Clinical & translational immunology. 2021, 10(10), e1350.
- Andersson, M.I., Arancibia-Carcamo, C.V., Auckland, K., Baillie, J.K., Barnes, E., Beneke, T., Bibi, S., Brooks, T., Carroll, M., Crook, D., Dingle, K., Dold, C., Downs, L.O., Dunn, L., Eyre, D.W., Gilbert Jaramillo, J., Harvala, H., Hoosdally, S., Ijaz, S., James, T., James, W., Jeffery, K., Justice, A., Klenerman, P., Knight, J.C., Knight, M., Liu, X., Lumley, S.F., Matthews, P.C., McNaughton, A.L., Mentzer, A.J., Mongkolsapaya, J., Oakley, S., Oliveira, M.S., Peto, T., Ploeg, R.J., Ratcliff, J., Robbins, M.J., Roberts, D.J., Rudkin, J., Russell, R.A., Screaton, G., Semple, M.G., Skelly, D., Simmonds, P., Stoesser, N., Turtle, L., Wareing, S. and Zambon, M. SARS-CoV-2 RNA detected in blood products from patients with COVID-19 is not associated with infectious virus. Wellcome open research, 2020, 5, 181.
- Ranucci, M., Ballotta, A., Di Dedda, U., Baryshnikova, E., Dei Poli, M., Resta, M., Falco, M., Albano, G. and Menicanti, L. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. Journal of thrombosis and haemostasis: JTH. 2020, 18(7), 1747-1751. [CrossRef]
- Fodor, A., Tiperciuc, B., Login, C., Orasan, O.H., Lazar, A.L., Buchman, C., Hanghicel, P., Sitar-Taut, A., Suharoschi, R., Vulturar, R. and Cozma, A. Endothelial Dysfunction, Inflammation, and Oxidative Stress in COVID-19-Mechanisms and Therapeutic Targets. Oxidative medicine and cellular longevity. 2021, 8671713.
- Xu, S., Ilyas, I. and Weng, J. Endothelial dysfunction in COVID-19: an overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacologica Sinica. 2023, 44(4), 695-709. [CrossRef]
- Miesbach, W. and Makris, M. COVID-19: Coagulopathy, Risk of Thrombosis, and the Rationale for Anticoagulation. Clinical and applied thrombosis/hemostasis: official journal of the International Academy of Clinical and Applied Thrombosis/Hemostasis. 2020, 26, 1076029620938149.
- Nguyen Dinh Cat, A., Montezano, A.C., Burger, D. and Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxidants & redox signaling. 2013, 19(10), 1110-1120.
- Iba, T., Connors, J.M. and Levy, J.H. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflammation research: official journal of the European Histamine Research Society ...[et al.]. 2020, 69(12), 1181-1189.
- Brandon Michael Henry, Maria Helena Santos de Oliveira, Isaac Cheruiyot, Justin L. Benoit, David S. Cooper, Giuseppe Lippi, Timothy D. Le Cras & Stefanie W. Benoit. Circulating level of Angiopoietin-2 is associated with acute kidney injury in coronavirus disease 2019 (Covid-19). Angiogenesis. 2021, 24, 403-406. [CrossRef]
- Lu, R.X.Z., Lai, B.F.L., Rafatian, N., Gustafson, D., Campbell, S.B., Banerjee, A., Kozak, R., Mossman, K., Mubareka, S., Howe, K.L., Fish, J.E. and Radisic, M. Vasculature-on-a-chip platform with innate immunity enables identification of angiopoietin-1 derived peptide as a therapeutic for SARS-CoV-2 induced inflammation. Lab on a chip. 2022, 22(6), 1171-1186.
- Del Valle, D.M., Kim-Schulze, S., Huang, H., Beckmann, N.D., Nirenberg, S., Wang, B., Lavin, Y., Swartz, T.H., Madduri, D., Stock, A., Marron, T.U., Xie, H., Patel, M., Tuballes, K., Van Oekelen, O., Rahman, A., Kovatch, P., Aberg, J.A., Schadt, E., Jagannath, S., Mazumdar, M., Charney, A.W., Firpo-Betancourt, A., Mendu, D.R., Jhang, J., Reich, D., Sigel, K., Cordon-Cardo, C., Feldmann, M., Parekh, S., Merad, M. and Gnjatic, S. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nature medicine. 2020, 26(10), 1636-1643. [CrossRef]
- Rendeiro, A.F., Ravichandran, H., Bram, Y., Chandar, V., Kim, J., Meydan, C., Park, J., Foox, J., Hether, T., Warren, S., Kim, Y., Reeves, J., Salvatore, S., Mason, C.E., Swanson, E.C., Borczuk, A.C., Elemento, O. and Schwartz, R.E. The spatial landscape of lung pathology during COVID-19 progression. Nature. 2021, 593(7860), 564-569. [CrossRef]
- Ramaiah, M.J. mTOR inhibition and p53 activation, microRNAs: The possible therapy against pandemic COVID-19. Gene reports. 2020, 20, 100765. [CrossRef]
- Lei, X., Shi, F., Basu, D., Huq, A., Routhier, S., Day, R. and Jin, W. Proteolytic processing of angiopoietin-like protein 4 by proprotein convertases modulates its inhibitory effects on lipoprotein lipase activity. The Journal of biological chemistry. 2011, 286(18), 15747-15756. [CrossRef]
- Bhatraju, P.K., Morrell, E.D., Stanaway, I.B., Sathe, N.A., Srivastava, A., Postelnicu, R., Green, R., Andrews, A., Gonzalez, M., Kratochvil, C.J., Kumar, V.K., Hsiang, T., Gale, M.J., Anesi, G.L., Wyles, D., Broadhurst, M.J., Brett-Major, D., Mukherjee, V., Sevransky, J.E., Landsittel, D., Hung, C., Altemeier, W.A., Gharib, S.A., Uyeki, T.M., Cobb, J.P., Liebler, J.M., Crosslin, D.R., Jarvik, G.P., Segal, L.N., Evans, L., Mikacenic, C. and Wurfel, M.M. 'Angiopoietin-Like4 Is a Novel Marker of COVID-19 Severity', Critical care explorations. 2022, 5(1), e0827. [CrossRef]
- Reyes, A., Corrales, N., Galvez, N.M.S., Bueno, S.M., Kalergis, A.M. and Gonzalez, P.A. Contribution of hypoxia inducible factor-1 during viral infections. Virulence. 2020, 11(1), 1482-1500. [CrossRef]
- Hadjadj, J., Yatim, N., Barnabei, L., Corneau, A., Boussier, J., Smith, N., Pere, H., Charbit, B., Bondet, V., Chenevier-Gobeaux, C., Breillat, P., Carlier, N., Gauzit, R., Morbieu, C., Pene, F., Marin, N., Roche, N., Szwebel, T., Merkling, S.H., Treluyer, J., Veyer, D., Mouthon, L., Blanc, C., Tharaux, P., Rozenberg, F., Fischer, A., Duffy, D., Rieux-Laucat, F., Kerneis, S. and Terrier, B. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science (New York, N.Y.). 2020, 369(6504), 718-724. [CrossRef]
- Vassiliou, A.G., Keskinidou, C., Jahaj, E., Gallos, P., Dimopoulou, I., Kotanidou, A. and Orfanos, S.E. ICU Admission Levels of Endothelial Biomarkers as Predictors of Mortality in Critically Ill COVID-19 Patients. Cells. 2021, 10(1), pp. 186. [CrossRef]
- Osama Abou Arab, Youssef Bennis, Pierre Gauthier, Herve Dupont, Said Kamel, Yazine Mahjoub. Association between inflammation, angiopoietins, and disease severity in critically ill COVID-19 patients: a prospective study. British Journal of Anaesthesia. 2021, 126. (3), 127-130 . [CrossRef]
Figure 1.
A schematic demonstrating the Angiopoietin 1 and 2 molecular signalling in a normal and abnormal vasculature. The binding of Angpt 1 and Tie 2 at junctional complexes receptor results in stimulation of PI3K/Akt, antiapoptotic protein, nitric oxide synthase 3 (eNOS) and that in turn promotes endothelial cell survival throughout normal vasculature. The inhibition of FKHR by Angpt1/Tie 2 receptor phosphorylation inhibits inflammatory molecules and support vascular stability. While, during the abnormal condition, the binding of Angpt 2 and Tie 2 result in pericyte loss, increased in inflammatory gene expression and vascular destabilisation.
Figure 1.
A schematic demonstrating the Angiopoietin 1 and 2 molecular signalling in a normal and abnormal vasculature. The binding of Angpt 1 and Tie 2 at junctional complexes receptor results in stimulation of PI3K/Akt, antiapoptotic protein, nitric oxide synthase 3 (eNOS) and that in turn promotes endothelial cell survival throughout normal vasculature. The inhibition of FKHR by Angpt1/Tie 2 receptor phosphorylation inhibits inflammatory molecules and support vascular stability. While, during the abnormal condition, the binding of Angpt 2 and Tie 2 result in pericyte loss, increased in inflammatory gene expression and vascular destabilisation.
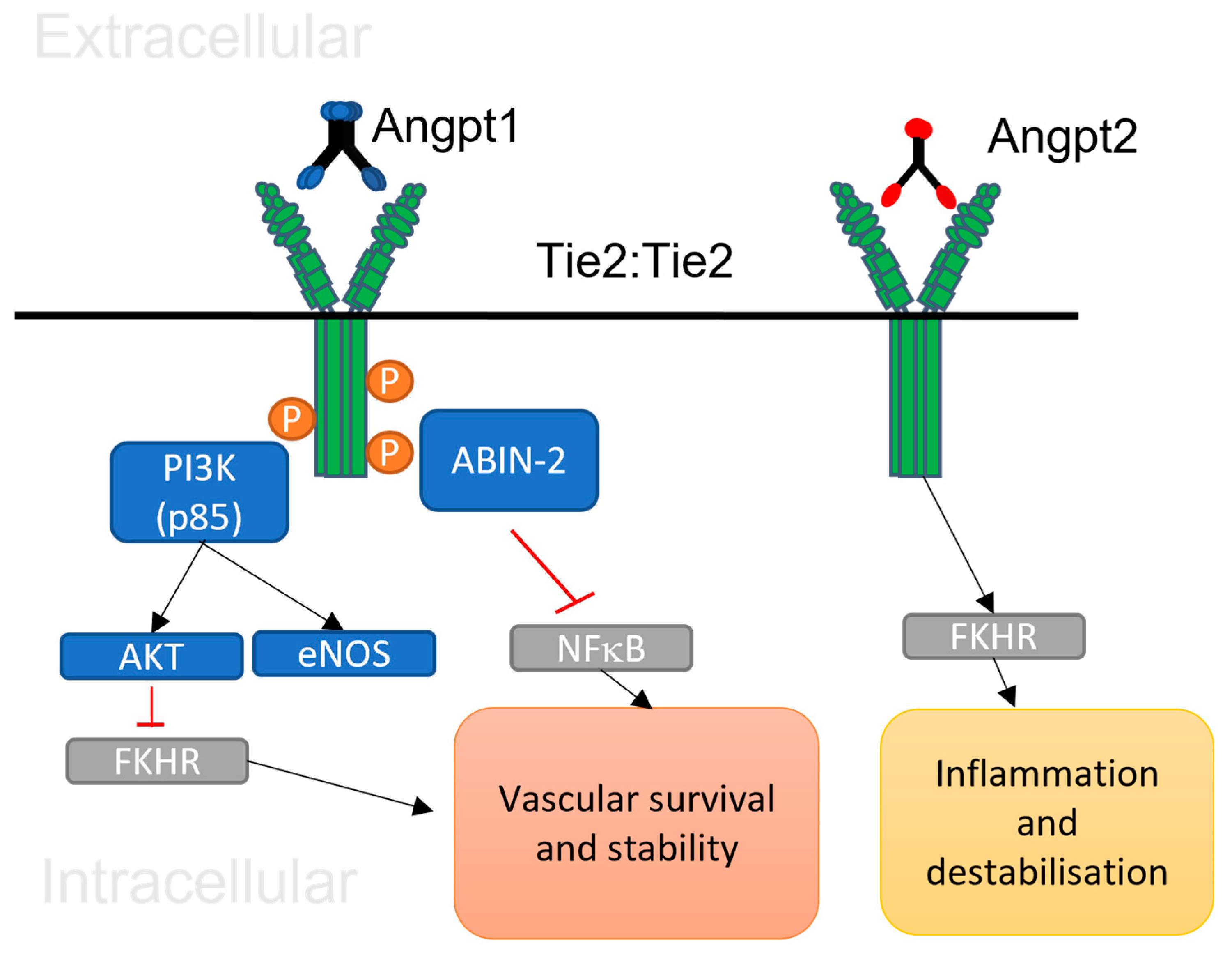
Figure 2.
A schematic demonstrating the molecular mechanisms of the effect of SARS-CoV-2 infection in endothelial cells. Virus infection affects endothelium and vascular function by several pathways. The inflammation response during this virus infection stimulates the Renin angiotensin system (RAS) by elevating angiotensin 1 or alternatively through decreasing the extracellular activation of angiotensin converting enzyme 2 (ACE2). SARS-CoV-2 infection elevates reactive oxygen species (ROS) and stimulates NFκB, and that in turn reduces nitric oxides and elevates inflammatory cytokines such as TNF-α and IL-6. This mechanism results in damaging the equilibrium amidst the vessel relaxation and narrowing, inflammation, blood clotting in arteries and capillaries, and thrombocytes stimulation.
Figure 2.
A schematic demonstrating the molecular mechanisms of the effect of SARS-CoV-2 infection in endothelial cells. Virus infection affects endothelium and vascular function by several pathways. The inflammation response during this virus infection stimulates the Renin angiotensin system (RAS) by elevating angiotensin 1 or alternatively through decreasing the extracellular activation of angiotensin converting enzyme 2 (ACE2). SARS-CoV-2 infection elevates reactive oxygen species (ROS) and stimulates NFκB, and that in turn reduces nitric oxides and elevates inflammatory cytokines such as TNF-α and IL-6. This mechanism results in damaging the equilibrium amidst the vessel relaxation and narrowing, inflammation, blood clotting in arteries and capillaries, and thrombocytes stimulation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



