
Submitted:
05 February 2024
Posted:
05 February 2024
You are already at the latest version
Abstract
Fibroblast activation protein (FAP) is a serine protease characterized by its high expression in cancer-associated fibroblasts (CAFs) and near absence in adult normal tissues and benign lesions. This unique expression pattern positions FAP as a prospective biomarker for targeted tumor radiodiagnosis and therapy. The advent of FAP-based radiotheranostics is anticipated to revolu-tionize cancer management. Among various types of FAP ligands, peptides and antibodies, have shown advantages over small molecules, exemplifying prolonged tumor retention in human volunteers. Within its scope, this review summarizes the recent research progress of the FAP radiopharmaceuticals based on antibodies and peptides, on tumor imaging and therapy. Additionally, it incorporates insights from recent studies, providing valuable perspectives on the clinical utility of FAP-targeted radiopharmaceuticals.
Keywords:
FAP
; Radiopharmaceuticals
; PET Imaging
; Theranostics
; Radionuclide therapy
1. Introduction
Cancer is a heterogeneous disease that develops in an incredibly complex microenvironment [1,2,3,4,5,6]. This microenvironment encompasses not only cancerous cells but also vital constituents like the extracellular matrix [7,8,9,10], stromal cells [11,12,13], and immune cells [14,15,16], forming the tumor microenvironment (TME) [17,18,19,20]. The TME, an intricate milieu unique to tumors, exerts profound influences on tumor progression, immune evasion, metastasis, and therapeutic resistance [4,21,22,23,24,25,26].
Among the various types of fibroblasts implicated in cancer, cancer-associated fibroblasts (CAFs) stand out prominently [27,28,29,30,31,32,33,34]. These CAFs constitute a staggering 80% of all fibroblasts within the TME, assuming a pivotal role as oncogenic regulators with far-reaching effects on tumor cell proliferation, migration, extracellular matrix remodeling, and immunosuppression [35,36,37,38,39,40,41]. Originating from fibroblasts in normal tissues [42,43,44,45], these cells undergo a reversible transformation akin to myofibroblasts after injury, actively participating in wound healing [27,46,47,48,49]. Myofibroblasts transition from their initial presence in granulation tissue to become the predominant cell type in the proliferative phase, ultimately diminishing as the wound healing process concludes [50,51,52]. In the context of cancer, fibroblasts, and other stromal cells orchestrate this transformation into CAFs by secreting transforming growth factors in the TME [27,46,53,54]. Notably, CAFs, in contrast to cancer cells, exhibit remarkable stability and resistance to drug resistance, underscoring their viability as a pivotal biological target for cancer diagnosis and therapy [55,56,57,58].
Identification of specific biomarkers on the surface of CAFs offers a strategic avenue for targeted radiological diagnostics and therapeutics [59,60,61,62,63,64]. Among these biomarkers, fibroblast activation protein (FAP) has gained widespread attention for its potential in CAF identification and targeting [65,66,67,68,69,70,71]. FAP, a member of the dipeptidyl peptidase 4 (DPP4) family, boasts a molecular weight of 170 kDa [72,73,74,75,76]. It assumes the guise of a type II transmembrane serine protease, typically existing as a homodimer [77,78,79]. Functionally, FAP exhibits dipeptidyl peptidase and endopeptidase activities [65,80,81,82], and its significance extends to normal embryonic development and tissue modeling [83,84,85]. Remarkably, FAP remains scarcely noticeable or entirely absent in normal adult tissues [86,87]. However, it undergoes marked upregulation during processes such as wound healing, atherosclerotic plaque formation, and fibrosis [88,89,90,91], and prominently features in over 90 % of human epithelial carcinomas [72,92,93,94,95,96,97,98,99]. The consistent presence and overexpression of FAP in CAFs across numerous epithelial tumors, including colon, pancreatic, hepatic, and ovarian cancers, have paved the way for targeted FAP approaches in tumor imaging and therapy [100,101,102,103,104,105].
As an extensively explored target, especially in conjunction with positron emission tomography/computerized tomography (PET/CT) [106,107], various strategies including small molecules, peptides, and antibodies have emerged to harness FAP for tumor imaging and treatment [108,109,110,111,112]. Small molecule FAP inhibitors (FAPIs) have entered a new milestone since UAMC-1110 developed by Jansen et al. in 2014 [113]. Numerous reviews on small molecule FAPIs have been summarized, such as the comprehensive review by Lan and Cai in 2022 [114]. While small molecule FAPIs have shown remarkable efficacy in tumor imaging, their therapeutic potential for tumors is limited by their short retention period in tumor tissues. Conversely, FAP inhibitors based on peptides and antibodies offer significantly prolonged tumor retention and are considered superior targeting vehicles for targeted radionuclide therapy. This focused review discusses the development and application of antibody- and peptide-based radiopharmaceuticals targeting FAP.
2. Antibody-based radiopharmaceuticals targeting FAP
2.1. Iodine-131-labeled monoclonal antibody F19
The discovery of FAP, a type II transmembrane serine protease, can be traced back to 1986 when it was initially identified as the F19 antigen during studies involving cultured fibroblasts and the monoclonal antibody (mAb) F19 in 1986 [115,116,117]. Subsequently, in 1994, the surface antigen expressed by F19 cells was officially named FAP [86,118]. In 1990, Garin-Chesa et al. proposed that in the context of cancer, epithelial cancer, F19+ fibroblasts, colloquially referred to as FAP, emerged as a consistent molecular trait of the reactive stroma. The role of mAbF19 in their identification was pivotal [94]. Human FAP, discerned through mAbF19, became a prominent cell surface antigen [119]. Due to its abundant presence within the tumor mesenchyme, FAP can serve as a target for radionuclide antibody conjugates in cancer patients [119,120,121]. Various radionuclide-labeled antibodies, designed for FAP-targeted imaging and therapy, have been developed (Table 1).
In 1988, Old et al. conducted a comprehensive examination of six human cell surface glycoproteins, each defined by mAb, with the intention of characterizing the surface phenotype of cultivated mesenchymal cells [92]. Among these antibodies, mAbF19 effectively identified glycoproteins with molecular weights of 120,000 and 95,000, expressed on cultivated fibroblasts and a proportion of sarcoma cell lines, respectively. This discovery marked mAbF19 as a superior antibody for these purposes compared with other candidates, such asmAbF24, G171, G253, and K117. Another antibody, S5, exhibited expression patterns similar to mAbF19 but had limited in vivo expression [92]. It became evident that the fibroblasts surrounding tumor cells offer effective targets for cancer immunolocalization or immunotherapy, owing to their recognition by mAbF19 [122,123,124].
The pioneering clinical study involving FAP-targeting radiopharmaceuticals utilized 131I-labeled mAbF19 for tumor imaging in patients with liver metastases from colon cancer [125]. Welt et al. conducted this study in 1994, wherein 17 patients scheduled for resection of localized metastases or regional chemotherapy received intravenous administration of 131I-mAbF19. Imaging results from this series of studies revealed that the tumor-to-normal tissue ratio reached its peak after 3-5 days of administration, enabling the visualization of lesions as small as 1 cm in diameter. Notably, colon cancer studies demonstrated specific localization of tumors and metastatic lesions. However, SPECT/CT results revealed slow kidney clearance, necessitating 3-5 days to achieve optimal imaging outcomes. This delayed renal clearance has implications for the imaging capabilities of 131I-mAbF19 (Figure 1) [125].
2.2. Iodine-131-labeled sibrotuzumab
mAbF19 stands out for its FAP-specific targeting capabilities, and scientists have endeavored to enhance its imaging potential as a radiopharmaceutical [131]. A study by Welt et al. established the positive expression of FAP in all 17 patients studied, and SPECT/CT imaging effectively facilitated precise tumor identification in humans [125]. This encouraged further investigation into the pharmacokinetic (PK) of sibrotuzumab, a humanized version of the murine anti-FAP mAbF19. A phase I&II clinical trial (NCT02198274) involving sibrotuzumab sought to assess its PK without radiolabeling in patients with FAP-positive malignancies, particularly advanced metastatic colorectal cancer patients. However, among the 17 patients enrolled who had undergone rigorous pretreatment, only two exhibited stable disease status, a count insufficient to meet the phase II trial criteria, which typically necessitate four patients in a stable condition or at least one patient in complete or partial remission. Consequently, the trial did not progress beyond phase II [132]. The PK study unveiled pertinent findings, including a mean clearance rate of 39.8 ± 19.8 mL/h and a terminal half-life of 5.3 ± 2.3 days for sibrotuzumab. Comparatively, mAbF19 displayed a mean clearance rate of 109 mL/h and a terminal half-life of 38 h. Sibutuzumab’s lower clearance rate and significantly protracted half-life rendered it a more amenable candidate for radioimmunotherapy. However, this study underscored the inadequacy of unlabeled sibrotuzumab in the treatment of advanced solid tumors, instigating investigations into sibrotuzumab radioisotope conjugates [132].
One such avenue of exploration encompassed a clinical phase I dose-escalation study of sibrotuzumab in patients with colorectal or non-small cell lung cancer (NCT02209727). This study, conducted in parallel with the aforementioned phase I/II trials, sought to evaluate biodistribution, PKs, immunogenicity, and safety profiles by incrementally administering sibrotuzumab intravenously to 26 patients. In tandem, two antecedent phase I studies employed 131I-mAbF19, with a focus on patients with hepatic metastases from colorectal cancer or soft tissue sarcoma [133]. These investigations delved into the PK parameters of the therapeutic mouse monoclonal antibody 131I-mAbF19, elucidating principles of selective tumor accumulation and stromal targeting in tumors through biodistribution imaging studies and biopsy analyses. These pivotal insights provided the foundation for the inaugural human clinical evaluation of sibrotuzumab.
In this human trial, patients received 8-10 mCi of 131I-labeled sibrotuzumab, administered concomitantly at weeks 1, 5, and 9 in a 12-week dosing cycle, with a focus on PK assessments. The ensuing analysis divulged that the mean clearance rate of 131I-sibrotuzumab amounted to 41.9 ± 16 mL/h, accompanied by a half-life of 4.9 days. Following a single cycle of 131I-sibrotuzumab treatment, two out of the 26 patients manifested stable disease conditions. The relatively abbreviated half-life of sibrotuzumab held promise for radioimmunotherapy. Regrettably, the trial did not yield definitive efficacy results for sibrotuzumab, prompting the discontinuation of further clinical development (Figure 1)[126]. Furthermore, it is noteworthy that despite considerations of alternative targets, the commercial viability of 131I-sibrotuzumab waned, ultimately leading to its discontinuation in 2014 [134].
2.3. 177Lu-ESC11/ESC14
The clinical application of 131I-mAbF19 and its humanized derivative, sibrotuzumab, has been hindered by their prolonged blood clearance and suboptimal therapeutic efficacy. To enhance the therapeutic potential of FAP-targeted antibodies, it is imperative to develop antibodies with enhanced attributes and utilize radiolabeling with more suitable radionuclides [135,136,137,138]. In addressing these considerations, Fischer et al. introduced a significant advancement with the development of 177Lu-ESC11 and 177Lu-ESC14 [127].
From the perspective of antibody discovery, ESC11 and ESC14, two antibodies noted for their selective accumulation within xenografted FAP-positive human melanoma and their capacity to impede tumor growth in vivo, were identified in the human FAP antibody library using the phage display technique. Subsequently, these antibodies underwent a transformation into IgG1 antibodies [127]. The phage display technique is a new technique predicated on specific affinity interactions, enabling the identification of proteins or peptides that exhibit particular binding properties. This technique is especially adept at discovering antibodies targeting challenging and intriguing molecules, making it a cornerstone in the quest for antibodies with precise attributes [139,140,141,142,143]. It enjoys widespread utilization in the quest for human antibody fragments boasting specific binding activity [143,144,145]. This technique facilitates the evolution and optimization of FAP-targeting antibodies, culminating in the selection of antibodies exhibiting robust affinity, rapid internalization, and propensity for tumor accumulation. Following their identification, ESC11 and ESC14 obtained by screening were labeled with lutetium-177 (177Lu), leading to a comprehensive investigation into their in vivo targeting capabilities. The outcomes of these investigations substantiated that both modified mAbs can selectively bind to human and mouse FAP, featuring affinities of 10 nM and 210 nM, respectively (Figure 1) [127].
From a radionuclide perspective, radiolabeling with iodine-131 is relatively straightforward for mAbs [146]. However, in the context of radioimmunotherapy, iodine-131 proves to be suboptimal due to its propensity for facile release from tumor sites after internalization of mAbs within cells [147,148]. A related drawback lies in the emission of high-energy (364 keV) γ-photons, accounting for a substantial 82% of its radiation output, thus posing concerns for radiation safety [149,150]. In stark contrast, the radiolanthanide 177Lu presents a more favorable profile, with a shorter emission range of 2 mm as opposed to iodine-131’s 3 mm. Additionally, 177Lu exhibits markedly improved physical radiation characteristics, featuring the emission of 208 keV γ-photons at a much lower abundance (11 %) [127]. As a result of these considerations, Fischer et al. chose 177Lu for antibody labeling. This decision culminated in the successful radiolabeling of ESC11 and ESC14 through their conjugation with CHX-A’’-DTPA.
These radiolabeled antibodies were subsequently administered to mice harboring SK-Mel-187 and SK-Mel-16 xenograft tumors to assess their tumor uptake. In this investigation, 177Lu-CHX-A’’-DTPA-vF19 and 177Lu-CHX-A’’-DTPA-A33 were included as control groups. Notably, SPECT/CT imaging conducted 72 h post-injection revealed a higher specific uptake of 177Lu-ESC11 in SK-MEL-187 tumors, whereas SK-MEL-16 xenografts exhibited lower uptake compared with the control group. This study, involving a comparative analysis of the in vivo targeting attributes of human-mouse chimeric antibodies, established that in mouse models characterized by higher levels of antigen expression, the cumulative tumor uptake of the nuclide-labeled antibody could reach levels corresponding to 50% of the administered dose per gram. Conclusive in vivo experiments in mice further corroborated that the ratio of tumor-to-organ uptake pertaining to 177Lu-labeled FAP mAbs ESC11 and ESC14 surpassed that of their first-generation radionuclide-labeled FAP-targeted antibodies [127]. The novel antibodies, ESC11 and ESC14, exhibited efficient internalization into FAP-expressing cells, thereby manifesting highly satisfactory in vivo targeting capabilities.
2.4. 89Zr-labeled F19 and B12 IgG
Clinical investigations involving first-generation FAP-targeting antibodies have provided novel insights by demonstrating the feasibility of modifying FAP-specific cancer targeting through the conjugation of toxins or chelators with FAP-specific antibodies[151,152]. As an illustrative example, Pandya et al. prepared the radiopharmaceutical antibody conjugate [89Zr]Zr-Df-Bz-F19 mAb for PET imaging by employing the bifunctional chelator Df-Bz-NCS to securely bind zirconium-89 (89Zr) (Figure 1) [128].
89Zr, a radionuclide, emits β+ particles at 902 keV with an abundance of 23% and possesses a half-life of 78.4 h [149,153]. Its attributes, characterized by high-resolution imaging, specific tissue binding, and strong signal contrast, render it a promising candidate for PET imaging applications [154,155]. Nonetheless, certain challenges persist, primarily the susceptibility to covalent bond breakage between the chelator and the protein, which can compromise stability [156]. To mitigate this concern, comprehensive in vitro characterization of [89Zr]Zr-Df-Bz-F19 mAb was conducted. The radiolabel displayed a remarkable radioactive purity exceeding 99.5% upon synthesis completion and retained its purity at levels greater than 99.1% in human serum after 7 days of incubation at 36℃-37℃. These findings affirm the robust stability of nucleoporin labeling and underscore the ability of [89Zr]Zr-Df-Bz-F19 to maintain its structural integrity following in vivo administration.
In vivo experiments in U87MG tumor-bearing mice unveiled the rapid clearance of [89Zr]Zr-Df-Bz-F19 from the bloodstream, with 51% of the radioactive drug eliminated from the blood within the timeframe spanning 2 to 72 h. Notably, 89Zr was observed to accumulate in bone tissues. Furthermore, the study revealed an increase in the average tumor-to-kidney (T/K) and tumor-to-blood uptake ratios over time, specifically at 2, 24, 48, and 72 h. After 72 h, the metabolism of the 89Zr labeled through bifunctional chelation of F19 became apparent. The exceptional radioactive purity, in vivo stability, and favorable tumor-to-tissue uptake ratio collectively position [89Zr]Zr-Df-Bz-F19 as a promising candidate for PET/CT imaging [128].
FAP expression is documented in multiple solid cancers, yet limited knowledge exists regarding its prevalence in metastatic castration-resistant prostate cancer (mCRPC) [157,158,159,160]. The evolving landscape of precision treatment strategies for prostate cancer was highlighted in the European Society of Medical Oncology 2022 report [161,162]. Advancements in CRPC therapies hinge on accurate imaging modalities, lesion visualization, disease staging, and informed therapeutic decision-making [149]. These findings underscore the ongoing demand for selective and sensitive imaging probes applicable to mCRPC patients.
To address the specific context of mCRPC, Hallie et al. employed a humanized antibody, initially identified by phage display, and labeled it with 89Zr. This antibody serves as a FAP-expressing tumor-selective imaging probe for PET/CT imaging in a preclinical prostate cancer xenograft model [129]. The investigative process commenced with genomic and immunohistochemistry assessments to determine the expression of FAP in prostate cancer [129,163,164]. Specifically, FAP localization in tissues and cells was determined via antigen-antibody binding reactions. Genomic analysis was predicated on RNA sequencing derived from primary prostate cancer patient sample or mCRPC bone and soft tissue tumor biopsies [165,166]. Concurrently, immunohistochemistry analyses were performed on cancer samples from a biological materials repository [129]. These comprehensive genomic and immunohistochemistry investigations unequivocally demonstrated FAP expression across transgenomic subtypes of metastatic disease and metastatic sites. Importantly, this affirmed FAP’s utility as a target imaging and therapeutic intervention within the prostate cancer tumor microenvironment [129,167].
Further validation entailed assessing the designed probe’s specificity for FAP-expressing tumors using near-infrared (NIR) dye-labeled B12 IgG. The outcomes affirmed that B12 IgG effectively and specifically detects cells expressing FAP by NIR fluorescence imaging [168]. This critical step established the foundation for the prospective development of B12 IgG as a contrast agent for PET imaging.
Subsequent PET/CT imaging evaluations were conducted in a mouse model established through the subcutaneous injection of CWR-R1FAP cells. Notably, [89Zr]Zr-B12 IgG demonstrated significantly enhanced tumor uptake in FAP-positive cells in mice bearing CWR-R1FAP relative to the control group injected with [89Zr]Zr-IC IgG. This heightened tumor accumulation is attributed to improved permeability and retention efficiency, leading to a sustained presence of [89Zr]Zr-IC IgG probe within the tumor tissue, unlike the control probe, which exhibited rapid clearance and near-invisibility at 72 h. Compared with that in the control group, there was heightened accumulation of [89Zr]Zr-B12 IgG in FAP-positive tumor cells (Figure 1) [129].
In a critical extension of this work, mice were subcutaneously injected with hPrCSC-44 (an immortalized human prostate cancer stromal cell line) in combination with DU145 (a FAP-null prostate cancer cell line) to establish a xenograft mouse model. Subsequent administration of [89Zr]Zr-B12 IgG or control [89Zr]Zr-IC IgG in mice bearing subcutaneous hPrCSC-44/DU145 xenografts, followed by serial imaging was performed at 24, 48, 72, 96, 120 and 144 h post-injection, allowed for comprehensive PET imaging evaluation. The results showed maximum tumor uptake of [89Zr]Zr-B12 IgG at 24 h, which surpassed control levels by 4-5 times. Additionally, pronounced [89Zr]Zr-B12 IgG uptake in the liver was observed. Over time, tumor and liver uptake exhibited parallel reductions, with diminished tumor uptake evident at 144 h after injection [129].
[89Zr]Zr-B12 IgG has the potential for noninvasive PET/CT imaging applications in mCRPC, serving as a specific imaging probe for FAP-expressing tumors. Notably, B12 IgG can be internalized by FAP-expressing cells, as evidenced by its robust tumor accumulation. Furthermore, it demonstrates an adeptness for preserving imaging signals and holds promise for immunoconjugate-based therapies. Importantly, B12 IgG facilitates facile nuclide exchange through chelator conjugation, rendering it a promising candidate for radioimmunotherapy applications [129].
2.5. Bispecific Antibodies
In the realm of tumor-targeting antibodies, an exciting development involves Hoffmann LaRoche’s bispecific antibodies RG7386 (FAP-DR5) and RO7300490 (FAP-CD40) (Figure 1) [130,169,170]. Currently in phase I clinical trials (NCT02558140, NCT04857138) [171,172], these antibodies represent a novel approach. One of these antibodies is engineered to target FAP for precise localization, while the other is designed to interact with molecules influencing tumor apoptosis or necrosis. These bispecific antibodies work in tandem, obstructing different signaling pathways simultaneously. Compared with monoclonal antibodies, bispecific antibodies possess two specific antigen-binding sites, which endow them with stronger specificity. Consequently, they precisely target tumor cells, minimize off-target toxicity, and can orchestrate immune cell-mediated tumor eradication through dual-target signal blockade [173,174,175,176,177,178,179,180]. This special structure confers a unique advantage to bispecific antibodies in the field of tumor therapy. Notably, LaRoche’s bispecific antibody, approved in 2017, achieved a revenue of $3.5 billion in 2021, establishing biphasic drugs as a prominent topic of interest following the success of PD-1 inhibitors.
The concept of bispecific antibodies also opens up new possibilities for radionuclide labeling. While no specific studies have explored the radionuclide labeling of bispecific antibodies targeting FAP, research into combination therapies involving radionuclides and bispecific antibodies targeting other antigens has been conducted [181,182]. For instance, Morris’ team combined radionuclides with bispecific antibodies (anti-CTLA-4 and anti-PD-L1). In a mouse model, this approach resulted in complete and enduring tumor remission, outperforming combinations involving monoclonal antibodies [183]. The potential to combine or label FAP-targeted bispecific antibodies is a promising avenue that introduces a fresh dimension to radiotherapy nuclide markers for FAP-targeted tumor therapy. Such innovations hold significant potential for advancing clinical FAP-targeted tumor therapy.
3. Peptide-based FAPIs
3.1. Peptide-based FAP radiopharmaceuticals
While radiolabeled antibodies against highly expressed FAPs on fibroblasts exhibit high affinity and specificity, their clinical utility in molecular imaging is hindered by prolonged circulation in the bloodstream and limited tumor penetration potential [184]. In contrast, small peptides typically exhibit favorable PK profiles characterized by rapid clearance from tissues and efficient tumor penetration [106,185,186,187,188,189,190]. As such, peptide-based radiopharmaceuticals have emerged as the most promising candidates for molecular imaging in the clinical setting [191,192,193,194,195,196,197,198,199,200,201,202,203,204]. Cyclic peptides, in particular, offer enhanced structural stability and resistance to enzymatic degradation compared with linear peptides, greatly enhancing their efficacy in radiological diagnosis and therapy [205,206,207,208,209,210,211,212,213]. FAP-targeted cyclic peptides have shown encouraging outcomes across numerous clinical trials.
3B Pharmaceuticals, for instance, introduced 263 different FAP-targeted peptide structures and identified peptides suitable for radiolabeling (Table 2). Notably, 3BP-3320, 3BP-3321, 3BP-3407, and 3BP-3554 exhibited exceptional radioactive purity. The selected polypeptides were labeled with indium-111 and subsequently injected into mice to evaluate the tissue-specific radioactive uptake. The findings highlighted that 111In-3BP-3554 displayed the highest tumor/tissue radioactivity uptake rate. Consequently, this tracer underwent in-depth in vivo PK evaluation in mice. Representative SPECT/CT imaging was performed three hours after injection of 111In-3BP-3554 in four different mouse sarcoma tumor models, consistently revealing significant radioactive uptake in all tumor types. Dose-response evaluations of 177Lu-3BP-3554 were further conducted in the Sarc4809 model. Compared with natLu-3BP-3554, 177Lu-3BP-3554 demonstrated marked inhibitory effects on tumors, with increasing doses amplifying this effect [214]. On September 21, 2019, Clovis Oncology secured an upfront payment of $12 million for exclusive rights to FAP-targeted radiolabeled peptidomimetics (FAP-2286) developed by 3B Pharmaceuticals, with the exception of Europe [215].
3.2. Structure of the selective and specific peptide FAPI
In their patent, 3B Pharmaceuticals has cataloged a range of peptide structures amenable to radiolabeling with radionuclides (Table 3 & Figure 2). Among these structures, 3BP-3554 emerged as a potential clinical candidate and was renamed FAP-2286.
Distinguishing features between FAP-2286 and other peptides within the same series manifest chiefly in the following facets. Firstly, the placement of the chelator dramatically affects both the radiopurity of the nuclide-labeled compound and its tumor uptake rate. When the chelator is linked to the tMeBn structure, which is instrumental in cyclization, it enhances radiopurity and results in a higher tumor uptake rate compared to compounds where the chelator is linked to the polypeptide chain. Moreover, different modifications of cyclic peptides can affect the compound’s specific affinity and polarity. For instance, altering only the cysteine substituent at the C-terminus of the cyclic peptide leads to corresponding changes in specific affinity. Moreover, extending the length of the chain segment impacts the compound’s polarity. In the absence of substituents, longer chain segments decrease molecule polarity, thereby diminishing polarity [216,217].
FAP-2286 demonstrates highly effective and specific binding to FAP of human and mouse origin, along with a pronounced inhibitory effect. The equilibrium dissociation constant (KD) values were 1.1 and 4.7 nM, and the half maximal inhibitory concentration (IC50) values were 3.2 and 22.1 nM for human and mouse FAP, respectively. The metal complexes of FAP-2286 exhibited potent binding to human and mouse FAP, featuring KD values of 0.2-1.4 nM and 1.9-7.7 nM, respectively. Importantly, these metal complexes did not compromise the inhibitory activity against FAP, affirming their exceptional FAP specificity. Mean IC50 values for FAP from human and murine sources were determined as 1.3-2.2 nM and 8.4-16.3 nM, respectively [216,218].
Furthermore, FAP-2286 has favorable stability, high radioactive purity after radionuclide labeling, and outstanding specific affinity. Consequently, Clovis Oncology and 3B Pharmaceuticals have embarked on collaborative initiatives to conduct preclinical imaging and therapeutic evaluation of FAP-2286. Concurrently, Clovis Oncology has initiated plans for clinical trials involving FAP-2286 [219,220].
3.3. 68Ga-FAP-2286 and 111In-FAP-2286 for nuclear imaging
Gallium-68-labeled FAP-2286 demonstrates remarkable efficacy in tumor imaging. 3B Pharmaceuticals directed their attention toward FAP-2286 and its chelation with natural nonradioactive metals (natGa-FAP-2286, natLu-FAP-2286, and natIn-FAP-2286), and proceeded to evaluate three radiotracers for their in vitro affinity and selectivity [216].
The excellent affinity, precision in targeting, and inherent stability of FAP-2286 and its complexes have been conclusively established, facilitating the coordination of gallium-68 and indium-111 radionuclides with FAP-2286 for use as PET or SPECT imaging agents, respectively [216]. In vivo SPECT imaging employing 111In-FAP-2286 in HEK-FAP tumor-bearing mice showcased stable accumulation within tumor tissues, accompanied by minimal uptake in non-tumor regions. Notably, the kidney displayed the highest nontargeted uptake, albeit the T/K ratio gradually declined over time, reaching its zenith at 48 h after treatment [216,218].
Compared with the small molecule FAPI series, cyclic peptide FAPIs manifested superior biological properties [221], including stronger receptor selectivity and binding affinity and longer tumor retention. Remarkably, 68Ga-FAP-2286 exhibited the same rapid renal clearance as 68Ga-FAPI-46, with no noticeable differences in tumor distribution between the two tracers [219]. Chen et al. performed a comparative uptake analysis of 68Ga-FAPI-46 and 68Ga-FAP-2286 in cancer patients to delineate the in vivo distribution patterns of different inhibitors [222]. 68Ga-FAP-2286 exhibited lower physiological uptake in muscles, salivary glands, thyroid, and pancreas than 68Ga-FAPI-46. Conversely, 68Ga-FAP-2286 displayed heightened uptake in the heart, kidneys, and liver relative to 68Ga-FAPI-46. Despite 68Ga-FAPI-46 undergoing clinical imaging investigations for diverse tumor models, its rapid blood clearance and limited tumor retention pose substantial limitations for diagnostic and therapeutic applications [223]. In preclinical studies, FAP-2286 demonstrated longer tumor retention and stronger antitumor activity over time than FAPI-46, maintaining consistent tumor uptake at 3 h post-injection. Key advantages inherent to FAP-2286 encompass elevated affinity for FAP binding, improved tumor accumulation, and prolonged tumor retention [222].
With its introduction into clinical practice, Clovis Oncology embarked on the radiolabeling of FAP-2286 with gallium-68 for clinical tumor imaging. In a phase I clinical trial (NCT04939610), 68Ga-FAP-2286 was used as a contrast agent for pretreatment PET scans in 30 patients with solid tumors. This approach will persist as a guide for pretreatment imaging and posttreatment evaluation in an ensuing phase II trial. Furthermore, Clovis Oncology has initiated a clinical evaluation of 68Ga-FAP-2286 (NCT04621435), which, as of June 2022, encompassed 48 patients with cancers of the breast, bladder, prostate, colon, head/neck, pancreas, sarcoma, cholangiocarcinoma, and lung [224]. Patients were subjected to 68Ga-FAP-2286 administration and imaged 64 ± 7 min after injection. Notably, cholangiocarcinoma exhibited the highest tumor uptake. PET imaging conducted on a 72-year-old patient with cholangiocarcinoma revealed peak tumor uptake at 120 min, with the tumor-to-background ratio progressively augmenting from 30 to 120 min. 68Ga-FAP-2286 PET emerges as a pivotal tool for staging patients across cancer types characterized by robust tumor uptake, renal metabolism, and negligible renal accumulation. Its application prospects are indeed promising [224].
3.4. 177Lu-FAP-2286 for radionuclide therapy
FAP-2286 can be radiolabeled with the radionuclide 177Lu for the treatment of clinical solid tumors. Prior to clinical trials, the therapeutic efficacy and specificity of this approach were evaluated in murine models. Notably, the administration of 177Lu-FAP-2286 to HEK293-FAP tumor-bearing mice and FAP-expressing xenografts mice from sarcoma patients did not result in significant weight loss. Furthermore, the tumor retention of 177Lu-FAP-2286 exceeded that of 177Lu-FAPI-46. Over the 24-72 h timeframe, the T/K ratio for 177Lu-FAP-2286 consistently increased, in contrast to the T/K ratio for 177Lu-FAPI-46 T/K ratio, which peaked at 24 h. Importantly, 177Lu-FAP-2286 exhibited commendable properties as an active targeting agent, characterized by potent and specific FAP binding, resulting in high tumor uptake, accumulation and demonstrable therapeutic effects [219].
Furthermore, clinical trials of FAP-2286 revealed prolonged tumor retention and superior tumor suppression compared with FAPI-46 [219]. Clovis Oncology initiated the LuMIERE phase I & II clinical trials of FAP-2286 (NCT04939610), enrolling patients with advanced solid tumors. Phase I focused on evaluating the safety and tolerability of 177Lu-FAP-2286, while phase II aims to determine the recommended dose of 177Lu-FAP-2286 and assess the objective response rate in patients. Phase I entails fixed-dose intravenous administration of 177Lu-FAP-2286 at six-week intervals, up to a maximum of six doses for patients exhibiting positive uptake of 68Ga-FAP-2286. The dose-escalation range, guided by Bayesian optimal interval design, spans between 3.7 and 9.25 GBq, encompassing four distinct doses (3.70 GBq/100 mCi, 5.55 GBq/150 mCi, 7.40 GBq/200 mCi, 9.25 GBq/250 mCi). Phase II involves intravenous administration of the recommended dose of FAP-2286 to a cohort of up to 40 patients with advanced solid tumors. As of October 2022, 177Lu-FAP-2286 is being examined with a 7.40 GBq metering regimen, with the LuMIERE study projected to conclude by June 1, 2026 [225].
The LuMIERE study has yielded noteworthy findings. In the phase I dose study, 11 patients underwent 68Ga-FAP-2286 imaging and received treatment with 177Lu-FAP-2286 across cohorts. Among these patients, three individuals with peritoneal pseudomucinous tumors or colorectal cancer received 3.70 GBq, while six patients with different solid tumors received 5.55 GBq, and two patients were administered 7.40 GBq of 177Lu-FAP-2286. Encouraging treatment outcomes were observed, with eight patients discontinuing treatment, and one patient, after completing six doses of 3.7 GBq 177Lu-FAP-2286 for more than 12 months, demonstrated stable disease without the requirement for subsequent anticancer interventions. Another with gallbladder adenocarcinoma, from the 5.55 GBq cohort, exhibited stable disease upon four doses. Notably, 177Lu-FAP-2286 exhibited a manageable safety profile with some preliminary evidence of antitumor activity [224].
Prof. Richard P. Baum released findings pertaining to the biodistribution and preliminary dosimetry from the first human trial of FAP-2286 in March 2022. Intravenous injection of 177Lu-FAP-2286, completed within 5-10 min, was performed on 11 patients with advanced adenocarcinoma, all of whom had undergone 68Ga-FAP-2286 or 68Ga-FAPI-04 PET/CT imaging. SPECT/CT imaging after treatment illustrated a significant uptake of 177Lu-FAP-2286 within the tumor lesions. Notably, Patient 4 with pancreatic cancer and liver, peripancreatic lymph node, and bone metastases, underwent PET/CT imaging before the intravenous injection of 177Lu-FAP-2286. The patient’s anterior and posterior SPECT/CT images 48 h post-injection illustrated a significant liver uptake of 177Lu-FAP-2286 (Figure 3a). Patient 6 with breast cancer, characterized by diffuse FAP-positive bone and bone marrow metastases and lymph node metastases in 68Ga-FAP-2286 PET/CT imaging, displayed regression of bone and bone marrow lesions within 10 days (Figure 3b) [220].
Coincidentally, in November 2022, Rao et al. administered [177Lu]Lu-FAP-2286 to a patient with systemic metastases from squamous cell carcinoma of the right lung[226]. This intervention was followed by meticulous pre- and post-treatment assessments through [68Ga]Ga-FAP-2286 PET/CT imaging. The MIP image revealed systemic metastases from the patient’s squamous cell carcinoma of the right lung, supraclavicular lymph nodes, irregular thickening on the right side, hypodense hepatic nodules, and osteolytic lesions in the left scapula. Following nine weeks of injection with [177Lu]Lu-FAP-2286 at a dose of 7.0 GBq, [68Ga]Ga-FAP-2286 PET/CT scans exhibited a reduction in the affected regions within the MIP images [226]. The above two investigations have provided clinical substantiation of FAP-2286’s efficacy in treating FAP-positive tumors, thereby implying its potential utility in addressing a spectrum of advanced primary tumors and metastases. However, both studies have certain limitations. Firstly, the patient cohort in these studies was relatively small and displayed heterogeneity. Secondly, the administration of 177Lu-FAP-2286 was performed as the last-line treatment, precluding a dose-escalation regimen. Finally, the assessment of the safety and efficacy of 177Lu-FAP-2286 hinged primarily on observational data. Therefore, it is imperative that comprehensive clinical trials are undertaken to thoroughly investigate the PK, safety profile, dosimetry, and therapeutic efficacy of 177Lu-FAP-2286 in patients with advanced solid tumors [149].
177Lu-FAP-2286 has demonstrated substantial therapeutic promise, with 177Lu serving as the radiolabeled nuclide pivotal in this therapeutic paradigm. Various 177Lu-labeled drugs targeting diverse molecular entities have received regulatory approval from the U.S. Food and Drug Administration (FDA) [227]. For example, 177Lu-PSMA-617, designated for treating male patients with PSMA-positive mCRPC, exhibited a noteworthy effective systemic half-life of 40 hours and entailed a mean absorbed red marrow dose of 0.03 GBq [227,228]. Similarly, 177Lu-DOTATATE, tailored for managing neuroendocrine tumors, boasted a prolonged effective systemic half-life of 55 h, accompanied by a mean absorbed red marrow dose of 0.04 GBq [227,229]. Comparatively, 177Lu-FAP-2286 manifested an effective systemic half-life of 35 h, coupled with a mean absorbed red marrow dose of 0.05 GBq. Importantly, the effective absorbed dose of 177Lu-FAP-2286 aligned closely with that of 177Lu-DOTATATE and 177Lu-PSMA-617, underscoring its compatibility for therapeutic application across a broad spectrum of malignancies. In addition, peptide-targeted radionuclide therapy with 177Lu-FAP-2286 relieves pain symptoms in invasive adenocarcinoma cases. Encouragingly, 177Lu-FAP-2286 also showed considerable potential in tumor remission and inhibition, shedding new light on FAP-targeted peptide-based radionuclide therapy. In the quest for improved efficacy modifying the radionuclide payload holds promise. For instance, while 177Lu emits gamma (γ) rays and β-particles, alternative radionuclides such as actinium-225 (225Ac) and radium-223 (223Re) emit α-rays capable of inducing DNA double-strand breaks, rendering them potentially more lethal than β-particles [149]. Presently, multiple enterprises are engaged in clinical investigations into FAP-2286, further underscoring its burgeoning status as an emerging therapeutic agent poised for future market introduction.
4. Conclusions
Since the seminal discovery by Garin-Chesa et al. in 1990, which revealed the high expression of FAP in most epithelial tumor cells[94], significant strides have been made in the realm of FAP-targeted therapeutics encompassing antibodies, and peptides. Radiolabeled antibodies have long stood as exemplars of high targeting affinity and specificity. Their high molecular weight translates to protracted circulation in the bloodstream and tumor retention time, but correspondingly sluggish clearance [230,231,232]. Peptide drugs are characterized by relatively low molecular weight and can offer better blood circulation and tumor retention. Peptides, owing to their potential for heightened receptor selectivity, binding affinity, and tumor retention, may outstrip other FAP inhibitors in the realms of diagnostic and therapeutic efficacy [233,234,235,236,237,238]. In conclusion, FAP has emerged as a prominent target in the pursuit of tumor-specific interventions, demonstrating promising outcomes in tumor imaging and therapy research. Nonetheless, the present study’s limitations, including a relatively small sample size and abbreviated follow-up duration, underscore the imperative for extensive future investigations and clinical trials to explore the diagnostic or therapeutic properties of FAP-targeted applications.
Author Contributions
Conceptualization, K.H.; Writing-original draft preparation, X.S., Y.W.; Writing-review and editing, X.K.W., X.G., S. Zh., R.L., K.H.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Nonprofit Central Research Institute Fund of Chinese Academy of Medical Sciences (No. 2022-RC350-04), the CAMS Innovation Fund for Medical Sciences (Nos. 2021-I2M-1-026, 2022-I2M-2-002-2, and 2021-I2M-3-001), Beijing Natural Science Fundation-Changping Innovation Joint Fund (L234044) and Beijing Nova Program to K. H. This work was also supported by the Projects of International Cooperation and Exchanges NSFC (No. 62211530446), the National Natural Science Foundation of China (Nos. 82372002, 62371051, 61971049 and 22278037), and the Discipline Construction of Material Science and Engineering (21090122014).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| CAF | Cancer-associated Fibroblast |
| CT | Computerized Tomography |
| DPP | Dipeptidyl Peptidase |
| FAP | Fibroblast Activation Protein |
| FAPI | FAP Inhibitor |
| FDA | Food and Drug Administration |
| IC50 | Half Maximal Inhibitory Concentration |
| ID/g | Injected Dose/Gram |
| MAb | Monoclonal Antibody |
| mCRPC | Metastatic Castration-resistant Prostate Cancer |
| MIP | Maximum Intensity Projection |
| NIR | Near-infrared |
| PET | Positron Emission Tomography |
| PK | Pharmacokinetic |
| SPECT | Single Photon Emission Computed Tomography |
| TME | Tumor Microenvironment |
| T/K | Tumor-to-kidney Ratio |
References
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2016, 387, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Dart, Tumour microenvironment: Radical changes. Nat Rev Cancer 2018, 18, 65. [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Katheder, N.S.; Khezri, R.; O’farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhász, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 1–23. [Google Scholar] [CrossRef]
- Gordon-Weeks, A.; Yuzhalin, A.E. Cancer Extracellular Matrix Proteins Regulate Tumour Immunity. Cancers 2020, 12, 3331. [Google Scholar] [CrossRef] [PubMed]
- Oudin, M.J.; Jonas, O.; Kosciuk, T.; Broye, L.C.; Guido, B.C.; Wyckoff, J.; Riquelme, D.; Lamar, J.M.; Asokan, S.B.; Whittaker, C.; et al. Tumor Cell–Driven Extracellular Matrix Remodeling Drives Haptotaxis during Metastatic Progression. Cancer Discov. 2016, 6, 516–531. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef]
- Berg, T.J.; Pietras, A. Radiotherapy-induced remodeling of the tumor microenvironment by stromal cells. Semin. Cancer Biol. 2022, 86, 846–856. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Galassi, C.; Galluzzi, L. Stress responses in stromal cells and tumor homeostasis. Pharmacol. Ther. 2019, 200, 55–68. [Google Scholar] [CrossRef]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xie, F.; Zhang, L.; Zhou, X.; Huang, J.; Wang, F.; Jin, J.; Zhang, L.; Zeng, L.; Zhou, F. Targeted Anti-Tumor Immunotherapy Using Tumor Infiltrating Cells. Adv. Sci. 2021, 8, 2101672. [Google Scholar] [CrossRef] [PubMed]
- Melssen, M.M.; Sheybani, N.D.; Leick, K.M.; Slingluff, C.L. Barriers to immune cell infiltration in tumors. J. Immunother. Cancer 2023, 11, e006401. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Yao, Y.; Tang, Y.; Xin, Z.; Wu, D.; Ni, C.; Huang, J.; Wei, Q.; Zhang, T. Radiation-induced tumor immune microenvironments and potential targets for combination therapy. Signal Transduct. Target. Ther. 2023, 8, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- CChen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 2015, 13, 1–14. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Jiang, J.; Mei, J.; Ma, Y.; Jiang, S.; Zhang, J.; Yi, S.; Feng, C.; Liu, Y.; Liu, Y. Tumor hijacks macrophages and microbiota through extracellular vesicles. Exploration 2022, 2. [Google Scholar] [CrossRef]
- Liu, H.; Sun, B.; Zhu, P.; Liu, C.; Zhang, G.; Wang, D.; Song, X.; Shi, J.; Yang, Y.; Lu, J. Preparation of Three-Dimensional Porous Graphene by Hydrothermal and Chemical Reduction with Ascorbic Acid and its Electrochemical Properties. ChemistryOpen 2022, 11, e202200161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Vu, L.T.; Ismail, N.N.; Le, M.T.; Grimson, A. Landscape of extracellular vesicles in the tumour microenvironment: Interactions with stromal cells and with non-cell components, and impacts on metabolic reprogramming, horizontal transfer of neoplastic traits, and the emergence of therapeutic resistance. Semin. Cancer Biol. 2021, 74, 24–44. [Google Scholar] [CrossRef]
- Ni, Y.; Zhou, X.; Yang, J.; Shi, H.; Li, H.; Zhao, X.; Ma, X. The Role of Tumor-Stroma Interactions in Drug Resistance Within Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Matsueda, S.; Saya, H. Significance of Cancer-Associated Fibroblasts in the Interactions of Cancer Cells with the Tumor Microenvironment of Heterogeneous Tumor Tissue. Cancers 2023, 15, 2536. [Google Scholar] [CrossRef]
- Li, Z.; Low, V.; Luga, V.; Sun, J.; Earlie, E.; Parang, B.; Ganesh, K.S.; Cho, S.; Endress, J.; Schild, T.; et al. Tumor-produced and aging-associated oncometabolite methylmalonic acid promotes cancer-associated fibroblast activation to drive metastatic progression. Nat. Commun. 2022, 13, 1–14. [Google Scholar] [CrossRef]
- Wen, Y.; Zhu, Y.; Zhang, C.; Yang, X.; Gao, Y.; Li, M.; Yang, H.; Liu, T.; Tang, H. Chronic inflammation, cancer development and immunotherapy. Front. Pharmacol. 2022, 13, 1040163. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Coller, H.A. Fibroblasts Prompt Tumors to Mobilize Their Glycogen Reserves. Trends Cell Biol. 2019, 29, 278–280. [Google Scholar] [CrossRef]
- M. Cully, Tumour microenvironment: Fibroblast subtype provides niche for cancer stem cells. Nat Rev Cancer 2018, 18, 136. [CrossRef]
- Roulis, M.; Kaklamanos, A.; Schernthanner, M.; Bielecki, P.; Zhao, J.; Kaffe, E.; Frommelt, L.-S.; Qu, R.; Knapp, M.S.; Henriques, A.; et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature 2020, 580, 524–529. [Google Scholar] [CrossRef]
- Park, D.; Sahai, E.; Rullan, A. SnapShot: Cancer-Associated Fibroblasts. Cell 2020, 181, 486–486. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2018, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Prakash, J. Cancer-Associated Fibroblasts: Perspectives in Cancer Therapy. Trends Cancer 2016, 2, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. Landmark Ed. 2010, 15, 166–179. [Google Scholar] [CrossRef]
- Álvarez-Teijeiro, S.; García-Inclán, C.; Villaronga, M. Á; Casado, P.; Hermida-Prado, F.; Granda-Díaz, R.; Rodrigo, J.P.; Calvo, F.; Del-Río-Ibisate, N.; Gandarillas, A.; et al. Factors Secreted by Cancer-Associated Fibroblasts that Sustain Cancer Stem Properties in Head and Neck Squamous Carcinoma Cells as Potential Therapeutic Targets. Cancers 2018, 10, 334. [Google Scholar] [CrossRef]
- Piper, M.; Mueller, A.C.; Karam, S.D. The interplay between cancer associated fibroblasts and immune cells in the context of radiation therapy. Mol. Carcinog. 2020, 59, 754–765. [Google Scholar] [CrossRef]
- Allam, A.; Yakou, M.; Pang, L.; Ernst, M.; Huynh, J. Exploiting the STAT3 Nexus in Cancer-Associated Fibroblasts to Improve Cancer Therapy. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol. Cancer 2021, 20, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2020, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Hausmann, C.; Zoschke, C.; Wolff, C.; Darvin, M.E.; Sochorová, M.; Kováčik, A.; Wanjiku, B.; Schumacher, F.; Tigges, J.; Kleuser, B.; et al. Fibroblast origin shapes tissue homeostasis, epidermal differentiation, and drug uptake. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.; Goumas, F.A.; Affeldt, M.; Sandtner, S.; Gehling, U.M.; Brilloff, S.; Walter, J.; Karnatz, N.; Lamszus, K.; Rogiers, X.; et al. Stromal Fibroblasts in Colorectal Liver Metastases Originate From Resident Fibroblasts and Generate an Inflammatory Microenvironment. Am. J. Pathol. 2007, 171, 1608–1618. [Google Scholar] [CrossRef]
- Li, A.; Chen, Y.-S.; Ping, X.-L.; Yang, X.; Xiao, W.; Yang, Y.; Sun, H.-Y.; Zhu, Q.; Baidya, P.; Wang, X.; et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017, 27, 444–447. [Google Scholar] [CrossRef]
- Sebastian, A.; Hum, N.R.; Martin, K.A.; Gilmore, S.F.; Peran, I.; Byers, S.W.; Wheeler, E.K.; Coleman, M.A.; Loots, G.G. Single-Cell Transcriptomic Analysis of Tumor-Derived Fibroblasts and Normal Tissue-Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers 2020, 12, 1307. [Google Scholar] [CrossRef] [PubMed]
- N.M. Anderson, M.C. Simon, The tumor microenvironment. Curr Biol 2020, 30, R921–R925. [CrossRef] [PubMed]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; López-Giráldez, F.; Dash, B.C.; Muñoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362, 909. [Google Scholar] [CrossRef]
- Yu, J.; Seldin, M.M.; Fu, K.; Li, S.; Lam, L.; Wang, P.; Wang, Y.; Huang, D.; Nguyen, T.L.; Wei, B.; et al. Topological Arrangement of Cardiac Fibroblasts Regulates Cellular Plasticity. Circ. Res. 2018, 123, 73–85. [Google Scholar] [CrossRef]
- Jiang, D.; Rinkevich, Y. Converting fibroblastic fates leads to wound healing without scar. Signal Transduct. Target. Ther. 2021, 6, 1–3. [Google Scholar] [CrossRef]
- Li, B.; Wang, J.H.-C. Fibroblasts and myofibroblasts in wound healing: Force generation and measurement. J. Tissue Viability 2011, 20, 108–120. [Google Scholar] [CrossRef]
- Hutchenreuther, J.; Leask, A. A tale of two orgins: do myofibroblasts originate from different sources in wound healing and fibrosis? Cell Tissue Res. 2016, 365, 507–509. [Google Scholar] [CrossRef]
- Foster, D.S.; Januszyk, M.; Yost, K.E.; Chinta, M.S.; Gulati, G.S.; Nguyen, A.T.; Burcham, A.R.; Salhotra, A.; Ransom, R.C.; Henn, D.; et al. Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef]
- H.F. Dvorak, Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986, 315, 1650–1659.
- Byun, J.S.; Gardner, K. Wounds That Will Not Heal: Pervasive Cellular Reprogramming in Cancer. Am. J. Pathol. 2013, 182, 1055–1064. [Google Scholar] [CrossRef]
- Koustoulidou, S.; Hoorens, M.W.H.; Dalm, S.U.; Mahajan, S.; Debets, R.; Seimbille, Y.; de Jong, M. Cancer-Associated Fibroblasts as Players in Cancer Development and Progression and Their Role in Targeted Radionuclide Imaging and Therapy. Cancers 2021, 13, 1100. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Wu, M.; Arcand, S.L.; Lavallée, S.; Hébert, J.; Tonin, P.N.; Basik, M. Breast Carcinoma–Associated Fibroblasts Rarely Contain p53 Mutations or Chromosomal Aberrations. Cancer Res 2010, 70, 5770–5777. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Chen, X.; Wan, Z.; Ge, M.; Ding, Y.; Gu, J.; Hua, J.; Guo, D.; Tan, M.; Xu, D. Identification of the novel therapeutic targets and biomarkers associated of prostate cancer with cancer-associated fibroblasts (CAFs). Front. Oncol. 2023, 13, 1136835. [Google Scholar] [CrossRef] [PubMed]
- C.D. van der Heide, S.U. Dalm. Radionuclide imaging and therapy directed towards the tumor microenvironment: a multi-cancer approach for personalized medicine. Eur J Nucl Med Mol Imaging 2022, 49, 4616–4641. [Google Scholar] [CrossRef] [PubMed]
- Imlimthan, S.; Moon, E.S.; Rathke, H.; Afshar-Oromieh, A.; Rösch, F.; Rominger, A.; Gourni, E. New Frontiers in Cancer Imaging and Therapy Based on Radiolabeled Fibroblast Activation Protein Inhibitors: A Rational Review and Current Progress. Pharmaceuticals 2021, 14, 1023. [Google Scholar] [CrossRef] [PubMed]
- Robb, M.A.; McInnes, P.M.; Califf, R.M. Biomarkers and Surrogate Endpoints. JAMA 2016, 315, 1107–1108. [Google Scholar] [CrossRef]
- Liu, D. Cancer biomarkers for targeted therapy. Biomark. Res. 2019, 7, 1–7. [Google Scholar] [CrossRef]
- Zhang, J.; Gu, C.; Song, Q.; Zhu, M.; Xu, Y.; Xiao, M.; Zheng, W. Identifying cancer-associated fibroblasts as emerging targets for hepatocellular carcinoma. Cell Biosci. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Hu, D.; Li, Z.; Zheng, B.; Lin, X.; Pan, Y.; Gong, P.; Zhuo, W.; Hu, Y.; Chen, C.; Chen, L.; et al. Cancer-associated fibroblasts in breast cancer: Challenges and opportunities. Cancer Commun. 2022, 42, 401–434. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.-W.; Qiu, S.-Q.; Zhang, G.-J. Molecular and functional imaging in cancer-targeted therapy: current applications and future directions. Signal Transduct. Target. Ther. 2023, 8, 1–32. [Google Scholar] [CrossRef]
- Hamson, E.J.; Keane, F.M.; Tholen, S.; Schilling, O.; Gorrell, M.D. Understanding fibroblast activation protein (FAP): Substrates, activities, expression and targeting for cancer therapy. Proteom. – Clin. Appl. 2014, 8, 454–463. [Google Scholar] [CrossRef]
- Brennen, W.N.; Rosen, D.M.; Wang, H.; Isaacs, J.T.; Denmeade, S.R. Targeting Carcinoma-Associated Fibroblasts Within the Tumor Stroma With a Fibroblast Activation Protein-Activated Prodrug. JNCI J. Natl. Cancer Inst. 2012, 104, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Choyke, P.L. PET of Fibroblast-Activation Protein for Cancer Staging: What We Know and What We Need to Learn. Radiology 2022, 304, 658–659. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, T.; Yin, R. Biomarkers for cancer-associated fibroblasts. Biomark. Res. 2020, 8, 1–8. [Google Scholar] [CrossRef]
- Mayola, M.F.; Thackeray, J.T. The Potential of Fibroblast Activation Protein-Targeted Imaging as a Biomarker of Cardiac Remodeling and Injury. Curr. Cardiol. Rep. 2023, 25, 515–523. [Google Scholar] [CrossRef]
- Ebert, L.M.; Yu, W.; Gargett, T.; Toubia, J.; Kollis, P.M.; Tea, M.N.; Ebert, B.W.; Bardy, C.; Hurk, M.v.D.; Bonder, C.S.; et al. Endothelial, pericyte and tumor cell expression in glioblastoma identifies fibroblast activation protein (FAP) as an excellent target for immunotherapy. Clin. Transl. Immunol. 2020, 9, e1191. [Google Scholar] [CrossRef]
- Rezaei, S.; Gharapapagh, E.; Dabiri, S.; Heidari, P.; Aghanejad, A. Theranostics in targeting fibroblast activation protein bearing cells: Progress and challenges. Life Sci. 2023, 329, 121970. [Google Scholar] [CrossRef]
- Levy, M.T.; McCaughan, G.W.; Abbott, C.A.; Park, J.E.; Cunningham, A.M.; Müller, E.; Rettig, W.J.; Gorrell, M.D. Fibroblast activation protein: A cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. J. Hepatol. 1999, 29, 1768–1778. [Google Scholar] [CrossRef]
- Juillerat-Jeanneret, L.; Tafelmeyer, P.; Golshayan, D. Fibroblast activation protein-α in fibrogenic disorders and cancer: more than a prolyl-specific peptidase? Expert Opin. Ther. Targets 2017, 21, 977–991. [Google Scholar] [CrossRef]
- Zhang, T.; Tong, X.; Zhang, S.; Wang, D.; Wang, L.; Wang, Q.; Fan, H. The Roles of Dipeptidyl Peptidase 4 (DPP4) and DPP4 Inhibitors in Different Lung Diseases: New Evidence. Front. Pharmacol. 2021, 12, 731453. [Google Scholar] [CrossRef]
- R. Han, X. Wang, W. Bachovchin, Z. Zukowska, J.W. Osborn, Inhibition of dipeptidyl peptidase 8/9 impairs preadipocyte differentiation. Sci Rep 2015, 5, 12348. [CrossRef]
- Brennen, W.N.; Isaacs, J.T.; Denmeade, S.R. Rationale Behind Targeting Fibroblast Activation Protein–Expressing Carcinoma-Associated Fibroblasts as a Novel Chemotherapeutic Strategy. Mol. Cancer Ther. 2012, 11, 257–266. [Google Scholar] [CrossRef]
- M. Zubal, B. Vymolova, I. Matrasova, P. Vymola, J. Veprkova, M. Syrucek, R. Tomas, Z. Vanickova, E. Krepela, D. Konecna, P. Busek, A. Sedo, Fibroblast activation protein as a potential theranostic target in brain metastases of diverse solid tumours. Pathology 2023, 55, 806–817.
- Wonganu, B.; Berger, B.W. A specific, transmembrane interface regulates fibroblast activation protein (FAP) homodimerization, trafficking and exopeptidase activity. Biochim. et Biophys. Acta (BBA) - Biomembr. 2016, 1858, 1876–1882. [Google Scholar] [CrossRef]
- Dendl, K.; Koerber, S.A.; Kratochwil, C.; Cardinale, J.; Finck, R.; Dabir, M.; Novruzov, E.; Watabe, T.; Kramer, V.; Choyke, P.L.; et al. FAP and FAPI-PET/CT in Malignant and Non-Malignant Diseases: A Perfect Symbiosis? Cancers 2021, 13, 4946. [Google Scholar] [CrossRef]
- Park, J.E.; Lenter, M.C.; Zimmermann, R.N.; Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Fibroblast Activation Protein, a Dual Specificity Serine Protease Expressed in Reactive Human Tumor Stromal Fibroblasts. J. Biol. Chem. 1999, 274, 36505–36512. [Google Scholar] [CrossRef]
- Kalaei, Z.; Manafi-Farid, R.; Rashidi, B.; Kiani, F.K.; Zarei, A.; Fathi, M.; Jadidi-Niaragh, F. The Prognostic and therapeutic value and clinical implications of fibroblast activation protein-α as a novel biomarker in colorectal cancer. Cell Commun. Signal. 2023, 21, 1–17. [Google Scholar] [CrossRef]
- Aertgeerts, K.; Levin, I.; Shi, L.; Snell, G.P.; Jennings, A.; Prasad, G.S.; Zhang, Y.; Kraus, M.L.; Salakian, S.; Sridhar, V.; et al. Structural and Kinetic Analysis of the Substrate Specificity of Human Fibroblast Activation Protein α. J. Biol. Chem. 2005, 280, 19441–19444. [Google Scholar] [CrossRef]
- Niedermeyer, J.; Garin-Chesa, P.; Kriz, M.; Hilberg, F.; Mueller, E.; Bamberger, U.; Rettig, W.J.; Schnapp, A. Expression of the fibroblast activation protein during mouse embryo development. The International journal of developmental biology 2001, 45, 445–447. [Google Scholar]
- Niedermeyer, J.; Kriz, M.; Hilberg, F.; Garin-Chesa, P.; Bamberger, U.; Lenter, M.C.; Park, J.; Viertel, B.; Püschner, H.; Mauz, M.; et al. Targeted Disruption of Mouse Fibroblast Activation Protein. Mol. Cell. Biol. 2000, 20, 1089–1094. [Google Scholar] [CrossRef]
- Brown, D.D.; Wang, Z.; Furlow, J.D.; Kanamori, A.; A Schwartzman, R.; Remo, B.F.; Pinder, A. The thyroid hormone-induced tail resorption program during Xenopus laevis metamorphosis. Proc. Natl. Acad. Sci. 1996, 93, 1924–1929. [Google Scholar] [CrossRef] [PubMed]
- Rettig, W.J.; Su, S.L.; Fortunato, S.R.; Scanlan, M.J.; Raj, B.K.M.; Garin-Chesa, P.; Healey, J.H.; Old, L.J. Fibroblast activation protein: Purification, epitope mapping and induction by growth factors. Int. J. Cancer 1994, 58, 385–392. [Google Scholar] [CrossRef]
- Dolznig, H.; Schweifer, N.; Puri, C.; Kraut, N.; Rettig, W.J.; Kerjaschki, D.; Garin-Chesa, P. Characterization of cancer stroma markers: in silico analysis of an mRNA expression database for fibroblast activation protein and endosialin. Cancer immunity 2005, 5, 10. [Google Scholar] [PubMed]
- Egger, C.; Cannet, C.; Gérard, C.; Suply, T.; Ksiazek, I.; Jarman, E.; Beckmann, N. Effects of the fibroblast activation protein inhibitor, PT100, in a murine model of pulmonary fibrosis. Eur. J. Pharmacol. 2017, 809, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Tillmanns, J.; Hoffmann, D.; Habbaba, Y.; Schmitto, J.D.; Sedding, D.; Fraccarollo, D.; Galuppo, P.; Bauersachs, J. Fibroblast activation protein alpha expression identifies activated fibroblasts after myocardial infarction. J. Mol. Cell. Cardiol. 2015, 87, 194–203. [Google Scholar] [CrossRef]
- S. Uitte de Willige, J.J. S. Uitte de Willige, J.J. Malfliet, H.L. Janssen, F.W. Leebeek, D.C. Rijken, Increased N-terminal cleavage of alpha-2-antiplasmin in patients with liver cirrhosis, J Thromb Haemost, 11 (2013) 2029-2036.
- Nagaraju, C.K.; Dries, E.; Popovic, N.; Singh, A.A.; Haemers, P.; Roderick, H.L.; Claus, P.; Sipido, K.R.; Driesen, R.B. Global fibroblast activation throughout the left ventricle but localized fibrosis after myocardial infarction. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rettig, W.J.; Garin-Chesa, P.; Beresford, H.R.; Oettgen, H.F.; Melamed, M.R.; Old, L.J. Cell-surface glycoproteins of human sarcomas: differential expression in normal and malignant tissues and cultured cells. Proc. Natl. Acad. Sci. 1988, 85, 3110–3114. [Google Scholar] [CrossRef]
- Bauer, S.; Jendro, M.C.; Wadle, A.; Kleber, S.; Stenner, F.; Dinser, R.; Reich, A.; Faccin, E.; Gödde, S.; Dinges, H.; et al. Fibroblast activation protein is expressed by rheumatoid myofibroblast-like synoviocytes. Arthritis Res. Ther. 2006, 8, R171–R171. [Google Scholar] [CrossRef]
- Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. USA 1990, 87, 7235–7239. [Google Scholar] [CrossRef] [PubMed]
- Mentlein, R.; Hattermann, K.; Hemion, C.; Jungbluth, A.A.; Held-Feindt, J. Expression and role of the cell surface protease seprase/fibroblast activation protein-α (FAP-α) in astroglial tumors. Biol. Chem. 2011, 392, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Brokopp, C.E.; Schoenauer, R.; Richards, P.; Bauer, S.; Lohmann, C.; Emmert, M.Y.; Weber, B.; Winnik, S.; Aikawa, E.; Graves, K.; et al. Fibroblast activation protein is induced by inflammation and degrades type I collagen in thin-cap fibroatheromata. Eur. Hear. J. 2011, 32, 2713–2722. [Google Scholar] [CrossRef]
- Wang, X.M.; Yu, D.M.T.; McCaughan, G.W.; Gorrell, M.D. Fibroblast activation protein increases apoptosis, cell adhesion, and migration by the LX-2 human stellate cell line. J. Hepatol. 2005, 42, 935–945. [Google Scholar] [CrossRef]
- Park, J.E.; Lenter, M.C.; Zimmermann, R.N.; Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Fibroblast Activation Protein, a Dual Specificity Serine Protease Expressed in Reactive Human Tumor Stromal Fibroblasts. J. Biol. Chem. 1999, 274, 36505–36512. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, M.J.; Raj, B.K.; Calvo, B.; Garin-Chesa, P.; Sanz-Moncasi, M.P.; Healey, J.H.; Old, L.J.; Rettig, W.J. Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc. Natl. Acad. Sci. USA 1994, 91, 5657–5661. [Google Scholar] [CrossRef] [PubMed]
- Henry, L.R.; Lee, H.-O.; Lee, J.S.; Klein-Szanto, A.; Watts, P.; Ross, E.A.; Chen, W.-T.; Cheng, J.D. Clinical Implications of Fibroblast Activation Protein in Patients with Colon Cancer. Clin. Cancer Res. 2007, 13, 1736–1741. [Google Scholar] [CrossRef]
- Cohen, S.J.; Alpaugh, R.K.; Palazzo, I.; Meropol, N.J.; Rogatko, A.; Xu, Z.; Hoffman, J.P.; Weiner, L.M.; Cheng, J.D. Fibroblast Activation Protein and Its Relationship to Clinical Outcome in Pancreatic Adenocarcinoma. Pancreas 2008, 37, 154–158. [Google Scholar] [CrossRef]
- Ju, M.-J.; Qiu, S.-J.; Fan, J.; Xiao, Y.-S.; Gao, Q.; Zhou, J.; Li, Y.-W.; Tang, Z.-Y. Peritumoral Activated Hepatic Stellate Cells Predict Poor Clinical Outcome in Hepatocellular Carcinoma After Curative Resection. Am. J. Clin. Pathol. 2009, 131, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, H.; Cai, J.; Zhang, T.; Guo, J.; Feng, D.; Wang, Z. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011, 303, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Teichgräber, V.; Monasterio, C.; Chaitanya, K.; Boger, R.; Gordon, K.; Dieterle, T.; Jäger, D.; Bauer, S. Specific inhibition of fibroblast activation protein (FAP)-alpha prevents tumor progression in vitro. Adv. Med Sci. 2015, 60, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Loktev, A.; Giesel, F.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of activated fibroblasts for imaging and therapy. EJNMMI Radiopharm. Chem. 2019, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Huangfu, Z.; Yang, J.; Wang, G.; Hu, K.; Gao, M.; Zhong, Z. Imaging-guided targeted radionuclide tumor therapy: From concept to clinical translation. Adv. Drug Deliv. Rev. 2022, 190, 114538. [Google Scholar] [CrossRef] [PubMed]
- Fonti, R.; Conson, M.; Del Vecchio, S. PET/CT in radiation oncology. Semin. Oncol. 2019, 46, 202–209. [Google Scholar] [CrossRef]
- Ostermann, E.; Garin-Chesa, P.; Heider, K.H.; Kalat, M.; Lamche, H.; Puri, C.; Kerjaschki, D.; Rettig, W.J.; Adolf, G.R. Effective Immunoconjugate Therapy in Cancer Models Targeting a Serine Protease of Tumor Fibroblasts. Clin. Cancer Res. 2008, 14, 4584–4592. [Google Scholar] [CrossRef]
- Chen, M.; Lei, X.; Shi, C.; Huang, M.; Li, X.; Wu, B.; Li, Z.; Han, W.; Du, B.; Hu, J.; et al. Pericyte-targeting prodrug overcomes tumor resistance to vascular disrupting agents. J. Clin. Investig. 2017, 127, 3689–3701. [Google Scholar] [CrossRef]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting Fibroblast Activation Protein in Tumor Stroma with Chimeric Antigen Receptor T Cells Can Inhibit Tumor Growth and Augment Host Immunity without Severe Toxicity. Cancer Immunol. Res. 2013, 2, 154–166. [Google Scholar] [CrossRef]
- Fu, H.; Guo, W.; Huang, J.; Wu, H.; Chen, H. Clinical applications of fibroblast activation protein-targeted theranostics in oncologic and nononcologic disease: Current status and future directions. iRADIOLOGY 2023, 1, 340–361. [Google Scholar] [CrossRef]
- M. Ying, Q. Yang, X. Xu, S. Wu, W. Yin, S. Liang, G. Pan, C. Zuo, Z. Guo, C. Cheng, S. Liu, Value of [68Ga]Ga-FAPI-04 PET imaging in acute coronary syndrome complicated by suspected gastrointestinal malignancies. Journal of Nuclear Medicine 2023, 64, 411.
- Jansen, K.; Heirbaut, L.; Verkerk, R.; Cheng, J.D.; Joossens, J.; Cos, P.; Maes, L.; Lambeir, A.-M.; De Meester, I.; Augustyns, K.; et al. Extended Structure–Activity Relationship and Pharmacokinetic Investigation of (4-Quinolinoyl)glycyl-2-cyanopyrrolidine Inhibitors of Fibroblast Activation Protein (FAP). J. Med. Chem. 2014, 57, 3053–3074. [Google Scholar] [CrossRef]
- Li, M.; Younis, M.H.; Zhang, Y.; Cai, W.; Lan, X. Clinical summary of fibroblast activation protein inhibitor-based radiopharmaceuticals: cancer and beyond. Eur. J. Nucl. Med. 2022, 49, 2844–2868. [Google Scholar] [CrossRef]
- Huang, R.; Pu, Y.; Huang, S.; Yang, C.; Yang, F.; Pu, Y.; Li, J.; Chen, L.; Huang, Y. FAPI-PET/CT in Cancer Imaging: A Potential Novel Molecule of the Century. Front. Oncol. 2022, 12, 854658. [Google Scholar] [CrossRef]
- Rettig, W.J.; Chesa, P.G.; Beresford, H.R.; Feickert, H.J.; Jennings, M.T.; Cohen, J.; Oettgen, H.F.; Old, L.J. Differential expression of cell surface antigens and glial fibrillary acidic protein in human astrocytoma subsets. Cancer research 1986, 46, 6406–12. [Google Scholar]
- Rettig, W.J.; Garin-Chesa, P.; Healey, J.H.; Su, S.L.; Ozer, H.L.; Schwab, M.; Albino, A.P.; Old, L.J. Regulation and heteromeric structure of the fibroblast activation protein in normal and transformed cells of mesenchymal and neuroectodermal origin. Cancer research 1993, 53, 3327–35. [Google Scholar] [PubMed]
- O’Brien, P.; O’Connor, B.F. Seprase: An overview of an important matrix serine protease. Biochim. et Biophys. Acta (BBA) - Proteins Proteom. 2008, 1784, 1130–1145. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.-C.; Chen, J.; Stefflova, K.; Warren, M.S.; Navab, R.; Bandarchi, B.; Mullins, S.; Tsao, M.; Cheng, J.D.; Zheng, G. Photodynamic Molecular Beacon Triggered by Fibroblast Activation Protein on Cancer-Associated Fibroblasts for Diagnosis and Treatment of Epithelial Cancers. J. Med. Chem. 2008, 52, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Peltier, A.; Seban, R.-D.; Buvat, I.; Bidard, F.-C.; Mechta-Grigoriou, F. Fibroblast heterogeneity in solid tumors: From single cell analysis to whole-body imaging. Semin. Cancer Biol. 2022, 86, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T. Fibroblast activation protein-α and dipeptidyl peptidase IV (CD26): Cell-surface proteases that activate cell signaling and are potential targets for cancer therapy. Drug Resist. Updat. 2005, 8, 51–58. [Google Scholar] [CrossRef]
- Privé, B.M.; Boussihmad, M.A.; Timmermans, B.; van Gemert, W.A.; Peters, S.M.B.; Derks, Y.H.W.; van Lith, S.A.M.; Mehra, N.; Nagarajah, J.; Heskamp, S.; et al. Fibroblast activation protein-targeted radionuclide therapy: background, opportunities, and challenges of first (pre)clinical studies. Eur. J. Nucl. Med. 2023, 50, 1906–1918. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Nam, E.H.; Park, J.Y.; Ghosh, P.; Kim, I.S. Identification of BR102910 as a selective fibroblast activation protein (FAP) inhibitor. Bioorganic Med. Chem. Lett. 2021, 37, 127846. [Google Scholar] [CrossRef]
- Aoyama, A.; Chen, W.T. A 170-kDa membrane-bound protease is associated with the expression of invasiveness by human malignant melanoma cells. Proc. Natl. Acad. Sci. 1990, 87, 8296–8300. [Google Scholar] [CrossRef] [PubMed]
- Welt, S.; Divgi, C.R.; Scott, A.M.; Garin-Chesa, P.; Finn, R.D.; Graham, M.; A Carswell, E.; Cohen, A.; Larson, S.M.; Old, L.J. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J. Clin. Oncol. 1994, 12, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wiseman, G.; Adjei, A.; Lee, F.-T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; Larson, S.M.; et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 2003, 9, 1639–1647. [Google Scholar]
- Fischer, E.; Chaitanya, K.; Wüest, T.; Wadle, A.; Scott, A.M.; van den Broek, M.; Schibli, R.; Bauer, S.; Renner, C. Radioimmunotherapy of Fibroblast Activation Protein Positive Tumors by Rapidly Internalizing Antibodies. Clin. Cancer Res. 2012, 18, 6208–6218. [Google Scholar] [CrossRef]
- Pandya, D.N.; Sinha, A.; Yuan, H.; Mutkus, L.; Stumpf, K.; Marini, F.C.; Wadas, T.J. Imaging of Fibroblast Activation Protein Alpha Expression in a Preclinical Mouse Model of Glioma Using Positron Emission Tomography. Molecules 2020, 25, 3672. [Google Scholar] [CrossRef] [PubMed]
- Hintz, H.M.; Gallant, J.P.; Griend, D.J.V.; Coleman, I.M.; Nelson, P.S.; LeBeau, A.M. Imaging Fibroblast Activation Protein Alpha Improves Diagnosis of Metastatic Prostate Cancer with Positron Emission Tomography. Clin. Cancer Res. 2020, 26, 4882–4891. [Google Scholar] [CrossRef]
- Brünker, P.; Wartha, K.; Friess, T.; Grau-Richards, S.; Waldhauer, I.; Koller, C.F.; Weiser, B.; Majety, M.; Runza, V.; Niu, H.; et al. RG7386, a Novel Tetravalent FAP-DR5 Antibody, Effectively Triggers FAP-Dependent, Avidity-Driven DR5 Hyperclustering and Tumor Cell Apoptosis. Mol. Cancer Ther. 2016, 15, 946–957. [Google Scholar] [CrossRef]
- E.E. Kim, Therapeutic Nuclear Medicine, Journal of nuclear medicine: official publication. Society of Nuclear Medicine 2016, 57, 163.
- Hofheinz, R.-D.; Al-Batran, S.-E.; Hartmann, F.; Hartung, G.; Jäger, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal Antigen Targeting by a Humanised Monoclonal Antibody: An Early Phase II Trial of Sibrotuzumab in Patients with Metastatic Colorectal Cancer. Onkologie 2003, 26, 44–48. [Google Scholar] [CrossRef]
- P. Tanswell, P. Garin-Chesa, W.J. Rettig, S. Welt, C.R. Divgi, E.S. Casper, R.D. Finn, S.M. Larson, L.J. Old, A.M. Scott, Population pharmacokinetics of antifibroblast activation protein monoclonal antibody F19 in cancer patients. British journal of clinical pharmacology 2001, 51, 177–180. [CrossRef]
- B. Ingelheim, Single Dose Escalation Study of 131I-Sibrotuzumab in Patients With Advanced or Metastatic Non-small Cell Lung Cancer, 2014-08-06.
- Jain, T.; Sun, T.; Durand, S.; Hall, A.; Houston, N.R.; Nett, J.H.; Sharkey, B.; Bobrowicz, B.; Caffry, I.; Yu, Y.; et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. 2017, 114, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Starr, C.G.; Tessier, P.M. Selecting and engineering monoclonal antibodies with drug-like specificity. Curr. Opin. Biotechnol. 2019, 60, 119–127. [Google Scholar] [CrossRef] [PubMed]
- J.-F. Chatal, C.A. Hoefnagel, Radionuclide therapy. Lancet 1999, 354, 931–935. [CrossRef]
- DeNardo, S.J.; DeNardo, G.L. Targeted radionuclide therapy for solid tumors: An overview. Endocrine 2006, 66, S89–S95. [Google Scholar] [CrossRef] [PubMed]
- Ledsgaard, L.; Ljungars, A.; Rimbault, C.; Sørensen, C.V.; Tulika, T.; Wade, J.; Wouters, Y.; McCafferty, J.; Laustsen, A.H. Advances in antibody phage display technology. Drug Discov. Today 2022, 27, 2151–2169. [Google Scholar] [CrossRef]
- Mimmi, S.; Maisano, D.; Quinto, I.; Iaccino, E. Phage Display: An Overview in Context to Drug Discovery. Trends Pharmacol. Sci. 2019, 40, 87–91. [Google Scholar] [CrossRef]
- Saw, P.E.; Song, E.-W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807. [Google Scholar] [CrossRef]
- Schofield, D.J.; Pope, A.R.; Clementel, V.; Buckell, J.; Chapple, S.D.; Clarke, K.F.; Conquer, J.S.; Crofts, A.M.; Crowther, S.R.; Dyson, M.R.; et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007, 8, R254–R254. [Google Scholar] [CrossRef]
- Frenzel, A.; Kügler, J.; Helmsing, S.; Meier, D.; Schirrmann, T.; Hust, M.; Dübel, S. Designing Human Antibodies by Phage Display. Transfus. Med. Hemotherapy 2017, 44, 312–318. [Google Scholar] [CrossRef]
- Nagano, K.; Tsutsumi, Y. Phage Display Technology as a Powerful Platform for Antibody Drug Discovery. Viruses 2021, 13, 178. [Google Scholar] [CrossRef]
- Peissert, F.; Plüss, L.; Giudice, A.M.; Ongaro, T.; Villa, A.; Elsayed, A.; Nadal, L.; Plaza, S.D.; Scietti, L.; Puca, E.; et al. Selection of a PD-1 blocking antibody from a novel fully human phage display library. Protein Sci. 2022, 31, e4486. [Google Scholar] [CrossRef]
- S. Gupta, S. Batra, M. Jain, Antibody Labeling with Radioiodine and Radiometals. Drug Delivery System 2014, 1141, 147–157.
- Stein, R.; Govindan, S.V.; Mattes, M.J.; Chen, S.; Reed, L.; Newsome, G.; McBride, B.J.; Griffiths, G.L.; Hansen, H.J.; Goldenberg, D.M. Improved iodine radiolabels for monoclonal antibody therapy. Cancer Res. 2003, 63, 111–118. [Google Scholar]
- Anderson, W.T.; Strand, M. Radiolabeled antibody: iodine versus radiometal chelates. NCI Monogr. 1987, 149–151. [Google Scholar]
- Sun, J.; Huangfu, Z.; Yang, J.; Wang, G.; Hu, K.; Gao, M.; Zhong, Z. Imaging-guided targeted radionuclide tumor therapy: From concept to clinical translation. Adv. Drug Deliv. Rev. 2022, 190, 114538. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, A.H.; Eerd, J.E.M.V.; Frielink, C.; Oosterwijk, E.; Oyen, W.J.G.; Corstens, F.H.M.; Boerman, O.C. Optimization of radioimmunotherapy of renal cell carcinoma: labeling of monoclonal antibody cG250 with 131I, 90Y, 177Lu, or 186Re. J. Nucl. Med. 2004, 45, 327–37. [Google Scholar] [PubMed]
- Hoffmann, R.M.; Mele, S.; Cheung, A.; Larcombe-Young, D.; Bucaite, G.; Sachouli, E.; Zlatareva, I.; Morad, H.O.J.; Marlow, R.; McDonnell, J.M.; et al. Rapid conjugation of antibodies to toxins to select candidates for the development of anticancer Antibody-Drug Conjugates (ADCs). Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Morais, M.; Ma, M.T. Site-specific chelator-antibody conjugation for PET and SPECT imaging with radiometals. Drug Discov. Today: Technol. 2018, 30, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Eriksson, L. Physics of pure and non-pure positron emitters for PET: a review and a discussion. EJNMMI Phys. 2016, 3, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Deri, M.A.; Zeglis, B.M.; Francesconi, L.C.; Lewis, J.S. PET imaging with 89Zr: From radiochemistry to the clinic. Nucl. Med. Biol. 2012, 40, 3–14. [Google Scholar] [CrossRef] [PubMed]
- De Feo, M.S.; Pontico, M.; Frantellizzi, V.; Corica, F.; De Cristofaro, F.; De Vincentis, G. 89Zr-PET imaging in humans: a systematic review. Clin. Transl. Imaging 2021, 10, 23–36. [Google Scholar] [CrossRef]
- Fischer, G.; Seibold, U.; Schirrmacher, R.; Wängler, B.; Wängler, C. 89Zr, a Radiometal Nuclide with High Potential for Molecular Imaging with PET: Chemistry, Applications and Remaining Challenges. Molecules 2013, 18, 6469–6490. [Google Scholar] [CrossRef] [PubMed]
- S. Sandhu, C.M. Moore, E. Chiong, H. Beltran, R.G. Bristow, S.G. Williams, Prostate cancer. Lancet 2021, 398, 1075–1090.
- Smith, M.R.; Hussain, M.; Saad, F.; Fizazi, K.; Sternberg, C.N.; Crawford, E.D.; Kopyltsov, E.; Park, C.H.; Alekseev, B.; Montesa-Pino, Á.; et al. Darolutamide and Survival in Metastatic, Hormone-Sensitive Prostate Cancer. New Engl. J. Med. 2022, 386, 1132–1142. [Google Scholar] [CrossRef]
- Hofman, M.S.; Emmett, L.; Sandhu, S.; Iravani, A.; Joshua, A.M.; Goh, J.C.; Pattison, D.A.; Tan, T.H.; Kirkwood, I.D.; Ng, S.; et al. [177Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): A randomised, open-label, phase 2 trial. Lancet 2021, 397, 797–804. [Google Scholar] [CrossRef]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. New. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef]
- ESMO Congress 2022, 2022-09.
- A.J.W. Murtha, E.; van der Eecken, K.; Kwan, E.; Herberts, C.; Sipola, J.; Ng, S.; Chen, E.; Fonseca, N.; Schönlau, E.; Bernales, C.; Donnellan, G.; Verbeke, S.; Lumen, N.; Van Dorpe, J.; De Laere, B.; Annala, M.; Vandekerkhove, G.; Ost, P.; Wyatt, A. W. 4MO - Multi-focal genomic dissection of synchronous primary and metastatic tissue from de novo metastatic prostate cancer.
- Lappalainen, T.; Scott, A.J.; Brandt, M.; Hall, I.M. Genomic Analysis in the Age of Human Genome Sequencing. Cell 2019, 177, 70–84. [Google Scholar] [CrossRef]
- De Smet, F.; Martinez, A.A.; Bosisio, F.M. Next-Generation Pathology by Multiplexed Immunohistochemistry. Trends Biochem. Sci. 2020, 46, 80–82. [Google Scholar] [CrossRef]
- Pflueger, D.; Terry, S.; Sboner, A.; Habegger, L.; Esgueva, R.; Lin, P.-C.; Svensson, M.A.; Kitabayashi, N.; Moss, B.J.; MacDonald, T.Y.; et al. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res. 2010, 21, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Bhanvadia, R.R.; VanOpstall, C.; Brechka, H.; Barashi, N.S.; Gillard, M.; McAuley, E.M.; Vasquez, J.M.; Paner, G.; Chan, W.-C.; Andrade, J.; et al. MEIS1 and MEIS2 Expression and Prostate Cancer Progression: A Role For HOXB13 Binding Partners in Metastatic Disease. Clin. Cancer Res. 2018, 24, 3668–3680. [Google Scholar] [CrossRef] [PubMed]
- Klarmann, G.J.; Hurt, E.M.; Mathews, L.A.; Zhang, X.; Duhagon, M.A.; Mistree, T.; Thomas, S.B.; Farrar, W.L. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin. Exp. Metastasis 2009, 26, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Hintz, H.M.; Cowan, A.E.; Shapovalova, M.; LeBeau, A.M. Development of a Cross-Reactive Monoclonal Antibody for Detecting the Tumor Stroma. Bioconjugate Chem. 2019, 30, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Sum, E.; Rapp, M.; Fröbel, P.; Le Clech, M.; Dürr, H.; Giusti, A.M.F.; Perro, M.; Speziale, D.; Kunz, L.; Menietti, E.; et al. Fibroblast Activation Protein α-Targeted CD40 Agonism Abrogates Systemic Toxicity and Enables Administration of High Doses to Induce Effective Antitumor Immunity. Clin. Cancer Res. 2021, 27, 4036–4053. [Google Scholar] [CrossRef]
- Labiano, S.; Roh, V.; Godfroid, C.; Hiou-Feige, A.; Romero, J.; Sum, E.; Rapp, M.; Boivin, G.; Wyss, T.; Simon, C.; et al. CD40 Agonist Targeted to Fibroblast Activation Protein α Synergizes with Radiotherapy in Murine HPV-Positive Head and Neck Tumors. Clin. Cancer Res. 2021, 27, 4054–4065. [Google Scholar] [CrossRef]
- A Dose Escalation Study of RO6874813 in Participants With Locally Advanced or Metastatic Solid Tumors, 2018.
- A Study to Evaluate Safety, Pharmacokinetics and Anti-Tumor Activity of RO7300490, as Single Agent or in Combination With Atezolizumab in Participants With Advanced Solid Tumors, (2022).
- Wang, F.; Tsai, J.C.; Davis, J.H.; Chau, B.; Dong, J.; West, S.M.; Hogan, J.M.; Wheeler, M.L.; Bee, C.; Morishige, W.; et al. Design and characterization of mouse IgG1 and IgG2a bispecific antibodies for use in syngeneic models. mAbs 2019, 12, 1685350. [Google Scholar] [CrossRef]
- Meermeier, E.W.; Welsh, S.J.; Sharik, M.E.; Du, M.T.; Garbitt, V.M.; Riggs, D.L.; Shi, C.-X.; Stein, C.K.; Bergsagel, M.; Chau, B.; et al. Tumor Burden Limits Bispecific Antibody Efficacy through T-cell Exhaustion Averted by Concurrent Cytotoxic Therapy. Blood Cancer Discov. 2021, 2, 354–369. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Theruvath, J.; Menard, M.; Smith, B.A.H.; Linde, M.H.; Coles, G.L.; Dalton, G.N.; Wu, W.; Kiru, L.; Delaidelli, A.; Sotillo, E.; et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat. Med. 2022, 28, 333–344. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [PubMed]
- P. Hingorani, M.D. Krailo, A. Buxton, P.R. Hutson, J. Davis, K.A. Janeway, R.G. Gorlick, M. Isakoff, Phase II study of antidisialoganglioside antibody, dinutuximab, in combination with GM-CSF in patients with recurrent osteosarcoma (AOST1421): A report from the Children’s Oncology Group. Journal of Clinical Oncology 2020, 38, 10508–10508.
- Edelman, M.; Dvorkin, M.; Laktionov, K.K.; Navarro, A.; Juan-Vidal, O.; Kozlov, V.; Golden, G.; Jordan, O.; Deng, C. The anti-disialoganglioside (GD2) antibody dinutuximab (D) for second-line treatment (2LT) of patients (pts) with relapsed/refractory small cell lung cancer (RR SCLC): Results from part II of the open-label, randomized, phase II/III distinct study. J. Clin. Oncol. 2020, 38, 9017–9017. [Google Scholar] [CrossRef]
- Sikic, B.I.; Narayanan, S.; Colevas, A.D.; Padda, S.K.; Fisher, G.A.; Supan, D.; Wakelee, H.A.; Aoki, R.; Pegram, M.D.; Villalobos, V.M.; et al. A first-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 2016, 34, 3019–3019. [Google Scholar] [CrossRef]
- F. Pharmaceuticals, Fusion Pharmaceuticals Announces IND Clearance For FPI-2068, A Jointly Developed Novel Targeted Alpha Therapy, 2023.
- Moores, S.L.; Chiu, M.L.; Bushey, B.S.; Chevalier, K.; Luistro, L.; Dorn, K.; Brezski, R.J.; Haytko, P.; Kelly, T.; Wu, S.-J.; et al. A Novel Bispecific Antibody Targeting EGFR and cMet Is Effective against EGFR Inhibitor–Resistant Lung Tumors. Cancer Res 2016, 76, 3942–3953. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Altunay, B.; Morgenroth, A.; Beheshti, M.; Vogg, A.; Wong, N.C.L.; Ting, H.H.; Biersack, H.-J.; Stickeler, E.; Mottaghy, F.M. HER2-directed antibodies, affibodies and nanobodies as drug-delivery vehicles in breast cancer with a specific focus on radioimmunotherapy and radioimmunoimaging. Eur. J. Nucl. Med. 2020, 48, 1371–1389. [Google Scholar] [CrossRef]
- K. Hu, X. Ma, L. Xie, Y. Zhang, M. Hanyu, H. Obata, L. Zhang, K. Nagatsu, H. Suzuki, R. Shi, W. Wang, M.-R. Zhang, Development of a Stable Peptide-Based PET Tracer for Detecting CD133-Expressing Cancer Cells. ACS Omega 2021, 7, 334–341.
- Hu, K.; Shang, J.; Xie, L.; Hanyu, M.; Zhang, Y.; Yang, Z.; Xu, H.; Wang, L.; Zhang, M.-R. PET Imaging of VEGFR with a Novel 64Cu-Labeled Peptide. ACS Omega 2020, 5, 8508–8514. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Wu, W.; Xie, L.; Geng, H.; Zhang, Y.; Hanyu, M.; Zhang, L.; Liu, Y.; Nagatsu, K.; Suzuki, H.; et al. Whole-body PET tracking of a d-dodecapeptide and its radiotheranostic potential for PD-L1 overexpressing tumors. Acta Pharm. Sin. B 2021, 12, 1363–1376. [Google Scholar] [CrossRef] [PubMed]
- L. Au - Zhang, S. Au - Zhang, W. Au - Wu, X. Au - Wang, J. Au - Shen, D. Au - Wang, K. Au - Hu, M.-R. Au - Zhang, F. Au - Wang, R. Au - Wang, Development of a 68Gallium-Labeled D-Peptide PET Tracer for Imaging Programmed Death-Ligand 1 Expression. J Vis Exp. 2023, e65047. [CrossRef]
- Zhang, L.; Zhang, S.; Wu, J.; Wang, Y.; Wu, Y.; Sun, X.; Wang, X.; Shen, J.; Xie, L.; Zhang, Y.; et al. Linear Peptide-Based PET Tracers for Imaging PD-L1 in Tumors. Mol. Pharm. 2023, 20, 4256–4267. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Xie, L.; Hanyu, M.; Zhang, Y.; Li, L.; Ma, X.; Nagatsu, K.; Suzuki, H.; Wang, W.; Zhang, M.-R. Harnessing the PD-L1 interface peptide for positron emission tomography imaging of the PD-1 immune checkpoint. RSC Chem. Biol. 2020, 1, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Okarvi, S. Peptide-based radiopharmaceuticals and cytotoxic conjugates: Potential tools against cancer. Cancer Treat. Rev. 2008, 34, 13–26. [Google Scholar] [CrossRef]
- Zhu, Z.; Miao, W.; Li, Q.; Dai, H.; Ma, Q.; Wang, F.; Yang, A.; Jia, B.; Jing, X.; Liu, S.; et al. 99mTc-3PRGD2 for Integrin Receptor Imaging of Lung Cancer: A Multicenter Study. J. Nucl. Med. 2012, 53, 716–722. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Z.; Lin, K.-S.; Lau, J.; Zeisler, J.; Colpo, N.; Perrin, D.M.; Bénard, F. Melanoma Imaging Using 18F-Labeled α-Melanocyte-Stimulating Hormone Derivatives with Positron Emission Tomography. Mol. Pharm. 2018, 15, 2116–2122. [Google Scholar] [CrossRef]
- H. Ma, S.Y. Liu, Z.W. Zhang, G.H. Tang, G.J. Yuan, J. Zhao, S. Su, Preliminary biological evaluation of Ga-68-labeled cyclic RGD dimer as an integrin alpha(v)beta(3)-targeting radiotracer for tumor PET imaging. Journal of Radioanalytical and Nuclear Chemistry 2019, 321, 857–865. [CrossRef]
- Sakai, K.; Passioura, T.; Sato, H.; Ito, K.; Furuhashi, H.; Umitsu, M.; Takagi, J.; Kato, Y.; Mukai, H.; Warashina, S.; et al. Macrocyclic peptide-based inhibition and imaging of hepatocyte growth factor. Nat. Chem. Biol. 2019, 15, 598–606. [Google Scholar] [CrossRef]
- R. Siitonen, E. Peuhu, A. Autio, H. Liljenback, E. Mattila, O. Metsala, M. Kakela, T. Saanijoki, I. Dijkgraaf, S. Jalkanen, J. Ivaska, A. Roivainen, (68)Ga-DOTA-E[c(RGDfK)]2 PET Imaging of SHARPIN-Regulated Integrin Activity in Mice. J Nucl Med 2019, 60, 1380–1387.
- Cooper, B.M.; Iegre, J.; Donovan, D.H.O.; Halvarsson, M..; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: peptide–drug conjugates (PDCs). Chem. Soc. Rev. 2020, 50, 1480–1494. [CrossRef]
- X. Feng, Y.P. Wang, D.H. Lu, X.X. Xu, X. Zhou, H.Y. Zhang, T. Zhang, H. Zhu, Z. Yang, F. Wang, N. Lie, Z.F. Liu, Clinical Translation of a Ga-68-Labeled Integrin alpha(v)beta(6)-Targeting Cyclic Radiotracer for PET Imaging of Pancreatic Cancer. Journal of Nuclear Medicine 2020, 61, 1461–1467.
- Y. Gai, Y. Jiang, Y. Long, L. Sun, Q. Liu, C. Qin, Y. Zhang, D. Zeng, X. Lan, Evaluation of an Integrin αvβ3 and Aminopeptidase N Dual-Receptor Targeting Tracer for Breast Cancer Imaging. Molecular Pharmaceutics 2020, 17, 349–358. [CrossRef] [PubMed]
- N. Pirooznia, K. Abdi, D. Beiki, F. Emami, S.S. Arab, O. Sabzevari, S. Soltani-Gooshkhaneh, yyy Lu-177-labeled cyclic RGD peptide as an imaging and targeted radionuclide therapeutic agent in non-small cell lung cancer: Biological evaluation and preclinical study. Bioorganic Chemistry 2020, 102, 104100.
- Uehara, T.; Sensui, A.; Ishioka, S.; Mizuno, Y.; Takahashi, S.; Takemori, H.; Suzuki, H.; Arano, Y. Manipulating Pharmacokinetics of Purification-Free 99mTc-Labeled Bivalent Probes for In Vivo Imaging of Saturable Targets. Mol. Pharm. 2020, 17, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- M. Zhao, J.N. Ding, Q.L. Mao, Y.Q. Zhang, Y.J. Gao, S.Y. Ye, H.N. Qin, H.B. Shi, A novel alpha(v)beta(3) integrin-targeted NIR-II nanoprobe for multimodal imaging-guided photothermal therapy of tumors in vivo. Nanoscale 2020, 12, 6953–6958. [CrossRef] [PubMed]
- Kwon, D.; Lozada, J.; Zhang, Z.; Zeisler, J.; Poon, R.; Zhang, C.; Roxin; Lin, K.-S.; Perrin, D.; Benard, F. High-Contrast CXCR4-Targeted 18F-PET Imaging Using a Potent and Selective Antagonist. Mol. Pharm. 2020, 18, 187–197. [CrossRef]
- Yang, Y.; Zhang, J.; Zou, H.; Shen, Y.; Deng, S.; Wu, Y. Synthesis and evaluation of 68Ga-labeled dimeric cNGR peptide for PET imaging of CD13 expression with ovarian cancer xenograft. J. Cancer 2021, 12, 244–252. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Q.; Wong, C.T.T.; Li, X. Chemoselective Peptide Cyclization and Bicyclization Directly on Unprotected Peptides. J. Am. Chem. Soc. 2019, 141, 12274–12279. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Meibohm, B. Pharmacokinetics and Pharmacokinetic–Pharmacodynamic Correlations of Therapeutic Peptides. Clin. Pharmacokinet. 2013, 52, 855–868. [Google Scholar] [CrossRef] [PubMed]
- K. Hu, H. Geng, Q. Zhang, Q. Liu, M. Xie, C. Sun, W. Li, H. Lin, F. Jiang, T. Wang, Y.-D. Wu, Z. Li, An In-tether Chiral Center Modulates the Helicity, Cell Permeability, and Target Binding Affinity of a Peptide. Angewandte Chemie International Edition 2016, 55, 8013–8017. [CrossRef]
- Hu, K.; Li, W.; Yu, M.; Sun, C.; Li, Z. Investigation of Cellular Uptakes of the In-Tether Chiral-Center-Induced Helical Pentapeptides. Bioconjugate Chem. 2016, 27, 2824–2827. [Google Scholar] [CrossRef]
- Hu, K.; Sun, C.; Li, Z. Reversible and Versatile On-Tether Modification of Chiral-Center-Induced Helical Peptides. Bioconjugate Chem. 2017, 28, 2001–2007. [Google Scholar] [CrossRef]
- K. Hu, W. Xiong, C. Sun, C. Wang, J. Li, F. Yin, Y. Jiang, M.-R. Zhang, Z. Li, X. Wang, Z. Li, Self-Assembly of Constrained Cyclic Peptides Controlled by Ring Size. CCS Chemistry 2020, 2, 42–51. [CrossRef]
- Li, W.; Hu, K.; Zhang, Q.; Wang, D.; Ma, Y.; Hou, Z.; Yin, F.; Li, Z. N terminal N-methylation modulates chiral centre induced helical (CIH) peptides’ biophysical properties. Chem. Commun. 2018, 54, 1865–1868. [Google Scholar] [CrossRef]
- K. Hu, F. Yin, M. Yu, C. Sun, J. Li, Y. Liang, W. Li, M. Xie, Y. Lao, W. Liang, Z.-g. Li, In-Tether Chiral Center Induced Helical Peptide Modulators Target p53-MDM2/MDMX and Inhibit Tumor Growth in Stem-Like Cancer Cell. Theranostics 2017, 7, 4566–4576. [CrossRef]
- F.Z.D. Osterkamp, E. Schneider, C. Haase, M. Paschke, A. Höhne, J. Ungewiss, C. Smerling, U. Reineke, B. A.;, Compounds comprising a fibroblast activation protein ligand and use thereof, 2021.
- Calais, J. FAP: The Next Billion Dollar Nuclear Theranostics Target? J. Nucl. Med. 2020, 61, 163–165. [Google Scholar] [CrossRef] [PubMed]
- F.Z.D. Osterkamp, E. Schneider, C. Haase, M. Paschke, A. Höhne, J. Ungewiss, C. Smerling, U. Reineke, Bredenbeck, A.Compounds Comprising a Fibroblast Activation Protein Ligand and Use Thereof., 2021.
- E Perico, M.; Chinol, M.; Nacca, A.; Luison, E.; Paganelli, G.; Canevari, S. The humoral immune response to macrocyclic chelating agent DOTA depends on the carrier molecule. J. Nucl. Med. 2001, 42, 1697–703. [Google Scholar]
- D. Trujillo-Benitez, M. Luna-Gutierrez, G. Ferro-Flores, B. Ocampo-Garcia, C. Santos-Cuevas, G. Bravo-Villegas, E. Morales-Avila, P. Cruz-Nova, L. Diaz-Nieto, J. Garcia-Quiroz, E. Azorin-Vega, A. Rosato, L. Melendez-Alafort, Design, Synthesis and Preclinical Assessment of Tc-99m-iFAP for In Vivo Fibroblast Activation Protein (FAP) Imaging. Molecules 2022, 27, 264.
- Zboralski, D.; Hoehne, A.; Bredenbeck, A.; Schumann, A.; Nguyen, M.; Schneider, E.; Ungewiss, J.; Paschke, M.; Haase, C.; von Hacht, J.L.; et al. Preclinical evaluation of FAP-2286 for fibroblast activation protein targeted radionuclide imaging and therapy. Eur. J. Nucl. Med. 2022, 49, 3651–3667. [Google Scholar] [CrossRef] [PubMed]
- R.P. Baum, C. Schuchardt, A. Singh, M. Chantadisai, F.C. Robiller, J. Zhang, D. Mueller, A. Eismant, F. Almaguel, D. Zboralski, F. Osterkamp, A. Hoehne, U. Reineke, C. Smerling, H.R. Kulkarni, Feasibility, Biodistribution, and Preliminary Dosimetry in Peptide-Targeted Radionuclide Therapy of Diverse Adenocarcinomas Using (177)Lu-FAP-2286: First-in-Humans Results. J Nucl Med 2022, 63, 415–423.
- Parker, M.F.; Blecha, J.; Rosenberg, O.; Ohliger, M.; Flavell, R.R.; Wilson, D.M. Cyclic 68Ga-Labeled Peptides for Specific Detection of Human Angiotensin-Converting Enzyme 2. J. Nucl. Med. 2021, 62, 1631–1637. [Google Scholar] [CrossRef]
- Y. Pang, L. Zhao, T. Meng, W. Xu, Q. Lin, H. Wu, J. Zhang, X. Chen, L. Sun, H. Chen, PET imaging of fibroblast activation protein in various types of cancers by using (68)Ga-FAP-2286: Comparison with (18)F-FDG and (68)Ga-FAPI-46 in a single-center, prospective study. Journal of Nuclear Medicine 2023, 64, 386–394.
- Meng, L.; Fang, J.; Zhao, L.; Wang, T.; Yuan, P.; Zhao, Z.; Zhuang, R.; Lin, Q.; Chen, H.; Chen, X.; et al. Rational Design and Pharmacomodulation of Protein-Binding Theranostic Radioligands for Targeting the Fibroblast Activation Protein. J. Med. Chem. 2022, 65, 8245–8257. [Google Scholar] [CrossRef] [PubMed]
- FAP-2286 and Clovis’ Targeted Radionuclide Therapy Development Program.
- C. Oncology, A Study of 177Lu-FAP-2286 in Advanced Solid Tumors (LuMIERE) (LuMIERE), 2022.
- Rao, Z.; Zhang, Y.; Liu, L.; Wang, M.; Zhang, C. [177Lu]Lu-FAP-2286 therapy in a case of right lung squamous cell carcinoma with systemic metastases. Eur. J. Nucl. Med. 2022, 50, 1266–1267. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Herrmann, K.; Schöder, H.; Scott, A.M.; Lewis, J.S. Radiotheranostics in oncology: current challenges and emerging opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 534–550. [Google Scholar] [CrossRef] [PubMed]
- Privé, B.M.; Peters, S.M.; Muselaers, C.H.; van Oort, I.M.; Janssen, M.J.; Sedelaar, J.M.; Konijnenberg, M.W.; Zámecnik, P.; Uijen, M.J.; Schilham, M.G.; et al. Lutetium-177-PSMA-617 in Low-Volume Hormone-Sensitive Metastatic Prostate Cancer: A Prospective Pilot Study. Clin. Cancer Res. 2021, 27, 3595–3601. [Google Scholar] [CrossRef]
- J.R. Strosberg, M.E. Caplin, P.L. Kunz, P.B. Ruszniewski, L. Bodei, A. Hendifar, E. Mittra, E.M. Wolin, J.C. Yao, M.E. Pavel, E. Grande, E. Van Cutsem, E. Seregni, H. Duarte, G. Gericke, A. Bartalotta, M.F. Mariani, A. Demange, S. Mutevelic, E.P. Krenning, 177Lu-Dotatate plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. The Lancet Oncology 2021, 22, 1752–1763.
- Larimer, B.M.; Wehrenberg-Klee, E.; Caraballo, A.; Mahmood, U. Quantitative CD3 PET Imaging Predicts Tumor Growth Response to Anti-CTLA-4 Therapy. J. Nucl. Med. 2016, 57, 1607–1611. [Google Scholar] [CrossRef]
- E. Jagoda, F. Basuli, M. Williams, A. Opina, K. Wong, S. Adler, A. Ton, L. Szajek, O. Vasalatiy, B. Xu, R. Swenson, P. Choyke, Immuno-PET imaging of the programmed cell death-1 ligand (PD-L1) using the therapeutic mAb, avelumab. Journal of Nuclear Medicine 2017, 58, 178.
- Geller, J. Yan, H. Guo, 89Zr-Df-anti-PD-L1 for evaluating in vivo PD-L1 level in lung cancer mouse model. Journal of Nuclear Medicine 2018, 59, 1114.
- Vermeulen, K.; Vandamme, M.; Bormans, G.; Cleeren, F. Design and Challenges of Radiopharmaceuticals. Semin. Nucl. Med. 2019, 49, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Lu, C.; Yang, Y.; Shi, X.; Wang, H.; Yang, N.; Yang, K.; Zhang, X. Current Status and Trends in Peptide Receptor Radionuclide Therapy in the Past 20 Years (2000–2019): A Bibliometric Study. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Minczeles, N.; Hofland, J.; de Herder, W.; Brabander, T. Strategies Towards Improving Clinical Outcomes of Peptide Receptor Radionuclide Therapy. Curr. Oncol. Rep. 2021, 23, 1–11. [Google Scholar] [CrossRef]
- Scott, A.; Bodei, L. Pharmacogenomics in Radionuclide Therapy: Impact on Response to Theranostics. J. Nucl. Med. 2020, 62, 884–885. [Google Scholar] [CrossRef]
- Wahl, R.L.; Chareonthaitawee, P.; Clarke, B.; Drzezga, A.; Lindenberg, L.; Rahmim, A.; Thackeray, J.; Ulaner, G.A.; Weber, W.; Zukotynski, K.; et al. Mars Shot for Nuclear Medicine, Molecular Imaging, and Molecularly Targeted Radiopharmaceutical Therapy. J. Nucl. Med. 2020, 62, 6–14. [Google Scholar] [CrossRef]
Figure 1.
Discovery and research progress of nuclide-tagged FAP-targeting antibodies [125,126,127,128,129,130].
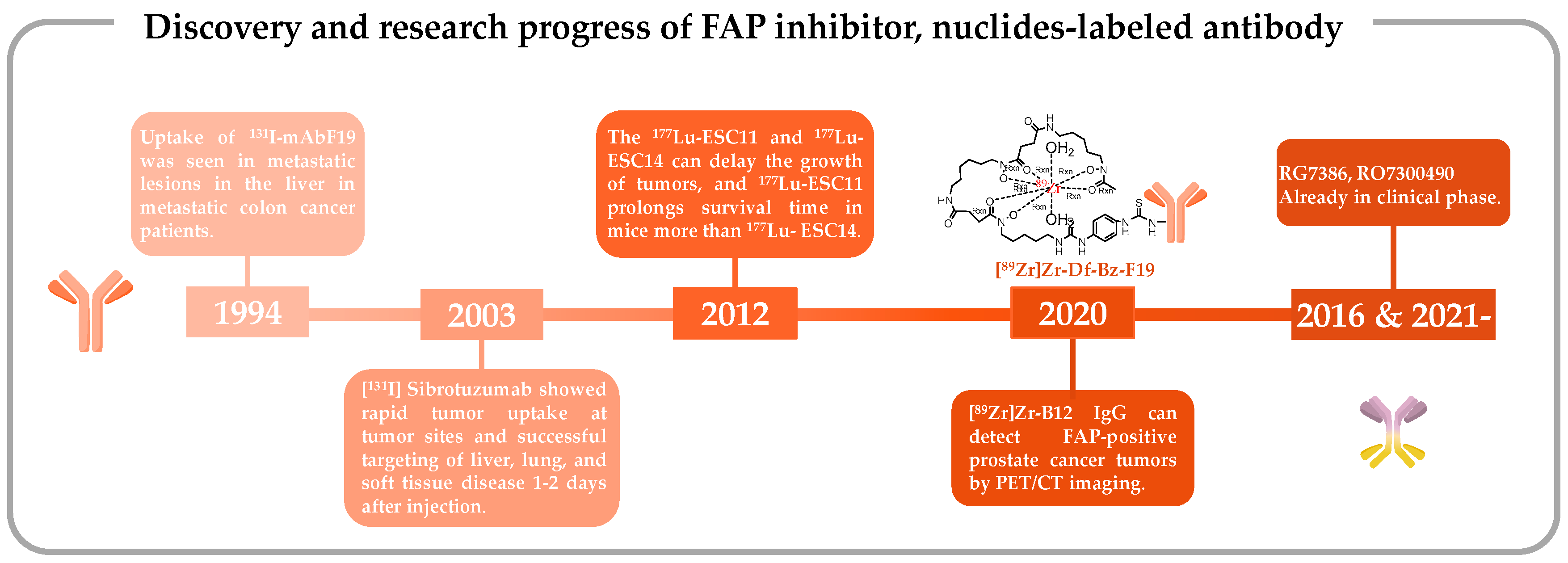
Figure 2.
Cyclic peptide compounds of 3B Pharmaceuticals.
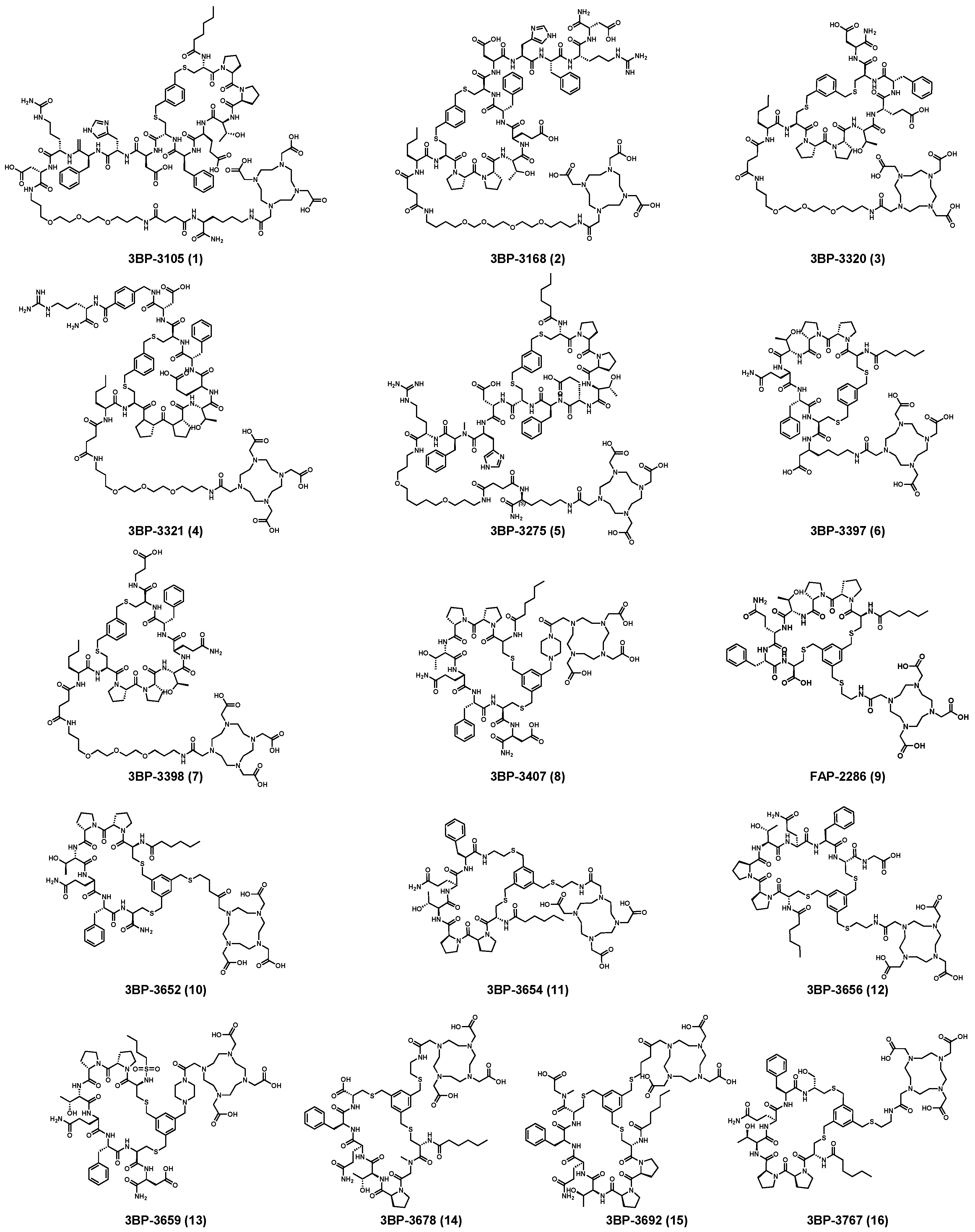
Figure 3.
Therapeutic comparison of 177Lu-labeled targets. (a) PET images and a transverse PET/CT image captured with 68Ga-FAP-2286 before treatment for the patients. (b) Anterior and posterior views and transverse SPECT/CT images at 48 h after 177Lu-FAP-2286 injection. Figures from Baum et al. [220].
Figure 3.
Therapeutic comparison of 177Lu-labeled targets. (a) PET images and a transverse PET/CT image captured with 68Ga-FAP-2286 before treatment for the patients. (b) Anterior and posterior views and transverse SPECT/CT images at 48 h after 177Lu-FAP-2286 injection. Figures from Baum et al. [220].
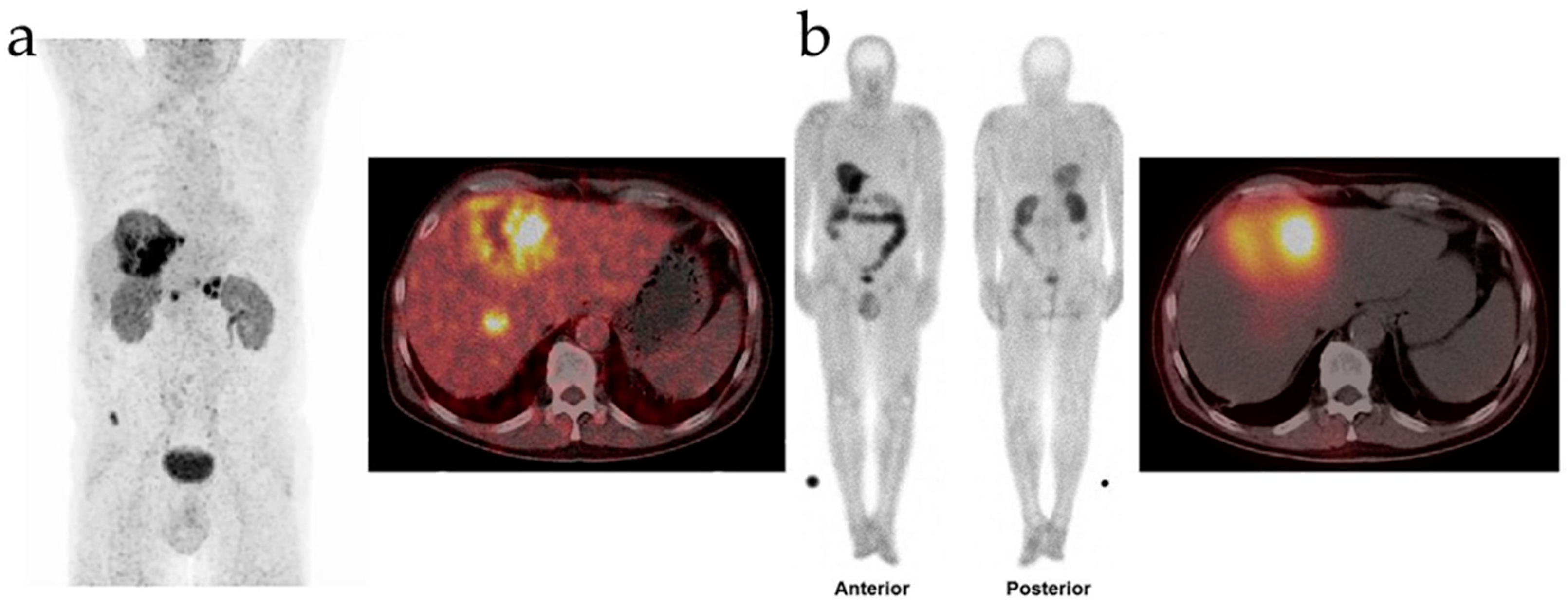

Table 1.
Results of an investigation of nuclide-labeled FAP-targeting antibodies.
| Developer | Name | In vitro Assessment | Tumor Model | Indication | Phase | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|---|
| Boehringer Ingelheim | F19 (BIBH-1) & 131I-BIBH-1 |
NA | Colorectal Cancer Metastatic Cancer |
SPECT/CT | Completed | NCT00005616 |
| Boehringer Ingelheim |
131I- Sibrotuzumab |
Radiochemical purity: ≥ 95 % | Patients of colorectal carcinoma and non-small cell lung cancer | SPECT | Terminated | NCT02209727 |
| Eliane Fischer | 177Lu-ESC11/ESC14 | Kd: 10 nM/ 210 nM |
Melanoma xenograft nude mouse | SPECT/CT | Preclinical | NA |
| Darpan N. Pandya | [89Zr]Zr-Df-Bz-F19 | Radiochemical purity: ≥ 99.5 % | U87MG tumor bearing mice | PET/CT | Preclinical | NA |
| Hallie M. Hintz | [89Zr]Zr- B12-IgG |
NA | Mice bearing intratibial CWR-R1FAP | PET/CT | Preclinical | NA |
Table 2.
HPLC analysis of the radiopurity for 177Lu-labeled compounds in a formulation buffer containing 100 mg/mL ascorbic acid and 5 mg/mL methionine.
Table 2.
HPLC analysis of the radiopurity for 177Lu-labeled compounds in a formulation buffer containing 100 mg/mL ascorbic acid and 5 mg/mL methionine.
| Name | Structural Unit | HPLC Retention Time (min) | HPLC Area% Day 0 Post End of Synthesis | HPLC Area% Day 6 Post End of Synthesis |
|---|---|---|---|---|
| 177Lu-3BP-3407 | Hex-[C(tMeBn(DOTA-PP))-P-P-T-Q-F-C]-D-NH2 | 7.5 | 95.7 | 94.0 |
| 177Lu-3BP-3554 | Hex-[C(tMeBn(DOTA-AET))-P-P-T-Q-F-C]-OH | 7.6 | 97.2 | 95.6 |
Table 3.
Structurally akin compounds to FAP-2286. (HEK-FAP Intake (Time) (%ID/g), ++++: 20-25, +++: 15-20, ++: 10-15, +: 6-10, -: <6, C (Cysteine, Cys), D (Aspartic acid, Asp), E (Glutamine Acid), F (Phenylalanine, Phe), G (Glycine, Gly), H (Histidine, His), K (Lysine, Lys), P (Proline, Pro), Q (Glutamine, Gln), R (Arginase, Arg), T (Threonine, Thr), The dot-circle structure represents, DOTA).
Table 3.
Structurally akin compounds to FAP-2286. (HEK-FAP Intake (Time) (%ID/g), ++++: 20-25, +++: 15-20, ++: 10-15, +: 6-10, -: <6, C (Cysteine, Cys), D (Aspartic acid, Asp), E (Glutamine Acid), F (Phenylalanine, Phe), G (Glycine, Gly), H (Histidine, His), K (Lysine, Lys), P (Proline, Pro), Q (Glutamine, Gln), R (Arginase, Arg), T (Threonine, Thr), The dot-circle structure represents, DOTA).
| Name | Molecular Formula |
Molecular Weight |
Structural Unit | Structures (Simple) | HPLC Retention Time (min) | HEK-FAP Intake (Time) in %ID/g |
|---|---|---|---|---|---|---|
|
3BP- 3105 |
C114H169N25O33S2 | 2481.171 | Hex-[C(3MeBn)-P-P-T-E-F-C]-D-H-F-R-D-Ttds-K(DOTA)-NH2 |  |
8.9 | + (6 h) |
|
3BP- 3168 |
C109H161N25O33S2 | 2412.110 | DOTA-Ttds-Nle-[C-(3MeBn)-P-P-T-E-F-C]-D-H-F-R-D-NH2 | 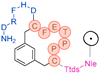 |
7.9 | + (3 h) |
|
3BP- 3320 |
C82H124N16O26S2 | 1812.841 | DOTA-Ttds-Nle-[C-(3MeBn)-P-P-T-E-F-C]-D-NH2 | 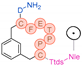 |
8.6 | + (1 h) |
|
3BP- 3321 |
C96H143N21O28S2 | 2101.993 | DOTA-Ttds-Nle-[C-(3MeBn)-P-P-T-E-F-C]-D-Pamb-R-NH2 | 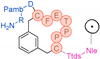 |
7.4 | + (3 h) |
|
3BP- 3275 |
C110H165N25O30S2 | 2379.820 | Hex-[C(3MeBn)-P-P-T-E-F-C]-D-H-Nmf-R-Ttds-K(DOTA)-NH2 | 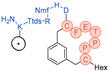 |
7.3 | + (3 h) |
|
3BP- 3397 |
C71H106N14O19S2 | 1522.720 | Hex-[C(3MeBn)-P-P-T-Q-F-C]-Bhk(DOTA)-OH | 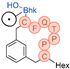 |
8.3 | ++ (24 h) |
|
3BP- 3398 |
C81H124N16O24S2 | 1768.842 | DOTA-Ttds-Nle-[C-(3MeBn)-P-P-T-Q-F-C]-Bal-OH | 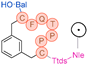 |
7.3 | + (3 h) |
|
3BP- 3407 |
C73H108N16O20S2 | 1592.737 | Hex-[C(tMeBn(DOTA-PP))-P-P-T-Q-F-C]-D-NH2 | 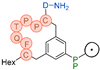 |
7.3 | ++ (1/24 h) |
|
3BP- 3554 |
C67H99N13O18S3 | 1469.640 | Hex-[C(tMeBn(DOTA-AET))-P-P-T-Q-F-C]-OH | 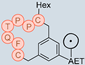 |
7.5 | ++++ (6 h) |
|
3BP- 3652 |
C67H100N14O17S3 | 1468.667 | Hex-[C(tMeBn(DOTA-AET))-P-Q-F-C]-NH2 |  |
7.3 | +++ (1 h) |
|
3BP- 3654 |
C66H99N13O16S3 | 1425.661 | Hex-[C(tMeBn(DOTA-AET))-P-P-T-Q-F-AET] | 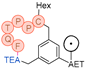 |
7.8 | + (1 h) |
|
3BP- 3656 |
C69H102N14O19S3 | 1526.665 | Hex-[C(tMeBn(DOTA-AET))-P-P-T-Q-F-C]-G-OH | 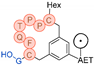 |
7.3 | - |
|
3BP- 3659 |
C70H104N14O19S3 | 1540.682 | Hex-[C(tMeBn(DOTA-AET))-P-P-T-Q-F-C]-Nmg-OH | 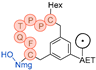 |
7.3 | - |
|
3BP- 3678 |
C65H97N13O18S3 | 1443.624 | Hex-[C(tMeBn(DOTA-AET))-Nmg-P-T-G-Q-C]-OH | 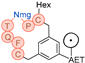 |
7.4 | + (1 h) |
|
3BP- 3692 |
C72H108N16O21S3 | 1628.706 | Pentyl-SO2-[C(tMeBn(DOTA-PP))-P-P-T-Q-F-C]-D-NH2 | 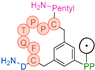 |
7.8 | + (3 h) |
|
3BP- 3767 |
C67H101N13O17S3 | 1455.666 | Hex-[C(tMeBn(DOTA-AET))-P-P-T-Q-F-Cysol] | 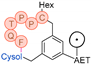 |
7.4 | ++ (1 h) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



