
Submitted:
03 February 2024
Posted:
05 February 2024
You are already at the latest version
Abstract
According to published descriptions, one of the most incapacitating forms of orofacial pain is trigeminal neuralgia (TN). At its core, trigeminal neuropathic pain (TN) is still a clinical diagnostic that has to be differentiated from other forms of TN and face pain linked to other neuralgias or headache disorders. Imaging can only help with the cause of the pain, whether it’s a vascular origin or not. It is becoming more and more evident that there is no one-size-fits-all medication or surgical procedure that can effectively treat every patient with trigeminal neuralgia (TN). This is probably because TN is a diverse collection of conditions that all present with face discomfort. Medical therapy using anticonvulsants like carbamazepine is still the first-line treatment for TN. If this doesn't work for the patient, surgical treatments are available. Microvascular decompression is often a safe, efficient operation with results that are both rapid and long-lasting. Patients who may benefit from percutaneous techniques such as radiofrequency ablation, glycerol ablation, balloon compression, or a combination, include those who cannot undergo general anesthesia or whose medical conditions prohibit a suboccipital craniectomy. Radiosurgery may be a great option for patients with bleeding diathesis brought on by blood-thinning medications who are not eligible for invasive procedures or who do not want to undergo open surgical procedures, as long as the patient is aware that achieving maximum pain relief may take several months. In conclusion, peripheral neurectomies persist in offering a low-cost and resource-saving substitute for pain management to those residing in areas with restricted financial and healthcare resources. In the end, developing a better understanding of the molecular processes behind trigeminal neuralgia will lead to new, less intrusive, and more effective treatments.
Keywords:
Trigeminal Neuralgia
; Treatment of TN
; Neurosurgery
; Radiosurgery
; Facial pain
1. Introduction
Trigeminal neuralgia, previously known as tic douloureux, is a chronic neuropathic pain syndrome, characterized by recurrent episodes of electric-shock-like pain in the trigeminal distribution. The episodes occur abruptly and usually without a trigger. Although in most cases the pain is unilateral, there have been reports of cases of bilateral trigeminal neuralgia (particularly in patients with multiple sclerosis). Some patients also experience autonomic symptoms with facial pain, such as conjunctival tearing [1,2].TN most commonly occurs sporadically without apparent risk factors. However, recently, some cases of familial TN have been seen, which raises concerns about the possible role of genetic and molecular basis in the pathogenesis of the condition [3,4,5].
It severely impairs the quality of life as it interferes with daily life activities like eating, talking, drinking, and touching the face [6]. There have been reports of increased rates of depression and anxiety in patients suffering from the condition, which signifies the importance of timely diagnosis and appropriate treatment [7,8]. Trigeminal neuralgia is more common in women than in men (F:M ratio 3:2) [9]. In most cases, the orofacial pain in TN is precipitated by innocuous stimuli like touching the face, talking, and brushing the teeth, however, a study has shown some unusual stimuli as well, i.e. turning the eyes, exposure to hot/cold food/water, flexing the trunk [10,11]. In most cases, the pain is in the maxillary and mandibular divisions of the trigeminal nerve, with only a few cases involving the ophthalmic branch [12].
1.1. Etiology:
TN is believed to occur due to vascular compression of the trigeminal nerve root in the area where it enters the brainstem, at the prepontine cistern within the Meckel’s cave [6]. This area is thought to be more vulnerable to damage by the compression since this is the transition zone between the peripherally myelinated nerve root (by Schwann cells) and the centrally myelinated nerve (by oligodendrocytes), i.e., the junction of the peripheral trigeminal nerve and root [8]. The demyelination could in most cases be due to a physical external compression by an artery or a vein (most common), or due to any space-occupying lesion like a vestibular schwannoma, meningioma, an aneurysm, or a cyst [9].
1.2. Classification:
In the most recent classification, published in 2018, by the ICHD-3, trigeminal neuralgia is classified into three types based on anatomical and electrophysiological findings, i.e., classical, secondary, and idiopathic. Classical TN is diagnosed when there is true neurovascular compression of the trigeminal nerve root causing morphological changes in it, seen on an MRI or during surgery [10]. Secondary TN is when TN is caused by an underlying neurovascular disease (except neurovascular compression) that is known to cause TN e.g., space-occupying lesions including cerebellopontine angle, AV malformation or fistula, skull-base bone deformity, connective tissue disease, and genetic causes of neuropathy [11]. Idiopathic TN, as the name suggests, is TN with no apparent cause (defined by ACHD-3 as symptomatic TN with neither MRI nor electrophysiological tests revealing significant abnormalities, suggesting TN without visible etiologies that are not fully understood yet). The idiopathic and classic types of TN are also subdivided based on the frequency of pain episodes [12]. There are two types: TN with pure paroxysmal pain and TN with concomitant continuous pain. The classification is aimed to help physicians delineate the underlying cause of the pain, and thereby, devise an appropriate treatment plan based on the cause [7].
2. Pathophysiology, Molecular Process:
TN is believed to occur due to vascular compression of the trigeminal nerve root in the area where it enters the brainstem, at the prepontine cistern within the Meckel’s cave [13]. This area is thought to be more vulnerable to damage by the compression since this is the transition zone between the peripherally myelinated nerve root (by Schwann cells) and the centrally myelinated nerve (by oligodendrocytes), i.e., the junction of the peripheral trigeminal nerve and root [14,15]. The demyelination could in most cases be due to a physical external compression by an artery or a vein (most common) or any space-occupying lesion like a vestibular schwannoma, meningioma, or an aneurysm, or a cyst [16,17,18]. The compression is thought to cause ischemic damage to the nerve root, which leads to demyelination. Therefore, it has long been thought that demyelination of the afferent trigeminal nerve root is the primary trigger of the pathogenesis and pathophysiology of TN [19]. However, the exact mechanism by which it clinically manifests is still not well understood. The demyelination causes a decrease in the excitatory threshold of the fast myelinated fibers, such that tactile signals can linger into the nearby slow nociceptive fibers, generating the pain paroxysms of TN [20,21,22].
Recently it has been seen that patients with TN have several molecular changes, channelopathies, and electrophysiological abnormalities in the trigeminal nerve [23]. Specifically, sodium channelopathies are hypothesized to have a significant role in the pathophysiological mechanisms of TN, since sodium channel blockers are effective in treating the condition [24]. This has been particularly backed by the study which showed abnormal expression of the voltage-gated sodium channels Nav1.7, NaV1.3, and Nav1.8 in patients with TN [5]. These channels are thought to be responsible for the occurrence and maintenance of the action potential. The abnormal constant pain that occurs in TN is shown to occur due to over-excitation of central sensory transmission since studies revealed abnormal nociceptive blink reflex and pain-related evoked potentials due to impairment of trigeminal nociception [6].
Studies have shown that the trigeminal nerve compression causes several structural changes to occur in the nerve root [25]. There are significant changes in the expression of voltage-gated channels that are caused by the downstream effects of nerve compression and subsequent ischemic damage. Among the various structural changes, some of the most significant are:
Changes in the expression of voltage-gated sodium (Nav) channels
Dysregulation of voltage-gated potassium channels
Downregulation of myelin-associated glycoprotein in Schwann cells
Among the sodium channel dysregulation, the most prominent ones are the upregulation of Nav1.1 and Nav1.3, and downregulation of Nav1.7. Nav1.3 is normally suppressed in adults, however, studies on TN patients have shown a considerable overexpression of the channel in the affected nerve [26]. Also, several neuropathic pain conditions are demonstrated to be linked with the overexpression of Nav1.3 [26, 27, 28]. In contrast, Nav1.7, which normally has a fast inactivation and slow recovery, is a channel that is resistant in its response to repetitive action potentials. It responds to graded potentials while it is in its prolonged close-gated inactivated state [27,29]. This makes it a threshold channel. The combined effect of these channelopathies causes an overall effect of increased neuronal excitability tendency [29]. This effect is amplified by the dysregulation of the resting membrane potentials due to impairment of the voltage-gated potassium channels, which leads to ectopic generation of action potentials due to hyper-excitability in the trigeminal neurons [30]. Additionally, there are prolonged after discharges in the demyelinated nerves which further increases the response to the trigeminal afferent inputs caused by normally innocuous stimuli. In addition, there is axonal sprouting that occurs due to the downregulation of myelin-associated glycoproteins, which are normally expressed by Schwann cells to inhibit axonal growth [31,32]. The axonal sprouting potentiates cross-talking between the hyper-excitable demyelinated nerves. This collective response caused by the decreased triggering threshold for activation of sensory nerve fibers of the trigeminal nerve is termed as “ignition hypothesis” by Devor et al. [33].
The amount of axonal loss is also in some way connected to the pain paroxysms in TN. A study backs this hypothesis. In the study, trigeminal nerves in patients with TN were analyzed using 3D MR imaging. The results were unique in that they showed that patients with TN with concomitant continuous pain had more severely atrophied trigeminal nerve roots than those with purely paroxysmal TN [34].
The histological studies of the affected trigeminal nerve root in patients with TN have provided a valuable insight for a better understanding of the disease's origins. The compression of the nerve induces inflammation in the nerve which leads to significant focal disintegration of the myelin sheath at the site of indentation [35]. This structural degradation initiates a cascade of demyelination and demyelination in the affected nerve root, similar to the pattern seen in cases of chronic nerve compression in animal models [36]. Additionally, a study has observed Schmidt-Lanterman incisures in trigeminal nerve root biopsies from TN patients, indicating a pathological increase in the metabolic demand for the growth and maintenance of the myelin sheath. This is the same phenomenon as observed in cases of chronic nerve compression [37]. The pathophysiology summary has been depicted in Figure 1.
Interestingly, the symptomatology of trigeminal neuralgia—classical, idiopathic, and secondary trigeminal neuralgia—is nearly identical in all cases. There is accumulating evidence of neurological disease at the root entrance zone as a result of a tumor or blood vessel compressing it [38]. This zone is where the myelination of central oligodendroglia myelinates from peripheral Schwann cells,35 which is why it is assumed that the entrance zone is more vulnerable to pressure [39]. Biopsy specimens taken from the compressed area during the procedure provide evidence for this theory, demonstrating demyelina tion, dysmyelination, and remyelination as well as the direct apposition of demyelinated axons [40].
Demyelinated afferents are known to become hyperexcitable and able to produce ectopic impulses that appear as pain that happens on their own [41]. Touch-evoked pain may be explained by haptic connections between demyelinated Aβ and Aδ fibers. According to one theory, touch-evoked sustained discharges from trigeminal ganglion cell somata that propagate from one cell to another are the cause of severe, almost explosive pain [42]. Neurophysiological investigations utilizing scalp far-field evoked potentials and QST are further evidence for the causal involvement of neurovascular compression at the root entry zone [43]. Both of these parameters begin to normalize following microvascular decompression [43].
Proposed causes for idiopathic trigeminal neuralgia include non-specific, non-multiple-sclerosis lesions in the brainstem, neural inflammation, and mutations causing a gain of function in neuronal voltage-gated ion channels. Concomitant continuous pain may be explained by ectopic impulse production, while other researchers have proposed that decreased descending inhibitory mechanisms or centrally mediated stimulation of nociceptive processing may also play a role. individuals with just paroxysmal pain and patients with concurrent chronic pain had decreased conditioned pain modulation, increased nociceptive blink reflexes, and brainstem-evoked potentials [44]. Ultimately, a blinded QST research was unable to discern differences between the two groups.
Given current understanding, a sizable fraction of these individuals also experienced compression injuries to the trigeminal root [45]. This was not recorded, most likely because trigeminal ganglionectomy does not remove the proximal portion of the root, where compression typically occurs [45]. We were unable to obtain TRG tissue from our patients for the same reason. If we had looked at our patients' TRGs, we most likely would have seen pathological alterations as well. Although the opposite is not true, root compression itself most likely results in retrograde alterations in the TRG [46]. The reported histological image of focal root disease limited to a zone directly next to a compressed blood artery would not have occurred in patients with original injury in the TRG. Instead, they would have displayed either anterograde (Wallerian) degeneration with axonal loss (if TRG somata or axons had been destroyed) or undamaged roots (if the TRG disease was largely demyelinating) [46].
These factors point to two possible causes of pain in TN patients who do not have a large amount of microvascular root compression [47]. First, even a small amount of root disease may cause severe discomfort in certain individuals who are prone to it. Second, and more commonly, TN patients with a primary problem in or close to the TRG may be those without root pathology [47]. MVD is the recommended treatment for people with a main root compression lesion, while there are other effective treatments as well. Partial rhizotomy is a sensible first choice for TN patients whose main lesion is located in the TRG [48]. The igniting theory states that both groups experience pain through the same process. Both the ganglion's axons and neuronal somata as well as the axons of the trigeminal root show the particular afferent pathophysiology that causes ignition [48]. The pathophysiology of multiple sclerosis and trigeminal neuralgia resulting from lesions that occupy space, such as tumors, aneurysms, or arteriovenous malformations, is likely similar to that of normal TN [48].
3. Diagnosis
Diagnostic criteria:
The International Classification of Headache Disorders edition 3 (ICHD-3) criteria for diagnosis of TN include [49]:
- A.
- Recurrent paroxysms of unilateral facial pain in the distribution(s) of one or more divisions of the trigeminal nerve, with no radiation beyond, and fulfilling criteria B and C.
- B.
- Pain has all of the following characteristics:
- Lasting from a fraction of a second to 2 min.
- Severe intensity.
- Electric shock-like shooting, stabbing, or sharp in quality.
- C.
- Precipitated by innocuous stimuli within the affected trigeminal distribution.
- D.
- Not better accounted for by another ICHD-3 diagnosis.
Since TN is mostly a clinical diagnosis, a diagnosis can be reached without the need for neuroimaging or laboratory testing. Patients who do not require more workup can be treated if they have a typical history and a normal neurologic examination other than discomfort [50, 51]. To rule out other potential causes of TN, such as inflammatory or mass lesions, all patients with TN often get elective imaging as part of their contemporary workup [52]. Magnetic resonance imaging (MRI) using high-resolution sequences at the skull base is often the modality of choice since computer tomography (CT) has limitations when examining the brain parenchyma, skull base nerves, and CSF cisterns [53,54]. According to a study by the Quality Standards Subcommittee of the European Federation of Neurological Societies and the American Academy of Neurology, up to 15% of TN patients have regular head imaging that detects nonvascular, structural causes of TN [54].
4. Pharmacological Treatment
Anticonvulsant medicine is used in medical care as a first-line therapy for TN. Patients may choose open surgery, radiation, transcutaneous, or percutaneous treatment if medication care is not successful for them because of ongoing discomfort or intolerable side effects [55]. Patients with type 1 TN often respond well to open surgical, radiosurgical, and percutaneous TN therapy [56]. Compared to individuals with type 1 TN, those with type 2 TN are more likely to experience pain recurrence and a shorter pain-free time. To relieve pain, patients with secondary TN (such as tumors) should get therapy for the underlying disease (such as decompression and tumor removal) [57,58]. For symptomatic relief, medication therapy of secondary TN may be recommended for individuals who are not surgical candidates [58]. Table 1 shows the different pharmacologic treatments used for trigeminal neuralgia.
5. Neurosurgical Treatments and Outcomes
5.1. Glycerol Injection
Glycerol is a viscous, colorless, and odorless liquid. Glycerol is extremely hypertonic at concentrations more than 99%, and it can induce neurolysis by rupturing myelin or by entering the perineurium directly [59]. A minimally invasive technique called percutaneous stereotactic radiofrequency rhizotomy (PSR) can reduce pain brought on by cluster headaches, glossopharyngeal neuralgia, and trigeminal neuralgia [60]. A fluoroscopy-visualized needle is inserted into the trigeminal cisterna during percutaneous glycerol rhizolysis. Under a local anesthetic, this is done. A 3.5" x 20 G spinal needle is usually placed in the skin next to the mouth and passed through the foramen ovale, a hole near the base of the skull [61]. A contrast dye can be injected into a sitting patient with their head flexed to measure the cisterna's size. Following the aspiration of the contrast dye, an equivalent amount of glycerol is administered [61]. According to Xu et al.'s account of their work with 3370 patients, 73% of them had pain reduction with a single injection. After four injections, the total success rate was 99.58%, rising with the number of injections [62]. Twenty-one percent of the 2,750 follow-up patients experienced a return of pain within five years. Throughout the trial, the total incidence of pain recurrence was 33%; other large case series show an initial pain alleviation rate of 70 to more than 90% [62].
Nevertheless, 20–40% of individuals relapse in pain 20–60 months following the operation. Glycerin rhizotomy complications typically involve face dysesthesia, hypoalgesia, and hyperesthesia [63]. Meningitis, hematoma, nausea/vomiting, optic nerve damage, and trigeminal motor paralysis are among the other perioperative problems. Keratitis ulcers and other issues about the eyes may arise from hypoesthesia of the superior trigeminal division [64]. A retrospective analysis of 53 operations at a single center over a 7-year period (2012-2018) when volume-maximized glycerol rhizolysis was used [65]. Analysis was done on the frequency, length of pain relief, and complications experienced over a median follow-up period of eight years and found that volume-maximized glycerol injection outcomes reported in the literature following conventional volume glycerol injections, the former is safer and more successful [66]. The length of pain freedom attained is greater than most research described in the literature, and the results of the hypoaesthesia are similar to those of other investigations [67]. Results for pain independence are better for patients who have post-procedure hypoaesthesia [67].
5.2. Balloon compression
Using a 14-gauge hollow metallic introducer is part of the procedure. Under fluoroscopic guidance, the introducer is advanced to the foramen ovale. Through the metallic introducer, a 4-French Fogarty balloon is inserted about 1 cm past the needle tip [68]. Under live fluoroscopy, contrast dye is introduced into the balloon, and compression is maintained for three to six minutes [69]. The balloon is inflated, and at the puncture site, the metallic introducer and the Fogarty balloon are extracted simultaneously with human pressure. In a rabbit model of balloon compression of the Gasserian ganglion, Brown et al. showed that tiny myelinated fibers were largely intact whereas big myelinated axons, which are implicated in the sensory trigger, were primarily destroyed [70]. According to a study by Skirving et al., 496 TN patients had 531 percutaneous balloon compressions of the trigeminal ganglion performed between 1980 and 1999 [71].126 All patients except one had timely pain relief following one of the 522 successful treatments. 19.2% of patients experienced a recurrence of pain within 5 years, and 31.9% did so during the research. According to some research, there can be an instant pain alleviation of 80% to over 90%, but there can also be a 15% to 50% chance of pain recurrence after two to five years [72]. The inflated balloon's form, the balloon's opening pressure the amount of contrast injected, and the compression duration are variables that have been demonstrated to impact balloon compression results [73]. In addition to face dysesthesias, cranial nerve palsies and abrupt variations in blood pressure and heart rate brought on by trigeminal cardiac reflexes are possible complications of balloon compression [74].
5.3. Ablation using Radiofrequency
Réthi initially invented electrocoagulation in 1913 to target the trigeminal nerve rootlets [75]. Serious side effects from the technique's early use included enucleation-requiring corneal ulcers, numerous cranial nerve palsies, carotid damage, cardiac arrest, meningitis, and even death [76]. The process is now safer thanks to advancements in electrode design, placement techniques, and temperature monitoring. The needle location is confirmed using fluoroscopic guidance, as previously mentioned. After removing the needle obturator, the electrode is inserted [77]. Usually, electric stimulation occurs between 0.2 and 1 V (50 Hz for 0.2 ms). After that, the electrode is swapped out for a thermocouple, and lesions are created for 30 to 120 seconds at a maximum voltage of 0.5 V and 75 cycles per second between 55 and 80°C. After that, the electrode and cannula are taken out [78]. Selective V2 or V3 targeting via the pterygopalatine fossa under ultrasonography guidance may be employed in some circumstances. Numerous extensive investigations have exhibited the effectiveness of radiofrequency ablation. According to Wu et al.'s results, out of 1860 patients, 79% had instant pain relief and 18% had lesser pain [79]. In the first year, 11.1% of patients experienced recurring pain; during the second year, 25% of patients experienced recurrent pain. The 25-year experience of Kanpolat et al. treating 1600 patients with 2138 radiofrequency ablation treatments was reported.168 Of the patients, 97.6% had acute pain alleviation [80]. Seven percent of the patients experienced an early pain recurrence within six months, while the other seventeen percent experienced a late pain recurrence [80]. At five years, 58% of patients who had a single surgery experienced complete pain alleviation; at ten and twenty years, this percentage dropped to 52% and 41%, respectively [80]. In rare cases, meningitis, carotid-cavernous fistulas, anesthesia dolorosa/trigeminal deafferentation discomfort, and CSF leaks may occur [81, 82].
To examine the safety and efficacy of trigeminal percutaneous radiofrequency ablation in classical refractory trigeminal neuralgia, a Randomized control study was conducted [83]. The research comprised thirty consecutive individuals with classical trigeminal neuralgia who had not responded to medication therapy. Two groups—one for thermal radiofrequency and the other for sham treatment—were randomly allocated to the patients. The mean Numerical Rating Scale score dropped after a month, from 8.9 to 5.8 in the sham group and from 9.2 to 0.7 in the radiofrequency group [83]. This noteworthy decrease was detectable from the first day following the treatment and persisted throughout the first month [83].
5.4. Transcutaneous Electrical Nerve Stimulation
Wall Street announced the groundbreaking finding in 1967 that nerve stimulation reduced the experience of pain. However, there is limited evidence that transcutaneous electrical nerve stimulation (TENS) is an effective treatment for TN. Stimulating electrodes are positioned in the regions innervated by the second and third branches of the trigeminal nerve, which is where patients usually suffer pain, as part of TENS treatment for TN [84]. Yameen et al. especially examined the effects of several forms of TENS therapy for TN that were either non-responsive or just partially responding to medication [85]. Continuous mode It has been demonstrated that TENS therapy, in which stimulation is administered continuously throughout sessions, is more efficacious than burst therapy, in which stimulation is administered in pulses [86]. After a three-week follow-up, 26 of the 31 TN patients were still responsive to TENS treatment [85]. There is no mention of long-term results. Singla et al.'s study, which had 30 TN patients receiving TENS for 20–40 days, provides support for these results [87]. After one month, the patient's total pain ratings decreased by 65%, and after three months, they decreased by 85% [87]. There are no long-term results available [87]. These results suggest that TENS may be a non-invasive therapy for TN; however, more research is required to examine the treatment's long-term effects and to confirm this effect in a blinded, randomized manner [88].
5.5. Peripheral Nerve Stimulation
Lead-based peripheral nerve stimulation therapy uses an implanted pulse generator (IPG) to provide electrical impulses from an electrode array to the Gasserian ganglion or branches of the trigeminal nerve [89]. Wall and Street reported on eight patients, one of whom had TN in the V2 distribution [90]. The patient had pain relief for 17 minutes following 5 minutes of infraorbital nerve stimulation; however, there is no long-term follow-up [90]. Three patients with typical trigeminal neuralgia in a V3 distribution had temporal craniotomies to insert platinum stimulator electrodes on V3211; the results of these procedures were reported by Shelden et al [91]. During the 3.4–5 month follow-up period, all three patients were pain-free, even though two of them had never had any nerve stimulation following implantation. The outcomes of Gasserian nerve stimulation with implanted electrodes in six patients—some of whom had atypical TN—were reported by Meyerson and Hakansson in 1980 [91]. Five patients reported full or partial pain alleviation at follow-up, which ranged from six to twenty-one months. Ellis et al. published a study on 35 individuals who had lead-based stimulation of the trigeminal branch more recently [92]. Type 1 TN was identified in two individuals, and type 2 TN in nine others. TN with symptoms was identified in one case [93]. Results showed that peripheral nerve stimulation often reduced pain, notwithstanding one incidence of superficial temporal artery pseudoaneurysm due to needle damage [93]. Trial stimulation was successful for the symptomatic TN patient, two of the nine type 2 TN patients, and both type 1 TN patients [93]. Thirteen of the thirty-five research participants reacted to the trial stimulation, and fifteen of them had permanent hardware implantation. At the latest follow-up, 11/15 individuals reported reduced pain, throughout a follow-up period of 15 months on average [94]. Which TN patients had permanent hardware implantation was not disclosed, and the patients' individual reactions were not mentioned. More research and clarity are needed to determine the long-term effects of peripheral nerve stimulation for the treatment of TN. Procedure-related complications include fracture or migration of the electrode and extension wire. For individuals with trigeminal neuropathic pain as compared to TN, peripheral nerve stimulation is typically limited by permanent lead implantation in the face and an unknown duration of effect [94].
5.6. Deep Brain Stimulation
Deep brain stimulation (DBS) was initially employed to treat chronic pain in the 1950s, but when two significant multicenter trials failed to show its effectiveness, the practice was abandoned in the late 1990s [95]. However, a significant amount of research suggests that the activation of the sensory thalamus, periventricular grey (PVG), and periaqueductal grey (PAG) may be targeted regions for pain [95]. Although the exact mode of action is yet unknown, endogenous opiate release might be involved in pain management. One patient with refractory TN was treated by DBS of the PVG and ventroposterolateral thalamic nucleus, according to Nandi et al [96]. Despite not having IPG implantation in the end, the patient had a 33% decrease in discomfort during the trial stimulation [96]. Tests for TN have also been conducted on other sites, including the posterior hypothalamus. Five patients with TN caused by multiple sclerosis were treated by Franzini et al [97]. They found that, after one to three years after surgery, all five patients had less pain, however only two of the patients did not need medication [97]. DBS is still a salvage technique for refractory TN because of its invasive nature and the requirement for permanent IPG implantation [98].
5.7. Stereotactic Radiosurgery
In 1951, Lars Leksell published the first account of stereotactic radiosurgery for TN [99]. Present-day radiation delivery techniques include CyberKnife (CK), Gamma Knife (GK), and linear accelerator (LINAC) [100]. Gamma Knife requires pins to be inserted percutaneously into the skull while under local anesthetic to achieve stereotactic head frame immobilization. stationary radioactive Gamma-emitting sources of cobalt-60 are then employed to target the treatment region from different directions [100]. When undergoing LINAC therapy, a face mask or head frame immobilization are the two options available. The intended radio surgical dosage is administered by rotating arks of LINAC X-ray-producing devices around the patient's head [101]. Wearing a face mask is part of the CyberKnife therapy regimen.
Here, radiation emitters are attached to a robotic arm and are capable of moving non-rotationally around the head of the patient. 6461 patients receiving GK, LINAC, or CK treatments were evaluated by Tuleasca et al [102]. Radiation doses for GK patients were recommended at the 100% isodose line and ranged from 60 to 97 Gy. those on LINAC were administered 50–90 Gy at the 80% isodose line, whereas those on CK were provided 66–90 Gy at the 90% isodose line [102]. Based on available level II data, the lowest possible effective dose is 70 Gy, whereas the highest possible effective dose is 90 Gy. While the effectiveness rate of radiosurgery does not change at doses more than 90 Gy, there is a higher incidence of problems. In contrast to other open or percutaneous surgical techniques, radiosurgery does not immediately relieve pain [102]. The majority of experts concur that the longest duration for pain relief is 180 days following therapy, while the timeframes to pain reduction vary from 0 to 480 days [103]. With or without TN drug supplementation, the average rates of pain relief were determined to be 86% for GK, 88% for LINAC, and 79% for CK [103]. Statistics did not show a difference in this. When just those patients who experienced no pain and didn't need any extra medicine are taken into consideration, radiosurgery's success rate in producing a pain-free response drops to 52% for GK, 43% for LINAC, and 58% for CK [102]. For GK, LINAC, and CK, the respective median recurrence rates for face discomfort were 23%, 29%, and 27%. For GK, the range of mean times to recurrence was 6 to 48 months; for LINAC, it was 7.5–20.4 months; and for CK, it was 9 months [102]. Long-term rates of pain control are seldom reported in research; nevertheless, at 7 years, rates of pain control vary from 22% to 60% [104]. Ten-year rates of pain management drop to much less than 50% [104]. Patients with multiple sclerosis have greater rates of pain recurrence and poorer rates of pain-free survival. Radiosurgery-related complications for TN include dry eye, keratitis/corneal damage from lack of feeling, face hypesthesia, and dysesthesia [104]. For GK, LINAC, and CK, the median rates of hypesthesia are 19%, 29%, and 19%, respectively. These problems might appear one to ninety-four months after they first occur. While further research is needed to determine the optimal time for radiosurgery following pain beginning, recent studies have demonstrated that conducting GK sooner in the course of TN pain development may enhance clinical results and minimize the need for drug therapy [104].
5.8. Microvascular Decompression (MVD)
The first trans-antral approach to the trigeminal ganglion was reported by Carnochan in 1856 [105]. Horsley (1891) reported a method of treating trigeminal neuralgia by sectioning the intradural nerve fibers of the trigeminal nerve [106]. An extradural method of trigeminal nerve rootlet sectioning at the foramen ovale and rotundum was reported by Hartley and Krause in 1892 [107]. Frazier and Spiller further refined this strategy by dividing certain nerve segments to reduce morbidity. Walter Dandy promoted partial trigeminal nerve sectioning with a posterior fossa technique in 1925 [108]. He saw during those surgeries that vascular structures were compressing the nerve, and he postulated in 1932 that TN was caused by vascular compression. This notion was verified in 1967 by Peter Jannetta using an operational microscope [109].
A suboccipital craniectomy and microsurgical dissection around the trigeminal nerve are two surgical procedures that go into microvascular decompression [110]. Once a compressive artery has been identified, the nerve and the problematic artery can be separated by using a pledget made of Teflon (polytetrafluoroethylene) or Ivalon (polyvinyl alcohol) [110]. Others have described arterial decompression as the construction of a sling using glue, sutures, or the tentorium. Bipolar cautery is used to split veins that are causing compression of the trigeminal nerve [110]. With systematic evaluations predicting better than 90% rates of first relief from pain, the results after MVD are positive. Of the 1185 patients who received microvascular decompression in the original seminal publication by Barker et al., 1155 were monitored for a year or longer [111]. The first two years following surgery were the most common times for postoperative pain recurrences. Eleven percent had further surgeries to address the recurrences. Seventy percent of the patients were pain-free ten years following surgery and did not need further medication for pain management [111]. A further 4% experienced sporadic discomfort for which long-term treatment was not necessary. According to the most current estimates of MVD outcomes, 80% of patients will still be pain-free after a year, 75% after three, and 73% after five [111]. Although major problems are uncommon, up to 20% of individuals may experience difficulties after MVD. Because cranial nerves IV through XII are exposed after surgery, issues connected to cranial nerves should be given particular attention in cases of motor neuropathy. Trigeminal nerve numbness and dysesthesias affect 5–10% of individuals. This is a large minority [110]. Facial nerve palsy is uncommon (<1%), and diplopia brought on by manipulation of the fourth or sixth nerve is frequently temporary [112]. The range of hearing loss is between 1 and 20%, based on subjective reports or audiometry [112]. When compared to other craniotomies, infections are uncommon and happen at a comparable incidence. Stroke, aseptic meningitis, and surgical bleeding are further uncommon side effects. It is believed that 0.2% of deaths are related to the surgery [112]. MVDs have a longer sustained rate of pain management and a lower rate of hypoesthesia and dysesthesia problems when compared to percutaneous procedures. As a result, once medicinal therapy fails, more and more neurosurgical professionals are considering MVD as a first-line surgical surgery [112].
6. Discussion
Trigeminal neuralgia is now diagnosed clinically; laboratory correlation or imaging is not necessary, while imaging can be useful in ruling out tumors and other conditions that may be associated with trigeminal pain. Many people appear with Type II TN pain, or Type I TN pain without evidence of nerve compression, even though most patients with Type I TN include neurovascular compression. It is still unknown why these people feel pain and what molecular processes differentiate Type I TN pain from Type II TN pain. It's also unclear if the differences in these unique processes originate from various stimuli that are distal to the root entrance zone or if they have to do with the core mechanisms of pain interpretation. Given that certain people with multiple sclerosis consistently experience reduced benefits from pharmacological and surgical therapies, it is plausible that neuroinflammation contributes to the pathophysiology and severity of the illness. Trigeminal neuralgia's neuronal-glial interaction will require further research to completely comprehend, and developing new and more potent treatments may become possible with a more accurate mechanistic knowledge of TN.
7. Conclusion
It is becoming more and more evident that there is no one-size-fits-all medication or surgical procedure that can effectively treat every patient with trigeminal neuralgia (TN). This is probably because TN is a diverse collection of conditions that all present with face discomfort. Anticonvulsants continue to be the first-line therapy for TN because of their lengthy history of use and manageable adverse effects. There are several surgical treatments accessible to patients who do not respond to medicinal therapy. Microvascular decompression is often a safe, efficient operation with results that are both rapid and long-lasting. Individuals who are not well enough to undergo general anesthesia or whose medical conditions prohibit a suboccipital craniectomy could benefit from percutaneous techniques such as radiofrequency ablation or glycerin ablation. If a patient is unwilling to undergo surgically invasive procedures or has iatrogenic bleeding diathesis from blood-thinning medications and is otherwise not eligible for any invasive procedure, radiosurgery may be a great option—as long as the patient is informed that achieving maximum pain relief may take several months. Finally, for the majority of patients in areas with few medical and economic resources, peripheral neurectomies remain an affordable and resource-saving alternative to pain treatment. In the end, developing a better understanding of the molecular processes behind trigeminal neuralgia will lead to the development of new, less intrusive, and more effective treatments.
Author Contributions
Conceptualization, S.S. and J.I.; methodology, S.S.; software, J.I. and M.K.; validation, S.S., J.I..; formal analysis, S.S.; investigation, S.S.; resources, J.I. and M.K.; data curation, S.S.; writing—original draft preparation, S.S. and M.K.; writing—review and editing, X.X.; visualization, X.X. supervision, X.X.; project administration, S.S., J.I..;. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
The appendix is an optional section that can contain details and data supplemental to the main text—for example, explanations of experimental details that would disrupt the flow of the main text but nonetheless remain crucial to understanding and reproducing the research shown; figures of replicates for experiments of which representative data is shown in the main text can be added here if brief, or as Supplementary data. Mathematical proofs of results not central to the paper can be added as an appendix.
Appendix B
All appendix sections must be cited in the main text. In the appendices, Figures, Tables, etc. should be labeled starting with “A”—e.g., Figure A1, Figure A2, etc.
References
- Katusic, S. , Beard C.M., Bergstralh E., Kurland L.T. Incidence and clinical features of trigeminal neuralgia, Rochester, Minnesota, 1945–1984. Ann. Neurol. 1990, 27, 89–95. [Google Scholar] [CrossRef]
- Cruccu, G. , Di Stefano, G., & Truini, A. Trigeminal Neuralgia. The New England journal of medicine 2020, 383, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Khawaja, S. N. , & Scrivani, S. J. Trigeminal Neuralgia. Dental clinics of North America 2023, 67, 99–115. [Google Scholar] [CrossRef]
- Headache Classification Subcommittee of The International Headache Society. The International classification of headache disorders. 38. 3rd edn. Cephalalgia, 2018: 1–211.
- Siqueira SRDT, Alves B, Malpartida HMG, et al. . Abnormal expression of voltage-gated sodium channels Nav1.7, NaV1.3 and Nav1.8 in trigeminal neuralgia. Neuroscience 2009, 164, 573–7. [Google Scholar] [CrossRef]
- Obermann M, Yoon M-S, Ese D, et al. . Impaired trigeminal nociceptive processing in patients with trigeminal neuralgia. Neurology 2007, 69, 835–41. [Google Scholar] [CrossRef]
- Zakrzewska JM, Wu J, Mon-Williams M, et al. . Evaluating the impact of trigeminal neuralgia. Pain 2017, 158, 1166–74. [Google Scholar] [CrossRef] [PubMed]
- Ferneini E., M. Trigeminal Neuralgia. Journal of oral and maxillofacial surgery : official journal of the American Association of Oral and Maxillofacial Surgeons 2021, 79, 2370–2371. [Google Scholar] [CrossRef]
- May, A. , & Hoffmann, J. Facial pain beyond trigeminal neuralgia. Current opinion in neurology 2021, 34, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A. , & Kondziolka, D. Trigeminal Neuralgia and Other Facial Neuralgias. Progress in neurological surgery 2019, 34, 273–278. [Google Scholar] [CrossRef]
- Liao, J. Y. , Zhou, T. H., Chen, B. K., & Liu, Z. X. Schwann cells and trigeminal neuralgia. Molecular pain 2020, 16, 1744806920963809. [Google Scholar] [CrossRef]
- Alwardian, M. , Chrysikos, D., Samolis, A., Papachristou, A., Spartalis, E., Piagkou, M., & Troupis, T. Trigeminal Neuralgia and Potential Correlations with Anatomical Variations of the Trigeminal Nerve. Acta medica academica 2021, 50, 292–299. [Google Scholar] [CrossRef]
- Bendtsen, L. , Zakrzewska, J. M., Abbott, J., Braschinsky, M., Di Stefano, G., Donnet, A., Eide, P. K., Leal, P. R. L., Maarbjerg, S., May, A., Nurmikko, T., Obermann, M., Jensen, T. S., & Cruccu, G. European Academy of Neurology guideline on trigeminal neuralgia. European journal of neurology 2019, 26, 831–849. [Google Scholar] [CrossRef]
- Di Stefano, G. , Maarbjerg, S., & Truini, A. Trigeminal neuralgia secondary to multiple sclerosis: from the clinical picture to the treatment options. The journal of headache and pain 2019, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J. L. , Domingo, R. A., Rowland, N. C., & Vandergrift Iii, W. A. Trigeminal Neuralgia Secondary to Meckel's Cave Meningoencephaloceles: A Systematic Review and Illustrative Case. Neurology India 2022, 70, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Gerwin, R. Chronic Facial Pain: Trigeminal Neuralgia, Persistent Idiopathic Facial Pain, and Myofascial Pain Syndrome-An Evidence-Based Narrative Review and Etiological Hypothesis. International journal of environmental research and public health 2020, 17, 7012. [Google Scholar] [CrossRef] [PubMed]
- Boeddinghaus, R. , & Whyte, A. Imaging of Trigeminal Neuralgia and Other Facial Pain. Neuroimaging clinics of North America 2021, 31, 485–508. [Google Scholar] [CrossRef] [PubMed]
- Ganz J., C. Trigeminal neuralgia and other cranial pain syndromes. Progress in brain research 2022, 268, 347–378. [Google Scholar] [CrossRef] [PubMed]
- Obermann, M. (2019). Recent advances in understanding/managing trigeminal neuralgia. F1000Research. [CrossRef]
- Dong, B. , Xu, R., & Lim, M. The pathophysiology of trigeminal neuralgia: a molecular review. Journal of neurosurgery 2023, 139, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Munoz, A. , Maxwell, C., Gofman, N., Liebman, K., & Veznedaroglu, E. The management of trigeminal neuralgia with triptans, a narrative review of the literature. Headache 2022, 62, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S. , Bansal, R. N., Singh Sodhi, S. P., & Brar, G. K. Animal models - Mimicking the pain of trigeminal neuralgia. Indian journal of pharmacology 2022, 54, 138–145. [Google Scholar] [CrossRef]
- Maltez, N. , Choi, M. Y., Troyanov, Y., Wang, M., Jantz, M., Fritzler, M. J., Baron, M., Hudson, M., & Canadian Scleroderma Research Group Trigeminal neuralgia in systemic sclerosis. Seminars in arthritis and rheumatism 2021, 51, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Kobata, H. , Kondo, A., Iwasaki, K., & Nishioka, T. Combined hyperactive dysfunction syndrome of the cranial nerves: trigeminal neuralgia, hemifacial spasm, and glossopharyngeal neuralgia: 11-year experience and review. Neurosurgery 1998, 43, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Chen Q, Yi DI, Perez JNJ, Liu M, Chang SD, Barad MJ, Lim M, Qian X. The Molecular Basis and Pathophysiology of Trigeminal Neuralgia. Int J Mol Sci. 2022, 23, 3604. [CrossRef]
- Liu, M.; Zhong, J.; Xia, L.; Dou, N.; Li, S. The expression of voltage-gated sodium channels in trigeminal nerve following chronic constriction injury in rats. Int. J. Neurosci. 2019, 129, 955–962. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, J.; Wang, Y.; Wang, L.; Wang, X. Changes in the expression of voltage-gated sodium channels Nav1.3, Nav1.7, Nav1.8, and Nav1.9 in rat trigeminal ganglia following chronic constriction injury. Neuroreport 2016, 27, 929–934. [Google Scholar] [CrossRef]
- Siqueira, S.R.; Alves, B.; Malpartida, H.M.; Teixeira, M.J.; Siqueira, J.T. Abnormal expression of voltage-gated sodium channels Nav1.7, Nav1.3 and Nav1.8 in trigeminal neuralgia. Neuroscience 2009, 164, 573–577. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. From genes to pain: Na v 1.7 and human pain disorders. Trends Neurosci. 2007, 30, 555–563. [Google Scholar] [CrossRef]
- Abd-Elsayed, A.A.; Ikeda, R.; Jia, Z.; Ling, J.; Zuo, X.; Li, M.; Gu, J.G. KCNQ channels in nociceptive cold-sensing trigeminal ganglion neurons as therapeutic targets for treating orofacial cold hyperalgesia. Mol. Pain 2015, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Rowshan, K.; Chao, T.; Mozaffar, T.; Steward, O. Chronic nerve compression induces local demyelination and remyelination in a rat model of carpal tunnel syndrome. Exp. Neurol. 2004, 187, 500–508. [Google Scholar] [CrossRef]
- Mackinnon, S.E.; Dellon, A.L.; Hudson, A.R.; Hunter, D.A. Chronic human nerve compression—A histological assessment. Neuropathol. Appl. Neurobiol. 1986, 12, 547–565. [Google Scholar] [CrossRef]
- Devor, M.; Amir, R.; Rappaport, Z.H. Pathophysiology of trigeminal neuralgia: The ignition hypothesis. Clin. J. Pain 2002, 18, 4–13. [Google Scholar] [CrossRef]
- Di Stefano G, De Stefano G, Leone C, et al. . Concomitant continuous pain in patients with trigeminal neuralgia is associated with trigeminal nerve root atrophy. Cephalalgia 2020, 40, 1502–10. [Google Scholar] [CrossRef]
- Peker, S.; Kurtkaya, O.; Uzun, I.; Pamir, M.N. Microanatomy of the central myelin-peripheral myelin transition zone of the trigeminal nerve. Neurosurgery 2006, 59, 354–359. [Google Scholar] [CrossRef]
- Prasad, S.; Galetta, S. Trigeminal neuralgia: Historical notes and current concepts. Neurologist 2009, 15, 87–94. [Google Scholar] [CrossRef]
- Berger, B.L.; Gupta, R. Demyelination secondary to chronic nerve compression injury alters Schmidt-Lanterman incisures. J. Anat. 2006, 209, 111–118. [Google Scholar] [CrossRef]
- Di Stefano, G. , Maarbjerg, S., Nurmikko, T., Truini, A., & Cruccu, G. Triggering trigeminal neuralgia. Cephalalgia : an international journal of headache 2018, 38, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska, J. M. , & Linskey, M. E. Trigeminal Neuralgia. American family physician 2016, 94, 133–135. [Google Scholar]
- Nurmikko, T. J. , & Eldridge, P. R. Trigeminal neuralgia--pathophysiology, diagnosis and current treatment. British journal of anaesthesia 2001, 87, 117–132. [Google Scholar] [CrossRef]
- Krafft R., M. Trigeminal neuralgia. American family physician 2008, 77, 1291–1296. [Google Scholar] [PubMed]
- Korczeniewska, O. A. , Kohli, D., Benoliel, R., Baddireddy, S. M., & Eliav, E. Pathophysiology of Post-Traumatic Trigeminal Neuropathic Pain. Biomolecules 2022, 12, 1753. [Google Scholar] [CrossRef]
- Stienen, M. N. , Cadosch, D., Seule, M. A., Fournier, J. Y., Hildebrandt, G., & Gautschi, O. P. Trigeminusneuralgie - Pathophysiologie, klinische Aspekte und Therapie [Trigeminal neuralgia - pathophysiology, clinical aspects and treatment]. Praxis 2010, 99, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Edlich, R. F. , Winters, K. L., Britt, L., & Long, W. B., 3rd. Trigeminal neuralgia. Journal of long-term effects of medical implants 2006, 16, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Nurmikko T., J. Pathophysiology of MS-related trigeminal neuralgia. Pain 2009, 143, 165–166. [Google Scholar] [CrossRef]
- Loeser, J. D. , Calvin, W. H., & Howe, J. F. Pathophysiology of trigeminal neuralgia. Clinical neurosurgery 1977, 24, 527–537. [Google Scholar] [CrossRef]
- Boto G., R. Neuralgia del trigémino [Trigeminal neuralgia]. Neurocirugia (Asturias, Spain) 2010, 21, 361–372. [Google Scholar] [CrossRef]
- Gardner W., J. Trigeminal neuralgia. Clinical neurosurgery 1968, 15, 1–56. [Google Scholar] [CrossRef]
- Zakrzewska, J. M. , & Linskey, M. E. Trigeminal neuralgia. BMJ (Clinical research ed.) 2015, 350, h1238. [Google Scholar] [CrossRef]
- Comi G, Filippi M, Rovaris M, Leocani L, Medaglini S, Locatelli T. Clinical, neurophysiological, and magnetic resonance imaging correlations in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1998, 64 (Suppl 1), S21–S25. [Google Scholar]
- uck K, Christensen H, Bazinski M. Systemic Lidocaine for the Treatment of Pain — Adult/Pediatric — Inpatient/Ambulatory/Emergency Department Clinical Practice Guideline, 2019.
- Linskey, ME. 143 pediatric trigeminal neuralgia (TN): results with early microvascular decompression (MVD). Neurosurgery. 2017, 64, 234. [Google Scholar] [CrossRef]
- Resnick DK, Levy EI, Jannetta PJ. Microvascular decompression for pediatric onset trigeminal neuralgia. Neurosurgery. 1998, 43, 804–807, discussion 807–808. [Google Scholar] [CrossRef]
- Bendtsen L, Zakrzewska JM, Abbott J, et al. European Academy of Neurology guideline on trigeminal neuralgia. Eur J Neurol. 2019, 26, 831–849. [Google Scholar] [CrossRef] [PubMed]
- Gambeta, E. , Chichorro, J. G., & Zamponi, G. W. Trigeminal neuralgia: An overview from pathophysiology to pharmacological treatments. Molecular pain 2020, 16, 1744806920901890. [Google Scholar] [CrossRef] [PubMed]
- Ruscheweyh, R. , Lutz, J., & Mehrkens, J. H. Trigeminusneuralgie : Moderne Diagnostik und Therapie [Trigeminal neuralgia : Modern diagnostic workup and treatment]. Schmerz (Berlin, Germany) 2020, 34, 486–494. [Google Scholar] [CrossRef]
- Wang, T. , Liu, L., Song, D., & Huang, D. Emerging roles of lncRNAs in the pathogenesis, diagnosis, and treatment of trigeminal neuralgia. Biochemical Society transactions 2022, 50, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, M. , & Di Stefano, G. Novel ways of approaching the pharmacologic treatment of trigeminal neuralgia. Headache 2022, 62, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Paranathala, M. P. , Ferguson, L., Bowers, R., & Mukerji, N. Percutaneous retrogasserian glycerol rhizotomy for trigeminal neuralgia: an alternative technique. British journal of neurosurgery 2018, 32, 657–660. [Google Scholar] [CrossRef] [PubMed]
- Piper, K. , Smith, T., Saez-Alegre, M., Jean, W., Bezchlibnyk, Y., & Van Loveren, H. Does Head Positioning After Percutaneous Glycerol Rhizotomy for Trigeminal Neuralgia Matter? World neurosurgery, 2023. [Google Scholar] [CrossRef]
- Joswig, H. , Staudt, M. D., MacDougall, K. W., & Parrent, A. G. Effect of Training on Percutaneous Glycerol Rhizotomy for Trigeminal Neuralgia: A Long-Term, Retrospective Comparison of Staff Neurosurgeon and Trainee Complications and Efficacy. World neurosurgery 2020, 134, e1001–e1007. [Google Scholar] [CrossRef] [PubMed]
- Xu-Hui W, Chun Z, Guang-Jian S, et al. Long-term outcomes of percutaneous retrogasserian glycerol rhizotomy in 3370 patients with trigeminal neuralgia. Turk Neurosurg. 2011, 21, 48–52. [Google Scholar]
- Piper, K. , George, Z., Gordon, J., Peto, I., Vakharia, K., & Van Loveren, H. Clival-Meckel's Cave Angle: A Predictor of Glycerol Displacement in Percutaneous Glycerol Rhizotomy for Trigeminal Neuralgia. Operative neurosurgery (Hagerstown, Md.), 2023. [Google Scholar] [CrossRef]
- Aljuboori, Z. , & Nauta, H. J. Multiple Recurrences of Trigeminal Neuralgia Caused by Deformation of the Trigeminal Nerve. Cureus 2019, 11, e6433. [Google Scholar] [CrossRef]
- Goel, A. , Kulkarni, G., Cotici, A., Paluzzi, A., Hayton, T., & Chelvarajah, R. Volume maximised glycerol rhizolysis for trigeminal neuralgia: a single centre analysis of outcomes. British journal of neurosurgery, 2023; 1–6. [Google Scholar] [CrossRef]
- Cordeiro, K. , Kim, J., Buckley, N., Kraemer, M., Pun, C., & Resnick, D. Pterygoid venous plexus anastomosis in trigeminal percutaneous glycerol rhizotomy: illustrative case. Journal of neurosurgery. Case lessons 2023, 6, CASE23173. [Google Scholar] [CrossRef]
- Krishnan, S. , Bigder, M., & Kaufmann, A. M. Long-term follow-up of multimodality treatment for multiple sclerosis-related trigeminal neuralgia. Acta neurochirurgica 2018, 160, 135–144. [Google Scholar] [CrossRef]
- Xia, Y. , Yu, G., Min, F., Xiang, H., Huang, J., & Leng, J. The Focus and New Progress of Percutaneous Balloon Compression for the Treatment of Trigeminal Neuralgia. Journal of pain research 2022, 15, 3059–3068. [Google Scholar] [CrossRef]
- Nascimento, R. F. V. , Pipek, L. Z., & de Aguiar, P. H. P. Is percutaneous balloon compression better than microvascular decompression to treat trigeminal neuralgia? A systematic review and meta-analysis. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia 2023, 109, 11–20. [Google Scholar] [CrossRef]
- Brown JA, Hoeflinger B, Long PB, et al. Axon and ganglion cell injury in rabbits after percutaneous trigeminal balloon compression. Neurosurgery. 1996, 38, 993–1003. [CrossRef]
- Skirving DJ, Dan NG. A 20-year review of percutaneous balloon compression of the trigeminal ganglion. J Neurosurg. 2001, 94, 913–917, discussion 1003–4. [Google Scholar] [CrossRef]
- Abdennebi B, Guenane L. Technical considerations and outcome assessment in retrogasserian balloon compression for treatment of trigeminal neuralgia. Series of 901 patients. Surg Neurol Int. 2014, 5, 118. [Google Scholar] [CrossRef]
- Leclerc, A. , Salkine, M. F., & Emery, E. Percutaneous balloon compression for trigeminal neuralgia: a how I do it. Acta neurochirurgica 2022, 164, 2939–2943. [Google Scholar] [CrossRef] [PubMed]
- Santiago, R. B. , Ali, A., Mandel, M., Muhsen, B., Adada, B., Borghei-Razavi, H., & Obrzut, M. Trigeminal Neuralgia-Step-by-Step DYNA-Computed Tomography-Assisted Balloon Compression Rhizotomy. World neurosurgery 2023, 171, 84. [Google Scholar] [CrossRef] [PubMed]
- Sweet WH, Wepsic JG. Controlled thermocoagulation of trigeminal ganglion and rootlets for differential destruction of pain fibers. 1. Trigeminal neuralgia. J Neurosurg. 1974, 40, 143–156. [CrossRef]
- Eskandar, E. , Kumar, H., Boini, A., Velasquez Botero, F., El Hunjul, G. N., Nieto Salazar, M. A., Quinonez, J., Dinh, B., & Mouhanna, J. E. The Role of Radiofrequency Ablation in the Treatment of Trigeminal Neuralgia: A Narrative Review. Cureus 2023, 15, e36193. [Google Scholar] [CrossRef]
- Abd-Elsayed, A. , Martens, J. M., Fiala, K. J., & Izuogu, A. Pulsed Radiofrequency for the Treatment of Trigeminal Neuralgia. Current pain and headache reports 2022, 26, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Orhurhu, V. , Sidharthan, S., Roberts, J., Karri, J., Umukoro, N., Hagedorn, J. M., Odonkor, C. A., & Abd-Elsayed, A. Radiofrequency Ablation for Craniofacial Pain Syndromes. Physical medicine and rehabilitation clinics of North America 2021, 32, 601–645. [Google Scholar] [CrossRef] [PubMed]
- Wu CY, Meng FG, Xu SJ, Liu YG, Wang HW. Selective percutaneous radiofrequency thermocoagulation in the treatment of trigeminal neuralgia: report on 1860 cases. Chin Med J (Engl). 2004, 117, 467–470. [CrossRef]
- Kanpolat Y, Savas A, Bekar A, Berk C. Percutaneous controlled radiofrequency trigeminal rhizotomy for the treatment of idiopathic trigeminal neuralgia: 25-year experience with 1600 patients. Neurosurgery. 2001, 48, 524–532, discussion 532–524. [CrossRef]
- Chakole, V. , Sharma, K., Tople, J., Akre, S., & Wanjari, M. B. Radiofrequency Ablation of Gasserian Ganglion in Trigeminal Neuralgia With Multiple Sclerosis: A Rare Clinical Case. Cureus 2022, 14, e32595. [Google Scholar] [CrossRef]
- Gusmao S, Oliveira M, Tazinaffo U, Honey CR. Percutaneous trigeminal nerve radiofrequency rhizotomy guided by computerized tomography fluoroscopy. Technical note. J Neurosurg. 2003, 99, 785–786. [CrossRef]
- Mansano, A. M. , Frederico, T. N., Valentin, R. E. B., Carmona, M. J. C., & Ashmawi, H. A. Percutaneous Radiofrequency Ablation for Trigeminal Neuralgia Management: A Randomized, Double-Blinded, Sham-Controlled Clinical Trial. Pain medicine (Malden, Mass.) 2023, 24, 234–243. [Google Scholar] [CrossRef]
- Zheng, Y. , Liu, C. W., Hui Chan, D. X., Kai Ong, D. W., Xin Ker, J. R., Ng, W. H., & Wan, K. R. Neurostimulation for Chronic Pain: A Systematic Review of High-Quality Randomized Controlled Trials With Long-Term Follow-Up. Neuromodulation : journal of the International Neuromodulation Society 2023, 26, 1276–1294. [Google Scholar] [CrossRef]
- Yameen, F. , Shahbaz, N. N., Hasan, Y., Fauz, R., & Abdullah, M. Efficacy of transcutaneous electrical nerve stimulation and its different modes in patients with trigeminal neuralgia. JPMA. The Journal of the Pakistan Medical Association 2011, 61, 437–439. [Google Scholar]
- Zayan, K. , Felix, E. R., & Galor, A. Transcutaneous Electrical Nerve Stimulation for Facial Pain. Progress in neurological surgery 2020, 35, 35–44. [Google Scholar] [CrossRef]
- Singla, S. , Prabhakar, V., & Singla, R. K. Role of transcutaneous electric nerve stimulation in the management of trigeminal neuralgia. Journal of neurosciences in rural practice 2011, 2, 150–152. [Google Scholar] [CrossRef]
- Motwani, M. , Fadnavis, A., & Dhole, A. Efficacy of transcutaneous electrical nerve stimulation (TENS) in the management of trigeminal neuralgia: A systematic review and meta-analysis. Journal of clinical and experimental dentistry 2023, 15, e505–e510. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y. , Yang, L., Han, R., Guo, G., Huang, S., Weng, L., Wang, X., Li, Z., Huang, D., Hu, R., & Zhou, H. Implantable Peripheral Nerve Stimulation for Trigeminal Neuropathic Pain: A Systematic Review and Meta-Analysis. Neuromodulation : journal of the International Neuromodulation Society 2021, 24, 983–991. [Google Scholar] [CrossRef]
- Wall PD, Sweet WH. Temporary abolition of pain in man. Science. 1967, 155, 108–109. [CrossRef]
- Meyerson BA, Hakansson S. Alleviation of atypical trigeminal pain by stimulation of the Gasserian ganglion via an implanted electrode. Acta Neurochir Suppl (Wien). 1980, 30, 303–309.
- Ellis JA, Mejia Munne JC, Winfree CJ. Trigeminal branch stimulation for the treatment of intractable craniofacial pain. J Neurosurg. 2015, 123, 283–288. [CrossRef]
- Klein, J. , Siepmann, T., Schackert, G., Ziemssen, T., & Juratli, T. A. Peripheral nerve field stimulation in medically refractory trigeminal neuralgia attributed to multiple sclerosis. Journal of neurosurgery 2020, 134, 1244–1250. [Google Scholar] [CrossRef]
- Chang M., C. Efficacy of Pulsed Radiofrequency Stimulation in Patients with Peripheral Neuropathic Pain: A Narrative Review. Pain physician 2018, 21, E225–E234. [Google Scholar] [CrossRef]
- Chung, M. , & Huh, R. Neuromodulation for Trigeminal Neuralgia. Journal of Korean Neurosurgical Society 2022, 65, 640–651. [Google Scholar] [CrossRef]
- Nandi D, Aziz T, Carter H, Stein J. Thalamic field potentials in chronic central pain treated by periventricular gray stimulation – a series of eight cases. Pain. 2003, 101, 97–107. [CrossRef]
- Franzini, A. , Messina, G., Cordella, R., Marras, C., & Broggi, G. Deep brain stimulation of the posteromedial hypothalamus: indications, long-term results, and neurophysiological considerations. Neurosurgical focus 2010, 29, E13. [Google Scholar] [CrossRef]
- Ben-Haim, S. , Mirzadeh, Z., & Rosenberg, W. S. Deep brain stimulation for intractable neuropathic facial pain. Neurosurgical focus 2018, 45, E15. [Google Scholar] [CrossRef]
- Leksell, L. Sterotaxic radiosurgery in trigeminal neuralgia. Acta chirurgica Scandinavica 1971, 137, 311–314. [Google Scholar]
- De La Peña, N. M. , Singh, R., Anderson, M. L., Koester, S. W., Sio, T. T., Ashman, J. B., Vora, S. A., & Patel, N. P. High-Dose Frameless Stereotactic Radiosurgery for Trigeminal Neuralgia: A Single-Institution Experience and Systematic Review. World neurosurgery 2022, 167, e432–e443. [Google Scholar] [CrossRef]
- Marchetti, M. , Pinzi, V., De Martin, E., Ghielmetti, F., & Fariselli, L. Radiosurgery for trigeminal neuralgia: the state of art. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology, 2019; 40, 153–157. [Google Scholar] [CrossRef]
- Tuleasca, C. , Régis, J., Sahgal, A., De Salles, A., Hayashi, M., Ma, L., Martínez-Álvarez, R., Paddick, I., Ryu, S., Slotman, B. J., & Levivier, M. Stereotactic radiosurgery for trigeminal neuralgia: a systematic review. Journal of neurosurgery 2018, 130, 733–757. [Google Scholar] [CrossRef]
- Tuleasca, C. , Régis, J., Sahgal, A., De Salles, A., Hayashi, M., Ma, L., Martínez-Álvarez, R., Paddick, I., Ryu, S., Slotman, B. J., & Levivier, M. Stereotactic radiosurgery for trigeminal neuralgia: a systematic review. Journal of neurosurgery 2018, 130, 733–757. [Google Scholar] [CrossRef]
- Matsuda, S. , Serizawa, T., Sato, M., & Ono, J. Gamma knife radiosurgery for trigeminal neuralgia: the dry-eye complication. Journal of neurosurgery, 2002; 97, 525–528. [Google Scholar] [CrossRef]
- Carnochan J., M. On Tic Douloureux: "The Painful Affection of the Face, Dolor Faciei Crucians," of Fothergill, with a New Operation for Its Cure. The American journal of dental science 1860, 10, 254–278. [Google Scholar] [PubMed]
- Tan, T. C. , & Black, P. M. Sir Victor Horsley (1857-1916): pioneer of neurological surgery. Neurosurgery 2002, 50, 607–612. [Google Scholar] [CrossRef]
- Patel, S. K. , & Liu, J. K. Overview and History of Trigeminal Neuralgia. Neurosurgery clinics of North America 2016, 27, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Dandy W., E. THE TREATMENT OF TRIGEMINAL NEURALGIA BY THE CEREBELLAR ROUTE. Annals of surgery 1932, 96, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A. M. , & Price, A. V. A history of the Jannetta procedure. Journal of neurosurgery 2019, 132, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Yang, L. , & Cheng, H. Surgical technique management of microvascular decompression for trigeminal neuralgia. Sebészi technikák a trigeminusneuralgia microvascularis dekompresszióval történő kezeléséhez. Ideggyogyaszati szemle 2022, 75, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Barker, F. G. , 2nd, Jannetta, P. J., Bissonette, D. J., Larkins, M. V., & Jho, H. D. The long-term outcome of microvascular decompression for trigeminal neuralgia. The New England journal of medicine 1996, 334, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, D. T. , Benedetto, N., & Perrini, P. Clinical outcome after microvascular decompression for trigeminal neuralgia: a systematic review and meta-analysis. Neurosurgical review 2022, 46, 8. [Google Scholar] [CrossRef]
Figure 1.
Pathophysiology of Trigeminal Neuralgia.
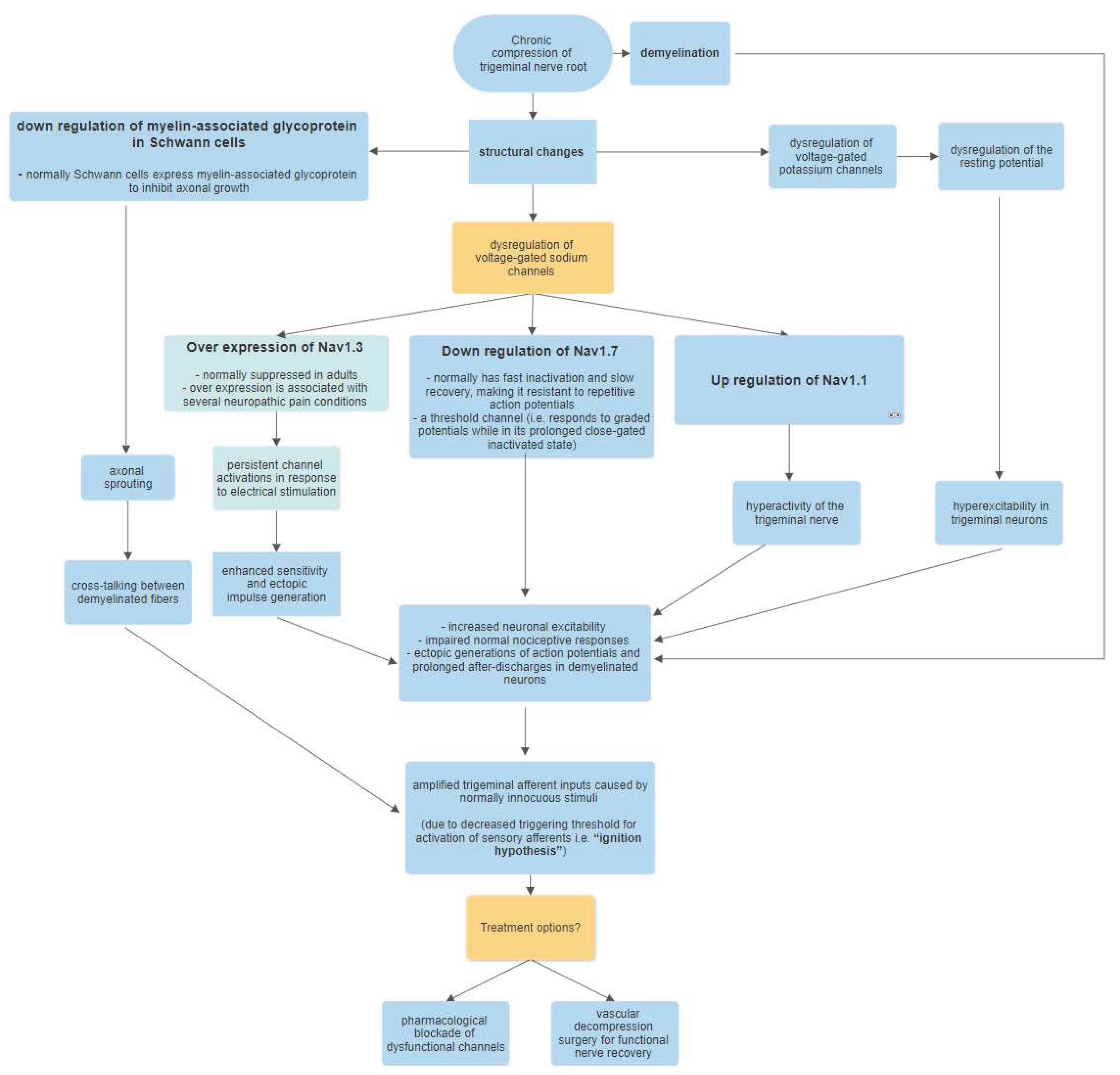

Table 1.
Pharmacologic Treatments For Trigeminal Neuralgia.
| Drug | Dose | Monitoring |
|---|---|---|
| Carbamazepine | 50 mg twice daily (for people over 65), 100 milligrams twice daily (for younger people) | Track baseline LFTs, CBC, and salt levels. |
| Oxcarbazepine | 150 mg twice daily (beginning) Twice daily, 300–600 mg | Monitor sodium, HLA-B*1502 variant screening |
| Levetiracetam | 3000–5000 mg per day, BID or TID | NA |
| Gabapentin | 300–1200 mg TID | NA |
| Valproate | 500–1500mg per day | Total and free valproate level, LFTs, CBC, ammonia |
| Lamotrigine | 100mg BID | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



