
Submitted:
02 February 2024
Posted:
06 February 2024
You are already at the latest version
Abstract
Salmonella Typhimurium infection in pigs is characterized by an intense inflammatory response in early infection which seems to dysregulate host physiology including the gut microbiota. Both, changes in host physiology and microbiome composition, must entail modifications within the gut environment. This study analyzes the gut metabolome in early infected pigs (48 hours after infection) and non-infected controls by untargeted metabolomics (LC–QTOF MS/MS and GC–TOF MS), supported with microbiome data (16S rRNA abundance and prediction functional analyses). The metabolomic profile in Salmonella-infected pigs differed from healthy controls in 39 of these metabolites, including eight amino acids, four bile acids and carboxylic acids, three carnitines and sugar derivates, eleven fatty acids and six other compounds. Changes in amino acids abundance and the observed decrease of proteolytic bacteria such as Lactobacillus or Bacteroides could favor the niche colonization by Desulfovibrionaceae. In addition, the decrease in short chain fatty acids and tricarboxylic acids cycle was correlated to a decrease in beneficial bacteria in infected pigs. Interestingly, we observed an increase of bile acids concentration and compounds such as succinic acid or pantothenic acid, which may boost the inflammatory response. Altogether, the results reveal how S. Typhimurium infection alters the gut microbiome composition and prompts changes in the metabolism with a potential interaction between intestinal metabolism and microbiota abundance.
Keywords:
gut
; untargeted metabolomics
; metagenomics
; microbiota
; swine
; zoonosis
; salmonellosis
1. Introduction
The pig is not only a natural host for Salmonella but also pork meat ranks among the three main sources of human salmonellosis in the European Union [1,2]. A few studies have characterized the pathogenicity of Salmonella Typhimurium infection in swine [3,4]. Their results revealed a high multiplication of the pathogen in the acute phase of infection, and severe intestinal epithelial damage, mainly in the ileum, where innate immune cells migrate to provoke a strong inflammatory response. Rather than limiting or controlling the infection, early gut inflammation appears to assist Salmonella in colonizing the intestine. This results in the creation of a distinct nutrient niche, fostering a more effective multiplication compared to other competitors [5]. Thus, Salmonella infection also promotes changes within the intestinal microbiota. In a previous study [6], we have reported that early S. Typhimurium infection alters gut microbiome composition, increasing richness variability, reducing the abundance of desirable bacteria such as Lactobacillus or Bifidobacterium, and increasing the abundance of pathobionts such as Citrobacter spp., which take advantage of the intestinal damage and alterations of the host equilibrium in the gut. The findings of this study have been supported by other microbiome studies performed on pigs infected with Salmonella [7,8,9]. Both, changes in the mucosal inflammatory state and microbiome composition, must entail modifications within metabolic behavior of the gut environment after S. Typhimurium infection. It is interesting to note that mechanisms of metabolic and immune control co-evolved, supporting the idea that the immune response is also regulated by metabolic pathways [10]. Furthermore, the immune response and the composition of the intestinal microbiota are closely linked to each other, resulting in a significant alteration of the metabolic profile of the infected tissue that may ultimately influence the outcomes of host-pathogen interactions. Understanding the metabolic implications of salmonellosis is crucial for a comprehensive characterization of the host’s response to infection. However, due to the intricate interconnections within metabolic pathways, a systems biology approach, specifically metabolomics, is necessary to evaluate and interpret these complex metabolic changes. [11]. In a previous study, we demonstrated that S. Typhimurium causes profound changes in the porcine intestinal microbiota [12]. Here, we used untargeted metabolomics to identify and measure the concentration of a broad spectrum of small molecules in a biological sample [11], coupled to metagenomic data from a previous study [6] and predicted functions of the microbiome, to explore the influence of Salmonella infection on the host’s metabolism. Unraveling the metabolic repercussions of bacterial infections is pivotal for gaining a thorough insight into inflammatory diseases. The intricate web of interlinked metabolic pathways across diverse organs, tissues, and cells necessitates a systems biology approach, such as metabolomics, to effectively assess and make sense of these dynamic metabolic changes.
2. Materials and Methods
Pig Samples
The samples used in this study were obtained from a previous challenge trial with S. Typhimurium performed with 4 weeks-old pigs. Further details can be found elsewhere [6]. Ileum content samples collected from three control Salmonella-free pigs and three Salmonella-infected pigs at day two post infection were used in the analyses performed in this study.
Metabolome Analyses
Detection of metabolites was performed by broad spectrum untargeted metabolomics [11]. Detection of metabolites was performed from 160 mg of the samples by liquid chromatography time-of-flight mass spectrometry (LC–QTOF MS/MS) run in Agilent 1200 Series LC system coupled to an Agilent 6540 UHD Accurate-Mass QTOF hybrid mass spectrometer (Santa Clara, CA, USA). Identification of the metabolites was supported on MS and MS/MS information and search in the METLIN MS and MS/MS databases (http://metlin.scripps.edu), the Human Metabolome Database (HMDB, 3.6 version) and the LIPID MAPS website ((http://www.lipidmaps.org), using in all cases the molecular features (MFs) obtained in the previous step. A table with the peak area of all identified compounds in the different samples injected was obtained as a result.
Gas chromatography time-of-flight mass spectrometry (GC–TOF MS) was performed by Agilent 7890A Series GC system coupled to an Agilent 7200 UHD Accurate-Mass QTOF hybrid mass spectrometer equipped with an electron impact (EI) source (Santa Clara, CA, USA). The list of MFs obtained for each analysis was exported as data files in compound exchange format (.cef files). Tentative identification of compounds was performed by searching each mass spectrum in the NIST 11 and Fiehn databases using the retention index or retention time value, respectively.
16S rRNA Microbiome Characterization
The characterization of the microbiota from the ileum digesta was performed by 16S rRNA sequencing. Details of DNA extraction, 16S rRNA sequencing and bioinformatic analyses of the sequences can be accessed in previous publications [12].
Functional Predictions
A functional prediction based on 16S rRNA marker gene sequences was performed using PICRUSt [13]. After excluding the unknown OTUs from the GreenGenes 13.5 reference database and normalizing by 16S rRNA gene copy number, functional metagenomes for each sample were predicted from the Kyoto Encyclopedia of Genes and Genomes (KEGG) catalogue and categorized to a specified KEGG level. In addition, Kegg orthology groups (KOs) were mapped to KEGG and visualized using the Interactive Pathway Explorer (iPath3.0) web-based tool [14].
Biostatistical Analyses
Metabolome and microbiome data were imported into R (version 3.5.1). For each metabolite, comparisons were made by the non-parametric Wilcoxon-test in R with a significant threshold of 0.05. Correlation between relative concentration of metabolites and microbiota abundance were estimated by Spearman correlation test and plots were prepared with corrplot package (version 0.84) with “hclust” method for hierarchical clustering of samples.
3. Results
3.1. Differentially Abundant Metabolites between Infected and Control Pigs
The combination of untargeted LC–QTOF MS/MS and GC–TOF MS detected more than 300 different compounds. The metabolomic profile in Salmonella-infected pigs differed from healthy controls in 39 of these metabolites (Table 1, Table S1). Among these compounds there were eight amino acids, four bile acids and carboxylic acids, three carnitines and sugar derivates, eleven fatty acids and six compounds not assigned to any of these groups.
3.2. Particular Changes Associated to Infection
Salmonella-infected pigs had significantly higher abundance of arginine, leucine, serine and tyrosine compared to controls (p<0.05). The abundance of these amino acids was associated to predictions in bacterial metabolism which highlighted higher abundance of bacteria associated to their biosynthesis (Figure 1A). Short-chain fatty acids (acetic, butyric and valeric) were significantly increased after infection, while medium chain (caprylic) and long chain fatty acids (linoleic, stearic and behenic) were higher in abundance in controls (Figure 1B). The last result contrast to predictions in microbial functions which revealed a higher abundance of KEGG orthologs (KOs) associated to biosynthesis of fatty acids in infected pigs. Abundance of aconitic, fumaric and pyruvic carboxylic acids were significantly reduced in infected pigs, a result which goes in line with the depletion in energy metabolism predicted by KOs abundance in infected pigs. However, there was no connection between metabolomic analysis and KO predictions when the tricarboxylic acids cycle was particularly evaluated (Figure 1C). The secondary bile acids taurocholic and glycocholic acid were significantly increased in abundance in the ileum from infected pigs. Some bile acids biosynthesis routes were also more abundant in the functional predictions in infected pigs (Figure 1D).
3.3. Significant Associations between Metabolome and Microbiome Changes
Metabolome and microbiome data were analyzed together to establish potential correlations among changes in abundance of metabolites and ileum microbiota (Figure 2). Amino acids and derivates and abundance of bacteria (Figure 2A,D), revealed positive correlations of creatine and tyrosine to some Pseudomonadota (former Proteobacteria) such as the familiae Peptostreptococcaceae (p=0.80), Desulfovibrionaceae (p=0.78) and Campylobacteraceae (p=0.77). In contrast, pyruvic carboxylic acid was negatively correlated to abundance of Desulfovibrionaceae, Enterobacteriaceae and Pasteurellaceae (p=-0.73; p=-0.63; p=-0.63 respectively; Figure 2D). Several positive correlations between members of Bacillota (former Firmicutes) and carboxylic acids were also observed. Succinic acid was positively associated to Turicibacteriaceae (p=0.97) and Clostridiaceae (ρ=0.93) while the family Lachnospiraceae was positively associated to the abundance of pyruvic acid (p=0.71) and aconitic acid to Streptococcaceae (p=0.65). The abundance of the short chain butyric acid and the presence of Desulfovibrionaceae were significantly associated (p=0.82) and medium chain caprylic acid correlated to the abundance of Verrucomicrobiaceae (p=0.88; Figure 2F). Among bile acids (Figure 2D,H), positive correlations were observed between glycocholic acid and abundance of Pseudomonadaceae (p=0.88), Ruminococaceae (p=0.81), Rickenellaceae (p=0.82) and Verrucomicrobiaceae (p=0.81).
4. Discussion
Our study revealed how S. Typhimurium infection alters the gut microbiome composition and prompts changes in the metabolism. Metabolomic analyses revealed many compounds linked to the changes in the microbiome and vice versa, changes in the microbiome which potentially explain the observed changes in the metabolome. Previous studies have used similar approaches in murine typhoid infection models [15], finding that hormone pathways were the most significantly affected.
The new “omic-era” is providing substantial progress in Salmonella infection in pigs, disclosing hidden details in the pathogenesis so far. In this respect, metagenomic studies have pictured the microbial disbalance occurring in S. Typhimurium infection [7,8,9]. The gut dysbiosis observed early after infection [12] together with dysregulation of host physiological processes [5,16] must entail changes in the gut metabolome. Understanding these changes is of paramount relevance to complete the factors influencing the success of non-typhoidal Salmonella serovars in swine salmonellosis.
The gut microbiota participates actively in a number of metabolic processes including vitamin and SCFA production, amino acids synthesis, bile acid biotransformation or hydrolisis and fermentation of non-digestible substrates [17]. We observed a good overlap between prediction of metabolic routes and metabolomic results in differentially abundant amino acids. The observed higher abundance of amino acids in the gut lumen from infected pigs could be associated to alterations in amino acids transportation by host intestinal epithelium, together with the observed decrease in abundance of proteolytic bacteria such as Lactobacillus and Bacteroides. This metabolic context may explain the proliferation of Desulfovibrio and Peptostreptococcus, two genera able to metabolize proteins [18], with less microbial competitors.
Thirteen fatty acids were identified, encompassing both short-chain and long-chain varieties, with no discernible pattern observed between the infected and control groups. In is noteworthy that KO predictions were linked to fatty acids biosynthesis in infected pigs. Regarding fatty acids correlations to particular groups of bacteria, the presence of higher abundance of octanoic or caprylic acid was linked to the family Desulfovibrionaceae, able to metabolize lipids. There were also differences in abundance of carboxylic acids such as aconitic, fumaric acid and pyruvic acid, which are involved in tricarboxylic acid cycle and the production of SCFA such as butyrate was decreased after infection. Kegg functional predictions revealed a higher abundance of functions linked to energy metabolism in non-infected pigs, fact which matched the abundance of the former compounds, although no specific connections between metabolomic analysis and KO predictions were established when the tricarboxylic acids cycle was particularly evaluated. Both at phylum and familiae levels, the abundance of these compounds was negatively associated with phyla Fusobacteriota (former Fusobacteria) and Pseudomonadota (former Proteobacteria). Abundance of two familiae from the last phylum, Enterobacteriaceae and Desulfovibrionaceae, were negatively associated with the abundance of these compounds, which are end products of the metabolism of beneficial gut microbes such as certain clusters of Clostridia (Veillonella, Megasphaera, Mitsuokella) and Saccharolytic bacteria (Prevotella, Lachnospira, Lactobacillus, Bacteroides etc.,) all decreased in abundance after Salmonella infection [12].
Increased concentrations of glycocholic and taurocholic acids are a proof-of-concept of the results described by Uribe et al. [16] where we observed a disruption of the bile acid absorption in enterocytes via farnesoid X receptor pathway. PICRUSt functional predictions also matched the results detected but we did not find any correlation to microbiome abundance, although the result must have an impact on abundance of microbial groups and needs further characterization.
Increased free radical production, decreased antioxidant capacity and excessive inflammation are well-known features in the pathogenesis of non-typhoidal Salmonella serovars. As stated in the introduction, Salmonella triggers inflammation to create a new nutrient- niche which contains substrates, like tetrathionate, on which the pathogen can grow faster than other members of the microbial community [5]. The present metabolomic analysis reveals a few other compounds which may be involved in the boost of the inflammation within the small intestine. For instance, succinate and pantothenic acid, both increased in abundance after infection, are known to limit the production of anti-inflammatory cytokines, particularly IL-10 [19] and to stimulate the expression of pro-inflammatory cytokines in epithelial cells [20] respectively. Thus, the release of succinic acid and pantothenic acid could favor the cytokine storm observed in acute infected pigs [21]. Similarly, an increase in mannitol abundance was observed; this sugar alcohol or polyalcohol acts as a quencher by pathogens in reactive oxygen species release, thus limiting their efficacy in pathogen control [22].
To our knowledge this is the first study approaching gut metabolomics and metagenomics in Salmonella infection in pigs. Here we have seen how S. Typhimurium infection alters the gut microbiome composition and prompts changes in the metabolism. Metabolomic analyses revealed many compounds linked to the changes in the microbiome and vice versa, changes in the microbiome which potentially explain the observed changes in the metabolome.
5. Conclusions
S. Typhimurium gastrointestinal infection altered abundance of amino acids, bile acids and carboxylic acids, carnitines and sugar derivates and fatty acids, among others. Correlation of amino acids abundance and decrease of Lactobacillus or Bacteroides could favor the niche colonization by Desulfovibrionaceae. Decrease in short chain fatty acids and tricarboxylic acids cycle was correlated to a decrease in beneficial bacteria. Additionally, we observed an increase of bile acid concentration and compounds such as succinic acid or pantothenic acid which may boost the inflammatory response. Altogether, the results reveal how S. Typhimurium infection alters the gut microbiome composition and prompts changes in the metabolism with a potential interaction between intestinal metabolism and microbiota abundance.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Raw data of metabolites identified in infected and control samples.
Author Contributions
CE and HA performed the experimental challenge study, sample collection and processing, statistical analyses and manuscript writing. MCS and FPC performed the metabolomics and the statistical analysis of the metabolome compounds. SZL and JJG designed the study, supervised the challenge study, helped in manuscript writing and revised and edited the final manuscript.
Funding
This work was supported by the Spanish Ministry of Economy and Competitiveness (AGL2017-87415-R), the Spanish Ministry of Science, Innovation and Universities (PID2022-142887OB-I00), and Operational Program FEDER 2014-2020/Consejería de Economía, Conocimiento, Empresas y Universidad de la Junta de Andalucía (Regional Ministry of Economy, Knowledge, Business and University of the Junta de Andalucía, Ref. UCO-FEDER 20 REF. 1380897-R).
Institutional Review Board Statement
All procedures involving animals were approved by the institutional bioethical committee of the University of Leon (reference number OEBA-ULE-009-2017 and registry number ES240890000172), and performed according to European regulations regarding animal welfare and protection of animals used for experimental and other scientific purposes.
Data Availability Statement
The full metagenomics data sets are accessible at NCBI Sequence Read Archive (SRA) under accession: SRP111505, (BioProject: PRJNA393762).
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflict of interest.
References
- EFSA; ECDC. The European Union One Health 2019 Zoonoses Report; 2021.
- Correia-Gomes, C.; Leonard, F.; Graham, D. Description of control programmes for Salmonella in pigs in Europe. Progress to date? Journal of Food Safety 2021, 41, e12916. [Google Scholar] [CrossRef]
- Collado-Romero, M.; Aguilar, C.; Arce, C.; Lucena, C.; Codrea, M.C.; Morera, L.; Bendixen, E.; Moreno, A.; Garrido, J.J. Quantitative proteomics and bioinformatic analysis provide new insight into the dynamic response of porcine intestine to Salmonella Typhimurium. Front Cell Infect Microbiol 2015, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Bellido-Carreras, N.; Argüello, H.; Zaldívar-López, S.; Jiménez-Marín, Á.; Martins, R.P.; Arce, C.; Morera, L.; Carvajal, A.; Garrido, J.J. Salmonella Typhimurium Infection Along the Porcine Gastrointestinal Tract and Associated Lymphoid Tissues. Vet Pathol 2019, 300985819843682. [Google Scholar] [CrossRef]
- Rivera-Chávez, F.; Bäumler, A.J. The Pyromaniac Inside You: Salmonella Metabolism in the Host Gut. Annu Rev Microbiol 2015, 69, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Argüello, H.; Estellé, J.; Zaldívar-López, S.; Jiménez-Marín, Á.; Carvajal, A.; López-Bascón, M.A.; Crispie, F.; O’Sullivan, O.; Cotter, P.D.; Priego-Capote, F.; et al. Early Salmonella Typhimurium infection in pigs disrupts Microbiome composition and functionality principally at the ileum mucosa. Sci Rep 2018, 8, 7788. [Google Scholar] [CrossRef] [PubMed]
- Bearson, S.M.; Allen, H.K.; Bearson, B.L.; Looft, T.; Brunelle, B.W.; Kich, J.D.; Tuggle, C.K.; Bayles, D.O.; Alt, D.; Levine, U.Y.; et al. Profiling the gastrointestinal microbiota in response to Salmonella: low versus high Salmonella shedding in the natural porcine host. Infect Genet Evol 2013, 16, 330–340. [Google Scholar] [CrossRef]
- Argüello, H.; Estellé, J.; Leonard, F.C.; Crispie, F.; Cotter, P.D.; O’Sullivan, O.; Lynch, H.; Walia, K.; Duffy, G.; Lawlor, P.G.; et al. Influence of the Intestinal Microbiota on Colonization Resistance to. mSystems 2019, 4. [Google Scholar] [CrossRef]
- Borewicz, K.A.; Kim, H.B.; Singer, R.S.; Gebhart, C.J.; Sreevatsan, S.; Johnson, T.; Isaacson, R.E. Changes in the Porcine Intestinal Microbiome in Response to Infection with Salmonella enterica and Lawsonia intracellularis. PLoS One 2015, 10, e0139106. [Google Scholar] [CrossRef]
- Patel, C.; Leone, R.; Horton, M.; Powell, J. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat Rev Drug Discov 2019, 18. [Google Scholar] [CrossRef]
- López-Bascón, M.A.; Calderón-Santiago, M.; Argüello, H.; Morera, L.; Garrido, J.J.; Priego-Capote, F. Comprehensive analysis of pig feces metabolome by chromatographic techniques coupled to mass spectrometry in high resolution mode: Influence of sample preparation on the identification coverage. Talanta 2019, 199, 303–309. [Google Scholar] [CrossRef]
- Arguello, H.; Estelle, J.; Zaldivar-Lopez, S.; Jimenez-Marin, A.; Carvajal, A.; Lopez-Bascon, M.A.; Crispie, F.; O’Sullivan, O.; Cotter, P.D.; Priego-Capote, F.; et al. Early Salmonella Typhimurium infection in pigs disrupts Microbiome composition and functionality principally at the ileum mucosa. Sci Rep 2018, 8, 7788. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Yamada, T.; Letunic, I.; Okuda, S.; Kanehisa, M.; Bork, P. iPath2.0: interactive pathway explorer. Nucleic Acids Res 2011, 39, W412–415. [Google Scholar] [CrossRef] [PubMed]
- Antunes, L.; Arena, E.; Menende, z.A.; Han, J.; Ferreira, R.; Buckner, M.; Lolic, P.; Madilao, L.; Bohlmann, J.; Borchers, C.; et al. Impact of salmonella infection on host hormone metabolism revealed by metabolomics. Infect Immun 2011, 79. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Uribe, J.; Collado-Romero, M.; Zaldivar-Lopez, S.; Arce, C.; Bautista, R.; Carvajal, A.; Cirera, S.; Claros, M.G.; Garrido, J.J. Transcriptional analysis of porcine intestinal mucosa infected with Salmonella Typhimurium revealed a massive inflammatory response and disruption of bile acid absorption in ileum. Vet Res 2016, 47, 11. [Google Scholar] [CrossRef]
- Vernocchi, P.; Del Chierico, F.; Putignani, L. Gut Microbiota Profiling: Metabolomics Based Approach to Unravel Compounds Affecting Human Health. Front Microbiol 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Wylensek, D.; Hitch, T.; Riedel, T.; Afrizal, A.; Kumar, N.; Wortmann, E.; Liu, T.; Devendran, S.; Lesker, T.; Hernández, S.; et al. A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity. Nat Commun 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; Kelly, B.; Logan, A.; Costa, A.; Varma, M.; Bryant, C.; Tourlomousis, P.; Däbritz, J.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Hu, S.; Du, X.; Wen, Q.; Zhong, X.; Zhou, X.; Zhou, C.; Xiong, W.; Gao, Y.; Zhang, S.; et al. Vitamin B5 Reduces Bacterial Growth via Regulating Innate Immunity and Adaptive Immunity in Mice Infected with Mycobacterium tuberculosis. Front Immunol 2018, 9. [Google Scholar] [CrossRef]
- Collado-Romero, M.; Arce, C.; Ramirez-Boo, M.; Carvajal, A.; Garrido, J.J. Quantitative analysis of the immune response upon Salmonella typhimurium infection along the porcine intestinal gut. Vet Res 2010, 41, 23. [Google Scholar] [CrossRef]
- Meena, M.; Prasad, V.; Zehra, A.; Gupta, V.; Upadhyay, R. Mannitol metabolism during pathogenic fungal-host interactions under stressed conditions. Front Microbiol 2015, 6. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Differentially abundant amino acids, (1A), fatty acids (1B), tricarboxylic acids (1C) and bile salts (1D) between infected and control pigs after an early infection (48h) by Salmonella Typhimurium. Metabolism routes reveal functions activated in infected (red) and control (green) pigs predicted by microbiota abundance in PICRUSt.
Figure 1.
Differentially abundant amino acids, (1A), fatty acids (1B), tricarboxylic acids (1C) and bile salts (1D) between infected and control pigs after an early infection (48h) by Salmonella Typhimurium. Metabolism routes reveal functions activated in infected (red) and control (green) pigs predicted by microbiota abundance in PICRUSt.
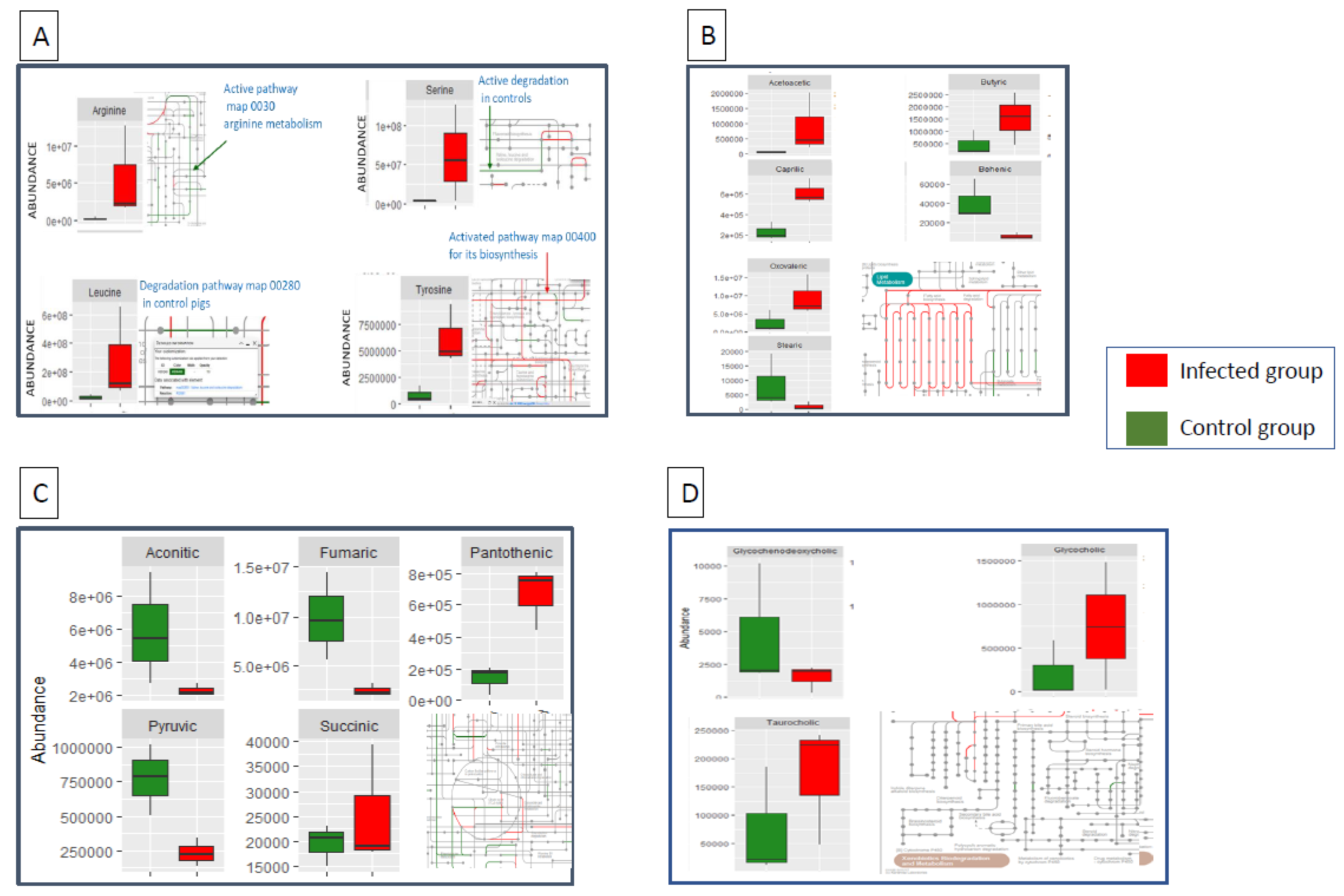
Figure 2.
Correlation between amino acids, (2A, 2E), tricarboxylic acids (2B, 2F) fatty acids (2C, 2G) and bile salts (2D, 2H) and significantly correlated microbiota (at phylum and familiae taxonomic levels) between infected and control pigs after an early infection (48h) by Salmonella Typhimurium. Dot size increases according to the correlation value while blue and red intensity varies according to positive and negative correlations respectively.
Figure 2.
Correlation between amino acids, (2A, 2E), tricarboxylic acids (2B, 2F) fatty acids (2C, 2G) and bile salts (2D, 2H) and significantly correlated microbiota (at phylum and familiae taxonomic levels) between infected and control pigs after an early infection (48h) by Salmonella Typhimurium. Dot size increases according to the correlation value while blue and red intensity varies according to positive and negative correlations respectively.
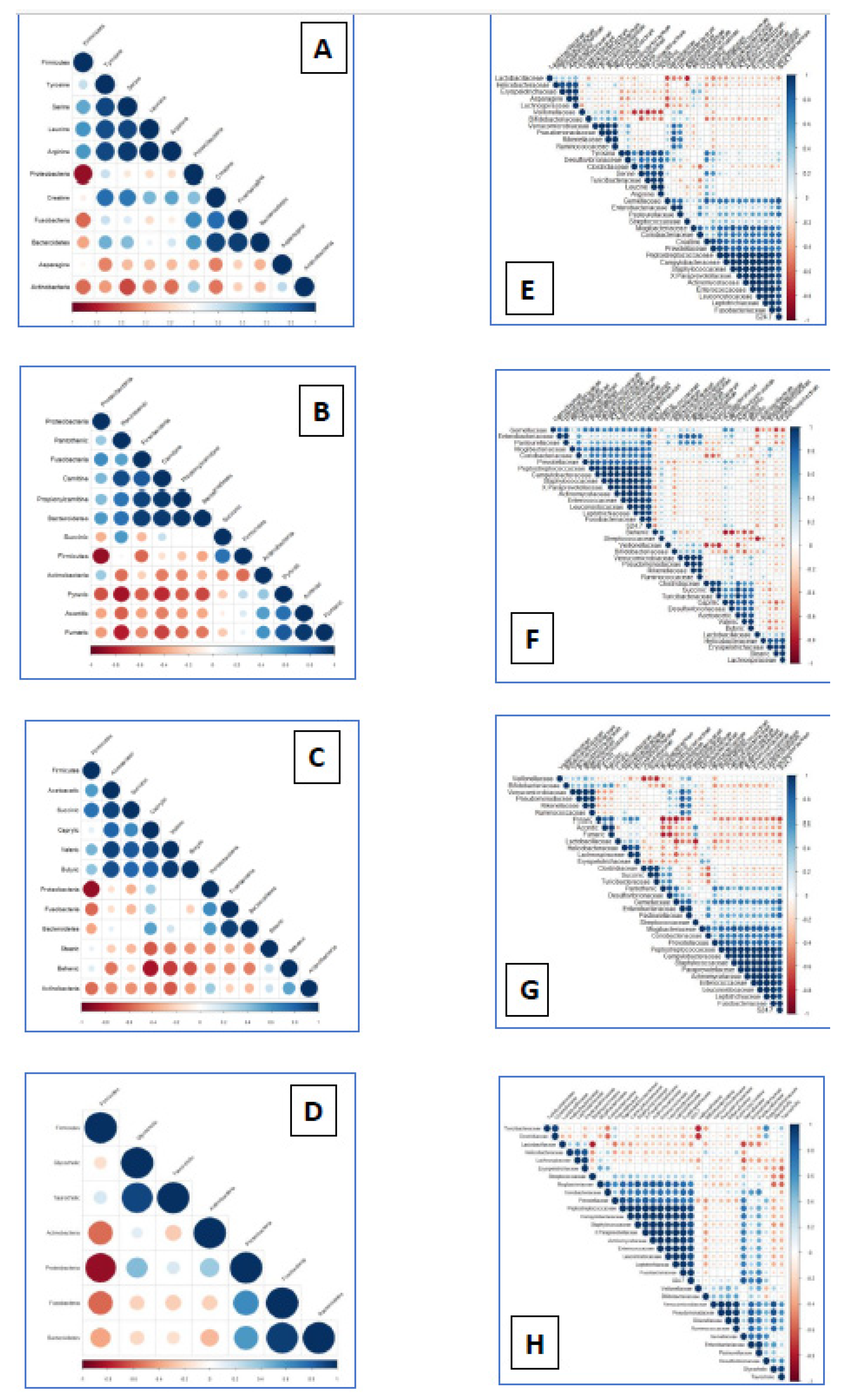

Table 1.
Differentially abundant metabolites (LC–QTOF MS/MS and GC–QTOF MS) detected by untargeted metabolomics between Salmonella Typhimurium infected and control pigs at two days post-infection.
Table 1.
Differentially abundant metabolites (LC–QTOF MS/MS and GC–QTOF MS) detected by untargeted metabolomics between Salmonella Typhimurium infected and control pigs at two days post-infection.
| Metabolite | Group | Abundance | P-value | |
|---|---|---|---|---|
| Mean Control | Mean Infected | |||
| Asparagine | Amino acids | 2,49E+05 | 3,10E+04 | <0.05 |
| Leucine | Amino acids | 2,23E+05 | 2,81E+06 | <0.01 |
| Phenylalanine | Amino acids | 1,06E+04 | 4,47E+03 | <0.01 |
| Serine | Amino acids | 3,25E+04 | 6,17E+05 | <0.01 |
| Tyrosine | Amino acids | 7,81E+03 | 6,20E+04 | <0.01 |
| Arginine | Amino acids | 2,15E+03 | 5,50E+04 | <0.01 |
| Creatine | Amino acids | 1,23E+04 | 6,84E+04 | <0.01 |
| Decenoylcarnitine | Amino acids | 6,72E+03 | 2,38E+03 | <0.01 |
| GCDCA | Bile acids | 4,68E+03 | 1,53E+03 | <0.05 |
| Glycocholic acid | Bile acids | 2,06E+05 | 7,45E+05 | <0.01 |
| Taurocholic acid | Bile acids | 7,30E+04 | 1,71E+05 | <0.05 |
| Tocopherol acetate | Bile acids | 1,29E+05 | 5,82E+04 | <0.05 |
| Fumaric acid | Carboxilic acids | 9,86E+04 | 2,40E+04 | <0.01 |
| Pantothenic acid | Carboxilic acids | 1,37E+03 | 6,67E+03 | <0.01 |
| Aconitic acid | Carboxylic acids | 5,87E+04 | 2,25E+04 | <0.05 |
| Pyruvic acid | Carboxylic acids | 7,72E+03 | 2,31E+03 | <0.01 |
| Succinic_acid | Carboxylic acids | 1,95E+04 | 2,54E+04 | <0.01 |
| Acetylcarnitine | Carnitines | 1,36E+04 | 5,58E+04 | <0.01 |
| Carnitine | Carnitines | 2,07E+04 | 4,85E+04 | <0.01 |
| Propionylcarnitine | Carnitines | 1,16E+04 | 4,70E+04 | <0.05 |
| Acetoacetic acid | Fatty acids | 6,44E+04 | 8,95E+05 | <0.01 |
| Butyric acid | Fatty acids | 4,56E+05 | 1,54E+06 | <0.01 |
| Oxovaleric acid | Fatty acids | 2,51E+06 | 9,47E+06 | <0.01 |
| Methylvaleric acid | Fatty acids | 6,57E+03 | 1,03E+04 | <0.05 |
| Caprylic acid | Fatty acids | 2,30E+05 | 6,13E+05 | <0.05 |
| Linoleic acid | Fatty acids | 4,85E+04 | 3,55E+04 | <0.01 |
| Stearic acid | Fatty acids | 8,60E+03 | 1,11E+03 | <0.01 |
| Behenic acid | Fatty acids | 4,16E+04 | 6,12E+03 | <0.01 |
| HODE_1 | Fatty acids | 5,19E+04 | 2,97E+03 | <0.01 |
| HODE_2 | Fatty acids | 3,65E+03 | 1,20E+04 | <0.01 |
| Hypoxanthine | Others | 3,77E+04 | 2,06E+05 | <0.01 |
| Choline | Others | 1,66E+06 | 3,66E+06 | <0.01 |
| Quinoline | Others | 5,71E+04 | 2,50E+03 | <0.01 |
| Sphinganine | Others | 1,00E+06 | 5,08E+05 | <0.01 |
| Sphingosine | Others | 3,02E+04 | 1,74E+05 | <0.01 |
| Stearoylethanolamide | Others | 6,05E+03 | 3,45E+03 | <0.01 |
| Glycericacid | Sugar derivates | 4,38E+04 | 7,66E+04 | <0.05 |
| Glycerol | Sugar derivates | 1,32E+04 | 3,28E+03 | <0.01 |
| Mannitol | Sugar derivates | 1,02E+05 | 9,71E+05 | <0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



