Submitted:
06 February 2024
Posted:
07 February 2024
You are already at the latest version
Abstract
Ion channels serve many cellular functions including ion homeostasis, volume regulation, signaling, nutrient acquisition, and developmental progression. Although the complex lifecycles of malaria parasites necessitate ion and solute flux across membranes, whole genome sequencing of the human pathogen, Plasmodium falciparum, revealed remarkably few orthologs of known ion channel genes. Contrasting with this, biochemical studies have implicated channel-mediated flux of ions and nutritive solutes across several membranes in infected erythrocytes. Here, I review advances in the cellular and molecular biology of ion channels in malaria parasites. These studies have implicated novel parasite genes in the formation of at least two ion channels, with additional ion channels likely present at various membranes and parasite stages. Computational approaches that rely on homology to known channel genes from higher organisms will not be very helpful in identifying the molecular determinants of these activities. Given their unusual properties, novel molecular and structural features, and essential roles in pathogen survival and development, parasite channels should be promising targets for therapy development.
Keywords:
ion channels
; nutrient uptake
; protein export
; transmembrane transport
; malaria
; Plasmodium falciparum
; antimalarial therapies.
1. Introduction
Malaria parasites are successful single-cell eukaryotic pathogens of humans, other primates, rodents, birds and reptiles. In humans, five Plasmodium species cause malaria and are responsible for significant morbidity and mortality; they also continue to exact a staggering toll on the global economy through reduced productivity and compromised childhood development.
The success of the most virulent human pathogen, P. falciparum, results in part from its complicated lifecycle, which allows cycles of exponential replication at multiple stages (Figure 1). A key feature of this lifecycle is the presence of extracellular and intracellular parasite forms in both the vertebrate host and the mosquito vector. In the host bloodstream, for example, the parasite invades and replicates within circulating erythrocytes. This intracellular habitat provides access to erythrocyte hemoglobin as an amino acid source; it also facilitates immune evasion as the sequestered parasite is hidden from immune cells and soluble effectors.
Another important feature apparent in the parasite lifecycle is the presence of multiple membrane barriers to ion and solute exchange with the extracellular environment. As with other eukaryotic cells, parasite growth and replication depends on this solute exchange. Uptake of extracellular nutrients such as amino acids, sugars, and precursors for nucleic acid and phospholipid biosynthesis is essential [1,2,3,4,5,6], especially in bloodstream forms as key nutrients are not adequately present in erythrocyte cytosol to fuel rapid parasite growth and replication (Figure 1). In addition to nutrient uptake, ions must also be transported across these various membranes. Ca++ uptake, for instance, is required for developmental progression and DNA replication in asexual parasite forms [7,8,9,10]; this divalent cation must be acquired in the face of an efficient PMCA Ca++ extrusion pump on the erythrocyte membrane that maintains a remarkably low intracellular [Ca++] [11,12]. Transmembrane flux of other ions such as Na+, K+, and Cl- are also essential for cell volume regulation, ion homeostasis, establishment of membrane potentials, and signaling [13]. These needs are further exacerbated for intracellular parasites because ions and other solutes must cross multiple membranes to permit survival and growth.
Because large-scale transport of ions and organic solutes is often mediated by ion channels, it was surprising and unexpected that the completion of the P. falciparum genome sequence revealed very few ion channel genes (Table 1). These genes were identified through homology to known channel genes from higher organisms [14]. Notably absent are conventional Ca++ channels despite the importance of Ca++ uptake in parasite growth and development; the absence of Cl- channels was also striking during genome assembly.
This review focuses on parasite ion channels and their genes; Table 1 does not include transporters and pumps, which are not considered further in this review. Channels are distinguished from transporters in that they allow higher rates of transport (up to 1010 ions/second). Channel-mediated flux is passive because ions and/or solutes move down their electrochemical gradients through a water-filled pore. While transporters can serve similar biological roles, their lower rates of transport make biochemical studies, such as with patch-clamp [15,16], more difficult.

Table 1.
P. falciparum ion channel genes identified by homology to channel genes in other organisms. For each gene, the number of predicted transmembrane domains (#TMD) was determined using DeepTMHMM [17].
Table 1.
P. falciparum ion channel genes identified by homology to channel genes in other organisms. For each gene, the number of predicted transmembrane domains (#TMD) was determined using DeepTMHMM [17].
| Gene | Annotation | # TMD | References |
|---|---|---|---|
| PF3D7_1227200 | potassium channel K1 | 8 | [18,19,20] |
| PF3D7_1465500 | potassium channel K2 | 8 | [19,21] |
| PF3D7_1107900 | small-conductance mechanosensitive ion channel (MscS) | 6 | [22] |
| PF3D7_1432100 | VDAC | 0 | [23] |
| PF3D7_1250200 | CSC1-like protein, putative | 11 | [24] |
| PF3D7_0810400 | aquaporin | 2 | [25] |
| PF3D7_1132800 | aquaglyceroporin | 6 | [26,27,28] |
| PF3D7_0408700 | perforin-like protein 1 | 0 | [29,30,31] |
| PF3D7_1216700 | perforin-like protein 2 | 0 | [29,32] |
| PF3D7_0923300 | perforin-like protein 3 | 0 | [33] |
| PF3D7_1473700 | nucleoporin NUP116/NSP116, putative | 0 | [34] |
Despite the paucity of ion channel genes uncovered by whole-genome sequencing of Plasmodium spp., patch-clamp and other transport assays have identified several unusual ion channels in blood-stage malaria parasites. Here, I review the discovery and properties of these channels and discuss insights into their molecular basis. These insights reveal that malaria parasites have unique channels encoded by genes absent from higher organisms. These unusual microbial channels serve essential roles in parasite biology and development and are, therefore, important targets for antimalarial therapies. Their study can also provide foundational insights into solute recognition and permeation.
2. The Plasmodial Surface Anion Channel (PSAC)
2.1. Background
The plasmodial surface anion channel (PSAC) is the prime example of an unusual ion channel present only in Plasmodium spp. This channel is on the host erythrocyte membrane and serves an essential role in nutrient acquisition for the intracellular parasite (Figure 2A). It accounts for the increased permeability of infected erythrocytes to a broad range of organic and inorganic solutes, as first identified some 75 years ago and characterized using tracer accumulation, osmotic fragility and other transport assays in numerous studies before 2000 [2,3,4,5].
These early studies had three key limitations, all of which arose because the transport methodologies depended on macroscopic flux measurements on populations of cells. First, the precise mechanism of solute uptake was unclear, with proposals including one or more parasite- or host-derived ion channels or transporters, lipid defects resulting from parasite invasion, fluid-phase endocytosis, and membranous ducts that could provide direct access to plasma [4,35,36,37]. Second, because endocytosis and membranous ducts were possible, macroscopic measurements could not determine the subcellular location of the solute flux across membranes. Finally, although there was substantial interest in identifying the molecular basis [38], these uncertainties prevented systematic studies aimed at gene identification.
A first advance in addressing these questions came from introduction of patch-clamp methods. The first patch-clamp recordings of infected human erythrocytes identified PSAC as an ion channel mechanism of transport [39]. Cell-attached patch-clamp revealed individual channel molecules on infected erythrocytes that were conserved in divergent Plasmodium spp. [39,40]. Because this method could limit the measurement of ion flux to a small “patch” of the erythrocyte plasma membrane [41], it also established the host cell membrane as the location of the transport activity. This study also reported whole-cell patch-clamp of infected cells, allowing a quantitative estimate of 1000-2000 functional channel molecules on a mature infected cell. When combined with noise analysis of single-channel and whole-cell recordings [42], the study also determined that PSAC was the predominant conductive pathway for ion flux on infected erythrocytes.
Interestingly, subsequent studies from other groups confirmed increased anion channel-mediated currents at the host membrane, but suggested multiple distinct channel types [43,44,45,46]. These studies also proposed various regulators of channel activity, including activation by oxidative stress [45], cyclic nucleotides [44], membrane stretch [47], and a link to various mammalian ion channels [46,48].
The completion of the Plasmodium falciparum whole-genome sequence failed to identify orthologs of known anion channels [14], raising questions about the molecular basis of the identified channel(s). Because synthesis, trafficking and insertion of parasite proteins at the host membrane was also considered overly complicated, nearly all workers assumed that the observed channels were host proteins that become activated or modified by the intracellular parasite [43,49]. There was already evidence for parasite modification of some erythrocyte membrane proteins [38,50,51], so this model appeared to be the most conservative one. It was, however, difficult to reconcile simple modification of preexisting host membrane proteins with formation of a channel having PSAC’s remarkable properties and with the observation that channel seems to be fine-tuned to permit parasite survival and replication.
2.2. Identification of the rhoph genes as PSAC determinants
These unusual properties prompted us to seek parasite genetic elements in PSAC formation. The first experimental evidence supporting parasite genes was the identification of differences in the channel’s voltage-dependent gating [52], a term that encompasses the process of opening and closing of the pore to ion flux. More compelling evidence included identification of distinct PSAC mutants with altered solute flux, selectivity, single channel gating, and pharmacology [53,54,55]; these mutants were identified through in vitro selection with blasticidin S and/or leupeptin, antiparasitic toxins that require PSAC-mediated uptake to reach their intracellular targets. These studies suggested that continuous cultivation with these toxins selected for outgrowth of mutants with reduced toxin uptake at the erythrocyte membrane.
Based on these findings, we screened a library of > 50,000 small molecules for PSAC inhibitors that produce differential block of channels associated with geographically divergent parasite clones. These high-throughput screens led to the identification of ISPA-28, a unique inhibitor that blocks channels associated with the Dd2 clone with 800-fold higher affinity than those on from other parasite clones such as HB3 [56]. An available Dd2 x HB3 genetic cross was then used to track inheritance in 34 progeny clones, revealing that most daughter parasites produced channels matching one or the other parental line and providing conclusive evidence for parasite genetic elements. Linkage analysis implicated a single locus near the 5’ end of parasite chromosome 3. Because none of the genes in this locus resembled known ion channel genes, we then used DNA transfection of the Dd2 line to produce merodiploid parasites expressing both the HB3 and Dd2 alleles of each of the 15 genes in this locus. While transfection with 13 of the genes did not change ISPA-28 affinity, complementation to add the HB3 allele of 2 related genes, clag3.1 and clag3.2, yielded an intermediate phenotype, as expected if both parental channel types are expressed on the host membrane [56]. Allelic exchange and a single nonsynonymous mutation at a highly conserved residue in the leupeptin-resistant PSAC mutant further supported a primary role of the encoded CLAG3 protein. CLAG3 localized to the erythrocyte membrane consistent with the site of PSAC activity. A small variant motif was found to be exposed at the host cell surface and later shown to account for the differential block by ISPA-28 and other clone-specific inhibitors [57,58,59]. Variation at this site strongly suggests selection against the exposed CLAG3 loop by host immune responses [60,61]. Immune selection is also supported by the high levels of anti-CLAG3 antibodies in endemic populations and by epigenetic silencing of clag genes [62,63,64,65,66].
Further evidence for a CLAG3 role in PSAC formation came from independent genetic mapping studies [58,67] and from epigenetic silencing of CLAG3 and CLAG2, a paralog encoded by a gene on parasite chromosome 2, in blasticidin S-resistant PSAC mutant [65,68]. These and other CLAG paralogs were known to associate with RhopH2 and RhopH3, two unrelated proteins encoded by single copy genes conserved in Plasmodium spp. [69,70,71]. We and others therefore examined these paralogs for their possible contributions to PSAC formation. While clag3-knockouts can be produced and propagated in nutrient-rich parasite culture media [58], rhoph2 and rhoph3 could not be disrupted using CRISPR-Cas9 with several high-scoring sgRNAs that efficiently cleave the genome to allow gene-editing [72]. Conditional knockdowns of these genes were therefore produced, revealing that both RhopH2 and RhopH3 are trafficked to the host membrane and are essential for PSAC formation [72,73,74]. RhopH3, but not CLAG3 or RhopH2, also contributes to host cell invasion. Biochemical studies suggest that each subunit is essential for PSAC formation, but that CLAG3 is dispensable because its paralogs—CLAG2, CLAG8 and CLAG9 in P. falciparum with at least two paralogs encoded by each examined Plasmodium species—can compensate for loss of CLAG3. Consistent with this model, RhopH2 and RhopH3 cannot be disrupted as they are encoded by single copy genes in all Plasmodium spp.
These studies have provided compelling evidence for parasite genetic elements and implicated the above gene products, which together form the RhopH complex. These findings are remarkable as workers had previously assumed this complex functioned in either cytoadherence or erythrocyte invasion [75,76,77,78]. They are also surprising because none of the subunits have homology to known channel proteins from higher organisms. Most fundamentally, they also lack the number of predicted transmembrane domains generally thought to be present in channel-forming proteins (Figure 2B). A tantalizing hint comes from the single amphipathic transmembrane domain detected in CLAG3, where helical wheel analysis reveals that polar residue side chains align at one face of the α-helical transmembrane domain with hydrophobic residues at the face [79]. This arrangement, confirmed by the subsequent determination of the RhopH complex structure by cryo-EM [80], is often found in pore-forming proteins, suggesting direct formation of the PSAC pore by these unusual proteins. Immunofluorescence studies as well as live-cell FRET reveals that all three members of the RhopH complex traffic together to the host membrane and that at least two of the subunits, RhopH2 and CLAG3, remain tightly associated after insertion at the host membrane [81], further supporting a model where these unusual parasite proteins form the ion channel.
At the same time, conclusive evidence for direct PSAC formation by the RhopH proteins is still missing. Biochemical studies reveal that the RhopH complex is manufactured as a soluble complex and that it is trafficked and eventually inserted at the host membrane [80]. The de novo structure of the soluble complex has been determined by cryo-EM microscopy using protein purified from an engineered P. falciparum line cultivated in human erythrocytes (Figure 2C, [80]), revealing that the single transmembrane domains of each subunit are buried in the trafficking complex. This finding suggests marked conformational changes associated with membrane insertion. These findings were also confirmed without purification of the RhopH complex using a novel cryo-ID method [82,83].
An important unanswered question is the structure of the membrane-embedded RhopH complex. This could directly implicate these proteins in PSAC formation, provide insights into how solutes permeate through the channel, and elucidate the mechanisms that underly the channel’s remarkable selectivity properties, as discussed below.
2.3. Unusual properties of the encoded channel
Paralleling the lack of homology with channel genes from other genera and the unusual structural properties of the RhopH proteins, the encoded channel has several unusual properties that distinguish it from all previously characterized ion channels. First, the single channel conductance, a measure of how many ions pass through an open pore per unit time, is remarkably small for a broad selectivity channel that passes bulky organic solutes. Generally, such channels have large pores to allow large solutes to navigate the pore, leading to high flux rates. Somehow, PSAC maintains a low rate of ion passage despite being able to accommodate large solutes of varying size, shape and charge [84]. This small single channel conductance necessitated use of patch-clamp solutions having molar [Cl-] and likely accounts for difficulties with patch-clamp detection of the channel in some laboratories [43]. Second and even more remarkable, PSAC stringently excludes the small Na+ ion despite passing much larger organic cations [84]; this Na+ exclusion is critical for intracellular parasite growth because a higher Na+ permeability would lead to osmotic lysis of infected cells in the bloodstream [85].
2.4. Essential role in nutrient uptake and a druggable target
Since its discovery, multiple studies have proposed various roles for the increased permeability of infected cells to diverse solutes. These include 1) nutrient acquisition for the developing intracellular parasite [1,3,4,5,86], 2) cation remodeling to raise [Na+] and lower [K+] in host cell cytosol [2,87,88], 3) volume regulation of infected cells by allowing efflux of excess amino acid production through hemoglobin digestion [89], 4) timed osmotic lysis of infected cells to allow daughter parasite egress from infected cells at the end of the intracellular cycle [88], and 5) a nonessential byproduct of infection and intracellular parasite metabolic activity [43]. Each of these proposals had some merit and was based in an understanding of parasite biology, but experimental evidence was missing and difficult to obtain prior to identification of the channel genes. Gene identification and experimental advances have now clarified the roles as discussed in this section.
A PSAC role in nutrient acquisition would be consistent with the channel’s high permeability to sugars, purines, key vitamins, and the essential amino acid isoleucine, all of which are required for parasite development and not available in adequate quantities within uninfected erythrocytes [1]. Although parasite killing by nonspecific PSAC inhibitors supported this and other proposed essential roles [90,91], uncertainties about mechanism of killing limited interpretation. Indeed, selection of a resistant mutant with unaltered PSAC activity and inhibitor affinity confirmed that phlorizdin, a commonly used inhibitor, kills parasites through action on unrelated targets [92]. To address this longstanding uncertainty, we developed a modified medium, termed PSAC growth inhibition medium (PGIM), with lower, more physiological concentrations of three key nutrients acquired via PSAC [67]. In contrast, the standard medium used in most labs, RPMI 1640 supplemented with a lipid source, has most nutrients present at concentrations > 10-fold higher than levels in plasma from healthy donors. We found that ISPA-28 had low potency against parasite growth in standard medium, but that it killed Dd2 parasites at nearly 800-fold lower concentrations than HB3 parasites in studies using PGIM, paralleling its clone-specific action against PSAC in these lines. Linkage analysis using the Dd2 x HB3 progeny clones and this difference in growth inhibitory activity mapped the clag3 locus, establishing that PSAC block accounts for killing by this uniquely specific inhibitor. Other PSAC inhibitors that do not exhibit differential activity against lab clones are also more effective against in vitro parasite growth in PGIM than in standard RPMI 1640-based media, whereas antimalarials acting on unrelated targets have indistinguishable IC50 values in these media [67]. Importantly, because these studies required use of a nutrient-optimized medium, they provided the first experimental evidence for an essential role in nutrient uptake.
The cation remodeling role for PSAC is based on the observation that infected cells gradually incur a concomitant increase in [Na+] and decrease in [K+] with intracellular parasite development because of the nonzero PSAC permeability to these cations [2,85,88,93]. The leak of these cations at the erythrocyte membrane dissipates the outward and inward gradients for these respective ions, as maintained by the host cell Na+/K+ ATPase pump [94]. This host cytosol cation remodeling was hypothesized to make the erythrocyte more hospitable for parasite growth, possibly by providing an inward Na+ gradient for coupled solute uptake at the intracellular parasite plasma membrane [87]. To explore this role, we designed and used a separate modified medium, 4suc:6KCl, that replaces the Na+ salts in the RPMI 1640-based medium with K+ salts and sucrose to preserve infected cell osmotic stability. Growth studies revealed that this medium supports unabated parasite growth without a need for adaptation, a remarkable finding in light of the marked changes in composition. Because it abolishes Na+ and K+ gradients across the erythrocyte membrane, parasite cultivation in 4suc:6KCl prevented PSAC-mediated cation leak and cation remodeling, as confirmed with infected erythrocyte ion content measurements [95]. This study revealed unexpectedly low Na+, K+, and Cl- requirements for parasite development; it also provided compelling evidence against Na+-coupled phosphate uptake at the parasite plasma membrane and excluded an essential role of K+ signaling in merozoite activation [96].
Alternate roles for PSAC have also been examined. Volume regulation of infected cells by allowing PSAC-mediated efflux of excess amino acids generated by hemoglobin digestion remains possible [89]. One prediction of this hypothesis is that potent PSAC inhibitors should lead to osmotic lysis of infected cells because of blocked amino acid efflux; as this has not been observed [67,97], this hypothesis should be considered with some caution. Another hypothesis, timed osmotic lysis of infected cells, proposes that gradual Na+ and K+ leak through PSAC leads to osmotic swelling and lysis ~ 44 hours after invasion; as this coincides with the duration of P. falciparum intracellular development, osmotic lysis may facilitate parasite egress at the end of the erythrocytic cycle [88]. This role is excluded by the normal developmental cycle in studies using 4suc:6KCl medium, where Na+ and K+ leak are abolished [95]; it is also inconsistent with studies implicating protein kinases in coordinated parasite egress [98,99]. Finally, proposals that PSAC is a nonessential byproduct of intracellular parasite development are excluded by rhoph2 and rhoph3 knockdown and by an advanced drug discovery and development project [72,73,74,97], both of which establish this target’s essentiality for bloodstream malaria parasites.
2.5. Drug discovery and development targeting PSAC
In addition to discovering isolate-specific inhibitors such as ISPA-28, high-throughput screens have identified multiple novel, potent PSAC inhibitors that sterilize in vitro parasite cultures. Iterations of medicinal chemistry and PSAC inhibition measurements with a robust transmittance-based assay have yielded improved target affinity with desirable pharmacokinetic properties for development of oral antimalarial drugs targeting this unexploited target [97]. The PSAC target has several desirable properties for therapy development. The unusual biochemical and molecular properties distinguish it from mammalian channels, permitting identification of specific inhibitors without activity against a battery of human channels and transporters and reducing the risk of drug side effects. PSAC’s surface location on infected cells is also a significant advantage for this target as it ensures target access by drugs in plasma; it also significantly reduces the risk of acquired resistance via drug efflux, a mechanism that has compromised effectiveness for several intracellular parasite targets [100,101]. Gene identification, DNA transfection studies, and cryo-EM structure determination should all help guide medicinal chemistry optimization of PSAC inhibitors to produce potent and specific derivatives that advance into clinical trials.
3. The PVM channel and PTEX translocon
3.1. Background
A second example of a unique parasite ion and solute channel localizes to the parasitophorous vacuole membrane (PVM, Figure 3A). The PVM is initially formed as an invagination of the erythrocyte membrane during invasion by the merozoite; it grows with the maturing pathogen through addition of lipids and proteins [102,103,104]. As it is an intracellular membrane for which there are not robust methods for isolation or purification, macroscopic transport methods such as tracer accumulation cannot be reliably used to characterize PVM transport properties. Patch-clamp methods, though complicated by the small size of the intracellular parasite, are therefore better suited for study of PVM transport. Indeed, they provided the first direct measurements of PVM ion transport as they identified a single ion channel type having a large conductance [105]. This PVM channel is present at high copy number and is primarily open at the resting membrane potential. It is also nonselective to ions and solutes of any charge and size up to 1400 dal in size, based on pore exclusion studies with polyethylene glycols [106]. Remarkably, fluorescent dye exclusion studies of fibroblasts infected with Toxoplasma gondii, a distantly related parasite that causes toxoplasmosis in humans and animals, revealed pores on that parasite’s PVM with a nearly identical size [107]; similar dye exclusion studies suggest that Eimeria nieschulzi, a distantly related parasite that produces intestinal disease in brown rats, also has such pores [108]. These observations indicate that the PVM is a molecular sieve for small solutes, a feature that appears to be highly conserved in divergent protozoan parasites with intracellular development.
The P. falciparum PVM must also mediate export of parasite proteins into host cytosol. These larger macromolecules serve various effector functions in the host cytosol, with some integrating into Maurer’s cleft and host erythrocyte membranes [109] [56,110,111]. Initially, study of this protein export proceeded independently of ion and solute the above PVM ion and solute flux studies. Our understanding of this process evolved from initial computational studies, revealing that most exported proteins carry a recessed PEXEL motif (RxLxE/D/Q) downstream of the ER signal sequence [112,113]. A pioneering study then used a GFP reporter protein fused to the mouse dihydrofolate reductase (mDHFR) protein to visualize protein export at the PVM and found that this chimeric reporter protein could not be exported upon addition of high-affinity folate analogs that prevent mDHFR unfolding [114]. These findings implicated a protein-conducting pore and revealed that proteins must be unfolded, presumably by one or more chaperones, to cross the PVM. This finding is also consistent with early studies showing that protein export is an ATP-dependent process [115].
3.2. Identification of PTEX components and other proteins invovled in export
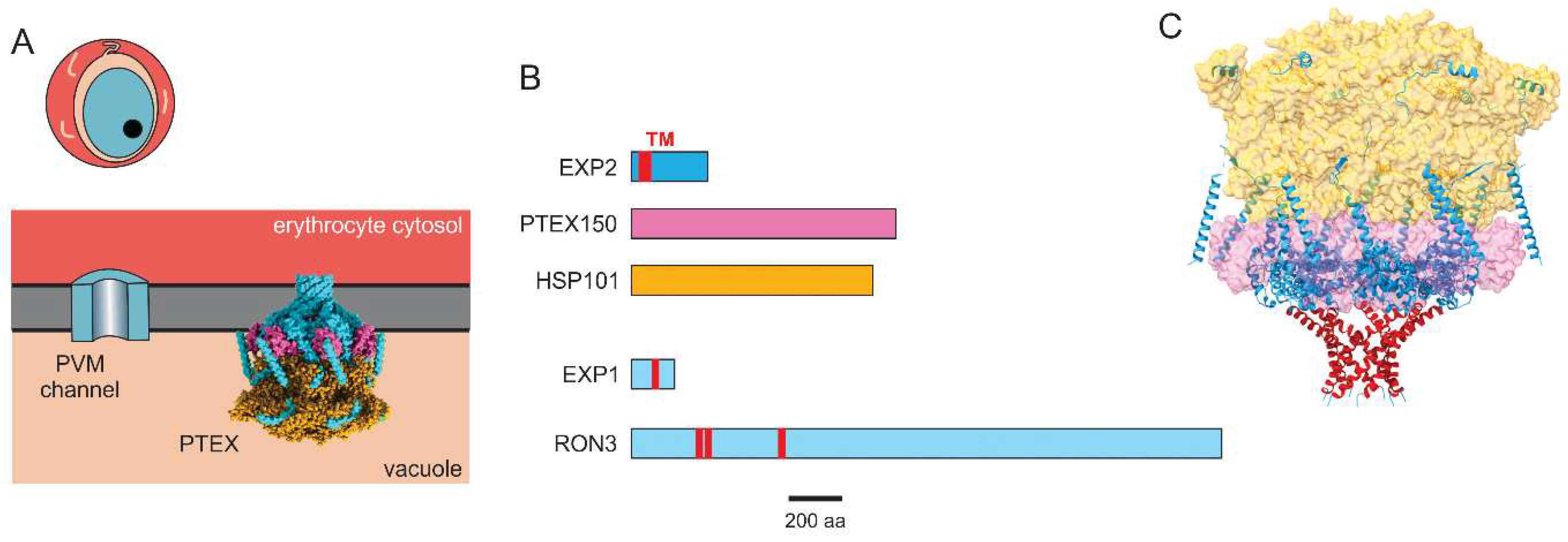
The molecular determinants of the protein export translocon were uncovered through a proteomic approach that capitalized on the above findings. Recognizing that an ATP-dependent chaperone is likely involved, de Koning-Ward et al. examined the detergent-resistant membrane proteome of immature infected cells to identified HSP101, a ClpA/B-like AAA+ ATPase chaperone protein [116]. Immunoprecipitation experiments were then used to identify associated proteins, PTEX150, EXP2, PTEX88, and TRX2. The interactions between these proteins were confirmed through reciprocal pull-down experiments. Importantly, this complex, termed the Plasmodium Translocon of EXported proteins (PTEX), was then shown to interact with exported proteins containing the PEXEL motif through mass spectrometry studies, providing compelling evidence for a role as a translocon. Based on biochemical studies showing that EXP2 is integral to membranes and that it has structural homology to pore-forming hemolysin E, EXP2 was proposed to be the pore-forming subunit of the putative translocon in this foundational study [116].
A direct functional link between the PTEX protein complex and export of effector parasite proteins then came through conditional knockdown studies of HSP101 and PTEX150. In one study [117], conditional knockdown of the P. falciparum HSP101 prevented export of various PEXEL-containing proteins into erythrocyte cytosol; several proteins that lack PEXEL domains but are known to be exported (termed PEXEL-negative exported proteins or PNEPs) were also blocked from export, indicating they also require an intact and functional PTEX translocon. Interestingly, CLAG3 export was found to be unaffected in this study but PSAC activity was abolished; a later study using the same antibodies and parasite line contradicted this, finding that export of CLAG3 and other RhopH proteins is abolished by PTEX knockdown, paralleling the failure to induce PSAC activity [72]. In the second study [118], HSP101 knockdown in the P. berghei rodent malaria parasite confirmed block of protein export and established the translocon’s importance under in vivo conditions. These workers also performed PTEX150 knockdown in P. falciparum, revealing that this subunit is also essential for translocon activity. Together, these two studies revealed failure of intracellular parasite maturation, inhibited in vitro and in vivo parasite growth and block of progression into gametocyte stages as required for transmission via mosquitoes.
In contrast to EXP2, PTEX150 and HSP101, the two other PTEX subunits identified by pull-down studies, TRX2 and PTEX88, can be genetically deleted. These knockout parasites exhibit slowed growth along with compromised sequestration and virulence in P. berghei-infected mice [119,120,121,122]. These findings may reflect accessory roles of these two components as some experiments reveal reduced export of proteins involved in infected cell cytoadherence [118,122]. Nevertheless, whether TRX2 and PTEX88 contribute to protein export or serve unrelated roles in intracellular parasite development remain to be conclusively elucidated.
Two proteins not detected in the initial PTEX component pull-down studies also appear to contribute to protein and solute transport at the PVM. The first of these, RON3, was discovered when its conditional knockout exhibited failed intracellular parasite maturation and blocked protein export at the PVM [123]. This study also used 2-NBDG, a labeled glucose analogue, to obtain indirect evidence linking RON3 to the PVM ion and nutrient uptake channel (Figure 3B). A subsequent study confirmed these findings and also implicated a RON3 role in erythrocyte invasion [124]; this study also reported compromised PSAC activity upon RON3 knockdown, presumably due to reduced export of RhopH proteins.
The second protein, EXP1, was also implicated through conditional knockout studies [125] (Figure 3B). In contrast to PTEX core components and RON3, EXP1 ablation did not compromise export of parasite proteins into host cytosol. At the same time, patch-clamp revealed near-complete loss of PVM channel activity; a reduced tolerance to amino acid deprivation suggested that this channel mediates nutrient uptake into the parasitophorous vacuole for parasite utilization. Subsequent studies revealed that EXP1 knockdown compromises PVM ultrastructure and EXP2 distribution on the PVM [126]; it also appears to reduce nutrient and drug uptake at the PVM in indirect transport measurements [127].
3.3. PTEX translocon structure
The de novo cryo-EM structure of the PTEX translocon, solved using protein complexes purified from blood cultures [128], then revealed a pore formed by seven EXP2 monomers. Immediately above the pore’s funnel, seven PTEX150 protomers were apparent with a hexameric HSP101 protein-unfolding motor at the top of the complex (Figure 3C). The accessory PTEX88 and TRX2 proteins were not visualized in the structure, adding to uncertainties about these proteins’ roles. Importantly, the complex was captured with unfolded cargo protein in transit through the pore. Two discrete conformations of this cargo-associated complex, designated as “engaged” and “resetting”, strongly implicate energy-driven threading of cargo through the PVM. These findings provide a structural mechanism for protein export and open the door to structure-guided therapy development.
3.4. Transport studies linking the translocon to the PVM channel
An important question has been whether the molecular basis of the PVM ion channel discovered in patch-clamp studies is the same as that of the PTEX translocon, studied primarily with protein imaging studies. To address this question, Garten et al. used engineered parasites carrying either EXP2 conditional knockdown or overexpression and performed patch-clamp [129]. Patch-clamp of these lines revealed a clear correlation between EXP2 expression and PVM ion channel abundance in cell-attached patch-clamp of released parasites with intact PVM, using methods similar to those used previously [72,105]. Patch-clamp of parasites carrying a C-terminal EXP2 truncation mutant revealed modest, but statistically signficant changes in the PVM channel’s voltage-dependence, suggesting a direct link between EXP2 and the functional PVM channel. Based on immunoprecipitation and stage-specific expression studies, this study proposed that EXP2 exists in two forms on the PVM: the PTEX protein translocon that maintains stable association with PTEX150 and HSP101 and a distinct heptameric pore not associated with other PTEX components that functions as the PVM ion and nutrient channel [129].
Support for this interesting model comes from studies of the PVM channel ortholog in Toxoplasma gondii [107]. Computatational analysis revealed two proteins, TgGRA17 and TgGRA23, with sequence homology to EXP2 [130]. Knockout of TgGRA17, but not TgGRA23, produced T. gondii parasites with abnormal PVM morphology in infected fibroblasts; this phenotype was rescued by complementation with the P. falciparum EXP2, supporting functional orthology. In contrast to the EXP2 knockdown, disruption of TgGRA17 or TgGRA23 did not compromise protein export by T. gondii. Dye uptake studies in infected fibroblasts and patch-clamp of Xenopus oocytes expressing TgGRA17 or TgGRA23 also supported a role for these proteins in formation of the Toxoplasma PVM channel, though these data were limited by incomplete effects of complementation and modest patch-clamp currents [130]. Also in partial support of this model, a recent study found that TgGRA17 expression in P. falciparum could not adequately complement EXP2 knockdown despite use of appropriate promoters and chimeric protein constructs that could allow association with other PTEX components [131].
Although these studies suggest that EXP2 forms the PVM channel, several uncertainties remain. In addition to the issues raised above, we can ask what role RON3 and EXP1 serve in formation of the PVM channel given their transmembrane domain topologies (Figure 3B), localization to the PVM, and transport studies suggesting involvement in ion and solute flux [123,125]. In light of technical challenges associated with PVM patch-clamp [41], limitations of DNA transfection studies, and the complex interactions between proteins at the PVM, new technologies may be required to conclusively define the roles served by these various proteins.
Regardless of their precise molecular determinants, the PVM channel and the PTEX protein export machinery are both exciting targets for antimalarial therapy development. Although direct inhibitors of these activities are presently unavailable, a potent chemical mimetic of the PEXEL motif that targets proteins for export, WEHI-842, effectively kills bloodstream parasites by preventing processing required for export [132], providing proof of concept for transport inhibition at this membrane as an important future direction for drug discovery and development.
4. Other channel activities that may be encoded by novel parasite-specific genes
In addition to the above relatively well-characterized parasite channels, there are additional P. falciparum transport activities suggestive of pathogen-specific channels. Increased Ca++ permeability at the host erythrocyte membrane is an important example identified through tracer flux [8,133,134]. 45Ca++ uptake measurements reveal that greater uptake by infected cells cannot be explained either by downregulation of the Ca++ extrusion pump or stimulation of the passive Ca++ carrier endogenous to erythrocyte membranes [135,136,137]. While PSAC is responsible for the increased permeability of other ions and solutes after infection, it does not account for increased Ca++ permeability as specific PSAC inhibitors do not reduce Ca++ transport at the infected erythrocyte membrane [9,10]. These observations suggest a distinct parasite-induced Ca++ transporter. Based on kinetic studies and rare events detected in patch-clamp, this transporter appears to be an ion channel [135]. It is not blocked by known blockers of mammalian Ca++ channels, but its activation after infection is compromised by PTEX knockdown [10]. Taken together, these findings suggest a parasite-derived channel that serves an essential role in Ca++ uptake and utilization by the intracellular pathogen [7].
Novel parasite ion channels are also likely present at other membrane barriers encountered throughout the parasite life cycle (Figure 1). Intracellular membranes, such as that of the parasite digestive vacuole or the multiple membranes surrounding the apicoplast [138,139], must have channels to mediate solute exchange and sustain organellar biochemical activities. Parasite stages that are not intracellular, such as bloodstream merozoites and ookinetes in the mosquito midgut, also require rapid exchange with their extracellular environment for signaling, motility, nutrient uptake, and metabolic waste removal.
The paucity of ion channel gene orthologs identified through whole-genome sequence of Plasmodium spp. is indeed surprising. Transport studies and directed gene identification strategies, as successfully used for PSAC and PTEX, should provide fundamental insights into parasite biology and uncover important targets for much-needed antimalarial therapies.
Funding
This research was funded by the Division of Intramural Research, NIAID, National Institutes of Health.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Divo, A.A.; Geary, T.G.; Davis, N.L.; Jensen, J.B. Nutritional requirements of Plasmodium falciparum in culture. I. Exogenously supplied dialyzable components necessary for continuous growth. J. Protozool 1985, 32, 59–64. [Google Scholar] [CrossRef]
- Overman, R.R. Reversible cellular permeability alterations in disease. In vivo studies on sodium, potassium and chloride concentrations in erythrocytes of the malarious monkey. Am. J. Physiol 1948, 152, 113–121. [Google Scholar] [CrossRef]
- Ginsburg, H.; Kutner, S.; Krugliak, M.; Cabantchik, Z.I. Characterization of permeation pathways appearing in the host membrane of Plasmodium falciparum infected red blood cells. Mol. Biochem. Parasitol 1985, 14, 313–322. [Google Scholar] [CrossRef]
- Kirk, K.; Horner, H.A.; Elford, B.C.; Ellory, J.C.; Newbold, C.I. Transport of diverse substrates into malaria-infected erythrocytes via a pathway showing functional characteristics of a chloride channel. J. Biol. Chem 1994, 269, 3339–3347. [Google Scholar] [CrossRef]
- Gero, A.M.; Wood, A.M. New nucleoside transport pathways induced in the host erythrocyte membrane of malaria and Babesia infected cells. Adv. Exp. Med. Biol 1991, 309A, 169–172. [Google Scholar]
- Bokhari, A.A.; Solomon, T.; Desai, S.A. Two distinct mechanisms of transport through the plasmodial surface anion channel. J. Membr. Biol 2008, 226, 27–34. [Google Scholar] [CrossRef]
- Wasserman, M.; Alarcon, C.; Mendoza, P.M. Effects of Ca++ depletion on the asexual cell cycle of Plasmodium falciparum. Am. J. Trop. Med. Hyg 1982, 31, 711–717. [Google Scholar] [CrossRef]
- Tanabe, K.; Mikkelsen, R.B.; Wallach, D.F. Calcium transport of Plasmodium chabaudi-infected erythrocytes. J. Cell Biol 1982, 93, 680–684. [Google Scholar] [CrossRef]
- Zipprer, E.M.; Neggers, M.; Kushwaha, A.; Rayavara, K.; Desai, S.A. A kinetic fluorescence assay reveals unusual features of Ca++ uptake in Plasmodium falciparum-infected erythrocytes. Malar. J 2014, 13, 184. [Google Scholar] [CrossRef]
- Kushwaha, A.K.; Apolis, L.; Ito, D.; Desai, S.A. Increased Ca(++) uptake by erythrocytes infected with malaria parasites: evidence for exported proteins and novel inhibitors. Cell Microbiol 2018, 20, e12853. [Google Scholar] [CrossRef]
- Stauffer, T.P.; Guerini, D.; Carafoli, E. Tissue distribution of the four gene products of the plasma membrane Ca2+ pump. A study using specific antibodies. J. Biol. Chem 1995, 270, 12184–12190. [Google Scholar] [CrossRef]
- Cali, T.; Brini, M.; Carafoli, E. Regulation of cell calcium and role of plasma membrane calcium ATPases. Int Rev Cell Mol Biol 2017, 332, 259–296. [Google Scholar] [CrossRef]
- Kirk, K. Ion regulation in the malaria parasite. Annu Rev Microbiol 2015, 69, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef]
- Lauger, P. Kinetic properties of ion carriers and channels. J Membr Biol 1980, 57, 163–178. [Google Scholar] [CrossRef]
- Hamill, O.P.; Marty, A.; Neher, E.; Sakmann, B.; Sigworth, F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch 1981, 391, 85–100. [Google Scholar] [CrossRef]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Almagro Armenteros, J.J.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. 2022. [Google Scholar] [CrossRef]
- Ellekvist, P.; Ricke, C.H.; Litman, T.; Salanti, A.; Colding, H.; Zeuthen, T.; Klaerke, D.A. Molecular cloning of a K+ channel from the malaria parasite Plasmodium falciparum. Biochem. Biophys. Res. Commun 2004, 318, 477–484. [Google Scholar] [CrossRef]
- Molbaek, K.; Tejada, M.; Ricke, C.H.; Scharff-Poulsen, P.; Ellekvist, P.; Helix-Nielsen, C.; Kumar, N.; Klaerke, D.A.; Pedersen, P.A. Purification and initial characterization of Plasmodium falciparum K+ channels, PfKch1 and PfKch2 produced in Saccharomyces cerevisiae. Microb Cell Fact 2020, 19, 183. [Google Scholar] [CrossRef]
- Waller, K.L.; McBride, S.M.; Kim, K.; McDonald, T.V. Characterization of two putative potassium channels in Plasmodium falciparum. Malar J 2008, 7, 19. [Google Scholar] [CrossRef]
- Ellekvist, P.; Mlambo, G.; Kumar, N.; Klaerke, D.A. Functional characterization of malaria parasites deficient in the K+ channel Kch2. Biochem Biophys Res Commun 2017, 493, 690–696. [Google Scholar] [CrossRef]
- Kenthirapalan, S.; Waters, A.P.; Matuschewski, K.; Kooij, T.W. Functional profiles of orphan membrane transporters in the life cycle of the malaria parasite. Nat Commun 2016, 7, 10519. [Google Scholar] [CrossRef]
- Anaguano, D.; Dedkhad, W.; Brooks, C.F.; Cobb, D.W.; Muralidharan, V. Time-resolved proximity biotinylation implicates a porin protein in export of transmembrane malaria parasite effectors. J Cell Sci 2023, 136. [Google Scholar] [CrossRef]
- Hou, C.; Tian, W.; Kleist, T.; He, K.; Garcia, V.; Bai, F.; Hao, Y.; Luan, S.; Li, L. DUF221 proteins are a family of osmosensitive calcium-permeable cation channels conserved across eukaryotes. Cell Res 2014, 24, 632–635. [Google Scholar] [CrossRef]
- Kenthirapalan, S.; Waters, A.P.; Matuschewski, K.; Kooij, T.W. Flow cytometry-assisted rapid isolation of recombinant Plasmodium berghei parasites exemplified by functional analysis of aquaglyceroporin. Int J Parasitol 2012, 42, 1185–1192. [Google Scholar] [CrossRef]
- Hansen, M.; Kun, J.F.; Schultz, J.E.; Beitz, E. A single, bi-functional aquaglyceroporin in blood-stage Plasmodium falciparum malaria parasites. J. Biol. Chem 2002, 277, 4874–4882. [Google Scholar] [CrossRef]
- Newby, Z.E.; O'Connell, J., 3rd; Robles-Colmenares, Y.; Khademi, S.; Miercke, L.J.; Stroud, R.M. Crystal structure of the aquaglyceroporin PfAQP from the malarial parasite Plasmodium falciparum. Nat Struct Mol Biol 2008, 15, 619–625. [Google Scholar] [CrossRef]
- Song, J.; Almasalmeh, A.; Krenc, D.; Beitz, E. Molar concentrations of sorbitol and polyethylene glycol inhibit the Plasmodium aquaglyceroporin but not that of E. coli: involvement of the channel vestibules. Biochim Biophys Acta 2012, 1818, 1218–1224. [Google Scholar] [CrossRef]
- Garg, S.; Agarwal, S.; Kumar, S.; Yazdani, S.S.; Chitnis, C.E.; Singh, S. Calcium-dependent permeabilization of erythrocytes by a perforin-like protein during egress of malaria parasites. Nat Commun 2013, 4, 1736. [Google Scholar] [CrossRef] [PubMed]
- Ishino, T.; Chinzei, Y.; Yuda, M. A Plasmodium sporozoite protein with a membrane attack complex domain is required for breaching the liver sinusoidal cell layer prior to hepatocyte infection. Cell Microbiol 2005, 7, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, K.; Camargo, N.; Coppens, I.; Morrisey, J.M.; Vaidya, A.B.; Kappe, S.H. A member of a conserved Plasmodium protein family with membrane-attack complex/perforin (MACPF)-like domains localizes to the micronemes of sporozoites. Mol. Biochem. Parasitol 2004, 133, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Wirth, C.C.; Glushakova, S.; Scheuermayer, M.; Repnik, U.; Garg, S.; Schaack, D.; Kachman, M.M.; Weissbach, T.; Zimmerberg, J.; Dandekar, T.; et al. Perforin-like protein PPLP2 permeabilizes the red blood cell membrane during egress of Plasmodium falciparum gametocytes. Cell Microbiol 2014, 16, 709–733. [Google Scholar] [CrossRef]
- Kadota, K.; Ishino, T.; Matsuyama, T.; Chinzei, Y.; Yuda, M. Essential role of membrane-attack protein in malarial transmission to mosquito host. Proc Natl Acad Sci U S A 2004, 101, 16310–16315. [Google Scholar] [CrossRef]
- Guizetti, J.; Martins, R.M.; Guadagnini, S.; Claes, A.; Scherf, A. Nuclear pores and perinuclear expression sites of var and ribosomal DNA genes correspond to physically distinct regions in Plasmodium falciparum. Eukaryot Cell 2013, 12, 697–702. [Google Scholar] [CrossRef]
- Ginsburg, H.; Stein, W.D. Biophysical analysis of a novel transport pathway induced in red blood cell membranes by the malaria parasite. Prog. Clin. Biol. Res 1988, 252, 317–322. [Google Scholar]
- Pouvelle, B.; Spiegel, R.; Hsiao, L.; Howard, R.J.; Morris, R.L.; Thomas, A.P.; Taraschi, T.F. Direct access to serum macromolecules by intraerythrocytic malaria parasites. Nature 1991, 353, 73–75. [Google Scholar] [CrossRef]
- Burns, E.R.; Pollack, S. P. falciparum infected erythrocytes are capable of endocytosis. In Vitro Cell Dev. Biol 1988, 24, 481–486. [Google Scholar] [CrossRef]
- Kutner, S.; Baruch, D.; Ginsburg, H.; Cabantchik, Z.I. Alterations in membrane permeability of malaria-infected human erythrocytes are related to the growth stage of the parasite. Biochim. Biophys. Acta 1982, 687, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.A.; Bezrukov, S.M.; Zimmerberg, J. A voltage-dependent channel involved in nutrient uptake by red blood cells infected with the malaria parasite. Nature 2000, 406, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Lisk, G.; Desai, S.A. The plasmodial surface anion channel is functionally conserved in divergent malaria parasites. Eukaryot. Cell 2005, 4, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Gezelle, J.; Saggu, G.; Desai, S.A. Promises and pitfalls of parasite patch-clamp. Trends Parasitol 2021, 37, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Wanke, E. Channel noise in nerve membranes and lipid bilayers. Q. Rev. Biophys 1975, 8, 451–506. [Google Scholar] [CrossRef]
- Staines, H.M.; Alkhalil, A.; Allen, R.J.; De Jonge, H.R.; Derbyshire, E.; Egee, S.; Ginsburg, H.; Hill, D.A.; Huber, S.M.; Kirk, K.; et al. Electrophysiological studies of malaria parasite-infected erythrocytes: current status. Int. J Parasitol 2007, 37, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Egee, S.; Lapaix, F.; Decherf, G.; Staines, H.M.; Ellory, J.C.; Doerig, C.; Thomas, S.L. A stretch-activated anion channel is up-regulated by the malaria parasite Plasmodium falciparum. J. Physiol 2002, 542, 795–801. [Google Scholar] [CrossRef]
- Duranton, C.; Tanneur, V.; Lang, C.; Brand, V.B.; Koka, S.; Kasinathan, R.S.; Dorsch, M.; Hedrich, H.J.; Baumeister, S.; Lingelbach, K.; et al. A high specificity and affinity interaction with serum albumin stimulates an anion conductance in malaria-infected erythrocytes. Cell Physiol Biochem 2008, 22, 395–404. [Google Scholar] [CrossRef]
- Verloo, P.; Kocken, C.H.; Van der Wel, A.; Tilly, B.C.; Hogema, B.M.; Sinaasappel, M.; Thomas, A.W.; De Jonge, H.R. Plasmodium falciparum-activated chloride channels are defective in erythrocytes from cystic fibrosis patients. J. Biol. Chem 2004, 279, 10316–10322. [Google Scholar] [CrossRef] [PubMed]
- Bouyer, G.; Egee, S.; Thomas, S.L. Three types of spontaneously active anionic channels in malaria-infected human red blood cells. Blood Cells Mol. Dis 2006, 36, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M.; Duranton, C.; Henke, G.; Van De, S.C.; Heussler, V.; Shumilina, E.; Sandu, C.D.; Tanneur, V.; Brand, V.; Kasinathan, R.S.; et al. Plasmodium induces swelling-activated ClC-2 anion channels in the host erythrocyte. J. Biol. Chem 2004, 279, 41444–41452. [Google Scholar] [CrossRef]
- Ginsburg, H.; Stein, W.D. How many functional transport pathways does Plasmodium falciparum induce in the membrane of its host erythrocyte? Trends Parasitol 2005, 21, 118–121. [Google Scholar] [CrossRef]
- Winograd, E.; Sherman, I.W. Malaria infection induces a conformational change in erythrocyte band 3 protein. Mol. Biochem. Parasitol 2004, 138, 83–87. [Google Scholar] [CrossRef]
- Ancelin, M.L.; Parant, M.; Thuet, M.J.; Philippot, J.R.; Vial, H.J. Increased permeability to choline in simian erythrocytes after Plasmodium knowlesi infection. Biochem. J 1991, 273, 701–709. [Google Scholar] [CrossRef]
- Alkhalil, A.; Cohn, J.V.; Wagner, M.A.; Cabrera, J.S.; Rajapandi, T.; Desai, S.A. Plasmodium falciparum likely encodes the principal anion channel on infected human erythrocytes. Blood 2004, 104, 4279–4286. [Google Scholar] [CrossRef]
- Hill, D.A.; Pillai, A.D.; Nawaz, F.; Hayton, K.; Doan, L.; Lisk, G.; Desai, S.A. A blasticidin S-resistant Plasmodium falciparum mutant with a defective plasmodial surface anion channel. Proc Natl Acad Sci U S A 2007, 104, 1063–1068. [Google Scholar] [CrossRef]
- Lisk, G.; Pain, M.; Gluzman, I.Y.; Kambhampati, S.; Furuya, T.; Su, X.Z.; Fay, M.P.; Goldberg, D.E.; Desai, S.A. Changes in the plasmodial surface anion channel reduce leupeptin uptake and can confer drug resistance in P. falciparum-infected erythrocytes. Antimicrob. Agents Chemother 2008, 52, 2346–2354. [Google Scholar] [CrossRef]
- Lisk, G.; Pain, M.; Sellers, M.; Gurnev, P.A.; Pillai, A.D.; Bezrukov, S.M.; Desai, S.A. Altered plasmodial surface anion channel activity and in vitro resistance to permeating antimalarial compounds. Biochim. Biophys. Acta 2010, 1798, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Nguitragool, W.; Bokhari, A.A.; Pillai, A.D.; Rayavara, K.; Sharma, P.; Turpin, B.; Aravind, L.; Desai, S.A. Malaria parasite clag3 genes determine channel-mediated nutrient uptake by infected red blood cells. Cell 2011, 145, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Nguitragool, W.; Rayavara, K.; Desai, S.A. Proteolysis at a specific extracellular residue implicates integral membrane CLAG3 in malaria parasite nutrient channels. PLoS. One 2014, 9, e93759. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bokhari, A.A.B.; Pillai, A.D.; Crater, A.K.; Gezelle, J.; Saggu, G.; Nasamu, A.S.; Ganesan, S.M.; Niles, J.C.; Desai, S.A. Complex nutrient channel phenotypes despite Mendelian inheritance in a Plasmodium falciparum genetic cross. PLoS Pathog 2020, 16, e1008363. [Google Scholar] [CrossRef] [PubMed]
- Alkhalil, A.; Pillai, A.D.; Bokhari, A.A.; Vaidya, A.B.; Desai, S.A. Complex inheritance of the plasmodial surface anion channel in a Plasmodium falciparum genetic cross. Mol Microbiol 2009, 72, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.S.; Kaewthamasorn, M.; Yahata, K.; Nakazawa, S.; Kaneko, O. Positive selection on the Plasmodium falciparum clag2 gene encoding a component of the erythrocyte-binding rhoptry protein complex. Trop. Med. Health 2011, 39, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.; Carret, C.; Kaneko, O.; Yim Lim, B.Y.; Ivens, A.; Holder, A.A. Epigenetic silencing of Plasmodium falciparum genes linked to erythrocyte invasion. PLoS. Pathog 2007, 3, e107. [Google Scholar] [CrossRef]
- Richards, J.S.; Arumugam, T.U.; Reiling, L.; Healer, J.; Hodder, A.N.; Fowkes, F.J.; Cross, N.; Langer, C.; Takeo, S.; Uboldi, A.D.; et al. Identification and prioritization of merozoite antigens as targets of protective human immunity to Plasmodium falciparum malaria for vaccine and biomarker development. J Immunol 2013, 191, 795–809. [Google Scholar] [CrossRef]
- Crowley, V.M.; Rovira-Graells, N.; de Pouplana, L.R.; Cortes, A. Heterochromatin formation in bistable chromatin domains controls the epigenetic repression of clonally variant Plasmodium falciparum genes linked to erythrocyte invasion. Mol Microbiol 2011, 80, 391–406. [Google Scholar] [CrossRef]
- Rovira-Graells, N.; Crowley, V.M.; Bancells, C.; Mira-Martinez, S.; Ribas de, P.L.; Cortes, A. Deciphering the principles that govern mutually exclusive expression of Plasmodium falciparum clag3 genes. Nucleic Acids Res 2015, 43, 8243–8257. [Google Scholar] [CrossRef] [PubMed]
- Mira-Martinez, S.; Rovira-Graells, N.; Crowley, V.M.; Altenhofen, L.M.; Llinas, M.; Cortes, A. Epigenetic switches in clag3 genes mediate blasticidin S resistance in malaria parasites. Cell Microbiol 2013, 15, 1913–1923. [Google Scholar] [CrossRef]
- Desai, S.A. Epigenetics of malaria parasite nutrient uptake, but why? Trends Parasitol 2022, 38, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A.D.; Nguitragool, W.; Lyko, B.; Dolinta, K.; Butler, M.M.; Nguyen, S.T.; Peet, N.P.; Bowlin, T.L.; Desai, S.A. Solute restriction reveals an essential role for clag3-associated channels in malaria parasite nutrient acquisition. Mol. Pharmacol 2012, 82, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Wollenberg, K.; Sellers, M.; Zainabadi, K.; Galinsky, K.; Moss, E.; Nguitragool, W.; Neafsey, D.; Desai, S.A. An epigenetic antimalarial resistance mechanism involving parasite genes linked to nutrient uptake. J. Biol. Chem 2013, 288, 19429–19440. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, O.; Yim Lim, B.Y.; Iriko, H.; Ling, I.T.; Otsuki, H.; Grainger, M.; Tsuboi, T.; Adams, J.H.; Mattei, D.; Holder, A.A.; Torii, M. Apical expression of three RhopH1/Clag proteins as components of the Plasmodium falciparum RhopH complex. Mol Biochem. Parasitol 2005, 143, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ling, I.T.; Florens, L.; Dluzewski, A.R.; Kaneko, O.; Grainger, M.; Yim Lim, B.Y.; Tsuboi, T.; Hopkins, J.M.; Johnson, J.R.; Torii, M.; et al. The Plasmodium falciparum clag9 gene encodes a rhoptry protein that is transferred to the host erythrocyte upon invasion. Mol Microbiol 2004, 52, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Vincensini, L.; Fall, G.; Berry, L.; Blisnick, T.; Braun, B.C. The RhopH complex is transferred to the host cell cytoplasm following red blood cell invasion by Plasmodium falciparum. Mol Biochem. Parasitol 2008, 160, 81–89. [Google Scholar] [CrossRef]
- Ito, D.; Schureck, M.A.; Desai, S.A. An essential dual-function complex mediates erythrocyte invasion and channel-mediated nutrient uptake in malaria parasites. Elife 2017, 6, e23485. [Google Scholar] [CrossRef]
- Sherling, E.S.; Knuepfer, E.; Brzostowski, J.A.; Miller, L.H.; Blackman, M.J.; van, O.C. The Plasmodium falciparum rhoptry protein RhopH3 plays essential roles in host cell invasion and nutrient uptake. Elife 2017, 6. [Google Scholar] [CrossRef]
- Counihan, N.; Chisholm, S.A.; Bullen, H.E.; Srivastava, A.; Sanders, P.R.; Jonsdottir, T.K.; Weiss, G.E.; Ghosh, S.; Crabb, B.S.; Creek, D.J.; et al. Plasmodium falciparum parasites deploy RhopH2 into the host erythrocyte to obtain nutrients, grow and replicate. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Trenholme, K.R.; Gardiner, D.L.; Holt, D.C.; Thomas, E.A.; Cowman, A.F.; Kemp, D.J. clag9: A cytoadherence gene in Plasmodium falciparum essential for binding of parasitized erythrocytes to CD36. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 4029–4033. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Valiyaveettil, M.; Achur, R.N.; Goyal, A.; Mattei, D.; Salanti, A.; Trenholme, K.R.; Gardiner, D.L.; Gowda, D.C. Dual stage synthesis and crucial role of cytoadherence-linked asexual gene 9 in the surface expression of malaria parasite var proteins. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 16643–16648. [Google Scholar] [CrossRef] [PubMed]
- Rungruang, T.; Kaneko, O.; Murakami, Y.; Tsuboi, T.; Hamamoto, H.; Akimitsu, N.; Sekimizu, K.; Kinoshita, T.; Torii, M. Erythrocyte surface glycosylphosphatidyl inositol anchored receptor for the malaria parasite. Mol Biochem. Parasitol 2005, 140, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Chugh, M.; Kumar, S.; Singh, S.; Kanodia, S.; Hossain, M.J.; Korde, R.; Grover, A.; Dhawan, S.; Chauhan, V.S.; et al. Proteome analysis reveals a large merozoite surface protein-1 associated complex on the Plasmodium falciparum merozoite surface. J. Proteome. Res 2011, 10, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Rayavara, K.; Ito, D.; Basore, K.; Desai, S.A. A CLAG3 mutation in an amphipathic transmembrane domain alters malaria parasite nutrient channels and confers leupeptin resistance. Infect. Immun 2015, 83, 2566–2574. [Google Scholar] [CrossRef] [PubMed]
- Schureck, M.A.; Darling, J.E.; Merk, A.; Shao, J.; Daggupati, G.; Srinivasan, P.; Olinares, P.D.B.; Rout, M.P.; Chait, B.T.; Wollenberg, K.; et al. Malaria parasites use a soluble RhopH complex for erythrocyte invasion and an integral form for nutrient uptake. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Manzella-Lapeira, J.; Saggu, G.; Ito, D.; Brzostowski, J.A.; Desai, S.A. Live-cell FRET reveals that malaria nutrient channel proteins CLAG3 and RhopH2 remain associated throughout their tortuous trafficking. mBio 2020, 11. [Google Scholar] [CrossRef]
- Ho, C.M.; Jih, J.; Lai, M.; Li, X.; Goldberg, D.E.; Beck, J.R.; Zhou, Z.H. Native structure of the RhopH complex, a key determinant of malaria parasite nutrient acquisition. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Ho, C.M.; Li, X.; Lai, M.; Terwilliger, T.C.; Beck, J.R.; Wohlschlegel, J.; Goldberg, D.E.; Fitzpatrick, A.W.P.; Zhou, Z.H. Bottom-up structural proteomics: cryoEM of protein complexes enriched from the cellular milieu. Nat Methods 2020, 17, 79–85. [Google Scholar] [CrossRef]
- Desai, S.A. Unique properties of nutrient channels on Plasmodium-infected erythrocytes. Pathogens 2023, 12. [Google Scholar] [CrossRef]
- Cohn, J.V.; Alkhalil, A.; Wagner, M.A.; Rajapandi, T.; Desai, S.A. Extracellular lysines on the plasmodial surface anion channel involved in Na+ exclusion. Mol. Biochem. Parasitol 2003, 132, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Saliba, K.J.; Horner, H.A.; Kirk, K. Transport and metabolism of the essential vitamin pantothenic acid in human erythrocytes infected with the malaria parasite Plasmodium falciparum. J. Biol. Chem 1998, 273, 10190–10195. [Google Scholar] [CrossRef] [PubMed]
- Saliba, K.J.; Martin, R.E.; Broer, A.; Henry, R.I.; McCarthy, C.S.; Downie, M.J.; Allen, R.J.; Mullin, K.A.; McFadden, G.I.; Broer, S.; Kirk, K. Sodium-dependent uptake of inorganic phosphate by the intracellular malaria parasite. Nature 2006, 443, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Staines, H.M.; Ellory, J.C.; Kirk, K. Perturbation of the pump-leak balance for Na(+) and K(+) in malaria- infected erythrocytes. Am. J. Physiol Cell Physiol 2001, 280, C1576–C1587. [Google Scholar] [CrossRef] [PubMed]
- Lew, V.L.; Macdonald, L.; Ginsburg, H.; Krugliak, M.; Tiffert, T. Excess haemoglobin digestion by malaria parasites: a strategy to prevent premature host cell lysis. Blood Cells Mol Dis 2004, 32, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Kutner, S.; Breuer, W.V.; Ginsburg, H.; Cabantchik, Z.I. On the mode of action of phlorizin as an antimalarial agent in in vitro cultures of Plasmodium falciparum. Biochem. Pharmacol 1987, 36, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Staines, H.M.; Dee, B.C.; O'Brien, M.; Lang, H.J.; Englert, H.; Horner, H.A.; Ellory, J.C.; Kirk, K. Furosemide analogues as potent inhibitors of the new permeability pathways of Plasmodium falciparum-infected human erythrocytes. Mol. Biochem. Parasitol 2004, 133, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.A.; Alkhalil, A.; Kang, M.; Ashfaq, U.; Nguyen, M.L. PSAC-independent phloridzin resistance in Plasmodium falciparum. J. Biol. Chem 2005, 280, 16861–16867. [Google Scholar] [CrossRef]
- Mauritz, J.M.; Seear, R.; Esposito, A.; Kaminski, C.F.; Skepper, J.N.; Warley, A.; Lew, V.L.; Tiffert, T. X-ray microanalysis investigation of the changes in Na, K, and hemoglobin concentration in Plasmodium falciparum-infected red blood cells. Biophys. J 2011, 100, 1438–1445. [Google Scholar] [CrossRef]
- Jorgensen, P.L.; Hakansson, K.O.; Karlish, S.J. Structure and mechanism of Na,K-ATPase: functional sites and their interactions. Annu. Rev. Physiol 2003, 65, 817–849. [Google Scholar] [CrossRef]
- Pillai, A.D.; Addo, R.; Sharma, P.; Nguitragool, W.; Srinivasan, P.; Desai, S.A. Malaria parasites tolerate a broad range of ionic environments and do not require host cation remodeling. Mol. Microbiol 2013, 88, 20–34. [Google Scholar] [CrossRef]
- Singh, S.; Alam, M.M.; Pal-Bhowmick, I.; Brzostowski, J.A.; Chitnis, C.E. Distinct external signals trigger sequential release of apical organelles during erythrocyte invasion by malaria parasites. PLoS. Pathog 2010, 6, e1000746. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.M.; Waidyarachchi, S.L.; Shao, J.; Nguyen, S.T.; Ding, X.; Cardinale, S.C.; Morin, L.R.; Kwasny, S.M.; Ito, M.; Gezelle, J.; et al. Optimized pyridazinone nutrient channel inhibitors are potent and specific antimalarial leads. Mol Pharmacol 2022, 102, 172–182. [Google Scholar] [CrossRef]
- Taylor, H.M.; McRobert, L.; Grainger, M.; Sicard, A.; Dluzewski, A.R.; Hopp, C.S.; Holder, A.A.; Baker, D.A. The malaria parasite cyclic GMP-dependent protein kinase plays a central role in blood-stage schizogony. Eukaryot Cell 2010, 9, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Dvorin, J.D.; Martyn, D.C.; Patel, S.D.; Grimley, J.S.; Collins, C.R.; Hopp, C.S.; Bright, A.T.; Westenberger, S.; Winzeler, E.; Blackman, M.J.; et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science 2010, 328, 910–912. [Google Scholar] [CrossRef]
- Cooper, R.A.; Lane, K.D.; Deng, B.; Mu, J.; Patel, J.J.; Wellems, T.E.; Su, X.; Ferdig, M.T. Mutations in transmembrane domains 1, 4 and 9 of the Plasmodium falciparum chloroquine resistance transporter alter susceptibility to chloroquine, quinine and quinidine. Mol Microbiol 2007, 64, 1139. [Google Scholar] [CrossRef]
- Wicht, K.J.; Mok, S.; Fidock, D.A. Molecular mechanisms of drug resistance in Plasmodium falciparum malaria. Annu Rev Microbiol 2020, 74, 431–454. [Google Scholar] [CrossRef] [PubMed]
- Dluzewski, A.R.; Mitchell, G.H.; Fryer, P.R.; Griffiths, S.; Wilson, R.J.; Gratzer, W.B. Origins of the parasitophorous vacuole membrane of the malaria parasite, Plasmodium falciparum, in human red blood cells. J Cell Sci 1992, 102, 527–532. [Google Scholar] [CrossRef]
- Bannister, L.H.; Hopkins, J.M.; Fowler, R.E.; Krishna, S.; Mitchell, G.H. A brief illustrated guide to the ultrastructure of Plasmodium falciparum asexual blood stages. Parasitol Today 2000, 16, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.E.; Zimmerberg, J. Hardly vacuous: the parasitophorous vacuolar membrane of malaria parasites. Trends Parasitol 2020, 36, 138–146. [Google Scholar] [CrossRef]
- Desai, S.A.; Krogstad, D.J.; McCleskey, E.W. A nutrient-permeable channel on the intraerythrocytic malaria parasite. Nature 1993, 362, 643–646. [Google Scholar] [CrossRef]
- Desai, S.A.; Rosenberg, R.L. Pore size of the malaria parasite's nutrient channel. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 2045–2049. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.C.; Beckers, C.J.; Joiner, K.A. The parasitophorous vacuole membrane surrounding intracellular Toxoplasma gondii functions as a molecular sieve. Proc. Natl. Acad. Sci. U. S. A 1994, 91, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Werner-Meier, R.; Entzeroth, R. Diffusion of microinjected markers across the parasitophorous vacuole membrane in cells infected with Eimeria nieschulzi (Coccidia, Apicomplexa). Parasitol Res 1997, 83, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Hviid, L.; Jensen, A.T. PfEMP1 - A parasite protein family of key importance in Plasmodium falciparum malaria immunity and pathogenesis. Adv. Parasitol 2015, 88, 51–84. [Google Scholar] [CrossRef] [PubMed]
- Kyes, S.A.; Rowe, J.A.; Kriek, N.; Newbold, C.I. Rifins: a second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 9333–9338. [Google Scholar] [CrossRef]
- Glenister, F.K.; Fernandez, K.M.; Kats, L.M.; Hanssen, E.; Mohandas, N.; Coppel, R.L.; Cooke, B.M. Functional alteration of red blood cells by a megadalton protein of Plasmodium falciparum. Blood 2009, 113, 919–928. [Google Scholar] [CrossRef]
- Marti, M.; Good, R.T.; Rug, M.; Knuepfer, E.; Cowman, A.F. Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science 2004, 306, 1930–1933. [Google Scholar] [CrossRef]
- Hiller, N.L.; Bhattacharjee, S.; van, O.C.; Liolios, K.; Harrison, T.; Lopez-Estrano, C.; Haldar, K. A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science 2004, 306, 1934–1937. [Google Scholar] [CrossRef]
- Gehde, N.; Hinrichs, C.; Montilla, I.; Charpian, S.; Lingelbach, K.; Przyborski, J.M. Protein unfolding is an essential requirement for transport across the parasitophorous vacuolar membrane of Plasmodium falciparum. Mol Microbiol 2009, 71, 613–628. [Google Scholar] [CrossRef]
- Ansorge, I.; Benting, J.; Bhakdi, S.; Lingelbach, K. Protein sorting in Plasmodium falciparum-infected red blood cells permeabilized with the pore-forming protein streptolysin O. Biochem J 1996, 315, 307–314. [Google Scholar] [CrossRef] [PubMed]
- de Koning-Ward, T.F.; Gilson, P.R.; Boddey, J.A.; Rug, M.; Smith, B.J.; Papenfuss, A.T.; Sanders, P.R.; Lundie, R.J.; Maier, A.G.; Cowman, A.F.; Crabb, B.S. A newly discovered protein export machine in malaria parasites. Nature 2009, 459, 945–949. [Google Scholar] [CrossRef]
- Beck, J.R.; Muralidharan, V.; Oksman, A.; Goldberg, D.E. PTEX component HSP101 mediates export of diverse malaria effectors into host erythrocytes. Nature 2014, 511, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Elsworth, B.; Matthews, K.; Nie, C.Q.; Kalanon, M.; Charnaud, S.C.; Sanders, P.R.; Chisholm, S.A.; Counihan, N.A.; Shaw, P.J.; Pino, P.; et al. PTEX is an essential nexus for protein export in malaria parasites. Nature 2014, 511, 587–591. [Google Scholar] [CrossRef]
- Matthews, K.; Kalanon, M.; Chisholm, S.A.; Sturm, A.; Goodman, C.D.; Dixon, M.W.; Sanders, P.R.; Nebl, T.; Fraser, F.; Haase, S.; et al. The Plasmodium translocon of exported proteins (PTEX) component thioredoxin-2 is important for maintaining normal blood-stage growth. Mol Microbiol 2013, 89, 1167–1186. [Google Scholar] [CrossRef] [PubMed]
- Matz, J.M.; Matuschewski, K.; Kooij, T.W. Two putative protein export regulators promote Plasmodium blood stage development in vivo. Mol Biochem Parasitol 2013, 191, 44–52. [Google Scholar] [CrossRef]
- Matz, J.M.; Ingmundson, A.; Costa Nunes, J.; Stenzel, W.; Matuschewski, K.; Kooij, T.W. In vivo function of PTEX88 in malaria parasite sequestration and virulence. Eukaryot Cell 2015, 14, 528–534. [Google Scholar] [CrossRef]
- Chisholm, S.A.; McHugh, E.; Lundie, R.; Dixon, M.W.; Ghosh, S.; O'Keefe, M.; Tilley, L.; Kalanon, M.; de Koning-Ward, T.F. Contrasting inducible knockdown of the auxiliary PTEX component PTEX88 in P. falciparum and P. berghei unmasks a role in parasite virulence. PLoS One 2016, 11, e0149296. [Google Scholar] [CrossRef]
- Low, L.M.; Azasi, Y.; Sherling, E.S.; Garten, M.; Zimmerberg, J.; Tsuboi, T.; Brzostowski, J.; Mu, J.; Blackman, M.J.; Miller, L.H. Deletion of Plasmodium falciparum protein RON3 affects the functional translocation of exported proteins and glucose uptake. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Kondo, Y.; Takashima, E.; Iriko, H.; Thongkukiatkul, A.; Torii, M.; Otsuki, H. Roles of the RON3 C-terminal fragment in erythrocyte invasion and blood-stage parasite proliferation in Plasmodium falciparum. Front Cell Infect Microbiol 2023, 13, 1197126. [Google Scholar] [CrossRef] [PubMed]
- Mesen-Ramirez, P.; Bergmann, B.; Tran, T.T.; Garten, M.; Stacker, J.; Naranjo-Prado, I.; Hohn, K.; Zimmerberg, J.; Spielmann, T. EXP1 is critical for nutrient uptake across the parasitophorous vacuole membrane of malaria parasites. PLoS Biol 2019, 17, e3000473. [Google Scholar] [CrossRef]
- Nessel, T.; Beck, J.M.; Rayatpisheh, S.; Jami-Alahmadi, Y.; Wohlschlegel, J.A.; Goldberg, D.E.; Beck, J.R. EXP1 is required for organisation of EXP2 in the intraerythrocytic malaria parasite vacuole. Cell Microbiol 2020, 22, e13168. [Google Scholar] [CrossRef] [PubMed]
- Mesen-Ramirez, P.; Bergmann, B.; Elhabiri, M.; Zhu, L.; von Thien, H.; Castro-Pena, C.; Gilberger, T.W.; Davioud-Charvet, E.; Bozdech, Z.; Bachmann, A.; Spielmann, T. The parasitophorous vacuole nutrient channel is critical for drug access in malaria parasites and modulates the artemisinin resistance fitness cost. Cell Host Microbe 2021, 29, 1774–1787. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.M.; Beck, J.R.; Lai, M.; Cui, Y.; Goldberg, D.E.; Egea, P.F.; Zhou, Z.H. Malaria parasite translocon structure and mechanism of effector export. Nature 2018, 561, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Garten, M.; Nasamu, A.S.; Niles, J.C.; Zimmerberg, J.; Goldberg, D.E.; Beck, J.R. EXP2 is a nutrient-permeable channel in the vacuolar membrane of Plasmodium and is essential for protein export via PTEX. Nat Microbiol 2018, 3, 1090–1098. [Google Scholar] [CrossRef]
- Gold, D.A.; Kaplan, A.D.; Lis, A.; Bett, G.C.; Rosowski, E.E.; Cirelli, K.M.; Bougdour, A.; Sidik, S.M.; Beck, J.R.; Lourido, S.; et al. The Toxoplasma dense granule proteins GRA17 and GRA23 mediate the movement of small molecules between the host and the parasitophorous vacuole. Cell Host Microbe 2015, 17, 642–652. [Google Scholar] [CrossRef]
- Pitman, E.L.; Counihan, N.A.; Modak, J.K.; Chowdury, M.; Gilson, P.R.; Webb, C.T.; de Koning-Ward, T.F. Dissecting EXP2 sequence requirements for protein export in malaria parasites. Front Cell Infect Microbiol 2023, 13, 1332146. [Google Scholar] [CrossRef]
- Hodder, A.N.; Sleebs, B.E.; Czabotar, P.E.; Gazdik, M.; Xu, Y.; O'Neill, M.T.; Lopaticki, S.; Nebl, T.; Triglia, T.; Smith, B.J.; et al. Structural basis for plasmepsin V inhibition that blocks export of malaria proteins to human erythrocytes. Nat Struct Mol Biol 2015, 22, 590–596. [Google Scholar] [CrossRef]
- Kramer, R.; Ginsburg, H. Calcium transport and compartment analysis of free and exchangeable calcium in Plasmodium falciparum-infected red blood cells. J. Protozool 1991, 38, 594–601. [Google Scholar] [PubMed]
- Rohrbach, P.; Friedrich, O.; Hentschel, J.; Plattner, H.; Fink, R.H.; Lanzer, M. Quantitative calcium measurements in subcellular compartments of Plasmodium falciparum-infected erythrocytes. J. Biol. Chem 2005, 280, 27960–27969. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.A.; McCleskey, E.W.; Schlesinger, P.H.; Krogstad, D.J. A novel pathway for Ca++ entry into Plasmodium falciparum-infected blood cells. Am. J. Trop. Med. Hyg 1996, 54, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Tiffert, T.; Lew, V.L. Kinetics of inhibition of the plasma membrane calcium pump by vanadate in intact human red cells. Cell Calcium 2001, 30, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.A.; Schlesinger, P.H.; Krogstad, D.J. Physiologic rate of carrier-mediated Ca2+ entry matches active extrusion in human erythrocytes. J. Gen. Physiol 1991, 98, 349–364. [Google Scholar] [CrossRef]
- Matz, J.M. Plasmodium's bottomless pit: properties and functions of the malaria parasite's digestive vacuole. Trends Parasitol 2022, 38, 525–543. [Google Scholar] [CrossRef]
- Kloehn, J.; Lacour, C.E.; Soldati-Favre, D. The metabolic pathways and transporters of the plastid organelle in Apicomplexa. Curr Opin Microbiol 2021, 63, 250–258. [Google Scholar] [CrossRef]
Figure 1.
Schematic showing the lifecycle of Plasmodium spp. in the vertebrate host and moquito vector. Notice the various intracellular and extracellular forms. During intracellular development, there are multiple membranous barriers to ion and nutrient exchange as highlighted with insets for infected erythrocytes and hepatocytes.
Figure 1.
Schematic showing the lifecycle of Plasmodium spp. in the vertebrate host and moquito vector. Notice the various intracellular and extracellular forms. During intracellular development, there are multiple membranous barriers to ion and nutrient exchange as highlighted with insets for infected erythrocytes and hepatocytes.
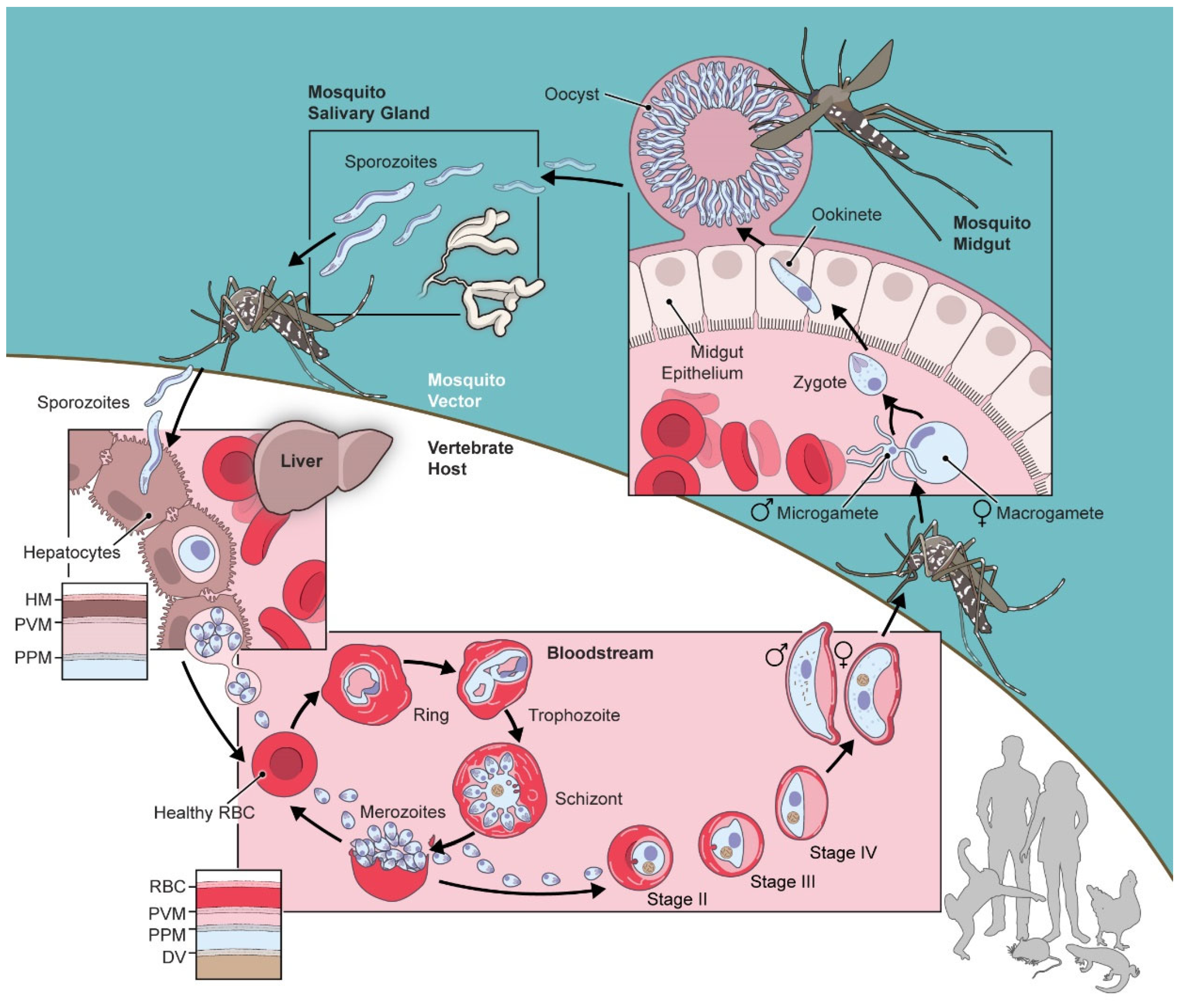
Figure 2.
The plasmodial surface anion channel (PSAC). A. Model for PSAC formation by subunits of the RhopH complex. CLAG3 is exposed at the erythrocyte surface, while RhopH2 and RhopH3 are endofacial proteins. B. Ribbon diagrams of RhopH proteins, drawn to scale. The positions of the single transmembrane domain predicted for each subunit are indicated in red; CLAG3 has a hypervariable region (HVR) exposed at the host cell surface. C. Cryo-EM reconstruction of the soluble RhopH complex prior to insertion in the host membrane. Subunit color scheme as in panel B, with α-helical transmembrane domains shown in red. PDB: 7KIY.
Figure 2.
The plasmodial surface anion channel (PSAC). A. Model for PSAC formation by subunits of the RhopH complex. CLAG3 is exposed at the erythrocyte surface, while RhopH2 and RhopH3 are endofacial proteins. B. Ribbon diagrams of RhopH proteins, drawn to scale. The positions of the single transmembrane domain predicted for each subunit are indicated in red; CLAG3 has a hypervariable region (HVR) exposed at the host cell surface. C. Cryo-EM reconstruction of the soluble RhopH complex prior to insertion in the host membrane. Subunit color scheme as in panel B, with α-helical transmembrane domains shown in red. PDB: 7KIY.
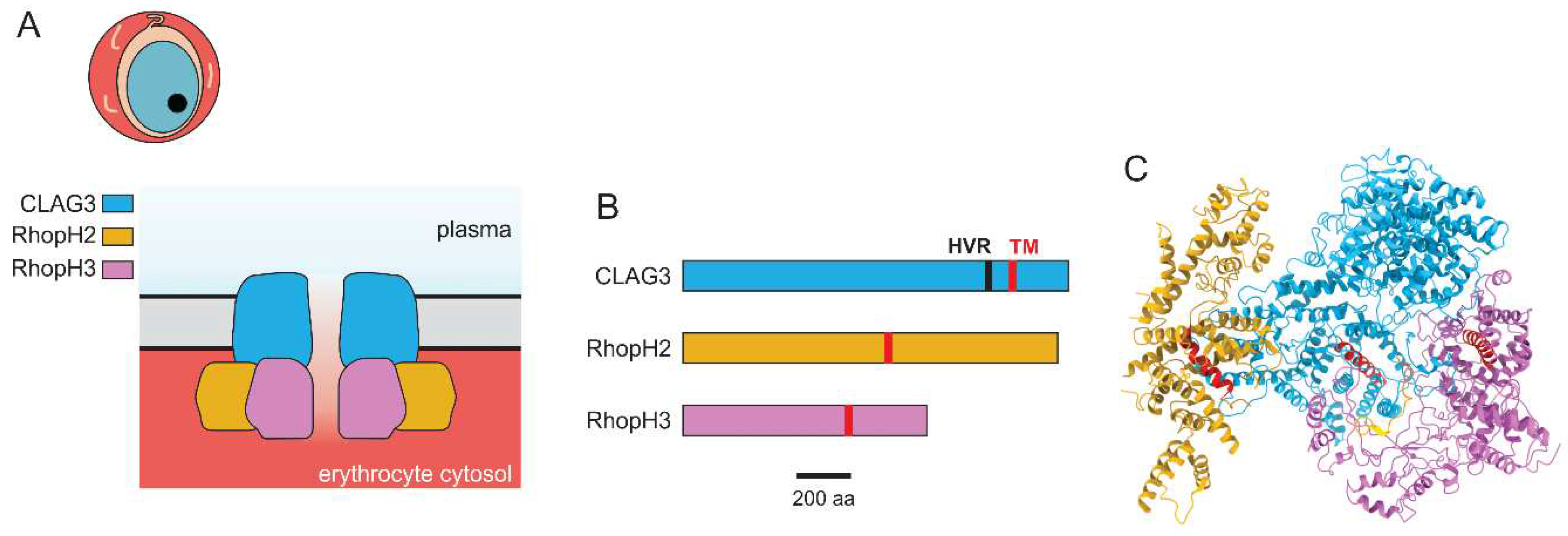
Figure 3.
The PVM channel and PTEX. A. Schematic showing the PVM channel and the PTEX translocon at the PVM. B. Ribbon diagrams of core PTEX components, EXP1, and RON3, drawn to scale. Predicted transmembrane domains indicated in red. C. Cryo-EM structure of the PTEX translocon, with core components color-coded as in panel B. The transmembrane domains (red) contributed by each of seven EXP2 monomers forms a pore. PDB: 6E11.
Figure 3.
The PVM channel and PTEX. A. Schematic showing the PVM channel and the PTEX translocon at the PVM. B. Ribbon diagrams of core PTEX components, EXP1, and RON3, drawn to scale. Predicted transmembrane domains indicated in red. C. Cryo-EM structure of the PTEX translocon, with core components color-coded as in panel B. The transmembrane domains (red) contributed by each of seven EXP2 monomers forms a pore. PDB: 6E11.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



