
Submitted:
06 February 2024
Posted:
08 February 2024
You are already at the latest version
Abstract
The ErbB/HER family of protein-tyrosine kinases (ErbB) and the phosphatidylinositol 3-kinase (PI3K) represent crucial targets in the treatment of head and neck squamous cell carcinoma (HNSCC). We previously reported that a combination therapy using Afatinib (ErbB inhibitor) and Copanlisib (PI3K inhibitor), both FDA-approved kinase inhibitors, suppressed the growth of HPV-positive HNSCC. In our current study, we further evaluated the efficacy and clinical potential of this combination therapy for treating HPV-negative HNSCC in vitro and in animal model. We found that the combination treatment of Afatinib and Copanlisib dramatically enhanced inhibition of cell proliferation and reduced cell survival when compared to treatment with either inhibitor in two HPV negative HNSCC cell lines. Notably, this combination led to significant inhibition of xenograft tumor growth in mice, without any apparent effects on body weight. Copanlisib alone effectively blocked PI3K/Akt signaling, but caused up-regulation of HER2 and HER3 phosphorylation as previously reported in other types of cancer. However, the combination treatment with Copanlisib and Afatinib completely blocked phosphorylation of the ErbB family (including HER3) and Akt, while also remarkedly increasing apoptosis. These results suggest that co-targeting the ErbB family and PI3K kinases by a combination treatment of Afatinib and Copanlisib can have clinical potential for patients.
Keywords:
HNSCC
; PI3K inhibitors
; ErbB inhibitor
; targeted therapy
1. Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most prevalent cancer globally, with 890,000 new cases and 450,000 deaths each year [1,2,3]. Based on etiological factors, HNSCC is classified into two types of disease: either HPV-positive or HPV-negative. The occurrence of HPV-negative HNSCC is associated with the use of tobacco and excessive consumption of alcohol, while HPV-positive HNSCC is related to human papillomavirus (HPV) infection [4,5,6]. Reports indict that patients with HPV-positive HNSCC have a higher 5-year survival rate (~80%) than HPV-negative patients (~50%) [7]. However, since HPV-positive HNSCC occurs mainly in oral and oropharyngeal tissues rather than other regions of head and neck, and may also progress to recurrent/metastatic disease in a significant portion of patients [8], there is an urgent need for new therapeutics to treat both HPV-positive and HPV-negative HNSCC [9, 10].
Classical therapies for HNSCC patients without distant metastasis typically include surgical resection, radiation therapy, chemotherapy, or a combination of these regimens. The specific therapeutic approach depends on various factors such as pre-existing clinical conditions, location of the cancer, and the TNM stages of the tumor. A combination of these treatments could reduce the rate of recurrence and distant metastasis for patients with local-regional disease. However, chemotherapy remains the primary option for patients with recurrent and distant metastatic HNSCC [11]. Cisplatin has been the most commonly used anticancer drug for treatment of advanced HNSCC, but, while many newly diagnosed patients with advanced HNSCC initially respond well to cisplatin-based chemotherapies, most patients either have intrinsic resistance or will eventually develop acquired resistance to cisplatin, leading to death within one year [12]. Immunotherapy has been recently introduced for refractory HNSCC, but its impact has been limited [12, 13]. Therefore, it remains of the utmost importance to find new therapeutic alternatives
The epidermal growth factor receptor (EGFR), a member of the ErbB kinases family, is notably overexpressed in 90-95% of HNSCC, and plays a crucial role in the cancer’s pathogenesis and clinical course [14,15,16]. EGFR controls the activation of several essential pathways such as PI3K/Akt/mTOR and RAS-RAF-MAPK (MEK)-ERK, which regulate cell proliferation, survival, and migration [17, 18]. In 2006, the monoclonal EGFR antibody cetuximab was approved by FDA for treatment of HNSCC in combination with the standard therapy [19,20,21]. However, the use of cetuximab resulted in very limited improvement in survival rates for patients undergoing cisplatin-based therapy [22]. In addition, small molecule kinase inhibitors like Gefitinib and Erlotinib, while effective in targeted therapies for Non-Small Cell Lung Cancer (NSCLC), have not demonstrated any benefits for HNSCC patients [23, 24].
Increasing evidence demonstrates the importance of the ErbB family, which contains EGFR, HER2, HER3, and HER4, in the carcinogenesis of HNSCC and its response to therapies. HER2 and HER3 form heterodimers with EGFR and play a role in PI3K/Akt activation. In addition, HER2 and HER3 are also associated with resistance to EGFR and PI3K inhibitors in cancer [25, 26]. These results indicate that targeting the ErbB family kinases could more effectively suppress HNSCC compared to solely using EGFR inhibitors [27, 28]. In fact, FDA-approved ErbB family inhibitor, Afatinib, has shown positive results in HNSCC clinical trials and is now listed on the National Comprehensive Cancer Network (NCCN) guidelines as a third-line single agent for HNSCC treatment [28,29,30,31,32]. Understanding the mechanisms behind resistance to Afatinib and exploring methods to avoid that resistance would be beneficial.
Phosphatidylinositol 3-Kinase (PI3K) is one of the most important downstream effectors of the EGFR/ErbB receptor family. The genes PIK3CA, PIK3CB, and PIK3CD encode three highly homologous catalytic isoforms of class IA PI3K, p110α, p110β, and p110δ, respectively. These isoforms associate with any of five regulatory isoforms: p85α, its splicing variants p55α and p50α, p85β, and p55γ [33]. The most important PI3K-p85 complex is PI3Kα/p85α. Recent studies demonstrate that mutations of PIK3CA, which codes for PI3Kα, are one of the most frequent mutations in HNSCC. In addition, PIK3CA/PI3Kα amplification or overexpression were also identified in HNSCC. Furthermore, PI3K/Akt signaling is activated in 34% of HPV-negative HNSCC tumors and 56% of HPV-positive tumors, which describe the prognosis of HNSCC [34,35,36,37]. PI3K activation in turn activates Akt, which then phosphorylates its substrates, such as TSC2, PRAS40, GSK3β, and FOXO, to regulate multiple cellular functions that consequently control cell proliferation, survival, and response to therapies. The tumor suppressor gene PTEN encodes the PTEN protein that dephosphorylates PIP3 to inhibit the PI3K pathway. Mutations that resulted in PTEN loss or deceased PTEN expression were frequently observed in HPV-positive and negative HNSCC [38, 39]. These alterations further result in the activation of PI3K/Akt [40]. Therefore, PI3Ks is one of the most attractive targets for the treatment of HNSCC [41,42,43].
We previously reported that co-targeting the ErbB family and PI3K, through a combination of Afatnib and Copanlisib, suppressed growth of HPV-positive HNSCC [44]. In this study, we further explored whether this combination is also effective at suppressing the growth of HPV-negative HNSCC. We found that the combination of Afatinib and Copanlisib led to significant inhibition of cell proliferation and induction of apoptosis in HPV-negative HNSCC. Furthermore, this combination suppressed tumor growth in xenograft models, while having no obvious effect on body weight loss in mice. These results highlight the feasibility of this combination for the treatment of HPV-negative HNSCC.
2. Materials and Methods
2.1. Cell Culture
HNSCC cell lines, Cal27, and FaDu were purchased from ATCC and were authenticated by short tandem repeat analysis (STR) and tested for mycoplasma contamination in the Translational Core Facility of the University of Maryland Marlene and Stewart Greenebaum Cancer Center. All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and 100 U/mL penicillin and streptomycin (Gibco).
2.2. Antibodies and Inhibitors
The following antibodies were purchased from Cell Signaling: phospho-Akt-S473 (CST-4508), phospho-Akt-T308 (CST-9275), Akt (CST-2938), phospho-S6K-T389 (CST-9205), S6K (CST-9202), phospho-HER2-Y1248 (CST-2247), HER2 (CST-4290), phospho-HER3-Y1289 (CST-2842), HER3 (CST-12708), C-caspase-3 (CST-xxx), and β-actin (CST-4967). Gefitinib, Erlotinib and Afatinib and all PI3K inhibitors were purchased from Selleck Chemicals.
2.3. Cell Lysis and Western Blot Analysis
2.4. Analyzing apoptosis by Annexin V/propidium iodide staining
2.5. Cell Viability Assay
2.6. Tumor Xenograft Formation in Mice
FaDu cells were subcutaneously injected on the right flank Nu/nu mice (Envigo, Frederick MD) at a density of 0.5 x106 cells/ml in the presence of 33% MatrigelTM (Fisher Scientific). When tumors reached approximately 200 mm3, mice were randomized to one of four treatment groups (7 mice/group): vehicle control, Copanlisib (6 mg/kg, IP), Afatinib (6 mg/kg, PO), or a combination of Copanlisib and Afatinib. Tumor volume was measured twice per week using electronic calipers and animals were weighed 5 days per week. Tumor volume was calculated as (L x W2)/2, where W is the smaller dimension and L is tumor length. Mice were euthanized on Day 32 of the treatment, and the tumors were excised, weighed, fixed, and frozen.
2.7. Statistical Analysis
All in vitro data are shown as mean ± SD and animal data was shown as mean ± SEM. Statistical analysis was performed using GraphPad Prism version 7.04 (GraphPad Software Inc.).
3. Results
3.1. HPV-Negative HNSCC Cell Lines are Sensitive to Afatinib.
We determine the EC50 values to EGFR/ErbB family inhibitors, Gefitinib, Erlotinib, and Afatinib in two HPV-negative HNSCC cell lines, Cal27 and FaDu. Both cell lines are relatively resistant to Erlotinib and sensitive to Gefitinib. However, they are very sensitive to Afatinib (Figure 1A and B). These results suggest that Afatinib is the more effective small molecule inhibitor for treatment of HPV negative HNSCC in comparison to EGFR inhibitors.
3.2. Copanlisib is the Most Effective PI3K Inhibitor to Suppress HPV-Negative HNSCC Proliferation.
To identify more effective PI3K inhibitors, we determined the EC50 values of six PI3K inhibitors, which included three pan-PI3K inhibitors, Copanlisib, BKM120, and GDC09410; the PI3Kα inhibitor BYL719; the PI3Kβ inhibitor AZD8186; and the PI3Kδ inhibitor Cal-101 (Figure 2A and B). Cal27 cells were strongly resistant to PI3Kδ, and relatively resistant to PI3Kα and PI3Kβ inhibitors. However, they were much more sensitive to Pan PI3K inhibitors, Copanlisib, GDC0941 and BKM120. Moreover, they showed much lower EC50 to Copanlisib in comparison to BKM120 and GDC0941. Similar results were found in FaDu cells (Supplementary Figure 1A and Figure 2B). These results suggest that Copanlisib is the most effective small molecule PI3K inhibitor for treatment of HPV-negative HNSCC.
3.3. Synergistic Inhibition of Cell Proliferation by a Combination of Afatinib and Copanlisib.
Our goal was to test whether simultaneous inhibition of ErbB family and PI3K pathways could more effectively inhibit HPV negative HNSCC proliferation. Based on the data described above, we selected Afatinib and Copanlisib for the combination therapy. Similar to the results in Figure 1 and Figure 2, Afatinib or Copanlisib alone inhibited cell proliferation, however, the combination caused enhanced inhibition of cell proliferation in both Cal27 (Figure 3A) and FaDu cells (Figure 3C). Furthermore, the related combination index (CI) value for each combination was calculated according to the Chou–Talalay method method [47] using CalcuSyn software. The CI values for all combinations were less than 1.0, which indicated a synergistic effect in the combination of Afatinib and Copanlisib (Figure 3B and D). These data indicate that Afatinib and Copanlisib synergistically inhibit HPV-negative HNSCC cell proliferation.
3.4. Synergistic Inhibition of Xenograft Tumor Growth by the Combination of Afatinib and Copanlisib in Mice.
It was important to evaluate the anti-tumor activity of the Afatinib and Copanlisib combination in vivo by using a mouse xenograft model. FaDu cells were inoculated into the mice, and when the tumors reached approximately 200 mm3, mice were randomized into four groups for treatment with vehicle control, Copanlisib, Afatinib, or the combination of both. We originally intended to treat the mice for more than 6-8 weeks, but we had to terminate the experiment on day 32 due to tumor necrosis in the majority of the control mice. The average tumor volume in groups treated with either Copanlisib or Afatinib was lower than that of the control group, but there were no significant differences in tumor volume among these three groups, which might be due to tumor necrosis in the control group (Figure 4A). However, tumor volumes in the combination treatment group were significantly lower than the control group by the end of the study (Figure 4A).
Similarly, after the tumors were excised and weighed at the end of the study, there was no significant difference in tumor weight (mg) between the groups treated with either Copanlisib or Afatinib (Figure 4B). However, tumor weight in the combination treatment group was significantly lower compared with single reagent treated groups (P<0.05, Figure 4B) and notably lower compared with control group (P<0.01, Figure 4B). In summary, while Afatinib or Copanlisib alone had modest inhibitory effects on tumor growth, it did not reach statistical significance, whereas the combination of the two drugs significantly inhibited tumor growth (Figure 4A and B).
More importantly, all doses of Copanlisib and Afatinib in single and combination treatment were well-tolerated, because there was no significant weight loss observed during the study (Figure 4C). These results demonstrate the feasibility of the Afatinib and Copanlisib combination in suppressing HPV-negative HNSCC.
3.5. A combination of Afatinib and Copanlisib Induces Apoptosis.
We tested whether a combination of Afatinib and Copanlisib could cause more apoptosis compared to either single treatment. Cal27 cells were treated with Afatinib (0.5 µM), Copanlisib (30 nM), or their combination for 48 hours before apoptosis assay was performed. Afatinib and Copanlisib alone induced cell apoptosis, whereas their combination significant increased cell apoptosis (Figure 5A and Supplementary Figure 2). Since FaDu cells showed higher IC50 to Afatinib and Copanlisib in comparison to Cal27 cells, we chose higher concentrations of Afatinib and Copanlisib for the treatments. Treatment with Copanlisib (125 nM) caused significant apoptosis, whereas treatment with Afatinib (1.0 µM) did not cause significantly increased apoptosis. However, a combination of Copanlisib (125 nM) and Afatinib (1.0 µM) led to significantly increased apoptosis compared to either of the single treatment (Figure 5B and Supplementary Figure 3). These data demonstrate that Afatinib and Copanlisib cooperate to induce apoptosis in HPV-negative HNSCC.
3.6. Combination of Afatinib and Copanlisib Completely Inhibits ErbB and PI3K Pathways Resulting in Induction of Caspase Cleavage.
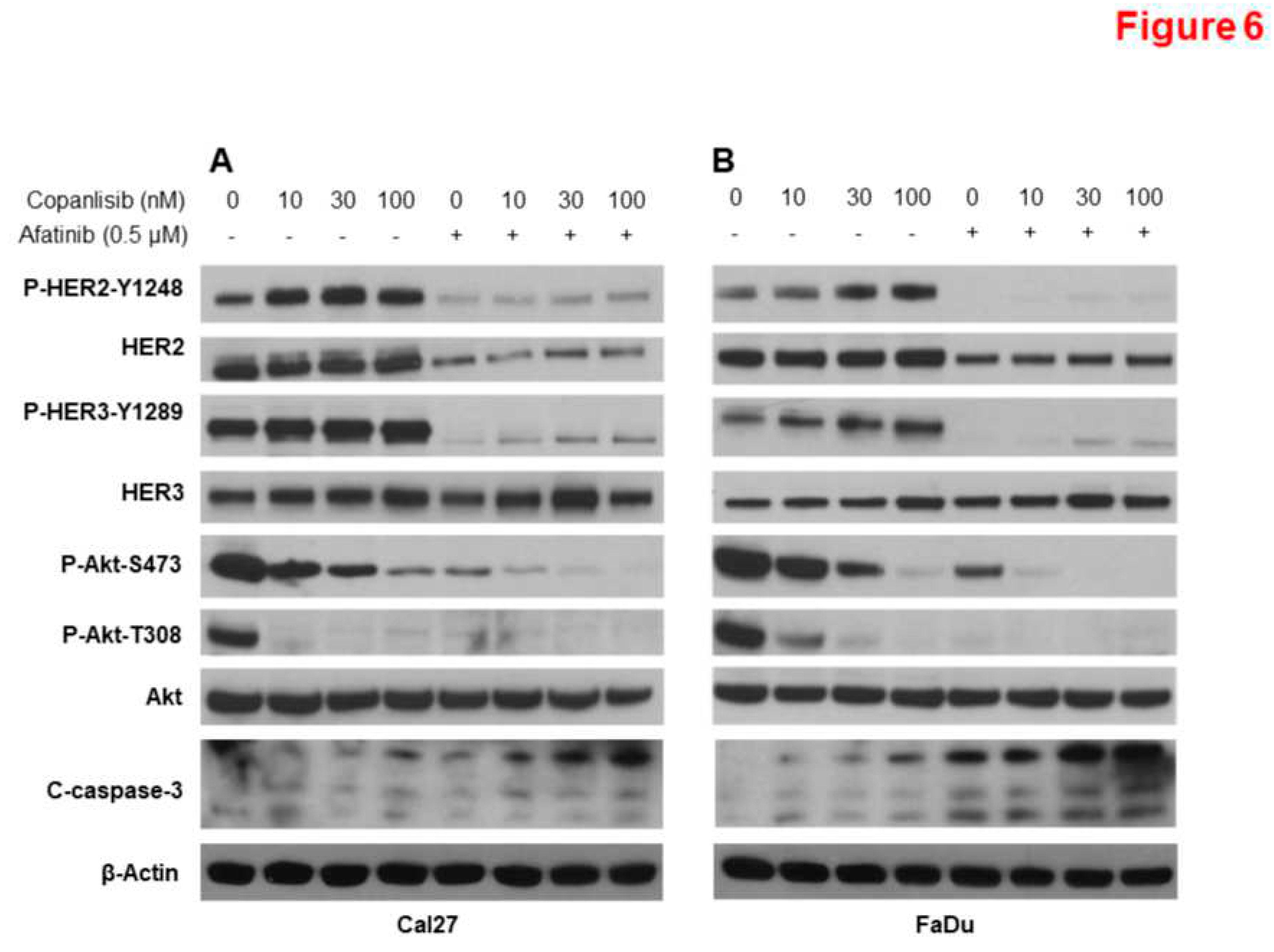
Previous studies demonstrated that PI3K inhibitors induced HER2 and HER3 phosphorylation, which thus conferred resistance to PI3K inhibitor [48, 49]. We recently showed that Copanlisib induced an increase of P-HER2 Y1248 and P-HER3 Y1289 in HPV+ HNSCC cells, while a combination of Afatinib and Copanlisib blocked phosphorylation of HER2 and HER3 [44]. We further tested whether Copanlisib induce P-HER2 Y1248 and P-HER3 Y1289 in HPV-negative Cal27 (Figure 6A) and FaDu cells (Figure 6B). Similarly, Copanlisib significantly inhibited phosphorylation of Akt and induced phosphorylation of P-HER2 Y1248 and P-HER3 Y1289, whereas the combination of Copanlisib and Afatinib completely blocked both phosphorylation of Akt, HER2 and HER3. In addition, increased caspase-3 cleavage was induced by the combination compared to the single treatment (Figure 6A and B).
4. Discussion
In this study, we tested the efficacy of co-inhibiting the ErbB family and PI3K through the combination of Afatinib and Copanlisib to inhibit HPV-negative HNSCC. Our results showed that the combination of Afatinib and Copanlisib caused dramatic inhibition of cell proliferation and suppressed cell survival in vitro in comparison to treatment with either Afatinib or Copanlisib alone. Notably, the combination led to significant inhibition of xenograft tumor growth without affecting the body weight of the mice. These results suggest that the combination of Afatinib and Copanlisib may have clinical potential for the treatment of HPV-negative HNSCC.
The EGFR/ErbB and PI3K/Akt/mTOR pathways have been the most attractive pathways to target for treatment of HNSCC due to over-expression or activating mutation of PIK3CA and loss function mutations of PTEN [6,50,51,52,53]. It has been reported that constitutive activation of PI3K/Akt/mTOR pathway due to the alterations in PIK3CA, PTEN, Akt or mTOR is associated with resistance to EGFR inhibitor [6]. Our data showed that treatment with Afatinib alone cannot completely block the phosphorylation of Akt (Figure 6). Furthermore, it has been reported that PI3K inhibition led to increased phosphorylation and total levels of HER3, which confer resistance to PI3K inhibitors [48, 49, 54,55,56]. Our data also showed that Copanlisib increased phospho-HER3 (Y1289), which was counteracted by the addition of Afatinib (Figure 6). Notably, the combination of Copanlisib and Afatinib induced significant caspase-3 cleavage in addition to the complete inhibition of ErbB and PI3K/Akt pathways (Figure 6). These results provide a rationale for the co-inhibition of ErbB and PI3K as a method to treat HNSCC.
We recently reported that the combination of Afatinib and Copanlisib effectively suppressed HPV-positive HNSCC. The combination therapy blocked both ErbB and PI3K/Akt pathways, which was accompanied by deceased E6 and E7, and the induction of Apoptosis, indicating increased efficacy of this combination in HPV+ HNSCC [44]. A publication by Milewska et al, reported that cell lines from multiple cancers, including HNSCC with PIK3CA mutations, are sensitive to the combination of Afatinib and Copanlisib [57]. As the basal level of PI3K/Akt is also high in HPV-negative HNSCC and plays essential roles in the regulation of growth, metastasis, and sensitivity to chemo- and targeted therapies [6, 42, 58, 59], it would be reasonable to predict that this combination would also be beneficial in HPV-negative HNSCC with upregulated PI3K/Akt signaling. Afatinib has shown positive results in HNSCC clinical trials and is now listed on the National Comprehensive Cancer Network (NCCN) guidelines as a third-line single agent for HNSCC treatment[28,29,30,31,32]. Our results indicate that the combination of Afatinib with Copanlisib would more effectively suppress HNSCC in patients with refractory disease.
Immunotherapy, including immune checkpoint blockade (ICB) targeting PD-L1/PD-1 using PD-1 inhibitor, Nivolumab or Pembrolizumab, was another important advancement in the treatment of advanced HNSCC. Afatinib modulates PD-L1 expression in multiple cancers, including gastric cancer [60]. In addition, it has been reported that PI3K inhibitors such as BKM120 deceased the expression of PD-L1 in HNSCC cells [61]. It would be interesting to determine the effects of Afatinib, Copanlisib, and their combination on the expression of PD-L1 in HNSCC and immune cells, such as T-cells, and the impact of the combination of Afatinib and Copanlisib on immunotherapy.
5. Conclusion
Our results suggest that co-targeting the ErbB family and PI3K kinases by a combination treatment of Afatinib and Copanlisib can have clinical potential for HNSCC patients.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Copanlisib more effectively inhibited cell proliferation compared to other PI3K inhibitors in FaDu cells, Figure S2: Combination of Copanlisib and Afatinib induced more apoptosis compared to either single treatment in Cal27 cells, Figure S3: A combination of Copanlisib and Afatinib induced more apoptosis compared to either single treatment in FaDu cells.
Author Contributions
Xinyan Geng: Data curation, Formal analysis, Writing – original draft. Shirin Azarbarzin: Data curation, Formal analysis. Zejia Yang: Data curation, Formal analysis. Rena G. Lapidus: Data curation, Formal analysis. Xiaoxuan Fan: Data curation, Formal analysis. Yong Teng: Formal analysis, Writing – review & editing. Ranee Mehra: Formal analysis, Writing – review & editing. Kevin J. Cullen: Conceptualization, Data curation, Formal analysis, Supervision, Writing – review & editing, Funding acquisition. Hancai Dan: Conceptualization, Data curation, Formal analysis, Supervision, Writing – review & editing, Funding acquisition.
Acknowledgments
We thank the Translational Core Facility and Flow Cytometry Shared Service of the University of Maryland Marlene and Stewart Greenebaum Cancer Center for analysis of data from Annexin V experiments that measured apoptosis, respectively. We also thank the Translational Laboratory Shared Services (TLSS) of Marlene & Stewart Greenebaum Comprehensive Cancer Center for animal work.
Grant Support
This research was supported, in part, by grants from the NIH National Cancer Institute (NCI) to H.D. (R00CA149178, R01CA212094 and R56DE030423) and the Orakowa Foundation. This research was also supported by funds through the National Cancer Institute - Cancer Center Support Grant (CCSG) – P30CA134274 and the Maryland Department of Health's Cigarette Restitution Fund Program – CH-649-CRF. The funding agency was not involved in the design of the study and collection, analysis, interpretation of data, or writing the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Farah, C.S. Molecular landscape of head and neck cancer and implications for therapy. Ann Transl Med 2021, 9, 915. [Google Scholar] [CrossRef]
- Barsouk, A.; Aluru, J.S.; Rawla, P.; Saginala, K.; Barsouk, A. Epidemiology, Risk Factors, and Prevention of Head and Neck Squamous Cell Carcinoma. Med Sci (Basel) 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Hashibe, M.; Boffetta, P.; Zaridze, D.; Shangina, O.; Szeszenia-Dabrowska, N.; Mates, D.; Fabiánová, E.; Rudnai, P.; Brennan, P. Contribution of tobacco and alcohol to the high rates of squamous cell carcinoma of the supraglottis and glottis in Central Europe. American journal of epidemiology 2007, 165, 814–820. [Google Scholar] [CrossRef]
- Hashibe, M.; Brennan, P.; Benhamou, S.; Castellsague, X.; Chen, C.; Curado, M.P.; Dal Maso, L.; Daudt, A.W.; Fabianova, E.; Fernandez, L.; et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Journal of the National Cancer Institute 2007, 99, 777–789. [Google Scholar] [CrossRef]
- Zaryouh, H.; De Pauw, I.; Baysal, H.; Peeters, M.; Vermorken, J.B.; Lardon, F.; Wouters, A. Recent insights in the PI3K/Akt pathway as a promising therapeutic target in combination with EGFR-targeting agents to treat head and neck squamous cell carcinoma. Med Res Rev 2022, 42, 112–155. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Li, S.; Henry, L.E.; Liu, S.; Sartor, M.A. Molecular Tumor Subtypes of HPV-Positive Head and Neck Cancers: Biological Characteristics and Implications for Clinical Outcomes. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Powell, S.F.; Vu, L.; Spanos, W.C.; Pyeon, D. The Key Differences between Human Papillomavirus-Positive and -Negative Head and Neck Cancers: Biological and Clinical Implications. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, P.T.; Westra, W.H.; Califano, J.A. Human papillomavirus and head and neck squamous cell carcinoma: recent evidence and clinical implications. J Dent Res 2009, 88, 300–306. [Google Scholar] [CrossRef]
- Ferreira, C.C. The relation between human papillomavirus (HPV) and oropharyngeal cancer: a review. PeerJ 2023, 11, e15568. [Google Scholar] [CrossRef]
- Milas, L.; Mason, K.A.; Liao, Z.; Ang, K.K. Chemoradiotherapy: emerging treatment improvement strategies. Head & neck 2003, 25, 152–167. [Google Scholar]
- Lee, Y.S.; Johnson, D.E.; Grandis, J.R. An update: emerging drugs to treat squamous cell carcinomas of the head and neck. Expert opinion on emerging drugs 2018, 23, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Ionna, F.; Longo, F.; Della Vittoria Scarpati, G.; De Angelis, C.; Ottaiano, A.; Botti, G.; Caponigro, F. Immune Response Against Head and Neck Cancer: Biological Mechanisms and Implication on Therapy. Translational oncology 2019, 13, 262–274. [Google Scholar] [CrossRef]
- Wen, Y.; Grandis, J.R. Emerging drugs for head and neck cancer. Expert opinion on emerging drugs 2015, 20, 313–329. [Google Scholar] [CrossRef]
- Park, B.J.; Chiosea, S.I.; Grandis, J.R. Molecular changes in the multistage pathogenesis of head and neck cancer. Cancer biomarkers : section A of Disease markers 2010, 9, 325–339. [Google Scholar] [CrossRef]
- Sharafinski, M.E.; Ferris, R.L.; Ferrone, S.; Grandis, J.R. Epidermal growth factor receptor targeted therapy of squamous cell carcinoma of the head and neck. Head & neck 2010, 32, 1412–1421. [Google Scholar]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nature reviews Drug discovery 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel) 2017, 9. [Google Scholar] [CrossRef]
- Alorabi, M.; Shonka, N.A.; Ganti, A.K. EGFR monoclonal antibodies in locally advanced head and neck squamous cell carcinoma: What is their current role? Critical reviews in oncology/hematology 2016, 99, 170–179. [Google Scholar] [CrossRef]
- Blaszczak, W.; Barczak, W.; Wegner, A.; Golusinski, W.; Suchorska, W.M. Clinical value of monoclonal antibodies and tyrosine kinase inhibitors in the treatment of head and neck squamous cell carcinoma. Medical oncology (Northwood, London, England) 2017, 34, 60. [Google Scholar] [CrossRef]
- Mehra, R.; Cohen, R.B.; Burtness, B.A. The role of cetuximab for the treatment of squamous cell carcinoma of the head and neck. Clinical advances in hematology & oncology : H&O 2008, 6, 742–750. [Google Scholar]
- Argiris A, Heron DE, Smith RP, Kim S, Gibson MK, Lai SY, Branstetter BF, Posluszny DM, Wang L, Seethala RR.; et al. Induction docetaxel, cisplatin, and cetuximab followed by concurrent radiotherapy, cisplatin, and cetuximab and maintenance cetuximab in patients with locally advanced head and neck cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2010, 28, 5294–5300. [CrossRef]
- Kang, J.J.; Ko, A.; Kil, S.H.; Mallen-St Clair, J.; Shin, D.S.; Wang, M.B.; Srivatsan, E.S. EGFR pathway targeting drugs in head and neck cancer in the era of immunotherapy. Biochim Biophys Acta Rev Cancer 2023, 1878, 188827. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; He, J.; Li, B.; Zheng, Y.; Li, K.; Zou, S.; Chen, L. Efficacy and Safety of Gefitinib in Patients with Advanced Head and Neck Squamous Cell Carcinoma: A Meta-Analysis of Randomized Controlled Trials. J Oncol, 6273. [Google Scholar]
- Sacco, A.G.; Worden, F.P. Molecularly targeted therapy for the treatment of head and neck cancer: a review of the ErbB family inhibitors. OncoTargets and therapy 2016, 9, 1927–1943. [Google Scholar] [PubMed]
- Rysman, B.; Mouawad, F.; Gros, A.; Lansiaux, A.; Chevalier, D.; Meignan, S. Human epidermal growth factor receptor 3 in head and neck squamous cell carcinomas. Head Neck 2016, 38 (Suppl 1), E2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, C.; Benvenuto, M.; Focaccetti, C.; Albonici, L.; Cifaldi, L.; Rufini, A.; Nardozi, D.; Angiolini, V.; Bei, A.; Masuelli, L.; et al. Recent findings on the impact of ErbB receptors status on prognosis and therapy of head and neck squamous cell carcinoma. Front Med (Lausanne) 2023, 10, 1066021. [Google Scholar] [CrossRef]
- Specenier, P.; Vermorken, J. Afatinib in squamous cell carcinoma of the head and neck. Expert opinion on pharmacotherapy 2016, 17, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Machiels, J.P.; Haddad, R.I.; Fayette, J.; Licitra, L.F.; Tahara, M.; Vermorken, J.B.; Clement, P.M.; Gauler, T.; Cupissol, D.; Grau, J.J.; et al. Afatinib versus methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): an open-label, randomised phase 3 trial. Lancet Oncol 2015, 16, 583–594. [Google Scholar]
- Kao, H.F.; Liao, B.C.; Huang, Y.L.; Huang, H.C.; Chen, C.N.; Chen, T.C.; Hong, Y.J.; Chan, C.Y.; Chia, J.S.; Hong, R.L. Afatinib and Pembrolizumab for Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma (ALPHA Study): A Phase II Study with Biomarker Analysis. Clinical cancer research : an official journal of the American Association for Cancer Research 2022, 28, 1560–1571. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ahn, M.J.; Chan, A.; Wang, C.H.; Kang, J.H.; Kim, S.B.; Bello, M.; Arora, R.S.; Zhang, Q.; He, X.; et al. Afatinib versus methotrexate as second-line treatment in Asian patients with recurrent or metastatic squamous cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 3): an open-label, randomised phase III trial. Ann Oncol 2019, 30, 1831–1839. [Google Scholar]
- Haddad, R.; Guigay, J.; Keilholz, U.; Clement, P.M.; Fayette, J.; de Souza Viana, L.; Rolland, F.; Cupissol, D.; Geoffrois, L.; Kornek, G.; et al. Afatinib as second-line treatment in patients with recurrent/metastatic squamous cell carcinoma of the head and neck: Subgroup analyses of treatment adherence, safety and mode of afatinib administration in the LUX-Head and Neck 1 trial. Oral Oncol 2019, 97, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Mellor, P.; Furber, L.A.; Nyarko, J.N.; Anderson, D.H. Multiple roles for the p85α isoform in the regulation and function of PI3K signalling and receptor trafficking. The Biochemical journal 2012, 441, 23–37. [Google Scholar] [CrossRef]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov 2013, 3, 761–769. [Google Scholar] [CrossRef]
- Tamburrino, A.; Molinolo, A.A.; Salerno, P.; Chernock, R.D.; Raffeld, M.; Xi, L.; Gutkind, J.S.; Moley, J.F.; Wells, S.A.; Jr Santoro, M. Activation of the mTOR pathway in primary medullary thyroid carcinoma and lymph node metastases. Clin Cancer Res 2012, 18, 3532–3540. [Google Scholar] [CrossRef] [PubMed]
- Hancox U, Cosulich S, Hanson L, Trigwell C, Lenaghan C, Ellston R, Dry H, Crafter C, Barlaam B, Fitzek M.; et al. Inhibition of PI3Kbeta signaling with AZD8186 inhibits growth of PTEN-deficient breast and prostate tumors alone and in combination with docetaxel. Molecular cancer therapeutics 2015, 14, 48–58. [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Poetsch, M.; Lorenz, G.; Kleist, B. Detection of new PTEN/MMAC1 mutations in head and neck squamous cell carcinomas with loss of chromosome 10. Cancer Genet Cytogenet 2002, 132, 20–24. [Google Scholar] [CrossRef]
- Sangale, Z.; Prass, C.; Carlson, A.; Tikishvili, E.; Degrado, J.; Lanchbury, J.; Stone, S. A robust immunohistochemical assay for detecting PTEN expression in human tumors. Appl Immunohistochem Mol Morphol 2011, 19, 173–183. [Google Scholar] [CrossRef]
- Squarize, C.H.; Castilho, R.M.; Abrahao, A.C.; Molinolo, A.; Lingen, M.W.; Gutkind, J.S. PTEN deficiency contributes to the development and progression of head and neck cancer. Neoplasia (New York, NY) 2013, 15, 461–471. [Google Scholar] [CrossRef]
- Psyrri, A.; Seiwert, T.Y.; Jimeno, A. Molecular pathways in head and neck cancer: EGFR, PI3K, and more. American Society of Clinical Oncology educational book American Society of Clinical Oncology Annual Meeting 2013, 246–255. [Google Scholar] [CrossRef]
- Marquard, F.E.; Jucker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochemical pharmacology 2020, 172, 113729. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, F.; Buonaguro, L.; Caponigro, F.; Ionna, F.; Starita, N.; Annunziata, C.; Buonaguro, F.M.; Tornesello, M.L. Update on Head and Neck Cancer: Current Knowledge on Epidemiology, Risk Factors, Molecular Features and Novel Therapies. Oncology 2015, 89, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liao, J.; Schumaker, L.; Carter-Cooper, B.; Lapidus, R.G.; Fan, X.; Gaykalova, D.A.; Mehra, R.; Cullen, K.J.; Dan, H. Simultaneously targeting ErbB family kinases and PI3K in HPV-positive head and neck squamous cell carcinoma. Oral Oncol 2022, 131, 105939. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liao, J.; Carter-Cooper, B.A.; Lapidus, R.G.; Cullen, K.J.; Dan, H. Regulation of cisplatin-resistant head and neck squamous cell carcinoma by the SRC/ETS-1 signaling pathway. BMC Cancer 2019, 19, 485. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.M.; Geng, X.; Bonazzi, V.F.; Ju, R.J.; Mahon, C.E.; Cummings, M.C.; Stephenson, S.A.; Pollock, P.M. PI3K Inhibitors Synergize with FGFR Inhibitors to Enhance Antitumor Responses in FGFR2(mutant) Endometrial Cancers. Mol Cancer Ther 2017, 16, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer research 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, A.; Sánchez, V.; Kuba, M.G.; Rinehart, C.; Arteaga, C.L. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proceedings of the National Academy of Sciences of the United States of America 2012, 109, 2718–2723. [Google Scholar] [CrossRef]
- Meister, K.S.; Godse, N.R.; Khan, N.I.; Hedberg, M.L.; Kemp, C.; Kulkarni, S.; Alvarado, D.; LaVallee, T.; Kim, S.; Grandis, J.R.; et al. HER3 targeting potentiates growth suppressive effects of the PI3K inhibitor BYL719 in pre-clinical models of head and neck squamous cell carcinoma. Scientific reports 2019, 9, 9130. [Google Scholar] [CrossRef]
- Zaryouh, H.; Van Loenhout, J.; Peeters, M.; Vermorken, J.B.; Lardon, F.; Wouters, A. Co-Targeting the EGFR and PI3K/Akt Pathway to Overcome Therapeutic Resistance in Head and Neck Squamous Cell Carcinoma: What about Autophagy? Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Zaryouh, H.; De Pauw, I.; Baysal, H.; Pauwels, P.; Peeters, M.; Vermorken, J.B.; Lardon, F.; Wouters, A. The Role of Akt in Acquired Cetuximab Resistant Head and Neck Squamous Cell Carcinoma: An In Vitro Study on a Novel Combination Strategy. Front Oncol 2021, 11, 697967. [Google Scholar] [CrossRef]
- Mock, A.; Plath, M.; Moratin, J.; Tapken, M.J.; Jäger, D.; Krauss, J.; Fröhling, S.; Hess, J.; Zaoui, K. EGFR and PI3K Pathway Activities Might Guide Drug Repurposing in HPV-Negative Head and Neck Cancers. Front Oncol 2021, 11, 678966. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Wang, Z.; Goto, Y.; Ando, T.; Wu, X.; Zhang, X.; Li, H.; Johnson, D.E.; Grandis, J.R.; Gutkind, J.S. Pathway-Specific Genome Editing of PI3K/mTOR Tumor Suppressor Genes Reveals that PTEN Loss Contributes to Cetuximab Resistance in Head and Neck Cancer. Mol Cancer Ther 2020, 19, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.S.; Garrett, J.T.; Sánchez, V.; Stanford, J.C.; Young, C.; Chakrabarty, A.; Rinehart, C.; Zhang, Y.; Wu, Y.; Greenberger, L.; et al. ErbB3 ablation impairs PI3K/Akt-dependent mammary tumorigenesis. Cancer research 2011, 71, 3941–3951. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Patel, H.; Alanazi, S.; Yuan, L.; Garrett, J.T. HER3 signaling and targeted therapy in cancer. Oncol Rev 2018, 12, 355. [Google Scholar] [CrossRef] [PubMed]
- Garrett, J.T.; Sutton, C.R.; Kurupi, R.; Bialucha, C.U.; Ettenberg, S.A.; Collins, S.D.; Sheng, Q.; Wallweber, J.; Defazio-Eli, L.; Arteaga, C.L. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110α inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer research 2013, 73, 6013–6023. [Google Scholar] [CrossRef] [PubMed]
- Milewska, M.; Cremona, M.; Morgan, C.; O'Shea, J.; Carr, A.; Vellanki, S.H.; Hopkins, A.M.; Toomey, S.; Madden, S.F.; Hennessy, B.T.; et al. Development of a personalized therapeutic strategy for ERBB-gene-mutated cancers. Ther Adv Med Oncol 2018, 10, 1758834017746040. [Google Scholar] [CrossRef] [PubMed]
- Akbari Dilmaghani, N.; Safaroghli-Azar, A.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/Akt/mTORC signaling axis in head and neck squamous cell carcinoma: Possibilities for therapeutic interventions either as single agents or in combination with conventional therapies. IUBMB Life 2021, 73, 618–642. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Kang, H.; Mehra, R. Targeting phosphoinositide 3-kinase (PI3K) in head and neck squamous cell carcinoma (HNSCC). Cancers of the head & neck 2018, 3, 3. [Google Scholar]
- Suh, K.J.; Sung, J.H.; Kim, J.W.; Han, S.H.; Lee, H.S.; Min, A.; Kang, M.H.; Kim, J.E.; Kim, J.W.; Kim, S.H.; et al. EGFR or HER2 inhibition modulates the tumor microenvironment by suppression of PD-L1 and cytokines release. Oncotarget 2017, 8, 63901–63910. [Google Scholar] [CrossRef]
- Fiedler M, Schulz D, Piendl G, Brockhoff G, Eichberger J, Menevse AN, Beckhove P, Hautmann M, Reichert TE, Ettl T.; et al. Buparlisib modulates PD-L1 expression in head and neck squamous cell carcinoma cell lines. Exp Cell Res 2020, 396, 112259. [CrossRef]
Figure 1.
Afatinib more effectively inhibited cell proliferation in HPV-negative HNSCC compared to Gefitinib and Erlotinib. A and B. Cal27 (A) and FaDu (B) cells were treated with DMSO or increasing concentrations of Gefitinib, Erlotinib, and Afatinib for 96 hours and cell proliferation was measured by SRB assay and EC50 were determined by GraphPad Prism version 7.04.
Figure 1.
Afatinib more effectively inhibited cell proliferation in HPV-negative HNSCC compared to Gefitinib and Erlotinib. A and B. Cal27 (A) and FaDu (B) cells were treated with DMSO or increasing concentrations of Gefitinib, Erlotinib, and Afatinib for 96 hours and cell proliferation was measured by SRB assay and EC50 were determined by GraphPad Prism version 7.04.
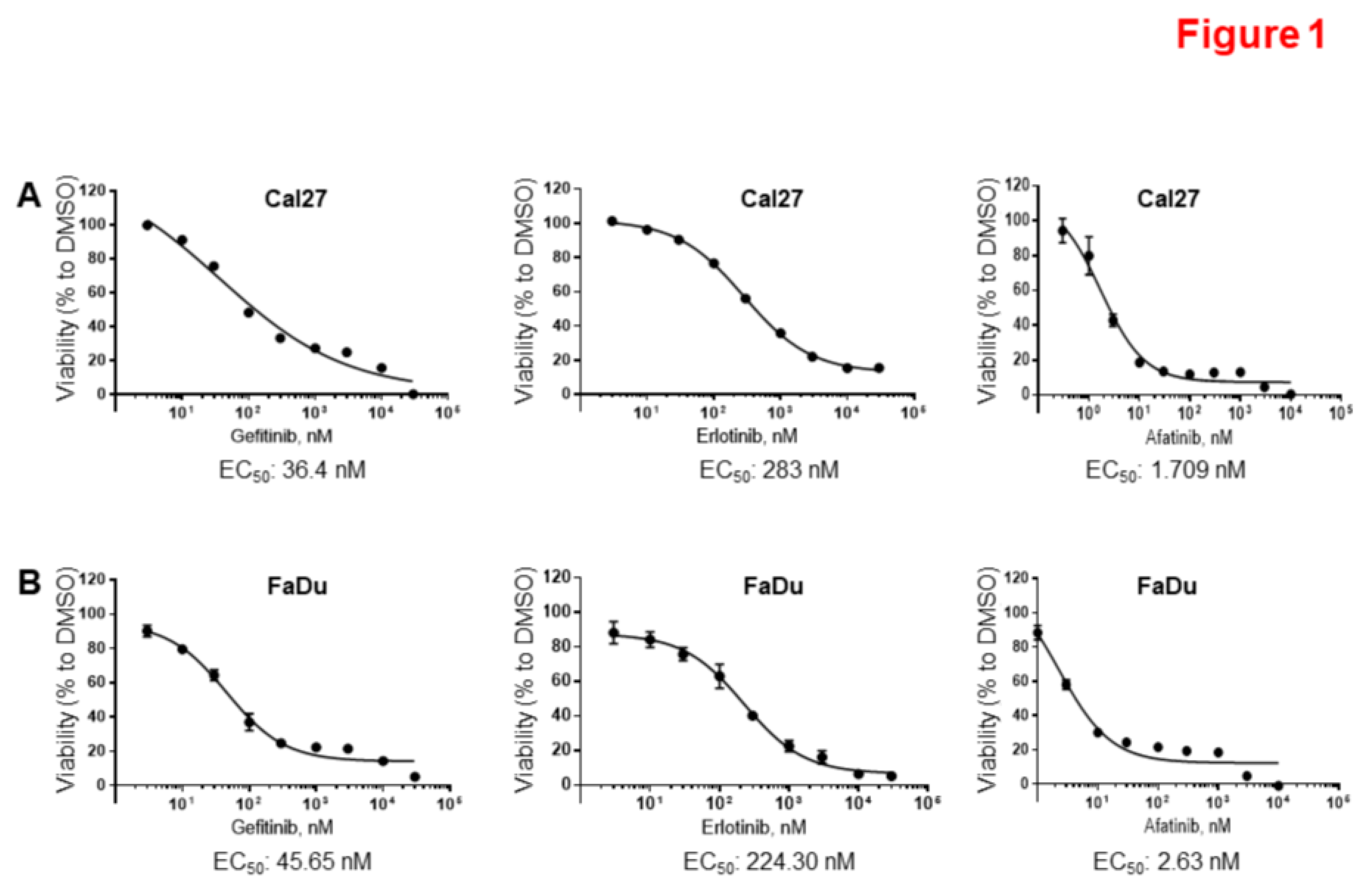
Figure 2.
Copanlisib more effectively inhibited cell proliferation compared to other PI3K inhibitors in Cal27 cells. A. Cal27 cells were treated with DMSO or increasing concentrations of Copanlisib and other PI3K inhibitors for 96 hours and cell proliferation was measured by SRB assay. The growth curves are shown. The experiments were performed in triplicate. The associated EC50 to the 6 inhibitors were determined by GraphPad Prism version 7.04. B. EC50 to six PI3k inhibitors in Cal27 and FaDu cells were listed.
Figure 2.
Copanlisib more effectively inhibited cell proliferation compared to other PI3K inhibitors in Cal27 cells. A. Cal27 cells were treated with DMSO or increasing concentrations of Copanlisib and other PI3K inhibitors for 96 hours and cell proliferation was measured by SRB assay. The growth curves are shown. The experiments were performed in triplicate. The associated EC50 to the 6 inhibitors were determined by GraphPad Prism version 7.04. B. EC50 to six PI3k inhibitors in Cal27 and FaDu cells were listed.
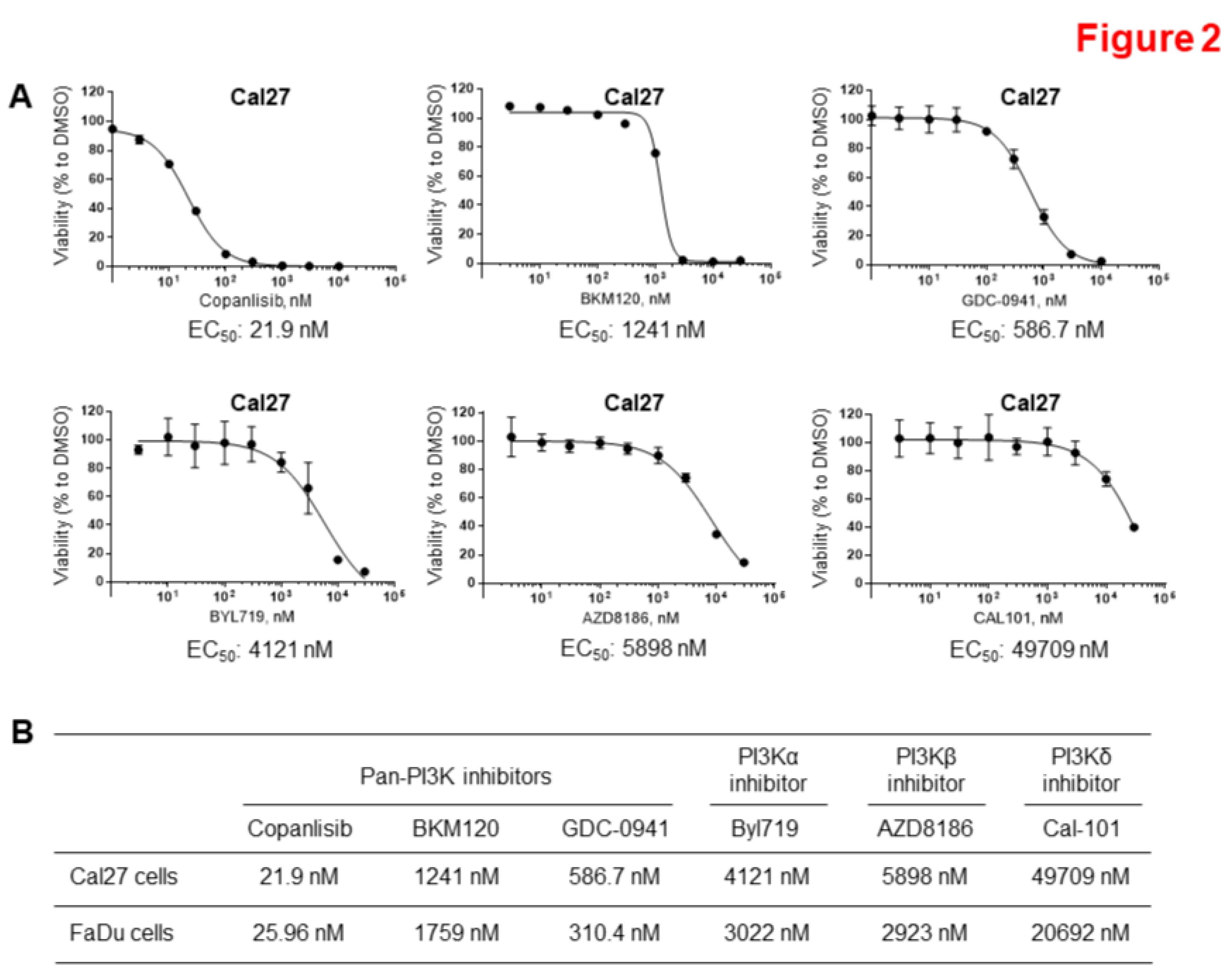
Figure 3.
Synergistic inhibition of cell proliferation by combination of Afatinib and Copanlisib in vitro. A and C. Cal27 (A) and FaDu (C) cells were treated with different concentrations of Afatinib, Copanlisib, or their combinations for 96 hours and cell proliferation was measured by SRB assay. The experiments were performed in triplicate. B and D. The combination index values (CI values) for different combinations were determined using CalcuSyn, Version 2.0 (C and D).
Figure 3.
Synergistic inhibition of cell proliferation by combination of Afatinib and Copanlisib in vitro. A and C. Cal27 (A) and FaDu (C) cells were treated with different concentrations of Afatinib, Copanlisib, or their combinations for 96 hours and cell proliferation was measured by SRB assay. The experiments were performed in triplicate. B and D. The combination index values (CI values) for different combinations were determined using CalcuSyn, Version 2.0 (C and D).
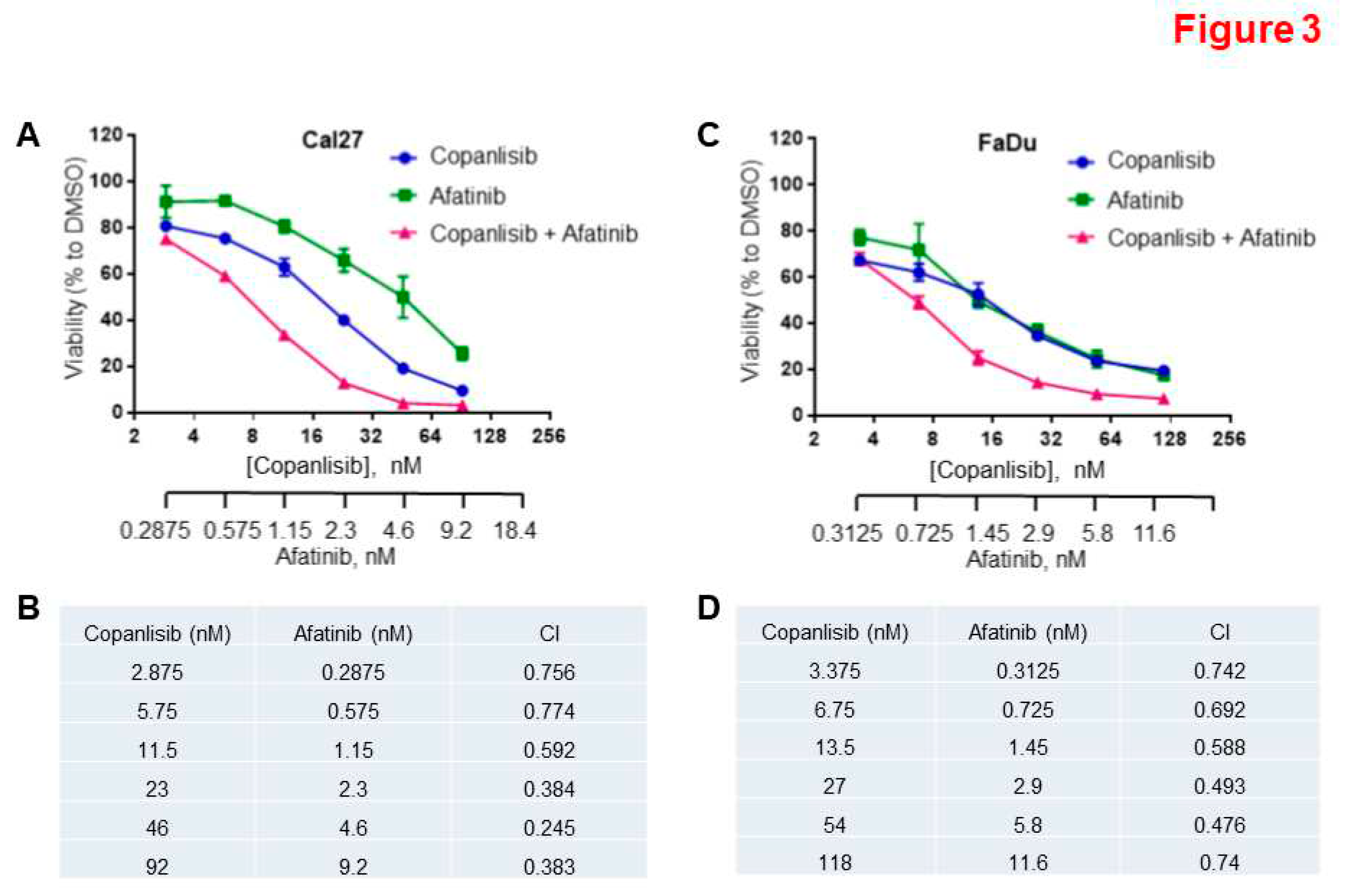
Figure 4.
Inhibition of HNSCC growth by combination of Afatinib and Copanlisib in vivo. A, B, and C. FaDu cells were inoculated into mice. When tumors reached approximately 200 mm3, mice were randomized to one of four treatment groups (7 mice/group): vehicle control, Copanlisib (6 mg/kg, IP), Afatinib (6 mg/kg, PO), or a combination of Copanlisib and Afatinib. The treatments were performed for 32 days. The xenograft tumor volumes (A), final average weights of tumors (B), and average body weights of mice (C) in each group were compared. (*P<0.05, **P<0.01).
Figure 4.
Inhibition of HNSCC growth by combination of Afatinib and Copanlisib in vivo. A, B, and C. FaDu cells were inoculated into mice. When tumors reached approximately 200 mm3, mice were randomized to one of four treatment groups (7 mice/group): vehicle control, Copanlisib (6 mg/kg, IP), Afatinib (6 mg/kg, PO), or a combination of Copanlisib and Afatinib. The treatments were performed for 32 days. The xenograft tumor volumes (A), final average weights of tumors (B), and average body weights of mice (C) in each group were compared. (*P<0.05, **P<0.01).
Figure 5.
A combination of Afatinib and Copanlisib increased cell apoptosis compared to Afatinib or Copanlisib treatment alone. A and B. Cal27CP (A) and FaDu (B) cells were treated with vehicle control, Copanlisib, Afatinib, or a combination for 48 hours, and cell apoptosis was analyzed by Annexin V/propidium iodide staining. The experiments were performed in triplicate, early and late-stage apoptotic, and dead cells were counted, and statistical analysis was performed. P values < 0.05 were considered to be statistically significant. Note: in A, (*P<0.05, **P<0.01, ***P<0.0001). .
Figure 5.
A combination of Afatinib and Copanlisib increased cell apoptosis compared to Afatinib or Copanlisib treatment alone. A and B. Cal27CP (A) and FaDu (B) cells were treated with vehicle control, Copanlisib, Afatinib, or a combination for 48 hours, and cell apoptosis was analyzed by Annexin V/propidium iodide staining. The experiments were performed in triplicate, early and late-stage apoptotic, and dead cells were counted, and statistical analysis was performed. P values < 0.05 were considered to be statistically significant. Note: in A, (*P<0.05, **P<0.01, ***P<0.0001). .
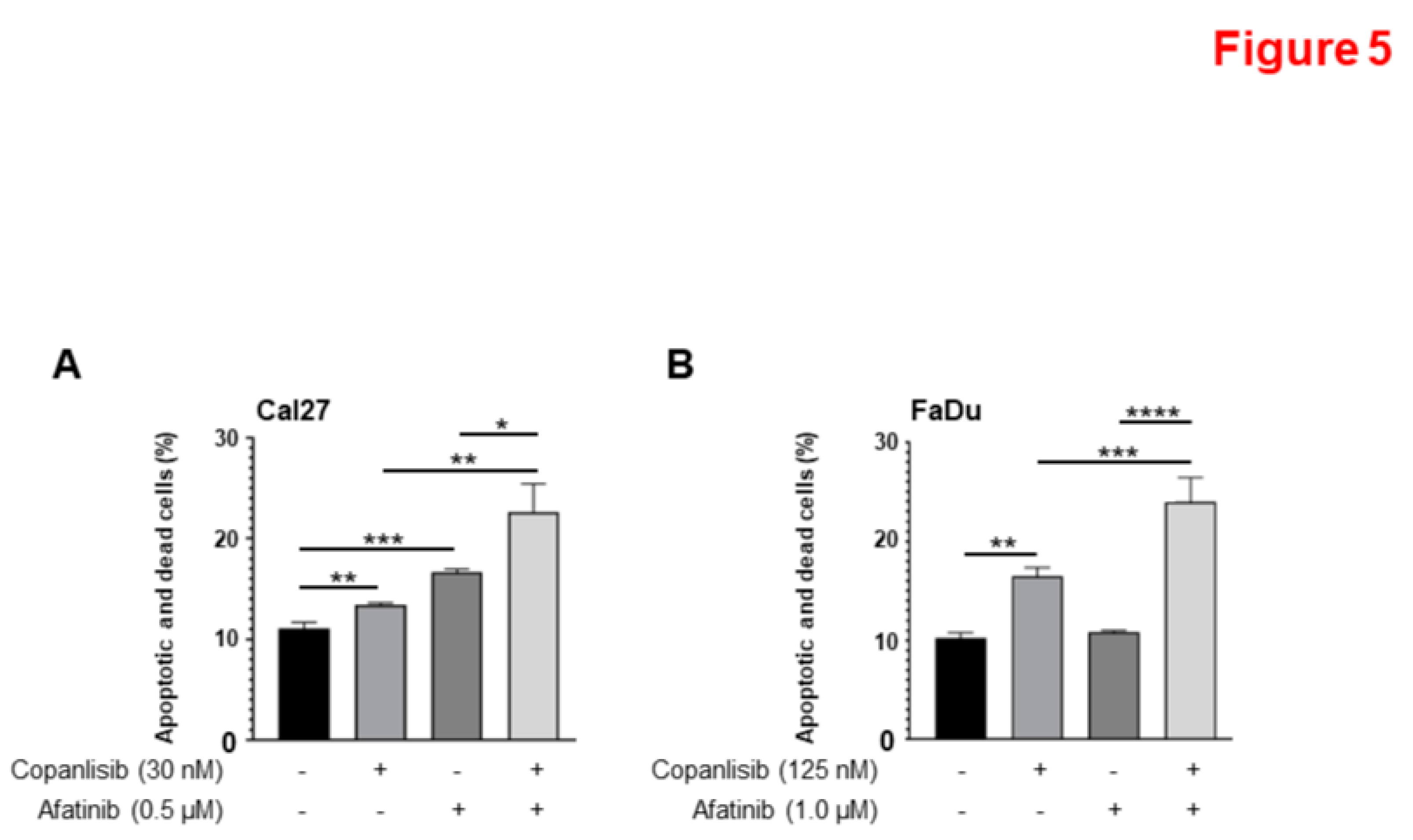
Figure 6.
Inhibition of both ErbB and PI3K/Akt pathways and induced caspase cleavage by combination of Afatinib and Copanlisib. A-B. Combination of Copanlisb and Afatinib effectively blocks the phosphorylation of HER2, HER3 and Akt and induces more caspase 3 cleavage. Cal27 (A), and FaDu (B) cells were treated with increasing concentrations of Copanlisib, Afatinib (0.5 μM), or their combination for 24 hours before lysed. The indicated proteins were detected by Western blot analysis.
Figure 6.
Inhibition of both ErbB and PI3K/Akt pathways and induced caspase cleavage by combination of Afatinib and Copanlisib. A-B. Combination of Copanlisb and Afatinib effectively blocks the phosphorylation of HER2, HER3 and Akt and induces more caspase 3 cleavage. Cal27 (A), and FaDu (B) cells were treated with increasing concentrations of Copanlisib, Afatinib (0.5 μM), or their combination for 24 hours before lysed. The indicated proteins were detected by Western blot analysis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



