
Submitted:
08 February 2024
Posted:
09 February 2024
You are already at the latest version
Abstract
Heterometallic rare-earth clusters have garnered much attention because of their interest-ing solid-state structures and versatile applications in catalysis and material and polymer chemistry over the last four decades. Particular interest is rare earth–transition metal co-ordination compounds that behave as single-molecule magnets (SMMs) because of their potential use in information storage, spintronic devices, and magnetic refrigeration sys-tems. Another implementation involves using lanthanide clusters in luminescent-based sensing for environmental protection and security screening or in photocatalytic energy conversion reactions. Heterometallic rare earth complexes offer the promise of catalytic performance of many organic reactions due to their high oxygenophilicity, Lewis acidity, high coordination numbers, and ability to change the coordination environment quickly. Therefore, they are commonly applied as catalysts in asymmetric synthesis, oxidation re-actions, and CO2 conversions or as initiators in ring-opening polymerization and copoly-merization of cyclic monomers. They are also attractive single-source molecular precur-sors for functional inorganic materials. This review focused on the unique features and properties of the heterometallic rare earth complexes, which are closely correlated with their molecular structure. Our summary is a valuable resource, providing researchers with an insightful exploration of the heterometallic rare-earth complexes that demonstrated multimetallic synergy and cooperativity effects in various applications.
Keywords:
heterometallic complexes
; rare-earth elements
; magnetism
; catalysis
; CO2
; energy conversion
; nanomaterials
1. Introduction
Rare-earth elements (REs), named for their scarcity in Earth’s crust, are the group of seventeen metals containing scandium, yttrium, and fifteen lanthanides. Natural RE deposits are infrequent, and separating compounds containing these elements is challenging due to their chemical similarities. Despite the nomenclature suggesting rareness, many of them are more abundant in the Earth’s upper crust than tin, molybdenum, silver, mercury, gold, or platinum. Remarkably, cerium, the most abundant rare-earth element, surpasses lead and copper in commonality. A notable observation is the greater abundance of rare-earth elements with even atomic numbers compared to those with off values, a phenomenon recognized as the Oddo-Harkins rule [1,2,3].
It is essential to highlight that promethium does not naturally occur due to the short half-lives of its isotopes. The elements from cerium to lutetium are collectively known as the 4f elements, reflecting the occupation of the 4f subshell. Yttrium and lanthanum are categorized under group 3 in the periodic table. Scandium stands apart from other rare-earth elements and is often regarded as a ferromagnesian trace element.
On the contrary, despite its atomic number and mass differences, yttrium exhibits numerous physical and chemical resemblances to the lanthanides, particularly those commonly categorized as heavy rare-earth elements (Gd through Lu). One noteworthy similarity lies in the size of their REIII cation radii, especially notable for YIII and HoIII. That and similar oxidation are stated to provide rare-earth metals with the capacity to substitute for one another within crystal lattices. Additionally, differences in cation radii lead to natural deposits usually being enriched in either heavy REs or light REs (La through Eu).
Rare-earth elements and their various derivatives exhibit numerous similarities [1,4], including high electrical conductivity, high coordination numbers, preferably binding with strongly electronegative elements, tarnishing of their surface when exposed to air (especially in humid conditions), and large stability of their oxides.
In the chemistry of rare-earths elements, a series of characteristics distinguish them from transition metals. Unlike the d-block elements, they do not form stable carbonyl complexes and exhibit almost no activity for the oxidation state of 0. The reactivity of these elements is more akin to alkaline earth metals. They commonly adopt the +3 oxidation state, especially in aqueous solutions. Some elements also exhibit the +2 oxidation state (e.g. Nd, Sm, Eu) or +4 oxidation state (e.g. Ce, Pr, Tb) to achieve a more stable electron configuration. Due to the presence of the 4f subshell, RE ions exhibit coordination numbers in the range of 7-12. Due to the predominantly electrostatic nature of 4f–ligand interactions, the coordination geometry of LnIII complexes is determined by repulsive interactions and steric effects. The 4f orbitals in the LnIII ion do not directly participate in bonding as the 5s2 and 5p6 orbitals will shield them. The general notion is that the lanthanides form predominantly ionic bonds, at least in their +3 oxidation state. The interconnection between the orbital overlap and energy-degeneracy-driven covalency and the subsequent impact on bond stability for lanthanide compounds is poorly understood [5]. Rare-earth ions are hard Lewis acids and readily form complexes with hard bases (strongly electron donating ligands) such as F-, Cl-, O2-, RO-, RCOO-, H2O. Due to their high hydration energy, REIII ions easily form aqua complexes [RE(H2O)9]3+ pictured in Figure 1 [6].
Though employed in relatively modest amounts, REs boast an extensive array of both current and potential applications. Their versatility is particularly pronounced in catalysis [7] and digital technologies [8], contributing to advancements in magnets [9], rechargeable batteries [10], lasers [11], coatings [12], phosphors [13], optical sensors [14], and even nuclear reactors [15]. Notably, certain RE ions possess the unique ability to emit visible luminescence.
One of the most cost-effective and straightforward methods for enhancing alloy properties involves microalloying and modification with rare-earth elements. Even a small addition of lanthanides or yttrium can significantly enhance the physicochemical characteristics of alloys by altering their microstructure. For instance, REs alloys with magnesium stand out as the most promising high-performance materials [16,17]. Unlimited solubility of REs in molten iron leads to an increase in molten iron viscosity and a reduction of surface tension [18]. Other examples include mischmetals or samarium-cobalt and neodymium-iron magnets, with the last ones being the strongest type of permanent magnet available commercially.
The majority of rare-earth oxides in nature exist in the form of sesquioxides, following the formula RE2O3. However, there are exceptions with Ce, Pr, and Tb. Cerium naturally forms CeO2, praseodymium oxide is Pr6O11, and terbium forms Tb4O7. Despite being non-sesquioxides, these compounds can, under specific conditions, adopt structures isostructural to other REs. Within catalysis, they stand out as highly active and selective catalysts, facilitating processes such as the oxidation coupling of CH4 to higher hydrocarbons [19], isomerization of 1-butene [20], acetone aldol addition [21], hydrogenation of 1,3-butadiene [22], and the dehydration of alkanediols into unsaturated alcohols [23]. The distinctive magnetic and optical properties of glasses derived from rare-earth metal oxides position them as ideal candidates for applications in optics and electronics. They have also been incorporated into electrochemical sensors for detecting humidity, toxins, drugs, and gases.
Rare-earth complexes are used in many practical applications as molecular probes, contrast agents, catalysts, molecular magnetic or luminescence materials, dopants, and modifiers for optical materials.
For this work, in the following sections, we discussed various methods for the synthesis of heterometallic rare earth compounds, then we described their magnetic properties, and we made a concise description of their catalytic applications in organic synthesis, polymerization of cyclic monomers or photocatalytic energy conversion processes. The last paragraph describes the use of heterometallic complexes as molecular precursors for synthesizing functional nanomaterials. Because luminescent properties were described thoroughly before [24,25,26], we have not discussed them in this review.
2. Heteromatallic rare-earth complexes, synthesis and applications
2.1. Synthesis routes
Usually, the synthesis of heterometallic transition metal-rare-earth complexes is performed by dissolution of RE(NO)3 and transition metal reagents (chlorides, nitrates, acetates) in the water-alcohol mixture as solvent, using amines to deprotonate the phenolic ligands. These reactions usually lead to thermodynamically stable crystalline products with molecular structure mostly depending on the reaction conditions, used reagents, or crystallization procedure. The more controlled synthesis method was used to prepare group 4 – rare earth oxo alkoxides by substituting the halogen atoms in RECl3 by K[Zr2(OiPr)9] in THF solution, leading to the formation of [REZr2(OiPr)9Cl2]x (REIII = YIII, NdIII, HoIII, CeIII for x = 2; ErIII, YbIII for x = 1) [27]. Alternative method claims the synthesis of titanium-lanthanide oxo clusters pictured in Figure 2, i.e., [RETi4(O)3(OiPr)2(OMc)11] (RE = La, Ce; OMc = methacrylate), [RE2Ti6O6(OMc)18(HOiPr)] (RE = La, Ce, Nd, Sm) and [RE2Ti4O4(OMc)14(HOMc)2] (RE = Sm, Eu, Gd, Ho) by the reaction of titanium isopropoxide, lanthanide acetate and methacrylic acid [28].
The direct route for the rare-earth complexes from the pure metals has not been considered as attractive because it requires activation by Hg, HgCl2, or I2 and prolonged heating [29].
Recently Sobota and coworkers reported the synthesis of heterometallic oxo-alkoxides by the reaction of metallic lanthanides with divalent transition metal chlorides MCl2 (MII = MnII, NiII or CoII) or group 4 metallocene dichlorides Cp2MCl2 (where MIV = TiIV, ZrIV, HfIV), using 2-methoxy ethanol or ethanol as solvents and reagents [30,31]. The reaction of RECl3 with the molar excess of alkaline or alkaline earth ligand precursors is used as the main method for synthesizing heterometallic rare earth – group 1/group 2 complexes [32,33].
Employing precursors with well-defined structures in synthesizing heterometallic complexes minimizes the risk of side reactions or the formation of multiple reaction products, ensuring a more controlled and selective outcome that enhances the synthesis efficiency and selectivity. The structurally authenticated precursors act as templates, guiding the coordination of specific ligands to metal sites to yield a predetermined complex. The advantages of this methodology are particularly evident in the enhanced reproducibility of heterometallic complexes and the ability to scale up the synthesis for practical applications. Moreover, the structurally authenticated precursors enable a deeper understanding of the reaction mechanism, facilitating the design of more sophisticated and tailored heterometallic rare-earth complexes with desired properties for diverse catalytic and material applications. For example, trinuclear ionic zinc compound based on [Zn3(OAc)2(L)2(H2O)2]2+ cation (where LH = 2-ethoxy-6-((pyridin-2-ylmethylimino)methyl)phenol)) [34] could be used as attractive molecular platform for the synthesis of a wide range heterometallic ZnIIYbIII clusters by the reaction with various ytterbium salts as shown in Figure 3.
The use of Yb(ClO4)3 led to the [Zn2Yb(L)2(OAc)4]ClO4, and Yb(NO3)3 led to [ZnYb(L)2(NO3)3]. The treatment of trinuclear zinc cation with Yb(OAc)3 allows the formation of [ZnYb(OAc)4(L)], but the reaction with YbCl3 led to the [Yb2(L)2(OAc)(H2O)Cl]2Cl4. The reversed sequence was applied in the reaction of [RE(L)(THF)n] (where REIII = YIII, SmIII, NdIII, LaIII; n == 1, 2) with Co(OAc)2 that as the results of ligand redistribution lead to the formation of [Co2RE2(OAc)4(L)2(THF)n] containing acetato bridged REIII dimers capped by CoII trisphenolato units, as seen in Figure 4 below [35].
2.2. Magnetism
Coordination compounds that behave as single-molecule magnets (SMMs), which exhibit slow magnetization reversal and distinct hysteresis in the absence of long-range magnetic order, are of particular interest because of their potential applications in high-density information storage [36]. Lanthanide complexes show great potential for SMMs because of their strong magnetic anisotropy and large-spin ground states, of which the best candidates are TbIII, DyIII, and HoIII. However, their combination with 3d metal ions is essential to suppress quantum tunneling effectively and establish a magnetic exchange between metal ions (3d–4f) [37,38,39]. The combination of different paramagnetic metal ions within the same molecular entity leads to a wide variety of magnetic properties of the heterometallic compounds. 3d–4f complexes have been intensively studied in order to reveal the factors governing the nature and magnitude of the exchange interaction between a 3d metal ion and various 4f metal ions. The comparison of magnetic properties of the series of [M3Ln2(opba)3] (for MII = CuII, NiII; opba4- = ortho-phenylenebis(oxamato)) based on a ladder-type structure with isostructural zinc compounds allow to determine the nature of interactions in LnIII−MII pairs. This led to the conclusion that the interaction is ferromagnetic for GdIII, TbIII, and DyIII and seems to be antiferromagnetic for CeIII, PrIII, NdIII, and ErIII [40,41]. For the series of trinuclear complexes of general formula [Cu2Ln(L)2(NO3)2]+ for (LH2 = 2,6-di(acetoacetyl)pyridine), the presence of an antiferromagnetic interaction for LnIII = CeIII, PrIII, NdIII and SmIII, and ferromagnetic for LnIII = GdIII, TbIII, DyIII, HoIII, and ErIII was observed [42]. The structures of those complexes can be found in Figure 5.
The nature of the NiII−LnIII exchange interaction within binuclear [NiLn(valpn)(CH3CN)2(NO3)3] (for LnIII = PrIII, NdIII, SmIII, EuIII, GdIII, ErIII) found in Figure 6, [NiLn(valpn)(CH3CN)(H2O)4(NO3)](NO3)2 (for LnIII = TbIII, DyIII) and [NiCe(valpn)(CH3CN)2(H2O)(NO3)3]·[NiCe(valpn)(CH3CN)(H2O)2(NO3)2](NO3) (for valpn2- = 1,3-propanediylbis(2-iminomethylene-6-methoxy-phenolato)) has been emphasized by comparison with the cryomagnetic behavior of the related [ZnIILnIII] derivatives. This route allowed for establishing that the interaction within these compounds is antiferromagnetic with the 4f ions of the beginning of the Ln series (CeIII – EuIII) and turns ferromagnetic from GdIII to ErIII. The [NiIIDyIII] complex shows slow relaxation processes of the magnetization close to 2 K [43].
For the heterodinuclear [CuLn(L)(NO3)3] (Figure 7) complexes (LnIII = CeIII−YbIII; LH2 = N,N‘-ethylenebis(3-ethoxysalicylaldiimine)), ferromagnetic interactions seem to be exhibited for the GdIII, TbIII, DyIII, HoIII, TmIII, and YbIII, while for CeIII, NdIII, and SmIII the interaction was antiferromagnetic. PrIII and EuIII analogs behaved as spin-uncorrelated systems, and no definite conclusions were reached for the ErIII complex [44]. Within the series of [CuLn(L)(Me2CO)(NO3)3] complexes pictured in Figure 8, the nature of the coupling between the CuII–LnIII ions was antiferromagnetic for CeIII, NdIII, SmIII, TmIII, and YbIII, and ferromagnetic for GdIII, TbIII, DyIII, HoIII, and ErIII, with CuII–Pr/EuIII pairs devoid of any significant interaction [45].
The first example of a 3d–4f SMM was [CuTb(hfac)2(L)]2 (LH3 = 1-(2-hydroxybenzamido)-2-(2-hydroxy-3-methoxy-benzylideneamino)-ethane; hfacH = hexafluoroacetylacetone), which has an effective energy barrier of 21 K, a relaxation time of 2.7 ×10–8 s, and an estimated blocking temperature of 1.2 K [46]. Since this discovery, combinations of TbIII , DyIII, ErIII, SmIII, YbIII, GdIII, or HoIII with transition metal ions such as CoII, MnII/III, FeII/III, NiII, and CuII have been extensively studied. SMM behavior has also been observed for TbIII and DyIII complexes within the series of trimetallic [CuLn(L)(C3H6O)(NO3)3] (where Ln = GdIII, TbIII, DyIII, HoIII, ErIII; L2- = 2,2'-((2,2-dimethylpropane-1,3-diyl)bis((nitrilo)methylylidene))bis(6-methoxyphenolato))) with activation energies of magnetization reversal equal to 42.3(4) and 11.5(10) K, respectively. The magnetic exchange couplings in CuII−LnIII display a monotonic decrease of ferromagnetic JLn–Cu in the order of the atomic number, from 64Gd to 68Er (JLn–CukB−1/K = 6.9 (Gd), ≥ 3.3 (Tb), 1.63(1) (Dy), 1.09(2) (Ho), 0.24(1) (Er)) [47].
The family of hetero-tri-metallic complexes [{CuTb(L)(H2O)4}{M(CN)6}]n (for MIII = CoIII, CrIII, FeIII; L2- = 2,2'-(propane-1,3-diylbis(nitrilomethylylidene))bis(6-methoxyphenolato), 2,2'-(ethane-1,2-diylbis(nitrilomethylylidene))bis(6-methoxyphenolato); n = 1, 2, ∞) are interesting examples that illustrate how SMM behavior of the [CuTb(L)]3+ moiety can be modulated via the control of intermolecular interactions with [M(CN)6]3− species [48].
When [Cr/Fe(CN)6]3− are used, weak antiferromagnetic interactions are responsible for the decrease of the SMM efficiency. The combination of the diamagnetic [Co(CN)6]3− with a [CuTb(L)]3+ moiety resulted in an improvement of the SMM properties compared to the reference [CuTb] complex, with a significantly longer relaxation time. The increase of the effective anisotropic barriers (Ueff) from 5–7 cm−1 present for the [{CuTb}Cr/Fe] compounds to 15–18 cm−1 for [{CuTb}Co] was also observed [49].
The heterometallic [MnLn2(QCl)8] (Ln = DyIII, TbIII; QCl- = 5-chloro-8-quinolinolate) display clearly resolved out-of-phase susceptibility maxima below 10 K originating from SMM behavior. Both of them are interesting examples demonstrating that the co-complexation of [Bu4N][Ln(QCl)4] with Mn(NO3)2 led to a structural transformation from mononuclear to trinuclear compounds enhancing single molecule magnetic (SMM) behavior. Molecular structure of [MnLn2(QCl)8] is presented in Figure 9 [50].
Heterometallic NiIIDyIII complexes generally exhibit field-induced single-molecule magnet behavior with an effective energy barrier of 6.6 - 16.9 K for reversal of the magnetization, i.e., [NiDy(valpn)(hfac)2(N3)(H2O)2] [51], [NiDy(L)(ArCOO)(NO3)2] (LH2 = N,N′,N″-trimethyl-N,N″-bis(2-hydroxy-3-methoxy-5-methylbenzyl)diethylenetriamine); Ar = benzyl, 9-anthracenyl) [52], [Ni2Dy2(CH3COO)3(HL)4(H2O)2](NO3)3 (LH-=2-methoxy-6-[(E)-2′-hydroxymethyl-phenyliminomethyl]-phenolate) [53], [Ni2Dy2(μ4-CO3)2(3-MeOsaltn)2(H2O)2(NO3)2] [54] (3-MeOsaltn- = N,N′-bis(3-methoxy-2-oxybenzylidene)-1,3-propanediaminato), [Ni2Dy2(L)4(NO3)2(MeOH)2] [55] (L2- = 2-(((2-hydroxyphenyl)imino)methyl)-6-methoxyphenolato), [R3NH]2[Ni2Dy2(μ3-OH)2(tBuCOO)10] [56] and many others [57,58,59]. Chosen NiIIDyIII complexes are presented in Figure 10.
The compounds [Zn2Ln(L)2(NO3)(SCN)2] (where LnIII = CeIII, NdIII; L2- = 2,2'-(ethane-1,2-diylbis(nitrilomethylylidene))bis(6-methoxyphenolato)) based on a linear ZnII–LnIII–ZnII motif with an axially stressed ligand field demonstrated the appearance of field-induced SMM behavior, which was correlated with the even-numbered Jz sublevels of Ce(III) and NdIII ions known as the Kramers system [60]. Field-induced SMM with an estimated Ueff barrier of 59K was [Zn2Dy(L)(NO3)3(OH)], despite the fact that mononuclear [Dy(H3L)(H2O)(NO3)](NO3)2 do not show slow relaxation of the magnetization [61]. An uncommon example of heterometallic clusters is [Zn2Dy4(HL)4(o-vanillin)2(OH)4(CH3OH)2](NO3)2] that shows typical ferromagnetic single molecule magnetic behavior with a slow zero-field relaxation [62].
In the first bifunctional SMM [ZnDy(NO3)3(L)(H2O)] (where LH2 = N,N’-bis(3-methoxysalicylidene)-1,2-diaminoethane), pictured in the Figure 11, diamagnetic zinc cation provides the compound with two significant profits: it increases the negative charge of the phenolates and it elevates rare-earth cations’ crystal field splitting without the suppression of the DyIII emission in the visible spectral window [63,64].
2.3. Catalysis, ring-opening polymerization and copolymerization of cyclic esters
Rare earth complexes offer the promise of catalytic performance of many organic reactions due to their high coordination numbers as well as their ability to change the coordination environment quickly. The catalytic applications of heterometallic rare earth complexes are mainly related to Shibasaki catalysts of the formula [M3RE(binol)3] (where MI is alkali metal ion, binol2- - binaphtholato as shown in Figure 12). Since the beginning of the 1990s, Shibasaki catalysts have been mainly used in asymmetric synthesis, and in particular in the Michael addition, [65] aldol [66]/nitro-aldol [67] condensation, cyclopropanation of enons, [68] aldol–Tishchenko reaction, [69] cyanation of aldehydes, [70] reduction of 1,4-benzoquinones, [71] Mannich reaction, [72] protonation, [73] Diels-Alder reaction, [74] hydrophosphonation of imines and aldehydes [75].
In asymmetric synthesis, the increase in enantioselectivity was mainly dependent on the type of MI, when the influence of the REIII was much lower. The effectiveness of these catalysts is due to their ability to behave as both a Lewis acid via REIII ions to activate electrophiles and Brönsted base via Mbinol to activate pronucleophiles, which is analogous to metalloenzyme reactions. The synergic action of the two metal centers allows for transformations that were not possible with conventional catalysts.
Schelter published the ethylzinc Shibasaki catalyst analogs [(EtZn)3RE(binol)3 (THF)3][76] (where RE = LaIII, PrIII, and EuIII) presented in Figure 13, which were used as catalysts for the enantioselective addition of ethyl groups to benzaldehyde in toluene at room temperature (yields 95% (LaIII), 89% (PrIII), 99% (EuIII). The greatest control over the process (ee = 70%) was achieved using an equimolar mixture of ZnII3EuIII complex, ZnEt2, and triphenylphosphine oxide.
Dual activity of [NiRE(L)3] [77] (where RE = LuIII, YIII, LaIII, L- = (iPr2PCH2NPh)-) was discovered in the hydrogenation of diphenylacetylene (DPA) to (E)-stilbene (4.6 atm H2 at 70 °C, 2.5 mol % of cat.). In the presence of DPA, [NiRE(L)3] compounds catalyze the production of (Z)-stilbene (99-98% conversion after 24 h). In the absence of DPA they are responsible for the cis to trans isomerization of stilbene, with trans:cis ratio: >99:1 (for LuIII and YIII) or 20:80 (for LaIII)), as presented in Figure 14. In performed reactions, electron-rich NiII centers were engaged in alkyne binding, while REIII ions provide an open, intramolecular coordination site to favor the hemilability of the phosphine ligand.
Another example was bimetallic rare-earth coordination polymers of formula {[Fe7RE(Hpmida)6]·2H2O}n [78] (where REIII = EuIII, DyIII, HoIII, YIII; H4pmida = N-(phosphonomethyl)iminodiacetic acid), which were useful as highly selective (95-98%) heterogenous catalysts for Knöevenagel condensation, an important C-C coupling reaction widely used for the synthesis of fine chemicals and pharmaceuticals. Depending on the reactants chosen, the conversion reached 20% to 57% (27 h, toluene, 60 °C), with the most active europium and dysprosium. In the experiments performed, it turned out that the presence of REIII improves the course of the reaction and significantly increases the conversion (from non-catalyzed 7-8%).
Heterometallic rare-earth complexes may also be used as initiators in the ring-opening polymerization (ROP) of cyclic esters or their copolymerization (ROCOP) with cyclic epoxides and anhydrides. ROP of cyclic esters initiated by metal catalysts permits the preparation of well-defined polyesters with strictly controlled molecular weight, dispersity, chain microstructure, and tacticity [79]. ROP and ROCOP initiators featuring f-block metals have been developed to take advantage of their Lewis acidity and large coordination spheres. The impact of REIII in monomer activation usually was observed for the alkali metal-rare earth initiators.
In the ε-caprolactone (ε-CL) polymerization studies initiated by [Na8Sm2(OCH2CH2NMe2)12(OH)2], [K12Na4Sm2(OCH2CH2NMe2)18(OH)4] and [K20RE4(OCH2CH2NMe2)26(OH)6] (where REIII = SmIII, NdIII, PrIII, YbIII) it has been shown that the interaction of heterobimetallic centers (Figure 15) allows for achieving within 1 min from 76 to 100% conversion of monomer at stoichiometry of ε-CL/I = 6.000 - 15.000, while homometallic alkoxides were inactive in the study process [80].
Another example is [LiY(C5Me4SiMe2NCH2CH2X)2] [81] (where X = OMe, NMe2), which in the ROP of ε-CL in toluene at room temperature reached monomer conversion of 75-94% in just 90 minutes. Poly(ε-caprolactone)s obtained were of high molecular weight (Mn > 30 kDa) with the highest values over 170 kDa (for ratio [ε-CL]/[initiator] = 188/1) and dispersity (Đ) < 2.0. Reported complexes provided higher conversion and lower Đ than [Y(N(SiMe3)2)3] for identical reaction conditions. Other examples of heterometallic initiators in ROP of ε-CL are mixed-metal allyl complexes of formula [RE(η3-C3H5)3(μ-C4H8O2)·Mg(η1-C3H5)2(μ-C4H8O2)1.5]n[82] (where REIII = LaIII, YIII). Both compounds are highly effective under mild conditions and reach monomer conversion of 99% (t = 0.3 min) and 61% (t = 1.3 min) with Mn values of 42.1 and 29.2 kDa with Đ ~1.4, respectively. The catalytic activity of both initiators was better than monometallic [Mg(η1-C3H5)2(BDI)(THF)] (BDIH = (2-(2,6-diisopropylphenyl)amino-4-(2,6-diisopropylphenyl)imino-2-pentene; [ε-CL]/[initiator] = 200/1, conversion 92% in 6 min, Mn = 13.6 kDa, Đ = 1.4). The interesting group of heterometallic complexes was obtained using Ferrocene-based tetradentate Schiff base ligands.[83] The key finding of this study was the use of [Ce(phosphen)(THF)2] (where phosphen = 1,10-di(2-tert-butyl-6-diphenylphosphiniminophenoxy)ferrocene) as redox control initiator for ROP of l-lactide (l-LA), which course was dependent on the oxidation state of the CeIII/IV ion [84]. The CeIII complex presented in Figure 16 was active in l-LA polymerization at 0 °C with a reagents stoichiometry of l-LA/Ce = 100/1, and it allowed to achieve 96% monomer conversion over 0.5 h, whereas under these conditions CeIV compound was inactive. In turn, the isostructural YIII compound was used as a model system to determine the influence of the oxidation state of Fe atoms in the ferrocene backbone on the YIII–OR activity in the l-LA polymerization. An initiator containing FeII atoms allowed to achieve 24% conversion of monomer in polymerization performed at a ratio of l-LA/Y = 100 through 1h at 25 ° C, while the complex containing FeIII centers was completely inactive in the studied reaction [85].
Series of heterometallic NiII-REIII complexes (where REIII = CeIII, NdIII, SmIII, EuIII, TbIII, HoIII, TmIII) with the acyclic Salen-type ligand (LH2 = N,N’-bis(3-methoxysalicylidene)ethylene-1,2-diamine), with the structure shown in Figure 17, were used as initiators for ROP of l-LA in bulk at 160 °C for 12 h (for ratio [l-LA][catalyst] = 1000/1). The presence of rare-earth ions was an influential factor because it effectively passivated the catalytic behaviors on the ROP, leading to increased molecular weights of obtained polymers (Mn = 28 961 – 31 555), and improved the polymerization control (Đ = 1.12 – 1.19). For the mentioned complexes, it was discovered that the catalytic activity is relative to the intramolecular NiII-REIII separations without the lanthanide contraction sequence [86].
Two-dimensional coordination polymer (Figure 18) [CuEr(pdc)2(Hpdc)(H2O)4]n [87] (where pdcH3 = 3,5-pyrazole dicarboxylic acid) was used as a heterogeneous catalyst in the cyclopropanation of styrene with ethyldiazoacetate at room temperature in dichloromethane, leading to achieving high diastereoselectivity (84%) at the 12% conversion rate after 24 hours.
A series of heterometallic {[Cu3Ln2(L)6(H2O)6]·10H2O}n [88] (where LnIII = LaIII, GdIII, YbIII, LuIII) coordination polymers were used for catalytic oxidation of olefins and aromatic benzylic substrates using tBuOOH or O2 as oxidants. The series of used compounds showed that their activity increases with the increase in the atomic number within the lanthanide group. The most active in the cyclohexene oxidation reaction (1,2-dichloroethane, tBuOOH (70%), 75 °C, 0.001 mol% [Cu]) was the CuII-LuIII complex (Figure 19), which after 24 h led to the achievement of 60% substrate conversion, while the remaining compounds allowed to achieve conversions of 48% (CuII-LaIII), 52% (CuII-GdIII), and 57% (CuII-YbIII). An identical trend in the reactivity of compounds was also maintained in the styrene oxidation reaction, in which the substrate conversion values were 75% for CuII-LuIII, 64% for CuII-LaIII, 69% for CuII-GdIII, and 73% for CuII-YbIII for the same reaction conditions.
2.4. CO2 conversion
In recent decades, rapid increases in atmospheric carbon dioxide concentrations, primarily driven by population growth, economic development, and energy consumption, have become a global concern. The elevated CO2 levels, mainly from fossil fuel use, contribute to ecological imbalances, including temperature growth, melting snow cover, permafrost thaw, and rising sea levels [89]. Consequently, mitigating CO2 emissions and reducing atmospheric levels have become crucial global objectives to combat climate change. Efforts have been directed toward capturing, utilizing, and storing CO2 to achieve decarbonization and emissions reduction goals. Carbon dioxide offers a broad spectrum of potential applications, ranging from direct uses in oil recovery, food processing, water treatment, fire retardants, coolants, and cleaning agents to chemical conversions into value-added products. CO2 offers an accessible, non-toxic, low-cost, renewable carbon feedstock for producing chemicals, fuels, plastics, and raw materials. However, due to its low reactivity, carbon dioxide conversion needs suitable reagents, catalysts, or high-energy sources.
Most published studies use highly reactive substrates and harsh reaction conditions to overcome the high thermodynamic stability and chemical inertness of CO2. Therefore, molecular catalysis has recently focused on developing more efficient systems that promote CO2 transformation, especially under mild conditions, in order to reduce production costs and energy consumption. Numerous metal-based catalysts, including main group elements, transition elements, and rare-earth elements, have been used for the chemical fixation of CO2 into value-added products. Both homogenous and heterogenous metal catalysts play a crucial role in CO2 conversion reactions, examples of which are shown in Figure 20. The synthesis of cyclic carbonates by cycloaddition of CO2 and epoxides is one of the most studied and significant reactions in green and sustainable chemistry. In this reaction, the Lewis acidic REIII site activates the epoxide molecule towards the nucleophilic attack of the Lewis base (X- = Cl-, Br-, I-), leading to the epoxide’s ring opening.
Then CO2 insertion occurs, forming a carbonate intermediate that undergoes intramolecular ring closure to release the cyclic carbonate. Lanthanum complex [La(L)(THF)] stabilized by tris(phenolato) ligand (L3- = 2,2’-[({2-[{[3,5-di-t-butyl-2-(hydroxy)phenyl]methyl}(methyl)amino]ethyl}azanediyl)bis(methylene)]bis(4,6-di-t-butylphenolato)}) is a rare example of catalyst that enables the cycloaddition of terminal epoxides with CO2 under mild conditions (i.e., 25 °C, 1 bar CO2, 0.3 mol% cat., TBAI 0.6 mol%), leading to cyclic carbonates with a yield of 49–99%, as seen in Figure 21 [90]. Several other studies have shown that synergistic interactions between different metal centers can improve the catalytic activity of the catalyst and the selectivity of this reaction [91]. The commonly accepted mechanism assumes simultaneous activation of epoxide and CO2 on adjacent metal centers.
For example, [ZnLa2(OBn)2(L)2] for L3- = 2,2’-[{[2-(2-oxidoethoxy)ethyl]azanediyl}bis(methylene)]bis(4,6-di-t-butylphenolato), a catalyst presented in Figure 22, showed 2 to 8 times better catalytic activity in the reaction of CO2 and 1,2-epoxyhexane (0.5 mol% cat., 1 mol% TBAB, 25 °C, 24 h, 1 atm CO2) than [La2(L)2(THF)2] or zinc aryloxide [Zn(OBn)2] [92].
Observed enhancement of the catalytic activity of heterometallic [EtZnY(L)(THF)] (L4- = N,N,N’,N'-tetrakis(3,5-di-t-butyl-2-oxybenzyl)ethane-1,2-diamine)), occurs as a result of the presence of ZnII centers, which allows for better delocalization of the electron density in the complex and drastically changes the energy barrier of the ring opening step by decreasing the electrostatic repulsion between yttrium center and bromine anion from the cocatalyst. Therefore, the epoxide conversion (0.2 mol% cat., 0.8 mol% TBAB, 40 °C, 18 h, 1 atm CO2) of 81% for heterometallic catalyst was much better than for [Y(HL)(THF)] amounting to 41% or Zn(OAc)2 16% [93]. Another example of catalysts with a synergistic interaction between REIII and ZnII Lewis acidic sites were [Zn4RE2(μ3-OH)2L4(OAc)2(NO3)2(DMF)2] (for REIII = DyIII, NdIII, TbIII; L2- = N-[(3-methoxy-2-oxidophenyl)methylidene]pyridine-3-carbohydrazonato), which showed higher catalytic activity than equivalent amounts of a ZnII salt, REIII salts, and a ligand mixture of each. The structure of the mentioned complexes can be found in Figure 23 [94].
Excellent catalytic activity during cycloaddition between CO2 and styrene oxide (0.01 mol% cat., 0.8 mol% TBAB, 80 °C, 1 bar CO2) was also reported for a series of heterometallic clusters [Zn2RE2(μ3-OH)2L4(NO3)4] (for REIII = EuIII, TbIII, ErIII, YbIII, NdIII; L- = 2-methoxy-6-(methoxycarbonyl)phenolato), which convert from 88 to 93% of epoxide within 14h.[95] There are also several examples of heterometallic lanthanide−zinc clusters that are less efficient than their homometallic counterparts due to the steric effect of the ligands and crowded coordination environments [96].
A reaction of great interest in the synthesis of cyclic carbonates is the oxidative carboxylation of olefins. However, so far, only a few lanthanide-based MOFs have been investigated as heterogeneous catalysts for this purpose. The application of [Nd2(BIPA-TC)1.5]n, [Eu(H2BIPA-TC)(BIPA-TC)0.5]n or [Tb(H2BIPA-TC)(BIPA-TC)0.5]n as catalysts in the reaction of styrene, tert-butyl hydroperoxide, and CO2, gave 80 – 87% cyclic carbonate after 10 h (0.18 mol % of MOF cat., 1 bar of CO2, 80 °C). The proposed mechanism for this reaction can be found in Figure 24 below [97].
Much attention has been paid to the construction of C–N bonds through CO2 fixation, but these reactions require high temperatures and pressures and the use of equivalent amounts of base. The bis(amidate) lanthanide complex (Figure 25) [Ln2L2(N(SiMe3)2)2(THF)2] (LnIII = EuIII, YbIII; L- = N-(2,6-diisopropylphenyl)benzenecarboximidato) showed good catalytic performance in the synthesis of 2,4-quinazolidinones from CO2 and 2-aminobenzonitriles (5 mol % cat., 5mol% DBU, 1 bar of CO2, 100 °C, 24h) leading to final product with a yield of 61 or 91% [98].
Other rare earth amides [RE2L2(N(SiMe3)2)2(THF)2] (REIII = LaIII, NdIII, YIII; LH2 = N, N’-(cyclohexane-1,2-diyl)bis(4-tert-butylbenzamide) turned out to be effective catalysts for the direct carboxylation of terminal alkynes at ambient pressure, leading to the formation of the C–C bond and the synthesis of acetylenic carboxylic acids with a yield of 80-89% (phenylacetylene (1.0 mmol), Cs2CO3 (3.0 mmol), cat. (0.04 mmol), 1 atm of CO2, 60 °C, DMSO (10 mL)) [99].
The transformation of CO2 into macromolecular compounds can be carried out by direct copolymerization of CO2 with epoxides/aziridines, polycondensation with amines, alcohols, and amino alcohols, or by the synthesis of CO2-based monomers that will be used in polymerization reactions dependent on the nature and functionality of the appropriate monomer. These synthetic routes enable the production of a wide range of polycarbonates (PC), polyurethanes (PU), polyureas (PUA), and polyesters using versatile polymerization techniques, including ring-opening polymerization (ROP), ring-opening copolymerization (ROCOP), polycondensation and terpolymerization. However, for synthesizing CO2-based polymers, the low reactivity of CO2 or its derivatives requires elevated temperatures, removal of by-products, and activation of the monomers by multiple metal centers to complete the reaction.
Direct polymerization routes to CO2-based polymers are mainly limited to the ROCOP of CO2 with epoxides or aziridines. CO2 is also used as a comonomer in the synthesis of multiblock copolymers by sequential monomer addition and tandem approach [100].
In the indirect approach, CO2 is first converted into linear or cyclic building blocks, which are then used to synthesize polymeric materials. Cyclic monomers (carbonates, carbamates) can be ring-opening polymerized to form the corresponding homopolymers (PCs, PUs). Cyclic 5-membered carbonates can also copolymerize with diols, diamines, diamines, and diols or cyclic ureas to form PCs, PUAs, or PUs [101]. Copolymerization of cyclic carbonates with lactones is also widely studied to obtain copolyesters.
Copolymerization of CO2 and epoxides is the most investigated method of synthesis of PCs [102]. High molecular weight polycarbonates exhibit properties suitable for replacing petrochemical polymers in sectors including packaging, coatings, rigid plastics, and medical materials. Recently, developing and understanding the catalytic performance of heterometallic catalysts that exhibit metal synergy are among the most frequently studied aspects of the synthesis of biodegradable polymers. It was shown that some heterometallic complexes having two different metal centers showed much higher activity than their homometallic counterparts. It is assumed that different metals play different roles in copolymerization; the stronger Lewis acid activates the epoxide, and the softer Lewis acid forms a labile bond with the carbonate of the propagating chain, making the carbonate more nucleophilic. Several heterometallic zinc-lanthanide complexes have been shown to initiate the copolymerization of CO2 and cyclohexene oxide (CHO).
For example, the heterometallic cluster pictured in Figure 26, [Zn2Nd2(μ-OBn)2(L)2] (for L2- = (((2-(bis(3,5-di-t-butyl-2-oxidobenzyl)amino)phenyl)amino)methyl)-4,6-di-t-butylphenolato)) gave polycarbonates with high molecular weight (Mn up to 295.8 kDa) with narrow dispersity (Đ = 1.65) and high selectivity (99%) at [CHO]:[cat.] = 2000:1 ratio, 25 °C, 12h and 7 bar CO2, and its reactivity was nine times greater than that of the isostructural yttrium catalyst [103]. Using [ZnLn(L)(L’)(NSiHMe2)2] (for L- = (cyclohexane-1,2-diylbis(azanylylidenemethylylidene))bis(6-methylphenolato); L’- = 2,2’-{[(2-methoxyethyl)azanediyl]bis(methylene)}bis(4,6-di-t-butylphenolato) as catalysts for CO2/CHO copolymerization, it was shown that the radius of the REIII ion significantly influences their activity. The most effective catalysts turned out to be ZnIIDyIII and ZnIISmIII complexes containing lanthanide ions with a moderate ionic radius, leading to polycarbonates with Mn = 148 kDa, Đ = 1.52–1.62, and selectivities of 99% (carbonate bonds). Another example was heterometallic Zn3RE clusters (REIII = LaIII, CeIII, PrIII, NdIII, SmIII, EuIII, GdIII, and DyIII) based on macrocyclic tri(salen) ligands, which showed a unique and rapid exchange of intra- and intermolecular acetate ligands. Lanthanide complexes with larger ionic radii (LaIII, CeIII, PrIII, NdIII) showed higher catalytic activity than those based on smaller lanthanides (SmIII, EuIII, GdIII, DyIII). The most active of them was Zn3Ce, which enables the synthesis of polycarbonates with Mn = 14 kDa, Đ = 1.3 at 100 °C within 3 h [104].
All the above-mentioned heterometallic catalysts for CO2 copolymerization operate via the chain-shuttling mechanism in which the Lewis acidic REIII enhances CHO coordination, and the unstable Zn-carbonate bond enhances nucleophilic attack to open the epoxide ring. The resulting RE-alkoxide and CO2 coordinated to ZnII form a carbonate complex, leading to the growth of the polycarbonate chain [105]. Introducing CoII instead of ZnII into heterometallic complexes led to the discovery of one of the most efficient multimetallic catalysts, Co3Nd, which had a turnover number (TON) of 13,000. This gave a polymer with >99% selectivity of carbonate bonds, Mn = 114 kDa and Đ = 1.05 at 2 MPa CO2, 130 °C after 8 h. Pathway of that reaction may be found in Figure 27 [106].
2.5. Catalysts for energy conversion processes
Recently, particular attention was placed on using 3d-4f clusters as promising catalysts for energy conversion processes like hydrogen evolution reaction (HER), oxygen evolution reaction (OER), overall water splitting, and CO2 reduction. The synergic cooperation between both REIII/MII centers led to their enhanced activity. For example, [Ni36Gd102(μ3-OH)132(L)18(L’)18(H2L’)24(OAc)84(SO4)18(NO3)18(H2O)30]Br6,(NO3)6 (Ni36Gd102, LH = 2-mercapto-5-methyl-1,3,4-thiadiazide; L’H3 = 2,2-dimethylol propionic acid) found in Figure 28 shows remarkable activity in photocatalytic CO2 reduction, providing a TON of 29700 and a turnover frequency (TOF) of 1.2 s−1 over 10 h with a selectivity of 90.2% for CO formation. This performance is much better than those of most homogeneous CO2-reduction catalysts because the Lewis-acidic GdIII modulates the electronic structure of the catalytic NiII centers, enhancing photocatalytic activity [107].
[Co12Eu36(μ4-O)6(μ3-OH)84(OAc)18(Cl)2(NO3)]33+ shows effective water oxidation activity under acidic conditions (TOF of 1.5 s-1 at 1.8 V) owing to the synergistic effect of EuIII and CoII ions on O–O bond formation [108]. Heterometallic cooperativity in water oxidation has also been reported for [Mn2RE2(O2CMe)6(pdmH)2(L)](NO3) (REIII = DyIII and GdIII; pdmH2 = 2,6-pyridine dimethanol; LH2 = (6-hydroxymethylpyridin-2-yl)-(6-hydroxymethylpyridin-2-ylmethoxy-methanol)) which structure can be found in Figure 29 [109].
The series [Co3RE(hmp)4(OAc)5(H2O)], where RE = HoIII, ErIII, TmIII, or YbIII (hmpH = 2-(hydroxymethyl)pyridine)), [110] and [NdCo3(btp)2(OAc)2(NO3)2](NO3) (btp = 2,6-bis(1,2,3-triazol-4-yl)pyridine) anchored in phospho-doped graphitic carbon nitride, [111] are further examples of water-oxidation catalysts, being mimetics of the {CaMn4O5} oxygen-evolution complex in photosystem II. [RE52Ni56(IDA)48(OH)154(H2O)38]18+ clusters (RE = PrIII, EuIII, GdIII; IDA = iminodiacetate) supported on CdS forms lanthanide–transition metal catalysts RE52Ni56-xCdx/CdS that show activity for photocatalytic hydrogen evolution. The high photocatalytic efficiency of 25353 μmol h−1 g−1 was achieved usingNi56-xCdxEu52/CdS, which enhances H2 production under visible-light irradiation (≥ 420 nm) owing to the formation of catalytic EuII centers by the transfer of photoexcited electrons from CdS to the LUMO of EuIII [112]. Furthermore, the iodide-templated 3D-coordination polymer {[Cu5Eu2(OH)2(pydc)6(H2O)8]·I8}n (pydc = pyridine-2,6-dicarboxylate) has been demonstrated to be an efficient photocatalyst for H2 production under UV irradiation, providing H2 evolution at a rate of 2262 μmol h−1 g−1 [113].
2.6. Molecular precursors of functional inorganic materials.
The use of heterometallic 3d−4f compounds as single-source molecular precursors for the synthesis of functional inorganic materials is also very limited. One of the few examples is the series of isostructural compounds [Fe2Ln2((OCH2)3CR)2(O2CtBu)6(H2O)4] (Ln = La, Gd and R = Me, Et), which were used to prepare lanthanide orthoferrite perovskites LnFeO3 [114]. Another example is the use of an equimolar mixture of (NH4)[Ln(EDTA)] (LnIII = PrIII, SmIII, EuIII, GdIII, DyIII, ErIII) and (NH4)3[V(O)2(EDTA)] at 800 °C for the preparation of lanthanide vanadates (LnVO4) [115]. Furthermore, the solid-phase thermal decomposition of [Ln(Mn(CO)3CpCOOH)2(OAc)(MeOH)]n (LnIII = NdIII, GdIII, DyIII), [Ln2(Mn(CO)3CpCOOH)4(OAc)2(H2O)4] (LnIII = HoIII, ErIII, TmIII), and [Ln2(Mn(CO)3CpCOOH)4(NO3)2(DME)2] (LnIII = EuIII, TbIII) at 670–900 °C has been investigated for the synthesis of metamagnetic LnMn2O5 [116,117].
Various synthesis methods have been explored in the literature to synthesize La2CuO4. One of them involves the thermal decomposition of an amorphous precursor [La2Cu(DTPA) 1.6]·6H2O[118] (H5DTPA = diethylenetriaminepentaacetic acid) initially at 450 °C to eliminate volatile organic constituents, and then by heat treatment at 650 °C yielded La2CuO4 with an average crystallite size of around 29 nm. An alternate approach to synthesize La2CuO4 included hydrolyzing a mixture of [La4(CO3)(O2CNBu2)10] and [Cu(O2CNBu2)(py)2] (py = 4-dimethylaminopyridine) in a toluene solution, resulting in the isolation of the compound [La2Cu(CO3)4]·5H2O [119]. The subsequent procedure involved heating the isolated precursor at 600°C, yielding a tetragonal polymorph with an average crystallite size of 15 nm. Above 850°C, the tetragonal form transformed into a rhombohedral phase with crystallites of sizes of 50 nm.
Recently, we have developed a simple and efficient synthetic strategy for the preparation of industrially important heterometallic perovskite-type materials of LaMnO3, GdMnO3, NdMnO3, Pr0.9MnO3 (Figure 30), and PrCoO3 by thermal decomposition of heterometallic 3d–4f alkoxides [Mn2Ln4(µ6-O)(µ3-OR)8(HOR)4Cl6] (Ln = LaIII, NdIII, GdIII); [Co2Pr4(µ6-O)(µ3-OR)8(HOR)2Cl6]; and [Mn2Pr4(µ3-OH)2(µ3-OR)4(µ-OR)4(µ-Cl)2(HOR)4Cl6], which molecular structures can be found in Figure 31, and external [MnCl2(HOR)]n or [Co4(µ3-OR)4(HOR)4Cl4] at 1100 °C [30]. When we performed the thermolysis of [Ni2Pr4(µ6-O)(µ3-OR)8(HOR)4Cl6] and [NiCl2(HOR)2] the formation of a mixture of the homo- and heterometallic oxides PrOCl, PrO2, NiO, PrNiO3, and Pr2NiO4 was observed. The representative PXRD pattern of Pr0.9MnO3 is presented in Figure 32. The synthesis of 3d–4f precursors was performed using an uncommon synthetic method involving the reaction of metallic lanthanides (LnIII = LaIII, PrIII, NdIII, GdIII) with divalent transition metal chlorides (MCl2, where M = MnII, NiII, or CoII) using 2-methoxyethanol (ROH) as the solvent and ligand precursor.
Another work reported that group 4–lanthanide ethoxides are attractive starting materials for the production of pyrochlore type phases Ln2M2O7 of considerable interest because of their use as materials for thermal coatings of turbine components for protection against hot and corrosive gas streams [120], high-temperature electrolytes in solid-oxide fuel cells [121], or radiation-resistant materials [122]. Thermal decomposition of the isostructural compounds [Ln2Ti4(µ4-O)2(µ3-OEt)2(µ-OEt)8(OEt)6(HOEt)2Cl2] (LnIII = LaIII, NdIII) at 950 °C gave La0.66TiO3 or a mixture of Nd4Ti9O24 and Nd0.66TiO3. Calcination of [M2La2(µ3-O)(µ-OEt)5(µ-Cl)(OEt)2(HOEt)4Cl4]n and [M4Nd4(µ3-O)2(µ-OEt)10(µ-Cl)4(OEt)8(HOEt)10Cl2] (MIV = ZrIV, HfIV) at 950–1500 °C led to the selective formation of heterometallic La2Zr2O7, La2Hf2O7, Nd2Zr2O7, and Nd2Hf2O7 phases, respectively [31].
3. Conclusions
Discussed here, heterometallic rare-earth-transition metal, alkaline earth, or alkali metal complexes are attractive molecular materials with a number of applications in energy storage and conversion process, molecular magnetism, organic synthesis, material and polymer engineering. This review focused on the correlation of molecular structures of heterometallic compounds with displayed magnetic, catalytic, or photocatalytic properties. The general routes for synthesizing heterometallic rare-earth complexes with various solid-state structures, efficiency, and reproducibility were also presented. We discussed catalytic applications in asymmetric synthesis, CO2 transformations, polymerization/copolymerization of heterocyclic monomers, water splitting, and hydrogen or oxygen evolution reactions in the context of multimetallic synergy and cooperativity effects. Furthermore, the transformative potential of these complexes into advanced nanomaterials with diverse applications underscores their significance in the field of materials science. The complexes contribute to understanding fundamental chemical principles and open new routes for applied research and technological advancements. The intricate synergy between metal ions in these complexes unravels a wealth of possibilities, inspiring many researchers to further studies. Heterometallic rare-earth complexes hold promise as key players in developing innovative materials and technologies.
Author Contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript. R. P. - conceptualization, project administration, funding acquisition, and original draft writing and editing (paragraphs 2.2, 2.4 - 2.6). A. K. - original draft writing (introduction, paragraphs 2.1 and 2.3, conclusions), visualization, technical editing.
Acknowledgments
The authors thank the Polish National Science Center for financial support, grant number 2017/26/D/ST5/01123 (RP). The work was also co-financed by a statutory activity subsidy from the Polish Ministry of Science and Education for the Faculty of Chemistry of Wrocław University of Science and Technology (AK).
Conflicts of Interest
There are no conflicts to declare.
References
- Castor, S.B. and Hedrick, L.B. Rare Earth Elements in Kogel, J.E, Trivedi, N.C., Barker, J.M., and Krukowski, S.T., ed., Industrial Minerals volume, 7th edition: Society for Mining, Metallurgy, and Exploration, Littleton, Colorado, United States, 2006; pp. 769–792.
- Wall, F. , in Gunn, G., ed., Critical Metals Handbook, John Wiley & Sons, Oxford, 2014; p. 312-339.
- Czerwiński, F. Critical Assessment 36: Assessing Differences between the Use of Cerium and Scandium in Aluminium Alloying. Mater. Sci. Technol. 2019, 36, 255–263. [Google Scholar] [CrossRef]
- Voncken, J. H. L. The Rare Earth Elements; 2016. [CrossRef]
- Vitova, T.; Roesky, P. W.; Dehnen, S. Open Questions on Bonding Involving Lanthanide Atoms. Commun. Chem. 2022, 5. [Google Scholar] [CrossRef]
- Cotton, S. Lanthanide and Actinide Chemistry; John Wiley & Sons, 2013.
- Wang, Z.; Guo, Y.; Gong, X.; Guo, Y.; Wang, Y.; Lu, G. Current Status and Perspectives of Rare Earth Catalytic Materials and Catalysis. Chin. J. Catal. 2014, 35, 1238–1250. [Google Scholar] [CrossRef]
- Van Gosen, B. S.; Verplanck, P. L.; Long, K. R.; Gambogi, J.; Seal, R. R. The Rare-Earth Elements: Vital to Modern Technologies and Lifestyles. Fact Sheet /. [CrossRef]
- Coey, J. M. D. Perspective and Prospects for Rare Earth Permanent Magnets. Engineering 2020, 6, 119–131. [Google Scholar] [CrossRef]
- Lucas, J.; Lucas, P.; Mercier, T. L.; Rollat, A.; Davenport, W. G. Rare Earths in Rechargeable Batteries. In Elsevier eBooks; 2015; pp 167–180. [CrossRef]
- Seddon, A. B.; Tang, Z.; Furniss, D.; Sujecki, S.; Benson, T. M. Progress in Rare-Earth-Doped Mid-Infrared Fiber Lasers. Opt. Express 2010, 18, 26704. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, M.; Murtaza, Q.; Kumar, P. Effect of Rare Earth Elements on Coatings Developed by Thermal Spraying Processes (TSP) – A Brief Review. Mater. Today: Proc. 2021, 44, 4053–4058. [Google Scholar] [CrossRef]
- Dubey, V.; Dubey, N.; Domanska, M. M.; Jayasimhadri, M.; Dhoble, S. J. Rare-Earth-Activated Phosphors: Chemistry and Applications; Elsevier, 2022.
- Hossain, Md. F.; Ahmed, M. H.; Khan, I.; Miah, Md. S.; Hossain, S. Recent Progress of Rare Earth Oxides for Sensor, Detector, and Electronic Device Applications: A Review. ACS Appl. Electron. Mater. 2021, 3, 4255–4283. [Google Scholar] [CrossRef]
- Hossain, M. K.; Raihan, G. A.; Akbar, M. A.; Rubel, M. H. K.; Ahmed, M. H.; Khan, I.; Hossain, S.; Sen, S. K.; Jalal, M. I. E.; El-Denglawey, A. Current Applications and Future Potential of Rare Earth Oxides in Sustainable Nuclear, Radiation, and Energy Devices: A Review. ACS Appl. Electron. Mater. 2022, 4, 3327–3353. [Google Scholar] [CrossRef]
- Wu, G.; Wang, C.; Sun, M.; Ding, W. Recent Developments and Applications on High-Performance Cast Magnesium Rare-Earth Alloys. J. Magnes. Alloys 2021, 9, 1–20. [Google Scholar] [CrossRef]
- Luo, Q.; Guo, Y.; Liu, B.; Feng, Y.; Zhang, J.; Li, Q.; Chou, K. Thermodynamics and Kinetics of Phase Transformation in Rare Earth–Magnesium Alloys: A Critical Review. J. Mater. Sci. Technol. 2020, 44, 171–190. [Google Scholar] [CrossRef]
- Смирнoв, Л. А.; Рoвнушкин, В. А.; Орыщенкo, А. С.; Kalinin, G. Yu.; Milyuts, V. G. Modification of Steel and Alloys with Rare-Earth Elements. Part 1. Metallurgist 2016, 59 (11–12), 1053–1061. [CrossRef]
- Zhao, K.; Gao, Y.; Wang, X.; Lis, B. M.; Liu, J.; Jin, B.; Smith, J.; Huang, C.; Gao, W.; Wang, X.; Wang, X.; Zheng, A.; Huang, Z.; Hu, J.; Schomaecker, R.; Wachs, I. E.; Li, F. Lithium Carbonate-Promoted Mixed Rare Earth Oxides as a Generalized Strategy for Oxidative Coupling of Methane with Exceptional Yields. Nat. Commun. 2023, 14. [Google Scholar] [CrossRef]
- Khodakov, Yu. S.; Nesterov, V. K.; Миначев, Х. М. Isomerization of 1-Butene on Oxides of Rare-Earth Elements. Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science 1975, 24, 1892–1894. [Google Scholar] [CrossRef]
- Wang, Z.; Fongarland, P.; Lu, G.; Wang, Z.; Essayem, N. Effect of Hydration on the Surface Basicity and Catalytic Activity of Mg-Rare Earth Mixed Oxides for Aldol Condensation. J. Rare Earths 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Imamura, H.; Ohmura, A.; Haku, E.; Tsuchiya, S. Rare Earth Metals as Hydrogenation Catalysts of Unsaturated Hydrocarbons. J. Catal. 1985, 96, 139–145. [Google Scholar] [CrossRef]
- Sato, S.; Sato, F.; Gotoh, H.; Yamada, Y. Selective Dehydration of Alkanediols into Unsaturated Alcohols over Rare Earth Oxide Catalysts. ACS Catal. 2013, 3, 721–734. [Google Scholar] [CrossRef]
- Bochkarev, M. N.; Pushkarev, A. P. Synthesis and Luminescence of Some Rare Earth Metal Complexes. Org. Photonics Photovolt. 2016, 4. [Google Scholar] [CrossRef]
- Armelao, L.; Quici, S.; Barigelletti, F.; Accorsi, G.; Bottaro, G.; Cavazzini, M.; Tondello, E. Design of Luminescent Lanthanide Complexes: From Molecules to Highly Efficient Photo-Emitting Materials. Coord. Chem. Rev. 2010, 254, 487–505. [Google Scholar] [CrossRef]
- Li, S.; Zhou, L.; Zhang, H. Investigation Progresses of Rare Earth Complexes as Emitters or Sensitizers in Organic Light-Emitting Diodes. Light-Sci. Appl. 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Evans, W. J.; Johnston, M. C.; Greci, M. A.; Ansari, M. A.; Brady, J. C.; Ziller, J. W. Synthesis of Arene-Soluble Mixed-Metal Zr/Ce, Zr/Y, and Related {[Zr2(OiPr)9]LnX2}n Complexes Using the Dizirconium Nonaisopropoxide Ligand. Inorg. Chem. 2000, 39, 2125–2129. [Google Scholar] [CrossRef]
- Artner, C.; Kronister, S.; Czakler, M.; Schubert, U. Ion-Size-Dependent Formation of Mixed Titanium/Lanthanide OXO Clusters. Eur. J. Inorg. Chem. 2014, 2014, 5596–5602. [Google Scholar] [CrossRef]
- Mashima, K.; Nakamura, A. Novel Synthesis of Lanthanoid Complexes Starting from Metallic Lanthanoid Sources. J. Chem. Soc.-Dalton Trans. 1999, 3899–3907. https://doi.org/10.1039/a905998i. (b) Deacon, G. B.; Hamidi, S.; Junk, P. C.; Kelly, R. L.; Wang, J. Direct Reactions of Iodine-Activated Rare-Earth Metals with Phenols of Varying Steric Bulk. Eur. J. Inorg. Chem. 2013, 2014, 460–468. Eur. J. Inorg. Chem. [CrossRef]
- Petrus, R.; Kowaliński, A.; Utko, J.; Matuszak, K.; Lis, T.; Sobota, P. Heterometallic 3D–4f Alkoxide Precursors for the Synthesis of Binary Oxide Nanomaterials. Inorg. Chem. 2023, 62, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- Petrus, R.; Chomiak, K.; Utko, J.; Bieńko, A.; Lis, T.; Sobota, P. Heterometallic Group 4–Lanthanide Oxo-Alkoxide Precursors for Synthesis of Binary Oxide Nanomaterials. Inorg. Chem. 2020, 59, 16545–16556. [Google Scholar] [CrossRef] [PubMed]
- Botta, M.; Casellato, U.; Scalco, C.; Tamburini, S.; Tomasin, P.; Vigato, P. A.; Aime, S.; Barge, A. Heterodinuclear LnNa Complexes with an Asymmetric Macrocyclic Compartmental Schiff Base. Chem. A Eur. J. 2002, 8, 3917–3926. [Google Scholar] [CrossRef]
- Xu, X.; Ma, M.; Yao, Y.; Zhang, Y.; Shen, Q. Synthesis, Characterisation of Carbon-Bridged (Diphenolato)Lanthanide Complexes and Their Catalytic Activity for Diels–Alder Reactions. Eur. J. Inorg. Chem. 2005, 2005, 676–684. [Google Scholar] [CrossRef]
- Zheng, Z.-P.; Ou, Y.-J.; Hong, X.; Wei, L.-M.; Wan, L.-T.; Zhou, W.; Zhan, Q.-G.; Cai, Y. Anion-Dependent Assembly of Four Sensitized Near-Infrared Luminescent Heteronuclear ZnII–YbIII Schiff Base Complexes from a Trinuclear ZnII Complex. Inorg. Chem. 2014, 53, 9625–9632. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yin, K.; Chen, Y.; Zhao, B.; Zhang, Y.; Zhu, X.; Yuan, D.; Yao, Y. Synthesis of Heterometallic Rare Earth(III)–Cobalt(II) Complexes and Their Application in Alternating Copolymerization of Cyclohexene Oxide and Carbon Dioxide. Chin. J. Chem. 2023, 41, 805–813. [Google Scholar] [CrossRef]
- Langley, S. K.; Le, C.; Ungur, L.; Moubaraki, B.; Abrahams, B. F.; Chibotaru, L. F.; Murray, K. S. Heterometallic 3D–4f Single-Molecule Magnets: Ligand and Metal Ion Influences on the Magnetic Relaxation. Inorg. Chem. 2015, 54, 3631–3642. [Google Scholar] [CrossRef] [PubMed]
- Piquer, L. R.; Sañudo, E. C. Heterometallic 3d–4f Single-Molecule Magnets. Dalton Trans. 2015, 44, 8771–8780. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Acharya, J.; Chandrasekhar, V. Heterometallic 3D–4F Complexes as Single-Molecule Magnets. Chem.-Asian J. 2019, 14, 4433–4453. [Google Scholar] [CrossRef]
- Chakraborty, A.; Goura, J.; Kalita, P.; Swain, A.; Rajaraman, G.; Chandrasekhar, V. Heterometallic 3d–4f Single Molecule Magnets Containing Diamagnetic Metal Ions. Dalton Trans. 2018, 47, 8841–8864. [Google Scholar] [CrossRef]
- Kahn, M. L.; Mathonière, C.; Kahn, O. Nature of the Interaction between LnIII and CuII Ions in the Ladder-Type Compounds {Ln2[Cu(Opba)]3}·S (Ln = Lanthanide Element; Opba = Ortho-Phenylenebis(Oxamato), S = Solvent Molecules). Inorg. Chem. 1999, 38, 3692–3697. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. L.; Lecante, P.; Verelst, M.; Mathonière, C.; Kahn, O. Structural Studies and Magnetic Properties of Polymeric Ladder-Type Compounds {Ln2[Ni(Opba)]3}·S (Ln = Lanthanide Element; Opba = o-Phenylenebis(Oxamato), S = Solvent Molecules). Chem. Mater. 2000, 12, 3073–3079. [Google Scholar] [CrossRef]
- Shiga, T.; Ohba, M.; Ōkawa, H. A Series of Trinuclear CuIILnIIICuII Complexes Derived from 2,6-Di(Acetoacetyl)Pyridine: Synthesis, Structure, and Magnetism. Inorg. Chem. 2004, 43, 4435–4446. [Google Scholar] [CrossRef] [PubMed]
- Păsătoiu, T. D.; Sutter, J.; Mădălan, A. M.; Fellah, F. Z. C.; Duhayon, C.; Andruh, M. Preparation, Crystal Structures, and Magnetic Features for a Series of Dinuclear [NIIILNIII] Schiff-Base Complexes: Evidence for Slow Relaxation of the Magnetization for the DYIII Derivative. Inorg. Chem. 2011, 50, 5890–5898. [Google Scholar] [CrossRef] [PubMed]
- Koner, R.; Lin, H.; Wei, H.; Mohanta, S. Syntheses, Structures, and Magnetic Properties of Diphenoxo-Bridged MIILnIII Complexes Derived from N,N‘-Ethylenebis(3-Ethoxysalicylaldiimine) (M = Cu or Ni; Ln = Ce−Yb): Observation of Surprisingly Strong Exchange Interactions. Inorg. Chem. 2005, 44, 3524–3536. [Google Scholar] [CrossRef]
- Costes, J.; Dahan, F.; Dupuis, A.; Laurent, J. Nature of the Magnetic Interaction in the (CU2+, LN3+) Pairs: An Empirical Approach Based on the Comparison between Homologous (CU2+, LN3+) and (NILS2+, LN3+) Complexes. Chem.-A Eur. J. 1998, 4, 1616–1620. [Google Scholar] [CrossRef]
- Osa, S.; Kido, T.; Matsumoto, N.; Re, N.; Pochaba, A. A.; Mroziński, J. A Tetranuclear 3d−4f Single Molecule Magnet: [CuIILTbIII(Hfac)2]2. Journal of the American Chemical Society 2003, 126, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tan, R.; Yi, T.; Gao, S.; Yan, C.; Cao, L. Preparation of Nanosized La2CuO4 Perovskite Oxide Using an Amorphous Heteronuclear Complex as a Precursor at Low-Temperature. Journal of Alloys and Compounds 2000, 311, 16–21. [Google Scholar] [CrossRef]
- Bridonneau, N.; Gontard, G.; Marvaud, V. A New Family of Hetero-Tri-Metallic Complexes [M(CuTb)]n (n = 1, 2, ∞; M = Co, Cr, Fe): Synthesis, Structure and Tailored Single-Molecule Magnet Behavior. Dalton Trans. 2015, 44, 5170–5178. [Google Scholar] [CrossRef]
- Bridonneau, N.; Gontard, G.; Marvaud, V. A New Family of Hetero-Tri-Metallic Complexes [M(CuTb)]n (n = 1, 2, ∞; M = Co, Cr, Fe): Synthesis, Structure and Tailored Single-Molecule Magnet Behavior. Dalton Trans. 2015, 44, 5170–5178. [Google Scholar] [CrossRef]
- Ghazali, N. F.; Vignesh, K. R.; Phonsri, W.; Murray, K. S.; Junk, P. C.; Deacon, G. B.; Turner, D. R. Efficient Synthetic Route to Heterobimetallic Trinuclear Complexes [Ln–Mn–Ln] and Their Single Molecule Magnetic Properties. Dalton Trans. 2022, 51, 18502–18513. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-Y.; Liu, Y.; Deng, X.; Zhu, Z.; Yao, M.; Jing, S. Synthesis, Structures and Magnetism of Heterodinuclear Ni–Ln Complexes: Field-Induced Single-Molecule Magnet Behavior in the Dysprosium Analogue. New J. Chem. 2015, 39, 3467–3473. [Google Scholar] [CrossRef]
- Colacio, E.; Ruíz, J.; Mota, A. J.; Palacios, M. A.; Cremades, E.; Ruiz, E.; White, F.; Brechin, E. K. Family of Carboxylate- and Nitrate-Diphenoxo Triply Bridged Dinuclear NIIILNIII Complexes (LN = EU, GD, TB, HO, ER, Y): Synthesis, Experimental and Theoretical Magneto-Structural Studies, and Single-Molecule Magnet Behavior. Inorg. Chem. 2012, 51, 5857–5868. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Das, C.; Vaidya, S.; Langley, S. K.; Murray, K. S.; Shanmugam, M. Nickel(II)-Lanthanide(III) Magnetic Exchange Coupling Influencing Single-Molecule Magnetic Features in {NI2LN2} Complexes. Chem.-A Eur. J. 2014, 20, 14235–14239. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Fujinami, T.; Nishi, K.; Matsumoto, N.; Mochida, N.; Ishida, T.; Sunatsuki, Y.; Re, N. Carbonato-Bridged NiII2LnIII2 (LnIII = GdIII, TbIII, DyIII) Complexes Generated by Atmospheric CO2 Fixation and Their Single-Molecule-Magnet Behavior: [(Μ4-CO3)2{NiII(3-MeOsaltn)(MeOH or H2O)LnIII(NO3)}2]·solvent [3-MeOsaltn = N,N′-Bis(3-Methoxy-2-Oxybenzylidene)-1,3-Propanediaminato]. Inorganic Chemistry 2013, 52, 7218–7229. [Google Scholar] [CrossRef] [PubMed]
- Mondal, K. C.; Kostakis, G. E.; Lan, Y.; Wernsdorfer, W.; Anson, C. E.; Powell, A. K. Defect-Dicubane NI2LN2 (LN = DY, TB) Single Molecule Magnets. Inorg. Chem. 2011, 50, 11604–11611. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Pineda, E.; Chilton, N. F.; Tuna, F.; Winpenny, R. E. P.; McInnes, E. J. L. Systematic Study of a Family of Butterfly-Like {M2LN2} Molecular Magnets (M = MGII, MNIII, COII, NIII, and CUII; LN = YIII, GDIII, TBIII, DYIII, HOIII, and ERIII). Inorg. Chem. 2015, 54, 5930–5941. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhao, L.; Xu, X.; Xu, G.; Guo, Y.; Tang, J.; Liu, Z. Heterometallic Cubanes: Syntheses, Structures, and Magnetic Properties of Lanthanide(III)−Nickel(II) Architectures. Inorg. Chem. 2011, 50, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
- Canaj, A. B.; Tzimopoulos, D. I.; Siczek, M.; Lis, T.; Inglis, R.; Milios, C. J. Enneanuclear [NI6LN3] Cages: [LNIII3] Triangles Capping [NIII6] Trigonal Prisms Including a [NI6DY3] Single-Molecule Magnet. Inorg. Chem. 2015, 54, 7089–7095. [Google Scholar] [CrossRef]
- Xiong, K.; Wang, X.; Jiang, F.; Gai, Y.; Xu, W.; Su, K.; Li, X.; Yuan, D.; Hong, M. Heterometallic Thiacalix[4]Arene-Supported Na2NiII12LnIII2 Clusters with Vertex-Fused Tricubane Cores (Ln = Dy and Tb). Chem. Commun. 2012, 48, 7456. [Google Scholar] [CrossRef]
- Takehara, C.; Then, P. L.; Kataoka, Y.; Nakano, M.; Yamamura, T.; Kajiwara, T. Slow Magnetic Relaxation of Light Lanthanide-Based Linear LnZn2 Trinuclear Complexes. Dalton Trans. 2015, 44, 18276–18283. [Google Scholar] [CrossRef] [PubMed]
- Fondo, M.; Corredoira-Vázquez, J.; Herrera-Lanzós, A.; García-Deibe, A. M.; Sanmartín-Matalobos, J.; Herrera, J. M.; Colacio, E.; Núñez, C. Improving the SMM and Luminescence Properties of Lanthanide Complexes with LnO9cores in the Presence of ZnII: An Emissive Zn2Dy Single Ion Magnet. Dalton Trans. 2017, 46, 17000–17009. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, P.; Wang, C.-Y.; Liu, Y.; Liu, W.; Zhang, M. Three Sandwich-Type Zinc(Ii)–Lanthanide(Iii) Clusters: Structures, Luminescence and Magnetic Properties. RSC Adv. 2017, 7, 22692–22698. [Google Scholar] [CrossRef]
- Long, J. Luminescent Schiff-Base Lanthanide Single-Molecule Magnets: The Association between Optical and Magnetic Properties. Front. Chem. 2019, 7. [Google Scholar] [CrossRef]
- Long, J.; Vallat, R.; Ferreira, R. a. S.; Carlos, L. D.; Paz, F. a. A.; Guari, Y.; Larionova, J. A Bifunctional Luminescent Single-Ion Magnet: Towards Correlation between Luminescence Studies and Magnetic Slow Relaxation Processes. Chem. Commun. 2012, 48, 9974. [Google Scholar] [CrossRef] [PubMed]
- Yamagiwa, N.; H, Q.; Matsunaga, S.; Shibasaki, M. Lewis Acid−Lewis Acid Heterobimetallic Cooperative Catalysis: Mechanistic Studies and Application in Enantioselective Aza-Michael Reaction. J. Am. Chem. Soc. 2005, 127, 13419–13427. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, N.; Kumagai, N.; Matsunaga, S.; Moll, G.; Ohshima, T.; Suzuki, T.; Shibasaki, M. Direct Catalytic Asymmetric Aldol Reaction: Synthesis of Either Syn- or Anti-α,β-Dihydroxy Ketones. J. Am. Chem. Soc. 2001, 123, 2466–2467. [Google Scholar] [CrossRef]
- Tosaki, S. Y.; Hara, K.; Gnanadesikan, V.; Morimoto, H.; Harada, S.; Sugita, M.; Yamagiwa, N.; Matsunaga, S.; Shibasaki, M. Mixed LA−LI Heterobimetallic Complexes for Tertiary Nitroaldol Resolution. J. Am. Chem. Soc. 2006, 128, 11776–11777. [Google Scholar] [CrossRef]
- Kakei, H.; Sone, T.; Sohtome, Y.; Matsunaga, S.; Shibasaki, M. Catalytic Asymmetric Cyclopropanation of Enones with Dimethyloxosulfonium Methylide Promoted by a La−Li3−(Biphenyldiolate)3 + NaI Complex. J. Am. Chem. Soc. 2007, 129, 13410–13411. [Google Scholar] [CrossRef]
- Gnanadesikan, V.; Horiuchi, Y.; Ohshima, T.; Shibasaki, M. Direct Catalytic Asymmetric Aldol-Tishchenko Reaction. J. Am. Chem. Soc. 2004, 126, 7782–7783. [Google Scholar] [CrossRef]
- Tian, J.; Yamagiwa, N.; Matsunaga, S.; Shibasaki, M. An Asymmetric Cyanation Reaction and Sequential Asymmetric Cyanation–Nitroaldol Reaction Using a [YLi3{tris(Binaphthoxide)}] Single Catalyst Component: Catalyst Tuning with Achiral Additives. Angew. Chem. Int. Ed. 2002, 41, 3636–3638. [Google Scholar] [CrossRef]
- Robinson, J. R.; Booth, C. H.; Carroll, P. J.; Walsh, P. J.; Schelter, E. J. Dimeric Rare-Earth BINOLate Complexes: Activation of 1,4-Benzoquinone through Lewis Acid Promoted Potential Shifts. Chem. - A Eur. J. 2013, 19, 5996–6004. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Harwood, S. J.; Gröger, H.; Shibasaki, M. The First Catalytic Asymmetric Nitro-Mannich-Type Reaction Promoted by a New Heterobimetallic Complex. Angew. Chem. Int. Ed. 1999, 38, 3504–3506. [Google Scholar] [CrossRef]
- Emori, E.; Arai, T.; Sasai, H.; Shibasaki, M. A Catalytic Michael Addition of Thiols to α,β-Unsaturated Carbonyl Compounds: Asymmetric Michael Additions and Asymmetric Protonations. J. Am. Chem. Soc. 1998, 120, 4043–4044. [Google Scholar] [CrossRef]
- Robinson, J. R.; Carroll, P. J.; Walsh, P. J.; Schelter, E. J. Uranium(IV) BINOLate Heterobimetallics: Synthesis and Reactivity in an Asymmetric Diels–Alder Reaction. Organometallics 2013, 32, 1493–1499. [Google Scholar] [CrossRef]
- Shibasaki, M.; Yoshikawa, N. Lanthanide Complexes in Multifunctional Asymmetric Catalysis. Chem. Rev. 2002, 102, 2187–2210. [Google Scholar] [CrossRef]
- Nieto, I.; Wooten, A. J.; Robinson, J. R.; Carroll, P. J.; Schelter, E. J.; Walsh, P. J. Synthesis and Catalytic Activity of Heterobimetallic Rare Earth–Zinc Ethyl BINOLate Analogues of Shibasaki’s Catalysts. Organometallics 2013, 32, 7431–7439. [Google Scholar] [CrossRef]
- Ramirez, B.; Lu, C. C. Rare-Earth Supported Nickel Catalysts for Alkyne Semihydrogenation: Chemo- and Regioselectivity Impacted by the Lewis Acidity and Size of the Support. J. Am. Chem. Soc. 2020, 142, 5396–5407. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Silva, A. R.; Fu, L.; Shi, F. Ionothermal Synthesis, Crystal Structure, Topology and Catalytic Properties of Heterometallic Coordination Polymers Constructed from N-(Phosphonomethyl) Iminodiacetic Acid. Dalton Trans. 2015, 44, 13745–13751. [Google Scholar] [CrossRef]
- Chisholm, M. H. Concerning the Ring-Opening Polymerization of Lactide and Cyclic Esters by Coordination Metal Catalysts. Pure Appl. Chem. 2010, 82, 1647–1662. [Google Scholar] [CrossRef]
- Sheng, H.; Shi, J.; Feng, Y.; Wang, H.; Jiao, Y.; Sheng, H.; Zhang, Y.; Shen, Q. Remarkable Effect of Alkali Metal on Polymerization of Cyclic Esters Catalyzed by Samarium–Alkali Metal Multinuclear Alkoxide Clusters. Dalton Trans. 2012, 41, 9232. [Google Scholar] [CrossRef] [PubMed]
- Hultzsch, K. C.; Spaniol, T. P.; Okuda, J. Chiral Lanthanocene Derivatives Containing Two Linked Amido−Cyclopentadienyl Ligands: Heterobimetallic Structure and Lactone Polymerization Activity. Organometallics 1997, 16, 4845–4856. [Google Scholar] [CrossRef]
- Sánchez-Barba, L. F.; Hughes, D. L.; Humphrey, S. M.; Bochmann, M. Ligand Transfer Reactions of Mixed-Metal Lanthanide/Magnesium Allyl Complexes with β-Diketimines: Synthesis, Structures, and Ring-Opening Polymerization Catalysis. Organometallics 2006, 25, 1012–1020. [Google Scholar] [CrossRef]
- Broderick, E. M.; Thuy-Boun, P. S.; Guo, N.; Vogel, C.; Sutter, J.; Miller, J. T.; Meyer, K.; Diaconescu, P. L. Synthesis and Characterization of Cerium and Yttrium Alkoxide Complexes Supported by Ferrocene-Based Chelating Ligands. Inorg. Chem. 2011, 50, 2870–2877. [Google Scholar] [CrossRef] [PubMed]
- Broderick, E. M.; Guo, N.; Wu, T.; Vogel, C.; Xu, C.; Sutter, J.; Miller, J. T.; Meyer, K.; Cantat, T.; Diaconescu, P. L. Redox Control of a Polymerization Catalyst by Changing the Oxidation State of the Metal Center. Chem. Commun. 2011, 47, 9897. [Google Scholar] [CrossRef] [PubMed]
- Broderick, E. M.; Guo, N.; Vogel, C.; Xu, C.; Sutter, J.; Miller, J. T.; Meyer, K.; Mehrkhodavandi, P.; Diaconescu, P. L. Redox Control of a Ring-Opening Polymerization Catalyst. J. Am. Chem. Soc. 2011, 133, 9278–9281. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. J.; Ding, L.; Chu, Z.; Chen, L.; Lü, X.; Zheng, X.; Song, J.; Fan, D. Controllable Bulk Solvent-Free Melt Ring-Opening Polymerization (ROP) of l-Lactide Catalyzed by Ni(II) and Ni(II)–Ln(III) Complexes Based on the Salen-Type Schiff-Base Ligand. J. Mol. Catal. A-Chem. [CrossRef]
- Yang, T.; Silva, A. R.; Shi, F. Six New 3d–4f Heterometallic Coordination Polymers Constructed from Pyrazole-Bridged CuIILnIII Dinuclear Units. Dalton Trans. 2013, 42, 13997. [Google Scholar] [CrossRef] [PubMed]
- Cancino, P.; Paredes-Garcı́A, V.; Torres, J.; Martínez, S.; Kremer, C.; Spodine, E. {[Cu3Lu2(ODA)6(H2O)6]·10H2O}n: The First Heterometallic Framework Based on Copper(Ii)/Lutetium(Iii) for the Catalytic Oxidation of Olefins and Aromatic Benzylic Substrates. Catal. Sci. Technol. 2017, 7, 4929–4933. [Google Scholar] [CrossRef]
- Gabrielli, P.; Gazzani, M.; Mazzotti, M. The Role of Carbon Capture and Utilization, Carbon Capture and Storage, and Biomass to Enable a Net-Zero-CO2 Emissions Chemical Industry. Ind. Eng. Chem. Res. 2020, 59, 7033–7045. [Google Scholar] [CrossRef]
- Xin, X.; Shan, H.; Tian, T.; Wang, Y.; Yuan, D.; You, H.; Yao, Y. Conversion of CO2 into Cyclic Carbonates under Ambient Conditions Catalyzed by Rare-Earth Metal Complexes Bearing Poly(Phenolato) Ligand. ACS Sustain. Chem. Eng. 2020, 8, 13185–13194. [Google Scholar] [CrossRef]
- Wang, L.; Xu, C.; Han, Q.; Tang, X.; Zhou, P.; Zhang, R.; Gao, G.; Xu, B.; Qin, W.; Liu, W. Ambient Chemical Fixation of CO2 Using a Highly Efficient Heterometallic Helicate Catalyst System. Chem. Commun. 2018, 54, 2212–2215. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; Hua, L.; Qu, L.; Yao, Q.; Wang, Y.; Yuan, D.; You, H.; Yao, Y. Heterobimetallic Rare Earth Metal–Zinc Catalysts for Reactions of Epoxides and CO2 under Ambient Conditions. Dalton Trans. 2021, 50, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Del Rosal, I.; Li, Q.; Wang, Y.; Yuan, D.; Yao, Y.; Maron, L. Efficient CO2 Transformation under Ambient Condition by Heterobimetallic Rare Earth Complexes: Experimental and Computational Evidences of a Synergistic Effect. Journal of CO2 Utilization 2019, 33, 413–418. [Google Scholar] [CrossRef]
- Gao, G.; Wang, L.; Zhang, R.; Xu, C.; Yang, H.; Liu, W. Hexanuclear 3d–4f Complexes as Efficient Catalysts for Converting CO2 into Cyclic Carbonates. Dalton Trans. 2019, 48, 3941–3945. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, L.; Xu, C.; Yang, H.; Chen, W.; Gao, G.; Liu, W. Anion-Induced 3d–4f Luminescent Coordination Clusters: Structural Characteristics and Chemical Fixation of CO2 under Mild Conditions. Dalton Trans. 2018, 47, 7159–7165. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.; Li, B.; Han, C.-T.; Gao, P.; Wang, Y.; Yuan, D.; Yao, Y. Synthesis of Homo- and Heteronuclear Rare-Earth Metal Complexes Stabilized by Ethanolamine-Bridged Bis(Phenolato) Ligands and Their Application in Catalyzing Reactions of CO2 and Epoxides. Inorg. Chem. 2019, 58, 8775–8786. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Tran, Y. B. N.; Nguyen, T. C.; Gándara, F.; Nguyen, P. L. T. A Series of Metal–Organic Frameworks for Selective CO2 Capture and Catalytic Oxidative Carboxylation of Olefins. Inorg. Chem. 2018, 57, 13772–13782. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, C.; Zhao, B.; Yao, Y. Synthesis and Characterization of Amidato Divalent Lanthanide Complexes and Their Use in Forming 2,4-Quinazolidinones from CO2 and 2-Aminobenzonitriles. European Journal of Organic Chemistry 2016, 2016, 2555–2559. [Google Scholar] [CrossRef]
- Cheng, H.; Zhao, B.; Yao, Y.; Lu, C. Carboxylation of Terminal Alkynes with CO2 Catalyzed by Bis(Amidate) Rare-Earth Metal Amides. Green Chem. 2015, 17, 1675–1682. [Google Scholar] [CrossRef]
- Bhat, G. A.; Darensbourg, D. J. Progress in the Catalytic Reactions of CO2 and Epoxides to Selectively Provide Cyclic or Polymeric Carbonates. Green Chem. 2022, 24, 5007–5034. [Google Scholar] [CrossRef]
- Grignard, B.; Gennen, S.; Jérôme, C.; Kleij, A. W.; Detrembleur, C. Advances in the Use of CO2 as a Renewable Feedstock for the Synthesis of Polymers. Chem. Soc. Rev. 2019, 48, 4466–4514. [Google Scholar] [CrossRef]
- Darensbourg, D. J. Making Plastics from Carbon Dioxide: Salen Metal Complexes as Catalysts for the Production of Polycarbonates from Epoxides and CO2. Chemical Reviews 2007, 107, 2388–2410. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Xu, B.; Zhang, Y.; Yuan, D.; Yao, Y. Cooperative Rare Earth Metal–Zinc Based Heterometallic Catalysts for Copolymerization of CO2 and Cyclohexene Oxide. Green Chem. 2016, 18, 4270–4275. [Google Scholar] [CrossRef]
- Nagae, H.; Aoki, R.; Akutagawa, S.; Kleemann, J.; Tagawa, R.; Schindler, T.; Choi, G.; Spaniol, T. P.; Tsurugi, H.; Okuda, J.; Mashima, K. Lanthanide Complexes Supported by a Trizinc Crown Ether as Catalysts for Alternating Copolymerization of Epoxide and CO2: Telomerization Controlled by Carboxylate Anions. Angew. Chem. Int. Ed. 2018, 57, 2492–2496. [Google Scholar] [CrossRef]
- Pan, Z.-H.; Weng, Z.-Z.; Kong, X.; Liu, L.; Zheng, L. Lanthanide-Containing Clusters for Catalytic Water Splitting and CO2 Conversion. Coord. Chem. Rev. 2022, 457, 214419. [Google Scholar] [CrossRef]
- Asaba, H.; Iwasaki, T.; Hatazawa, M.; Deng, J.; Nagae, H.; Mashima, K.; Nozaki, K. Alternating Copolymerization of CO2 and Cyclohexene Oxide Catalyzed by Cobalt–Lanthanide Mixed Multinuclear Complexes. Inorg. Chem. 2020, 59, 7928–7933. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liao, P.; Jin, P.; Zhang, L.; Ling, B.; Wang, S.; Chan, Y.; Chen, X.; Zheng, Y. The Gigantic {NI36GD102} Hexagon: A Sulfate-Templated “Star-of-David” for Photocatalytic CO2 Reduction and Magnetic Cooling. J. Am. Chem. Soc. 2020, 142, 4663–4670. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chen, C.; Du, M.; Wang, X.; Wang, C.; Liu, L.; Kong, X.; Zheng, L. Soluble Lanthanide-Transition-Metal Clusters Ln36Co12 as Effective Molecular Electrocatalysts for Water Oxidation. Chem. Commun. 2021, 57, 3611–3614. [Google Scholar] [CrossRef]
- Lan, T.; Gao, W.-S.; Chen, C.; Wang, H.; Wang, M.; Yu, F. Two Tetranuclear 3d–4f Heterometal Complexes Mn2Ln2 (Ln = Dy, Gd): Synthesis, Structure, Magnetism, and Electrocatalytic Reactivity for Water Oxidation. New J. Chem. 2018, 42, 5798–5805. [Google Scholar] [CrossRef]
- Evangelisti, F.; Moré, R.; Hodel, F. H.; Luber, S.; Patzke, G. R. 3D–4F {COII3LN(OR)4} Cubanes as Bio-Inspired Water Oxidation Catalysts. J. Am. Chem. Soc. 2015, 137, 11076–11084. [Google Scholar] [CrossRef]
- Chen, R.; Zhuang, G.; Wang, Z.; Gao, Y.; Li, Z.; Wang, C.; Yang, Z.; Du, M.; Zeng, S.; Liu, L.; Kong, X.; Zheng, L. Integration of Bio-Inspired Lanthanide-Transition Metal Cluster and P-Doped Carbon Nitride for Efficient Photocatalytic Overall Water Splitting. Natl. Sci. Rev. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yan, Z.; Kong, X.; Liu, L.; Zheng, L. Integration of Lanthanide–Transition-Metal Clusters onto CdS Surfaces for Photocatalytic Hydrogen Evolution. Angew. Chem. Int. Ed. 2018, 57, 16796–16800. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Sun, C.; Qin, C.; Wang, X.; Wang, H. N.; Zhou, E. L.; Li, W. E.; Su, Z. Iodine-Templated Assembly of Unprecedented 3d–4f Metal–Organic Frameworks as Photocatalysts for Hydrogen Generation. Chem. Commun. 2013, 49, 3564. [Google Scholar] [CrossRef]
- Alsowayigh, M. M.; Timco, G. A.; Borilović, I.; Alanazi, A. M.; Vitórica-Yrezábal, I. J.; Whitehead, G. F. S.; McNaughter, P. D.; Tuna, F.; O’Brien, P.; Winpenny, R. E. P.; Lewis, D. J.; Collison, D. Heterometallic 3D–4F Complexes as Air-Stable Molecular Precursors in Low Temperature Syntheses of Stoichiometric Rare-Earth Orthoferrite Powders. Inorg. Chem. 2020, 59, 15796–15806. [Google Scholar] [CrossRef] [PubMed]
- Deligne, N.; Gonze, V.; Bayot, D.; Devillers, M. Yttrium, Lanthanide and Mixed Y-Ln Vanadates Prepared from Molecular Precursors Based on EDTA. Eur. J. Inorg. Chem. 2008, 2008, 896–902. [Google Scholar] [CrossRef]
- Koroteev, P. S.; Dobrokhotova, Z. V.; Ilyukhin, A. B.; Ефимoв, Н. Н.; Kirdyankin, D. I.; Tyurin, A. V.; Гаврикoв, А. В.; Нoвoтoрцев, В. М. Polymeric Lanthanide Acetates with Peripheral Cymantrenecarboxylate Groups – Synthesis, Magnetism and Thermolysis. Polyhedron 2015, 85, 941–952. [Google Scholar] [CrossRef]
- Дoбрoхoтoва, Ж. В.; Koroteev, P. S.; Kirdyankin, D. I.; Кискин, М. А.; Kovba, M. L.; Ефимoв, Н. Н.; Гаврикoв, А. В.; Tyurin, A. V.; Нoвoтoрцев, В. М. Synthesis of Lanthanide Manganites LnMnO3 and LnMn2O5 from Individual Molecular Precursors. Russ. J. Inorg. Chem. 2015, 60, 1433–1443. [Google Scholar] [CrossRef]
- Zhu, Y.; Tan, R.; Yi, T.; Gao, S.; Yan, C.; Cao, L. Preparation of Nanosized La2CuO4 Perovskite Oxide Using an Amorphous Heteronuclear Complex as a Precursor at Low-Temperature. Journal of Alloys and Compounds 2000, 311, 16–21. [Google Scholar] [CrossRef]
- Dell’Amico, D. B.; Di Giacomo, A.; Falchi, L.; Labella, L.; Marelli, M.; Evangelisti, C.; Lezzerini, M.; Marchetti, F.; Samaritani, S. A Convenient Preparation of La2CuO4 from Molecular Precursors. Polyhedron 2017, 123, 33–38. [Google Scholar] [CrossRef]
- Xu, Q.; Pan, W.; Jing-Dong, W.; Wan, C.; Liu, Q.; Miao, H.; Mori, K.; Torigoe, T. Rare-Earth Zirconate Ceramics with Fluorite Structure for Thermal Barrier Coatings. J. Am. Ceram. Soc. 2005, 89, 340–342. [Google Scholar] [CrossRef]
- Liu, Z. -g.; Ouyang, J.; Sun, K. Improvement of Electrical Conductivity of Trivalent Rare-earth Cation-doped Neodymium Zirconate by Co-doping Gadolinium and Ytterbium. Fuel Cells 2010, 10, 1050–1056. [Google Scholar] [CrossRef]
- Zuniga, J. P.; Gupta, S. K.; Abdou, M.; Mao, Y. Effect of Molten Salt Synthesis Processing Duration on the Photo- and Radioluminescence of UV-, Visible-, and X-Ray-Excitable LA2HF2O7:EU3+ Nanoparticles. ACS Omega 2018, 3, 7757–7770. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Molecular structure of [RE(H2O)9]3+ complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms were omitted for clarity.
Figure 1.
Molecular structure of [RE(H2O)9]3+ complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms were omitted for clarity.
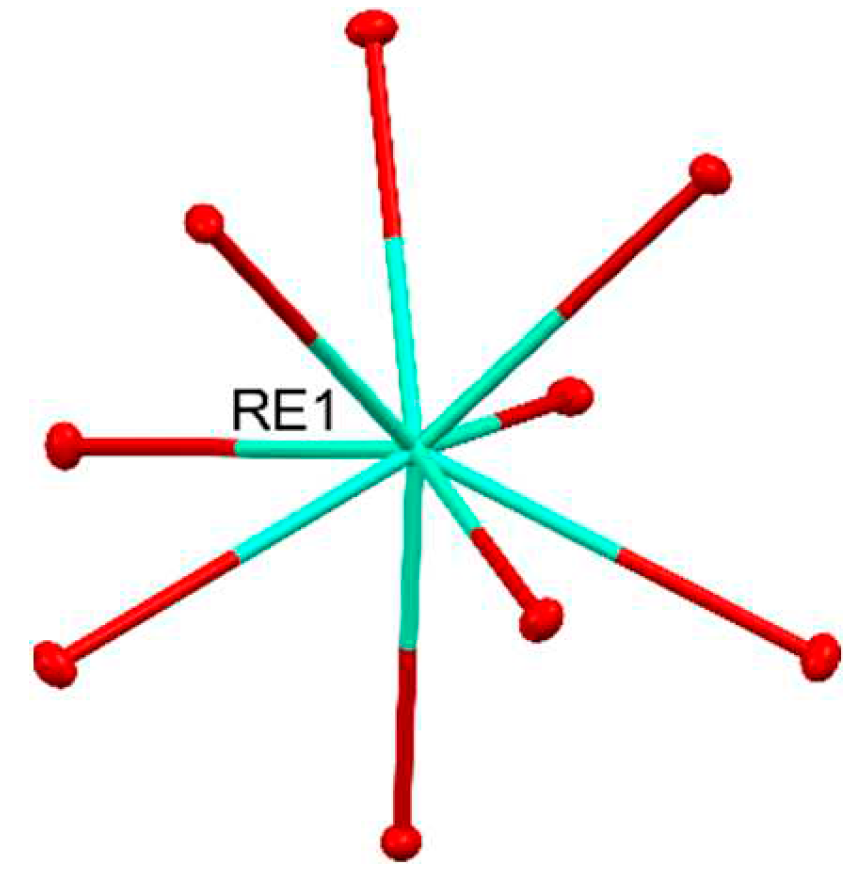
Figure 2.
Molecular structures of [RETi4(O)3(OiPr)2(OMc)11] (a), [RE2Ti6O6(OMc)18(HOiPr)] (b) and [RE2Ti4O4(OMc)14(HOMc)2] (c). Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 2.
Molecular structures of [RETi4(O)3(OiPr)2(OMc)11] (a), [RE2Ti6O6(OMc)18(HOiPr)] (b) and [RE2Ti4O4(OMc)14(HOMc)2] (c). Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
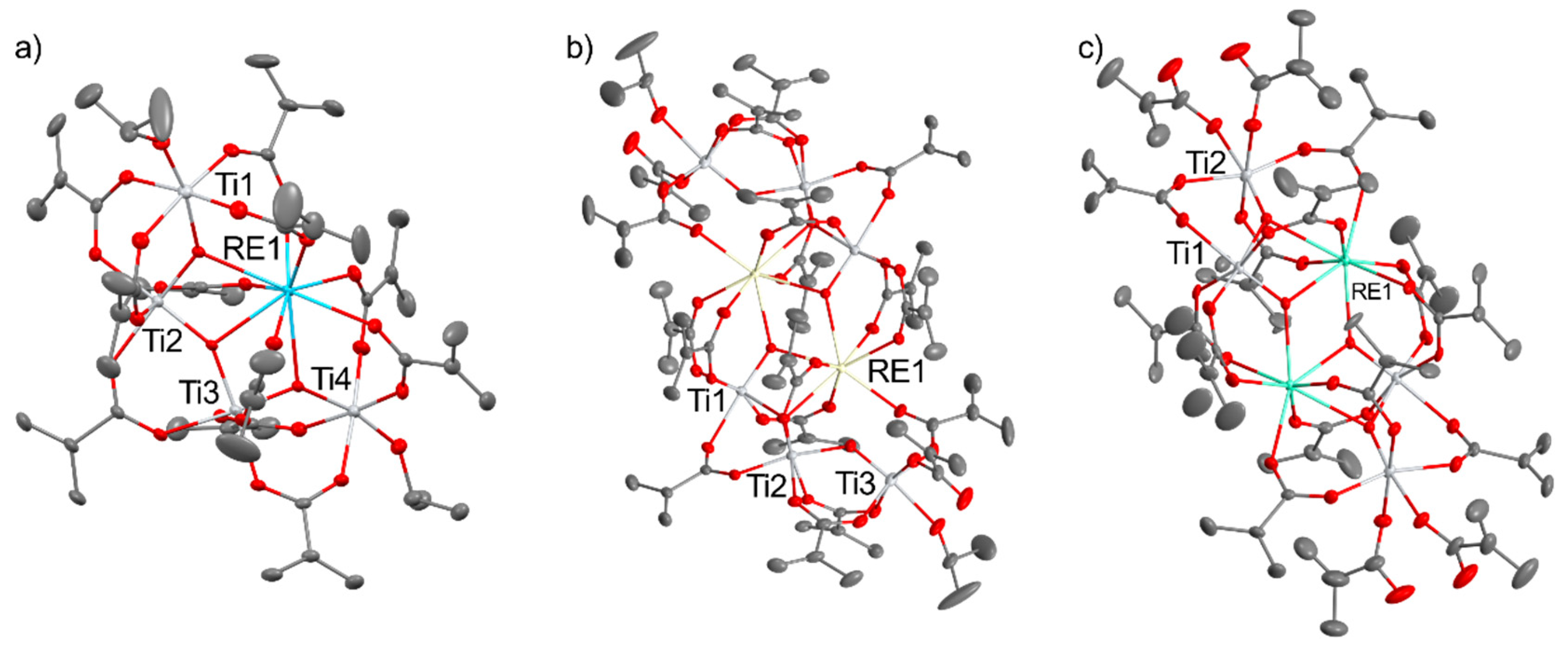
Figure 3.
Synthesis of various ytterbium complexes using [Zn3(OAc)2(L)2(H2O)2]2+ and various ytterbium salts.
Figure 3.
Synthesis of various ytterbium complexes using [Zn3(OAc)2(L)2(H2O)2]2+ and various ytterbium salts.
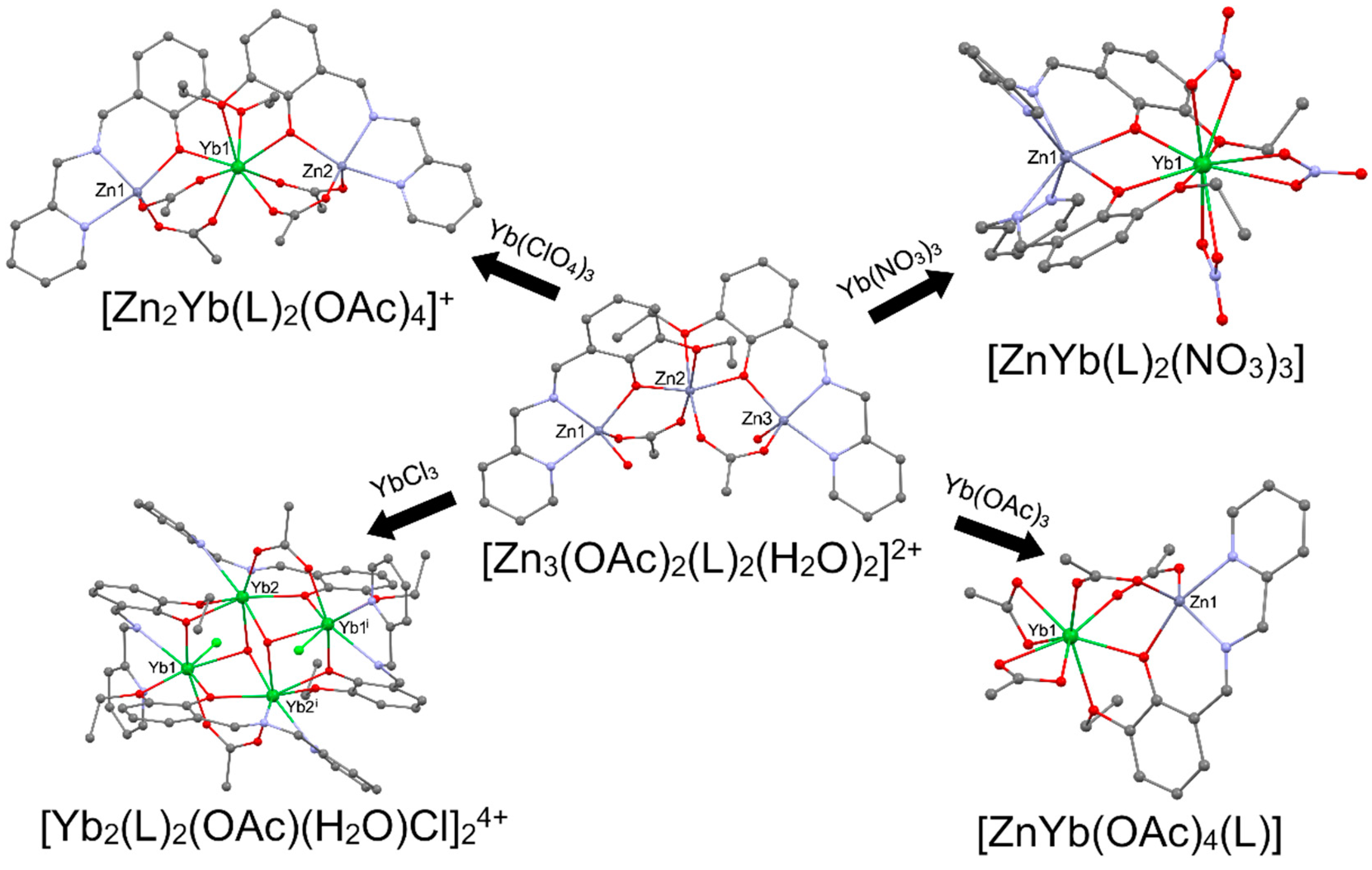
Figure 4.
Molecular structure of [Co2RE2(OAc)4(L)2(THF)n] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 4.
Molecular structure of [Co2RE2(OAc)4(L)2(THF)n] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
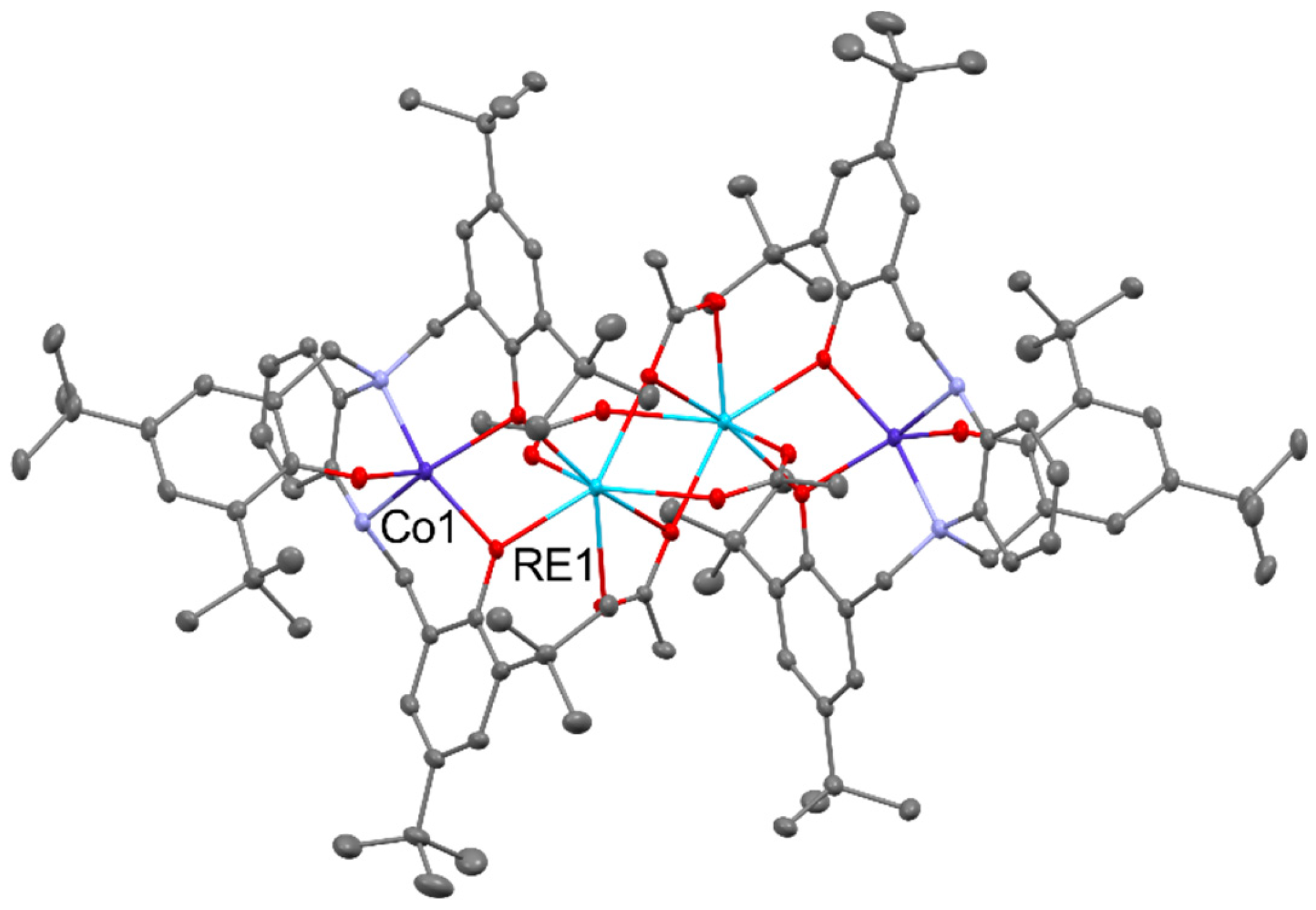
Figure 5.
Molecular structure of [Cu2Ln(L)2(NO3)2]+ cation. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 5.
Molecular structure of [Cu2Ln(L)2(NO3)2]+ cation. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
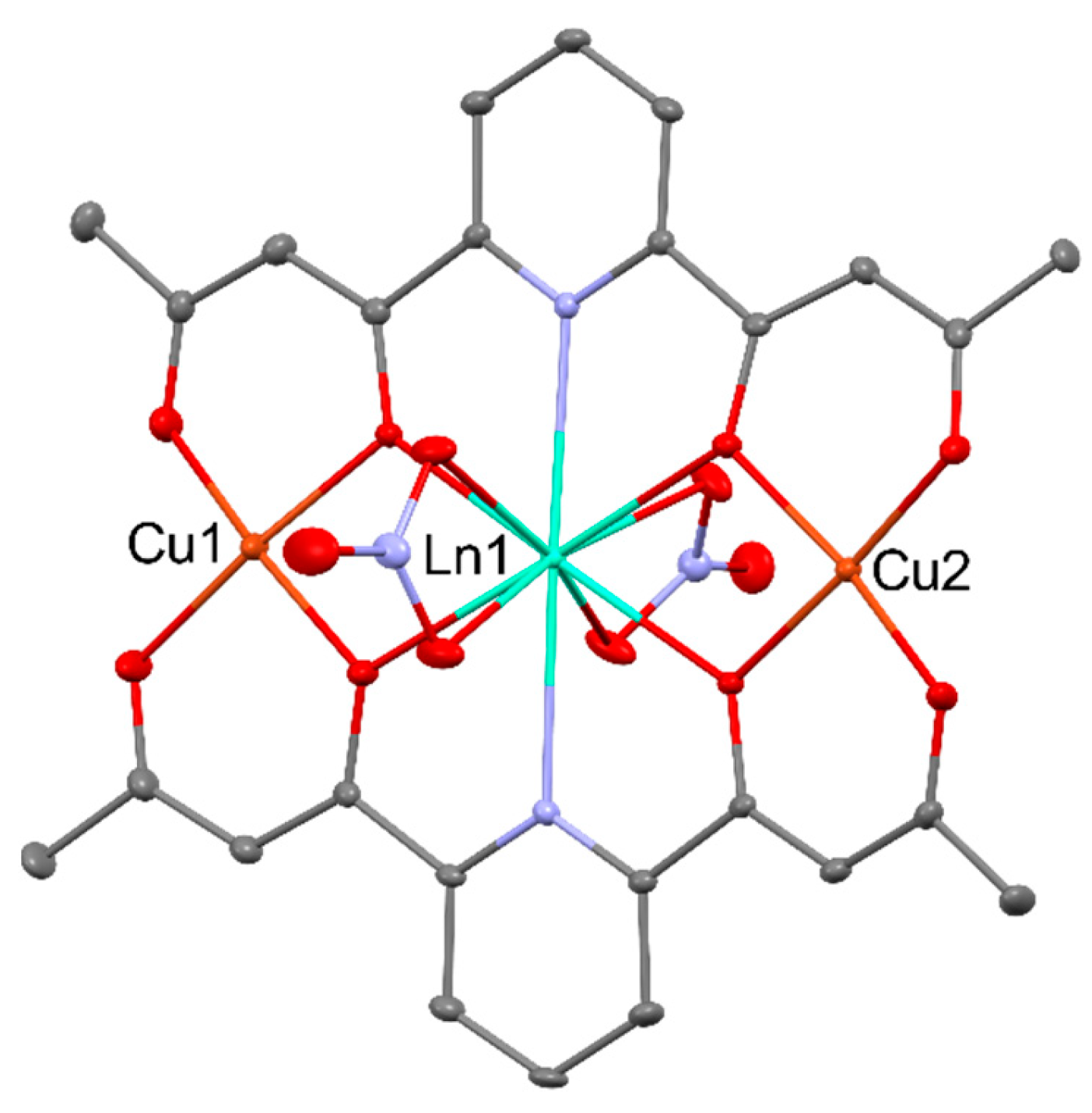
Figure 6.
Molecular structure of [NiLn(valpn)(CH3CN)2(NO3)3] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 6.
Molecular structure of [NiLn(valpn)(CH3CN)2(NO3)3] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
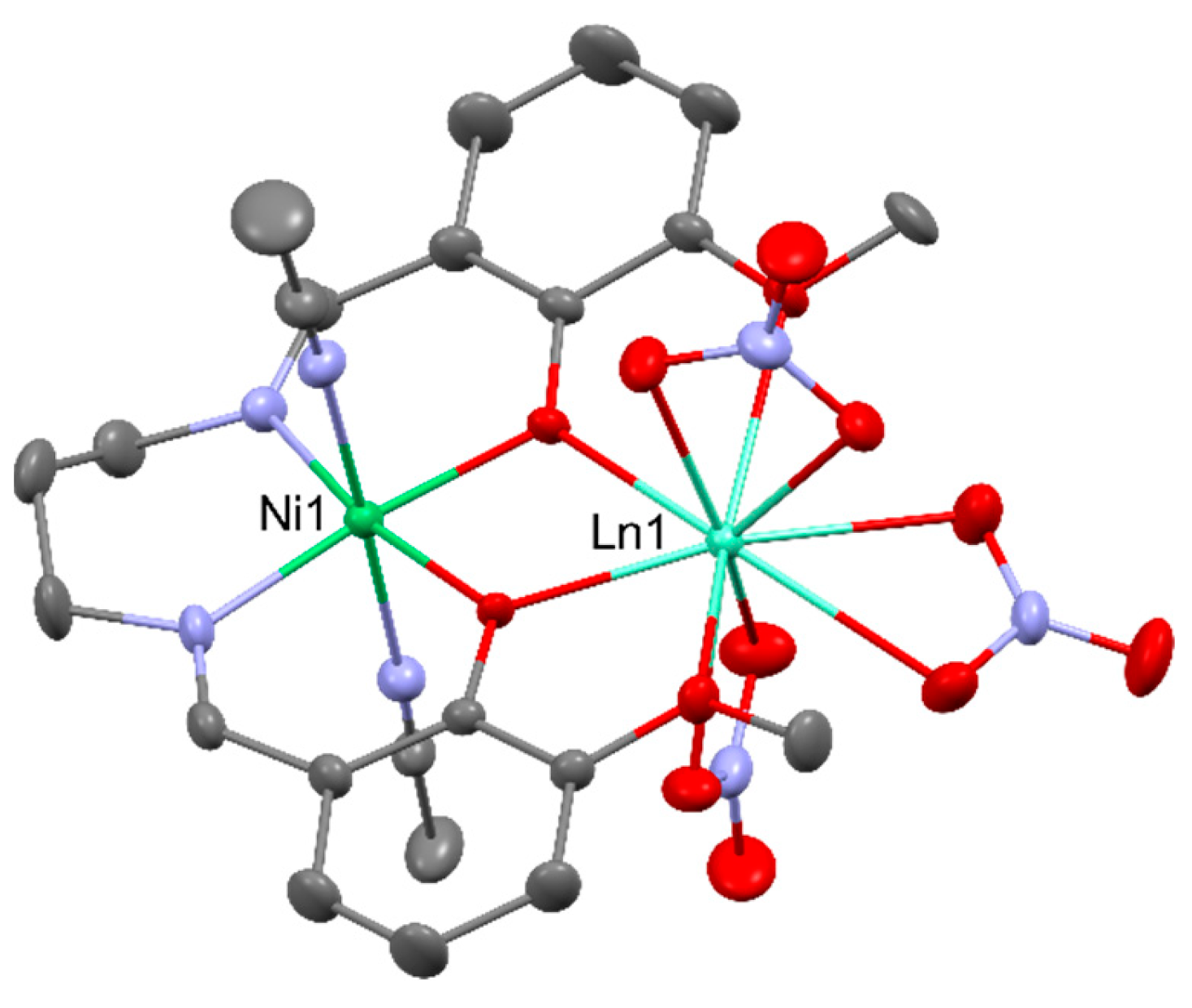
Figure 7.
Molecular structure of [CuLn(L)(NO3)3] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 7.
Molecular structure of [CuLn(L)(NO3)3] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
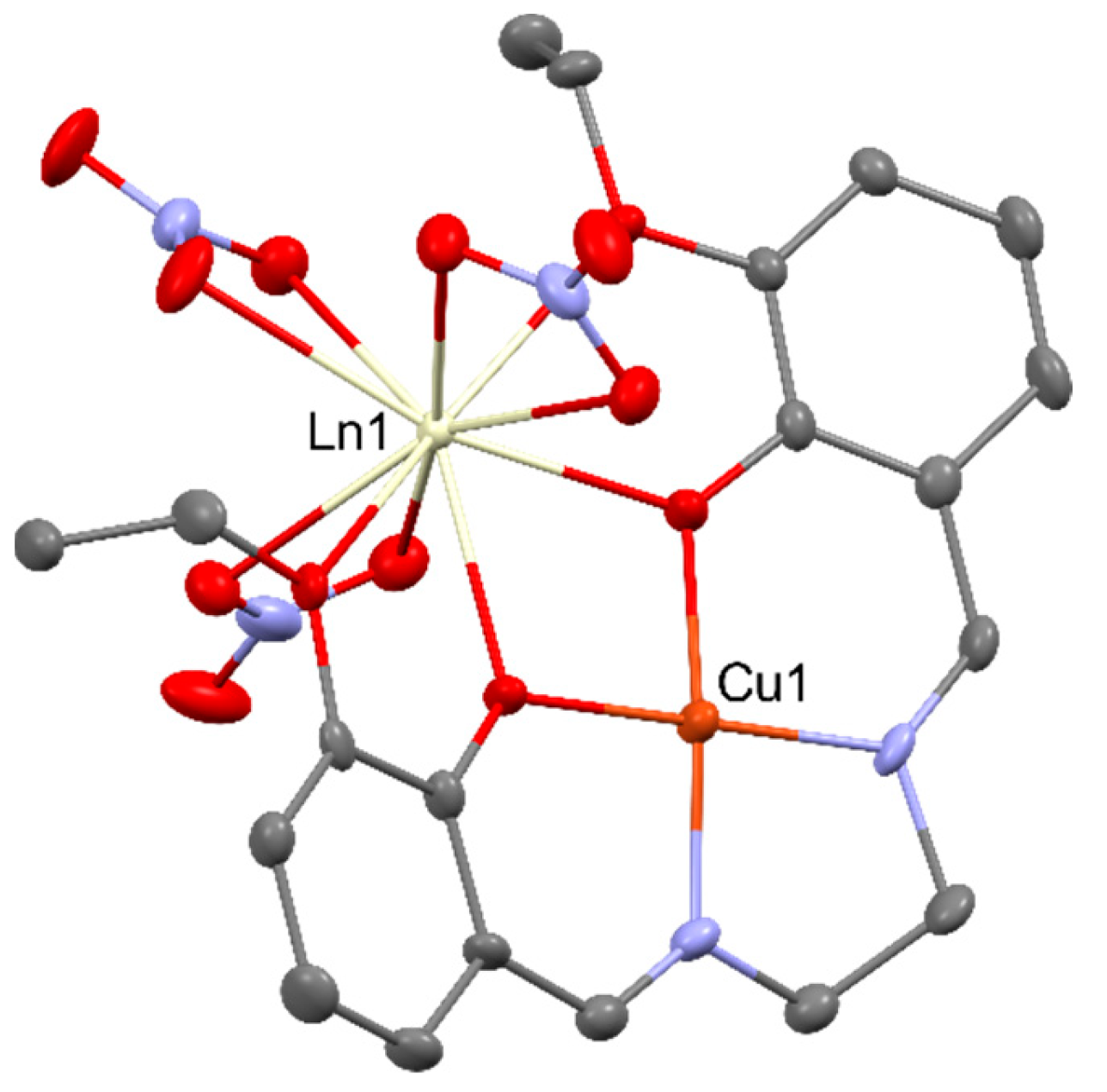
Figure 8.
Molecular structure of [CuLn(L)(Me2CO)(NO3)3] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 8.
Molecular structure of [CuLn(L)(Me2CO)(NO3)3] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
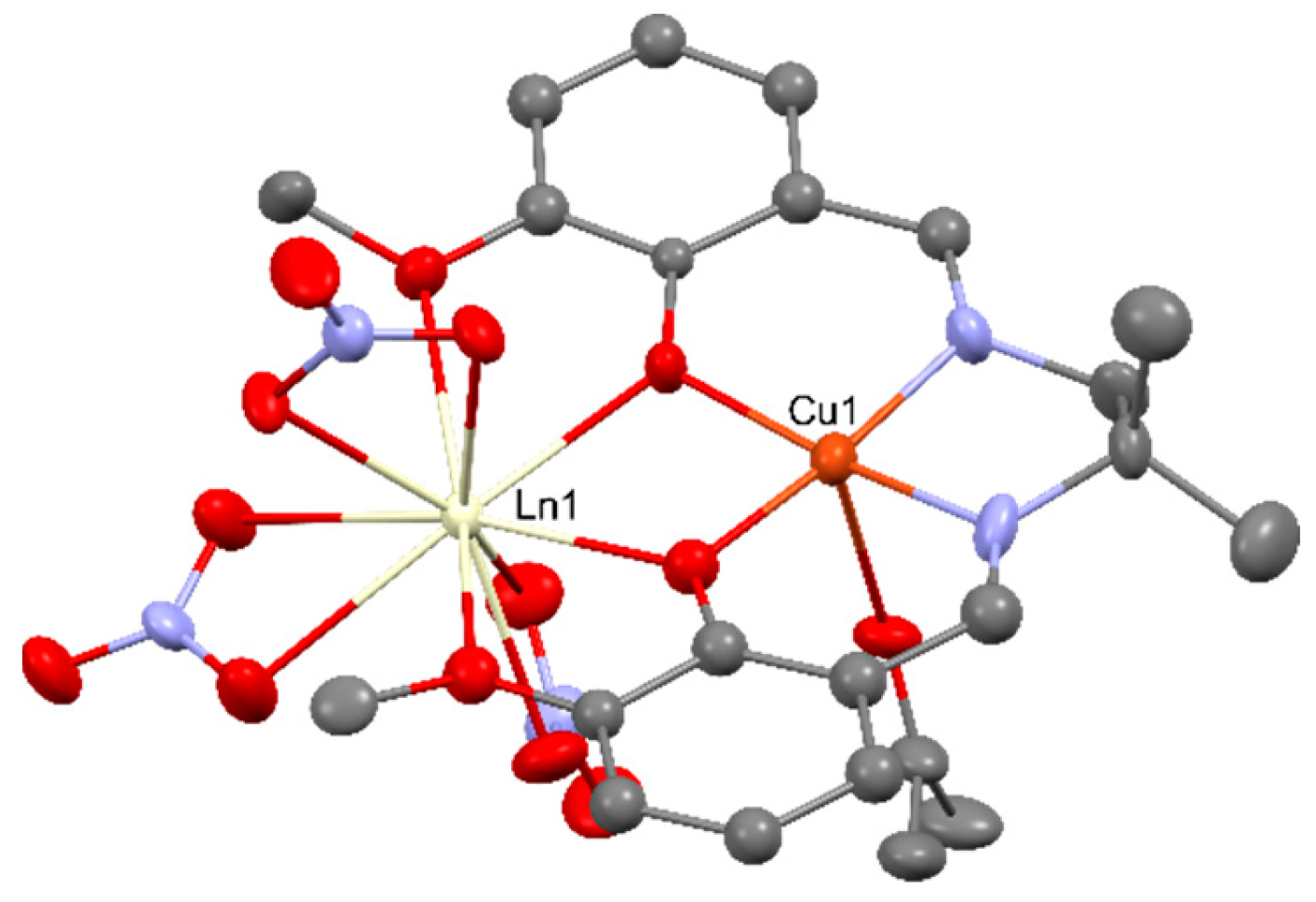
Figure 9.
Molecular structure of [MnLn2(QCl)8] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 9.
Molecular structure of [MnLn2(QCl)8] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
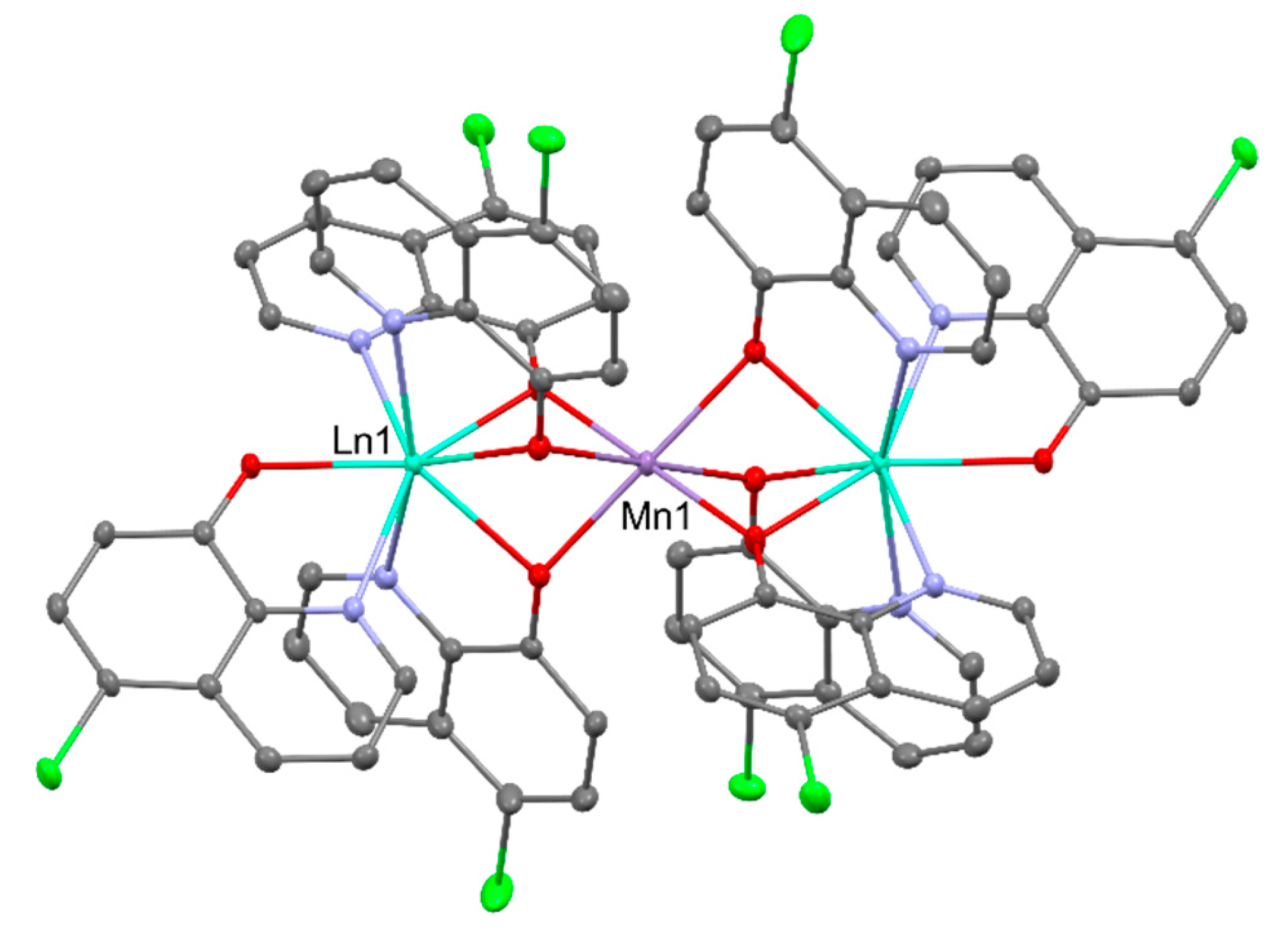
Figure 10.
Molecular structures of [NiDy(valpn)(hfac)2(N3)(H2O)2] (a), [NiDy(L)(ArCOO)(NO3)2] (b), [Ni2Dy2(μ4-CO3)2(3-MeOsaltn)2(H2O)2(NO3)2] (c) and [Ni2Dy2(L)4(NO3)2(MeOH)2] (d). Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 10.
Molecular structures of [NiDy(valpn)(hfac)2(N3)(H2O)2] (a), [NiDy(L)(ArCOO)(NO3)2] (b), [Ni2Dy2(μ4-CO3)2(3-MeOsaltn)2(H2O)2(NO3)2] (c) and [Ni2Dy2(L)4(NO3)2(MeOH)2] (d). Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
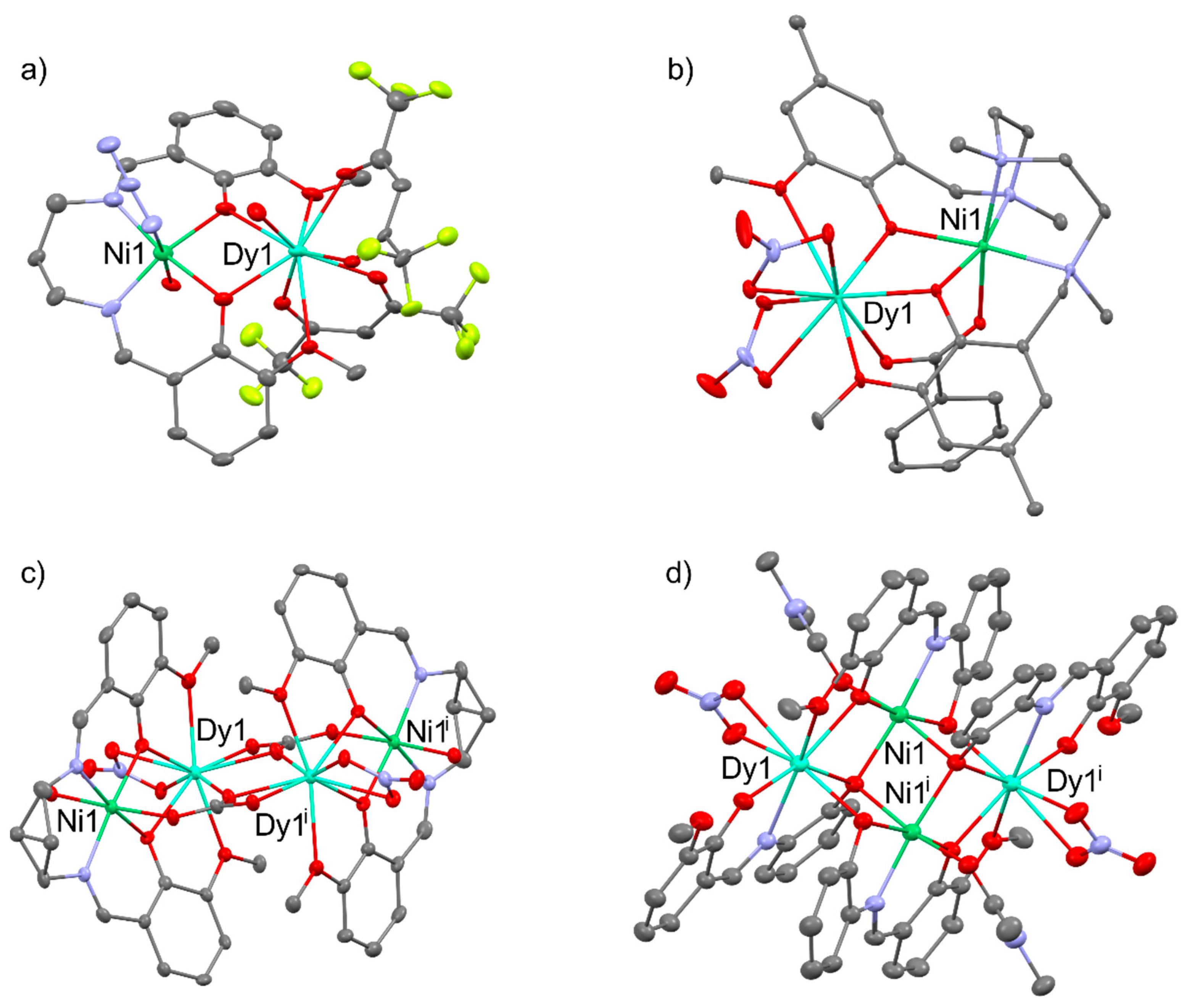
Figure 11.
Molecular structure of [ZnDy(NO3)3(L)(H2O)]. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 11.
Molecular structure of [ZnDy(NO3)3(L)(H2O)]. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
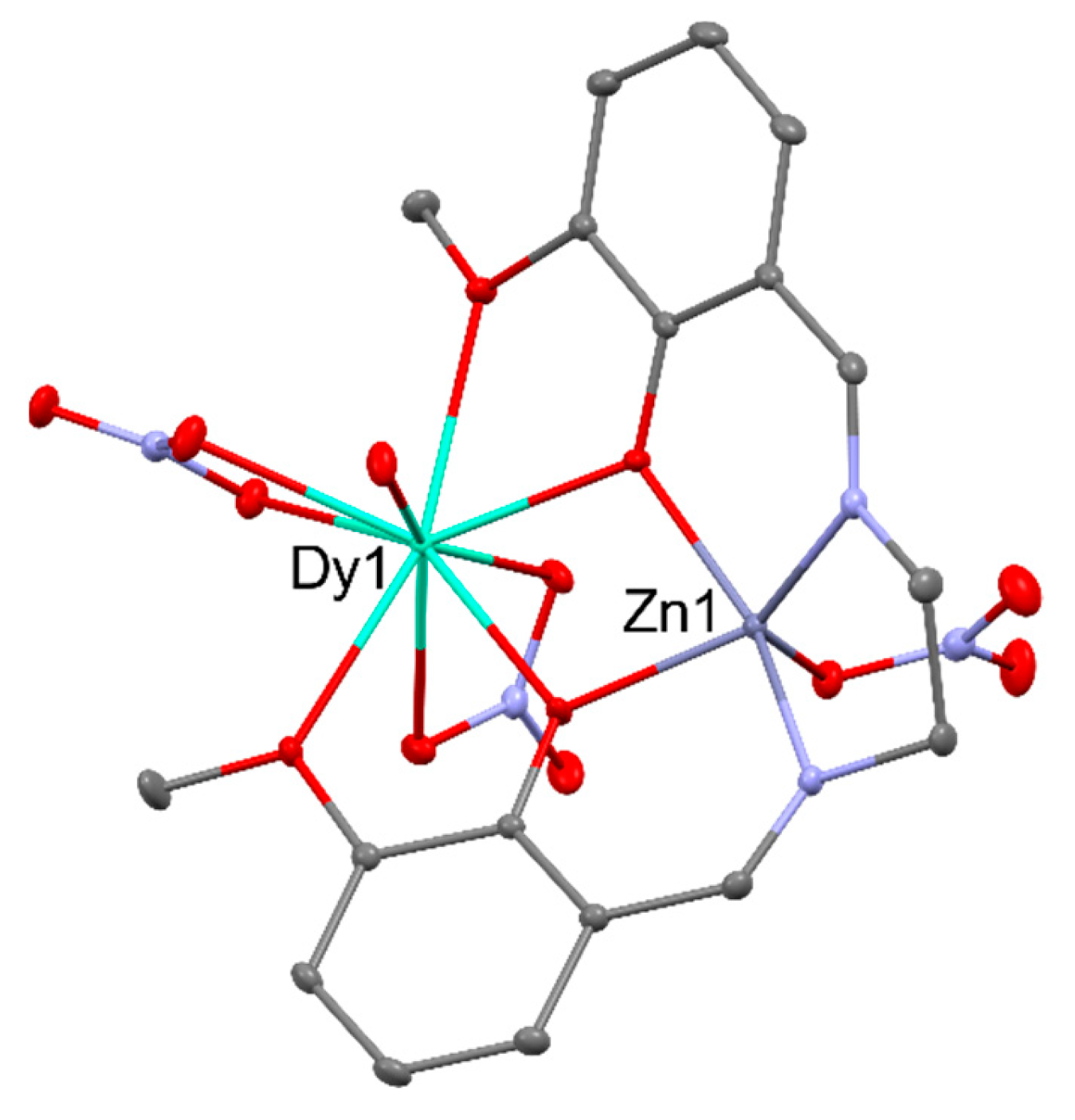
Figure 12.
General structure of Shibasaki catalysts of formula [M’3RE(binol)3].
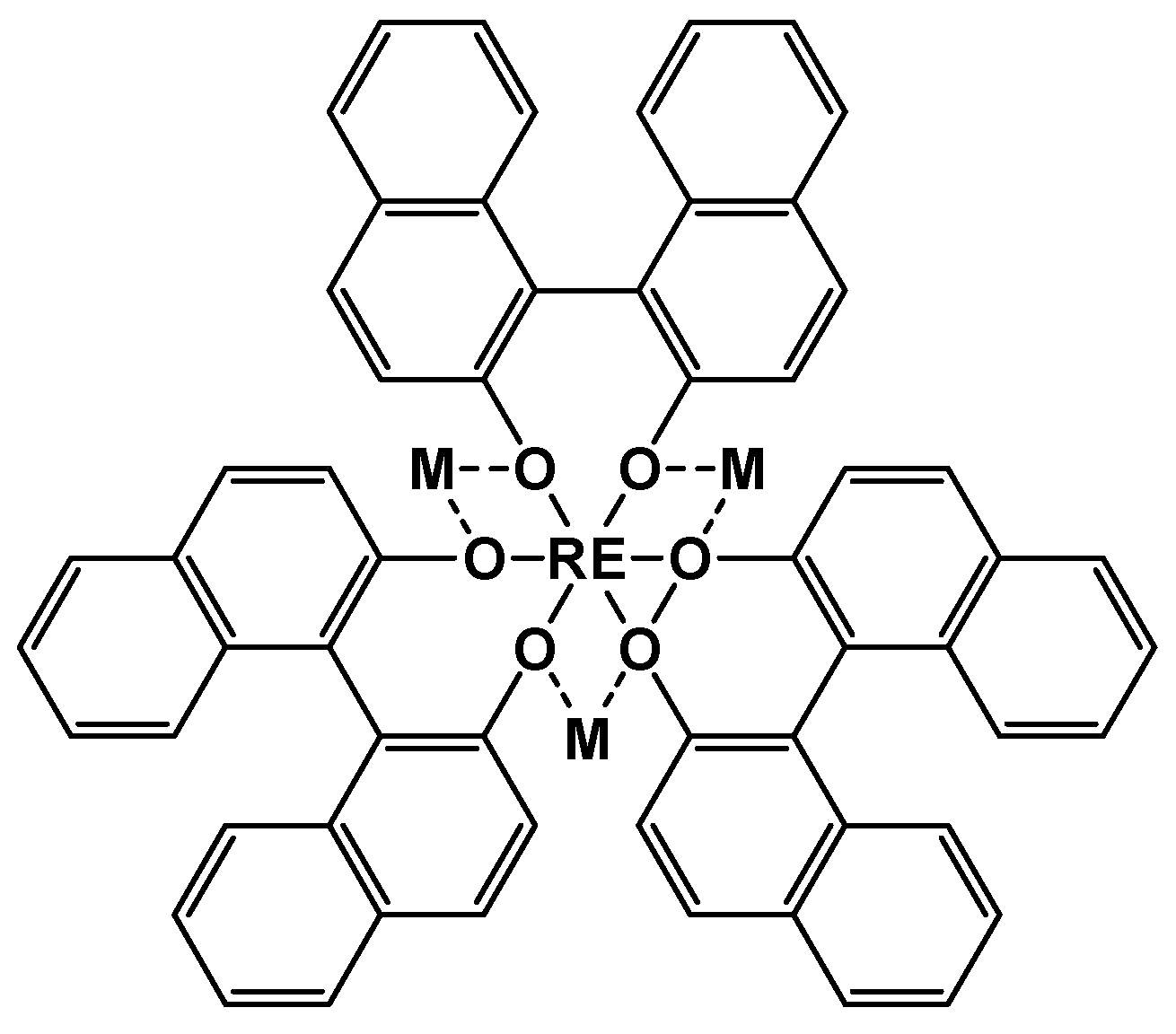
Figure 13.
Molecular structure of [(EtZn)3RE(binol)3(THF)3]. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 13.
Molecular structure of [(EtZn)3RE(binol)3(THF)3]. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
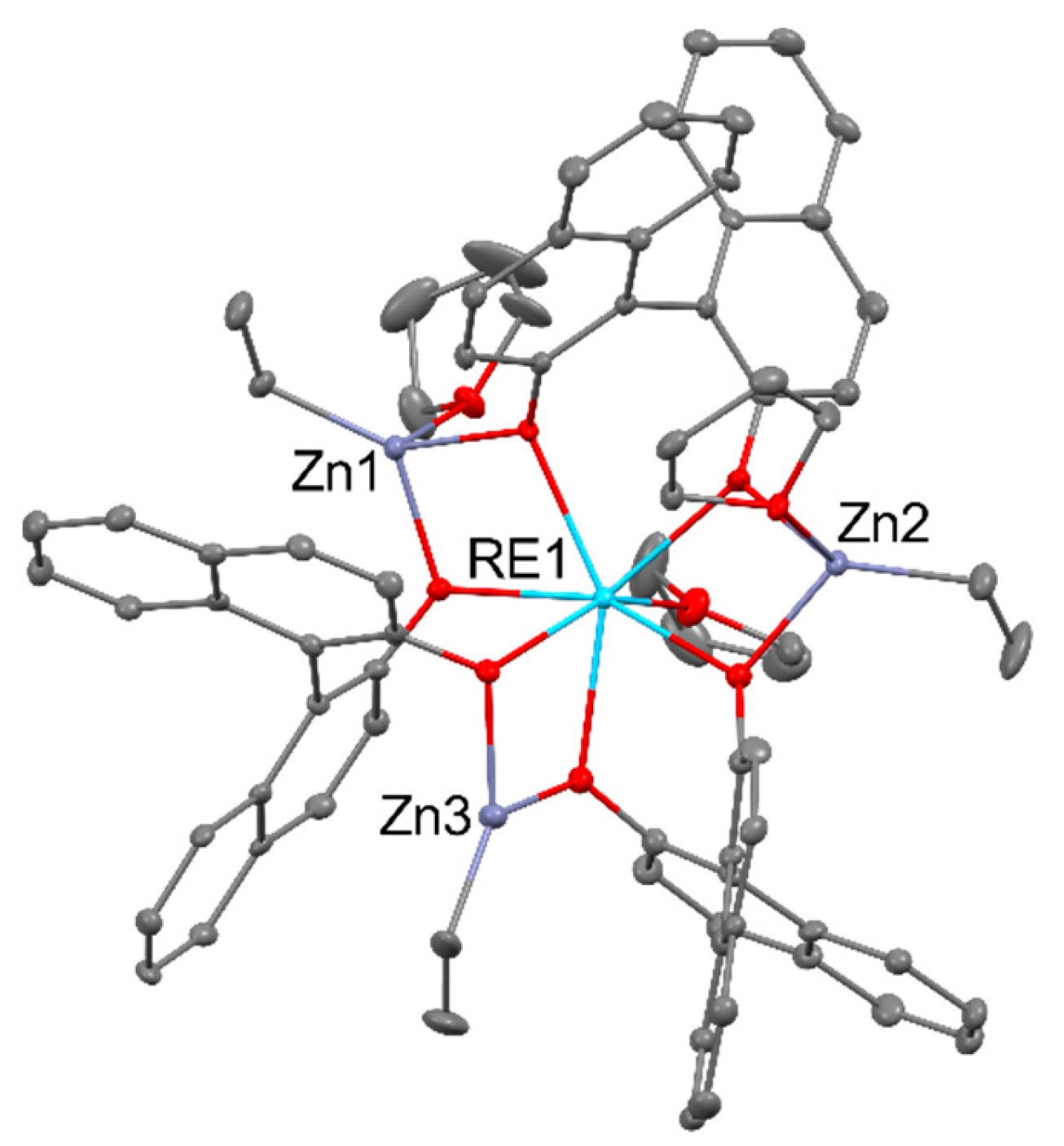
Figure 14.
Proposed catalytic cycle for [NiRE(L)3]-catalyzed hydrogenation of DPA and isomerization of stillbene. Reprinted with permission from [77] Ramirez, B.; Lu, C. C. Rare-Earth Supported Nickel Catalysts for Alkyne Semihydrogenation: Chemo- and Regioselectivity Impacted by the Lewis Acidity and Size of the Support. Journal of the American Chemical Society 2020, 142 (11), 5396–5407. https://doi.org/10.1021/jacs.0c00905. Copyright © 2020 American Chemical Society.
Figure 14.
Proposed catalytic cycle for [NiRE(L)3]-catalyzed hydrogenation of DPA and isomerization of stillbene. Reprinted with permission from [77] Ramirez, B.; Lu, C. C. Rare-Earth Supported Nickel Catalysts for Alkyne Semihydrogenation: Chemo- and Regioselectivity Impacted by the Lewis Acidity and Size of the Support. Journal of the American Chemical Society 2020, 142 (11), 5396–5407. https://doi.org/10.1021/jacs.0c00905. Copyright © 2020 American Chemical Society.
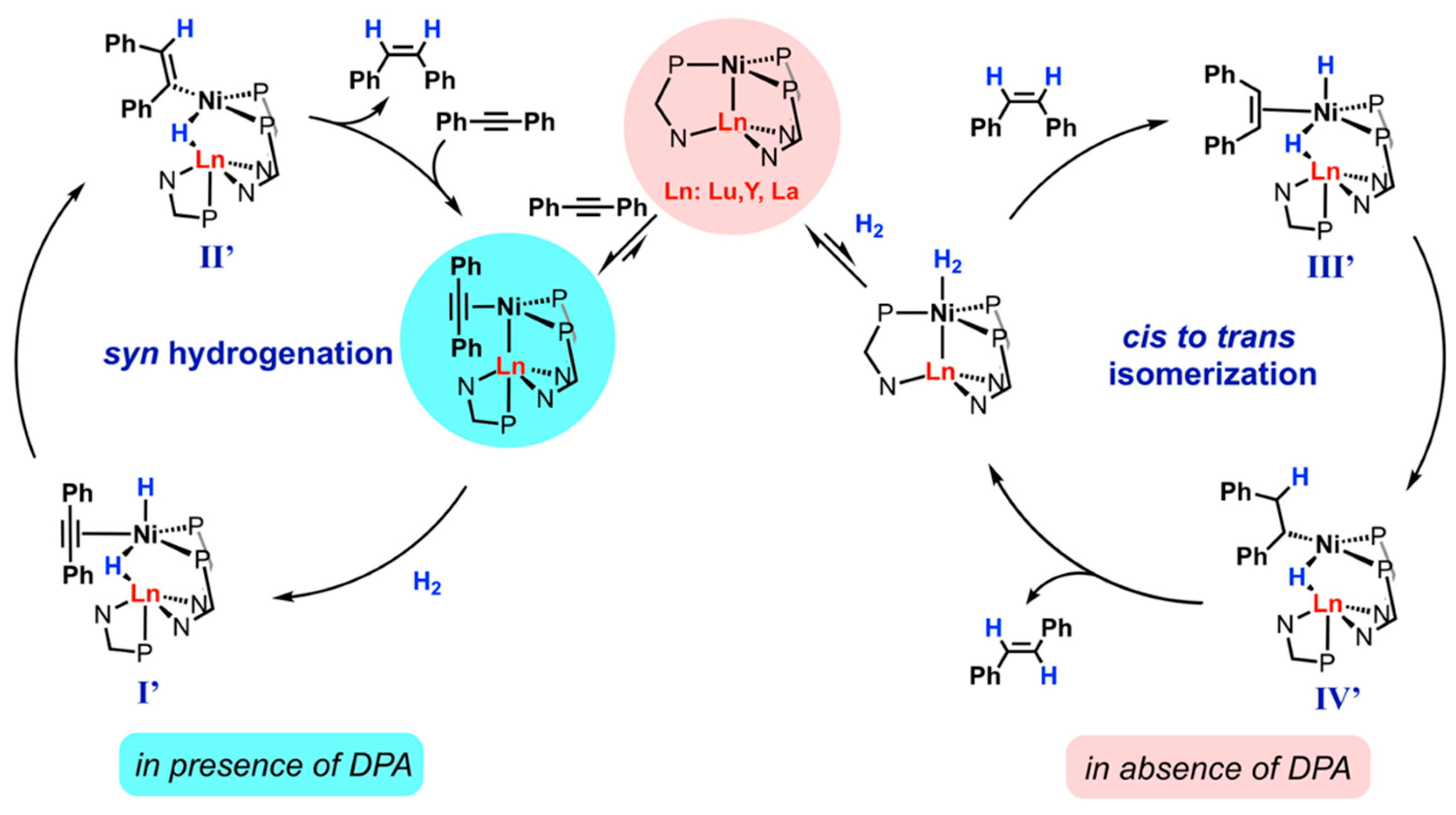
Figure 15.
Synergic cooperation of metallic centers in ROP of ε-CL initiated by [Na8Sm2(OCH2CH2NMe2)12(OH)2], [K12Na4Sm2(OCH2CH2NMe2)18(OH)4] and [K20Sm4(OCH2CH2NMe2)26(OH)6] compounds. Reproduced from [80] Sheng, H.; Shi, J.; Feng, Y.; Wang, H.; Jiao, Y.; Sheng, H.; Zhang, Y.; Shen, Q. Remarkable Effect of Alkali Metal on Polymerization of Cyclic Esters Catalyzed by Samarium–Alkali Metal Multinuclear Alkoxide Clusters. Dalton Transactions 2012, 41 (30), 9232. https://doi.org/10.1039/c2dt30677h. with permission from the Royal Society of Chemistry.
Figure 15.
Synergic cooperation of metallic centers in ROP of ε-CL initiated by [Na8Sm2(OCH2CH2NMe2)12(OH)2], [K12Na4Sm2(OCH2CH2NMe2)18(OH)4] and [K20Sm4(OCH2CH2NMe2)26(OH)6] compounds. Reproduced from [80] Sheng, H.; Shi, J.; Feng, Y.; Wang, H.; Jiao, Y.; Sheng, H.; Zhang, Y.; Shen, Q. Remarkable Effect of Alkali Metal on Polymerization of Cyclic Esters Catalyzed by Samarium–Alkali Metal Multinuclear Alkoxide Clusters. Dalton Transactions 2012, 41 (30), 9232. https://doi.org/10.1039/c2dt30677h. with permission from the Royal Society of Chemistry.
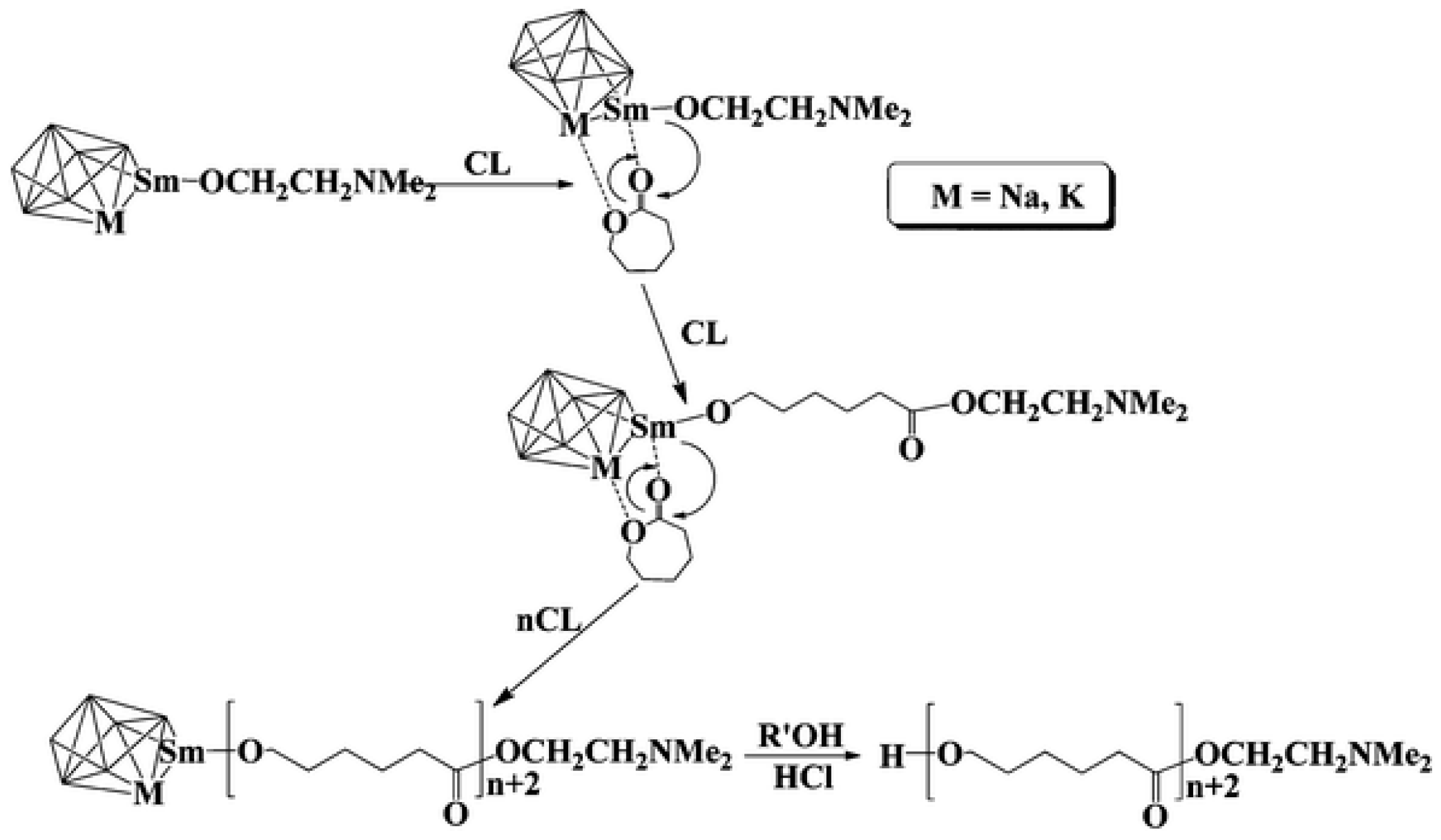
Figure 16.
Complexes of CeIII and CeIV with Ferrocene-based tetradentate Schiff base ligand. Reprinted with permission from [85] Broderick, E. M.; Guo, N.; Vogel, C.; Xu, C.; Sutter, J.; Miller, J. T.; Meyer, K.; Mehrkhodavandi, P.; Diaconescu, P. L. Redox Control of a Ring-Opening Polymerization Catalyst. Journal of the American Chemical Society 2011, 133 (24), 9278–9281. https://doi.org/10.1021/ja2036089. Copyright © 2011 American Chemical Society.
Figure 16.
Complexes of CeIII and CeIV with Ferrocene-based tetradentate Schiff base ligand. Reprinted with permission from [85] Broderick, E. M.; Guo, N.; Vogel, C.; Xu, C.; Sutter, J.; Miller, J. T.; Meyer, K.; Mehrkhodavandi, P.; Diaconescu, P. L. Redox Control of a Ring-Opening Polymerization Catalyst. Journal of the American Chemical Society 2011, 133 (24), 9278–9281. https://doi.org/10.1021/ja2036089. Copyright © 2011 American Chemical Society.
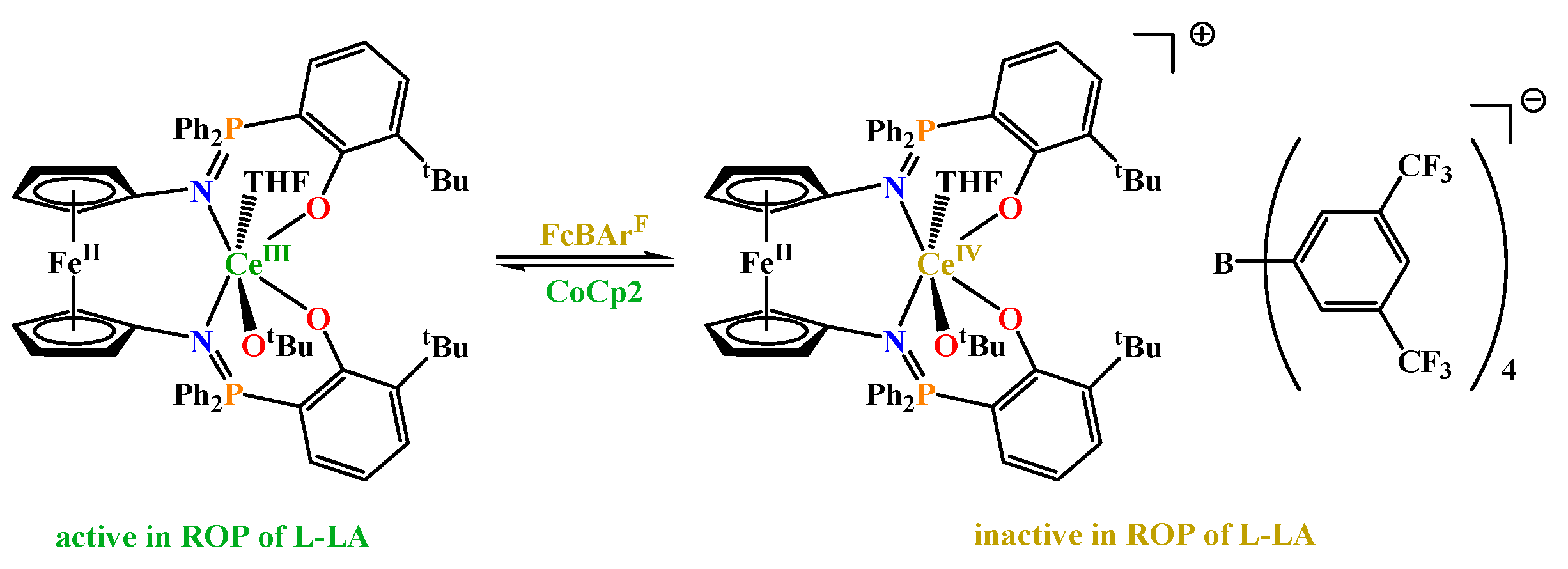
Figure 17.
Molecular structure of NiII-REIII complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms were omitted for clarity.
Figure 17.
Molecular structure of NiII-REIII complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms were omitted for clarity.
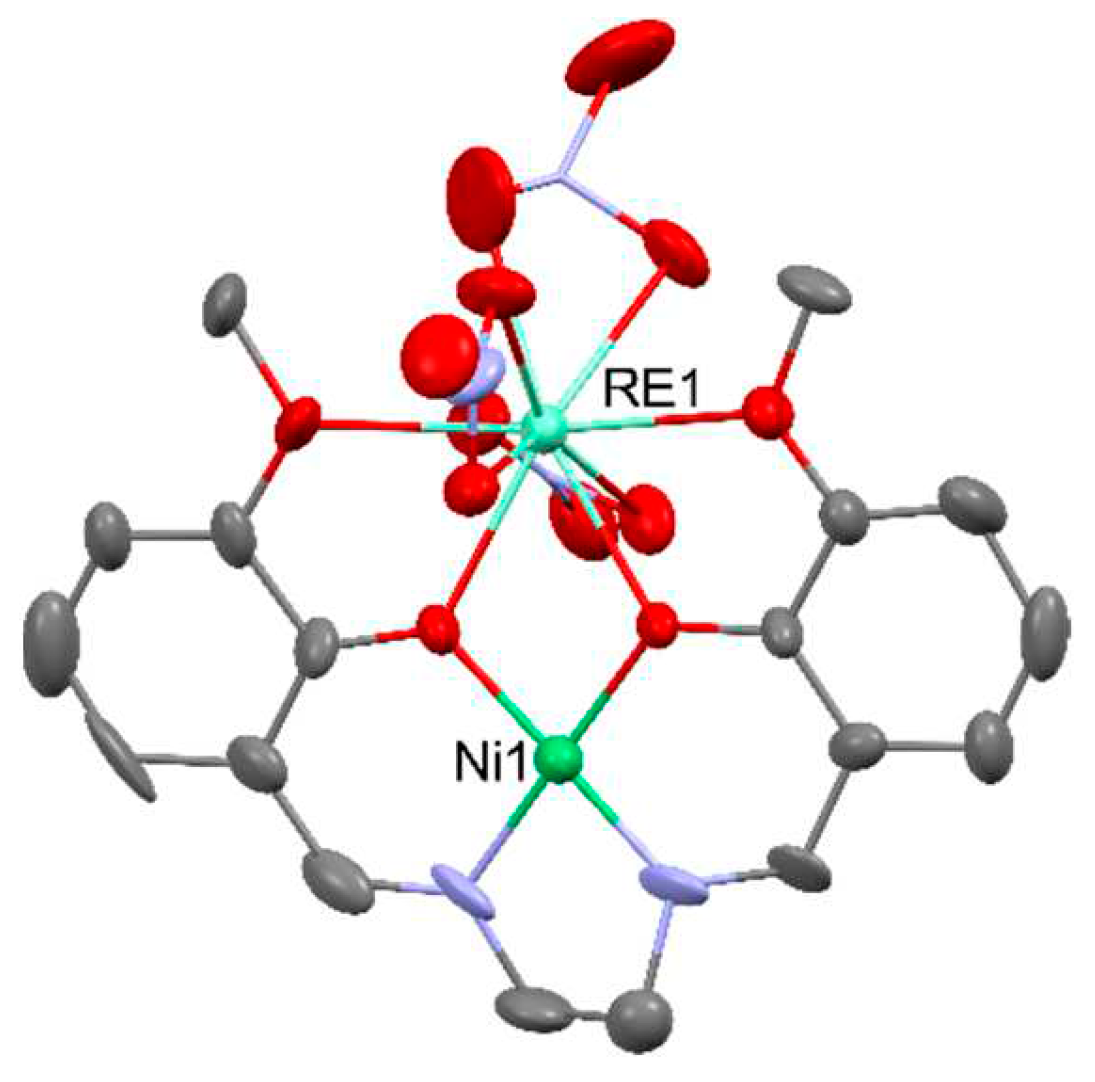
Figure 18.
Molecular structure of [CuEr(pdc)2(Hpdc)(H2O)4]n. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 18.
Molecular structure of [CuEr(pdc)2(Hpdc)(H2O)4]n. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
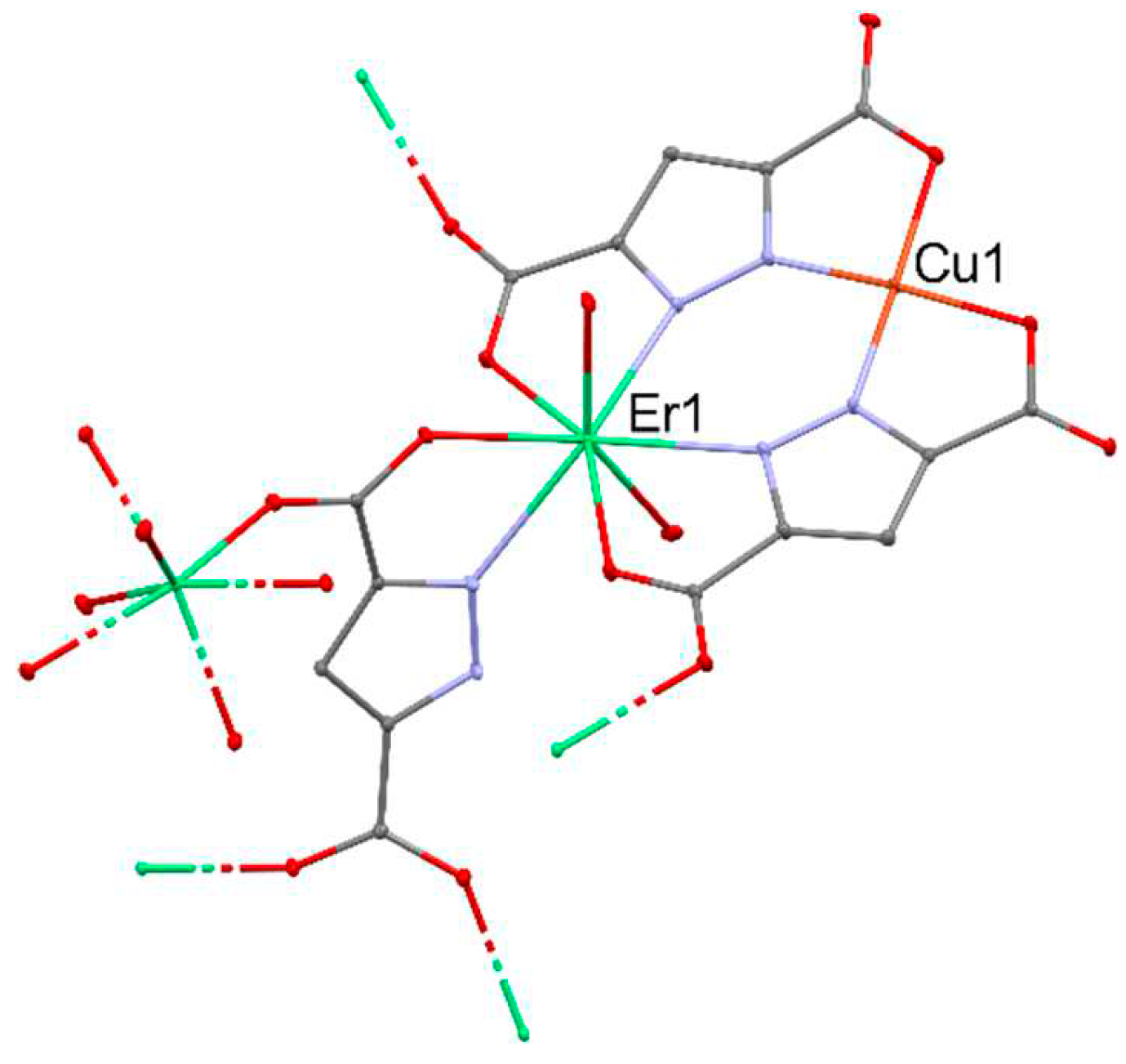
Figure 19.
Molecular structure of {[Cu3Lu2(L)6(H2O)6]·10H2O}n. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 19.
Molecular structure of {[Cu3Lu2(L)6(H2O)6]·10H2O}n. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
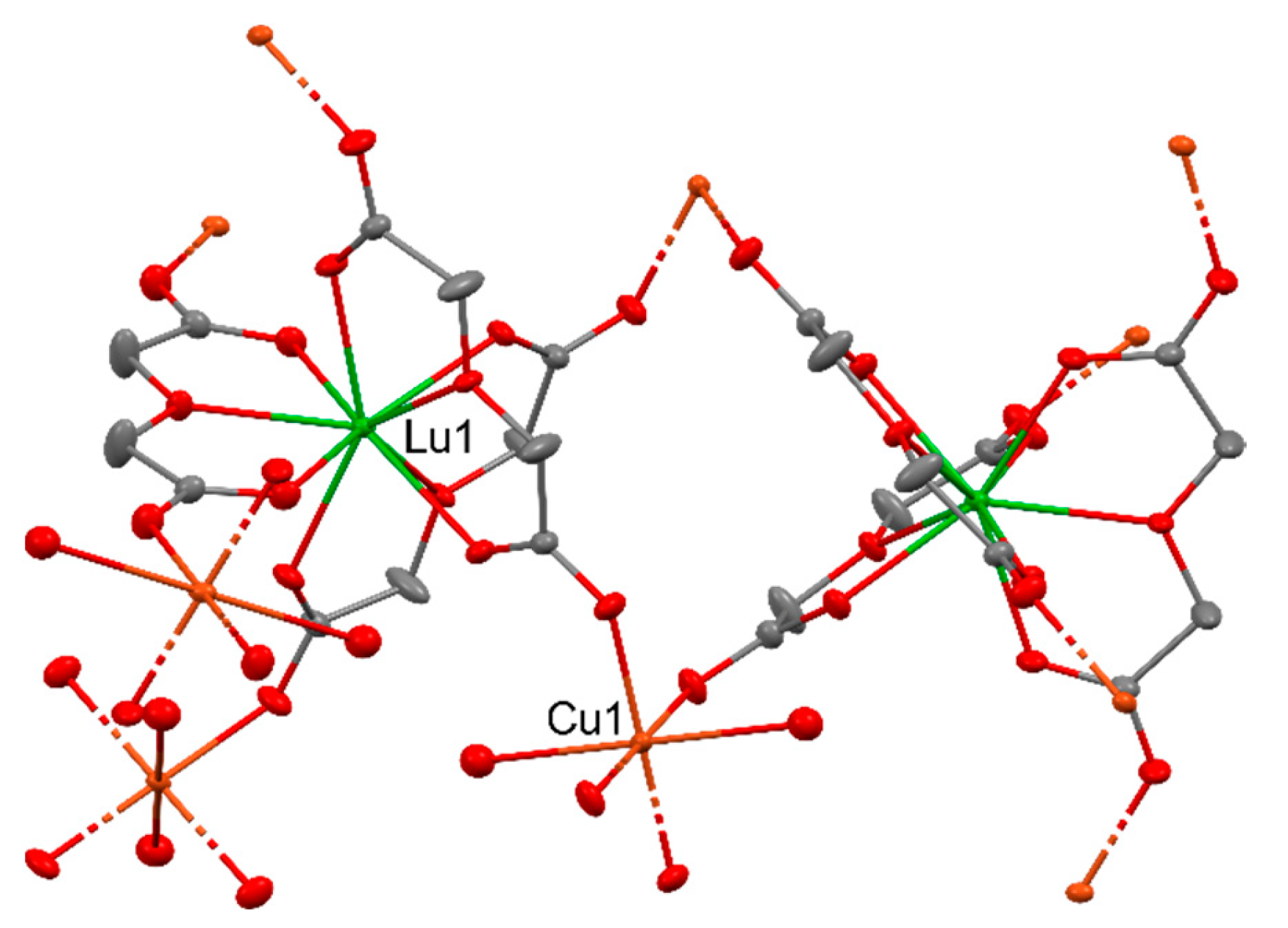
Figure 20.
Possible conversions of CO2 in synthetic chemistry.
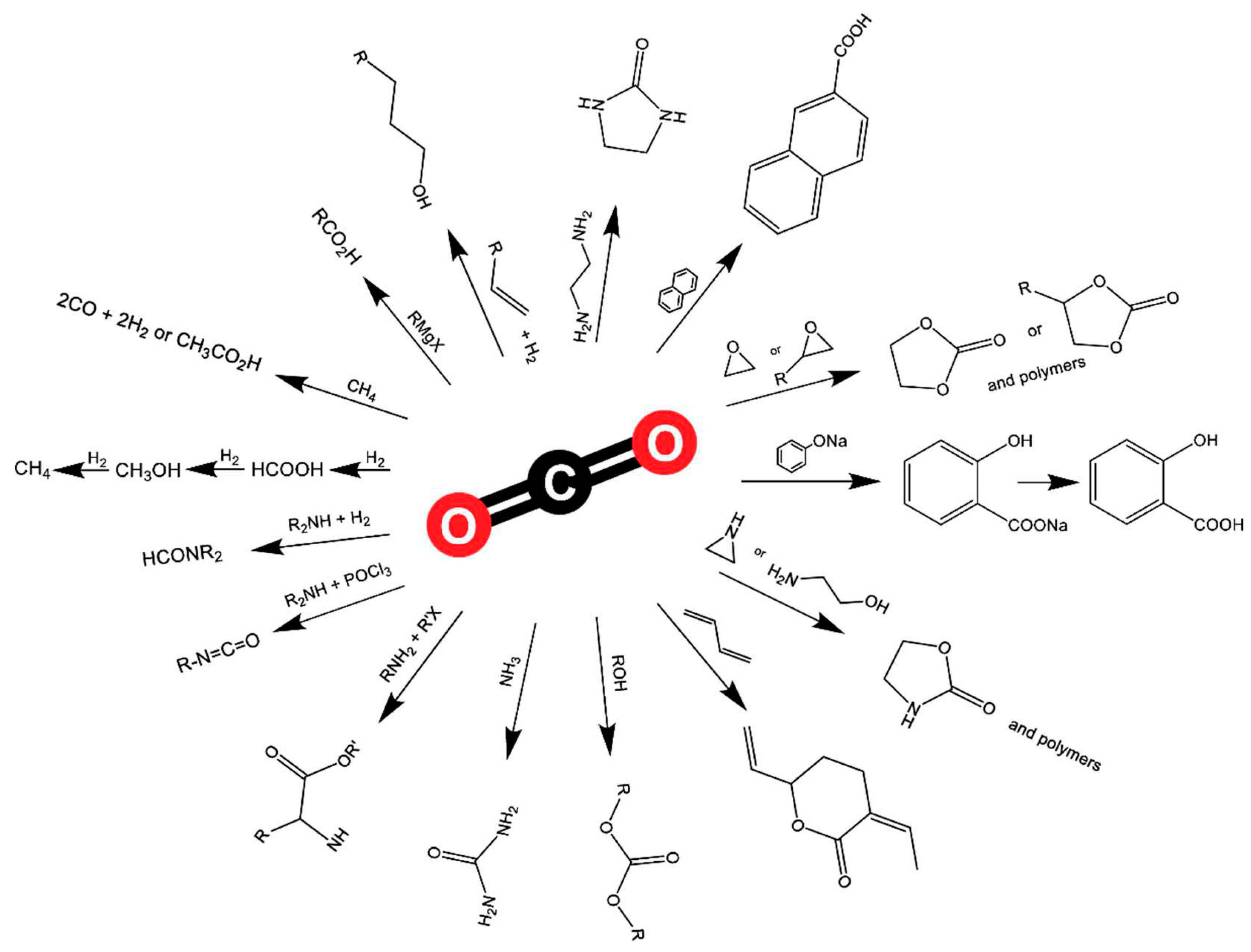
Figure 21.
Cycloaddition of terminal epoxides and CO2 catalyzed by [La(L)(THF)] (L3- = 2,2’-(({2-({(3,5-di-t-butyl-2-(hydroxy)phenyl]methyl}(methyl)amino)ethyl}azanediyl)bis(methylene))bis(4,6-di-t-butylphenolato)}). Reprinted with permission from [90] Xin, X.; Shan, H.; Tian, T.; Wang, Y.; Yuan, D.; You, H.; Yao, Y. Conversion of CO2 into Cyclic Carbonates under Ambient Conditions Catalyzed by Rare-Earth Metal Complexes Bearing Poly(Phenolato) Ligand. ACS Sustainable Chemistry & Engineering 2020, 8 (35), 13185–13194. https://doi.org/10.1021/acssuschemeng.0c01736. Copyright © 2020 American Chemical Society.
Figure 21.
Cycloaddition of terminal epoxides and CO2 catalyzed by [La(L)(THF)] (L3- = 2,2’-(({2-({(3,5-di-t-butyl-2-(hydroxy)phenyl]methyl}(methyl)amino)ethyl}azanediyl)bis(methylene))bis(4,6-di-t-butylphenolato)}). Reprinted with permission from [90] Xin, X.; Shan, H.; Tian, T.; Wang, Y.; Yuan, D.; You, H.; Yao, Y. Conversion of CO2 into Cyclic Carbonates under Ambient Conditions Catalyzed by Rare-Earth Metal Complexes Bearing Poly(Phenolato) Ligand. ACS Sustainable Chemistry & Engineering 2020, 8 (35), 13185–13194. https://doi.org/10.1021/acssuschemeng.0c01736. Copyright © 2020 American Chemical Society.
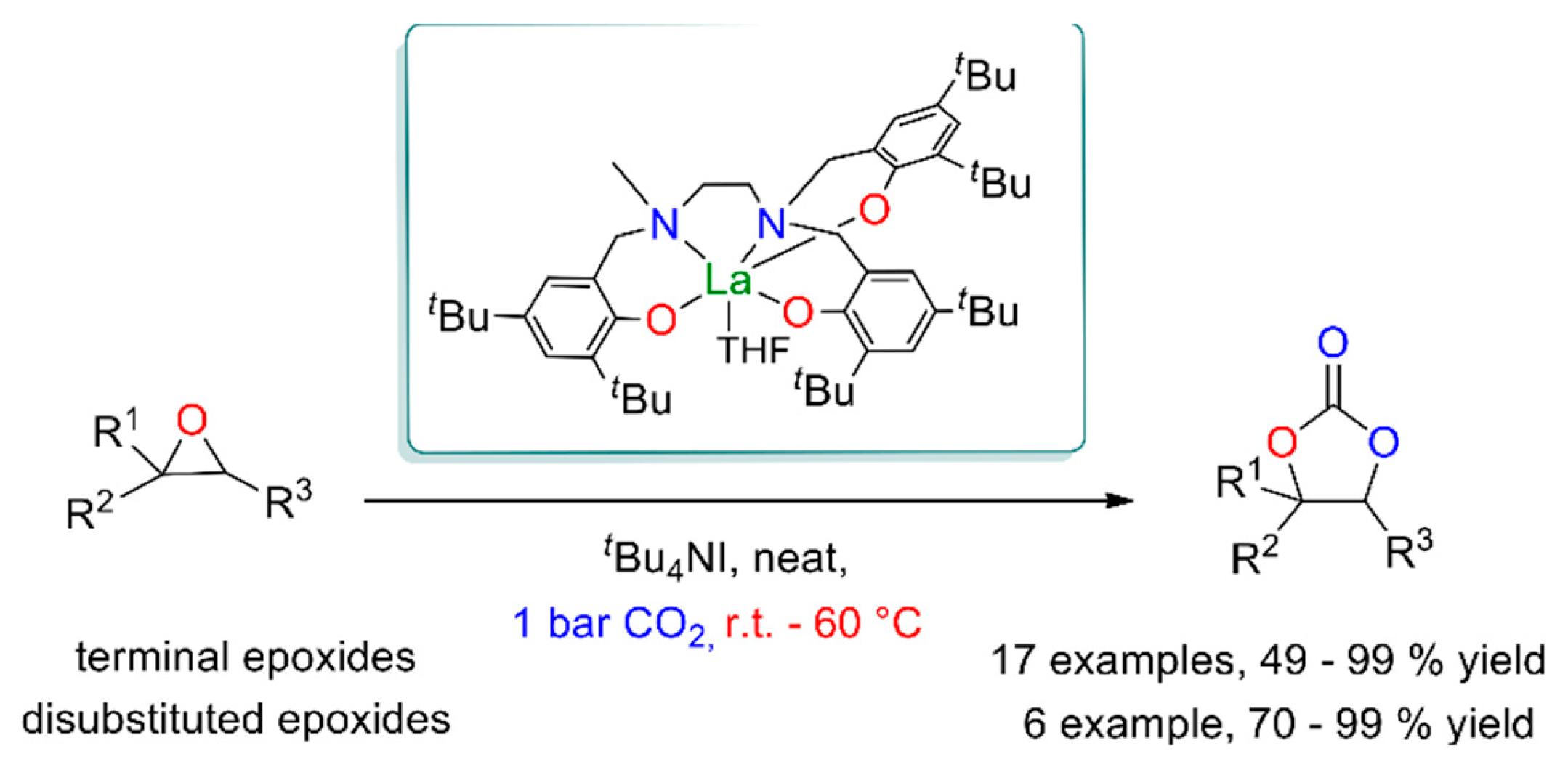
Figure 22.
Molecular structure of [ZnLa2(OBn)2(L)2]. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 22.
Molecular structure of [ZnLa2(OBn)2(L)2]. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
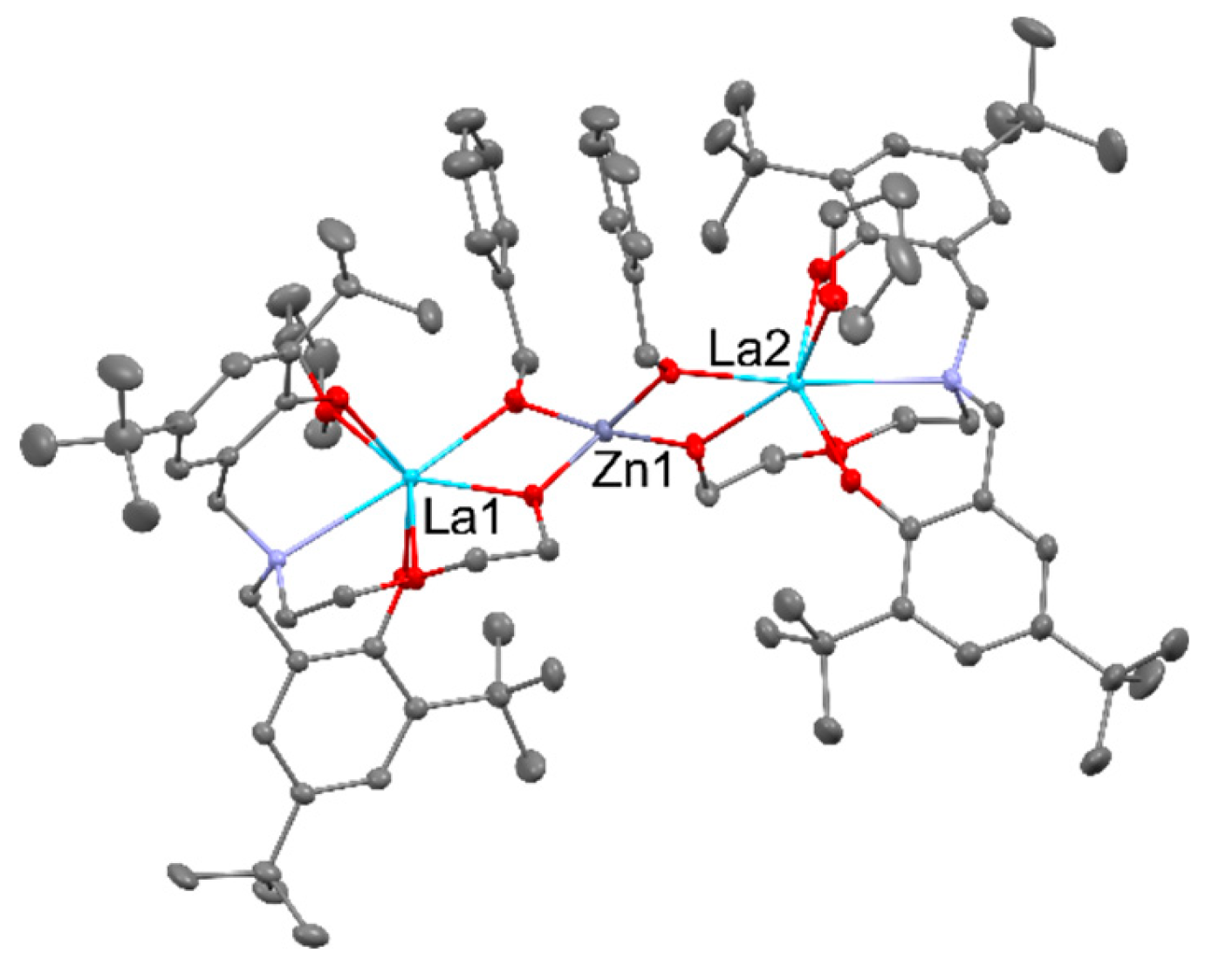
Figure 23.
Molecular structure of [Zn4RE2(μ3-OH)2L4(OAc)2(NO3)2(DMF)2] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 23.
Molecular structure of [Zn4RE2(μ3-OH)2L4(OAc)2(NO3)2(DMF)2] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
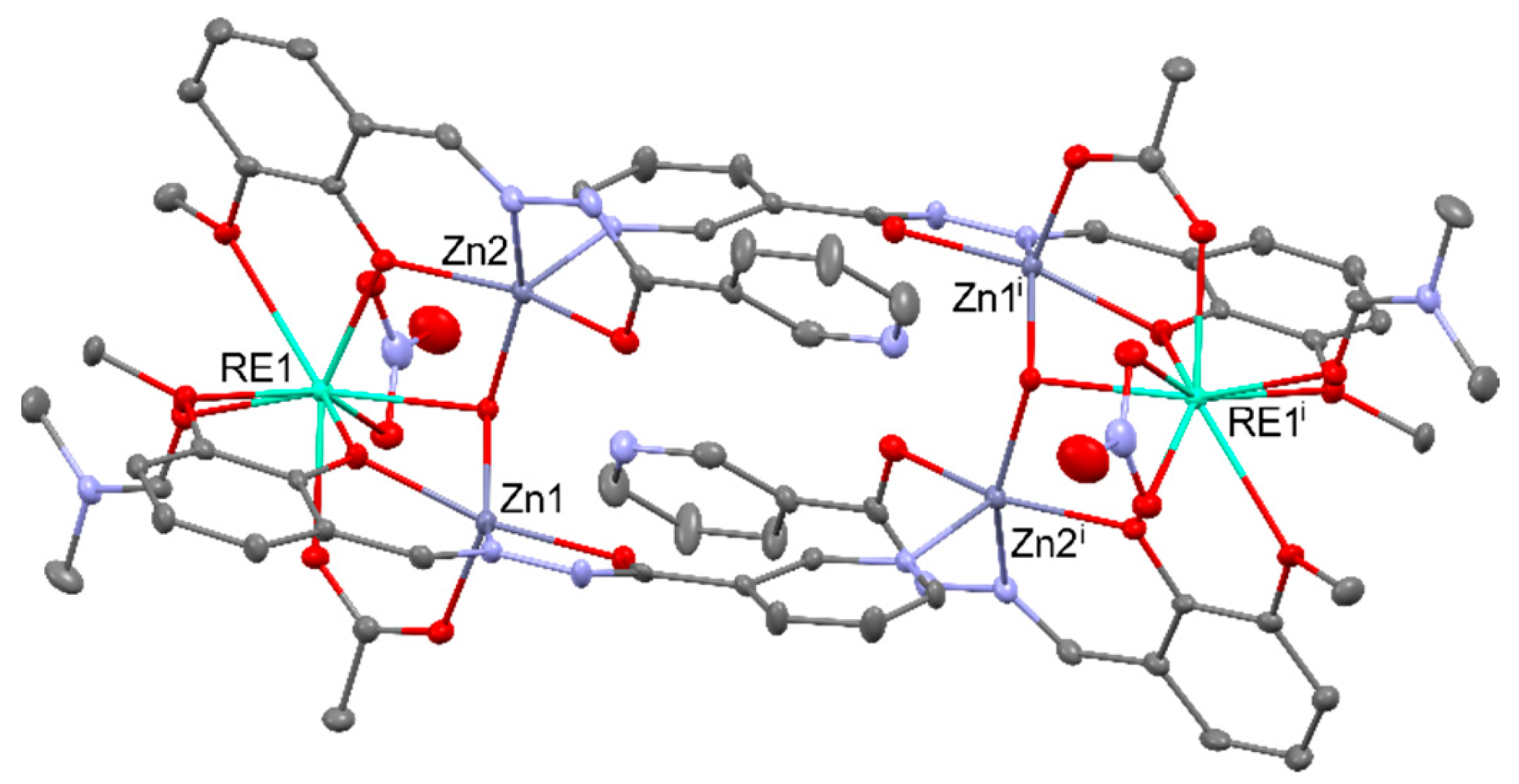
Figure 24.
Proposed mechanism for the oxidative carboxylation of Styrene and CO2 catalyzed by [Nd2(BIPA-TC)1.5]n. Reprinted with permission from [97] Nguyen, H.; Tran, Y. B. N.; Nguyen, T. C.; Gándara, F.; Nguyen, P. L. T. A Series of Metal–Organic Frameworks for Selective CO2 Capture and Catalytic Oxidative Carboxylation of Olefins. Inorganic Chemistry 2018, 57 (21), 13772–13782. https://doi.org/10.1021/acs.inorgchem.8b02293. Copyright © 2018 American Chemical Society.
Figure 24.
Proposed mechanism for the oxidative carboxylation of Styrene and CO2 catalyzed by [Nd2(BIPA-TC)1.5]n. Reprinted with permission from [97] Nguyen, H.; Tran, Y. B. N.; Nguyen, T. C.; Gándara, F.; Nguyen, P. L. T. A Series of Metal–Organic Frameworks for Selective CO2 Capture and Catalytic Oxidative Carboxylation of Olefins. Inorganic Chemistry 2018, 57 (21), 13772–13782. https://doi.org/10.1021/acs.inorgchem.8b02293. Copyright © 2018 American Chemical Society.
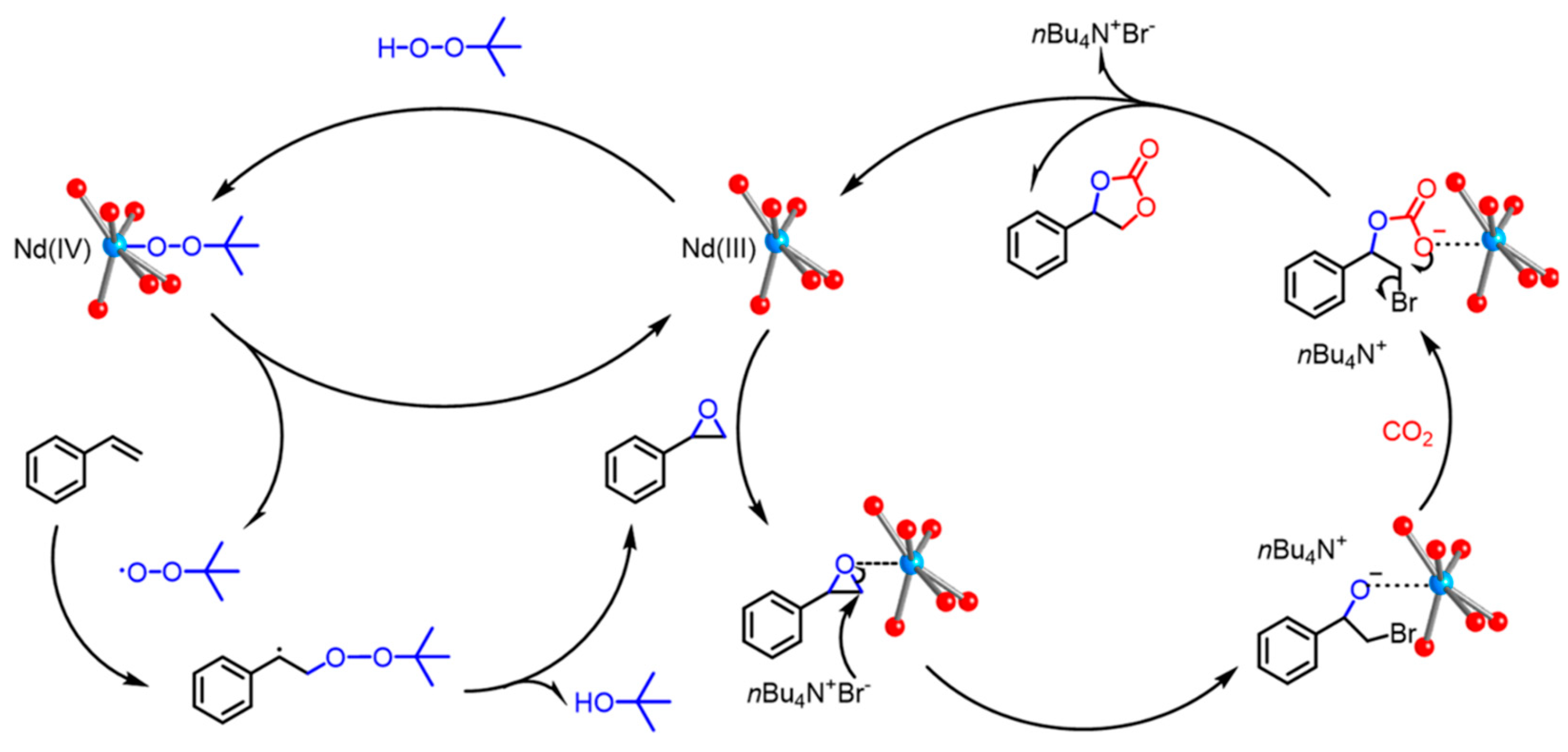
Figure 25.
Molecular structure of [Zn4RE2(μ3-OH)2L4(OAc)2(NO3)2(DMF)2] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 25.
Molecular structure of [Zn4RE2(μ3-OH)2L4(OAc)2(NO3)2(DMF)2] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
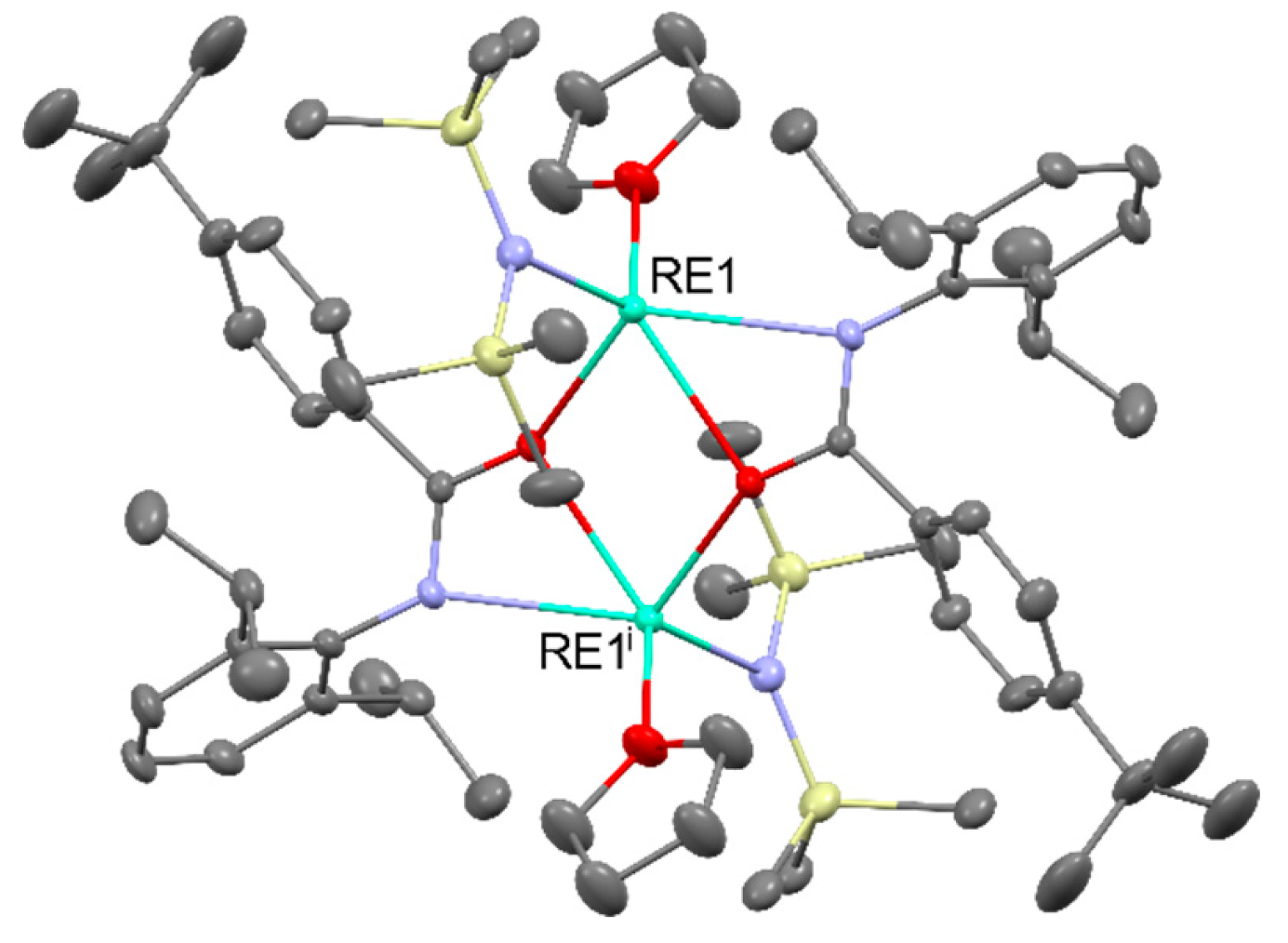
Figure 26.
Molecular structure of [Zn4RE2(μ3-OH)2L4(OAc)2(NO3)2(DMF)2] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 26.
Molecular structure of [Zn4RE2(μ3-OH)2L4(OAc)2(NO3)2(DMF)2] complexes. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
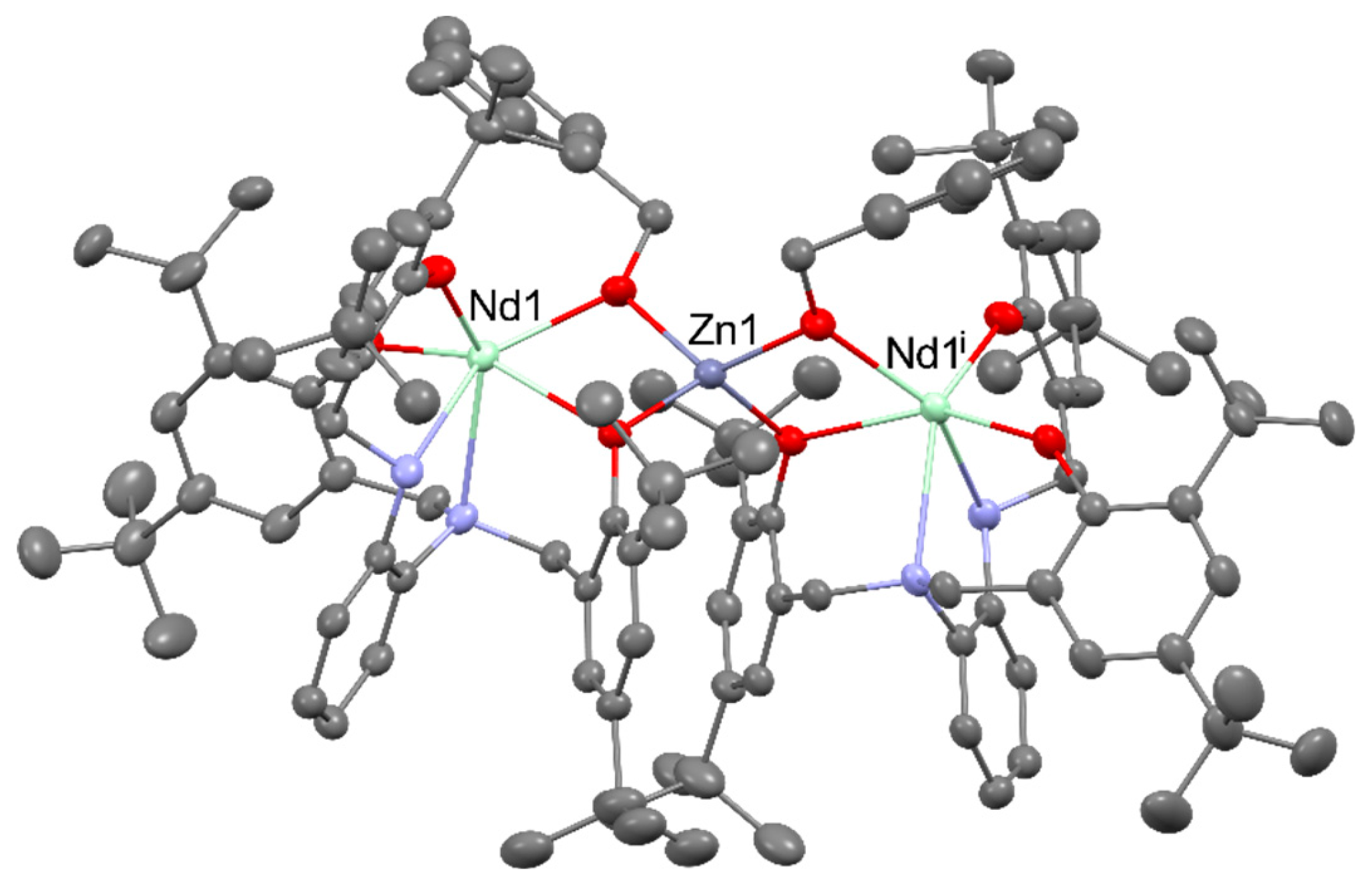
Figure 27.
CO2 and CHO copolymerization pathway catalyzed by NdCo3. Reprinted with permission from [106] Asaba, H.; Iwasaki, T.; Hatazawa, M.; Deng, J.; Nagae, H.; Mashima, K.; Nozaki, K. Alternating Copolymerization of CO2 and Cyclohexene Oxide Catalyzed by Cobalt–Lanthanide Mixed Multinuclear Complexes. Inorganic Chemistry 2020, 59 (12), 7928–7933. https://doi.org/10.1021/acs.inorgchem.0c01156. Copyright © 2020 American Chemical Society.
Figure 27.
CO2 and CHO copolymerization pathway catalyzed by NdCo3. Reprinted with permission from [106] Asaba, H.; Iwasaki, T.; Hatazawa, M.; Deng, J.; Nagae, H.; Mashima, K.; Nozaki, K. Alternating Copolymerization of CO2 and Cyclohexene Oxide Catalyzed by Cobalt–Lanthanide Mixed Multinuclear Complexes. Inorganic Chemistry 2020, 59 (12), 7928–7933. https://doi.org/10.1021/acs.inorgchem.0c01156. Copyright © 2020 American Chemical Society.
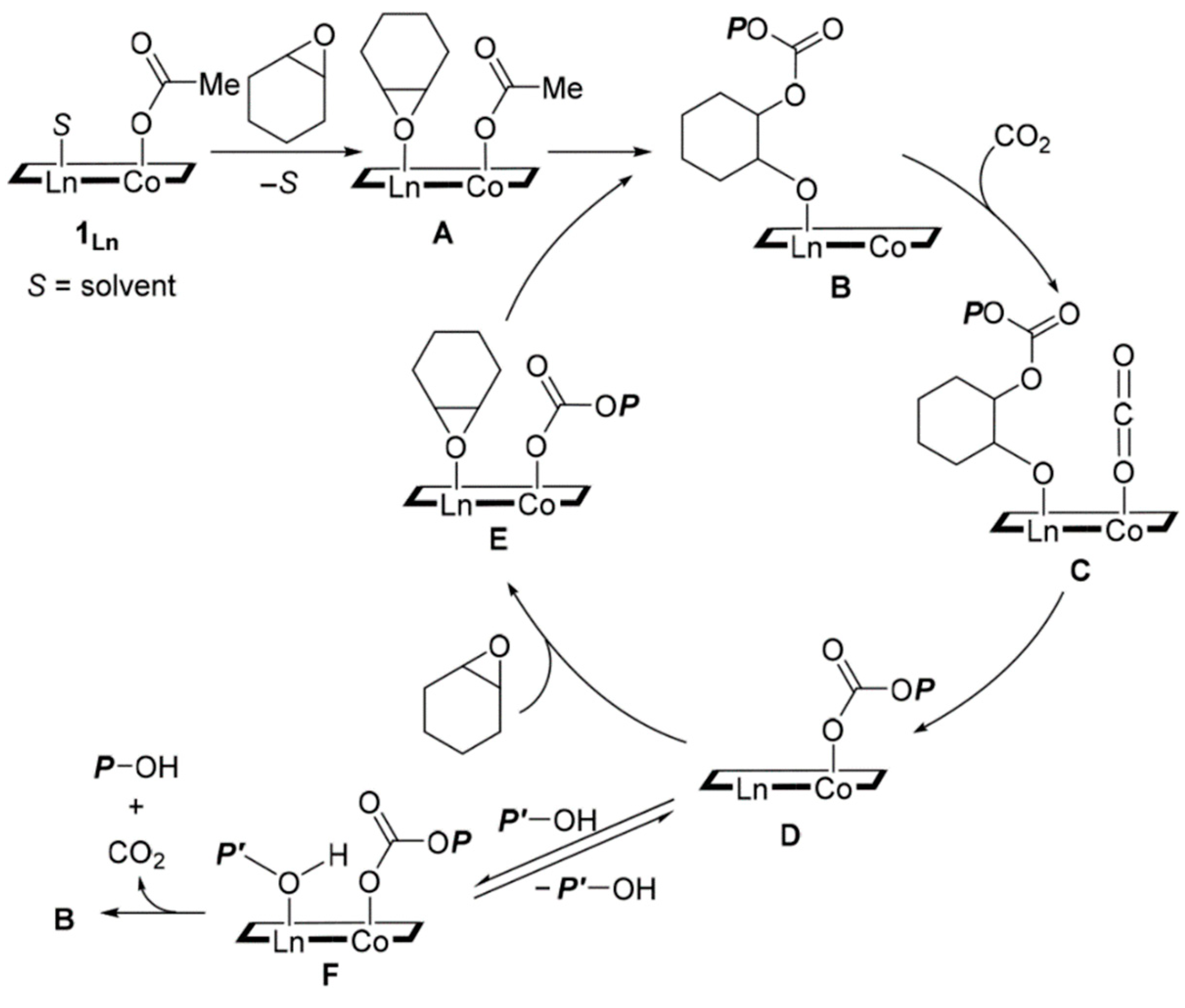
Figure 28.
Perspective view of the hexagonal molecule (a) and the arrangement of 138 metal ions in {Ni36Gd102} core (b). Color codes (the same in the following pictures): Gd, purple; Ni, blue; S, yellow; N, cyan; O, orange; C, gray; H, white.. Reprinted with permission from [107] Chen, W.; Liao, P.; Jin, P.; Zhang, L.; Ling, B.; Wang, S.; Chan, Y.; Chen, X.; Zheng, Y. The Gigantic {NI36GD102} Hexagon: A Sulfate-Templated “Star-of-David” for Photocatalytic CO2 Reduction and Magnetic Cooling. Journal of the American Chemical Society 2020, 142 (10), 4663–4670. https://doi.org/10.1021/jacs.9b11543. Copyright © 2020 American Chemical Society.
Figure 28.
Perspective view of the hexagonal molecule (a) and the arrangement of 138 metal ions in {Ni36Gd102} core (b). Color codes (the same in the following pictures): Gd, purple; Ni, blue; S, yellow; N, cyan; O, orange; C, gray; H, white.. Reprinted with permission from [107] Chen, W.; Liao, P.; Jin, P.; Zhang, L.; Ling, B.; Wang, S.; Chan, Y.; Chen, X.; Zheng, Y. The Gigantic {NI36GD102} Hexagon: A Sulfate-Templated “Star-of-David” for Photocatalytic CO2 Reduction and Magnetic Cooling. Journal of the American Chemical Society 2020, 142 (10), 4663–4670. https://doi.org/10.1021/jacs.9b11543. Copyright © 2020 American Chemical Society.
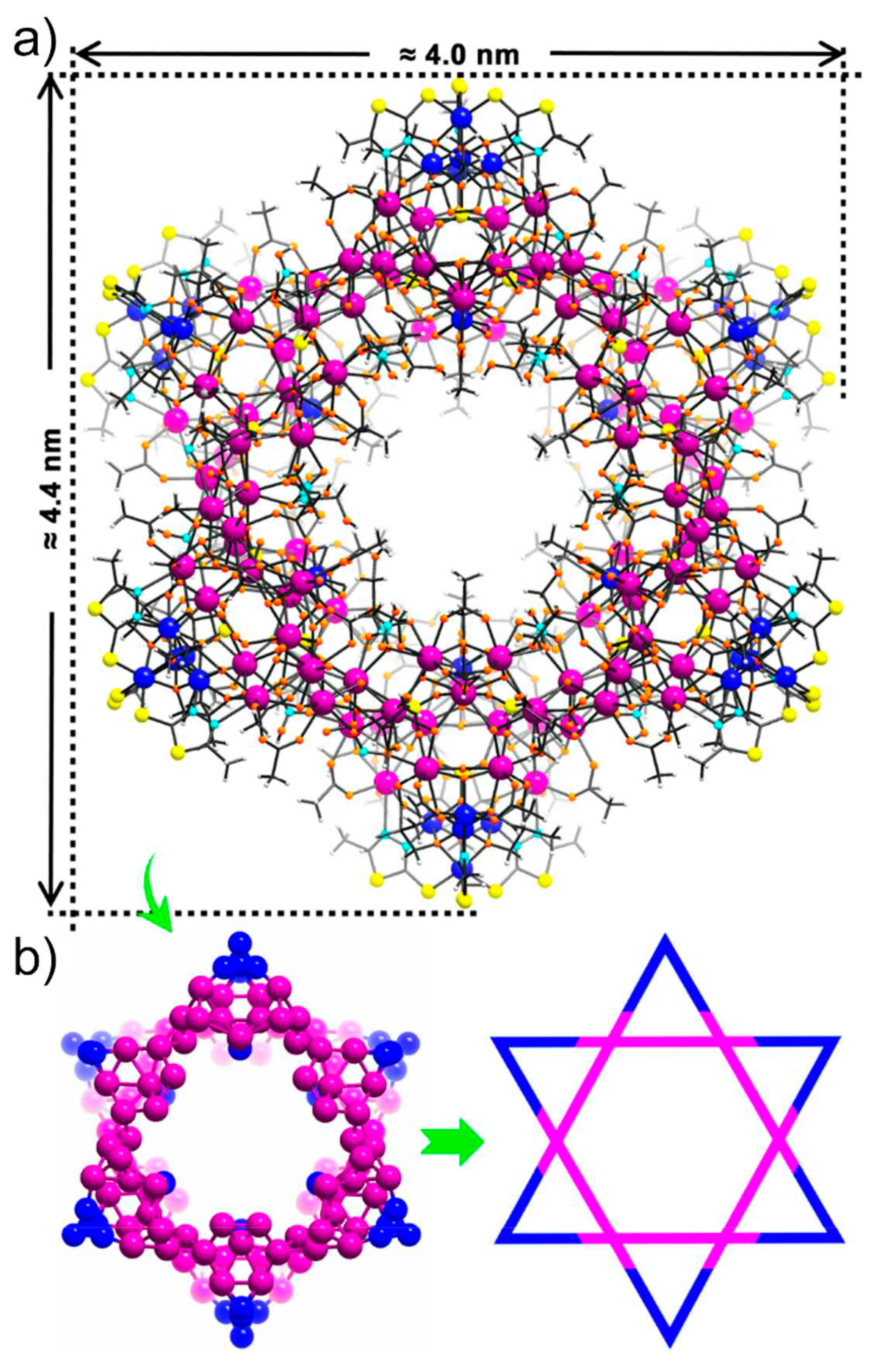
Figure 29.
Molecular structure of [Mn2RE2(O2CMe)6(pdmH)2(L)]+ cation. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 29.
Molecular structure of [Mn2RE2(O2CMe)6(pdmH)2(L)]+ cation. Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
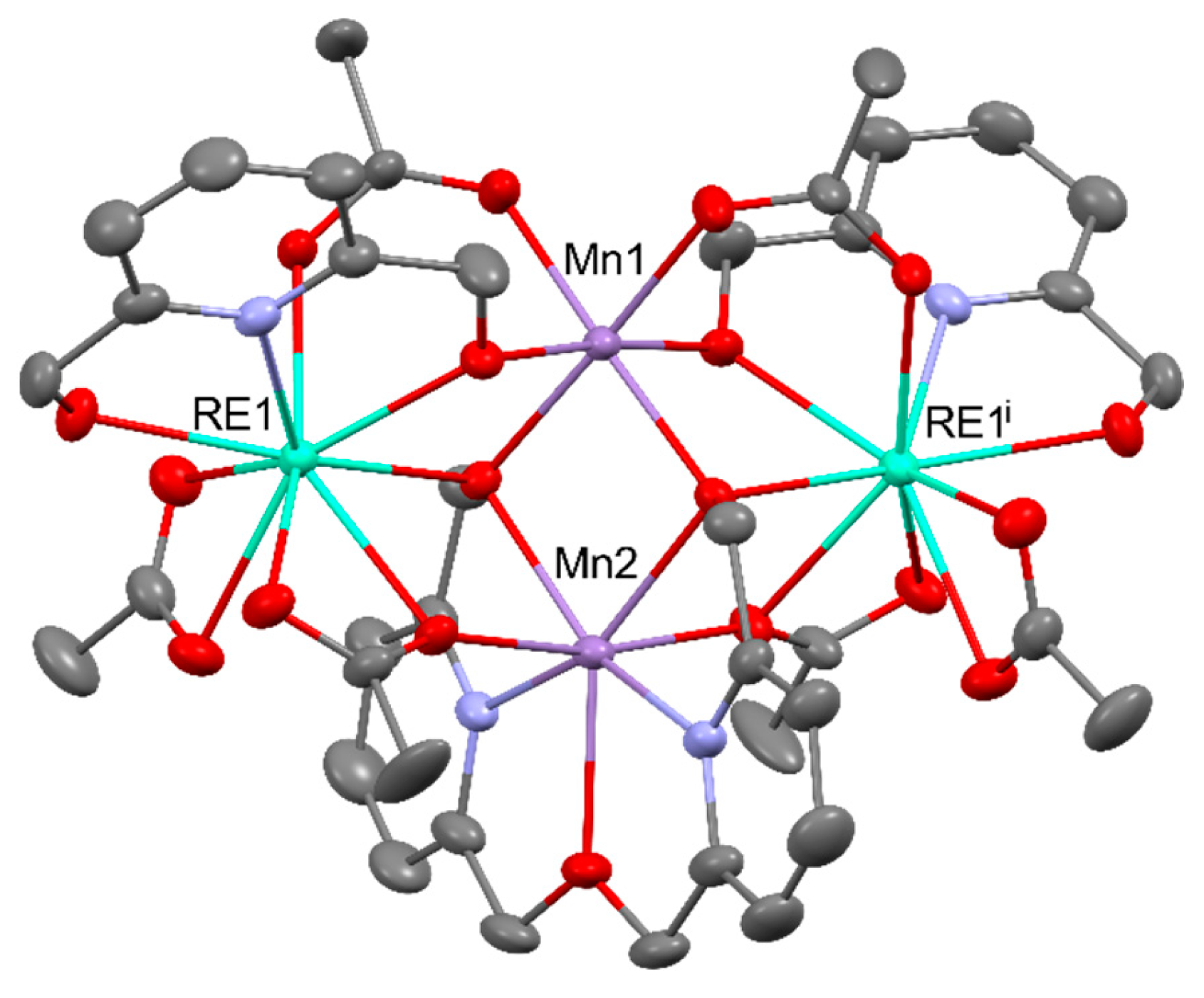
Figure 30.
TEM images of oxide materials (Pr0.9MnO3) prepared by calcination of [Mn2Pr4(µ3-OH)2(µ3-OR)4(µ-OR)4(µ-Cl)2(HOR)4Cl6] and [MnCl2(HOR)]n at 1100 °C.
Figure 30.
TEM images of oxide materials (Pr0.9MnO3) prepared by calcination of [Mn2Pr4(µ3-OH)2(µ3-OR)4(µ-OR)4(µ-Cl)2(HOR)4Cl6] and [MnCl2(HOR)]n at 1100 °C.
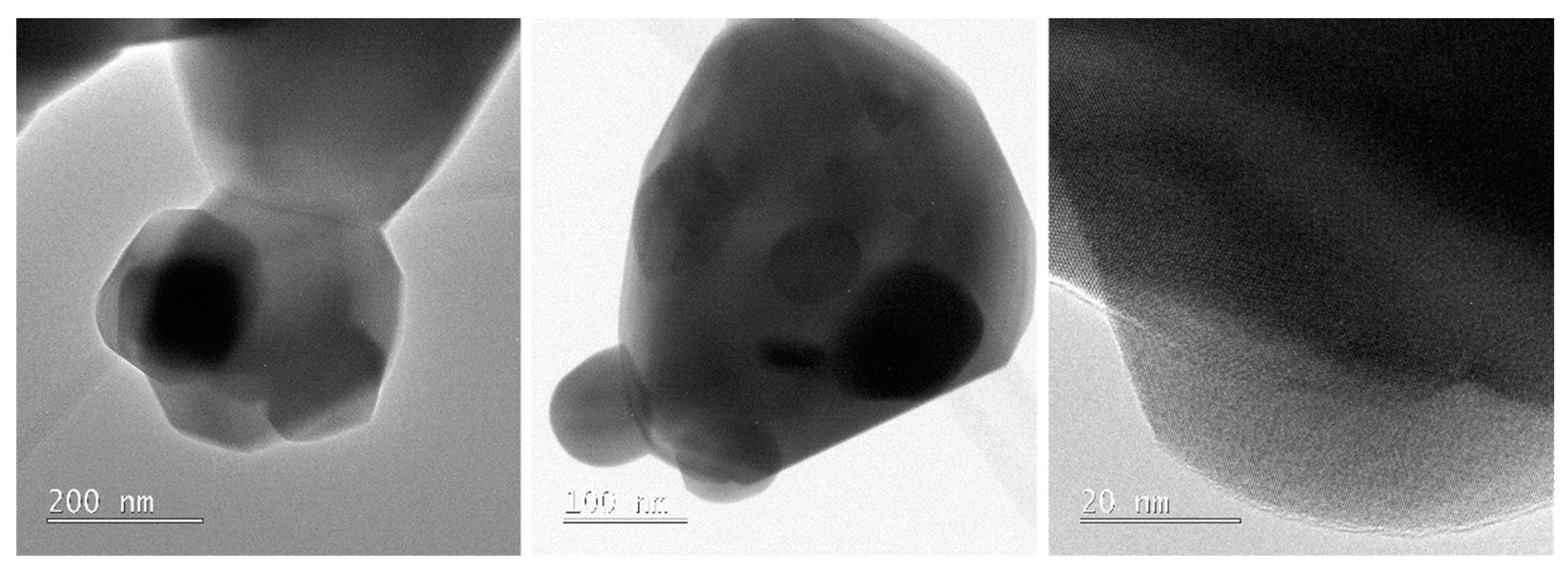
Figure 31.
Molecular structures of [Mn2Ln4(µ6-O)(µ3-OR)8(HOR)4Cl6] (a), [Co2Pr4(µ6-O)(µ3-OR)8(HOR)2Cl6] (b), and [Mn2Pr4(µ3-OH)2(µ3-OR)4(µ-OR)4(µ-Cl)2(HOR)4Cl6] (c). Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
Figure 31.
Molecular structures of [Mn2Ln4(µ6-O)(µ3-OR)8(HOR)4Cl6] (a), [Co2Pr4(µ6-O)(µ3-OR)8(HOR)2Cl6] (b), and [Mn2Pr4(µ3-OH)2(µ3-OR)4(µ-OR)4(µ-Cl)2(HOR)4Cl6] (c). Displacement ellipsoids are drawn at the 25% probability level. Hydrogen atoms and solvent molecules were omitted for clarity.
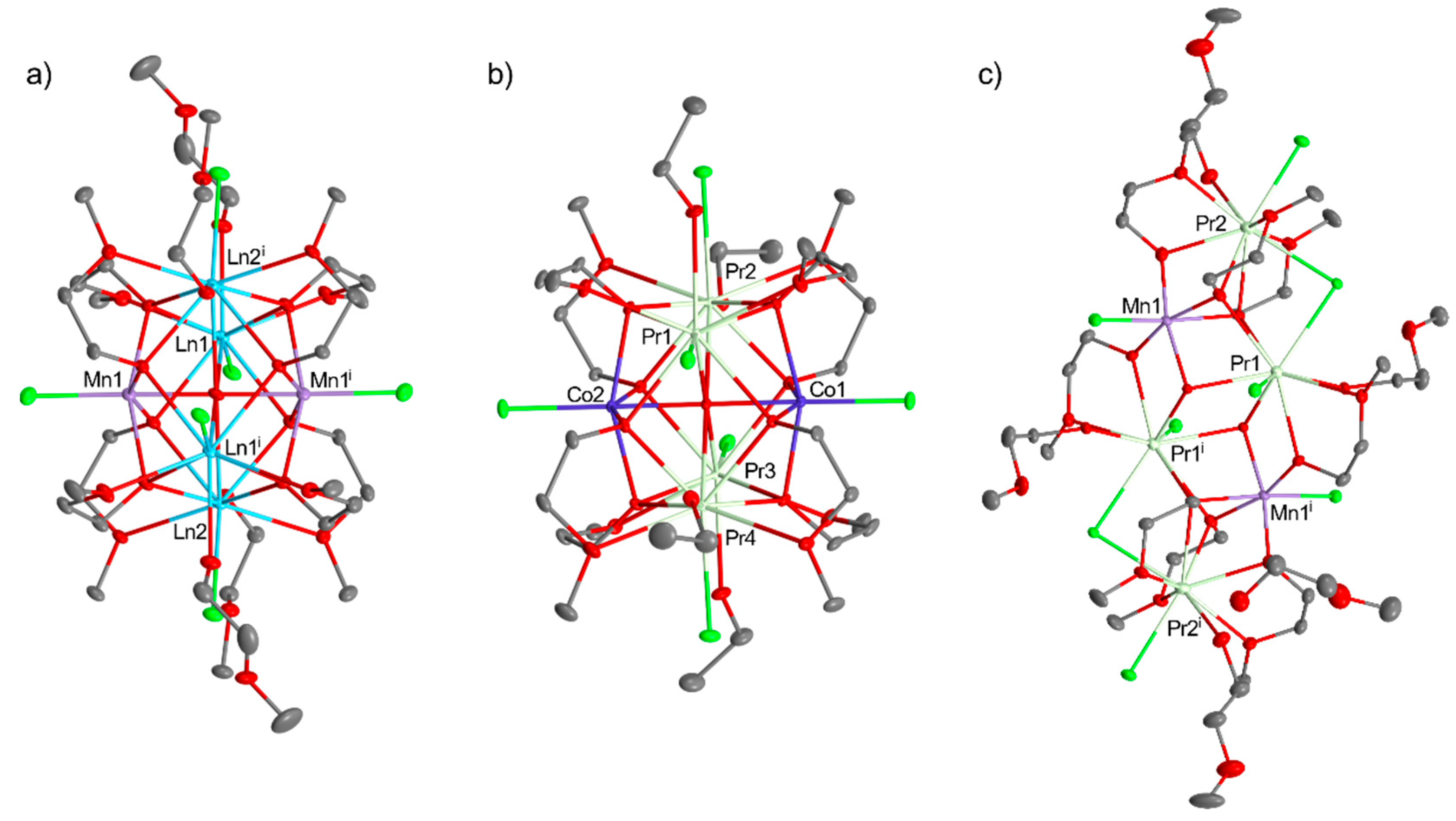
Figure 32.
PXRD patterns of oxide materials prepared by calcination of [Mn2Pr4(µ3-OH)2(µ3-OR)4(µ-OR)4(µ-Cl)2(HOR)4Cl6] and [MnCl2(HOR)]n at 1100 °C (a), and PXRD pattern of Pr0.9MnO3 [1526086] (b).
Figure 32.
PXRD patterns of oxide materials prepared by calcination of [Mn2Pr4(µ3-OH)2(µ3-OR)4(µ-OR)4(µ-Cl)2(HOR)4Cl6] and [MnCl2(HOR)]n at 1100 °C (a), and PXRD pattern of Pr0.9MnO3 [1526086] (b).
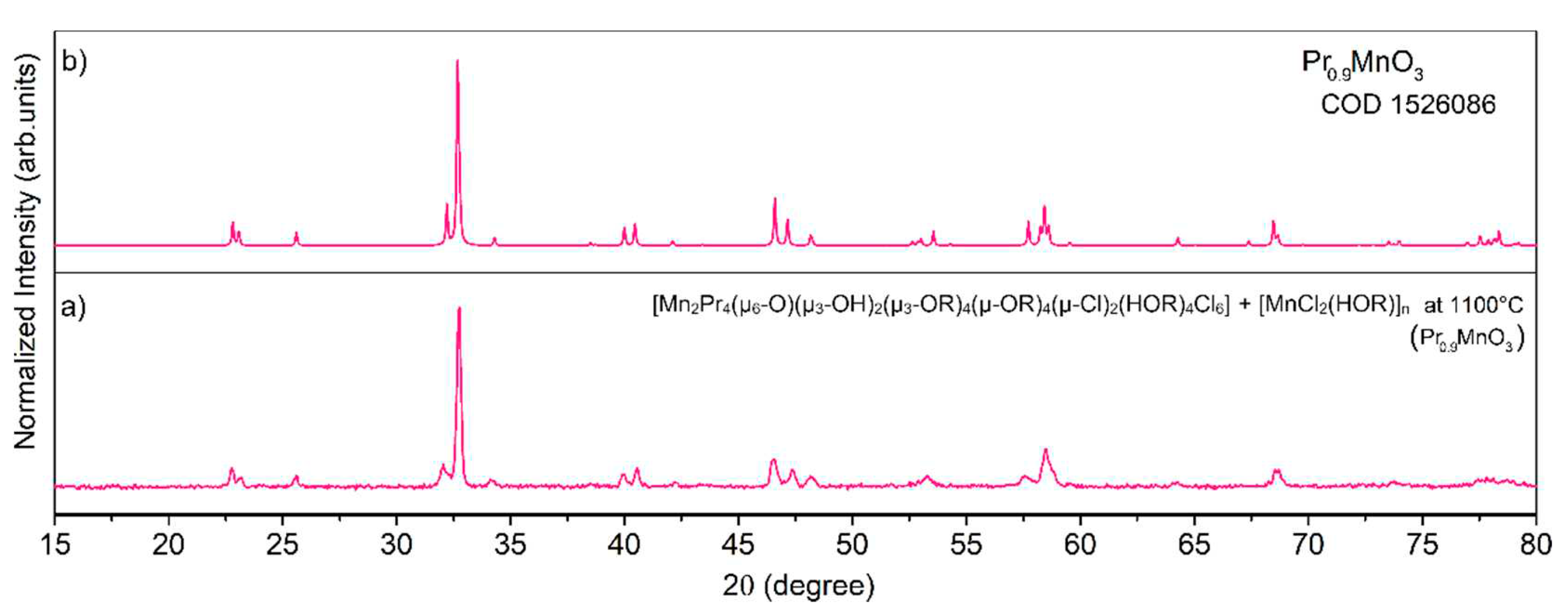
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



