
Submitted:
09 February 2024
Posted:
12 February 2024
You are already at the latest version
Abstract
Drug repurposing, rebranding an existing drug for a new therapeutic indication, is deemed a beneficial approach for a quick and cost-effective drug discovery process by skipping preclinical, Phase 1 trials and pharmacokinetic studies. Several psychotropic drugs including selective serotonin reuptake inhibitors (SSRIs) and tricyclic antide-pressants (TCAs) were studied for their potential application in different diseases es-pecially in cancer therapy. Fluoxetine (FLX) is one of the most prescribed psychotropic agents from SSRIs class for the treatment of several neuropsychiatric disorders with a favourable safety profile. FLX exhibited different oncolytic effects via mechanisms dis-tinct from its main serotonergic activity. Taking advantage of its ability to rapidly pen-etrate the blood-brain barrier, FLX could be particularly useful in brain tumors. This was proved by different in vitro and in vivo experiments using FLX as a monotherapy or combination with temozolomide (TMZ) or radiotherapy. In this review of literature, we summarize the potential pleiotropic oncolytic roles of FLX against different cancers highlighting the multifaceted activities of FLX and its ability to interrupt cancer prolif-eration via several molecular mechanisms and even surmount multidrug resistance (MDR). We elaborated on the successful synergistic combinations such as FXR/temozolomide and FXR/raloxifene for the treatment of glioblastoma and breast cancer, respectively. We showcased beneficial pharmaceutical trials to load FLX on car-riers to enhance its safety and efficacy on cancer cells. This is the first review article ex-tensively summarizing all previous FLX repurposing studies for the management of cancer.
Keywords:
Fluoxetine
; Cancer
; Repurposing
; multidrug resistance (MDR)
1. Introduction
Drug repurposing or repositioning is an attractive modality for finding new applications of old drugs [1,2]. This kind of drug recycling introduces a batch of pros including a shorter production time by eliminating Phase I clinical trials and a lower cost as a consequence in comparison to de novo drug discovery [1]. A pronounced merit of repurposing is that the pharmacokinetic and toxicity profiles of the repurposed entities were already identified. Previously, repurposing was dependent on serendipity, however recently it is based on advanced omics technologies and computational tools [3]. One of the most outstanding examples is aspirin repurposing from a non-steroidal anti-inflammatory drug to anti platelet aggregation drug. Concomitantly, owing to the potential relationship between COX-2 and cancer, aspirin could be further reused for cancer therapy [4]. The substantial role of drug repurposing in modern drug discovery emerged during endeavors to rapidly contain the COVID-19 pandemic using libraries of existing drugs [5,6,7,8].
Cancer is still a major cause of death globally, accounting for one-sixth of global mortality [9,10,11]. Available drugs encounter resistance and sometimes possess intolerable undesirable effects [12]. The scientific community is usually urged to pursue alternative cancer chemotherapeutic agents to address the issues and resistance of existing drugs. One obstacle is the lengthy period of time required for developing one new drug in addition to the huge cost and high possibility to end up failing clinical trials or facing pharmacokinetic issues [13].
There are continuous trials demonstrating repurposing non-oncology drugs towards cancer therapy on both basic and clinical levels. Figure 1 demonstrates the remarkable increment of publications connecting cancer to repurposing as found by search in Web of Science database. Sildenafil, a phosphodiesterase-5 inhibitor designed for treatment of ischemic heart diseases and repurposed to treat erectile dysfunction. Having said that, sildenafil is a sensitizer of cancer cells toward chemotherapy and radiation therapy [14,15]. Metformin, the widely used antidiabetic drug, exhibited anticancer and chemosensitization properties in preclinical and clinical studies [16,17].
The antimalarial drug quinacrine was found to be a dual target antiproliferative agent by inhibition of Topo II and Hsp90 [18]. In addition, quinacrine was repurposed for managing cancer by other several mechanisms [19,20,21]. We have successful stories in the field of drug repurposing in cancer field. Recently, we repositioned anti-HIV substituted benzimidazole derivatives for cell migration inhibition targeting heterogeneous nuclear ribonucleoprotein-M (hnRNP-M) [22]. Additionally, we repurposed S-trityl L-cysteine and S-trityl cysteamine derivatives from kinesin Eg5 inhibitors to Sirtuins 2 inhibitors [23,24]. A batch of repurposed non-oncology drugs toward cancer management was extensively reviewed elsewhere [13,25].
Antidepressant drugs have remarkable role in therapy of cancer patients who are prone to depression disorders [26,27]. Early observations showed conflicting findings on antidepressants effect on cancer promotion and growth [26,28]. Later studies revealed a great potential of antidepressant drugs including tricyclic antidepressants (TCAs) and selective serotonin reuptake inhibitors (SSRIs) for repurposing to cancer therapy via several mechanisms of action [2,8,27,29,30]. In fact, among all classes of antidepressants, SSRIs have the highest repurposing potential for managemnt of cancer [2,31].
Owing to their favourable safety profile, SSRIs are the most prescribed antidepressant drugs, and they are used as adjuvant therapy for treatment other neuropsychiatric disorders [32,33]. Basically, a three-year study on larger sample of patients using SSRIs ruled out any breast cancer risk due to their administration [34]. Concomitantly several SSRIs proved oncopreventive and/or oncolytic properties against cancers of lung [35], colorectal [36,37], breast [38]. Accumulating evidences confirmed that SSRIs’ oncolytic activity is mainly through independent actions of their primary serotonergic-mediated mechanisms [30].
Indeed, all SSRIs showed various oncolytic activities except for vilazodone [2]. For example, sertraline (Zoloft®), induces apoptosis in colon cancer cells [39], suppresses tumor growth by blocking 5’ adenosine monophosphate-activated protein kinase /mammalian target of rapamycin (AMPK/mTOR) pathway and promoting autophagic flux in non-small cells lung cancer (NSCLC) cells [40], shows synergistic effects with sorafenib against hepatocellular carcinoma (HCC) cells proliferation [41], and reduces breast cell growth by interrupting serine/glycine synthesis [42,43].
Paroxetine (Paxil®) induces apoptosis in NSCLC via ROS-MAPK pathway [44], in colon cancer cells by suppressing MET and HER3 kinases [45], and in MCF-7 by increasing extracellular Ca+2 and p38 [31]. Citalopram (CeleXA®) has proapoptotic effect on acute myeloid leukemia (AML) via caspase-3 activation. Notably, it lowers invasion and metastasis of colon cancer cells by inhibition of transforming growth factor-β (TGF-β) signaling pathway. The S-(+)-enantiomer of citalopram, escitalopram (Lexapro®), induces apoptosis and autophagy in glioblastoma [46] and NSCLC [47] and suppresses breast cancer cell growth [48].
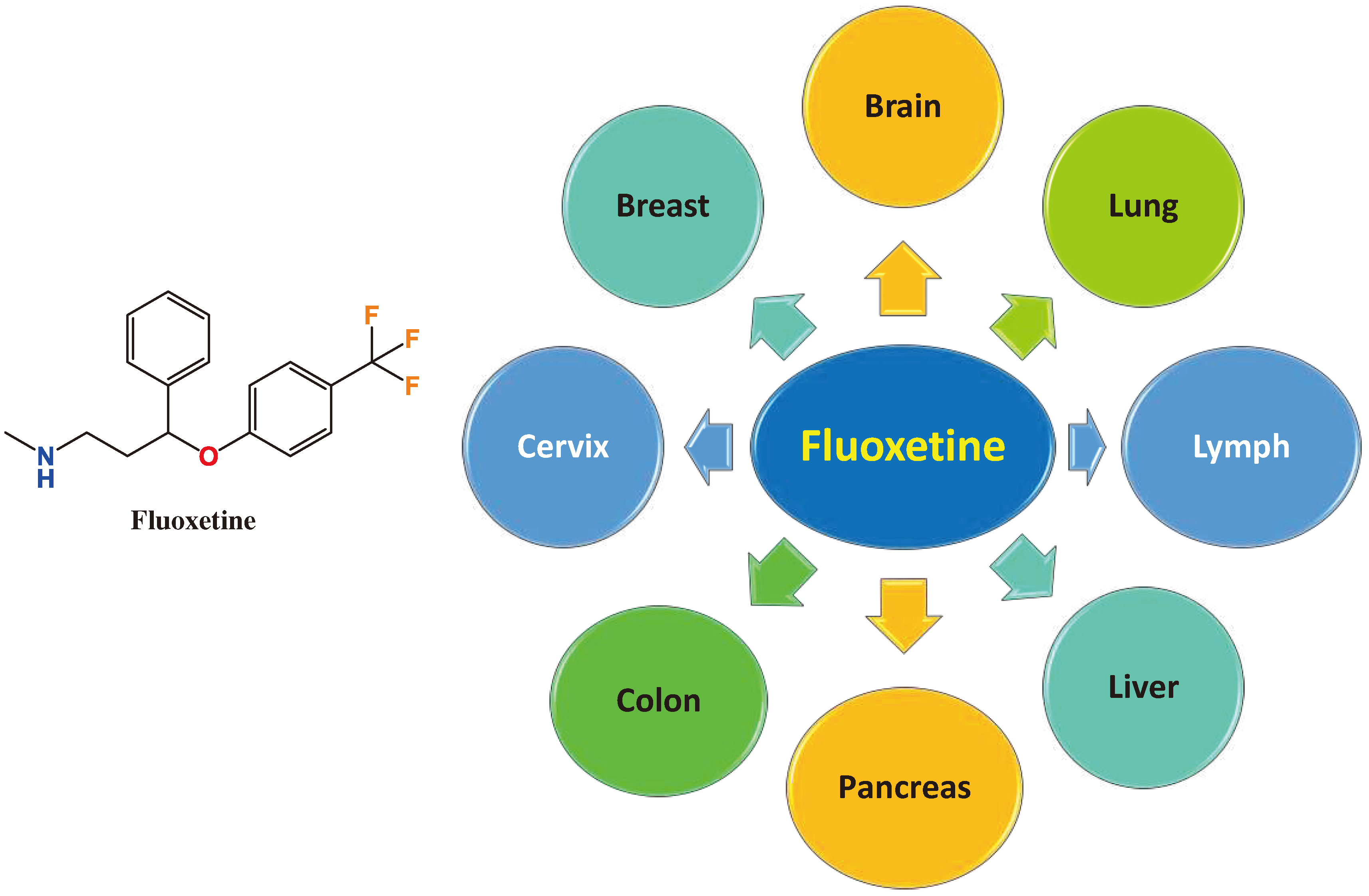
Fluoxetine (FLX) (Prozac®), Figure 1, is the first approved SSRI and still one of the most prescribed antidepressants worldwide. The literature is full of interesting studies on the potential rebranding of FLX for management of different cancer types. Among different antidepressants, only FLX improved overall survival of patients receiving FLX/PD-1/L1 immunotherapy compared to only PD-1/L1 according to a cohort study on cancer patients using checkpoint inhibitors [49]. For the first time, we introduce a review article to emphasize the potential role of FLX in management of cancers either solely or in combination with other chemotherapeutic agents. Additionally, the role of FLX in overcoming multidrug resistance (MDR) is discussed. Figure 2 summarizes the different cancer types that can be modulated by of FLX and explained in this article. The review further includes pharmaceutical trials to load FLX on carriers to control its cellular release and enhance its efficacy.
2. Materials and Methods
We explored the literature in PubMed, Google Scholar, and Web of Science databases using the three keywords fluoxetine, repurposing, and cancer to conduct a comprehensive search. Our search was carried out without years limitations because there was no previous review article on this particular topic. The outcome was around 92 research and review articles of which 49 were extensively considered for the current review article. The residual articles were not investigated thoroughly because they mainly focused on repurposing of other antidepressants or antipsychotics. We divided the results into sections based on the target cancer type.
3. Results
3.1. FLX in Brain Tumors
For brain tumors like glioblastoma multiforme (GBM), blood-brain barrier (BBB) may hinder the development of some drugs that efficiently stop GBM cell proliferation and trigger apoptosis in vitro [30,50]. In other words, the efficacy of GBM drugs is often faced by poor drug delivery due to the presence of BBB [51]. As an antidepressant drug, FLX has favourable physicochemical and pharmacokinetic properties to bypass BBB, hence its repurposing for GBM therapy is substantially useful [52]. De facto, medical records from insurance databases showed remarkably enhanced life expectancy in GBM patients taking fluoxetine but not other SSRIs [53].
FLX exhibited in vitro apoptotic effect on human GBM cells viz., U87 and GMB8401 by increasing the intracellular Ca+2 concentration, damaging mitochondrial membranes, and releasing apoptogenic factors. This effect was reversed by the coadministrations of (2,3-dioxo-6-nitro-7-sulfamoyl-benzo[f]quinoxaline) (NBQX), a blocker of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) which is highly expressed in GBM. In silico calculations supported by experimental tools emphasized that FLX bind to GLUR1 subunit of AMPAR [54,55]. The apoptotic effect was consistent in vivo in a tumor xenograft using U87 GBM cells. Of note, the apoptotic effect was significant in brain tumor but not normal brain cells of mice reflecting a favourable safety profile of FLX [51].
Temozolomide (TMZ) is one of the most effective chemotherapeutics for GBM. Due to its common resistance, Wang group studied FLX activity in GBM and its synergistic effect with TMZ. Only FLX was able to inhibit growth of different rat and human GBM cells including C6, U87-MG, U373 and U251 in a dose dependent manner. C6 were the most sensitive cells toward FXL with IC50 14.7 µM. FLX clearly enhanced C6 apoptosis by increasing caspase-3 concentration and endoplasmic reticulum stress (ERS). Mechanistically, FXL elevated the expression of C/EBP homologous protein (CHOP) and autophosphorylation of its upstream and downstream signaling pathways as identified by western blotting. Notably, FLX sensitized C6 cells to TMZ treatment showing a synergistic antiproliferative affect at lower inhibitory concentrations. The latter effect is also mediated by CHOP pathway as the knockdown of CHOP omitted FXR/TMZ synergism [56].
Considering that the TMZ resistance is mainly caused by DNA repair O6-methylguanine-DNA methyltransferase (MGMT), Song et al., studied the role of FLX in suppressing MGMT expression. The authors concluded that FLX significantly reduced MGMT concentrations via disrupting NF-kB/p65 signaling and hence sensitizing GBM cells to TMZ. Those in vitro results were further validated in vivo in murine subcutaneous xenograft model of human GBM cells U-138 MG [57].
An elegant research work by Bi et al. in 2021 extensively showed the effect of FLX on signaling and metabolism of GBM. They identified acid sphingomyelinase (ASM; sphingomyelin phosphodiesterase 1 [SMPD1]) as an attractive GBM target that is necessary for cancer cell survival. In GBM cells, fluoxetine inhibited SMPD1 enzymatic activity- formation of ceramide from sphingomyelin- resulting in a dose dependent death in three different types of GBM cells. Consistently, this effect was reproduced in orthotopic xenografts implanted in nude mice brain. Interestingly, this effect is accompanied by lysosomal stress and suppression of the overexpressed oncogenic epidermal growth factor receptor VIII (EGFRvIII), a constitutively active form mutation of EGFR [53].The authors confirmed in vitro and in vivo FLX/TMZ synergism in inducing DNA damage and cell death in multiple GBM models. This is in accordance with the recorded higher survival rates of GBM patients who co-administer FLX/TMZ [53].
Owing to its favourable safety profile, a research group studied the possibility of FLX repurposing for the fatal neuroblastoma in children which accounts for 7% of cancer diagnosis of children under 15-year-old. In neuroblastoma, the Myc oncoprotein amplification is used as a negative prognostic marker and is a hallmark of a high-risk disease. Myc regulates CKS1/SKP2/p27kip1 axis which can be interrupted by FLX. The latter inhibited CKS1, increased p27Kip1 expression and triggered neuroblastoma cell death. Furthermore, small doses of FLX reduced the invasiveness and metastasis of neuroblastoma cells. This effect was further validated in a xenograft mice model [58].
FLX is not only effective as a monotherapy or combination therapy with TMZ but also it could sensitize GBM cells (U-87 MG) to radiotherapy. Surprisingly, FLX showed a radioprotective effect on normal fibroblast cells HFFF2 in vitro [59]. Conclusively, FLX represents the best SSRI in terms of brain cancer management. Its effect is remarkable when combined with TMZ or radiotherapy.
3.2. FLX in Breast Cancer
Breast cancer is the most common invasive cancer in women with potential fatal metastasis to bones, liver, lungs, and brain [60,61]. The literature is loaded with considerable results showing the sensitivity of different types of breast cancer to FLX. The group of Chan take the advantage of FLX to circumvent MDR in the MCF-7/adr (doxorubicin-resistant human breast carcinoma) cells; they used stealth liposome co-encapsulation of doxorubicin and FLX for prolonged circulation half-life and improved safety in vivo. The used formulation demonstrated promising anticancer, under both in vitro and in vivo conditions, capable of effective reversal of doxorubicin resistance [62].
The mechanism of FLX for reversing MDR was not fully understood. Thus, another group studied the synergism of FLX with adriamycin (ADM) and paclitaxel (PXL). FLX-ADM combination enhanced apoptosis significantly in MCF-7/ADM resistant cells but not MCF-7 cells. The authors found that the chemo-sensitizing effect of FLX occurs via simultaneous upregulation of the tumor suppressor protein p53 and downregulation of B-cell lymphoma 2 (Bcl-2) [63].
Raloxifene (RAL) is widely used for the treatment of estrogen receptor positive breast cancer with less side effects than the parent tamoxifen. Kabel et al., investigated the potential synergistic effect in case of using FLX/RAL combination in experimentally 7,12-Dimethyl Benzanthracene (DMBA)-induced breast cancer in female Wistar rats. They found that either FLX or RAL is effective proapoptotic agent, but RAL/FLX combination had a better outcome than either of them as shown by the tumor volume size. RAL/FLX administration improved tumor antioxidant status by elevation of catalase (CAT) and superoxide dismutase (SOD) and reduction of malondialdehyde (MDA) in tumor tissue compared to the control group. The titled combination reduced tissue concentration of the pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) and suppressed TGF-β [64].
A complementary study to the above-mentioned one regarding FLX/RAL combination showed that it can suppress invasion, metastasis, and angiogenesis of (DMBA)-induced breast cancer. The combination lowered mammary tissue vascular endothelial growth factor (VEGF), macrophage colony-stimulating factor (M-CSF), and matrix metalloproteinase-9 (MMP-9) levels as determined by enzyme-linked immunosorbent assay (ELISA). In accordance with the previous study, FLX/RAL oncolytic effect significantly surpasses each sole drug [65].
According to Duarte et al., the IC50 of FLX against MCF-7 cells is 7.78 µM as detected by MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) assay. When combined with PXL, a significant synergistic antiproliferative effect was observed using both MTT and SRB assays [66]. The same research group found significant synergism for combination of FXL and doxorubicin against MCF-7 in another study [67]. They also found that co-treatment of FXL and honeybee venom demonstrated better cytotoxic effect than either of them [68].
In a study testing different antidepressants including amitriptyline, bupropion, FLX, paroxetine, and tianeptine, as a monotherapy for induction of apoptotic cell death in breast cancer cells MCF-7, only paroxetine followed by FLX demonstrated significant activity in a dose-dependent manner [31]. Another study benchmarked standalone FLX on triple-negative breast cancer (TNBC) in vivo. Surprisingly, FLX suppressed tumor growth by downregulating STAT3 signaling transduction and triggering caspase-mediated apoptotic pathway [69].
By virtue of the aggressiveness of TNBC, Bowie et al., extensively studied the effect of FLX in TNBC cells SUM149PT. They found that treatment with FLX led to induced apoptosis, enhanced ER stress and autophagy. This is accompanied by cell cycle arrest at G1 phase and caspase-7 mediated cell death. Those effects are less prominent in the non-transformed MCF-10A cells reflecting a favourable safety profile [70].
Another research group studied FLX effect on other TNBC cells, MDA-MB-231 and MDA-MB-436. Sun et al., revealed that FLX induced both apoptosis and autophagic cell death in the titled cell lines. The apoptotic effect is attributed to upregulation of the expression levels of caspase-3 and caspase-8 and poly (ADP-ribose) polymerase (PARP). In addition, FLX significantly reduced the phosphorylation of eukaryotic elongation factor-2 kinase (eEF2K) which is overexpressed in different cancers and plays an indispensable role in the crosstalk between apoptosis and autophagy in TNBC. Furthermore, FLX modulates autophagic proteins; it decreases phosphorylation of mTOR, activates AMPK, and increases ULK1 phosphorylation [71].
3.3. FLX in Hepatocellular Carcinoma (HCC)
Hepatocellular carcinoma (HCC) is the most common form of primary liver malignancy with yearly 906,000 new cases and 830,000 deaths worldwide, making it the third leading cause of cancer death [72,73]. Several SSRIs were tested for their possible cytotoxic effect on HCC [74]. FLX reduced the viability of human HCC cell line Hep3B and induced apoptosis as detected by MTT assay and staining, respectively. Disruption of mitochondrial membrane potential (MMP) is an early indicator to reactive nitrogen species-induced apoptosis. The authors found that treatment of Hep3B with FLX resulted in loss of MMP and enhanced formation of reactive oxygen species (ROS). This was accompanied with suppression of anti-apoptotic phosphorylated extracellular signal-regulated kinase 1/2 (pERK1/2) protein and increase in proapoptotic c-JUN N-terminal kinase (c-JNK) and p38 mitogen-activated protein kinases (MAPK) [75].
In vivo study showed that FLX significantly suppressed tumor growth of Hep3B cells in a xenograft mice model without induced liver pathology or general toxicity at 10 mg/kg FLX dose. The oncolytic effect is attributed to upregulation of the extrinsic and intrinsic apoptosis-associated proteins including caspase-3, caspase-8, and caspase-9 and suppression of metastasis-associated protein VEGF, MMP-9, urokinase-type plasminogen activator (uPA), and Cyclin- D1, as validated by immunohistochemistry (IHC) staining. This indicated that FLX could reduce Hep3B angiogenesis and cells invasion. Additionally, FLX attenuated phosphorylation of nuclear factor-kappa B (NF-κB) p65 on ser276, AKT and ERK in the titled cells as validated by IHC [76]. A similar action was induced by FLX treatment of HCC SK-Hep1 cells in another study for the same research group [77].
Hend et al., prepared FLX-loaded hexosome (HEX) using the hot emulsification method to prolong the release and enhance the activity of FXR on HCC cells HepG2. Successfully, the optimized HEX was able to prolong FLX release, where only 19.5 % released in PBS pH 7.4 after 24 h which was highly increased in acidic medium pH 5.5 which imitates cancer microenvironment. Accordingly, HEX improved enhanced cellular delivery and cytotoxic activity of FLX against HepG2 compared to the drug solution [78].
3.4. FLX in Colon Cancer
Colorectal cancer is ranked as the third most common cancer in the United States [79,80]. MTT assay showed significant antiproliferative effect of FLX on colon cancer cells HT-29 with IC50 value of 6.12 µM. However, co-treatment of FLX and 5- fluorouracil (5-FU) (IC50 3.79 µM) did not show synergistic effect at different concentrations [66]. Nevertheless, the combination of fluoxetine and honeybee venom demonstrated a significant synergistic effect against HT-29 cells [68]. Another study showed that FLX remarkably augmented the antiproliferative effect of either cisplatin or carboplatin but not oxaliplatin on colon cancer cells HCT116. The authors attributed this synergistic effect to calmodulin inhibition by FLX [81].
Another effective combination of metformin/Efavirenz/FLX against HCT116 human colon cancer cells was recently reported by Kang et al. [82]. The three-drug combination triggered a massive increase in ROS levels and exhibited dramatic antiproliferative effect on HCT116 but not human dermal fibroblasts (HDF) cells. The treated HCT116 cells showed increased DNA damage, apoptosis, autophagy, and necroptosis-related factors as detected by western blotting. In HCT116 xenograft mice model, the titled combination showed consistent results by reducing tumor weight and size compared to the control group [82].
Another validating study showed that FLX induced concentration-dependent apoptosis and DNA fragmentation in HCT116+/+ and p53 gene-depleted HCT116-/- human colorectal cancer cells with respective IC50 values 3.19 ± 0.23 μM and 4.73 ± 0.5 μM and less effect on normal cells. Mechanistically, FLX treatment resulted in cell cycle arrest at Sub-G1 and G0/G1 phases in both cell lines as detected by FACS analysis [83]. In sum, the molecular mechanism of FLX cytotoxicity against HCT116 cells is independent of p53 modulation.
3.5. FLX in Cervical Cancer
Cervical cancer is one of the most spreading cancers in female worldwide [10]. Resistance of cervical cancer cells to cisplatin is a real challenge that leads to decline of the survival rate [84]. In cervical cancer HeLa cells, FLX reduced viability by energy depletion and increase in increase cytosolic Ca+2 concentration by emptying the endoplasmic reticulum (ER) through the translocon, an ER Ca2+ leakage structure [85].
In a dose-dependent manner, FLX promoted HeLa cellular chemo-sensitivity to cisplatin in vitro and in xenograft mice model compared to FLX or cisplatin alone. The titled combination triggered G0/G1 phase arrest (73%) compared with FLX (58%) or cisplatin (60%) as observed by FACS analysis. Additionally, the percentage of apoptotic cells was 48.3% compared with FLX (38.1%) or cisplatin (31.5%). In vivo assay showed that FXR, cisplatin, and FXR/cisplatin combination effectively reduced the tumor weight with inhibition rates of 10.9%, 46.6%, and 53.7%, respectively. Mechanistically, FXR/cisplatin upregulated apoptotic proteins caspase-9, p17 and suppressed MDR proteins glutathione S-transferase π (GSTπ) and P-gp [86].
Naz et al., developed FLX-dextran nanoparticles conjugates for better efficacy of FLX. Dextran was oxidized by sodium iodate to form the corresponding aldehyde which was readily reacted with FLX forming a Schiff base [87,88]. FLX-dextran nanoparticles were stable at physiological blood circulation and normal tissues with remarkable higher release in acidic environment, i. e., pH 5 which resulted in high specificity toward cancer cells and reduced systemic undesirable effects. Indeed, only FLX showed higher toxicity to normal mouse embryo fibroblast cells 3T3 than FLX-dextran nanoparticles. However, the later showed similar anticancer effect on HeLa cells at 30 µM concentration [87].
3.6. FLX in NSCLC
NSCLC represents 85% of lung cancer cases and it is characterized with invasive and metastatic nature; the acquired resistance to receptor tyrosine kinases inhibitors is a major therapeutic obstacle [89]. A research group has extensively studied the effect of FLX on NSCLC cells CL1-5-F4. FLX reduced their viability via induction of apoptosis in a dose-dependent manner with IC50 40 µM as shown by MTT assay. Western blot analysis revealed that FLX significantly reduced the level of DNA repair-associated proteins including levels of mediator of DNA damage checkpoint 1 (MDC1), O6-methylguanine DNA methyltransferase MGMT, and 14-3-3. Reporter gene assay showed that FLX remarkably inhibited NF-ĸB activation in CL1-5-F4 cells at the IC50 concentration of FLX. The related level of metastasis associated proteins MMP-2, MMP-9, uPA, and VEGF was suppressed. Accordingly, CL1-5-F4 metastasis and invasion were attenuated [90].
In highlight of the above data, Hsua et al., benchmarked FLX in vivo using CL1-5-F4 bearing mice model. FLX significantly suppressed tumor growth and size in the established model without general toxicity at 10 mg/kg FLX dose. The anticancer effect is ascribed to promoting the level of extrinsic and intrinsic apoptosis-associated proteins including caspase-3, caspase-8, and caspase-9 and reduction of metastasis associated protein VEGF, MMP-9, urokinase-type plasminogen activator (uPA), and Cyclin-D1. This validated the previous conclusion of FLX ability to attenuate NSCLC angiogenesis and invasion [90]. Additionally, FLX attenuated phosphorylation of nuclear factor- kappa B (NF-κB) p65 on ser276, AKT and ERK [76].
3.7. FLX in Pancreatic Cancer
Pancreatic cancer is a relatively uncommon cancer that is characterized by a complex microenvironment. Its incidence is increasing and it is expected to become the second-leading cause of cancer-related death by 2030 [91]. As a hallmark of FLX safety to normal pancreatic cells, its administration did not induce acute pancreatitis compared to citalopram and other SSRIs [92]. A rough in vivo study on subcutaneous xenograft model of human pancreatic cancer cell line SW1990 in mice revealed that FLX did not induce significant reduction of the tumor growth [93].
However, another impressive study by Schneider et al., showed a promising role of FLX in management of pancreatic cancer via serotonergic pathway. De facto, Platelet-derived peripheral serotonin enhances growth of murine pancreatic cancer cells Panc02. This is mediated by enhanced expression of PD-L1 on mouse and human cancer cells in vitro via serotonylation reaction by covalently binding to glutamine amino acid, resulting in activation of small G proteins. In turn, this impairs accumulation of immune defense cells CD8+T and upregulate PD-L1 expression in pancreatic cancer tumor microenvironment. Inhibition of serotonin cargo in platelets by FLX or TPH1 inhibitor telotristat reduced colon and pancreatic tumor growth in established C57BL/6 mice model by increasing CD8+T accumulation within tumor [94]. The author concluded that combining FLX with anti–PD-1 therapy could be a potential way for treatment of solid tumors [94].
3.8. FLX in Lymphoma
Lymphomas are a heterogeneous group of malignancies which attributed to inadequate proliferation of lymphocytes at different maturation stages and accounts for around 5% of malignances [95,96]. Human Burkitt lymphoma (BL) is an aggressive form due to the rapid proliferation rate and it need immediate intervention with strongly efficient chemotherapeutics [97]. FLX exhibited quick apoptotic mediated cytotoxicity against chemosensitive BL cells MUTU-I accompanied with insignificant effect on normal blood cells. Independent from the serotonergic effect of FLX, it induced apoptosis by caspase pathway, DNA cleavage, and PARP cleavage. [98].
Later on, the same group explored the detailed mechanism using chemoresistant BL cell line DG-75 alongside with MUTU-I. The former lacks Bax and Bak, making it difficult for any chemotherapy to induce apoptosis. FLX induced autophagy programmed cell death in DG-75 independently from caspase pathway, DNA cleavage, and PARP cleavage. In DG-75 cells, western blot showed upregulation of the autophagy marker Beclin 1 and the cytotoxic effect of FLX was reversed by co-treatment with the autophagy inhibitor 3-Methyladenine in contrary to MUTU-I cells. Extracellular Ca+2 influx was found to be crucial in FLX effect on DG-75 but not MUTU-I cells. Furthermore, the authors ruled out ROS involvement in BL cell death. Conclusively, in the chemo-sensitive cells MUTU-I, FLX elicits classic type I cell death apoptosis, nevertheless, it triggers type II cell death autophagy in the chemo-resistant cells DG-75 [99].
In EL4 lymphoma C57BL/6J mice model, FLX attenuated tumor growth accelerated by mice exposure to chronic stress conditions. Cell cycle regulators cyclins A2, D1, and D3 were elevated whereas FLX treatment restored their mRNA expression levels to control values. Furthermore, FLX reduced invasiveness and metastasis to liver and kidney. Once again, FLX reversed the effect of chronic stress by reducing MMP-2 and MMP-9 and increasing tissue inhibitors of metalloproteases TIMPs levels. Intriguingly, treatment with FLX promoted the antitumor immune response in animals [100].
3.8. FLX in Multidrug Resistance (MDR)
MDR operated by extrusion pumps such as P-glycoprotein is one of the main reasons for chemotherapy failure [101,102,103]. One of the earliest studies found that FLX works as a highly effective chemo-sensitizer by elevating cytotoxicity of the anticancer drugs doxorubicin, mitomycin C, vinblastine, and paclitaxel in drug-resistant cancer cells. FLX modulated MDR in vitro an in vivo by slowing down the efflux rate of the titled chemotherapeutics [104].
Recent reports showcased that MDR is caused by the overexpression of transporters on cancer cell membrane that expel chemotherapy out of the tumor cells. These pumps are members of ATP-Binding Cassette (ABC) transporters superfamily [105]. There are seven subfamilies belonging to ABC viz., ABCA—ABCG, where ABCC includes 13 proteins, 9 of which are indicated as MDR proteins (MRPs). Recently, ABCC1/MRP1, and ABCC10/MRP7 were identified as the main drug transporters in MDR and their inhibition is a potential tool for reversing MDR [106,107].
An interesting new study by Kanner et al., assessed three SSRIs including FLX for binding and inhibition of MRP1 and MRP7 in silico. They found that FLX is well adopted in the binding site of MRP1 and MRP7 with slightly higher affinity toward MRP7. Experimentally, they found that FLX reversed MDR resistance of MRP1-over expressing human epidermoid carcinoma cell line KB/CV60 to vincristine, and doxorubicin. In parallel, the same sensitizing effect was observed for MRP7-overexpressing human ovarian adenocarcinoma cell line SKOV3/MRP7 which is resistant to paclitaxel [108]. Conclusively, FLX could be an efficient chemo-sensitizer for overcoming MDR.
4. Conclusions
Drug repurposing represents an indispensable tool for modern drug discovery that paves the way for a short-time and cost-effective bench-to-bedside drug transition. Several SSRIs have been efficiently benchmarked against a multitude of cancer types. FLX is distinguished with a highly favourable safety profile; compared to other SSRIs it is unlikely to cause pancreatitis. Among different antidepressants, only FLX enhanced the overall survival of patients receiving antidepressant/PD-1/L1 immunotherapy combination compared to PD-1/L1 alone. Of note, FLX is characterized by high repurposing potential, especially against GBM and neuroblastoma cells which it can access readily due to its rapid BBB penetration ability. FLX exhibited cytotoxic effects against cancers from different origins including breast, liver, colon, cervix, lung, pancreas, and lymph system cancers. We showed that FLX/TMZ combination action significantly surpasses monotherapy against GBM on both in vitro and in vivo levels. Similarly, FLX/RAL combination outperforms monotherapy for the treatment of DMBA-induced breast cancer. Additionally, a combination of cisplatin with fluoxetine could be a judicious choice for the treatment of cervical cancer with a better outcome than cisplatin alone.
In this review, we further explained that FLX is a promising chemo-sensitizer that can circumvent MDR of chemotherapy. We also discussed pharmaceutical preparation that loaded fluoxetine on carriers to improve its delivery and selectivity towards cancer cells which have characteristic acidic environment. During writing this article, a new study was released showcasing the ability of FLX to inhibit cell proliferation, invasion, metastasis, and angiogenesis of osteaosarcoma cells by suppressing phosphorylation of signal transducer and activator of transcription 3 (STAT3) [109]. Taken together, FLX repurposing could be a potential avenue for the cotreatment of cancer patients.
Supplementary Materials
Not applicable.
Author Contributions
Conceptualization, M.O.R.; methodology, S.F.K., A.M.S.A., M.O.R.; software, S.F.K., A.M.S.A., M.O.R; investigation S.F.K., A.M.S.A, resources S.F.K., A.M.S.A.; data curation S.F.K., A.M.S.A.; writing—original draft preparation, S.F.K., M.O.R.; writing—review and editing, S.F.K., M.O.R.; visualization, S.F.K., A.M.S.A.; supervision, M.O.R.; project administration, M.O.R.; funding acquisition, S.F.K., A.M.S.A. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are thankful to the Deanship of Scientific Research at the University of Bisha for supporting this work through the Fast-Track Research Support Program.
Conflicts of Interest
All the authors declare that they have no conflict of interest of any kind.
References
- De Oliveira, E.A.M.; Lang, K.L. Drug Repositioning: Concept, Classification, Methodology, and Importance in Rare/Orphans and Neglected Diseases. J. Appl. Pharm. Sci. 2018, 8, 157–165. [Google Scholar] [CrossRef]
- Song, Y.; Yang, X.; Yu, B. Repurposing Antidepressants for Anticancer Drug Discovery. Drug Discov. Today 2022, 27, 1924–1935. [Google Scholar] [CrossRef]
- Rascol, O.; Fabbri, M.; Poewe, W. Amantadine in the Treatment of Parkinson’s Disease and Other Movement Disorders. Lancet Neurol. 2021, 20, 1048–1056. [Google Scholar] [CrossRef]
- Menter, D.G.; Schilsky, R.L.; DuBois, R.N. Cyclooxygenase-2 and Cancer Treatment: Understanding the Risk Should Be Worth the Reward. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 1384–1390. [Google Scholar] [CrossRef]
- Sezer, A.; Halilović-Alihodžić, M.; Vanwieren, A.R.; Smajkan, A.; Karić, A.; Djedović, H.; Šutković, J. A Review on Drug Repurposing in COVID-19: From Antiviral Drugs to Herbal Alternatives. J. Genet. Eng. Biotechnol. 2022, 20, 78. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.; Bento Cunha, R.; Vassilevskaia, T.; Viveiros, M.; Cunha, C. Drug Repurposing for COVID-19: A Review and a Novel Strategy to Identify New Targets and Potential Drug Candidates. Mol. Basel Switz. 2022, 27, 2723. [Google Scholar] [CrossRef]
- Hijikata, A.; Shionyu, C.; Nakae, S.; Shionyu, M.; Ota, M.; Kanaya, S.; Shirai, T. Current Status of Structure-Based Drug Repurposing against COVID-19 by Targeting SARS-CoV-2 Proteins. Biophys. Physicobiology 2021, 18, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Lenze, E.J.; Reiersen, A.M.; Zorumski, C.F.; Santosh, P.J. Beyond “Psychotropic”: Repurposing Psychiatric Drugs for COVID-19, Alzheimer’s Disease, and Cancer. J. Clin. Psychiatry 2023, 84, 22r14494. [Google Scholar] [CrossRef]
- Radwan, M.O.; Abd-Alla, H.I.; Alsaggaf, A.T.; El-Mezayen, H.; Abourehab, M.A.S.; El-Beeh, M.E.; Tateishi, H.; Otsuka, M.; Fujita, M. Gypsogenin Battling for a Front Position in the Pentacyclic Triterpenes Game of Thrones on Anti-Cancer Therapy: A Critical Review—Dedicated to the Memory of Professor Hanaa M. Rady. Molecules 2023, 28, 5677. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–386. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, A.K.; Sakamoto, T.; Toma, T.; Sakamoto, M.; Abourehab, M.A.S.; Otsuka, M.; Fujita, M.; Tateishi, H.; Radwan, M.O. New Insights into the Structural Requirements of Isatin-Derived Pro-Apoptotic Agents against Acute Myeloid Leukemia. Pharm. 2022 Vol 15 Page 1579 2022, 15, 1579. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming Cancer Therapeutic Bottleneck by Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Cruz-Burgos, M.; Losada-Garcia, A.; Cruz-Hernández, C.D.; Cortés-Ramírez, S.A.; Camacho-Arroyo, I.; Gonzalez-Covarrubias, V.; Morales-Pacheco, M.; Trujillo-Bornios, S.I.; Rodríguez-Dorantes, M. New Approaches in Oncology for Repositioning Drugs: The Case of PDE5 Inhibitor Sildenafil. Front. Oncol. 2021, 11, 627229. [Google Scholar] [CrossRef] [PubMed]
- Di, X.; Gennings, C.; Bear, H.D.; Graham, L.J.; Sheth, C.M.; White, K.L.; Gewirtz, D.A. Influence of the Phosphodiesterase-5 Inhibitor, Sildenafil, on Sensitivity to Chemotherapy in Breast Tumor Cells. Breast Cancer Res. Treat. 2010, 124, 349–360. [Google Scholar] [CrossRef] [PubMed]
- He, X.-K.; Su, T.-T.; Si, J.-M.; Sun, L.-M. Metformin Is Associated With Slightly Reduced Risk of Colorectal Cancer and Moderate Survival Benefits in Diabetes Mellitus: A Meta-Analysis. Medicine (Baltimore) 2016, 95, e2749. [Google Scholar] [CrossRef]
- Urpilainen, E.; Puistola, U.; Boussios, S.; Karihtala, P. Metformin and Ovarian Cancer: The Evidence. Ann. Transl. Med. 2020, 8, 1711. [Google Scholar] [CrossRef]
- Pan, X.; Mao, T.-Y.; Mai, Y.-W.; Liang, C.-C.; Huang, W.-H.; Rao, Y.; Huang, Z.-S.; Huang, S.-L. Discovery of Quinacrine as a Potent Topo II and Hsp90 Dual-Target Inhibitor, Repurposing for Cancer Therapy. Mol. Basel Switz. 2022, 27, 5561. [Google Scholar] [CrossRef]
- Oien, D.B.; Pathoulas, C.L.; Ray, U.; Thirusangu, P.; Kalogera, E.; Shridhar, V. Repurposing Quinacrine for Treatment-Refractory Cancer. Semin. Cancer Biol. 2021, 68, 21–30. [Google Scholar] [CrossRef]
- Gurova, K.V.; Hill, J.E.; Guo, C.; Prokvolit, A.; Burdelya, L.G.; Samoylova, E.; Khodyakova, A.V.; Ganapathi, R.; Ganapathi, M.; Tararova, N.D.; et al. Small Molecules That Reactivate P53 in Renal Cell Carcinoma Reveal a NF-kappaB-Dependent Mechanism of P53 Suppression in Tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 17448–17453. [Google Scholar] [CrossRef]
- Park, S.; Oh, A.-Y.; Cho, J.-H.; Yoon, M.-H.; Woo, T.-G.; Kang, S.-M.; Lee, H.-Y.; Jung, Y.-J.; Park, B.-J. Therapeutic Effect of Quinacrine, an Antiprotozoan Drug, by Selective Suppression of p-CHK1/2 in P53-Negative Malignant Cancers. Mol. Cancer Res. MCR 2018, 16, 935–946. [Google Scholar] [CrossRef]
- Radwan, M.O.; Toma, T.; Arakaki, Y.; Kamo, M.; Inoue, N.; Koga, R.; Otsuka, M.; Tateishi, H.; Fujita, M. New Insight into the Bioactivity of Substituted Benzimidazole Derivatives: Repurposing from Anti-HIV Activity to Cell Migration Inhibition Targeting hnRNP M. Bioorg. Med. Chem. 2023, 117294–117294. [Google Scholar] [CrossRef]
- Radwan, M.O.; Ciftci, H.I.; Ali, T.F.S.; Ellakwa, D.E.; Koga, R.; Tateishi, H.; Nakata, A.; Ito, A.; Yoshida, M.; Okamoto, Y.; et al. Antiproliferative S-Trityl-l-Cysteine -Derived Compounds as SIRT2 Inhibitors: Repurposing and Solubility Enhancement. Molecules 2019, 24, 3295. [Google Scholar] [CrossRef]
- Radwan, M.O.; Ciftci, H.I.; Ali, T.F.S.; Koga, R.; Tateishi, H.; Nakata, A.; Ito, A.; Yoshida, M.; Fujita, M.; Otsuka, M. Structure Activity Study of S-Trityl-Cysteamine Dimethylaminopyridine Derivatives as SIRT2 Inhibitors: Improvement of SIRT2 Binding and Inhibition. Bioorg. Med. Chem. Lett. 2020, 30, 127458–127458. [Google Scholar] [CrossRef]
- Singhal, S.; Maheshwari, P.; Krishnamurthy, P.T.; Patil, V.M. Drug Repurposing Strategies for Non-Cancer to Cancer Therapeutics. Anticancer Agents Med. Chem. 2022, 22, 2726–2756. [Google Scholar] [CrossRef] [PubMed]
- Sternbach, H. Are Antidepressants Carcinogenic? A Review of Preclinical and Clinical Studies. J. Clin. Psychiatry 2003, 64, 1153–1162. [Google Scholar] [CrossRef]
- Bielecka, A.M.; Obuchowicz, E. Antidepressant Drugs as a Complementary Therapeutic Strategy in Cancer. Exp. Biol. Med. Maywood NJ 2013, 238, 849–858. [Google Scholar] [CrossRef]
- Tutton, P.J.; Barkla, D.H. Influence of Inhibitors of Serotonin Uptake on Intestinal Epithelium and Colorectal Carcinomas. Br. J. Cancer 1982, 46, 260–265. [Google Scholar] [CrossRef]
- Zheng, Y.; Chang, X.; Huang, Y.; He, D. The Application of Antidepressant Drugs in Cancer Treatment. Biomed. Pharmacother. Biomedecine Pharmacother. 2023, 157, 113985. [Google Scholar] [CrossRef] [PubMed]
- Radin, D.P.; Patel, P. A Current Perspective on the Oncopreventive and Oncolytic Properties of Selective Serotonin Reuptake Inhibitors. Biomed. Pharmacother. Biomedecine Pharmacother. 2017, 87, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-W.; Kim, E.-J.; Nyiramana, M.; Shin, E.-J.; Jin, H.; Ryu, J.; Kang, K.; Lee, G.-W.; Kim, H.; Han, J.; et al. Paroxetine Induces Apoptosis of Human Breast Cancer MCF-7 Cells through Ca2+-and P38 MAP Kinase-Dependent ROS Generation. Cancers 2019, 11, 64. [Google Scholar] [CrossRef]
- Gillman, P.K. Tricyclic Antidepressant Pharmacology and Therapeutic Drug Interactions Updated. Br. J. Pharmacol. 2007, 151, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Edinoff, A.N.; Akuly, H.A.; Hanna, T.A.; Ochoa, C.O.; Patti, S.J.; Ghaffar, Y.A.; Kaye, A.D.; Viswanath, O.; Urits, I.; Boyer, A.G.; et al. Selective Serotonin Reuptake Inhibitors and Adverse Effects: A Narrative Review. Neurol. Int. 2021, 13, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Ashbury, J.E.; Lévesque, L.E.; Beck, P.A.; Aronson, K.J. A Population-Based Case-Control Study of Selective Serotonin Reuptake Inhibitors (SSRIs) and Breast Cancer: The Impact of Duration of Use, Cumulative Dose and Latency. BMC Med. 2010, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Toh, S.; García Rodríguez, L.A.; Hernández-Díaz, S. Use of Antidepressants and Risk of Lung Cancer. Cancer Causes Control 2007, 18, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Tamim, H.; Shapiro, S.; Stang, M.R.; Collet, J.-P. Use of Antidepressants and Risk of Colorectal Cancer: A Nested Case-Control Study. Lancet Oncol. 2006, 7, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Coogan, P.F.; Strom, B.L.; Rosenberg, L. Antidepressant Use and Colorectal Cancer Risk. Pharmacoepidemiol. Drug Saf. 2009, 18, 1111–1114. [Google Scholar] [CrossRef] [PubMed]
- Wernli, K.J.; Hampton, J.M.; Trentham-Dietz, A.; Newcomb, P.A. Antidepressant Medication Use and Breast Cancer Risk. Pharmacoepidemiol. Drug Saf. 2009, 18, 284–290. [Google Scholar] [CrossRef]
- Gil-Ad, I.; Zolokov, A.; Lomnitski, L.; Taler, M.; Bar, M.; Luria, D.; Ram, E.; Weizman, A. Evaluation of the Potential Anti-Cancer Activity of the Antidepressant Sertraline in Human Colon Cancer Cell Lines and in Colorectal Cancer-Xenografted Mice. Int. J. Oncol. 2008, 33, 277–286. [Google Scholar] [CrossRef]
- Jiang, X.; Lu, W.; Shen, X.; Wang, Q.; Lv, J.; Liu, M.; Cheng, F.; Zhao, Z.; Pang, X. Repurposing Sertraline Sensitizes Non–Small Cell Lung Cancer Cells to Erlotinib by Inducing Autophagy. JCI Insight 3 e98921. [CrossRef]
- Ozunal, Z.G.; Cakil, Y.D.; Isan, H.; Saglam, E.; Aktas, R.G. Sertraline in Combination with Sorafenib: A Promising Pharmacotherapy to Target Both Depressive Disorders and Hepatocellular Cancer. Biol. Futura 2019, 70, 341–348. [Google Scholar] [CrossRef]
- Geeraerts, S.L.; Kampen, K.R.; Rinaldi, G.; Gupta, P.; Planque, M.; Louros, N.; Heylen, E.; De Cremer, K.; De Brucker, K.; Vereecke, S.; et al. Repurposing the Antidepressant Sertraline as SHMT Inhibitor to Suppress Serine/Glycine Synthesis–Addicted Breast Tumor Growth. Mol. Cancer Ther. 2021, 20, 50–63. [Google Scholar] [CrossRef]
- Baldissera, A.B.; Boia-Ferreira, M.; Basílio, A.B.C.; Resende, J.S. de S.; Castro, M.A.A.; Chaim, O.M.; Gremski, L.H.; Veiga, S.S.; Senff-Ribeiro, A. Sertraline as a Potential Cancer Therapeutic Approach: Biological Relevance of TCTP in Breast Cancer Cell Lines and Tumors. Adv. Med. Sci. 2023, 68, 227–237. [Google Scholar] [CrossRef]
- Wang, K.; Gong, Q.; Zhan, Y.; Chen, B.; Yin, T.; Lu, Y.; Zhang, Y.; Wang, H.; Ke, J.; Du, B.; et al. Blockage of Autophagic Flux and Induction of Mitochondria Fragmentation by Paroxetine Hydrochloride in Lung Cancer Cells Promotes Apoptosis via the ROS-MAPK Pathway. Front. Cell Dev. Biol. 2019, 7, 397. [Google Scholar] [CrossRef]
- Jang, W.-J.; Jung, S.K.; Vo, T.T.L.; Jeong, C.-H. Anticancer Activity of Paroxetine in Human Colon Cancer Cells: Involvement of MET and ERBB3. J. Cell. Mol. Med. 2019, 23, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.C.-H.; Hsieh, Y.-H.; Chen, L.-J.; Hsu, T.-C.; Tzang, B.-S. Escitalopram Oxalate Induces Apoptosis in U-87MG Cells and Autophagy in GBM8401 Cells. J. Cell. Mol. Med. 2018, 22, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Yuan, I.; Horng, C.-T.; Chen, V.C.-H.; Chen, C.-H.; Chen, L.-J.; Hsu, T.-C.; Tzang, B.-S. Escitalopram Oxalate Inhibits Proliferation and Migration and Induces Apoptosis in Non-Small Cell Lung Cancer Cells. Oncol. Lett. 2018, 15, 3376–3382. [Google Scholar] [CrossRef] [PubMed]
- Bavadekar, S.; Panchal, P.; Hanbashi, A.; Vansal, S. Cytotoxic Effects of Selective Serotonin- and Serotonin-Norepinephrine Reuptake Inhibitors on Human Metastatic Breast Cancer Cell Line, MCF-7 (842.3). FASEB J. 2014, 28, 842–3. [Google Scholar] [CrossRef]
- Magagnoli, J.; Narendran, S.; Pereira, F.; Cummings, T.H.; Hardin, J.W.; Sutton, S.S.; Ambati, J. Association between Fluoxetine Use and Overall Survival among Patients with Cancer Treated with PD-1/L1 Immunotherapy. Pharmaceuticals 2023, 16, 640. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.I.; Radwan, M.O.; Sever, B.; Hamdy, A.K.; Emirdağ, S.; Ulusoy, N.G.; Sozer, E.; Can, M.; Yayli, N.; Araki, N.; et al. EGFR-Targeted Pentacyclic Triterpene Analogues for Glioma Therapy. Int. J. Mol. Sci. 2021, 22, 10945. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-H.; Yang, S.-T.; Lin, Y.-K.; Lin, J.-W.; Lee, Y.-H.; Wang, J.-Y.; Hu, C.-J.; Lin, E.-Y.; Chen, S.-M.; Then, C.-K.; et al. Fluoxetine, an Antidepressant, Suppresses Glioblastoma by Evoking AMPAR-Mediated Calcium-Dependent Apoptosis. Oncotarget 2014, 6, 5088–5101. [Google Scholar] [CrossRef]
- You, F.; Zhang, C.; Liu, X.; Ji, D.; Zhang, T.; Yu, R.; Gao, S. Drug Repositioning: Using Psychotropic Drugs for the Treatment of Glioma. Cancer Lett. 2022, 527, 140–149. [Google Scholar] [CrossRef]
- Bi, J.; Khan, A.; Tang, J.; Armando, A.M.; Wu, S.; Zhang, W.; Gimple, R.C.; Reed, A.; Jing, H.; Koga, T.; et al. Targeting Glioblastoma Signaling and Metabolism with a Re-Purposed Brain-Penetrant Drug. Cell Rep. 2021, 37, 109957. [Google Scholar] [CrossRef]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The Glutamate Receptor Ion Channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar] [PubMed]
- Sobolevsky, A.I.; Rosconi, M.P.; Gouaux, E. X-Ray Structure, Symmetry and Mechanism of an AMPA-Subtype Glutamate Receptor. Nature 2009, 462, 745–756. [Google Scholar] [CrossRef]
- Ma, J.; Yang, Y.-R.; Chen, W.; Chen, M.-H.; Wang, H.; Wang, X.-D.; Sun, L.-L.; Wang, F.-Z.; Wang, D.-C. Fluoxetine Synergizes with Temozolomide to Induce the CHOP-Dependent Endoplasmic Reticulum Stress-Related Apoptosis Pathway in Glioma Cells. Oncol. Rep. 2016, 36, 676–684. [Google Scholar] [CrossRef]
- Song, T.; Li, H.; Tian, Z.; Xu, C.; Liu, J.; Guo, Y. Disruption of NF-κB Signaling by Fluoxetine Attenuates MGMT Expression in Glioma Cells. OncoTargets Ther. 2015, 8, 2199–2208. [Google Scholar] [CrossRef]
- Bibbo’, S.; Lamolinara, A.; Capone, E.; Purgato, S.; Tsakaneli, A.; Panella, V.; Sallese, M.; Rossi, C.; Ciufici, P.; Nieddu, V.; et al. Repurposing a Psychoactive Drug for Children with Cancer: p27Kip1-Dependent Inhibition of Metastatic Neuroblastomas by Prozac. Oncogenesis 2020, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hosseinimehr, S.J.; Najafi, S.H.; Shafiee, F.; Hassanzadeh, S.; Farzipour, S.; Ghasemi, A.; Asgarian-Omran, H. Fluoxetine as an Antidepressant Medicine Improves the Effects of Ionizing Radiation for the Treatment of Glioma. J. Bioenerg. Biomembr. 2020, 52, 165–174. [Google Scholar] [CrossRef]
- Patanaphan, V.; Salazar, O.M.; Risco, R. Breast Cancer: Metastatic Patterns and Their Prognosis. South. Med. J. 1988, 81, 1109–1112. [Google Scholar] [CrossRef]
- Nelson, H.D.; Smith, M.E.B.; Griffin, J.C.; Fu, R. Use of Medications to Reduce Risk for Primary Breast Cancer: A Systematic Review for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2013, 158, 604–614. [Google Scholar] [CrossRef]
- Ong, J.C.-L.; Sun, F.; Chan, E. Development of Stealth Liposome Coencapsulating Doxorubicin and Fluoxetine. J. Liposome Res. 2011, 21, 261–271. [Google Scholar] [CrossRef]
- Zhou, T.; Duan, J.; Wang, Y.; Chen, X.; Zhou, G.; Wang, R.; Fu, L.; Xu, F. Fluoxetine Synergys with Anticancer Drugs to Overcome Multidrug Resistance in Breast Cancer Cells. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2012, 33, 1299–1306. [Google Scholar] [CrossRef]
- Kabel, A.M.; Elkhoely, A.A. Ameliorative Potential of Fluoxetine/Raloxifene Combination on Experimentally Induced Breast Cancer. Tissue Cell 2016, 48, 89–95. [Google Scholar] [CrossRef]
- Tatar, O.; Ilhan, N.; Ilhan, N.; Susam, S.; Ozercan, I.H. Is There Any Potential Anticancer Effect of Raloxifene and Fluoxetine on DMBA-induced Rat Breast Cancer? J. Biochem. Mol. Toxicol. 2019, 33, e22371. [Google Scholar] [CrossRef]
- Duarte, D.; Cardoso, A.; Vale, N. Synergistic Growth Inhibition of HT-29 Colon and MCF-7 Breast Cancer Cells with Simultaneous and Sequential Combinations of Antineoplastics and CNS Drugs. Int. J. Mol. Sci. 2021, 22, 7408. [Google Scholar] [CrossRef]
- Duarte, D.; Rêma, A.; Amorim, I.; Vale, N. Drug Combinations: A New Strategy to Extend Drug Repurposing and Epithelial-Mesenchymal Transition in Breast and Colon Cancer Cells. Biomolecules 2022, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Falcão, S.I.; El Mehdi, I.; Vilas-Boas, M.; Vale, N. Honeybee Venom Synergistically Enhances the Cytotoxic Effect of CNS Drugs in HT-29 Colon and MCF-7 Breast Cancer Cell Lines. Pharmaceutics 2022, 14, 511. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.-A.; Chu, P.-Y.; Tan, Z.-L.; Hsu, F.-T.; Lee, Y.-C.; Wu, H.-J. STAT3 Inactivation and Induction of Apoptosis Associate With Fluoxetine-Inhibited Epithelial-Mesenchymal Transition and Growth of Triple-Negative Breast Cancer In Vivo. Anticancer Res. 2022, 42, 3807–3814. [Google Scholar] [CrossRef]
- Bowie, M.; Pilie, P.; Wulfkuhle, J.; Lem, S.; Hoffman, A.; Desai, S.; Petricoin, E.; Carter, A.; Ambrose, A.; Seewaldt, V.; et al. Fluoxetine Induces Cytotoxic Endoplasmic Reticulum Stress and Autophagy in Triple Negative Breast Cancer. World J. Clin. Oncol. 2015, 6, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhu, L.; Zhao, Y.; Jiang, Y.; Chen, L.; Yu, Y.; Ouyang, L. Fluoxetine Induces Autophagic Cell Death via eEF2K-AMPK-mTOR-ULK Complex Axis in Triple Negative Breast Cancer. Cell Prolif. 2018, 51, e12402. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA. Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, J.; Yamada, T.; Egashira, N.; Ueda, M.; Zukeyama, N.; Ushio, S.; Masuda, S. Comparison of the Anti-Tumor Effects of Selective Serotonin Reuptake Inhibitors as Well as Serotonin and Norepinephrine Reuptake Inhibitors in Human Hepatocellular Carcinoma Cells. Biol. Pharm. Bull. 2015, 38, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Mun, A.-R.; Lee, S.-J.; Kim, G.-B.; Kang, H.-S.; Kim, J.-S.; Kim, S.-J. Fluoxetine-Induced Apoptosis in Hepatocellular Carcinoma Cells. Anticancer Res. 2013, 33, 3691–3697. [Google Scholar] [PubMed]
- Hsu, L.-C.; Tu, H.-F.; Hsu, F.-T.; Yueh, P.-F.; Chiang, I.-T. Beneficial Effect of Fluoxetine on Anti-Tumor Progression on Hepatocellular Carcinoma and Non-Small Cell Lung Cancer Bearing Animal Model. Biomed. Pharmacother. 2020, 126, 110054. [Google Scholar] [CrossRef]
- Chen, W.-T.; Hsu, F.-T.; Liu, Y.-C.; Chen, C.-H.; Hsu, L.-C.; Lin, S.-S. Fluoxetine Induces Apoptosis through Extrinsic/Intrinsic Pathways and Inhibits ERK/NF-κB-Modulated Anti-Apoptotic and Invasive Potential in Hepatocellular Carcinoma Cells In Vitro. Int. J. Mol. Sci. 2019, 20, 757. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Bar, H.M.; Khater, S.E.; Ghorab, D.M.; Al-mahallawi, A.M. Hexosomes as Efficient Platforms for Possible Fluoxetine Hydrochloride Repurposing with Improved Cytotoxicity against HepG2 Cells. ACS Omega 2020, 5, 26697–26709. [Google Scholar] [CrossRef]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal Cancer Statistics, 2023. CA. Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and Number of Cancer Cases and Deaths Attributable to Potentially Modifiable Risk Factors in the United States. CA. Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef]
- Engelmann, B.J.; Ryan, J.J.; Farrell, N.P. Antidepressants and Platinum Drugs. Anticancer Res. 2014, 34, 509–516. [Google Scholar]
- Kang, B.-G.; Shende, M.; Inci, G.; Park, S.-H.; Jung, J.-S.; Kim, S.B.; Kim, J.H.; Mo, Y.W.; Seo, J.-H.; Feng, J.-H.; et al. Combination of Metformin/Efavirenz/Fluoxetine Exhibits Profound Anticancer Activity via a Cancer Cell-Specific ROS Amplification. Cancer Biol. Ther. 2023, 24, 2161803. [Google Scholar] [CrossRef]
- Marcinkute, M.; Afshinjavid, S.; Fatokun, A.A.; Javid, F.A. Fluoxetine Selectively Induces P53-Independent Apoptosis in Human Colorectal Cancer Cells. Eur. J. Pharmacol. 2019, 857, 172441. [Google Scholar] [CrossRef]
- Minagawa, Y.; Kigawa, J.; Itamochi, H.; Kanamori, Y.; Shimada, M.; Takahashi, M.; Terakawa, N. Cisplatin-resistant HeLa Cells Are Resistant to Apoptosis via P53-dependent and -independent Pathways. Jpn. J. Cancer Res. Gann 1999, 90, 1373–1379. [Google Scholar] [CrossRef]
- Charles, E.; Hammadi, M.; Kischel, P.; Delcroix, V.; Demaurex, N.; Castelbout, C.; Vacher, A.-M.; Devin, A.; Ducret, T.; Nunes, P.; et al. The Antidepressant Fluoxetine Induces Necrosis by Energy Depletion and Mitochondrial Calcium Overload. Oncotarget 2016, 8, 3181–3196. [Google Scholar] [CrossRef]
- Liu, Y.; Li, T.; Xu, M.; Che, X.; Jiang, X. Fluoxetine Enhances Cellular Chemosensitivity to Cisplatin in Cervical Cancer.
- Naz, A.; Hashim, F.; Ali, S.A.; Badshah, M. Fabrication, Characterization and Therapeutic Evaluation of Fluoxetine-Dextran Nanoparticles. ChemistrySelect 2023, 8, e202204110. [Google Scholar] [CrossRef]
- Sagnella, S.M.; Duong, H.; MacMillan, A.; Boyer, C.; Whan, R.; McCarroll, J.A.; Davis, T.P.; Kavallaris, M. Dextran-Based Doxorubicin Nanocarriers with Improved Tumor Penetration. Biomacromolecules 2014, 15, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-Y.; Lin, S.-S.; Hsu, F.-T.; Chung, J.-G. Fluoxetine Inhibits DNA Repair and NF-ĸB-Modulated Metastatic Potential in Non-Small Cell Lung Cancer. Anticancer Res. 2018, 38, 5201–5210. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Aakjær, M.; Kristiansen, S.B.; Pape, K.; Sessa, M.; Dalhoff, K.P.; De Bruin, M.L.; Andersen, M. Investigation of the Potential Association between the Use of Fluoxetine and Occurrence of Acute Pancreatitis: A Danish Register-Based Cohort Study. Int. J. Epidemiol. 2022, 51, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Shang, Y.-Y.; Li, Y.-Y. Effect of Antidepressants on Body Weight, Ethology and Tumor Growth of Human Pancreatic Carcinoma Xenografts in Nude Mice. World J. Gastroenterol. 2008, 14, 4377–4382. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.A.; Heeb, L.; Beffinger, M.M.; Pantelyushin, S.; Linecker, M.; Roth, L.; Lehmann, K.; Ungethüm, U.; Kobold, S.; Graf, R.; et al. Attenuation of Peripheral Serotonin Inhibits Tumor Growth and Enhances Immune Checkpoint Blockade Therapy in Murine Tumor Models. Sci. Transl. Med. 2021, 13, eabc8188. [Google Scholar] [CrossRef] [PubMed]
- Matasar, M.J.; Zelenetz, A.D. Overview of Lymphoma Diagnosis and Management. Radiol. Clin. North Am. 2008, 46, 175–198. [Google Scholar] [CrossRef]
- Mugnaini, E.N.; Ghosh, N. Lymphoma. Prim. Care 2016, 43, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Faderl, S.; O’Brien, S.; Bueso-Ramos, C.; Cortes, J.; Garcia-Manero, G.; Giles, F.J.; Verstovsek, S.; Wierda, W.G.; Pierce, S.A.; et al. Chemoimmunotherapy with Hyper-CVAD plus Rituximab for the Treatment of Adult Burkitt and Burkitt-Type Lymphoma or Acute Lymphoblastic Leukemia. Cancer 2006, 106, 1569–1580. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Drozgowska, A.; Fayne, D.; Williams, D.C. The Antidepressants Maprotiline and Fluoxetine Have Potent Selective Antiproliferative Effects against Burkitt Lymphoma Independently of the Norepinephrine and Serotonin Transporters. Leuk. Lymphoma 2010, 51, 523–539. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Williams, D.C. The Antidepressants Maprotiline and Fluoxetine Induce Type II Autophagic Cell Death in Drug-Resistant Burkitt’s Lymphoma. Int. J. Cancer 2011, 128, 1712–1723. [Google Scholar] [CrossRef]
- Di Rosso, M.E.; Sterle, H.A.; Cremaschi, G.A.; Genaro, A.M. Beneficial Effect of Fluoxetine and Sertraline on Chronic Stress-Induced Tumor Growth and Cell Dissemination in a Mouse Model of Lymphoma: Crucial Role of Antitumor Immunity. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Tan, B.; Piwnica-Worms, D.; Ratner, L. Multidrug Resistance Transporters and Modulation. Curr. Opin. Oncol. 2000, 12, 450–458. [Google Scholar] [CrossRef]
- Emran, T.B.; Shahriar, A.; Mahmud, A.R.; Rahman, T.; Abir, M.H.; Siddiquee, Mohd.F.-R.; Ahmed, H.; Rahman, N.; Nainu, F.; Wahyudin, E.; et al.Multidrug Resistance in Cancer: Understanding Molecular Mechanisms, Immunoprevention and Therapeutic Approaches. Front. Oncol. 2022, 12. [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug Resistance in Cancer: Role of ATP–Dependent Transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Dekel, Y.; Melikhov, D.; Margalit, R. Fluoxetine Inhibits Multidrug Resistance Extrusion Pumps and Enhances Responses to Chemotherapy in Syngeneic and in Human Xenograft Mouse Tumor Models. Cancer Res. 2004, 64, 7562–7569. [Google Scholar] [CrossRef] [PubMed]
- Wilkens, S. Structure and Mechanism of ABC Transporters. F1000Prime Rep. 2015, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Peña-Solórzano, D.; Stark, S.A.; König, B.; Sierra, C.A.; Ochoa-Puentes, C. ABCG2/BCRP: Specific and Nonspecific Modulators. Med. Res. Rev. 2017, 37, 987–1050. [Google Scholar] [CrossRef] [PubMed]
- Stefan, S.M.; Wiese, M. Small-Molecule Inhibitors of Multidrug Resistance-Associated Protein 1 and Related Processes: A Historic Approach and Recent Advances. Med. Res. Rev. 2019, 39, 176–264. [Google Scholar] [CrossRef]
- Bin Kanner, Y.; Teng, Q.-X.; Ganoth, A.; Peer, D.; Wang, J.-Q.; Chen, Z.-S.; Tsfadia, Y. Cytotoxicity and Reversal Effect of Sertraline, Fluoxetine, and Citalopram on MRP1- and MRP7-Mediated MDR. Front. Pharmacol. 2023, 14. [Google Scholar] [CrossRef]
- Wt, C.; Yh, T.; P, T.; Ft, H.; Hd, W.; Wc, L.; Fh, L.; Ct, W. Fluoxetine Inhibits STAT3-Mediated Survival and Invasion of Osteosarcoma Cells. Anticancer Res. 2023, 43. [Google Scholar] [CrossRef]
Figure 1.
Number of scientific publications comprising scientific output linking repursposing studies with cancer over the period 2005-2023. Data was retrieved from the Web of Science database by uusing the keywords repursposing and cancer.
Figure 1.
Number of scientific publications comprising scientific output linking repursposing studies with cancer over the period 2005-2023. Data was retrieved from the Web of Science database by uusing the keywords repursposing and cancer.
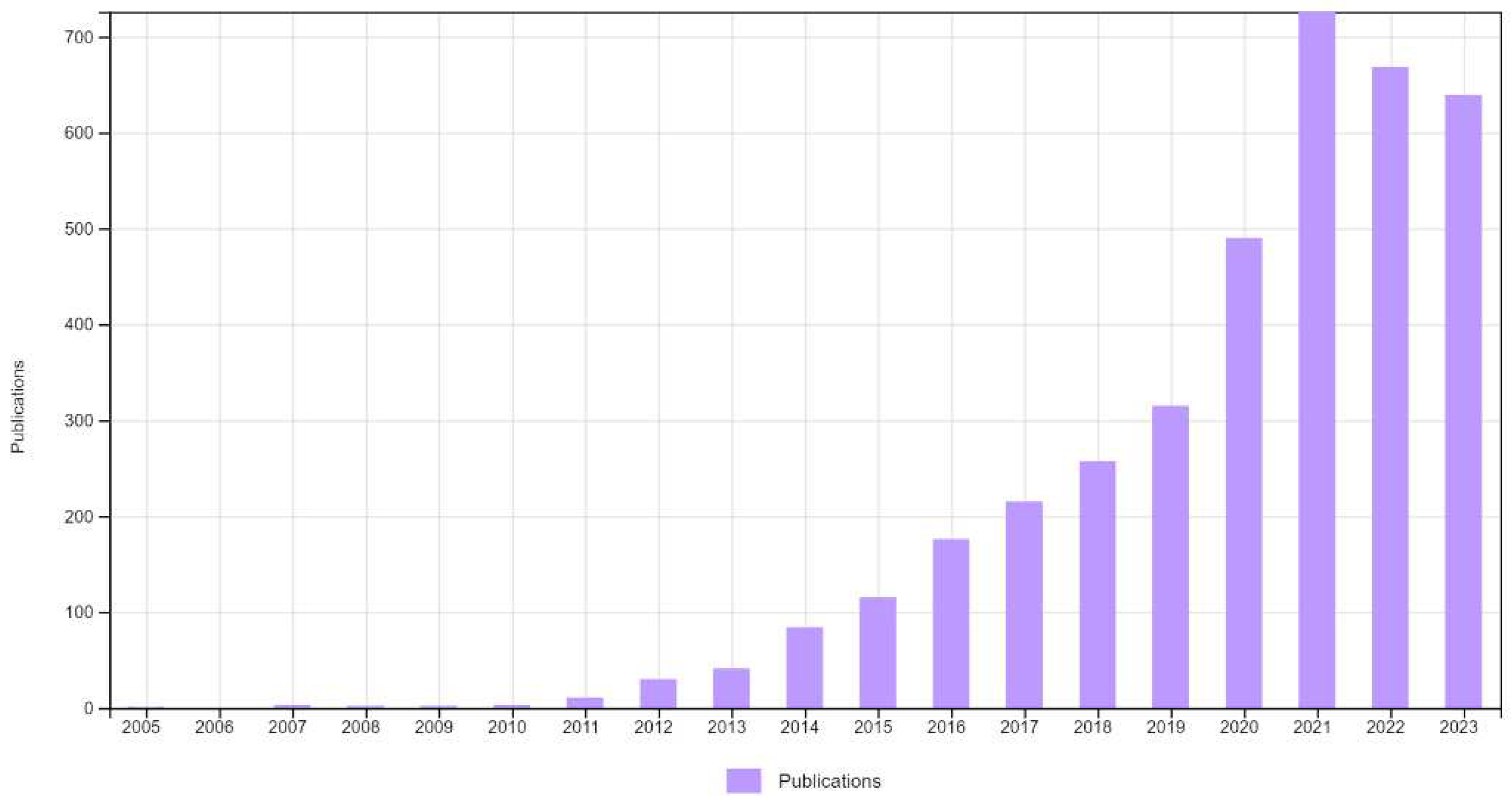
Figure 2.
Chemical structure of fluoxetine (FLX) and summary of different sensitive cancers to fluoxetine treatment.
Figure 2.
Chemical structure of fluoxetine (FLX) and summary of different sensitive cancers to fluoxetine treatment.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



