
Submitted:
10 February 2024
Posted:
13 February 2024
You are already at the latest version
Abstract
Cornelia de Lange syndrome (CdLS) is a complex genetic disorder with a distinct set of facial features, growth limitations, and limb anomalies. Its clinical spectrum is broad and presents significant challenges in pediatric diagnosis and management. The variable presentation of the disorder, due to cohesin complex mutations, requires extensive research to refine care and improve outcomes.
This article provides a case series review of pediatric CdLS patients, alongside a comprehensive literature review, exploring clinical variability and the relationship between genotypic changes and phenotypic outcomes. It also discusses the evolution of diagnostic and therapeutic techniques, with an emphasis on innovations in genetic testing, including the detection of mosaicism and novel genetic variations.
The aim is to synthesise case studies with current research to advance our understanding of CdLS and the effectiveness of management strategies in pediatric healthcare. This work highlights the need for an integrated, evidence-based approach to diagnosis and treatment, and aims to fill existing research gaps and advocate for holistic care protocols and tailored treatment plans for CdLS patients, ultimately improving their quality of life.
Keywords:
Cornelia de Lange syndrome
; pediatric genetics
; phenotypic variability
; cohesinopathies
; rare diseases
1. Introduction
Cornelia de Lange syndrome (CdLS, OMIM 122470) is an essential model of pediatric congenital anomalies that poses multidimensional challenges to clinicians and researchers. This genetic disorder, characterized by distinctive facial features, growth retardation, and limb defects, is attracting attention because of its clinical heterogeneity and underlying genetic complexity. Impairments can affect multiple systems, including the cardiac, gastrointestinal, craniofacial, genitourinary, musculoskeletal, and central nervous systems, with developmental abilities ranging from profound intellectual disability to average intelligence with learning difficulties [1,2].
The cohesin complex is a critical regulator of genomic stability and gene expression. It is frequently implicated in CdLS [3]. Mutations in its component genes reveal the pathophysiological basis of the disorder. CdLS is most likely caused by mutations in one of the cohesin complex genes, including NIPBL, which accounts for about 50–60% of cases; SMC1A and HDAC8, linked to the X chromosome; and SMC3 and RAD21. Mutations can be spontaneous, autosomal dominant, or X-recessive [2,4]. However, the diagnosis is made clinically [1,2].
CdLS is estimated to occur in 1 in 10,000 to 30,000 live births, making it a significant concern in pediatric healthcare. The spectrum of the syndrome, from mild to severe forms, and the variety of possible presentations require astute clinical acumen for diagnosis [1,2]. Particularly challenging are the milder phenotypes, which can result from mosaic genetic patterns and require advanced genetic analysis to confirm the diagnosis.
This article aims to provide a methodological review of pediatric case series and a comprehensive review of the literature on CdLS. Our review is twofold. Firstly, we delve into the case series to examine the clinical presentations, management strategies, and outcomes observed in pediatric patients with CdLS. This will enable us to appreciate the full range of phenotypic expression and the intricacies of care required in this population. Second, we are reviewing the extensive literature to synthesize findings from genetic studies, clinical reports, and therapeutic trials. Such synthesis is essential to identify the current state of knowledge and practice and to illuminate the way forward for pediatric CdLS care.
In addition, this article aims to highlight the importance of mosaicism and its implications for the diagnosis of CdLS, its presentation, and the genetic counseling of families. The relationship between an individual’s genetic makeup and symptoms, especially with the added layer of mosaicism, represents a valuable area for research.
In essence, we aim to provide a resource that will enhance pediatricians’ understanding of CdLS and thus improve the care of those affected. We hope this work will advance the clinical approach to CdLS and inspire further research into pediatric genetic disorders. This will lead to a more holistic understanding of the syndrome.
2. Case Series
This case series presents an insightful examination of four different cases of Cornelia de Lange Syndrome (CdLS) managed at the Department of Neonatology, Gynaecology, and Obstetrics Hospital, Medical University of Poznan Poland, over the period 2018–2023. The focus is on the clinical management and progression of the condition, following the journey from birth through the various stages of hospital care. Our narrative is dedicated to the key decisions, therapeutic interventions, and follow-up approaches that embody the essence of CdLS management. Together, these four cases provide a comprehensive overview of the clinical course of the syndrome, highlighting the actionable aspects of paediatric care that are critical to improving outcomes for affected patients.
Case 1:
A baby girl weighing 1880g (fetal growth restriction: FGR 1 point) was delivered by cesarean section (due to life-threatening symptoms) in severe general condition at 37 weeks’ gestation. The postnatal examination revealed hypotrophy (1 point), microcephaly (1 point), and numerous facial dysmorphic features, including a high forehead, synophrys (2 points), hypertelorism, narrow eyelid fissures, a short nose with an upturned tip (2 points), narrow red lips (2 points), an elongated philtrum (2 points), problems opening the mouth, trismus, short contracted fingers, and small hands and feet (1 point). The baby had a small lower jaw and a shortened tongue frenulum, which prevented weight gain. She had a percutaneous endoscopic gastrostomy-jejunostomy (PEG-PEJ) to help with feeding. However, gastro-oesophageal reflux (GERD) continued to interfere with weight gain. The baby also required a tracheostomy due to tracheomalacia. Respiratory support was required, and weaning from the ventilator was intolerable. In addition, imaging studies revealed a ventricular septal defect (VSD), a horseshoe kidney, and multiple renal pelvicoceles. Microarray-based comparative genomic hybridization (aCGH) analysis showed the presence of an interstitial deletion in the 2q13 region, including the NPHP1 gene, but this was unrelated to the patient’s phenotype. The baby was diagnosed with classic CdLS based on clinical features typical of CdLS, with a score of 12 points. After completing her initial hospitalization in the neonatal unit, the infant diagnosed with Cornelia de Lange Syndrome (CdLS) was transferred to a specialist pediatric care and treatment facility. Unfortunately, there is currently no updated information on the patient’s progress following the transfer.
Case 2:
A male newborn weighing 1820g (FGR = 1 point) was born at 40 weeks gestation by vaginal delivery due to severe asphyxia. Prenatal aCGH was performed because of suspected renal and brain developmental defects. The result was within the resolution limits of the test (100kb). Postnatal examination revealed numerous dysmorphic features: microcephaly (1 point), square face, high and broad forehead, synophrys (2 points), coloboma oculi of the left eye, asymmetric and dysplastic auricles, flat and broad nasal bridge (2 points), bilateral choanal atresia, trismus, micrognathia, hypertelorism, short fifth fingers (1 point), syndactyly of the 2nd and third toes (2 points), widely spaced nipples, and contractures of the limbs. Further imaging studies revealed the presence of VSD, diaphragmatic hernia (2 points), dysplasia of the right kidney, and cryptorchidism (Figure 1). The transfontanel ultrasound (TFUI) showed abnormalities of brain structure: cortical-subcortical atrophy, numerous post-hemorrhagic cysts, slightly dilated lateral ventricles, hypoplasia of the corpus callosum, and enlargement of the choroid plexuses. Other impairments in the boy included growth retardation (1 point), arterial hypertension, congenital hypertonia, external auditory canal stenosis, mixed hearing loss, congenital pneumonia, GERD, and dysplastic acetabulum. The boy required mechanical ventilation and a gastrostomy for feeding. The child’s karyotype was evaluated to rule out the presence of a reciprocal break translocation in the 8q12.2 region related to the CHD7 gene. CHARGE syndrome was initially suspected. Whole exome sequencing (WES) was performed and revealed a de novo HDAC8 mutation (c.883C>T variant). Based on clinical diagnostic criteria (the boy scored 11 points), CdLS was diagnosed (Figure 1).
Follow-up: At four years of age, the boy presented with psychomotor retardation and growth restriction. He sat but did not walk but was able to hold objects correctly in his hands. He underwent mandibular distraction and gingival plasty (Figure 1).
Case 3:
A 1555-gram female newborn (fifth pregnancy 5, birth 1, 36 weeks gestation) was delivered by cesarean section (due to life-threatening fetal symptoms) with moderate birth asphyxia. The general condition of the newborn was described as average. The mother had gestational diabetes type 1. Prenatally, oligohydramnios, FGR (1 point), and upper limb defects (2 points) were noted. The newborn required non-invasive respiratory support. After birth, the physical examination revealed hypotrophy (1 point) and facial dysmorphic features: low hairline on forehead and nape of the neck, low forehead, synophrys (2 points), short nose (2 points), comprehensive, asymmetric, low set auricles, thin upper lip (2 points), underdeveloped mandible and tempomandibular join (TMJ), cleft hard palate, short, broad neck, marble skin, hirsutism (1 point), widely spaced nipples. In addition, radiographic and orthopedic examination revealed deformed, shortened upper limbs, broad shoulder girdle, bilateral absence of ulna bones, flexion contracture of elbow and wrist joints, tiny feet (1 point), limited hip abduction, narrow hip girdle, and numerous other finger defects (Figure 4). The TFUI showed hypoplasia of the cerebellar vermis. In contrast, the TTE (transthoracic echocardiography) showed a patent foramen ovale, a VSD, and a bicuspid aortic valve with trace regurgitation. A tracheostomy was performed on the third day of life. The girl required mechanical ventilation until the 22nd day of life, followed by passive oxygen therapy. The girl also had hematological abnormalities such as coagulopathy and microcytic anemia (oral iron supplementation and erythropoietin treatment). The girl was fed partially parenterally and enterally; on the fourth day of life, stimulation of the sucking and swallowing reflexes was started, but without the expected effect. She was discharged from the hospital under the care of a nutrition clinic for intragastric feeding. At eight months of age, the above cardiological diagnoses were confirmed. Trace mitral regurgitation was also diagnosed, but cardiac dimensions and contractility were within the reference range. Weight gain in the first months of life was unsatisfactory (875 g at five months). She also had a global developmental delay (1 point) and hypotonia. The patient was diagnosed with classic CdLS based on clinical features typical of CdLS, with a score of 11 points. Karyotype analysis excluded chromosomal aberrations. A percutaneous endoscopic gastrostomy (PEG) was placed at eight months of age. The girl suffered from GERD.
Case 4:
A 1600-gram baby girl (FGR-1 point) was born at 36 weeks’ gestation by vaginal delivery with meconium-stained amniotic fluid and a two-vessel umbilical cord. Initially, the newborn required cardiopulmonary resuscitation, but by the time she was transferred to the neonatal unit, her circulatory and respiratory systems were functional. The mother had gestational diabetes type 1. Genetic amniocentesis was performed during pregnancy on suspicion of FGR, but the result was in the normal range. Postnatal physical examination revealed numerous dysmorphic features (hirsutism (1 point), synophrys (2 points), long eyelashes, short nose with concave nasal ridge (2 points), thin upper lip vermilion (2 points), long philtrum (2 points), microcephaly (1 point), shortened forearms, broad hands with conical fingers, sunken chest, marble skin, hypertonic peripheral muscles, and weak signs of primitive neonatal reflexes). TTE showed a bicuspid aortic valve. The girl was fed an intragastric tube (foremilk and breastmilk supplement) from the fifth day of life due to a lack of sucking reflex (Figure 2). Classic CdLS was diagnosed based on clinical features typical of the syndrome (11 points in the clinical diagnostic classification). The girl had an intragastric feeding tube because of her intolerance to foremilk and pacifier feeding (presence of frequent regurgitation, abdominal pain, difficulty sucking, and lack of expected weight gain) (Figs. 4, 5). After discharge from the neonatal unit, the infant, diagnosed with Cornelia de Lange Syndrome (CdLS), was placed under the supervision of specialist outpatient clinics, including cardiology, genetics, audiology, and nutrition. At one year of life, the child was found to have significant deficits in body mass and growth, gastroesophageal reflux disease (GERD), and delayed psychomotor development (Figure 2).
4. Discussion
Clinical Manifestations
Cornelia de Lange Syndrome (CdLS) is a paradigm in pediatric genetics that provides insight into the spectrum of congenital anomalies. The 2018 International Consensus provides a framework for classifying CdLS as either classical or non-classical, depending on clinical manifestations and genetic markers within chromatin-regulating genes, such as those constituting the cohesin complex [1,2].
The diagnostic scale quantifies major (2 points) and minor (1 point) clinical features to facilitate a structured approach to diagnosis. Major features contributing to this scale include synophrys and thick eyebrows; an elongated, smooth fossa; a short, convex nose; a thin upper lip and lower corners of the mouth; oligodactyly or adactyly; and congenital diaphragmatic hernia [5]. Additional considerations include delayed intellectual development, intrauterine growth retardation, microcephaly, small hands, and feet, a short fifth toe, and hirsutism [Table 1].
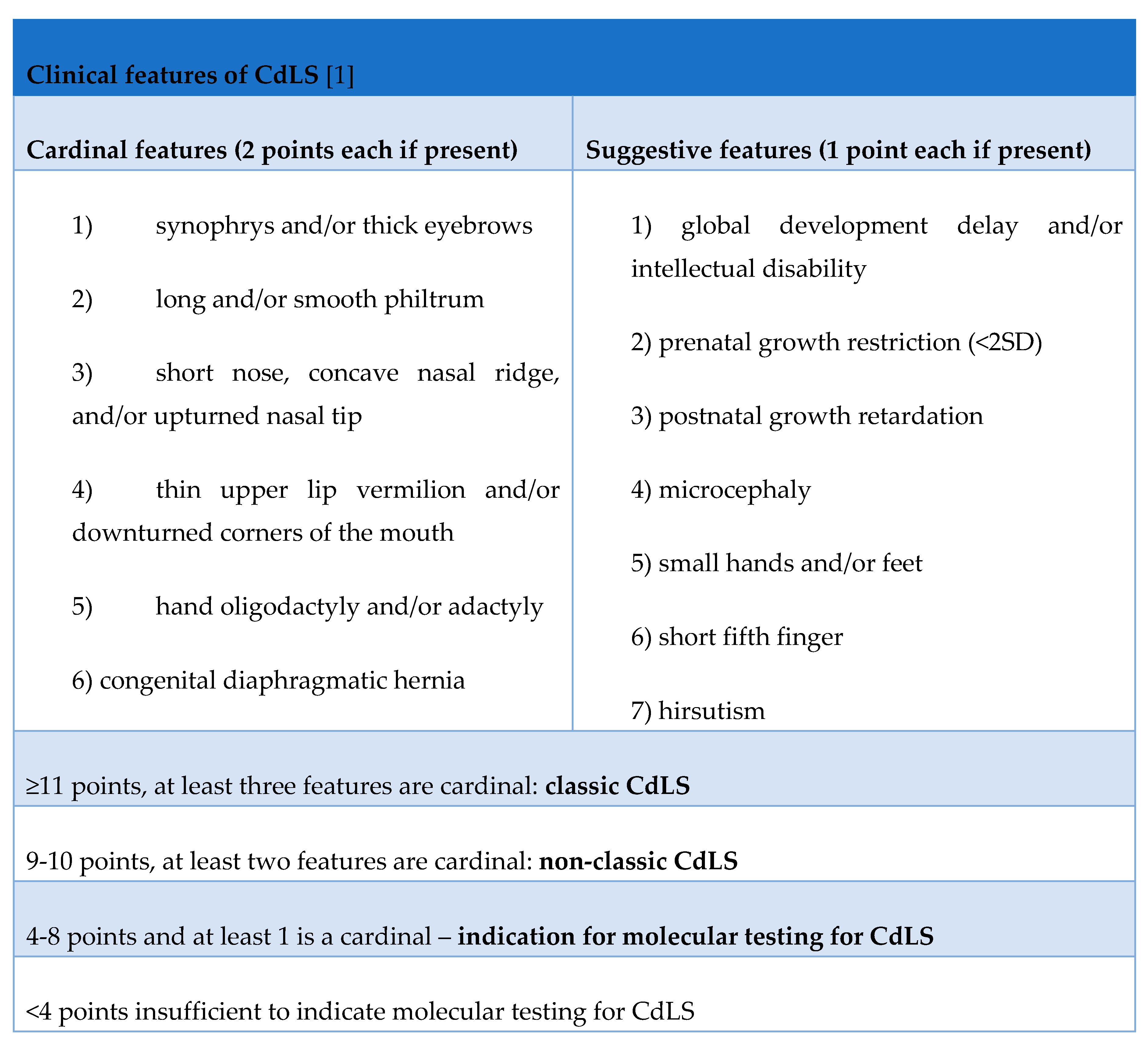
A cumulative score of 11 or more supports a diagnosis of classic CdLS, whereas a score of 9 to 10 indicates a non-classical variant. Scores in the intermediate range require molecular testing. If the patient scores less than 4, there is no indication for molecular testing for CdLS [1].
Suspicion of CdLS is usually raised immediately after birth because of the characteristic phenotype [1,2]. A prominent philtrum with thin, sunken lips, narrow palpebral fissures, low-set ears, a depressed flat nasal bridge, anteverted nostrils, well-defined arched eyebrows with synophrys, long eyelashes, a cleft palate, microcephaly (small head), and micrognathia are some of the characteristic facial features associated with CDLS [6].
CdLS is characterized by psychomotor retardation and intellectual disability. Learning disabilities range from mild to severe [7].
One of the skin symptoms of CdLS is generalized hirsutism. This is excessive and coarse hair growth, particularly on the face, back, and extremities.
Children with CdLS often have musculoskeletal abnormalities in the upper extremities, ranging from small hands and feet to clinodactyly of the fifth finger to a missing forearm with digits. Although less common in the lower limbs, tiny feet and syndactyly of the toes may be seen. CdLS is also associated with hip dysplasia, scoliosis, pectus excavatum, early development of bunions, and cervical spine abnormalities [8].
It should also be noted that, in addition to the clinical diagnostic criteria, other characteristic gastrointestinal symptoms or behavioral disturbances are common in patients with CdLS. Common gastrointestinal findings in Cornelia de Lange syndrome patients include gastroesophageal reflux (GERD), volvulus, rumination, chronic constipation, submucous cleft palate, chronic sinusitis, and nasal polyps. Reflux may only present as difficulty swallowing, wheezing, vomiting, or aspirating food into the lungs. Silent reflux may be expected in CdLS. If there are no symptoms of reflux, it may be helpful to look for behavioral signs or signs of pain and discomfort to help diagnose [9]. Other findings may include Barrett’s esophagus, gastrointestinal problems such as the risk of volvulus, rumination, chronic constipation, and renal and urinary tract abnormalities [10,11].
One of CdLS’s most prominent behavioral features is self-injurious behavior (SIB), including head banging, hitting, or biting, which can be of considerable concern to carers. SIB has been reported in up to 70% of people with CdLS. It is considered a maladaptive coping mechanism and may be associated with frustration, difficulty communicating needs, sensory sensitivity, hyperactivity, impulsivity, and attention deficit [12]. Symptoms of attention deficit hyperactivity disorder (ADHD) have been observed in a significant proportion of people with CdLS [13].
In addition, individuals with CdLS may exhibit engagement (e.g., hand flapping), rigid adherence to routines, and repetitive vocalizations that interfere with adaptive functioning and social engagement [14,15]. Understanding and managing behavioral problems in CdLS requires a multidisciplinary approach, including behavioral interventions, occupational therapy, and pharmacological management as appropriate.
Congenital heart defects are present in approximately 25–30% of children with CdLS. Neurological disorders such as epilepsy, particularly partial epilepsy, are seen in up to 20% of cases of CdLS. People with CdLS may also have peripheral neuropathy, which can cause an abnormally high threshold for pain. Finally, structural abnormalities of the kidneys or urinary tract, such as vesicoureteral reflux, pelvic dilatation, and renal dysplasia, affect up to 40% of people with CdLS. There have also been reports of bicornuate or septate uteri, hypospadias, cryptorchidism, genital hypoplasia, and irregular menstruation [10].
Perioperative Care
Individuals with CdLS require special care when undergoing anesthesia due to the potential for increased perioperative stress, airway difficulties, and cardiovascular compromise. A physical examination of the airway and facial structures is critical to identifying potential difficulties that may affect the administration of anesthesia, such as cranial anomalies and micrognathia. In some cases, imaging techniques such as CT or MRI may help identify new or unknown airway abnormalities that may affect the administration of anesthesia.
Inhalational anesthetics provide a smooth induction of anesthesia and avoid the added stress of needle insertion. Alternatively, intravenous induction with careful titration of drugs such as propofol or etomidate can provide better airway control, protection against seizures, and reduced emergence agitation if dosed appropriately. Percutaneous tracheostomy may be a chronic airway management strategy [16,17].
Genetic Diagnosis
The list of genes that are functionally related to the cohesin apparatus and whose haploinsufficiency results in situations that either belong to or overlap with the CdLS spectrum has recently been expanded with the availability of genotype-driven studies [20].
The molecular basis of CdLS is based on mutations in genes of the cohesin complex, which is responsible for chromatin stabilization, chromosome segregation, regulation of gene expression, and DNA repair [21,22]. Seven genes are associated with CdLS (NIPBL, SMC1A, SMC3, BRD4, HDAC8, RAD21, and ANKRD11), and several genes on different chromosomes contribute to its complex development [23,24,25,26,27]. The Nipped-B-like (NIPBL) protein on chromosome 5 accounts for approximately 60% of cases, with the remaining genes accounting for 10% [2,28]. The remaining thirty percent of CdLS patients are classified as idiopathic. Almost all cases of the condition are sporadic (de novo heterozygous mutations). However, an X-linked dominant pattern or autosomal dominant inheritance has been reported [29]. The exact pathomechanism of CdLS is not yet fully understood.
The basis for genetic diagnosis in the classic phenotype is next-generation sequencing (NGS). Genetic testing should target the NIPBL gene for sequencing if this is unavailable. In non-classical CdLS, gene selection for sequencing is possible. As a first step in genetic diagnosis, whole exome sequencing (WES) or whole genome sequencing (WGS) may be performed [1]. Difficulties in identifying mutations may arise from mosaicism or the coexistence of other genetic diseases [30]. In the absence of mutations in CdLS-specific genes, mutations may be present in different cell lines (mosaicism) derived from tissues other than blood, such as fibroblasts. MLPA (multiplex ligation-dependent probe amplification) or aCGH (array comparative genomic hybridization) tests can also be done to look for duplications or deletions in the NIPBL gene [1,31].
Genotype-Phenotype Correlation
The complexity of CdLS far exceeds the scope of clinical diagnostic criteria, and knowledge of the location of a specific mutation may offer opportunities to predict the future of a given patient [1]. Typical of classical CdLS is a mutation in the NIPBL gene, and non-classical CdLS mutations are in the SMC1A, RAD21, and ANKRD11 genes [13,26,32]. In contrast, patients with a mutation in the HDAC8 gene most commonly present with non-classical CdLS, but cases of patients fulfilling the criteria for the classical syndrome have been described [33], and mutations in the BRD4 and SMC3 genes cause an atypical phenotype [25,27]. Of course, some features of dysmorphia (mainly craniofacial) are similar or even the same regardless of genotype, but there are several differences. Patients with a mutation in the NIPBL gene have the most severe phenotypic features of classic CdLS. These include intrauterine growth restriction, short stature, microcephaly, low frontal hairline, thick eyebrows, synophrys, long eyelashes, concave nasal root, shallow fossa, thin upper lip, lowered corners of the mouth, widely spaced teeth, micrognathia, small hands, short fifth toe, tiny feet, hirsutism, or intellectual disability [24,34]. The SMC1A gene mutation causes a short nasal root and a round face, similar to Rett syndrome (OMIM 312750). Patients with the NIPBL gene mutation also have facial dysmorphic features that meet the clinical diagnostic criteria for CdLS. These include a low frontal hairline, synophrys, thick eyebrows, long eyelashes, shallow fossa, thin upper lip, lower corners of the mouth, small hands, clinodactyly (short fifth toe), tiny feet, hirsutism, and intellectual disability. These symptoms can also be seen in people with changes in the BRD4, RAD21, or ANKRD11 genes, although they are not as severe or common [13]. A mutation in the SMC3 gene is a rare cause of CdLS, and patients with this mutation have numerous congenital malformations in addition to intellectual disability and short stature [25,35]. Constipation and GERD are more common in patients with mutations in the NIPBL and SMC1A genes, visual impairment in NIPBL, SMC1A, and HDAC8, seizures in SMC1A, structural brain abnormalities in NIPBL, and sleep problems in NIPBL and SMC1A [10,32,33,36].
The Problem of Germinal and Somatic Mosaicism
Mosaicism in Cornelia de Lange syndrome (CdLS) is a major diagnostic and clinical challenge, reflecting the complex interplay of genetics in developmental disorders.
Germline mosaicism is a scenario in which a fraction of an individual’s gametes sperm or eggs carry a genetic mutation. The somatic cells are unaffected. This phenomenon is central to genetic counseling because it can result in the inheritance of a genetic disorder by offspring despite the absence of phenotypic manifestations in the parent due to transmission through the mutant gametes.
On the other hand, somatic mosaicism results from postzygotic mutations that lead to a divergence of the genetic landscape within an individual. This results in multiple genetically distinct cell lines within the same organism. The clinical implications of somatic mosaicism are profound, contributing to the heterogeneous expression of genetic disorders depending on the distribution and proportion of affected cells in different tissues.
The degree of mosaicism, the tissues affected, and the specific genetic alterations contribute to the variability in clinical presentation. Unlike individuals with a uniform genetic mutation in all cells, those with mosaicism may present with a milder or less typical phenotype. This variability can complicate accurate diagnosis, as the phenotypic expression in mosaic cases may not fit neatly with established diagnostic criteria for CdLS. It is essential to consider mosaicism as a potential cause in cases where clinical suspicion of CdLS remains despite negative genetic test results. Evidence of mosaicism has been reported in up to 20 percent of people with typical features of CdLS [8,30].
Mosaicism in CdLS has been observed in several genes associated with the syndrome, most notably in the NIPBL gene and other cohesin complex genes such as SMC1A and HDAC8. Somatic mosaicism, where the genetic change occurs after fertilization, has been shown to play a significant role in the causality of CdLS [37,38].
The presence of mosaicism in CdLS also has implications for genetic counseling and relapse risk for affected families. Unlike germline mutations with a more predictable inheritance pattern, mosaic mutations can occur spontaneously and unpredictably. This unpredictability requires a nuanced approach to counseling affected families [39].
Prenatal Diagnosis
The diagnosis of CdLS after birth is predominantly clinical, based on the physical features of the syndrome. While a comparable antenatal approach is impossible, detailed fetal anatomy can be captured using high-resolution ultrasound. However, it is unrealistic to expect general sonographers to be familiar with such a rare condition [23].
Cardinal and suggestive features include distinct facial features such as thick, meeting eyebrows (synophrys), a short nose with a depressed nasal bridge, a long philtrum, a thin upper lip, and downturned mouth corners. Limb abnormalities such as oligodactyly, absence of bones, and congenital diaphragmatic hernia are also indicative, as are fetal growth restriction and microcephaly.
Advances in ultrasound technology, mainly 3D volumetric imaging, have improved the detection of these facial features. Even standard 2D ultrasound can detect abnormalities of the nose and philtrum. Limb defects, ranging from minor to complete absences, are detectable and often asymmetrical in CdLS. Congenital diaphragmatic hernia, classified as a cardinal feature, and growth restriction, including microcephaly, can be observed prenatally and are essential indicators of CdLS. In addition, a short fifth finger, often associated with trisomy 21, can also be seen in CdLS.
Other ultrasound findings in CdLS include increased nuchal translucency, long eyelashes, and facial micrognathia. Cardiac malformations, although not exclusive to CdLS, are also noted and are essential for prognosis and management. The text concludes by emphasizing the role of volumetric ultrasound in identifying the distinct phenotype of classic CdLS [40,41].
Differential Diagnosis
The diagnostic process for Cornelia de Lange syndrome (CdLS) involves a comprehensive evaluation to distinguish it from other conditions with similar clinical features. This complex and challenging process relies on clinical assessment, genetic testing, and expert knowledge to arrive at an accurate diagnosis. Coffin-Siris syndrome (OMIM 135900) should be considered in the differential diagnosis. This condition shares similarities with CdLS, such as intellectual disability and facial dysmorphism. However, Coffin-Siris syndrome has additional features such as sparse hair, an absent or underdeveloped fifth finger or toenails, and feeding difficulties. CdLS and Rubinstein-Taybi syndrome (OMIM 180849) have overlapping symptoms, such as intellectual disability and distinctive facial features. However, Rubinstein-Taybi syndrome is typically associated with broad thumbs and toes, distinctive facial features, and an increased risk of tumors. Smith-Lemli-Opitz syndrome (OMIM 270400) also has facial dysmorphism and intellectual disability. However, other features such as microcephaly, feeding difficulties, and limb abnormalities can distinguish it from CdLS. Although CdLS and Seckel syndrome (OMIM 210600) share some features, such as intellectual disability and growth retardation, Seckel syndrome is characterized by severe short stature, microcephaly, and distinctive facial abnormalities. Wolf-Hirschhorn syndrome (OMIM 194190) may present with facial dysmorphism similar to CdLS and intellectual disability. However, Wolf-Hirschhorn syndrome is distinguished by its specific pattern of facial features, such as the “Greek warrior helmet” appearance, seizures, and growth retardation [20,28].
Chung-Jansen syndrome is a recently identified condition (CHUJANS OMIM 617991) characterized by global developmental delay, intellectual disability, behavioral problems, obesity, and facial dysmorphism. Facial dysmorphism may include full eyebrows, synophrys, an upturned nose, large ears, tapered fingers, and bilateral clinodactyly of the fifth finger. Behavioral problems may include ADHD, autistic features, mood disorders, and anxiety disorders [42].
Adolescent and Adult CdLS Problems
Diagnosing CdLS in adults with milder involvement can be challenging as it can resemble other conditions such as fetal alcohol spectrum disorder, Rubinstein-Taybi syndrome, and an autism spectrum disorder. Recognizable adult facial features of CdLS include synophrys (joined eyebrows), narrow downward slanting palpebral fissures, a short nose with an anteverted nasal bridge, a prominent philtrum, thin down-turned lips, and a square chin.
CdLS’s average adult growth parameters for weight, height, and head circumference are lower than standard growth curves. Most individuals were below the 5th percentile for growth in all parameters and had microcephaly. Some patients were also obese or had a low weight for their height [43].
Facial features in CdLS develop over time, becoming slightly coarser and longer. Typical facial features include synophrys, narrow palpebral fissures, a prominent philtrum, a short nose with an anteverted nasal bridge, jaw abnormalities, and down-turned corners of the mouth. Gastrointestinal manifestations in CdLS include gastroesophageal reflux, oesophagitis, gastritis, duodenitis, oesophageal stricture, hiatal hernia, and gastrointestinal malformations. Other problems, such as constipation, chronic diarrhea, dysphagia, lactose intolerance, and milk protein allergy, are also reported [44,45].
The most common medical complication in CdLS is gastroesophageal reflux disease (GERD), which can manifest without apparent signs or symptoms. The study mentions a higher incidence of Barrett’s esophagus, a complication of GERD, in CdLS patients compared to the general population. This finding raises the possibility of silent reflux and the need for regular gastrointestinal evaluation in CdLS patients to address complications such as oesophageal metaplasia and Barrett’s esophagus [46].
65% had sensorineural hearing loss, usually mild to moderate. Dental problems included crowding, delayed eruptions, missing teeth, dental caries, and bruxism [47].
41% of patients had ptosis (drooping of the eyelid), most of which was unilateral and did not require surgery. 71% of patients had blepharitis, an inflammation of the eyelids [48].
Scoliosis was documented in 39% of patients, and thoracic kyphosis in 7%.
80% of patients had hypertrichosis (excessive hair growth), and 61% had cutis marmarata (a marbled appearance of the skin).
The delayed onset of puberty was seen in both males and females.
Sleep problems were reported in more than two-thirds of the families. These included frequent waking during the night, minimal sleep requirements (only 2-4 hours per night), and the ability to go without sleep for extended periods (maximum three days) [49].
Neurodevelopmental manifestations included high pain tolerance, seizures, and varying degrees of mental retardation.
Behavioral manifestations included self-injury, aggression, attention deficit disorder, wandering behavior, anxiety, depression, obsessive-compulsive tendencies, autistic-like behavior, and substance abuse problems [13,14,50].
It emphasizes the need for consistent medical care and stresses the role of a balanced diet and regular exercise. Transitioning from pediatric care to internal medicine or family practice is also crucial. Regular check-ups by ophthalmologists and dentists are recommended, especially to monitor conditions such as high myopia. For gastroesophageal reflux disease (GERD), proactive management and continuous gastrointestinal (GI) surveillance are recommended. An upper GI series is suggested to rule out malrotation, and any evidence of bowel obstruction or volvulus warrants immediate emergency department intervention, possibly requiring surgery. Hormone therapy is recommended for both contraception and menstrual regulation [9,11,46].
In Poland, these patients are cared for by a multidisciplinary team working at the Medical University of Gdansk for many years [51].
Carer and Family of a CDLS Patient
Carers of people with Cornelia de Lange syndrome (CdLS) face significant challenges, mainly due to the complex medical profile of the syndrome. They often have to deal with the intensive management of multiple health problems, including gastrointestinal complications, cardiac anomalies, and developmental delays that require specialized medical care [54]. Behavioral difficulties, such as self-injury and aggression, require constant vigilance and specialized behavioral management strategies. The need for ongoing care and navigation of multiple medical systems can lead to carer fatigue [55]. In addition, ensuring effective communication with healthcare providers and advocating for the individual’s medical needs is a constant challenge, adding to these carers’ emotional and logistical demands [51].
5. Conclusions
Cornelia de Lange syndrome is a rare genetic disorder with a wide range of clinical manifestations and genetic causes. This comprehensive article provides an overview of CdLS, focusing on its clinical features, genetic basis, and management strategies. Although considerable progress has been made in understanding CdLS, further research is needed to define the genotype-phenotype correlations better and to develop targeted therapeutic interventions. A collaborative and multidisciplinary approach involving health professionals, researchers, and carers is essential to improving the diagnosis, management, and quality of life of people with CdLS.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, KGS and KW.; methodology, EDN,JD,KW,JM,MSB and KGS.; validation, KGS,KW,JM and MSB formal analysis, KGS,KW,JM investigation, EDN,JD,KW resources, KW,JM,KGS.; data curation, KW,JM,KGS; writing—original draft preparation, KGS,EDN,JD,KW; writing—review and editing, JM,MSB,KGS.; visualization, EDN,JD supervision, JM,KGS.; project administration, JM. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent was obtained from the parents of the patient(s) to publish photographs of the baby in this article.
Data Availability Statement
The data presented in this study are available on request form the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| CdLS | Cornelia de Lange Syndrome |
| FGR | fetal growth restriction |
| PEG-PEJ | percutaneous endoscopic gastrostomy-jejunostomy |
| GERD | gastro-oesophageal reflux |
| VSD | ventricular septal defect |
| aCGH | array comparative genomic hybridization |
| NGS | next generation sequencing |
| MLPA | multiplex ligation-dependent probe amplification |
| WES | whole exome sequencing |
| WGS | whole genome sequencing |
| TFU | transfontanel ultrasound |
| TMJ | tempomandibular join |
| TTE | transthoracic echocardiography |
| PEG | percutaneous endoscopic gastrostomy |
| SIB | self-injurious behavior |
| ADHD | attention deficit hyperactivity disorder |
| PACU | post-anesthesia care unit |
References
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet 2018, 19, 649–666. [Google Scholar] [CrossRef] [PubMed]
- CORNELIA DE LANGE SYNDROME 1; CDLS1. Available online: https://www.omim.org/entry/122470 (accessed on 25.12.2023).
- Bose, T.; Gerton, J.L. Cohesinopathies, gene expression, and chromatin organization. J Cell Biol 2010, 189, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Mfarej, M.G.; Hyland, C.A.; Sanchez, A.C.; Falk, M.M.; Iovine, M.K.; Skibbens, R.V. Cohesin: an emerging master regulator at the heart of cardiac development. Molecular biology of the cell 2023, 34, rs2–rs2. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, S.; Clark, D.; Kline, A.D.; Jackson, L.G.; Pie, J.; Siu, V.; Ramos, F.J.; Krantz, I.D.; Deardorff, M.A. Facial diagnosis of mild and variant CdLS: Insights from a dysmorphologist survey. American journal of medical genetics. Part A 2010, 152A, 1641–1653. [Google Scholar] [CrossRef] [PubMed]
- Collis, L.; Moss, J.; Jutley, J.; Cornish, K.; Oliver, C. Facial expression of affect in children with Cornelia de Lange syndrome. Journal of intellectual disability research : JIDR 2008, 52, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Cereda, A.; Mariani, M.; Rebora, P.; Sajeva, A.; Ajmone, P.F.; Gervasini, C.; Russo, S.; Kullmann, G.; Valsecchi, G.; Selicorni, A. A new prognostic index of severity of intellectual disabilities in Cornelia de Lange syndrome. American journal of medical genetics. Part C, Seminars in medical genetics 2016, 172, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Huisman, S.A.; Redeker, E.J.W.; Maas, S.M.; Mannens, M.M.; Hennekam, R.C.M. High rate of mosaicism in individuals with Cornelia de Lange syndrome. Journal of medical genetics 2013, 50, 339–344. [Google Scholar] [CrossRef]
- Luzzani, S.; Macchini, F.; Valadè, A.; Milani, D.; Selicorni, A. Gastroesophageal reflux and Cornelia de Lange syndrome: typical and atypical symptoms. American journal of medical genetics. Part A 2003, 119A, 283–287. [Google Scholar] [CrossRef]
- Gillis, L.A.; McCallum, J.; Kaur, M.; DeScipio, C.; Yaeger, D.; Mariani, A.; Kline, A.D.; Li, H.-h.; Devoto, M.; Jackson, L.G.; Krantz, I.D. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. American journal of human genetics 2004, 75, 610–623. [Google Scholar] [CrossRef]
- Clermidi, P.; Abadie, V.; Campeotto, F.; Irtan, S. Sigmoid Volvulus: An Underestimated Cause of Intestinal Obstruction in Cornelia de Lange Syndrome. The Journal of pediatrics 2015, 167, 941–941. [Google Scholar] [CrossRef]
- Bell, L.; Oliver, C.; Wittkowski, A.; Moss, J.; Hare, D. Attenuated behaviour in Cornelia de Lange and fragile X syndromes. Journal of intellectual disability research : JIDR 2018, 62, 486–495. [Google Scholar] [CrossRef]
- Moss, J.; Penhallow, J.; Ansari, M.; Barton, S.; Bourn, D.; FitzPatrick, D.R.; Goodship, J.; Hammond, P.; Roberts, C.; Welham, A.; Oliver, C. Genotype-phenotype correlations in Cornelia de Lange syndrome: Behavioral characteristics and changes with age. American journal of medical genetics. Part A 2017, 173, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Grados, M.A.; Alvi, M.H.; Srivastava, S. Behavioral and psychiatric manifestations in Cornelia de Lange syndrome. Current opinion in psychiatry 2017, 30, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Nelson, L.; Powis, L.; Waite, J.; Richards, C.; Oliver, C. A Comparative Study of Sociability in Angelman, Cornelia de Lange, Fragile X, Down and Rubinstein Taybi Syndromes and Autism Spectrum Disorder. American journal on intellectual and developmental disabilities 2016, 121, 465–486. [Google Scholar] [CrossRef] [PubMed]
- Stevic, M.; Milojevic, I.; Bokun, Z.; Simic, D. Unpredictable drug reaction in a child with Cornelia de Lange syndrome. International journal of clinical pharmacy 2015, 37, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Moretto, A.; Scaravilli, V.; Ciceri, V.; Bosatra, M.; Giannatelli, F.; Ateniese, B.; Mariani, M.; Cereda, A.; Sosio, S.; Zanella, A.; et al. Sedation and general anesthesia for patients with Cornelia De Lange syndrome: A case series. American journal of medical genetics. Part C, Seminars in medical genetics 2016, 172, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Orphan Anesthesia CdLS. Available online: https://www.orphananesthesia.eu/en/rare-diseases/published-guidelines/cornelia-de-lange-syndrome/285-cornelia-de-lange-syndrome/file.html (accessed on 5 December 2023).
- Passport CdLS. Available online: https://www.cdlsworld.org/xwiki/bin/download/cdlsPublications/medicalPassport/worldCarecard2018.pdf?rev=1.1 (accessed on 10.12.2023).
- Avagliano, L.; Parenti, I.; Grazioli, P.; Di Fede, E.; Parodi, C.; Mariani, M.; Kaiser, F.J.; Selicorni, A.; Gervasini, C.; Massa, V. Chromatinopathies: A focus on Cornelia de Lange syndrome. Clin Genet 2020, 97, 3–11. [Google Scholar] [CrossRef]
- Michaelis, C.; Ciosk, R.; Nasmyth, K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 1997, 91, 35–45. [Google Scholar] [CrossRef]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef]
- Avagliano, L.; Bulfamante, G.P.; Massa, V. Cornelia de Lange syndrome: To diagnose or not to diagnose in utero? Birth defects research 2017, 109, 771–777. [Google Scholar] [CrossRef]
- Krantz, I.D.; McCallum, J.; DeScipio, C.; Kaur, M.; Gillis, L.A.; Yaeger, D.; Jukofsky, L.; Wasserman, N.; Bottani, A.; Morris, C.A.; et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nature genetics 2004, 36, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Kaur, M.; Yaeger, D.; Rampuria, A.; Korolev, S.; Pie, J.; Gil-Rodríguez, C.; Arnedo, M.; Loeys, B.; Kline, A.D.; et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. American journal of human genetics 2007, 80, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Wilde, J.J.; Albrecht, M.; Dickinson, E.; Tennstedt, S.; Braunholz, D.; Mönnich, M.; Yan, Y.; Xu, W.; Gil-Rodríguez, M.C.; et al. RAD21 mutations cause a human cohesinopathy. American journal of human genetics 2012, 90, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Olley, G.; Ansari, M.; Bengani, H.; Grimes, G.R.; Rhodes, J.; von Kriegsheim, A.; Blatnik, A.; Stewart, F.J.; Wakeling, E.; Carroll, N.; et al. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange-like syndrome. Nature genetics 2018, 50, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Deardorff Ma, N.S.E.K.I.D.C.d.L.S.S.I.A.M.P.F.J.M.G.M.; et al. GeneReviews® [Internet]; University of Washington, 1993-2024. [Google Scholar]
- Musio, A.; Selicorni, A.; Focarelli, M.L.; Gervasini, C.; Milani, D.; Russo, S.; Vezzoni, P.; Larizza, L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nature genetics 2006, 38, 528–530. [Google Scholar] [CrossRef]
- Ansari, M.; Poke, G.; Ferry, Q.; Williamson, K.; Aldridge, R.; Meynert, A.M.; Bengani, H.; Chan, C.Y.; Kayserili, H.; Avci, S.; et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. Journal of medical genetics 2014, 51, 659–668. [Google Scholar] [CrossRef]
- Ratajska, M.; Wierzba, J.; Pehlivan, D.; Xia, Z.; Brundage, E.K.; Cheung, S.W.; Stankiewicz, P.; Lupski, J.R.; Limon, J. Cornelia de Lange syndrome case due to genomic rearrangements including NIPBL. European journal of medical genetics 2010, 53, 378–382. [Google Scholar] [CrossRef]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.-M.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes and genotypes in individuals with SMC1A variants. American journal of medical genetics. Part A 2017, 173, 2108–2125. [Google Scholar] [CrossRef]
- Kaiser, F.J.; Ansari, M.; Braunholz, D.; Concepción Gil-Rodríguez, M.; Decroos, C.; Wilde, J.J.; Fincher, C.T.; Kaur, M.; Bando, M.; Amor, D.J.; et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Human molecular genetics 2014, 23, 2888–2900. [Google Scholar] [CrossRef]
- Jackson, L.; Kline, A.D.; Barr, M.A.; Koch, S. de Lange syndrome: a clinical review of 310 individuals. American journal of medical genetics 1993, 47, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Gil-Rodríguez, M.C.; Deardorff, M.A.; Ansari, M.; Tan, C.A.; Parenti, I.; Baquero-Montoya, C.; Ousager, L.B.; Puisac, B.; Hernández-Marcos, M.; Teresa-Rodrigo, M.E.; et al. De novo heterozygous mutations in SMC3 cause a range of Cornelia de Lange syndrome-overlapping phenotypes. Human mutation 2015, 36, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, M.T.; Nagaraj, U.D.; Pearl, P.L. Neuroimaging features of Cornelia de Lange syndrome. Pediatric radiology 2015, 45, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Pellicer, A.; Gil-Salvador, M.; Parenti, I.; Lucia-Campos, C.; Trujillano, L.; Marcos-Alcalde, I.; Arnedo, M.; Ascaso, Á.; Ayerza-Casas, A.; Antoñanzas-Pérez, R.; et al. Clinical relevance of postzygotic mosaicism in Cornelia de Lange syndrome and purifying selection of NIPBL variants in blood. Scientific reports 2021, 11, 15459–15459. [Google Scholar] [CrossRef]
- Slavin, T.P.; Lazebnik, N.; Clark, D.M.; Vengoechea, J.; Cohen, L.; Kaur, M.; Konczal, L.; Crowe, C.A.; Corteville, J.E.; Nowaczyk, M.J.; et al. Germline mosaicism in Cornelia de Lange syndrome. American journal of medical genetics. Part A 2012, 158A, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Krawczynska, N.; Wierzba, J.; Wasag, B. Genetic Mosaicism in a Group of Patients With Cornelia de Lange Syndrome. Frontiers in pediatrics 2019, 7, 203–203. [Google Scholar] [CrossRef]
- Hague, J.; Twiss, P.; Mead, Z.; Park, S.-M. Clinical Diagnosis of Classical Cornelia de Lange Syndrome Made From Postmortem Examination of Second Trimester Fetus With Novel NIPBL Pathogenic Variant. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society 2019, 22, 475–479. [Google Scholar] [CrossRef]
- Panaitescu, A.M.; Duta, S.; Gica, N.; Botezatu, R.; Nedelea, F.; Peltecu, G.; Veduta, A. A Broader Perspective on the Prenatal Diagnosis of Cornelia de Lange Syndrome: Review of the Literature and Case Presentation. Diagnostics (Basel, Switzerland) 2021, 11. [Google Scholar] [CrossRef]
- Conti, B.; Rinaldi, B.; Rimoldi, M.; Villa, R.; Iascone, M.; Gangi, S.; Porro, M.; Ajmone, P.F.; Colli, A.M.; Mosca, F.; Bedeschi, M.F. Chung-Jansen syndrome can mimic Cornelia de Lange syndrome: Another player among chromatinopathies? American journal of medical genetics. Part A 2023, 191, 1586–1592. [Google Scholar] [CrossRef]
- Kline, A.D.; Barr, M.; Jackson, L.G. Growth manifestations in the Brachmann-de Lange syndrome. American journal of medical genetics 1993, 47, 1042–1049. [Google Scholar] [CrossRef]
- Nelson, L.; Moss, J.; Oliver, C. A longitudinal follow-up study of affect in children and adults with Cornelia de Lange syndrome. American journal on intellectual and developmental disabilities 2014, 119, 235–252. [Google Scholar] [CrossRef]
- Wierzba, J.; Wierzba, T.; Mazurkiewicz-Bełdzińska, M.; Szyca, R.; Kozłowski, J.; Banach, P.; Potaż, P.; Limon, J. Dorosły z rzadkim schorzeniem genetycznym - diagnostyka i terapia zespołu Cornelii de Lange. Forum Medycyny Rodzinnej 2010, 4, 273–280. [Google Scholar]
- Mariani, M.; Decimi, V.; Bettini, L.R.; Maitz, S.; Gervasini, C.; Masciadri, M.; Ajmone, P.; Kullman, G.; Dinelli, M.; Panceri, R.; et al. Adolescents and adults affected by Cornelia de Lange syndrome: A report of 73 Italian patients. American journal of medical genetics. Part C, Seminars in medical genetics 2016, 172, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Janek, K.C.; Smith, D.F.; Kline, A.D.; Benke, J.R.; Chen, M.-L.; Kimball, A.; Ishman, S.L. Improvement in hearing loss over time in Cornelia de Lange syndrome. International journal of pediatric otorhinolaryngology 2016, 87, 203–207. [Google Scholar] [CrossRef]
- Avgitidou, G.; Cursiefen, C.; Heindl, L.M. [Ophthalmological manifestations of Cornelia de Lange syndrome: Case report and review of the literature]. Der Ophthalmologe : Zeitschrift der Deutschen Ophthalmologischen Gesellschaft 2015, 112, 455–458. [Google Scholar] [CrossRef]
- Zambrelli, E.; Fossati, C.; Turner, K.; Taiana, M.; Vignoli, A.; Gervasini, C.; Russo, S.; Furia, F.; Masciadri, M.; Ajmone, P.; et al. Sleep disorders in Cornelia de Lange syndrome. American journal of medical genetics. Part C, Seminars in medical genetics 2016, 172, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Kline, A.D.; Grados, M.; Sponseller, P.; Levy, H.P.; Blagowidow, N.; Schoedel, C.; Rampolla, J.; Clemens, D.K.; Krantz, I.; Kimball, A.; et al. Natural history of aging in Cornelia de Lange syndrome. American journal of medical genetics. Part C, Seminars in medical genetics 2007, 145C, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Wierzba, J.; Mazurkiewicz-Bełdzińska, M.; Jabłońska-Brudło, J.; Potaż, P.; Banach, P. Challenges of caring for a patient with a rare disease--as demonstrated by Cornelia de Lange Syndrome. Developmental period medicine 2015, 19, 511–515. [Google Scholar]
- Cacioppo, C.N.; Conway, L.J.; Mehta, D.; Krantz, I.D.; Noon, S.E. Attitudes about the use of internet support groups and the impact among parents of children with Cornelia de Lange syndrome. American journal of medical genetics. Part C, Seminars in medical genetics 2016, 172, 229–236. [Google Scholar] [CrossRef]
- CdLS world. Available online: https://www.cdlsworld.org/xwiki/bin/view/Main/ (accessed on 10.12.2023).
- January, K.; Conway, L.J.; Deardorff, M.; Harrington, A.; Krantz, I.D.; Loomes, K.; Pipan, M.; Noon, S.E. Benefits and limitations of a multidisciplinary approach to individualized management of Cornelia de Lange syndrome and related diagnoses. American journal of medical genetics. Part C, Seminars in medical genetics 2016, 172, 237–245. [Google Scholar] [CrossRef]
- Wulffaert, J.; van Berckelaer-Onnes, I.; Kroonenberg, P.; Scholte, E.; Bhuiyan, Z.; Hennekam, R. Simultaneous analysis of the behavioural phenotype, physical factors, and parenting stress in people with Cornelia de Lange syndrome. Journal of intellectual disability research : JIDR 2009, 53, 604–619. [Google Scholar] [CrossRef]
Figure 1.
Clinical features of patients with Cornelia de Lange Syndrome-case 2 (A) patient at 2 months of life, patient at 3 year of age (C,D).
Figure 1.
Clinical features of patients with Cornelia de Lange Syndrome-case 2 (A) patient at 2 months of life, patient at 3 year of age (C,D).
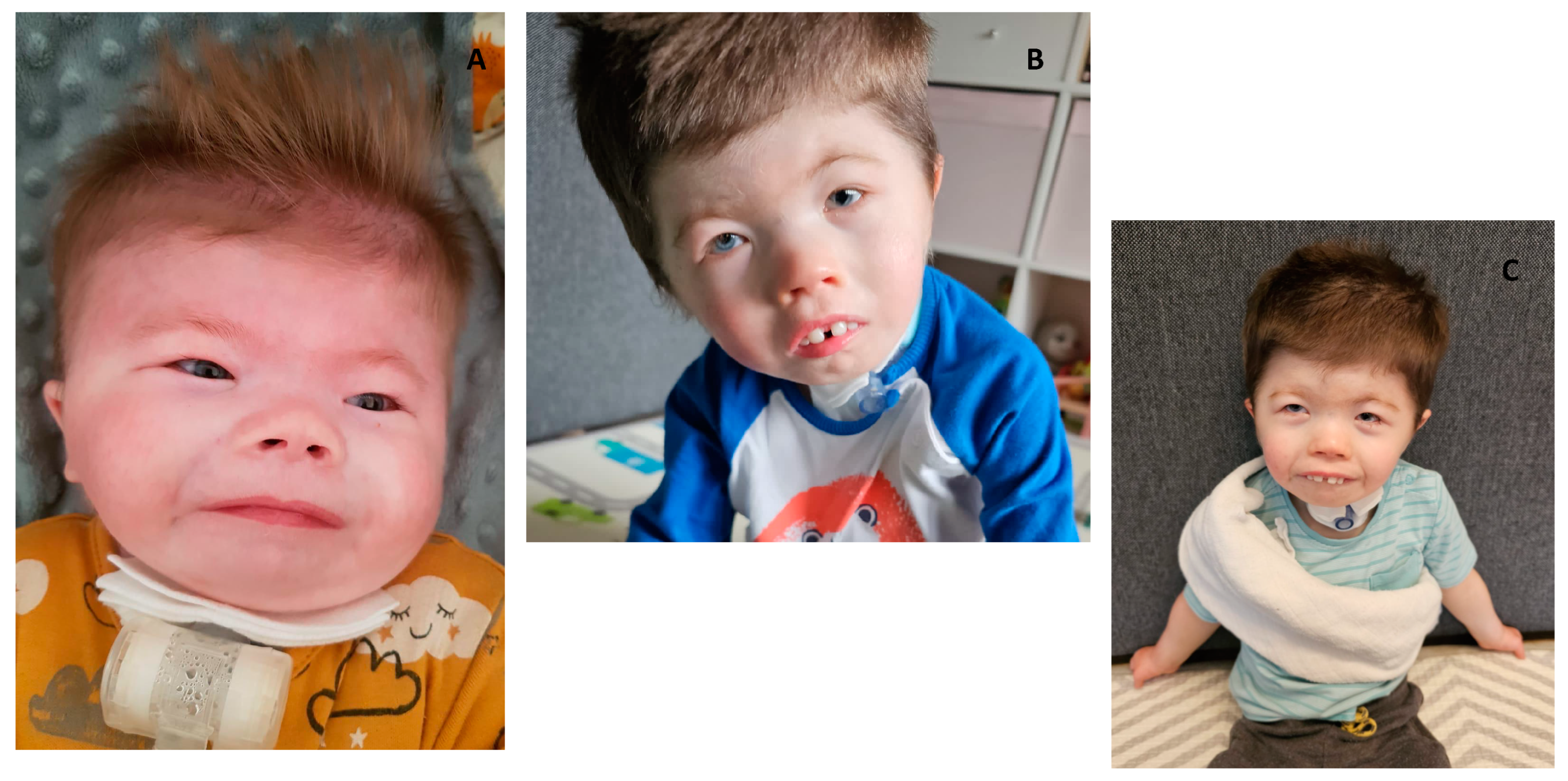
Figure 2.
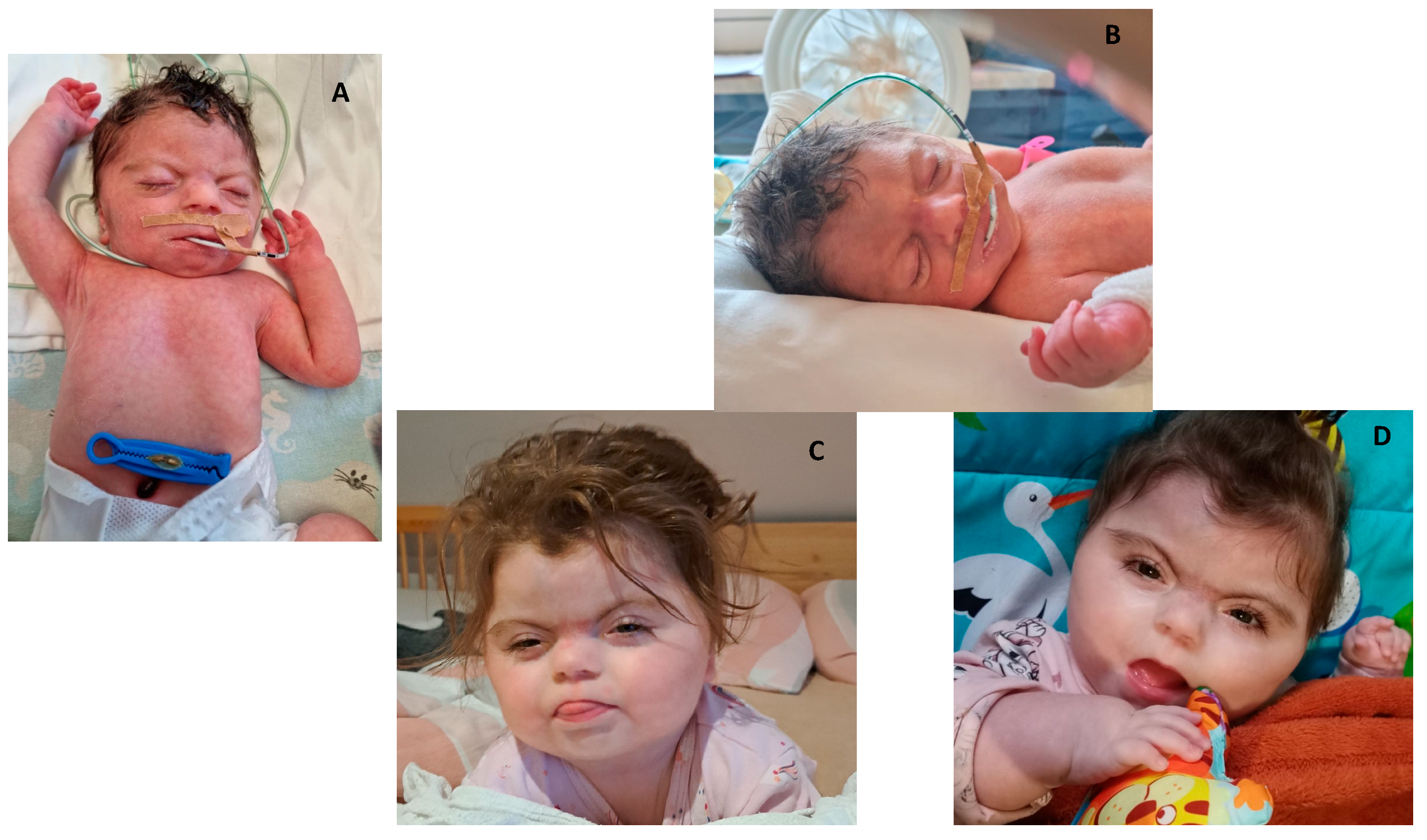
Clinical features of patients with Cornelia de Lange Syndrome-case 4 (A,B), patient at 5 day of life, patient at 13 months of age (C,D).
Figure 2.
Clinical features of patients with Cornelia de Lange Syndrome-case 4 (A,B), patient at 5 day of life, patient at 13 months of age (C,D).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



