
Submitted:
09 February 2024
Posted:
12 February 2024
Read the latest preprint version here
Abstract
Objective: We report the case of a patient who developed acute pancytopenia and mixed-pattern liver injury after concomitant administration of low-dose methotrexate (MTX), and high-dose esomeprazole and metamizole. Case summary: A 71-year-old female patient with chronic systemic idiopathic erosive arthritis was admitted for conservative treatment with analgesia and physiotherapy for a pelvic ring fracture. Due to the arthritis, she received treatment with MTX 15 mg/week, hydroxychloroquine 200 mg/day and prednisone 7.5 mg/day. On admission, she had no infectious, hematologic or oncologic diseases. She received analgesic therapy with metamizole 4000 mg/day and oxycodone/naloxone 30/15 mg/day. After hospital admission, she received MTX 15 mg as planned in addition to esomeprazole 80 mg/day and torasemide 10 mg/day. Subsequent laboratory tests revealed pancytopenia and elevated liver enzymes. This condition persisted and worsened during the first two weeks of hospitalization. The follow-up clinical examinations were unremarkable, with the exception of sub-febrile temperatures and an exacerbation of pre-existing mouth ulcers. Further investigations were unable to determine a definitive etiology. Due to the suspicion of methotrexate-induced hematotoxicity and hepatotoxicity, antagonizing therapy with calcium folinate 7.5 mg 4 times/day was administered. After an expert pharmacological evaluation, the concomitant administration of metamizole and esomeprazole was discontinued, after which the blood counts and liver parameters normalized. Discussion: After MTX administration, a new onset leukopenia (2.34 G/L), thrombocytopenia (46 G/L), worsened anemia (83 g/L) and elevated liver enzymes were detected. Presumably, a methotrexate-induced bone marrow suppression was already in progress. Due to the administration of high-dose metamizole immediately before the administration of low-dose MTX, the pre-existing hematotoxic pharmacodynamic effect of MTX was acutely enhanced by that of metamizole, although folic acid was administered preemptively. In addition, the concomitant administration of high-dose esomeprazole and normal dose torasemide resulted in a pharmacokinetic interaction with MTX by decreasing its renal secretion and elimination, further enhancing the concentration-dependent hematotoxic and hepatotoxic side effects of MTX. Conclusions: The relatively high demand for analgesics in patients with chronic rheumatic diseases already being treated with MTX, proton pump inhibitors, and loop diuretics necessitates that healthcare providers consider drug-drug interactions as potential causes of acute hematotoxicity and hepatotoxicity after the administration of metamizole or nonsteroidal anti-inflammatory drugs. In this category of patients, it is strongly recommended to switch to analgesics of other classes, consider dose adjustment of relevant concomitant medications and closely monitor laboratory parameters to preempt any adverse drug reactions.
Keywords:
pharmacokinetic-pharmacodynamic interactions
; low-dose methotrexate
; proton pump inhibitors
; analgesics
; pancytopenia
; hepatotoxicity
1. Clinical Case
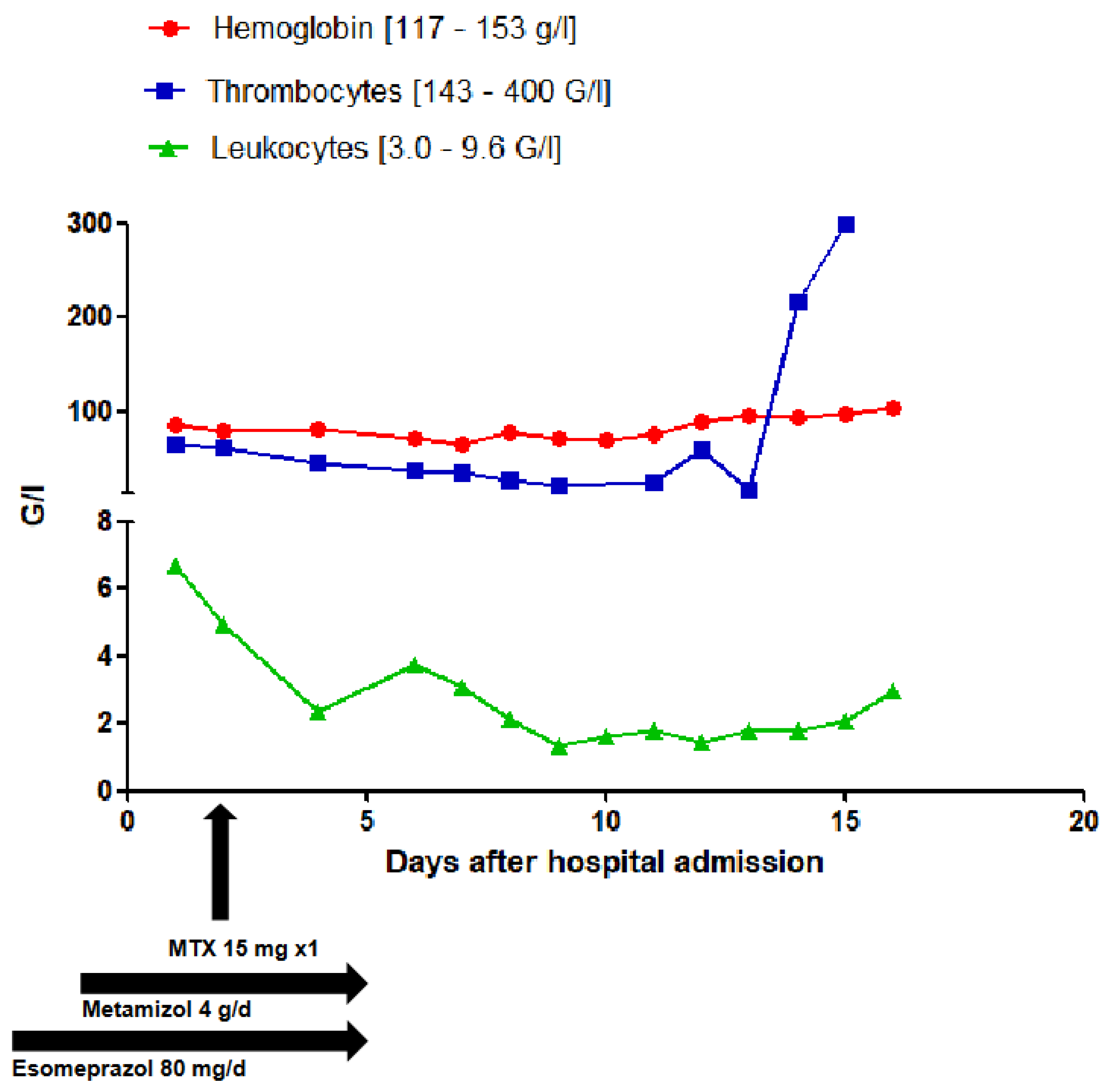
A 71-year-old patient presented to the emergency department with acute onset of severe movement aggravated pain in her left pubic bone. After the diagnosis of a pelvic ring fracture type IIIb (Rommens classification) she was hospitalized for further conservative treatment with analgesia and physiotherapy. The primary diagnosis of the patient was chronic systemic idiopathic erosive arthritis (initial manifestation in 1957) that was at the time of admission treated with methotrexate (MTX) 15 mg/week, hydroxychloroquin 200 mg/day and prednisone 7.5 mg/day. She had a positive history of the following diseases: symptomatic osteoporosis, monoclonal gammopathy of undetermined significance, urinary tract infection, heart failure with coronary heart disease, peripheral artery disease, chronic obstructive pulmonary disease, recurrent depressive disorder, chronic cervico- and lumbo-spondylogenic syndrome and ischemic colitis. Most importantly, she had no known underlying hematologic or oncologic conditions at the time of hospital admission. Her medication list just before admission is summarized in Table 1. She was administered an analgesic therapy comprising of metamizole 4000 mg/day, paracetamol 2000 mg/day and oxycodon/naloxone 30/15 mg/day. The analgesia was augmented with oxycodon 5 mg for a maximum of 10x/day as needed. Apart from aspirin that was stopped on the day of admission, all her previous medications were continued. During the first few days of hospitalization, new medications were added to the preexisting medication list as shown in Table 2. Two days after admission, the patient received MTX 15 mg as planned. The day before and after the administration of MTX, she received 5 mg of folic acid. Apart from preexisting mild normocytic normochromic anemia with hemoglobin level ranging from 106 g/L to 87 g/L. Baseline laboratory parameters prior to hospital admission revealed normal leucocyte and thrombocyte counts. In the full blood count monitoring carried out the next day after the administration of MTX, pancytopenia and elevated liver enzymes were detected. This persisted and aggravated over the subsequent days as depicted in the overview of the laboratory findings in Table 3 and the evolution of liver parameters and blood count levels in Figure 2 and Figure 3. Parallel to the pancytopenia, a new onset derangement of all liver parameters was evident. Follow-up clinical examinations were unremarkable except for sub-febrile temperatures and an aggravation of preexisting oral ulcers. After various microbiological sampling were undertaken, no pathogenic cause was detected. An empirical antiviral treatment with valtrex® (acyclovir) 1000 mg 2x/day was initiated. Subsequently, she was transfused a total of three units of erythrocyte concentrates. Further investigations by the departments of hematology, rheumatology, oncology and infectious diseases could not ascertain any definitive etiology. On account of high index of suspicion of a MTX induced pancytopenia, an antagonizing therapy with calium/folinic acid 7.5 mg 4x/day was administered over a cumulative period of 3 days and an expert pharmacological consultation was requested.
1.1. Results of Laboratory Investigations
Figure 1.
Evolution of the estimated glomerular filtration rate.
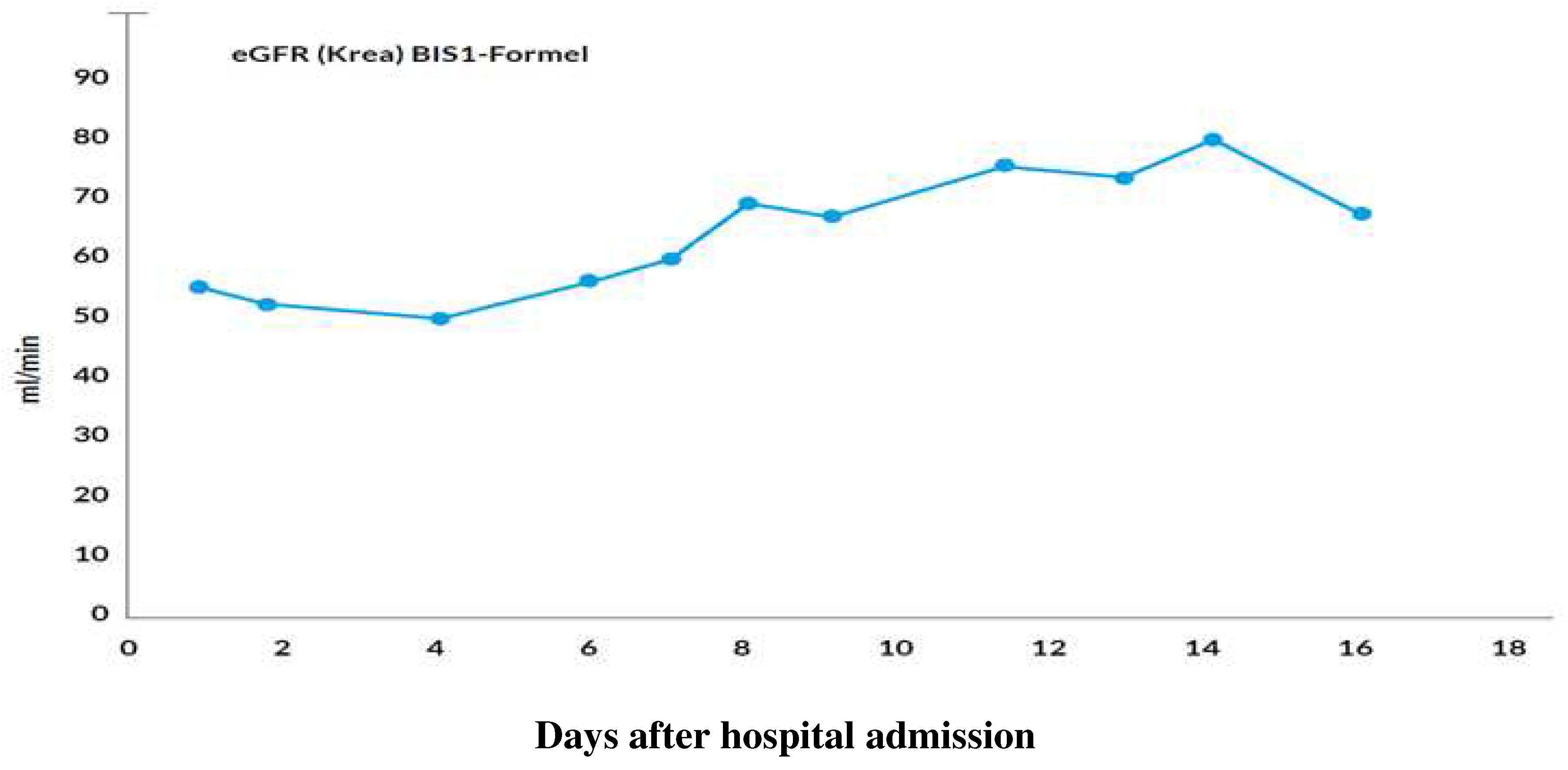
Figure 2.
Evolution of the estimated ALT and ALP values.
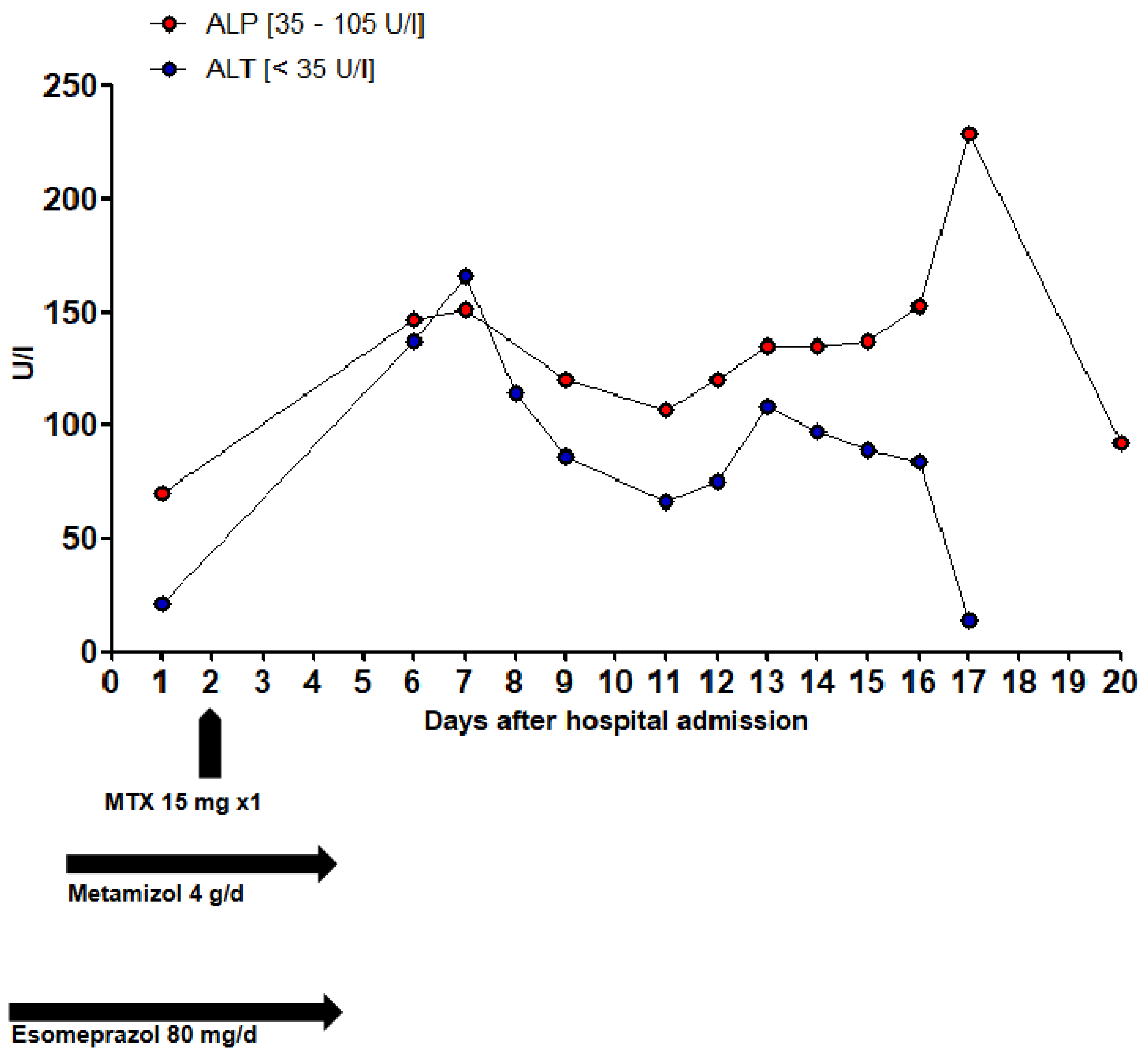
Figure 3.
Evolution of the full blood count levels.
2. Pharmacology of Methotrexate
Methotrexate is an antimetabolite of the enzyme dihydrofolate reductase (DHF reductase) [1,2]. MTX is taken up into cells by an active transport mechanism. Intracellularly it exerts its effects mainly during the "S-phase" of cell division by competitive inhibition of DHF reductase [3]. Dihydrofolates are usually reduced by DHF reductase to tetrahydrofolates, which are required for the transfer of methyl groups, particularly in the formation of thymidine and purine bases [1,2,3]. MTX thus inhibits DNA/RNA synthesis, repair, and cell proliferation. The affinity of dihydrofolate reductase for MTX is much greater than the affinity for folic or dihydrofolic acid [3]. Actively proliferating tissues such as malignant cells, bone marrow, fetal cells, oral and intestinal mucosa, hair matrix, and the cells of the urinary bladder are generally more sensitive to the effects of MTX [3]. Due to the non-specific mechanism of action of MTX, rapidly dividing healthy cells are usually affected. This is why the specific antidote, folinic acid, is often administered after high dose therapy, whilst folic acid is substituted after 24 hours of the application of the once-weekly low dose therapy [3]. The primary pharmacologic action of MTX correlates with its ability of exerting cytostatic, apototic and immunosuppressive effects [1]. However, the exact mechanism of action of low dose MTX in its immunosuppressive effect is not fully elucidated [1,2,3]. It also possesses analgesic, antiphlogistic and anti-inflammatory properties [2,3]. It is not entirely clear whether the efficacy of MTX in the treatment of inflammatory diseases is due to its anti-inflammatory or immunosuppressive effects [3]. Intracellularly, MTX is converted to MTX polyglutamate. MTX polyglutamate accumulates because it gets entrapped and cannot passively diffuse to the extracellular compartment [4,5,6]. Intracellular accumulation of these MTX polyglutamates occurs primarily in the liver, kidney and spleen, which may persist for weeks to months [3,6]. MTX can however be transported outside the cell by the drug efflux transporter P-glycoprotein (P-gp) [7,8]. P-gp inhibitors can therefore prolong the intracellular retention of MTX [7]. The plasma protein binding of MTX is approximately 50% [3]. Approximately 10% of MTX is metabolized in the liver [3]. The cytochrome P450 (CYP) system has not been shown to be involved in its metabolism. 5-20% of MTX and 1-5% of the major metabolite, 7-hydroxymethotrexate (7-OH-MTX), are eliminated through the biliary tree, with some portion undergoing significant enterohepatic recycling [3,4]. The elimination of MTX occurs mainly in unchanged form through the kidneys via glomerular filtration and active tubular secretion [4,5]. The metabolite 7-OH-MTX is 3 to 5 times less soluble than the parent compound [3]. Although this metabolite accounts for only a small proportion of the total drug, significant accumulation may occur at high doses. Elimination is significantly delayed in impaired renal function. The terminal half-life averages 6-7 hours and shows considerable inter-individual variations (about 3-17 hours) [3]. In patients with a third distribution space (pleural and pericardial effusion or ascites), the half-life may be prolonged up to fourfold [3]. Very severe degree of dehydration, especially in old patients, can potentially increase the toxicity of MTX [3,9]. The Swiss drug information of MTX describes hematotoxicity (leukocytopenia, anemia and thrombocytopenia) as a common ADR with a prevalence of about 1% to 10% and hepatic derangement as a very common ADR with prevalence of up to 70%.
2.1. Pharmacodynamic Interactions of Methotrexate with Other Drugs
The concomitant administration of MTX and metamizole can result in a significant increase in the hematotoxic effects of both drugs in a synergistic way. Under the heading "Precautions and Warnings" of the Swiss drug information, reference is made to agranulocytosis or neutropenia caused by a mono therapy with metamizole. Agranulocytosis under metamizole is described as an allergic immune reaction [3,4,5]. This adverse drug reaction (ADR) is however unrelated to the dose administered, and may occur at any time during treatment in an idiosyncratic process [3,4]. According to the American databanks, Micromedex™ and UpToDate™, agranulocytosis occurs very commonly during the administration of metamizole. When left untreated, agranulocytosis under metamizole can sometimes lead to fatal outcomes [4,5]. Drug-induced agranulocytosis was most commonly associated with metamizole in a 20-year descriptive study using data from the drug safety unit of the Dutch inspectorate for health care [10]. In some retrospective analyses, the onset of agranulocytosis was usually unpredictable and fatal cases occurred after short-term, intermittent or long-term administration [11,12,13]. In many instances, a hypersensitivity mechanism is postulated [14]. Due to the inherent hematotoxicity of both drugs, especially in elderly patients, concomitant use of MTX and metamizole must be avoided and alternative analgesics should be considered.
The concomitant administration of drugs that cause folate deficiency such as sulfonamides, trimethoprim, pyrimethamine, triamterene, aminopterin can result in increased toxicity of MTX [15]. Cases of severe MTX toxicity have been reported in patients receiving a combination of trimethoprim with MTX [16,17,18]. Although trimethoprim exerts a relatively lower affinity for the human dihydrofolate reductase [19], it can potentiate the hematotoxic adverse reactions of MTX, particularly in the presence of pre-existing risk factors such as folic acid deficiency, advanced age, hypoalbuminemia, renal impairment, and reduced bone marrow reserves [3]. Furthermore, trimethoprim can increase the concentration of free MTX by about 30% and decrease its excretion by about 50% [20]. Pyrimethamine is an antiprotozoal agent from the diaminopyrimidine group indicated for the treatment of toxoplasmosis. It interferes primarily with the folic acid metabolism of protozoan parasites by competitively inhibiting dihydrofolate reductase [21,22]. Although the substance has a relatively higher affinity for the parasitic enzyme than the human enzyme, it can significantly contribute in antagonizing the human enzyme when co-administered with MTX [22].
2.2. Pharmacokinetic Interactions of Methotrexate with Other Drugs
Proton pump inhibitors (PPIs) at particularly high doses can potentially reduce the renal elimination of MTX, which can result in increased effective serum concentration with corresponding increase in the toxicity of MTX [3]. A competition for organic anion transport proteins (OAT) in the kidney has been shown to underlie this pharmacokinetic interaction [23]. In vitro pharmacokinetic investigations showed that the inhibition of renal tubular OAT3 by PPIs was directly responsible for the impaired renal elimination of MTX [24,25]. In another pharmacokinetic study, the H2-recepter blocker, famotidine demonstrated a relatively weak inhibitory effect on OAT3-mediated MTX uptake compared to that of PPIs [26]. The clinical significance of this interaction has been shown particularly in patients taking relatively high dosages of MTX and PPIs [27,28,29]. In a patient who was administered a combination of low-dose MTX with pantoprazole 20 mg/d, an inhibition of the renal elimination of the metabolite 7-OH-MTX with severe myalgia has been reported [30]. Probenecid, nonsteroidal anti-inflammatory drugs, salicylates and some weak organic acids such as loop diuretics can potentially decrease the renal excretion of MTX by competitively inhibiting renal OAT leading to higher serum concentrations and increased hematologic toxicity of MTX [31,32,33,34,35,36,37,38]. Previous pharmacokinetic studies have demonstrated significant increase in the serum concentrations of MTX after co-administration with probenecid [39,40]. Non-steroidal anti-inflammatory drugs (NSAIDS) can potentially decrease the effective renal excretion of MTX by decreasing renal perfusion at the afferent arterioles and by inhibiting diverse transport proteins in the renal tubules that are directly involved in the secretion of MTX [41,42,43]. In some clinical and pharmacoepidemiologic studies, diverse NSAIDs have been implicated in increasing the toxicity of MTX in patients that were otherwise methotrexate-stable [44,45,46,47]. The results from these studies need to be further validated with higher patient populations.
Penicillin antibiotics have been shown to compete with MTX for excretion sites via the organic anion transporters in the proximal renal tubules [3,41]. This pharmacokinetic interaction can increase the likelihood of MTX retention which can subsequently lead to its toxicity. The importance of this interaction is expected to increase with increasing penicillin or MTX dose [3]. In pharmacokinetic studies involving laboratory animals, the concomitant administration of piperacillin and MTX resulted in significant increase in the area under the curve of MTX and its metabolite 7-OH-MTX respectively [42,43,44]. In vitro and in vivo pharmacokinetic investigations carried in rhesus and cynomolgus monkeys concluded that penicillin competitively inhibits MTX uptake in the renal tubular cells resulting in reduced secretion and elimination of MTX [45]. Clinically significant MTX-toxicities due to concomitant administration with penicillin antibiotics have been reported particularly after high dosages of both drugs [46,47]. The concomitant administration of cephalosporins and MTX can potentially aggravate the toxic effects of MTX. Since MTX is mainly eliminated through OAT1/3 and cephalosporins primarily through OAT3 [48,49], competition at the binding site of OAT3 can lead to reduced elimination and subsequent increased accumulation of MTX [5,41]. In a case report, two patients with joint infection developed severe neutropenia after co-administration of low-dose MTX and ceftriaxone [50]. In relation to fluoroquinolones, several cases of acute renal failure triggered by ciprofloxacin have led to increased plasma exposure of MTX, with potential risk of increased MTX toxicity [51,52,53,54]. However, a clinically relevant interaction with MTX is not to be expected in the absence of significant renal insufficiency [5]. In another case report, the co-administration of MTX and levofloxacin resulted in delayed MTX elimination due to competition for tubular secretion between MTX and levofloxacin [55].
3. Case Evaluation
The decisive factor for the assessment of the causal relationship between the administration of a drug and an observed adverse reaction is dependent on the intrinsic evidence (drug exposure in the framework of a dechallenge and rechallenge, the exclusion of other unexplained etiologic factors and pathophysiological plausibility) and the extrinsic evidence. The latter is the causality judgment based on well-documented comparable cases in the summary of product characteristics (SmPC), pharmacovigilance databases, as well as from systematic clinical or epidemiological studies, and case reports.
Pancytopenia is defined as an absolute neutrophil granulocyte count of < 1.8 G/l or an absolute leukocyte count of < 3.0 G/l, anemia with an hemoglobin level of < 130 g/l and a thrombocyte count of < 150 G/l [5]. Drug-induced pancytopenia is usually idiosyncratic, thus independent of the dose of the causative agents. However, there are also dose-dependent forms, as observed in patients receiving drugs such as azathioprine, MTX and other chemotherapeutic or antineoplastic substances. In principle, drug-induced pancytopenia is a diagnosis of exclusion. Primarily immunological, hematological as well as infectious causes should be considered for differential diagnosis. A relatively short latency period between drug exposure and the onset of pancytopenia or immune-related hematotoxicity is to be expected especially if sensitization may have already occurred as a result of previous therapy with the same drug. In principle, a longer latency period of more than 2 weeks is, however, also possible.
Drug-induced liver injury (DILI) is a diagnosis of exclusion, as no specific biomarkers are yet clinically available [63]. According to the pattern of injury, DILI can be classified into three categories: hepatocellular, cholestatic and mixed pattern. It is predominantly idiosyncratic and independent of the dose of the drugs administered [63,64]. More rarely, hepatotoxicity is dose-dependent, such as with paracetamol. Intrinsic (temporal relationship) and extrinsic (plausibility) factors are also decisive in the causality classification of DILI. The pattern of injury can be determined by the R-value, which is derived from the alanine aminotransferase (ALT) and alkaline phosphatase (AP) level [R = ALT/ULN_ALT/ALP/ULN_ALP]. An R value ≥ 5 is indicative of hepatocellular jury and < 2 of a cholestatic injury [56]. Based on the documented ALT and AP values of the patient three days after the administration of MTX, an R value of 3.3 was calculated, which is indicative of a mixed pattern liver injury. Drug-induced liver injuries are most frequently reported as adverse drug reactions (ADRs) with paracetamol, esomeprazole, amoxicillin/clavulanic acid and atorvastatin [65].
4. Conclusion
The causal relationship between the administration of a drug and an observed ADR can only be established as a diagnosis of exclusion after ruling out other organic or pathophysiologic etiologies such as underlying immunologic, hematologic or infectious diseases. Based on the positive timeframe correlation and the documentation in the SmPC, the literature and international databases, a drug-related etiological cause of the pancytopenia and acute liver injury in this patient was adjudged probable due to the synergistic pharmacokinetic and pharmacodynamic effects of MTX, metamizole, esomeprazole and torasemide.
The patient had a leucopenia of 2.34 G/l, thrombocytopenia of 46 G/l and markedly elevated liver parameters, one day after the last administration of MTX. However, a mild anemia was already detected prior to the hospital admission. Most probably, a methotrexate-induced bone marrow suppression was already underway. Due to the administration of high-dose metamizole just before the administration of the low-dose MTX, the pre-existing hematotoxic pharmacodynamic adverse effect of MTX may have been acutely aggravated by that of metamizole despite the preemptive administration of folic acid on a day before and after the administration of MTX. The co-administration of high-dose esomeprazole and normal dose torasemide further reduced the renal elimination of MTX, thereby further enhancing the hematotoxic and hepatotoxic adverse effects of MTX.
Studies explicitly describing the duration of methotrexate-induced adverse effects after a dechallenge do not exist. The most important measure in the case of a suspected drug-induced hematotoxicity and hepatotoxicity is the immediate discontinuation of the suspected culprit drugs. Drug-induced blood disorders are reversible in many cases, but may persist in rare instances. Unfortunately, for individual patients, the potential of recovery and the time course cannot be estimated with accuracy. In dose-dependent leucopenia or thrombocytopenia, as observed in the use of chemotherapeutic agents, the lowest blood count values are usually recorded after 7 - 10 days. Immunologically related cytopenias usually normalize 1-2 weeks after discontinuation of drug exposure. In general, the recurrence of hematotoxic and hepatotoxic adverse effects after re-administration of the same drug under which an immunologically induced myelosuppressive reaction occurred is very likely. In this case a re-exposure of the culprit drugs in the framework of a re-challenge cannot be recommended.
The relatively high demand of analgesics in patients with chronic rheumatologic conditions already under treatment with MTX, proton pump inhibitors and loop diuretics require that health care providers consider drug-drug interactions as potential causes of acute hematotoxicity and hepatotoxicity after the administration of metamizole or non-steroidal anti-inflammatory drugs. In this category of patients, it is highly recommended to resort to other classes of analgesics, consider dose adjustment of relevant concomitant medications and monitor laboratory parameters closely for timely intervention of any adverse drug reactions.
Funding
This case report received no external funding.
Institutional Review Board Statement
Ethical approval was not required.
Informed Consent Statement
A written informed consent statement from the patient is available.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Genestier L, Paillot R, Fournel S, Ferraro C, Miossec P, Revillard JP. Immunosuppressive properties of methotrexate: apoptosis and clonal deletion of activated peripheral T cells. J Clin Invest. 1998 Jul 15;102(2):322-8. [CrossRef]
- Chan ES, Cronstein BN. Methotrexate--how does it really work? Nat Rev Rheumatol. 2010 Mar;6(3):175-8. [CrossRef]
- Swiss Drug Information. Available online: www.swissmedicinfo.ch (accessed on 6 August 2023).
- Micromedex™. Available online: www.micromedexsolutions.com (accessed on 6 August 2023).
- UpToDate™. Available online: www.uptodate.com (accessed on 6 August 2023).
- Chabner BA, Allegra CJ, Curt GA, Clendeninn NJ, Baram J, Koizumi S, Drake JC, Jolivet J. Polyglutamation of methotrexate. Is methotrexate a prodrug? J Clin Invest. 1985 Sep;76(3):907-12. [CrossRef]
- García-Carrasco M, Mendoza-Pinto C, Macías-Díaz S, Etchegaray-Morales I, Méndez-Martínez S, Soto-Santillán P, Pérez-Romano B, Jiménez-Herrera EA, Guzmán-Ruiz O, Ruiz-Argüelles A. Clinical relevance of P-glycoprotein activity on peripheral blood mononuclear cells and polymorphonuclear neutrophils to methotrexate in systemic lupus erythematosus patients. Clin Rheumatol. 2017 Oct;36(10):2267-2272. [CrossRef]
- De Graaf D, Sharma RC, Mechetner EB, Schimke RT, Roninson IB. P-glycoprotein confers methotrexate resistance in 3T6 cells with deficient carrier-mediated methotrexate uptake. Proc Natl Acad Sci U S A. 1996 Feb 6;93(3):1238-42. [CrossRef]
- Riquelme-Mc Loughlin C, Giavedoni P, Mascaró JM Jr. More than skin deep. Am J Emerg Med. 2018 Sep;36(9):1719.e3-1719.e4. [CrossRef]
- Van der Klauw MM, Wilson JHP, & Stricker BHC: Drug-associated agranulocytosis: 20 years of reporting in the netherlands (1974-1994). Am J Hematol 1998; 57:206-211. [CrossRef]
- Hoffmann F, Bantel C, Jobski K. Agranulocytosis attributed to metamizole: An analysis of spontaneous reports in EudraVigilance 1985-2017. Basic Clin Pharmacol Toxicol. 2020 Feb;126(2):116-125. [CrossRef]
- Blaser LS, Tramonti A, Egger P, Haschke M, Krähenbühl S, Rätz Bravo AE. Hematological safety of metamizole: retrospective analysis of WHO and Swiss spontaneous safety reports. Eur J Clin Pharmacol. 2015 Feb;71(2):209-17. [CrossRef]
- Blanchet E, Beau P, Frat JP. Bone marrow aplasia following dipyrone treatment in a patient with Crohn's disease receiving long-term methotrexate. Gastroenterol Clin Biol. 2004 May;28(5):502-3. French. [CrossRef]
- Trautmann A, Brockow K, Stoevesandt J. Metamizole-induced reactions as a paradigm of drug hypersensitivity: Non-allergic reactions, anaphylaxis, and delayed-type allergy. Clin Exp Allergy. 2020 Sep;50(9):1103-1106. [CrossRef]
- Stone SR, Morrison JF. Mechanism of inhibition of dihydrofolate reductases from bacterial and vertebrate sources by various classes of folate analogues. Biochim Biophys Acta. 1986 Feb 14;869(3):275-85. [CrossRef]
- Hamid M, Lashari B, Ahsan I, Micaily I, Sarwar U, Crocetti J. A deadly prescription: combination of methotrexate and trimethoprim-sulfamethoxazole. J Community Hosp Intern Med Perspect. 2018 Jun 12;8(3):149-151. [CrossRef]
- Thomas DR, Dover JS, Camp RD. Pancytopenia induced by the interaction between methotrexate and trimethoprim-sulfamethoxazole. J Am Acad Dermatol. 1987 Dec;17(6):1055-6. [CrossRef]
- Groenendal H, Rampen FH. Methotrexate and trimethoprim-sulphamethoxazole--a potentially hazardous combination. Clin Exp Dermatol. 1990 Sep;15(5):358-60. [CrossRef]
- Cody V, Pace J, Makin J, Piraino J, Queener SF, Rosowsky A. Correlations of inhibitor kinetics for Pneumocystis jirovecii and human dihydrofolate reductase with structural data for human active site mutant enzyme complexes. Biochemistry. 2009 Mar 3;48(8):1702-11. [CrossRef]
- Ferrazzini G, Klein J, Sulh H, Chung D, Griesbrecht E, Koren G. Interaction between trimethoprim-sulfamethoxazole and methotrexate in children with leukemia. J Pediatr. 1990 Nov;117(5):823-6. [CrossRef]
- Kümpornsin K, Modchang C, Heinberg A, Ekland EH, Jirawatcharadech P, Chobson P, Suwanakitti N, Chaotheing S, Wilairat P, Deitsch KW, Kamchonwongpaisan S, Fidock DA, Kirkman LA, Yuthavong Y, Chookajorn T. Origin of robustness in generating drug-resistant malaria parasites. Mol Biol Evol. 2014 Jul;31(7):1649-60. [CrossRef]
- Liu H, Qin Y, Zhai D, Zhang Q, Gu J, Tang Y, Yang J, Li K, Yang L, Chen S, Zhong W, Meng J, Liu Y, Sun T, Yang C. Antimalarial Drug Pyrimethamine Plays a Dual Role in Antitumor Proliferation and Metastasis through Targeting DHFR and TP. Mol Cancer Ther. 2019 Mar;18(3):541-555. [CrossRef]
- Ueda H, Narumi K, Sato Y, Furugen A, Kobayashi M, Iseki K. Evaluation of possible pharmacokinetic interaction between methotrexate and proton pump inhibitors in rats. Pharmacol Rep. 2020 Oct;72(5):1426-1432. [CrossRef]
- Ueda H, Narumi K, Sato Y, Furugen A, Kobayashi M, Iseki K. Evaluation of possible pharmacokinetic interaction between methotrexate and proton pump inhibitors in rats. Pharmacol Rep. 2020 Oct;72(5):1426-1432. [CrossRef]
- Chioukh R, Noel-Hudson MS, Ribes S, Fournier N, Becquemont L, Verstuyft C. Proton pump inhibitors inhibit methotrexate transport by renal basolateral organic anion transporter hOAT3. Drug Metab Dispos. 2014 Dec;42(12):2041-8. [CrossRef]
- Narumi K, Sato Y, Kobayashi M, Furugen A, Kasashi K, Yamada T, Teshima T, Iseki K. Effects of proton pump inhibitors and famotidine on elimination of plasma methotrexate: Evaluation of drug-drug interactions mediated by organic anion transporter 3. Biopharm Drug Dispos. 2017 Dec;38(9):501-508. [CrossRef]
- Santucci R, Levêque D, Kemmel V, Lutz P, Gérout AC, N'guyen A, Lescoute A, Schneider F, Bergerat JP, Herbrecht R. Severe intoxication with methotrexate possibly associated with concomitant use of proton pump inhibitors. Anticancer Res. 2010 Mar;30(3):963-5.
- Santucci R, Levêque D, Lescoute A, Kemmel V, Herbrecht R. Delayed elimination of methotrexate associated with co-administration of proton pump inhibitors. Anticancer Res. 2010 Sep;30(9):3807-10.
- Boerrigter E, Crul M. A non-interventional retrospective cohort study of the interaction between methotrexate and proton pump inhibitors or aspirin. Ann Pharm Fr. 2017 Sep;75(5):344-348. [CrossRef]
- Tröger U, Stötzel B, Martens-Lobenhoffer J, Gollnick H, Meyer FP. Drug points: Severe myalgia from an interaction between treatments with pantoprazole and methotrexate. BMJ. 2002 Jun 22;324(7352):1497. [CrossRef]
- Uwai Y, Saito H, Inui K. Interaction between methotrexate and nonsteroidal anti-inflammatory drugs in organic anion transporter. Eur J Pharmacol. 2000 Dec 1;409(1):31-6. [CrossRef]
- Maeda A, Tsuruoka S, Kanai Y, Endou H, Saito K, Miyamoto E, Fujimura A. Evaluation of the interaction between nonsteroidal anti-inflammatory drugs and methotrexate using human organic anion transporter 3-transfected cells. Eur J Pharmacol. 2008 Oct 31;596(1-3):166-72. [CrossRef]
- Iwaki M, Shimada H, Irino Y, Take M, Egashira S. Inhibition of Methotrexate Uptake via Organic Anion Transporters OAT1 and OAT3 by Glucuronides of Nonsteroidal Anti-inflammatory Drugs. Biol Pharm Bull. 2017;40(6):926-931. [CrossRef]
- Uwai Y, Suzuki R, Iwamoto K. [Effect of nonsteroidal anti-inflammatory drugs on pharmacokinetics of methotrexate: a meta-analysis]. Yakugaku Zasshi. 2011;131(5):853-61. Japanese. [CrossRef]
- Dupuis LL, Koren G, Shore A, Silverman ED, Laxer RM. Methotrexate-nonsteroidal antiinflammatory drug interaction in children with arthritis. J Rheumatol. 1990 Nov;17(11):1469-73.
- Liang CA, Su YC, Lin SJ, Tsai TH. Risk factors for acute kidney injury after high-dose methotrexate therapy: a single-center study and narrative review. Eur J Clin Pharmacol. 2023 Jun;79(6):789-800. [CrossRef]
- Stewart CF, Fleming RA, Germain BF, Seleznick MJ, Evans WE. Aspirin alters methotrexate disposition in rheumatoid arthritis patients. Arthritis Rheum. 1991 Dec;34(12):1514-20. [CrossRef]
- Liegler DG, Henderson ES, Hahn MA, Oliverio VT. The effect of organic acids on renal clearance of methotrexate in man. Clin Pharmacol Ther. 1969 Nov-Dec;10(6):849-57. [CrossRef]
- Aherne GW, Piall E, Marks V, Mould G, White WF. Prolongation and enhancement of serum methotrexate concentrations by probenecid. Br Med J. 1978 Apr 29;1(6120):1097-9. [CrossRef]
- Lilly MB, Omura GA. Clinical pharmacology of oral intermediate-dose methotrexate with or without probenecid. Cancer Chemother Pharmacol. 1985;15(3):220-2. [CrossRef]
- El-Sheikh AA, van den Heuvel JJ, Koenderink JB, Russel FG. Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J Pharmacol Exp Ther. 2007 Jan;320(1):229-35. [CrossRef]
- Maeda A, Tsuruoka S, Ushijima K, Kanai Y, Endou H, Saito K, Miyamoto E, Fujimura A. Drug interaction between celecoxib and methotrexate in organic anion transporter 3-transfected renal cells and in rats in vivo. Eur J Pharmacol. 2010 Aug 25;640(1-3):168-71. [CrossRef]
- Statkevich P, Fournier DJ, Sweeney KR. Characterization of methotrexate elimination and interaction with indomethacin and flurbiprofen in the isolated perfused rat kidney. J Pharmacol Exp Ther. 1993 Jun;265(3):1118-24.
- Thyss A, Milano G, Kubar J, Namer M, Schneider M. Clinical and pharmacokinetic evidence of a life-threatening interaction between methotrexate and ketoprofen. Lancet. 1986 Feb 1;1(8475):256-8. [CrossRef]
- Tracy TS, Krohn K, Jones DR, Bradley JD, Hall SD, Brater DC. The effects of a salicylate, ibuprofen, and naproxen on the disposition of methotrexate in patients with rheumatoid arthritis. Eur J Clin Pharmacol. 1992;42(2):121-5. [CrossRef]
- Dupuis LL, Koren G, Shore A, Silverman ED, Laxer RM. Methotrexate-nonsteroidal antiinflammatory drug interaction in children with arthritis. J Rheumatol. 1990 Nov;17(11):1469-73.
- Kremer JM, Petrillo GF, Hamilton RA. Pharmacokinetics and renal function in patients with rheumatoid arthritis receiving a standard dose of oral weekly methotrexate: association with significant decreases in creatinine clearance and renal clearance of the drug after 6 months of therapy. J Rheumatol. 1995 Jan;22(1):38-40.
- Takeda M, Khamdang S, Narikawa S, Kimura H, Hosoyamada M, Cha SH, Sekine T, Endou H. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J Pharmacol Exp Ther. 2002 Aug;302(2):666-71. [CrossRef]
- Najjar TA, Abou-Auda HS, Ghilzai NM. Influence of piperacillin on the pharmacokinetics of methotrexate and 7-hydroxymethotrexate. Cancer Chemother Pharmacol. 1998;42(5):423-8. [CrossRef]
- Iven H, Brasch H. The effects of antibiotics and uricosuric drugs on the renal elimination of methotrexate and 7-hydroxymethotrexate in rabbits. Cancer Chemother Pharmacol. 1988;21(4):337-42. [CrossRef]
- Iven H, Brasch H. Influence of the antibiotics piperacillin, doxycycline, and tobramycin on the pharmacokinetics of methotrexate in rabbits. Cancer Chemother Pharmacol. 1986;17(3):218-22. [CrossRef]
- Williams WM, Chen TS, Huang KC. Effect of penicillin on the renal tubular secretion of methotrexate in the monkey. Cancer Res. 1984 May;44(5):1913-7.
- Ronchera CL, Hernández T, Peris JE, Torres F, Granero L, Jiménez NV, Plá JM. Pharmacokinetic interaction between high-dose methotrexate and amoxycillin. Ther Drug Monit. 1993 Oct;15(5):375-9. [CrossRef]
- Titier K, Lagrange F, Péhourcq F, Moore N, Molimard M. Pharmacokinetic interaction between high-dose methotrexate and oxacillin. Ther Drug Monit. 2002 Aug;24(4):570-2. [CrossRef]
- Ueo H, Motohashi H, Katsura T, Inui K. Human organic anion transporter hOAT3 is a potent transporter of cephalosporin antibiotics, in comparison with hOAT1. Biochem Pharmacol. 2005 Oct 1;70(7):1104-13. [CrossRef]
- Takeda M, Babu E, Narikawa S, Endou H. Interaction of human organic anion transporters with various cephalosporin antibiotics. Eur J Pharmacol. 2002 Mar 8;438(3):137-42. Erratum in: Eur J Pharmacol. 2002 Aug 16;450(1):111. [CrossRef]
- Bubik RJ, Osmon DR, Oravec CP, Rivera CG. Two cases of severe neutropenia in patients on low-dose methotrexate and ceftriaxone. Am J Health Syst Pharm. 2019 May 17;76(11):804-809. [CrossRef]
- Jarfaut A, Santucci R, Levêque D, Herbrecht R. Severe methotrexate toxicity due to a concomitant administration of ciprofloxacin. Med Mal Infect. 2013 Jan;43(1):39-41. [CrossRef]
- Dalle JH, Auvrignon A, Vassal G, Leverger G. Interaction between methotrexate and ciprofloxacin. J Pediatr Hematol Oncol. 2002 May;24(4):321-2. [CrossRef]
- Aouinti I, Gaïes E, Trabelsi S, Salouage I, Jebabli N, Charfi R, Lakhal M, Klouz A. Delayed elimination of methotrexate in a patient receiving ciprofloxacin. Therapie. 2013 May-Jun;68(3):175-7. [CrossRef]
- Kamangar F, Berger TG, Fazel N, Koo JY. Methotrexate toxicity induced by ciprofloxacin leading to psoriatic plaque ulceration: a case report. Cutis. 2013 Sep;92(3):148-50.
- Urata S, Yoshikawa N, Saito K, Tazaki T, Ohno R, Takeshima H, Ikeda R. Delayed methotrexate elimination in a patient with primary central nervous system lymphoma: A case report. J Clin Pharm Ther. 2021 Dec;46(6):1796-1799. [CrossRef]
- Livertox® (www.livertox.nih.gov). Available online: www.swissmedicinfo.ch (accessed on 6 August 2023).
- Weiler S, Merz M, Kullak-Ublick GA. Drug-induced liver injury: the dawn of biomarkers? F1000Prime Rep. 2015 Mar 3;7:34. [CrossRef]
- Ortland I, Mirjalili M, Kullak-Ublick GA, Peymani P. Drug-induced liver injury in Switzerland: an analysis of drug-related hepatic disorders in the WHO pharmacovigilance database VigiBase from 2010 to 2020. Swiss Med Wkly. 2021 May 12;151:w20503. [CrossRef]

Table 1.
List of medications before admission.
| Medication and Dosage | Dose Interval * | Remarks |
|---|---|---|
| Acetylsalicyclic acid 100 mg | 1-0-0-0 | |
| Atorvastatin 40 mg | 0-0-1-0 | |
| Bisoprolol 2.5 mg | 0.5-0-1-0 | |
| Escitalopram 10 mg | 1-0-0-0 | |
| Esomeprazole 40 mg | 2-0-0-0 | |
| Folic acid 5 mg | 1-0-0-0 | Saturdays at 8 am |
| Hydroxychloroquin 200 mg | 1-0-0-0 | |
| Iron, Folic acid 80/0.38 mg | 1-0-0-0 | before meals |
| Levothyroxin 0.05 mg | 1-0-0-0 | every other day |
| Methotrexat 15 mg | 1-0-0-0 | |
| Mirtazapin 30 mg | 0.5-0-0-0 | |
| Roflumilast 500 mcg | 0.5-0-0-0 | |
| Prednison 5 mg | 1.5-0-0-0 | |
| Temazepam 10 mg | 0-0-0-1 | |
| Torasemid 10 mg | 1-0-0-0 |
* The number pattern is a standardized way of summarizing the number of times a medication is taken during the four time phases within a day: morning-afternoon-evening-night.
Table 2.
List of medications during the first phase of hospitalization.
| Medication and Dosage | Dose Interval * | Remarks |
|---|---|---|
| Atorvastatin 40 mg | 0-0-1-0 | |
| Betamethasone 0.5 mg | 1-1-1-1 | started on admission day |
| Bisoprolol 2.5 mg | 0.5-0-1-0 | |
| Cholecalciferol 4000 EI/ml | 1-0-0-0 | started on day 4 |
| Chlorhexidine, Lidocaine 2 mg/ml | 1-1-1-1 | started on admission day |
| Dalteparin 5000 EI | 0-0-1-0 | started on admission day |
| Escitalopram 10 mg | 1-0-0-0 | |
| Esomeprazole 40 mg | 1-0-0-0 | |
| Folic acid 5 mg | 1-0-0-0 | 1 day before and after MTX |
| Hydroxychloroquin 200 mg | 1-0-0-0 | |
| Isopropanol, Isopropylalcohol 0.2 % | 1-1-1-1 | started on admission day |
| Levothyroxin 0.05 mg | 1-0-0-0 | every other day |
| Magnesium 10 mmol | 0-1-0-0 | started on admission day |
| Metamizol 500 mg | 2-2-2-2 | started on admission day |
| Methotrexat 15 mg | 1-0-0-0 | Fridays at 8 am |
| Mirtazapin 30 mg | 0.5-0-0-0 | |
| Oxycodon, Naloxone 15/7.5 mg | 1-0-1-0 | started on admission day |
| Paracetamol 500 mg | 1-1-1-1 | started on admission day |
| Potassium chloride 10 mmol | 2-2-2-0 | started on admission day |
| Roflumilast 500 mcg | 0.5-0-0-0 | |
| Prednison 5 mg | 1.5-0-0-0 | |
| Temazepam 10 mg | 0-0-0-1 | |
| Torasemid 10 mg | 1-0-0-0 | |
* The number pattern is a standardized way of summarizing the number of times a medication is taken during the four time phases within a day: morning-afternoon-evening-night.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



