
Submitted:
13 February 2024
Posted:
14 February 2024
You are already at the latest version
Abstract
Necrotizing enterocolitis (NEC) is a major cause of neonatal morbidity and mortality. Although the specific etiology is unknown, previous research shows that maternal environment may be a contributing factor. Gestational stress may lead to the development of NEC by interfering with the barrier function of the intestinal epithelial barrier. The migration of cells from the crypts to the tips of the villi is crucial in maintaining the integrity of the epithelial barrier. Previously, the Martin lab found that pups from psychologically stressed pregnant dams were more susceptible to NEC-like injury. We have also shown that investigated how stress signaling and zinc impact intestinal epithelial wound healing. Through a scratch assay, human Caco-2 cell migration was studied over 24 hours, and the percent wound healing was calculated. We found that only hydrocortisone at the 30 ug/ml concentration led to an inhibition of wound healing, and that zinc was unable to rescue hydrocortisone mediated inhibition.
Keywords:
Wound healing
; Necrotizing enterocolitis
; Stress signaling
; Intestinal Injury
Introduction
Necrotizing Enterocolitis (NEC) is the leading cause of newborn intestinal disease. Not only is NEC the leading cause of neonatal emergency from a gastrointestinal disease, but it also has a mortality rate as high as 50% [1]. NEC is characterized by mucosal injury, necrosis, and perforation of the gastrointestinal system in newborns. In NEC, intestinal inflammation occurs due to impaired barrier function and lack of GI defense mechanisms such as gastric acid, digestive enzymes, and mucus production [2]. Under normal immune development, the gastrointestinal epithelial lining functions as a barrier between the antigen in the lumen of the intestine and the rest of the body. However, when the gastrointestinal epithelial barrier function is impaired in diseases like NEC, antigen crossing the epithelium in an uncontrolled manner can lead to a switch to a proinflammatory state [3]. Eventually the inflammation can become systemic, which ultimately causes death in neonates [4]. Other multifactorial etiologies of NEC include intestinal immaturity, premature birth, formula feeding, and abnormal intestinal microbiota [4]. A better understanding of what impacts the development and prevention of NEC is crucial to reduce the severity and prevalence of NEC. Although knowledge about severe NEC has advanced, treatment strategies have not been impacted in the same manner. Therefore, developing treatments to prevent NEC is crucial to improving patient outcomes.
Previous research demonstrates that neonates diagnosed with NEC have an altered microbiome composition compared to controls at birth [5,6]. This suggests that factors during gestation may lead to the development of NEC. Infants are more likely to be born perinatal complications that come from stressful environments. Such stressful environmental conditions include barriers to access prenatal health care and previous complications with pregnancy [7]. Previous work has shown that gestational psychological stress on pregnant mice leads to offspring with significantly decreased intestinal surface area with reduced villi height and crypt depth[8]. This research supports the idea that gastrointestinal structural development is impaired due to increased cortisol from stress, however, further research is needed to determine if there is a functional difference.
Cortisol is a naturally occurring stress hormone that increases by two to four-fold during pregnancy.[8] While it is needed for fetal development, cortisol levels are tightly regulated across the placenta. The enzyme 11beta-hydroxysteroid dehydrogenase type 2 (HSD11B2) deactivates cortisol across the placenta, but excess maternal cortisol can still lead to increased fetal exposure to cortisol [7]. Longer exposures to high fetal cortisol correlate to lower birth weights and shorter gestation time, which are both factors in NEC development. In turn, abnormal increases in cortisol during gestation due to environmental stressors may overwhelm the HSD11B2 regulatory mechanism, resulting in impaired fetal gastrointestinal development.
In efforts to better understand the mechanism of NEC and stress, we have shown that following stress, there is increased serum zinc and no change in gastrointestinal tissue zinc levels. Zinc has previously shown to aid in the maintenance of the gastrointestinal epithelium structure and function. Deficiencies in zinc cause increased tight junction permeability in Caco-2 cells and a pro-inflammatory state [9]. Increased permeability of the epithelial lining and further inflammation can lead to further development of NEC.
The gastrointestinal epithelial layer, a major component of maintaining the barrier consists of stem cells in the crypts that undergo differentiation which then migrate into the villus until they are released into the lumen. Specifically, LGR5 represents a discernible marker of intestinal stem cells, predominantly situated within the intestinal crypts. Upon the provision of suitable stimuli, LGR5-positive cells undergo differentiation into epithelial cell lineages and subsequently traverse the crypt-villus axis, thereby contributing to the preservation of an unimpaired intestinal barrier. [8,10] This migration leads to continuously renewing and healing the gastrointestinal barrier. Stress signaling leads to disruption of gastrointestinal stem cell differentiation and could lead to impaired wound healing. Intestinal injury found in NEC could be impacted from impaired wound healing and lead to further severity of the disease.
Zinc is an indispensable ion, possessing paramount importance in the preservation of physiological homeostasis. Zinc deficiency manifests across a spectrum of clinical outcomes, encompassing delayed wound healing, compromised immune function, and perturbations in various sensory modalities. Moreover, zinc exerts a pivotal regulatory influence over each stage of the wound healing process, spanning from membrane restoration, management of oxidative stress, facilitation of coagulation, modulation of inflammatory responses and immune defenses, promotion of tissue re-epithelialization, angiogenesis, to the ultimate formation of fibrosis or scar tissue. Additionally, it is noteworthy that zinc exerts a discernible impact on platelet activity and aggregation, a phenomenon mediated via Protein kinase C (PKC)-orchestrated tyrosine phosphorylation of platelet proteins. [11] This initiates the inflammatory phase of wound healing. Zinc acts as a first responder to tissue injury with neutrophils by enhancing neutrophilic phagocytosis of opsonized particles.[11] During the proliferative stage of wound resolution, zinc allows for granulation tissue to form and enhanced re-epithelialization. [11,12] The effects of Zinc can be translated to its role in gastrointestinal wound healing in stressed conditions. This study provides insight to better understanding the implications of zinc and stress signaling on gastrointestinal wbarrier injury, which could be contributing factors for the development and the treatment of NEC respectively.
Methods
Caco-2 Cell Culture
In this experiment, the gastrointestinal epithelial lining was modeled by adherent human colon carcinoma (Caco-2) cells to evaluate the impact of stress signaling and zinc levels on wound healing. DMEM media with 10% fetal bovine serum (FBS) and 1% pen strep was used to provide nutrients for cell growth and was changed every 2-3 days. The cells were passaged every three to four days at about 80% confluency. After removing the media, 10 mL of phosphate buffered saline (PBS) was used to wash the cells to remove any excess FBS so that the trypsin enzyme used to detach the cells would not be inactivated. The cells were administered with 3 mL of 0.05% trypsin and incubated for three minutes. 7 mL of media was dispensed into the flask to match the volume in a 10 mL conical tube. The cell suspension was transferred to a 10 mL conical tube and centrifuged for 5 minutes at 1000 rpm. The supernatant was aspirated off, which left the pellet of cells at the bottom of the conical tube. The tube was thwacked to break up the pellet, and the cells were resuspended in 10 mL of media. After counting the cell density with a hemocytometer, cells were seeded into T75 flasks with about 0.6 x 10^6 cells or 0.5 x 10^6 cells depending on the next projected passaging date.
Scratch Assay
To investigate how wound healing is impacted by stress signaling, a scratch assay model with Caco-2 cells was used. After passaging the cells in the procedure described above, 24-well plates were seeded at 0.07 x 10^6 cells per well with 2 mL per well. A monolayer was allowed to form over a period of three days. In order to ensure that the wound closure was due to cell migration, not proliferation, the cells were serum starved overnight before the scratch was performed. The initial circular wound was formed in the middle of each well with a P200 pipette tip without a filter attached to a vacuum in the cell culture hood. After the initial wound was formed, the media was replaced with media containing the different concentrations of stress signaling hormones cortisol and hydrocortisone, as well as zinc.
Gap Wound Healing Analysis
Using ImageJ, images of the scratch assay wound were taken at 0 and 24 hours to measure the surface area of the gap over time. The wound healing percentage was calculated by Equation (1). The data was then analyzed using GraphPad independent t test for significance.
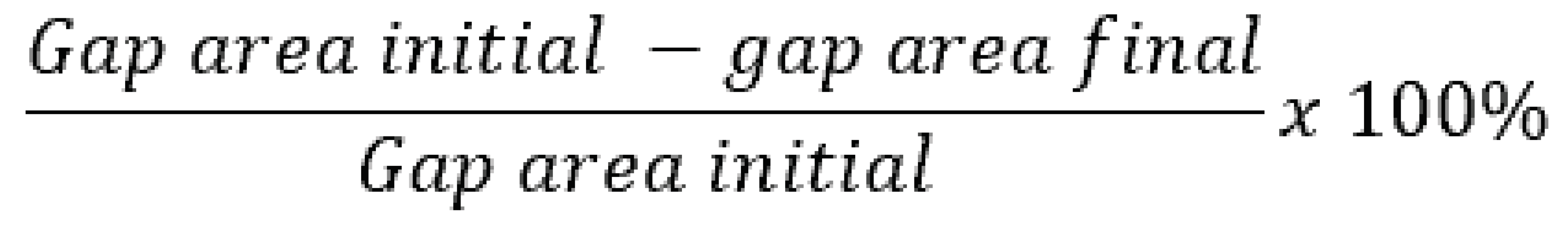
Equation (1) Wound healing percentage calculation.
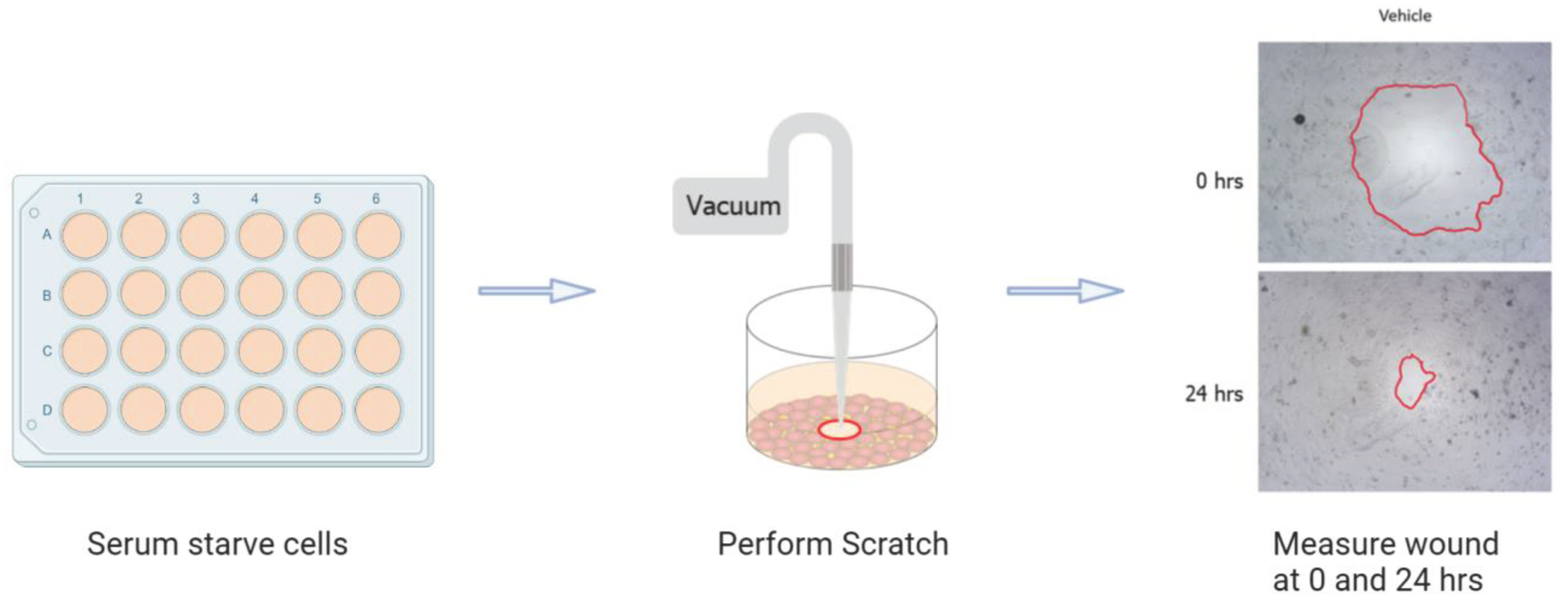
Figure 1.
Scratch assay methodology is shown here. After a monolayer of Caco-2 cells was formed on a 24-well plate, it was serum-starved overnight to synchronize cell cycles. The cells were scratched with a pipette tip attached to a vacuum to form a circular wound that was imaged at 0 and 24 hours.
Figure 1.
Scratch assay methodology is shown here. After a monolayer of Caco-2 cells was formed on a 24-well plate, it was serum-starved overnight to synchronize cell cycles. The cells were scratched with a pipette tip attached to a vacuum to form a circular wound that was imaged at 0 and 24 hours.
RNA Isolation for qPCR
RNA from Caco-2 cells in different stress signaling conditions was isolated for PCR analysis. First, Caco-2 cells were grown on 6 well plates. The media was aspirated and the cells were washed with 1-2 mls of ice cold PBS. After PBS was removed, 1 ml of TRIzol was added. The plate was scraped to detach the cells, and the TRIzol/cell lysate was transferred into a 1.5 ml Eppendorf tube. After leaving the lysate at room temperature for 5 minutes, 250 ul of chloroform as added and the tube was shaken vigorously for 15 seconds. After letting the mixture sit at room temperature for another five minutes, the lysate was centrifuged at 10,000 rpm for 5 minutes. The middle aqueous phase was removed with a pipette and placed in another 1.5 ml Eppendorf tube. 500 ul of isopropanol was added to the aqueous phase and mixed gently. After leaving it at room temperature for another five minutes, it was centrifuged at 14,000 rpm for twenty minutes. The samples were then placed on ice. The isopropanol was poured off and replaced with 1 ml of 75% ethanol in DEPC treated H2O. After mixing gently, it was recentrifuged at 9,500 rpm for 5 min. The ethanol was then poured off and the pellets were allowed to air dry. Then 15-25 ul of DEPEC treated TE buffer was added to the RNA pellet. The absorbance was measured to confirm a ratio greater than 1.8. Then the samples were sent to the Translational Research in Normal and Disordered Development (TReNDD) Genotyping Core for qPCR.
Statistical Analyses
For all data, a two-tailed unpaired Student’s t-test was performed between groups to calculate group differences. Calculations were performed using GraphPad Prism software version 5.0. A p value of <0.05 was considered statistically significant. For the figures in this paper, one star corresponds to a p-value less than 0.05, two stars corresponds to less than 0.01, and three stars corresponds to less than 0.001.
Results
This study’s aim was to determine how stress signaling affects gastrointestinal wound healing since excess stress during pregnancy may be a contributing factor towards the development of NEC in neonates. Additionally, we wanted to determine if zinc could potentially be used as a therapeutic to protect against NEC development by promoting wound healing function. In order to examine this, a scratch assay model was used to observe wound healing in Caco-2 cells over a 24-hour period.
Caco-2 Scratch Assay
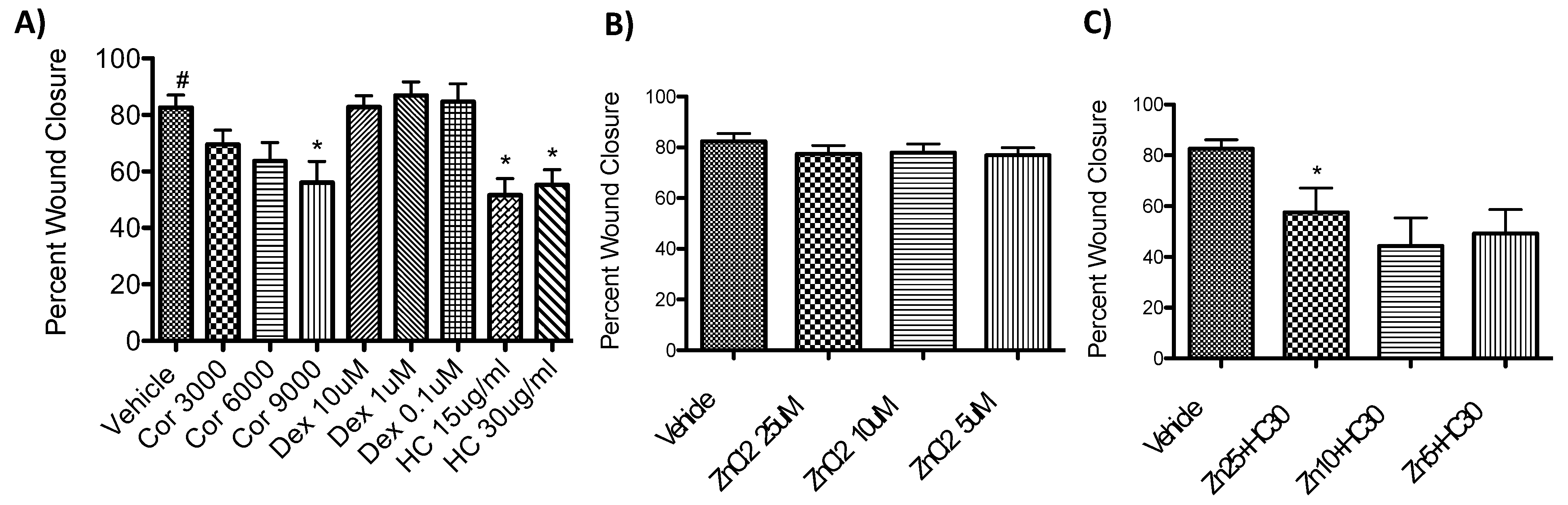
Initially, the percent of wound healing closure was examined in response to cortisol, which is a naturally occurring hormone that is highly regulated during pregnancy. As shown in Figure 2A, even at supraphysiological concentrations of cortisol at 3000 and 6000 nM, there was not a significant difference in wound healing. There was a significant inhibition (p<0.05) of wound healing at 9000 nM cortisol, however, since this is not a concentration applicable to humans, we wanted to explore how synthetic hormones that bound to the same glucocorticoid receptor would potentially impact wound healing.
Hydrocortisone was found to significantly inhibit wound healing for both the 15 ug/ml concentration (p<0.05) and the 30 ug/ml concentration (p<0.05, Figure 2A). We wanted to determine if using dexamethasone, an even more potent synthetic hormone, would lead to a greater inhibition of wound healing. T-tests determined that none of the dexamethasone trials were significantly different. Instead, when repeating the hydrocortisone at 30 ug/ml, it was confirmed that it significantly inhibited wound healing (p <0.001, Figure 2B).
Upon confirming that hydrocortisone at 30 ug/ml lead to a significant inhibition of wound healing, the effects of zinc on wound healing was studied. As shown in Figure 2C, zinc did not have any negative effect on wound healing percentage. Then to determine if zinc could potentially rescue the inhibition of wound healing caused by hydrocortisone at 30 ug/ml, a trial was done with both zinc and hydrocortisone in the scratch assay. However, there was still a significant inhibition of wound healing with all the different zinc concentrations with the hydrocortisone at 30 ug/ml.
Figure 2.
Wound healing percentages from the Caco-2 scratch assays. A) Percent wound healing in response to different concentrations of cortisol and hydrocortisone. B) Percent wound healing in response to different concentrations of zinc. C) Percent wound healing of different concentrations of zinc with hydrocortisone at 30 ug/ml.
Figure 2.
Wound healing percentages from the Caco-2 scratch assays. A) Percent wound healing in response to different concentrations of cortisol and hydrocortisone. B) Percent wound healing in response to different concentrations of zinc. C) Percent wound healing of different concentrations of zinc with hydrocortisone at 30 ug/ml.
Caco-2 qPCR
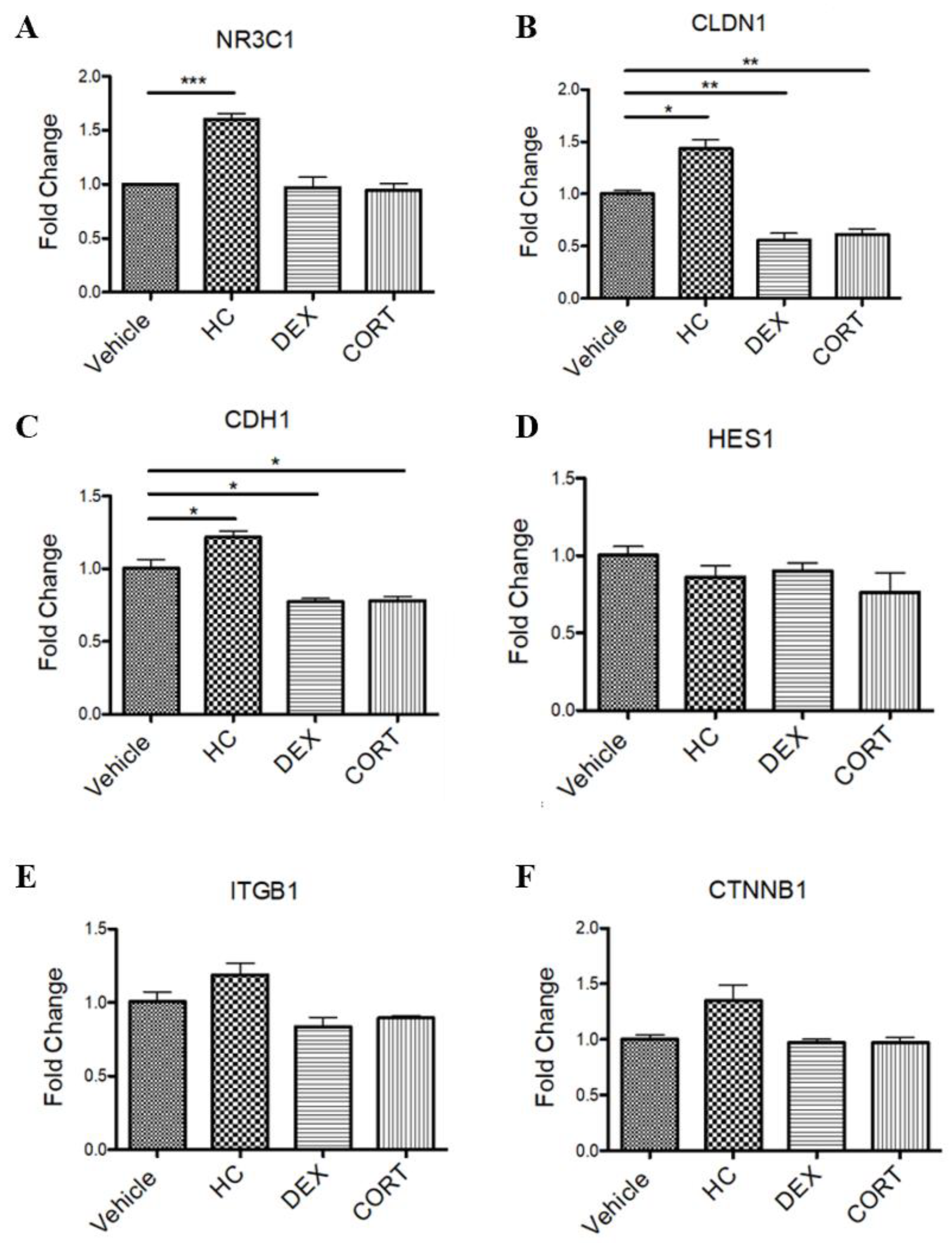
After determining that in this Caco-2 scratch assay model only hydrocortisone led to a significant inhibition of wound healing, mRNA was isolated from the different stress signaling conditions to perform a qPCR that would be able to determine differences in mRNA expression. Only in the hydrocortisone condition was there a significant increase (p=0.0003) in expression of the glucocorticoid receptor NRC1 (Figure 3A). For claudin 1 mRNA expression, hydrocortisone had a significant increase (p=0.0113), while dexamethasone and cortisol had a significant decrease in expression at p=0.0037 and p=0.0030 respectively (Figure 3B). In a similar pattern, for E-cadherin-1, hydrocortisone again had a significant increase (p=0.0439), while dexamethasone and cortisol had a significant decrease in expression at p=0.0239 and p=0.0309 respectively (Figure 3C). Primers for HES1, ITGB1, and CTNNB1 were used for the qPCR. There was no significant difference for any stress signaling hormone condition (Figure 3D–F).
Figure 3.
qPCR results of Caco-2 cell mRNA expression in response to stress signaling.
Discussion
Despite the increase in knowledge, NEC pathogenesis can be difficult to discern due to multifactorial causes. Intestinal immaturity seen in NEC leads to a disrupted intestinal epithelial barrier, an underdeveloped immune defense, and reduced mesenteric perfusion leading to poor vascular development and tone.[13] Furthermore, the compromised epithelial barrier and underdeveloped immune system, when exposed to luminal microbiota can lead to intestinal inflammation and poor wound healing. These damaging effects to the immune system during NEC can lead to bacterial translocation and systemic sepsis.
In the scratch assay on Caco-2 cells, this study found that stress signaling with solely hydrocortisone led to decreased wound healing potentially due to hydrocortisone having a higher potency than cortisol. The study hypothesized that the more potent and longer acting dexamethasone would lead to a greater inhibition of wound healing. However, we found this hypothesis to not be proven possibly due to dexamethasone’s inability to exhibit mineralocorticoid potency. Therefore, the altering of salt-balance could have led to an inhibition of wound healing from hydrocortisone’s mineralocorticoid effects. The main purpose of this study was to determine if zinc could potentially rescue an inhibition of wound healing caused by stress signaling. In this model no evidence was found to support that. In the future, different models could study the interaction between zinc and gut development.
Scratch assays are a useful model to study wound healing, however, they do have some limitations. Since scratch assays are done on a cell monolayer on a well plate, it is a two-dimensional surface, which does not model for the three-dimensional anatomy in the actual gastrointestinal system. However, future studies could evaluate barrier integrity in response to stress signaling, since if stress impairs barrier integrity, it could lead to a greater risk of NEC development. in a transwell model. To monitor barrier integrity, we plan to use transepithelial electrical resistance measurements (TEER), which measures the resistance across the apical and basolateral compartments of a transwell. Transwells involve an insert into a typical well plate, which allows for an apical and basolateral layer about the cell monolayer and models for a three-dimensional environment. This will more accurately model the barrier structure found in vivo.
In this Caco-2 scratch assay model, hydrocortisone was the only stress signaling hormone found to inhibit wound healing function. Additionally, hydrocortisone was the only stress signaling molecule that led to increased expression of the glucocorticoid receptor and cell adhesion proteins. This provides further suggestion that NEC and gut function may be impacted by cortisol signaling. In the current study zinc did not prevent intestinal repair. Future investigations will be dedicated to the exploration of the mechanistic impact of cortisol on the integrity and functionality of the intestinal barrier. These inquiries will employ the transepithelial electrical resistance (TEER) technique as a pivotal analytical tool to assess alterations in barrier properties. Similarly, the examination will extend to the influence of zinc, an essential micronutrient, on TEER measurements, with the aim of uncovering its potential regulatory role in bolstering or compromising the integrity of the intestinal barrier.
Author Contributions
H.S., J.S., C.M. experimental design, writing and editing of the manuscript. T.J.: Experimental design.
Acknowledgements
All authors have no financial and personal relationships with other people or organizations that could potentially and inappropriately influence (bias) their work and conclusions.
References
- Thakkar, H.S.L.; K. Necrotizing Enterocolitis. Surgery (United Kingdom) 2022, (40), 713–716. [Google Scholar] [CrossRef]
- Nino, D.F.; Sodhi, C.P.; Hackam, D.J. Necrotizing enterocolitis: new insights into pathogenesis and mechanisms. Nat Rev Gastroenterol Hepatol 2016, 13(10), 590–600. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Tan, H.; Kaiko, G.E. Role of the Intestinal Epithelium and Its Interaction With the Microbiota in Food Allergy. Front Immunol 2020, 11, 604054. [Google Scholar] [CrossRef] [PubMed]
- Tanner, S.M.; et al. Pathogenesis of necrotizing enterocolitis: modeling the innate immune response. Am J Pathol 2015, 185(1), 4–16. [Google Scholar] [CrossRef] [PubMed]
- Morowitz, M.J.; et al. Redefining the role of intestinal microbes in the pathogenesis of necrotizing enterocolitis. Pediatrics 2010, 125(4), 777–85. [Google Scholar] [CrossRef] [PubMed]
- Heida, F.H.; et al. A Necrotizing Enterocolitis-Associated Gut Microbiota Is Present in the Meconium: Results of a Prospective Study. Clin Infect Dis 2016, 62(7), 863–870. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, C.T.; Carter, S.R.; Martin, C.A. Impact of maternal factors, environmental factors, and race on necrotizing enterocolitis. Semin Perinatol 2023, 47(1), 151688. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.; et al. The Effects of Gestational Psychological Stress on Neonatal Mouse Intestinal Development. J Surg Res 2019, 235, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, W.; Fukada, T. Contribution of Zinc and Zinc Transporters in the Pathogenesis of Inflammatory Bowel Diseases. J Immunol Res 2019, 2019, 8396878. [Google Scholar] [CrossRef] [PubMed]
- McCabe, L.R.; Parameswaran, N. Recent Advances in Intestinal Stem Cells. Curr Mol Biol Rep 2017, 3(3), 143–148. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.A.; Pugh, N. The contribution of zinc to platelet behaviour during haemostasis and thrombosis. Metallomics 2016, 8(2), 144–55. [Google Scholar] [CrossRef] [PubMed]
- Tenaud, I.; et al. Zinc, copper and manganese enhanced keratinocyte migration through a functional modulation of keratinocyte integrins. Exp Dermatol 2000, 9(6), 407–16. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; et al. Animal models of gastrointestinal and liver diseases. Animal models of necrotizing enterocolitis: pathophysiology, translational relevance, and challenges. Am J Physiol Gastrointest Liver Physiol 2014, 306(11), G917–28. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



