Submitted:
12 February 2024
Posted:
14 February 2024
You are already at the latest version
Abstract
The origin of neurodegenerative diseases is linked to the abnormal folding of disease-associated proteins. The traditional amyloid hypothesis has been a focal point of research, but recent findings suggest a more complex picture. This review explores the role of physiological conformation of aggregation-prone domain (also known as prion-like low complexity domain) and multiple co-misfolded proteins in neurodegenerative diseases. We highlight the importance of targeting multi-heterotypic misfolded proteins for effective treatments and emphasize the irreversible neurodegeneration stemming from the loss of physiological folding in prion-like disease-causing proteins underscores the criticality of refolding. Additionally, we introduce the concept of ReproFold Therapeutics, a novel approach aimed at restoring the cellular prion-like folding of disease-causing proteins to rebuild normal cross-β interactomes and mitigate neurodegeneration. Baicalein, a potential drug candidate for this approach, shows promise in vitro and in animal models. Moreover, prion-like low complexity domains implicated in neurodegeneration are also associated with other disorders such as diabetes, cancers, and mental illnesses. Understanding the core pathology of neurodegenerative diseases and the broader implications of prion-like LC domains could revolutionize healthcare approaches and policies worldwide.
Keywords:
Neurodegenerative diseases
; prion-like low complexity domain
; amyloid
; SMN1
; TDP-43
; physiological cross-β interactome
; baicalein
; ReproFold.
1. Core Pathology of Neurodegenerative Diseases: Multiple Prions and Aberrant Physiological Cross-β Networks
1.1. Amyloid hypothesis and multiple prions
Neurodegenerative diseases pose significant challenges to healthcare systems worldwide. A characteristic feature of neurodegenerative diseases is the abnormal accumulation of misfolded proteins in the brain, contributing to the associated symptoms (1). The accumulation of misfolded proteins occurs early in the disease process and becomes more pronounced as the severity of the condition worsens (2). Genetic studies have linked mutated proteins, including amyloid beta, alpha-synuclein, tau, TDP-43, and SOD1, to form protein aggregates and the development of these disorders (3,4,5,6,7). The prion-like behavior of misfolded alpha-synuclein, TDP-43, and tau plays a crucial role in neurodegenerative diseases, challenging the notion of localized aggregation. Understanding these dynamic processes is paramount for a comprehensive comprehension of disease advancement. Researchers have long explored the amyloid hypothesis, which suggests that amyloid deposition initiates a cascade of events, including neuroinflammation, oxidative stress, and synaptic dysfunction, ultimately leading to nerve cell degeneration and cognitive impairment (8). Protein conformational diseases involve aggregate formation due to destabilized α-helical structures and simultaneous development of β-sheet structures. These β-sheets form through hydrogen bonding between pleated structures of alternating peptide strands, requiring a pleated peptide sequence and a β-sheet capable of accepting an additional strand. Advanced imaging techniques offer a nuanced view of amyloid aggregates, emphasizing the need to consider distinct conformations. Meanwhile, the recognition of soluble oligomers challenges the conventional understanding that only large fibrillar deposits contribute to neurodegeneration. Recent clinical trials targeting amyloid β proteins for removal through specific antibody treatments have shown limited efficacy in improving cognitive function, prompting a reassessment of the amyloid hypothesis's significance (9).
The FDA's approval of Alzheimer's drugs like lecanemab and aducanumab, which act by reducing amyloid plaques, has ignited widespread debate. The core of the controversy lies in the observation that despite these drugs' effectiveness in decreasing amyloid levels, significant improvements in patients' cognitive abilities and daily functions remain unproven. Clinical trials have not shown these drugs to meet or surpass the Minimum Clinically Important Difference (MCID), suggesting that while statistical significance may be observed in scores like the Mini Mental State Examination (MMSE), these do not equate to meaningful clinical benefits for patients. Notably, critical aspects such as effects on functional dependence, caregiver burden, and behavioral issues have been underemphasized, despite their importance in assessing the full impact of treatment. This situation underscores the ongoing debate about the appropriateness of using amyloid reduction as the primary benchmark for Alzheimer's drug approval, raising questions about the true benefit of such treatments in real-world scenarios. Recently, Mark H. Ebell and colleagues from the University of Georgia conducted a meta-analysis that reviewed 19 studies with 23,202 participants, focusing on eight anti-amyloid monoclonal antibodies for Alzheimer's disease (10). This analysis, covering randomized controlled trials against placebos, revealed that these treatments do not offer clinically meaningful benefits. Importantly, they carry significant risks, notably amyloid-related imaging abnormalities (ARIA), such as edema and hemorrhage, with notable increased risks (NNH for ARIA-edema = 9, symptomatic ARIA-edema = 86, ARIA-hemorrhage = 13). The study underscores that the risks and high costs of these antibodies outweigh their clinical benefits. The evolving understanding of neurodegenerative diseases highlights the need for comprehensive therapeutic strategies.
We propose two factors that could influence these results. Firstly, the presence of co-misfolded protein pathology should be considered (11). Most individuals with neurodegenerative conditions exhibit multiple coexisting pathologies involving various misfolded proteins. For instance, patients with Alzheimer's disease (AD) may have deposits of amyloid beta, tau, synuclein, and/or TDP-43 (12). Additionally, accumulating evidence suggests that, apart from self-propagation, these misfolded proteins associated with neurodegeneration can act as prions and cross-seed each other, a phenomenon known as heterotypic propagation in vitro (13, 14, 15). For instance, oligomeric tau has been shown to induce TDP-43 mislocalization, further complicating the disease's underlying mechanisms (16). Focusing on the removal of a single type of aggregate may lead to less efficient outcomes and prove ineffective in the long run, as the remaining misfolded proteins can continue to spread and aggregate, forming pathological inclusions. Future therapeutics should consider targeting the multi-heterotypic misfolded proteins for effective treatment. Secondly, neuron loss is largely irreversible, as the brain's ability to regenerate new neurons is limited in adult humans (17). In patients with AD or PD, the lost neurons are essential for memory formation, information processing, and decision-making (18). While there are treatments and interventions that can help manage symptoms and slow the progression of the disease, they cannot reverse the extensive neuron loss that has occurred by the time of diagnosis. Early detection and intervention remain critical in improving the quality of life for individuals affected by neurodegeneration. Treatments would be more effective when initiated in the preclinical stages of the diseases.
1.2. Aberrant physiological cross-β Networks
Proteins known to cause neurodegeneration, such as tau, TDP-43, and synuclein, possess a prion-like low-complexity (LC) domain (19, 20, 21). This domain is responsible for promoting protein condensation in both normal and pathological conditions. Mutations associated with inherited or sporadic cases of neurodegeneration often occur in this LC domain. The prion-like domain is a specific group of molecules characterized by their lack of a fixed or ordered three-dimensional structure, known as intrinsic disorder (22). These intrinsically disordered proteins (IDPs) exhibit weak but highly cooperative and dynamic interactions, allowing for adaptability and flexibility (23). Upon binding to specific targets, unstructured proteins undergo a transformation to more ordered states. In the case of prion-like proteins, their structural disorder in the bound state can be either static or dynamic, resembling a prion-like cross-β configuration (24). Within cells, the prion-like LC domains can undergo phase separation, leading to the formation of membraneless structures like stress granules, paraspeckles, and nucleoids (25, 26). These prion-like LC domains serve essential biological functions, including DNA repair, chromosome organization, splicing, and transport of messenger RNA (mRNP) through homotypic and heterotypic cross-β polymerization (25, 26, 27, 28). However, during disease progression, it can also form toxic aggregates. Notably, unlike misfolded protein aggregates, the physiological prion-like condensation is reversible, temporary, and crucial for cell survival and regulated by post-modification and RNA.
Our latest research has shown that disrupting the physiological prion-like cross-β interactions of SMN1 can lead to the formation of aberrant ribonucleoproteins (RNPs), aggregates of PFN1, and mislocalization of TDP-43 in spinal muscular atrophy (SMA) (29). These findings indicate that removing a single prion-like domain can trigger rearrangements in prion-like cross-β interactomes, resulting in aberrant RNP formation, and the collapse of prion-like cross-β interactomes, leading to aggregation of partner proteins such as TDP-43 and PFN1. These findings shed light on the intricate nature of prion-like interactions in normal cells and, crucially, their role in the onset and progression of neurodegenerative diseases. Figure 1 presents a model that delineates three cellular physiological abnormalities arising from the disruption of cross-β interactions of low complexity proteins, specifically prion-like protein A and protein B, in cells. These disruptions are caused by changes in the proteins' structure and can result in: (1). Amyloid cascade: This cascade is triggered when protein A undergoes a structural change and morphs into a pathological form, labeled as A'. This transformation sets off the amyloid cascade, a process that is widely recognized. (2). Mixed proteinopathies: Typically, prion-like proteins A and B form either heterotypic (AA) or homotypic (AB) cross-β interactions, essential for certain biological processes like the assembly of nuclear bodies through liquid phase separation. When protein A's normal structure is lost, and it transitions into A', protein B loses its regular interaction partner. This causes protein B to initiate a secondary amyloid cascade. The resulting misfolded structures, whether B’B’ or even A’B’, together with the primary cascade's A’A’, lead to mixed proteinopathies. (3). Cellular homeostasis dysfunction: Any remaining protein B may undergo degradation or potentially interact with other proteins, such as prion-like protein C or self-interact. Such interactions produce aberrant prion-like interactomes, for instance, aberrant hnRNPs. This shift culminates in cellular homeostasis dysfunction. To summarize, the three pathological processes, initiated by structural changes in the proteins involved, collectively result in the symptoms observed in neurodegenerative diseases.
2. Baicalein's ReproFold: Beyond Clearance of Misfolded Proteins to Restoration of Cellular Cross-β Interactome in Neurodegenerative Diseases
Recently, in spinal muscular atrophy (SMA), we demonstrated that the loss of a prion-like domain of SMN1 leads to abnormal assembly of ribonucleoproteins (RNPs), multiple proteinopathies, and motor neurodegeneration due to the disruption of heterotypic cross-β interactions (29). This finding suggests that clearing existing misfolded protein alone is not enough to significantly improve patients' quality of life (30). Instead, it becomes essential to restore the cellular prion-like folding of disease-causing proteins and rebuild the normal prion-like cross-β interactome to bring the neurons back to a healthy state. Addressing neurodegenerative diseases effectively demands a strategy that not only addresses misfolded proteins but redirects them to restore prion-like folding and cross-β interactions. We have named this therapeutic approach ‘ReproFold’.
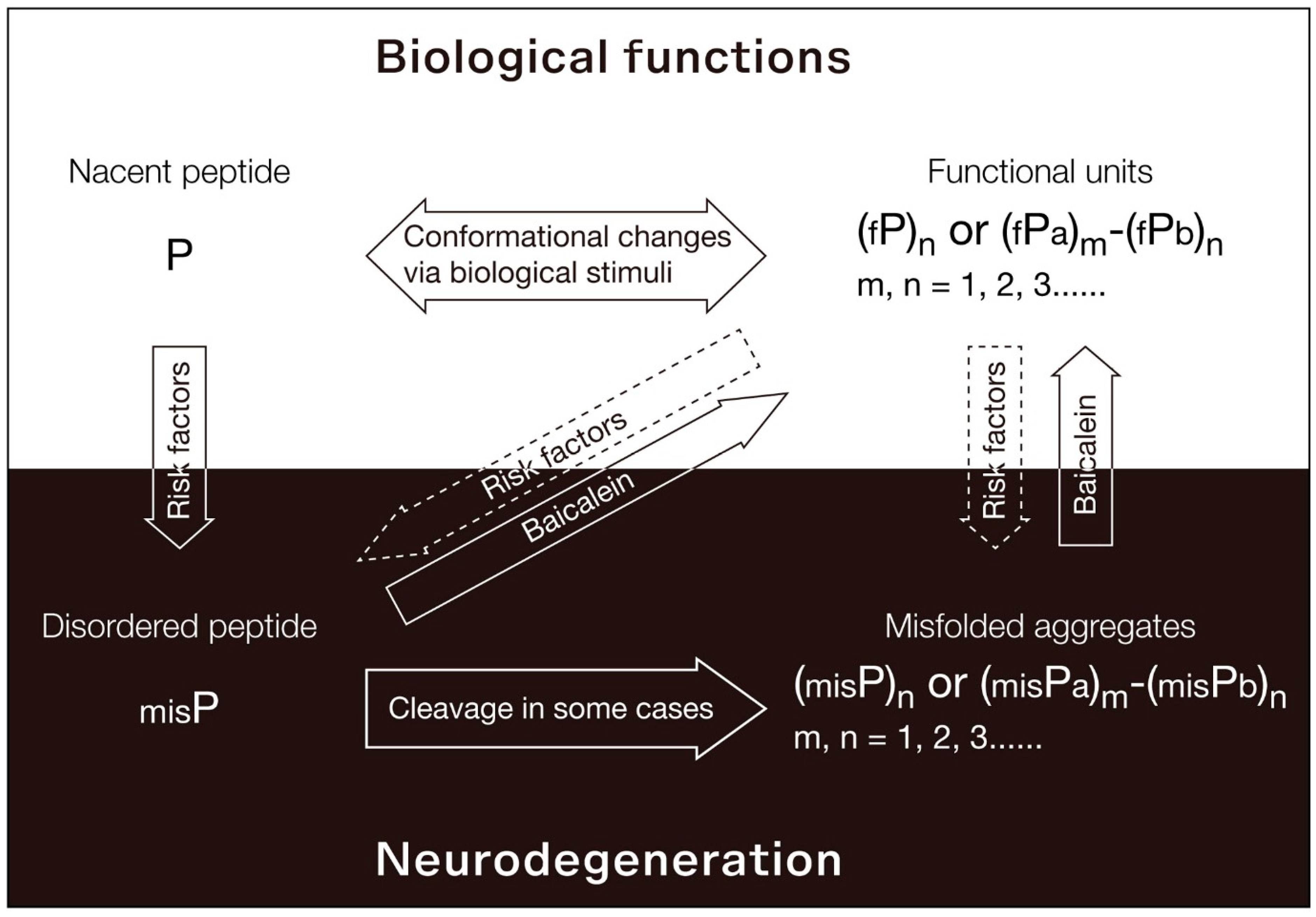
For many years, it was believed that addressing the misfolding and guiding these proteins back to their native cellular structure at early stage could potentially offer a groundbreaking solution for curing neurodegenerative diseases. However, the lack of identified compounds capable of achieving this has hindered the validation of this hypothesis. We recently investigated baicalein as one such molecule, serving to validate the viability of this therapeutic avenue. We found baicalein remodels existing TDP-43 aggregates into an oligomeric state in vitro and functionally restores the bioactivity of misfolded TDP-43 proteins in cell-based models of ALS and genetic premature aging (31). From the data we've gathered, we propose the functional PLD (fPLD)-based therapy, as illustrated in Figure 2. Under normal physiological conditions, nascent polypeptides (P) evolve into functional, structured units that are perfectly suited to meet biological requirements. These units can exist as monomers, oligomers, or even complexes with other proteins, represented as (fP)n or (fPa)m-(fPb)n. The conformation of these polypeptides or functional units is dynamically influenced by various biological stimuli. However, when exposed to risk factors like aging, ROS, heavy metals, or pathogens, the natural folding and aggregation processes of these polypeptides or functional units become compromised. This leads to the creation of misfolded polypeptides (misP) and the pathological aggregates seen in neurodegenerative diseases, denoted as (misP)n or (misPa)m-(misPb)n. To target TDP-43-related issues in such diseases, we've developed the “ReproFold” method, aiming to retain the functionality of oligomeric TDP-43 from misfolded TDP-43 via baicalein." Unfortunately, the existing mouse models for testing baicalein's effects on TDP-43 proteinopathies have two major drawbacks: they only involve the overexpression of mutated TDP-43, requiring excessively high doses of baicalein to remove surplus proteins, and this persistent overexpression can obstruct the drug's action, possibly leading to misleading results regarding its efficacy.
Alternatively, in SMA study, we found that baicalein can revitalize the prion-like structure of SMN2. This action not only restores the formation of gems and RNPs but also eliminates the aggregates of its interaction partners, TDP-43 and PFN1, thereby improving motor neuron functions in SMA mice (29). This makes baicalein a promising drug candidate for restoring prion-like activity and targeting disease-causing proteins in neurodegenerative disorders. For optimal results in treating prion-like protein diseases, it's essential to refold these proteins early, ideally during the presymptomatic phase. Identifying early biomarkers is crucial for timely intervention and offers a chance to prevent, not just halt, neurodegeneration.
In addition to its effects on TDP-43 and SMN, baicalein has demonstrated potential in disaggregating misfolded α-synuclein, a protein associated with Parkinson’s diseases, with studies showing promising results both in vitro and in mice (32,33). This suggests that baicalein could serve as a universal chaperone for a broad spectrum of misfolded proteins often found in neurodegenerative diseases, establishing it as a viable therapeutic strategy to tackle multi-prion disorders and mixed neurodegeneration. While biological chaperones also hold promise in achieving this objective, their implementation in clinical practice is hampered by the complex interplay of multiple substrates and the reciprocal interactions that occur between the chaperones and biological systems. Conversely, chemical chaperones appear to be more effective, bypassing some of these challenges. Moreover, baicalein stands out due to its oral bioavailability, minimal side effects, and affordability. These attributes position it as an exceptionally promising option for widespread use during asymptomatic stages, potentially extending to prophylactic use in healthy individuals.
Baicalein, extracted from the traditional Chinese herb Scutellaria baicalensis and known as Huang Qin in Traditional Chinese Medicine, is renowned for its antioxidant and anti-inflammatory properties. This flavonoid, with a deep history in herbal medicine, is now recognized for its potential in treating neurodegenerative diseases such as Alzheimer's and Parkinson's due to these properties (34, 35). Our research has identified a novel application of baicalein as a pharmacological chaperone for prion-like low complexity proteins, including TDP-43 and SMN, which are critical in neurodegenerative diseases. Operating at micromolar concentrations, baicalein effectively refolds misfolded proteins, preventing their degradation and stabilizing them in their functional conformations. This innovation aligns with precision medicine strategies by targeting specific molecular mechanisms underlying conformational diseases, presenting a promising pathway for therapies aimed at correcting loss of function or mitigating the gain of toxic function in misfolded proteins. Furthermore, the success of pharmacological chaperones in treating diseases caused by protein misfolding, such as Gaucher's disease and transthyretin (TTR) amyloidosis—where Miglustat stabilizes the glucocerebrosidase enzyme to improve symptoms and reduce substrate accumulation, and Tafamidis prevents amyloid formation by stabilizing TTR tetramers—underscores the potential of baicalein as a groundbreaking approach in the treatment of diseases characterized by protein misfolding (36, 37).
3. Prion-like LC Domains: Disease Hubs and Beyond
Prion-like LC domains are dynamic, intrinsically disordered regions of proteins that exhibit properties reminiscent of prion proteins, characterized by self-templating aggregation and propagation of misfolding. These domains are implicated in the onset and progression of various neurodegenerative diseases. However, emerging evidence suggests that their involvement extends beyond the realm of neurodegeneration, encompassing disorders such as diabetes, cancers, and mental illness.
Several studies have revealed a link between dysregulation of prion-like LC domains and diabetes. In particular, Insulin-degrading enzyme-Associated Peptide (IAP) aggregation has been observed in patients with diabetes (38). In T2D patients, islet amyloid deposits composed mainly of misfolded and aggregated IAPP are commonly observed (39). These amyloid deposits have been linked to the loss of β cell mass, further exacerbating the insulin secretion defects (40). In vitro and in vivo experiments using isolated islet cultures and transgenic mouse models overexpressing human IAPP have provided compelling evidence for the prion-like behavior of IAPP aggregates (41). Administration of pancreatic tissue homogenates containing preformed IAPP aggregates induced IAPP aggregation and diabetic pathology in both ex vivo and in vivo (42). The disease-associated alterations were absent when the aggregates were removed using specific antibodies targeting IAPP (43). We deduced that these misfolded IAP prions may trigger a cascade of misfolded proteins, potentially leading to multi-proteinopathies in brain (44, 45). This point offers a new perspective on diabetes-associated complications like dementia.
Cancer research has uncovered prion-like behavior in certain tumor suppressor proteins, with p53 being a prominent example (46). In tumor tissues, mutant p53 has been found to form amyloid aggregates, which impair its function and can lead to the progression of cancer (47). One promising strategy is the reactivation of mutant p53 in order to reinstate its crucial downstream activities. There are small compounds being developed that aim to stabilize the native conformation of mutant p53. This would help prevent its misfolding and aggregation (48). Nevertheless, there is a pressing need for further research to fine-tune these molecules for specific p53 mutant variants and to evaluate their long-term safety and effectiveness.
Insoluble aggregates of Disrupted-in-schizophrenia 1(DISC1) have been observed in human post-mortem brain tissue (49). Large DISC1 aggregates had pathological consequences within neurons, causing disruptions in the intracellular transport of critical organelles, such as mitochondria (50). Additionally, DISC1 aggregates exhibited prion-like behavior by being cell-invasive, a phenomenon observed in purified aggresomes and recombinant DISC1 fragments (51). These aggregates were internalized into cells with an efficiency comparable to that of α-synuclein. As a result of these groundbreaking discoveries, researchers have proposed a new classification for DISC1-dependent brain disorders, referring to them as "DISC1opathies" (52). This classification highlights the significance of protein conformational disorders in the pathogenesis of chronic mental diseases.
Beyond neurodegenerative diseases, diabetes, cancers, and mental illness, prion-like LC domains have been linked to cardiovascular diseases as well. Misfolded proteins, including immunoglobulin light chain protein and transthyretin (wild-type or mutated), deposit in cardiac tissue, leading to amyloid cardiomyopathy and heart failure (53, 54). The toxicity arising from the random binding of misfolded proteins contributes to the development of cardiovascular diseases.
These specific regions of the prion-like LC proteins play a crucial role in mediating numerous interactions. Remarkably, even a single mutation within the prion-like domain is sufficient to initiate neurodegeneration, strongly bolstering the hypothesis that aberrant conformational conversion in this domain lie at the core of the disease's origin. Understanding this mechanism could pave the way for proactive interventions to prevent neurodegeneration and alleviate the burden on families and healthcare facilities. ReproFold, with its focus on early intervention, holds the potential to revolutionize the treatment of neurodegenerative diseases, offering hope for a cure.
4. Discussion
The proposed model, which emphasizes the role of multiple prions and aberrant physiological cross-β networks in neurodegenerative diseases, challenges the traditional amyloid hypothesis. While the amyloid hypothesis has provided valuable insights into the pathological processes involved in neurodegeneration, the limitations of targeting a single misfolded protein are increasingly apparent. The presence of co-misfolded protein pathology and the phenomenon of heterotypic propagation highlight the complexity of the disease mechanism. This discussion underscores the importance of considering a broader approach in therapeutic strategies, targeting multiple misfolded proteins for more effective treatment outcomes.
The irreversible nature of neuron loss in neurodegenerative diseases underscores the imperative for early detection and intervention. Given the current treatments' limitations to reverse significant neuron damage, there is a pressing need to adopt proactive approaches during the preclinical stages of these diseases. Early diagnosis and intervention are crucial in preserving neuronal function and enhancing life quality for those impacted by neurodegenerative conditions. Future research is called upon to identify reliable biomarkers for early detection and to devise innovative intervention strategies that tackle the underlying pathology before irreversible harm is done. Additionally, ARIA further underscore the importance of early intervention in conditions marked by amyloid-beta (Aβ) accumulation, such as Alzheimer's disease. ARIA likely results from the immunotherapy-induced release of Aβ from plaques, which then accumulates in the basement membranes of blood vessels, leading to blockages, inflammation, and vascular damage (55). The significance of neuropathological data in understanding ARIA, pointing out that Aβ or Aβ-antibody complexes near blood vessels could trigger perivascular macrophages to emit cytokines and free radicals, thereby damaging vessels and causing leakage (56). Although advancements in MRI technology for detecting microbleeds offer a precautionary measure, finding an effective treatment for severe ARIA cases remains challenging. While steroids are commonly used, their efficacy is not uniformly observed. The emergence of ARIA stresses the need to prevent the accumulation of misfolded proteins in the brain and spinal cord from the earliest stages. It suggests that any therapeutic approach focused on clearing misfolded protein aggregates, be it antibody-based or chemical, could inadvertently raise the risk of ARIA. This highlights the critical need for developing therapeutic strategies that not only aim at clearing amyloid aggregates but also meticulously weigh the benefits against the risk of triggering ARIA, thereby ensuring a balanced approach in treating amyloid-associated diseases. Treatment in the early preclinical stages, especially at the onset of protein misfolding, is crucial.
5. Conclusions
In conclusion, the review presents a comprehensive perspective on the core pathology of neurodegenerative diseases, emphasizing the role of multiple prions and aberrant physiological cross-β networks. The limitations of the amyloid hypothesis and the need to consider co-misfolded protein pathology are highlighted. The irreversibility of neuron loss underscores the urgency of early detection and intervention. The proposed model offers a nuanced understanding of the intricate nature of prion-like interactions and their implications for disease onset and progression. The discussion calls for a paradigm shift in therapeutic approaches, advocating for strategies that target multi-heterotypic misfolded proteins for improved treatment efficacy.
Our work endeavors to shed light on the intricate web of neurodegenerative diseases, focusing on the catastrophic impact of misfolded proteins in the brain. Historically, the widely accepted amyloid hypothesis has served as the lodestar, guiding the trajectories of most empirical investigations in this domain. Our review, however, offers a paradigmatic shift. We've highlighted the dire consequences of of the disruption of physiological folding in the aggregation-prone domain of disease-causative proteins (also known as prion-like low complexity (LC) domains), and underscored the pressing need to refold these proteins, a step that has often been overlooked. Importantly, we addressed the pathological consequences arising from the loss of “heterotypic interactions” of disease-causative proteins with their prion-like partners.
Central to our review is the introduction of "ReproFold Therapeutics", a groundbreaking strategy using baicalein. This compound not only refolds misfolded proteins back to their original structures but also aids in reconstructing a healthy cross-β interactome within cells. Such actions alleviate protein misfolding stress, reinvigorate cellular balance, and staunch the relentless progression of neurodegenerative processes. Critically, the efficacy of baicalein extends from its potential in vitro to demonstrable success in animal models, emphasizing its promise as a therapeutic intervention.
Moreover, our review explores prion-like low-complexity (LC) domains extensively. Although primarily associated with neurodegenerative diseases, these domains have wider implications across a range of disorders, including diabetes, cancer, and mental illnesses. This insight could pave the way for groundbreaking healthcare strategies and policies worldwide.
6. Future Directions
The emerging concept of ReproFold as a therapeutic approach opens new avenues for research and development. Further investigation into the potential of baicalein and similar compounds in restoring cellular prion-like folding and cross-β interactions is warranted. Validating the ReproFold method in diverse neurodegenerative conditions and expanding its application to other proteinopathies will be crucial for establishing its efficacy as a broad-spectrum treatment strategy. Additionally, the exploration of prion-like LC domains beyond neurodegenerative diseases into realms such as diabetes, cancers, mental illness, and cardiovascular diseases presents a fascinating area for future research. Understanding the commonalities and differences in the behavior of prion-like LC domains across diverse disorders could unveil shared mechanisms and therapeutic targets. Further development of early biomarkers for various conditions linked to prion-like LC domains would be instrumental in advancing timely interventions.
A highly accurate blood test capable of real-time detection of conformational changes in misfolded proteins, such as α-synuclein and TDP-43, represents a significant breakthrough. These changes, transitioning from functional cross-β structures in healthy cells to misfolded and aggregated states, mark the beginning of pathogenesis in neurodegenerative diseases. The capacity to identify these shifts in peripheral systems is crucial, facilitating the development of accessible diagnostic tools for neurodegenerative conditions like AD, ALS and PD at asymptomatic stage. Baicalein, recognized for its ability to refold these misfolded proteins and its low toxicity, emerges as an ideal candidate for use in the asymptomatic phase, in conjunction with asymptomatic blood tests. With affordable, accurate asymptomatic diagnostics and a low-toxicity therapeutic agent, we foresee a shift towards early prevention and intervention within a decade, moving away from the conventional model of symptom management after diagnosis. This innovative approach seeks to prevent symptom onset, representing a significant departure from the traditional symptom management model post-diagnosis. The impact of intervening before symptoms appear is profound, enhancing individual autonomy and quality of life by preserving cognitive functions, slowing disease progression, and averting symptom onset, thereby delivering considerable benefits to society at large.
Acknowledgments
This research was funded by Garage Brain Science (ND-006), and YEEFAN Med Inc.
Conflicts of Interest Statement
H.-Y. Chang and I.-F Wang are financially compensated as employees of Garage Brain Science Co., Ltd. in Taiwan and YEEFAN MED INC. in CA.
References
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature. 1997, 388, 839–40. [Google Scholar] [CrossRef]
- Goedert, M.; Wischik, C.M.; Crowther, R.A.; Walker, J.E.; Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988, 85, 4051–4055. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J; Schuck, T.; Grossman, M.; Clark, C.M.; McCluskey, L.F.; Miller, B.L.; Masliah, E.; Mackenzie, I.R.; Feldman, H.; Feiden, W.; Kretzschmar, H.A.; Trojanowski, J.Q.; Lee, V.M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006, 314, 130–133. [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O'Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Ebell, M.H.; Barry, H.C.; Baduni, K.; Grasso, G. Clinically Important Benefits and Harms of Monoclonal Antibodies Targeting Amyloid for the Treatment of Alzheimer Disease: A Systematic Review and Meta-Analysis. The Annals of Family Medicine January 2024, 22, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.; Kovacs, G. Current Concepts of Mixed Pathologies in Neurodegenerative Diseases. Canadian Journal of Neurological Sciences 2023, 50, 329–345. [Google Scholar] [CrossRef]
- Spina, S.; La Joie, R.; Petersen, C.; Nolan, A.L.; Cuevas, D.; Cosme, C.; Hepker, M.; Hwang, J.H.; Miller, Z.A.; Huang, E.J.; et al. Comorbid neuropathological diagnoses in early versus late-onset Alzheimer's disease. Brain. 2021, 144, 2186–2198. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser G, Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009, 11, 909–913. [CrossRef]
- Polymenidou, M.; Cleveland, D.W. The seeds of neurodegeneration: prion-like spreading in ALS. Cell 2011, 147, 498–508. [Google Scholar] [CrossRef]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: the propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and Tau Oligomers in Alzheimer's Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol Dis. 2020, 146, 105130. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Wang, I.F.; Chang, H.Y.; Hou, S.C.; Liou, G.G.; Way, T.D.; Shen, C.K. The self-interaction of native TDP-43 C terminus inhibits its degradation and contributes to early proteinopathies. Nat Commun. 2012, 3, 766. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Vladimir, N. Uversky. Introduction to Intrinsically Disordered Proteins (IDPs). Chemical Reviews 2014, 114, 6557–6560. [Google Scholar]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012 May 11;149(4):753-67. 11 May. [CrossRef]
- Kwon, I.; Kato, M.; Xiang, S.; Wu, L.; Theodoropoulos, P.; Mirzaei, H.; Han, T.; Xie, S.; Corden, J.L.; McKnight, S.L. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013, 2013 155, 049–1060. [Google Scholar] [CrossRef]
- Xiang, S.; Kato, M.; Wu, L.C.; Lin, Y.; Ding, M.; Zhang, Y.; Yu, Y.; McKnight, S.L. The LC Domain of hnRNPA2 Adopts Similar Conformations in Hydrogel Polymers, Liquid-like Droplets, and Nuclei. Cell. 2015, 163, 829–839. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, 6357. [Google Scholar] [CrossRef]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.H.; Tsai, L.K.; Chang, H.Y.; Lai, H.Y.; Chen, C.L.; Wang, I.F. Prion-like Conformational Editing of SMN2 Proteins Rescues Spinal Muscular Atrophy. [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen S.; et al. Lecanemab in Early Alzheimer's Disease. N Engl J Med. 2023, 388, 9–21. [CrossRef] [PubMed]
- Chang, H.Y.; Wang, I.F. Reversal of Disease Phenotypes in ALS and Genetic Premature Aging Cells by Redirecting Misfolded TDP-43 Proteins into Physiological TDP-43 Oligomers. [CrossRef]
- Zhu, M.; Rajamani, S.; Kaylor, J.; Han, S.; Zhou, F.; Fink, A.L. The flavonoid baicalein inhibits fibrillation of alpha-synuclein and disaggregates existing fibrils. J Biol Chem. 2004, 279, 26846–26857. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Zhuang, X.; Lu, J. Neuroprotective effects of baicalein in animal models of Parkinson's disease: A systematic review of experimental studies. Phytomedicine. 2019, 55, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Obregon, D.; Ehrhart, J.; Deng, J.; Tian, J.; Hou, H.; Giunta, B.; Sawmiller, D.; Tan, J. Baicalein reduces β-amyloid and promotes nonamyloidogenic amyloid precursor protein processing in an Alzheimer's disease transgenic mouse model. J Neurosci Res. 2013, 91, 1239–1246. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, J.; Hölscher, C. Therapeutic Potential of Baicalein in Alzheimer's Disease and Parkinson's Disease. CNS Drugs. 2017, 31, 639–652. [Google Scholar] [CrossRef]
- Abian, O.; Alfonso, P.; Velazquez-Campoy, A.; Giraldo, P.; Pocovi, M.; Sancho, J. Therapeutic strategies for Gaucher disease: miglustat (NB-DNJ) as a pharmacological chaperone for glucocerebrosidase and the different thermostability of velaglucerase alfa and imiglucerase. Mol Pharm. 2011, 8, 2390–2397. [Google Scholar] [CrossRef]
- Lamb, Y.N. Tafamidis: A Review in Transthyretin Amyloid Cardiomyopathy. Am J Cardiovasc Drugs. 2021, 21, 113–121. [Google Scholar] [CrossRef]
- Westermark, P.; Engström ,U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci U S A. 1990, 87, 5036–5040. [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef] [PubMed]
- Hull, R.L.; Westermark, G.T.; Westermark, P.; Kahn, S.E. Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 3629–3643. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Soeller, W.C.; Roche, P.C.; Nelson, R.T.; Torchia, A.J.; Kreutter, D.K.; Butler, P.C. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci U S A. 1996, 93, 7283–7288. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, C.A.; Toukatly, M.N.; Fligner, C.L.; Udayasankar, J.; Subramanian, S.L.; Zraika, S.; Aston-Mourney, K.; Carr, D.B.; Westermark, P.; Westermark, G.T.; et al. β-cell loss and β-cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am J Pathol. 2011, 178, 2632–4260. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.S.; Roesti, E.S.; Mohsen, M.O.; Leonchiks, A.; Vogel, M.; Bachmann, M.F. Anti-IAPP Monoclonal Antibody Improves Clinical Symptoms in a Mouse Model of Type 2 Diabetes. Vaccines (Basel). 2021, 9, 1316. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.; Raimundo, A.F.; Menezes, R.; Martins, I.C. Islet amyloid polypeptide & amyloid beta peptide roles in Alzheimer's disease: two triggers, one disease. Neural Regen Res. 2021, 16, 1127–1130. [Google Scholar]
- Raimundo, A.F.; Ferreira, S.; Martins, I.C.; Menezes, R. Islet Amyloid Polypeptide: A Partner in Crime with Aβ in the Pathology of Alzheimer's Disease. Front Mol Neurosci. 2020, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Ano Bom, A.P.; Rangel, L.P.; Costa, D.C.; de Oliveira, G.A.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.; Cepeda, A.O.; Stumbo, A.C.; et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. J Biol Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell. 2016, 29, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Leliveld, S.R.; Bader, V.; Hendriks, P.; Prikulis, I.; Sajnani, G.; Requena, J.R.; Korth, C. Insolubility of disrupted-in-schizophrenia 1 disrupts oligomer-dependent interactions with nuclear distribution element 1 and is associated with sporadic mental disease. J Neurosci. 2008, 28, 3839–3845. [Google Scholar] [CrossRef]
- Ottis, P.; Bader, V.; Trossbach, S.V.; Kretzschmar, H.; Michel, M.; Leliveld, S.R.; Korth, C. Convergence of two independent mental disease genes on the protein level: recruitment of dysbindin to cell-invasive disrupted-in-schizophrenia 1 aggresomes. Biol Psychiatry. 2011, 70, 604–610. [Google Scholar] [CrossRef]
- Bader, V.; Ottis, P.; Pum, M.; Huston, J.P.; Korth, C. Generation, purification, and characterization of cell-invasive DISC1 protein species. J. Vis. Exp. 2012, 66, e4132. [Google Scholar]
- Korth, C. Aggregated proteins in schizophrenia and other chronic mental diseases: DISC1opathies. Prion 2012, 6, 134–141. [Google Scholar] [CrossRef]
- Iadanza, M.G.; Silvers, R.; Boardman, J.; Smith, H.I.; Karamanos, T.K.; Debelouchina, G.T.; Su, Y.; Griffin, R.G.; Ranson, N.A.; Radford, S.E. The structure of a β2-microglobulin fibril suggests a molecular basis for its amyloid polymorphism. Nat Commun. 2018 Oct 30;9(1):4517. [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Solopova, E.; Romero-Fernandez, W.; Harmsen, H.; Ventura-Antunes, L.; Wang, E.; Shostak, A.; Maldonado, J.; Donahue, M.J.; Schultz, D.; Coyne, T.M.; et al. Fatal iatrogenic cerebral β-amyloid-related arteritis in a woman treated with lecanemab for Alzheimer's disease. Nat Commun. 2023, 14, 8220. [Google Scholar] [CrossRef] [PubMed]
- Taylor, X.; Clark, I.M.; Fitzgerald, G.J.; Oluoch, H.; Hole, J.T.; DeMattos, R.B.; Wang, Y.; Pan, F. Amyloid-β (Aβ) immunotherapy induced microhemorrhages are associated with activated perivascular macrophages and peripheral monocyte recruitment in Alzheimer's disease mice. Mol Neurodegener. 2023, 18, 59. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Conceptual Model of Key Pathological Mechanisms in Neurodegenerative Diseases. This model emphasizes the critical role that compromised structural integrity of proteins with prion-like LC domains plays in the onset and progression of neurodegenerative diseases. It particularly sheds light on the pathological consequences arising from the loss of heterotypic interactions with prion-like partners. The model delineates three intersecting pathways: amyloid cascade, mixed proteinopathies and cellular homeostasis dysfunction. Collectively, these pathways provide insights into the overlapping pathological features observed in neurodegenerative disorders, such as AD, PD, and ALS. Dashed lines represent support from in vitro experiments.
Figure 1.
Conceptual Model of Key Pathological Mechanisms in Neurodegenerative Diseases. This model emphasizes the critical role that compromised structural integrity of proteins with prion-like LC domains plays in the onset and progression of neurodegenerative diseases. It particularly sheds light on the pathological consequences arising from the loss of heterotypic interactions with prion-like partners. The model delineates three intersecting pathways: amyloid cascade, mixed proteinopathies and cellular homeostasis dysfunction. Collectively, these pathways provide insights into the overlapping pathological features observed in neurodegenerative disorders, such as AD, PD, and ALS. Dashed lines represent support from in vitro experiments.
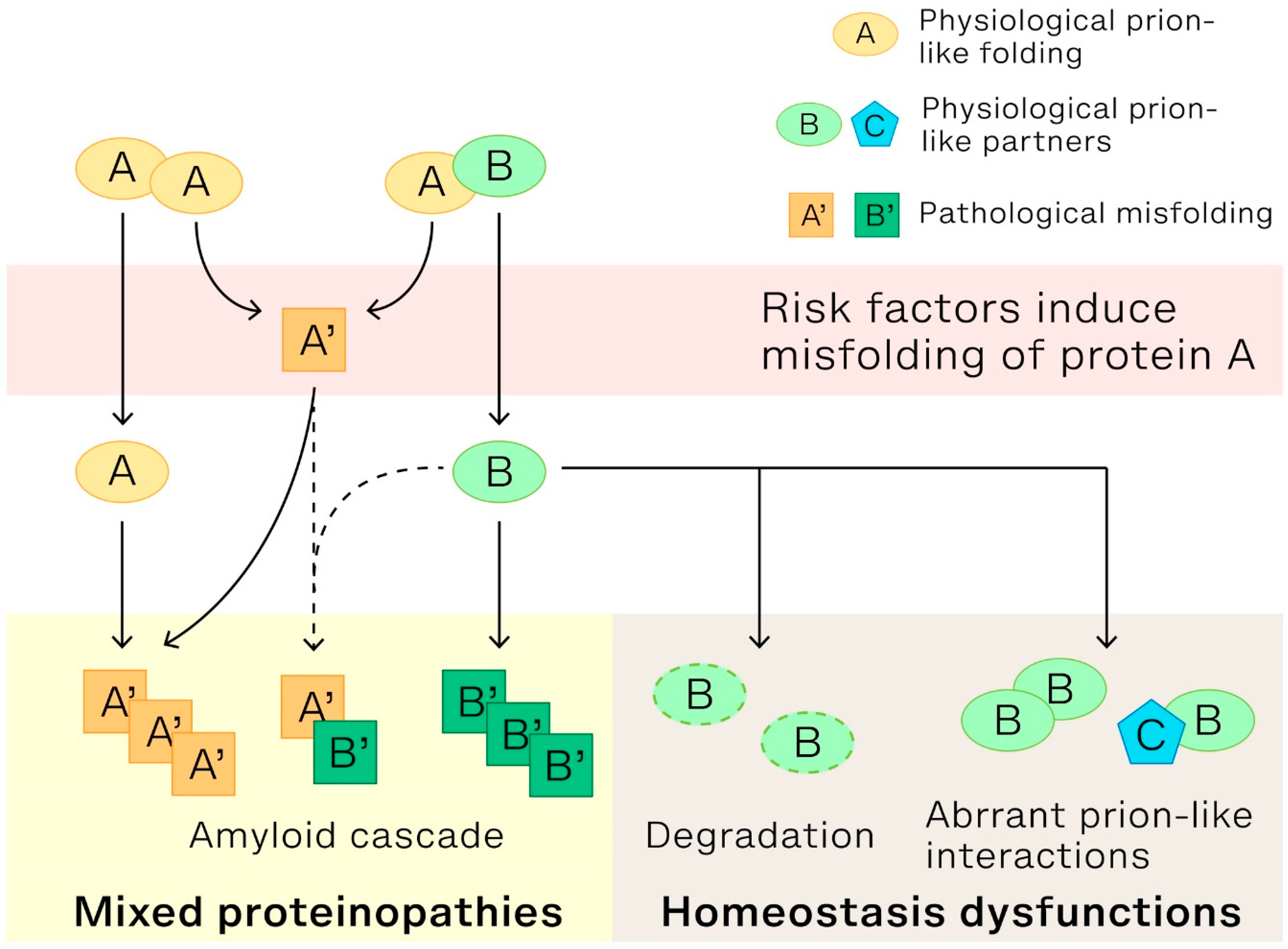
Figure 2.
The dependent origination of proteinopathies and fPLD-based therapeutic strategy. The key focus of the fPLD-based therapeutic strategy is to restore the normal physiological functions of pathological proteins. Only through this strategy, loss-of- function and gain-of-cytotoxicity result from misfolded pathological proteins can be simultaneously addressed.
Figure 2.
The dependent origination of proteinopathies and fPLD-based therapeutic strategy. The key focus of the fPLD-based therapeutic strategy is to restore the normal physiological functions of pathological proteins. Only through this strategy, loss-of- function and gain-of-cytotoxicity result from misfolded pathological proteins can be simultaneously addressed.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



