
Submitted:
23 July 2024
Posted:
25 July 2024
You are already at the latest version
Abstract
Hypertension is a frequent risk factor for cardiovascular diseases and the prevalence rate is continuously rising. Multiple pathophysiological mechanisms are responsible for hypertension, necessitating a combination of drugs from different classes for best management. Combination therapy is five times more effective in decreasing blood pressure compared to escalating the dose of a single agent. As per the ACCOMPLISH trial analysis, coupling a renin-angiotensin system inhibitor with a calcium channel blocker is more effective than the combination of a renin-angiotensin system inhibitor with diuretics in reducing major cardiovascular events, cardiovascular-related mortality, and the progression of chronic kidney disease. The renin-angiotensin system can be suppressed by inhibiting the angiotensin-converting enzyme or angiotensin type 1 receptor (AT1R), resulting, in decreased vascular smooth muscle contraction, heart performance, and reduced aldosterone synthesis. Calcium channel blockers work by inhibiting L-type calcium channels, reducing heart performance via negative inotropic, chronotropic, and bathmotropic effects, along with reducing vascular smooth muscle contraction. Diuretics exert their antihypertensive impact by changing body fluid volume and electrolytes. Initially, hypertension treatment commences with a single medication but often, different classes of two or more antihypertensive medications are necessary for achieving the target blood pressure goal. Sometimes, for Resistant hypertension four different classes of antihypertensive medications are necessary to reach the target blood pressure goal.
Keywords:
Hypertension
; calcium channel blocker
; combination therapy
; thiazide diuretics
; Renin-angiotensin-aldosterone system inhibitor
1. Introduction
Hypertension is the most prevalent and major risk factor for cardiovascular diseases (CVD) [1,2], which competitively increases CVD risk three times more than normotensive persons the same age [3]. The most common CVD is coronary disease [4,5,6], left ventricular hypertrophy [7], valvular heart disease [8], cardiac arrhythmia causes fibrillation of the atrium [9,10], cerebral stroke [11,12], and renal failure [13,14,15,16]. The prevalence rate of Hypertension is comparatively less in higher-income countries, about 349 million, than in lower-middle-income countries, nearly 1.04 billion, while the prevalence rate increased by 7.7% from 2000 to 2010 [17]. The USA prevalence rate was estimated at 29% [18]. Every year, globally, around 7.5 million (12.8% of the world population) deaths occur associated with hypertension [19,20]. Hypertension means chronic enhancement of systemic arterial pressure [21,22,23,24]. According to the National Institute for Health and Care Excellence (NICE) guidelines [25], clinically measured blood pressure (BP) ≥140/90 mmHg is diagnosed as hypertension. The Eighth Joint National Committee (JNC-8) [26] recommended the target blood pressure goal <150/90 in the general population aged 60 or older and others aged <140/90 who suffer from either diabetes [27,28] or chronic kidney disease [29,30,31] or both. BP is controlled primarily by renal sodium excretion, which interferes with total plasma and body volume, cardiac performance [32,33], and blood vessel muscle tone [34,35]. Some factors, including salt intake, insulin resistance, obesity, and endothelial dysfunction influence normal BP [36]. The genetic mutation also may cause Hypertension, such as the M235T polymorphism of the angiotensinogen gene [37]. Multiple pathophysiological mechanisms are responsible for hypertension [36]. Using a single medication to achieve the objective blood pressure goal is often impossible because compensatory response stimulates another mechanism. Approximately 75% of patients fail to meet the desired blood pressure goal while utilizing monotherapy [38,39]. Monotherapy approximately controls arterial pressure in only 50-60% of all hypertensive patients [40]. Combination therapy promotes tolerability by counteracting the side effects of each drug. The adherence rates increase by about 26% when patients get a single-pill combination [41]. Single-pill combination means the lower doses of two or more drugs combined into one tablet, considerably enhancing patient compliance [42,43,44]. Combining complementary classes of medication is five times more effective in decreasing blood pressure compared to raising the dose of a single agent [45]. So, combining therapy to achieve maximum therapeutic effect is logical. Various classes of antihypertensive drug combinations are available, including angiotensin receptor blocker (ARB)-Diuretic, β-Adrenoceptor Antagonist-Diuretic, angiotensin-converting enzyme inhibitor (ACEI)-Diuretic, ACEI-CCB (calcium channel blocker), ARB-CCB, ACEI-ARB, Centrally Acting Agents-Diuretic [46], in which currently, most preferred combination is renin-angiotensin system inhibitor plus diuretics or a calcium channel blocker [47]. This paper’s goal is a comparative analysis of the most frequently used combination: RAAS inhibitor with diuretics or a calcium channel blocker.
2. Rationale for Usage
2.1. Renin Angiotensin Aldosterone System
Renin-angiotensin aldosterone system (RAAS) [48,49] is a complex system comprising many peptides, receptors, and enzymes [50]. RAAS is considered a key element in maintaining the hemostatic of the body fluid, electrolytes, and arterial blood pressure [51]. The juxtaglomerular epithelioid cells [52,53], are found in the wall of renal afferent arterioles near the entrance of glomerular capillary networks, where most of the renin is synthesized. It functions as an aspartyl protease and is released from these cells in response to negative feedback for renal perfusion and blood pressure. The renin is encoded by chromosome 1 (1q32) containing the gene. It first produces a 406 amino acid pre-prorenin protein, which penetrates the rough endoplasmic reticulum. Here, a pre-part, 23 amino acids (signal peptide) are broken down and converted into prorenin (386 amino acids). Prorenin then moves to the Golgi apparatus where it is glycosylated. This prorenin is transformed into renin (340 amino acids) by prohormone convertases and the enzyme cathepsin B [54,55]. Angiotensinogen [56,57,58] is mostly produced by hepatocytes, adipocytes, proximal tubule epithelial cells, and astrocytes [59,60]. It is then quickly released into the extracellular space. The angiotensinogen gene, which has five exons and four introns, is located on chromosome 1 (1q42.2). Angiotensinogen protein has 485 amino acids, composed of a mature protein of 452 amino acids and a signal peptide of 33 amino acids [61].
Angiotensinogen is the only substrate for renin in the RAAS. Renin converts angiotensinogen into the decapeptide angiotensin I by cleaving the first ten amino acids from the N-terminal by proteolytic activity [62]. Subsequently, it undergoes conversion into the bioactive molecule angiotensin II by dicarboxylic peptidase angiotensin-converting enzyme (ACE), which cleaves two amino acids from the C-terminal of angiotensin I (Figure 1) [63] and simultaneously breaks down and inactivate the vasodilator bradykinin [64,65]. ACE is abundantly present on the luminal surface of vascular endothelia of the lung and less found in the kidney. angiotensin I is biologically inactive and serves as a precursor for angiotensin II [66,67,68]. Since angiotensin I is formed from angiotensinogen, whose activation is prompted by the rising plasma renin. So, renin is the first and rate-limiting step in RAAS [69]. Active Angiotensin II interacts with the plasma membrane receptors known as angiotensin type 1 (AT1) and type 2 (AT2) subtypes [70,71,72]. AT2 is mainly expressed in the brain, and adrenal glands, and has lower expression in other body organs. Activation of AT2 contributes to the suppression of vascular smooth muscle development and smooth muscle relaxation. In adult coronary arteries and the aorta, the presence of low-level AT2 receptors may aid in protecting tissues against ischemia. AT2 largely has a role in organ morphogenesis and prenatal development [73,74,75].
However, most of the physiological effects are mediated by the AT1 receptors. The AT1 receptor, present in the smooth muscle of the vascular wall, elicits considerable muscular contraction through the Gq class of G protein-coupled receptor (GPCR) signaling pathways [76,77]. When angiotensin II binds to the AT1 receptor found in smooth muscle, it initiates the activation of phospholipase C, responsible for hydrolyzing phosphoinositide into diacylglycerol (DAG) and inositol trisphosphate (IP3). IP3 activates both endoplasmic reticulum and cell membrane calcium channels, leading to a rise in intracellular calcium concentration. calcium binds to calmodulin, generating a calcium-calmodulin complex that activates myosin light chain kinase (MLCK). Consequently, MLCK phosphorylates the myosin light chain (MLC), resulting in the contraction of smooth muscle (as depicted in Figure 3) [78,79].
Previously, we mentioned that ACE inactivates the vasodilator bioactive product, Bradykinin. In normal physiology, Bradykinin interacts with endothelial-expressed bradykinin receptor type 2 (B2R) [80,81]. This interaction activates Gαq/11 signaling pathways, consequently, increasing cytoplasmic calcium concentration. The Calcium interacts with calmodulin, forming a calcium-calmodulin complex, that activates endothelial nitric oxide synthase (eNOS). eNOS is responsible for the conversion of L-arginine to L-citrulline, in this process, nitric oxide (NO) is reduced as a byproduct, which subsequently flows to the vascular smooth muscle [82]. In the smooth muscle, NO activates soluble guanylyl cyclase, which converts guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). This activation subsequently activates cGMP-dependent protein kinase I (cGKⅠ). The activated cGKI is responsible for the closing of the endoplasmic reticulum calcium channel and the dephosphorylation of the myosin light chain. Consequently, this allows smooth muscular relaxation (as depicted in Figure 3) [83,84].
Angiotensin II also stimulates the synthesis and release of aldosterone, a steroid hormone generated from cholesterol within the mitochondria of the zona glomerulosa cells in the adrenal cortex [85]. AT1Rs, present in these adrenal zona glomerulosa cells, belong to the Gq/11 family of GPCR-type proteins. The binding of angiotensin II on this receptor initiates the activation of phospholipase C-β (PLCβ), which in turn hydrolyzes phosphoinositide into diacylglycerol (DAG) and inositol trisphosphate (IP3) [86]. IP3 binds to the endoplasmic reticulum receptors, notably IP3R1, IP3R2, and IP3R3, resulting in the opening of calcium channels and increasing the cytoplasmic calcium concentration. This rise in calcium concentration causes the acceleration of aldosterone production and exocytosis of aldosterone vesicles via the activation of calcium/calmodulin-dependent kinases (CaMKs). Activated CaMKs stimulate the production of aldosterone synthetase CYP11B2 in cells, promoting the conversion of corticosterone to aldosterone. Simultaneously, diacylglycerol (DAG) activates protein kinase C (PKC), commencing a sequence of events where PKC and CaMKs collectively increase the amounts of steroidogenic acute regulatory protein (StAR). StAR possesses a cholesterol-binding site. following numerous intermediary stages, cholesterol ultimately undergoes conversion into aldosterone (as depicted in Figure 2) [87,88].
Aldosterone acts on the principal cells, located on the late distal tubule and collecting ducts of nephrons. Aldosterone is responsible for enhancing sodium reabsorption, promoting water retention, and increasing potassium excretion. Aldosterone is a mineralocorticoid, that quickly crosses the cell membrane and binds with intracellular mineralocorticoid receptors (MR) [89]. As a result, the production of serine/threonine protein kinase 1 (SGK 1) is increased and converted to active form through phosphorylation. Once active, SGK 1 phosphorylates ubiquitin ligase Nedd4-2, commencing its interaction with 14-3-3 proteins. The free ubiquitin ligase Nedd4-2 has a crucial role in suppressing the surface expression of the endothelial sodium channel (ENaC). Consequently, the density of ENaC on the apical membrane increases approximately 2-5 times. The high concentration of luminal Na+ flowing through ENaC generates a negative charge into the lumen, forcing the potassium ion secretion through the renal outer medullary potassium channel (ROMK) and chloride absorption (as depicted in Figure 2). Moreover, aldosterone also increases the transcription of the α and ß subunits of the Na1-K1-ATPase, accelerating the counter-transport mechanism for Na+ and K+ over the basolateral side [90]. In mice, the angiotensin AT1 receptor is expressed in the organum vasculosum of the lamina terminalis (OVLT) and the subfornical organ (SFO). In these areas most of the neurons are glutaminergic, their axon terminated to the paraventricular nucleus (PVN) of the hypothalamus. Within the PVN, magnocellular neurons are responsible for the synthesis of arginine vasopressin (AVP), that are stored in the posterior pituitary gland.
Additionally, the PVN contains parvocellular neurons whose axonal extensions regulate the autonomic nervous system. Upon angiotensin binding to the AT1 receptor, increases the production of arginine vasopressin and simultaneously stimulates the sympathetic outflow [91]. Besides, substantial morphological evidence demonstrates that the axonal efferent from SFO is projected to the supraoptic nucleus. Intraventricular (i.v.t.) injection of Angiotensin II binds to both SFO and OVLT expressed AT1 receptor, activates vasopressin production, and considerably elevates AVP in plasma [92]. in summary, angiotensin Ⅱ strongly increases sympathetic outflow and AVP in plasma. The released vasopressin binds to the Gs class GPCR type V2 receptor in the principal cells of the distal tubule and collecting duct of the kidney. This binding results in a rise of cyclic adenosine monophosphate (cAMP) concentration through the activation of adenyl cyclase. Subsequently, cAMP stimulates protein kinase A (PKA), which phosphorylates intracellular storage vesicle Aquaporin 2 (AQP-2), enabling the migration and insertion of AQP-2 into the apical membrane. Water passage via AQP-2 raises blood volume, potentially contributing to elevated blood pressure. Additionally, vasopressin binds to the Gq class GPCR type V1 receptor in vascular smooth muscle. This interaction stimulates the activation of protein kinase C (PKC). The resultant signaling cascade ultimately leads to blood vessel constriction and heightened peripheral resistance (Figure 3) [93,94,95].
Angiotensin Ⅱ stimulates the thirst center, causing feeling thirsty and increasing water intake, so high blood volume may increase BP [96,97]. The overactivation of the sympathetic nervous system (SNS) leads to increased secretion of nor-epinephrine from postganglionic sympathetic fibers [98,99,100]. Nor-epinephrine binds largely to myocyte-expressed β-adrenergic receptors, particularly beta-1 receptors [101,102]. This binding increases intracellular cAMP concentration and activates PKA by adenyl cyclase. Consequently, a positive ionotropic effect led to an increase in heart rate. Additionally, it stimulates the secretion of calcium ions from the sarcoplasmic reticulum, resulting in more forceful contractions of the myocyte, potentially increasing stroke volume and blood pressure [103,104,105].
Besides, nor-epinephrine (NE) also binds to arteries, arterioles, and veins smooth muscle expressing α1 and α2 receptors, beginning downstream signaling cascades. This process, over many steps, activates calmodulin-dependent myosin light chain kinase, which phosphorylates myosin light chain. Subsequently, the binding between actin and myosin filaments causes smooth muscle contraction. Postganglionic sympathetic nerves also release other vasoconstrictor neurotransmitters such as neuropeptide Y (NPY) or ATP [106]. The axon of the postganglionic sympathetic nerve has angiotensin receptors. When angiotensin II binds to these receptors, it suppresses the reuptake of nor-epinephrine and further magnifies peripheral resistance [107]. The Beta-adrenergic receptor expresses the zona glomerulosa cell. Sympathetic stimulation of this receptor also triggers renin secretion and further accelerates the renin-angiotensin-aldosterone system [108]. All the way, directly and indirectly, the overactivation of the angiotensin aldosterone system contributes to high blood pressure.
Various pharmacological treatments are designed to target specific components within the RAAS (as depicted in Figure 1). Angiotensin-converting enzyme inhibitors act by preventing the synthesis of angiotensin II, suppressing the renin-angiotensin-aldosterone system. This action lowers downstream effects mediated by the AT1 receptor, such as vascular muscle constriction, salt and water retention, and sympathetic outflow [109]. However, angiotensin Ⅱ synthesis is not fully controlled because angiotensin Ⅱ also synthesized by various additional ACE-independent alternative pathways in tissues such as chymase, kallikrein, cathepsin G, and elastase- 2 system [110]. Although ACEI is tolerated well in patients, it induces 5% to 35% dry cough and 0.2% to 0.7% angioedema [111]. ACEI inhibits the enzymatic cleavage of Bradykinin, increased plasma concentration of Bradykinin, activating the B2 receptor leads to vasodilation. most common ACEI are Benazepril, Captopril, Cilazapril, Enalapril, Fosinopril, Lisinopril, Moexipril, Perindopril, Quinapril, Ramipril, Tradolapril, Delapril and Imidapril [112]. AT1 receptor blocker is considered an effective and well-tolerated antihypertensive drug; selective inhibition of the AT1 receptor not only suppresses vasoconstriction effect and vascular hypertrophy but also increases angiotensin Ⅱ through compensatory response. The highly concentrated angiotensin Ⅱ may provide further pharmacological advantages by activating the AT2 receptor subtype. most common AT1 receptor blockers are Losartan, Valsartan, Irbesartan, Eprosartan, Telmisartan, and Candesartan [113]. Angiotensin receptor blockers (ARBs) don’t affect ACE. As a result, ARBs offer the advantage of bypassing the side effects created by ACE inhibitors (ACEIs) and avoiding the bradykinin-changing effect. Aliskiren (ALI) only drug that directly inhibits the renin. This medicine became clinically available as an orally active antihypertensive treatment and was authorized in 2007 [114]. By inhibiting the primary rate-limiting phase of RAAS, the activity of renin in plasma declines, preventing the synthesis of angiotensin II and decreasing aldosterone levels without interfering with bradykinin metabolism or the AT1 receptor. ACE inhibitors (ACEIs) and ARBs produce a compensatory response and increase renin levels in plasma, which can lead to inadequate inhibition of the RAAS system. ACE inhibition increases the angiotensin I level, which might then undergo conversion to angiotensin II via ACE-independent mechanisms. ARBs enhance the levels of Angiotensin II, potentially competing for receptor occupancy with the medication [115].
2.2. Calcium Channel Blocker
The calcium channels, biochemically well-defined a complex protein, comprising a pore-forming alpha (α1) 1 subunit, one out of ten, alongside four unique auxiliary subunits: a beta (β), α2, a delta (δ), and a gamma (γ) subunit [116]. The biggest α1 subunit (190-250 kDα) forms a Ca2+ permeable pore. This α1 subunit of the calcium channel is arranged into four repeating homologous domains, known as (I-IV), with each domain including tightly packed six transmembrane segments (S1-S6). Within the S4 region sits the gating charges, responsible for starting conformational alterations that open the pores in response to changes in the electric field. Meanwhile, the pore loop between the transmembrane segments, S5 and S6 of each domain, modulates ion conductance and selectivity [117]. The β subunit primarily contacts with the inner surfaces of domains I and II of the α1 subunit, positioned on the cytoplasmic side. In contrast, the γ subunit spans the cell membrane, including four transmembrane regions that largely interact with domain IV of the α1 subunit. Additionally, the proteolytically processed α2δ subunit, positioned extracellularly and linked by disulfide connections, largely interfaces with the extracellular surface of domains I–III of the alpha 1 subunit [118]. The alpha 1 subunit functions not only as the pore formation but also determines the channel type.
Voltage-gated calcium channels are classified into three types: Cav1, Cav2, and Cav3. These channels obtained their names from the chemical symbol of the principal permeating ion (Ca). The main physiological regulator (voltage) is indicated by a subscript (CaV). The number represents the CaV channel α1 subunit gene subfamily (varying from 1 to 3) and the sequence in which the alpha 1 subunit was discovered within that subfamily (from 1 to n) [119]. To date, ten different α1 subunits, encoded by the CACNA1x genes, have been found. The Cav1 subfamily (CaV1.1 to CaV1.4), represents L-type channels (where ‘L’ stands for Long-lasting). The CaV2 subfamily (CaV2.1 to CaV2.3), includes P/Q-type and N-type channels. Finally, the Cav3 subfamily (CaV3.1 to CaV3.3), represents T-type channels [120].
In which the L-type calcium channel is highly expressed on the vascular smooth muscle, myocyte, and nodal tissue in the heart. The influx of calcium ions through this channel helps maintain the membrane potential more positively, releasing calcium ions from the endoplasmic reticulum (calcium-induced calcium release) and stimulating smooth muscle and cardiac myocytes to contract [121]. All L-type channels have the same pharmacological profile, and their conformational shift is susceptible to numerous drugs that selectively block L-type calcium channels. This medication allosterically binds to three separate receptor sites on the L-type calcium channel, inhibiting calcium influx into the cytoplasm (as depicted in Figure 4) [122].
From a pharmacological and clinical perspective, the calcium channel blocker is classified into dihydropyridines and non-dihydropyridines. The dihydropyridines (e.g., amlodipine, nifedipine, felodipine, nimodipine, lacidipine) selectively bind with L-type calcium channel, that expressed on the vascular smooth muscle, resulting in reduced peripheral resistance due to relaxing vascular smooth muscle (pronounce vasodilation). Non-dihydropyridine (verapamil, diltiazem) is less tissue-specific; it binds both cardiac and vascular smooth muscle-expressed L-type calcium channels, resulting in diminished cardiac output due to negative ionotropic, chronotropic, and dromotropic effects. Although non-dihydropyridines decrease heart rate dihydropyridine is prone to increase heart rate due to sympathetic activation by pronounced vasodilation [123,124]. Some of the most typical side effects associated with dihydropyridine calcium channel blockers are peripheral edema, lightheadedness, flushing, headaches, and gingival hyperplasia, as well as the treatment with non-dihydropyridine calcium channel blockers associated with constipation (25%) and bradycardia. In some cases, CCB increases the tendency of gastrointestinal bleeding, probably in older patients due to inhibition of platelet aggregation [125]. The calcium channel blocker is used in not only the treatment of Hypertension but also some other vasospasm-related diseases such as Raynaud’s phenomenon, migraine and cluster headaches, high-altitude pulmonary edema, and even premature labor due to its vasodilation effect [126].
2.3. Diuretic
Diuretics are the most frequently used to treat Hypertension [127]. It reduces renal Na+ and water reabsorption (increase Na+ and water excretion) through direct action at various tubular sites of the nephrons [128]. As the class of antihypertensive drugs, diuretics are the second most prescribed drug [129]. JNC-8 [26] recommends diuretics as first-line therapy for Hypertension, particularly emphasizing thiazide-type diuretics. Most recent European and American guidelines also recommend thiazide-type and thiazide-like diuretics as the first-line drugs in treating Hypertension [130]. The European Society of Cardiology and European Society of Hypertension (ESC/ESH) [42] guidelines recommend thiazide diuretics should be regarded as equally effective to beta-blockers, calcium antagonists, ACE inhibitors, and angiotensin receptor blockers for the initiation and maintenance of antihypertensive treatment. for more than 50 years, thiazide diuretics have been the mainstay treatment option for Hypertension. In the USA and Western Europe, Thiazides are among the most often prescribed antihypertensive medications, accounting for around 30% of prescriptions [130].
A thiazide diuretic (Chlorothiazide, Hydrochlorothiazide (HCTZ), Bendroflumethiazide (BDTZ), Polythiazide, Methyclothiazide) contain both benzothiadiazole ring and sulfonamide moiety. in contrast, thiazide like diuretics (Chlorthalidone (CLTD), Metolazone, indapamide (INDA), Xipamide) do not present benzothiazole core but contain sulfonamide moiety [131]. Thiazides act as diuretics due to the inhibition of sodium reabsorption by blocking Na+ /Cl- cotransporter (NCC) located on the apical membrane in the renal distal convoluted tubule where approximately 5-10 % filtered loaded sodium reabsorption occurs [132,133]. The exact mechanism of thiazide diuretic by which chronically lower blood pressure remains unclear despite investigation of more than 50 years. Hypothetically, it is believed that thiazide’s antihypertensive effect is shown by decreasing total peripheral resistance by its endothelial or vascular smooth muscle-mediated direct vasodilatory effect via opening the calcium-activated potassium (KCA) channel [134]. In Comparison, CLTD is 1.5-2 times as potent as HCTZ [135]. CLTN prolongs the action, on average, for 2 to 3 days due to the reservoir into red blood cells [129]. BDTZ 1.25 mg/day showed a 24-hour antihypertensive effect by 11/7 mmHg with no clinically significant effects on potassium and urate [136]. meta-analysis of dose-response relationships of HCTZ, BDTZ, and CLTD conducted in 4683 subjects in over 53 comparison arms found that the potency series was BDTZ > CLTD > HCTZ and to reduce serum potassium by 0.4mmol/L, the doses needed were 4.2, 11.9 and 40.5 mg respectively [137].
Thiazide like a diuretic, INDA reduces systolic blood pressure about 54% more than HCTZ without evidence for greater adverse effects. Along with its diuretic properties, INDAP also shows an antihypertensive effect via a calcium antagonist like vasorelaxant effect [130]. The excretion of calcium ions is reduced by 40%-50% with long-term thiazide therapy [127]. In addition to their antihypertensive effect, thiazide prevents the development of osteoporosis and bone fractures [138]. Thiazide diuretics may raise LDL cholesterol levels by 5% to 15%, total cholesterol level by 12%, and triglyceride by 10%; high-density lipoprotein (HDL) cholesterol usually remains stable.
Thiazide induces more frequent Impotence in contrast with other diuretics [139,140]. The two most frequent side effects associated with thiazide diuretics are hypokalemia and hyponatremia [141]. Thiazide diuretics have been associated with other side effects, such as hyperuricemia, which may increase the risk of developing gout, Hyperglycemia leads to impaired glucose tolerance and may cause the induction of new onset of diabetic and some allergic experiences such as headaches, rash, hives, swelling of the mouth and lips, wheezing or trouble breathing, asthma attack, and anaphylaxis due to sulfonamide moiety [142]. Prolonged use of thiazide diuretics was associated with skin cancer risk [143]. All adverse effects are dose-dependent and can be minimized by using lower doses [130].
Loop diuretics produce diuresis and natriuresis by blocking the Na+-K+-2Cl− cotransporter (NKCC2) on the apical side of epithelial cells in the loop of Henle [144]. In vitro study shows that the diuretics frusemide does not show direct arterial vasodilator or anti-vasoconstrictor but has a direct venodilator effect, mediated by local vascular prostaglandin synthesis [145]. Loop diuretics example is furosemide (formerly frusemide), bumetanide, torsemide, piretanide, azosemide, ethacrynic acid, indacrinone, muzolimine, ozolinone, xipamide, and tienilic acid. Although loop diuretics show modest blood pressure diminishing efficacy, the estimated was systolic 7.9 (-10.4 to -5.4) mmHg and diastolic -4.4 (-5.9 to -2.8) mmHg [146]. Still, JNC 8 [26], the European Society of Hypertension, and the 2018 European Society of Cardiology guidelines (ESH/ESC) [42], recommend that Loop diuretics are not often the preferred choice for treating hypertension, due to limited outcome evidence. They are regarded as less effective than thiazide diuretics in decreasing blood pressure. Therefore, its principal therapeutic application lies in treating patients with edema-associated illnesses such as congestive heart failure (HF), cirrhosis with ascites, and nephritic edema [128,147,148]. “Hyponatremia, Hypochloremia, Hypomagnesemia, metabolic alkalosis, Hypokalemia, prerenal azotemia, dehydration, Hypercholesterolemia, postural Hypotension, Hyperuricemia, gout, Hypertriglyceridemia headache, restlessness, Dizziness, vertigo, and syncope are the common adverse effect of loop diuretics therapy. Other adverse reactions include skin photosensitivity, myalgias, interstitial nephritis, tinnitus, ototoxicity, deafness, and muscle soreness in patients with renal failure who receive high doses” [149]
Potassium-sparing diuretics, such as eplerenone and spironolactone, function as antagonists’ mineralocorticoid receptors (MRs), which are found in the principal cells of nephrons. Both drugs show antihypertensive efficacy in patients with low-renin or resistant Hypertension. In resistant hypertensive patients with or without primary aldosteronism, adding a low dose of Spironolactone (12.5-25 mg/day) to a multidrug regimen that includes an ACEI or ARB, produces a mean decrease in Systolic/diastolic BP 21/10 and 25/12 mmHg at 6 weeks and 6 months of treatment, respectively [128]. Despite these benefits, an important limiting factor for the long-term utility of Spironolactone has been its poor tolerability, especially in male patients at higher doses. The incidence of gynecomastia with concomitant sexual dysfunction in men increases from 10% to 30% with daily doses of 25 mg and higher [150].
3. Clinical Trial Evaluation
3.1. Perindopril/Indapamide vs. Placebo (PROGRESS TRAIL)
The perindopril protection against recurrent stroke study (PROGRESS) [151], randomized 6105 patients (mean age 64 years, male 70%, Asian 39%, history of ischemic stroke 71%, previous Cerebral hemorrhage 11%, transient ischemic attack 22%, Diabetes 12.5%, Coronary heart disease 16%, Current smoker 20% and Hypertension 48%) with a history of stroke or transient ischemic attack, of whom 3051 patients received perindopril (4mg/day) associate with indapamide (2-2.5 mg/day) in 58% of patient and 3054 patients received placebo. After a mean follow-up 3.9 years, Combination therapy with perindopril plus indapamide reduced blood pressure by 12/5 mm Hg while perindopril along blood pressure reduced by 5/3 mm Hg. The active treatment shows a 28% relative risk reduction in the primary endpoint of stroke (10% in active vs. 14% in placebo, HR 0.72, [95% CI 17-38%]; p<0·0001). The active treatment also showed 26% relative risk reduction in the secondary endpoint of the major vascular event (15% in active vs. 20 % in placebo, HR 0.74, [95% CI 16 to 34%]; p<0.001) and 4% relative risk reduction of death (41% in active vs. 43% in placebo, HR 0.91, [95% CI –12-18%]).
3.2. Perindopril/Indapamide vs. Placebo (ADVANCE TRAIL)
The Action in Diabetes and Vascular Disease: preterAx and diamicroN-MR Controlled Evaluation (ADVANCE) [152], randomized 11140 patients (mean age 66 years, male 57%, History of major macrovascular disease 32%, History of myocardial infarction 12%, History of major microvascular disease10%, History of stroke 9%, current smoker 15%, Microalbuminuria 26% and Hypertension 69%) with type 2 diabetes, of whom 5569 patients received perindopril (2-4 mg/d) plus indapamide (0.625-1.25 mg/d) and 5571 patients received placebo. After a mean follow-up 4.3 years, the active therapy had a mean reduction in blood pressure 5·6/2.2mm Hg. The relative risk of a major macrovascular or microvascular event was reduced by 9% (15·5% in active vs. 16·8% in placebo, HR 0·91, [95% CI 0·83–1·00]; p=0·04). The relative risk of death from cardiovascular disease was reduced by 18% (3·8% active vs. 4·6% placebo; HR 0·82, [95% CI 0·68-0·98]; p=0·03) and death from any cause was reduced by 14% (7·3% in active vs. 8·5% in placebo, HR 0·86, [95% CI 0·75-0·98], p=0·03). the relative risk of the total coronary event was reduced by 14% (8.4% in active vs. 9.6% in placebo, HR 0.86, [95% CI 0.76-0.98]; P=0.02). The relative risk of development of microalbuminuria was reduced by 21% (19.6% in active vs. 23.6% in placebo, HR 0.79, [95%CI 0.73-0.86]; P<0.0001), and the relative risk of Visual deterioration was reduced by 5% (43.9% in active vs. 45.1% in placebo, [95% CI 0.9-1.1]; P=0.1).
3.3. Perindopril/Indapamide vs. Placebo (HYVET TRAIL)
The Hypertension in the Very Elderly Trial (HYVET) [153], randomized 3845 elderly patients (mean age 84 years, male 40%, history of cardiovascular disease 12%, diabetes 7%, current smoker 7%, Hypertension 89.9% and heart failure 2.9%) with Hypertension, of whom 1933 patients received sustain release indapamide (1.5 mg/d) associated with perindopril (2-4 mg/d) in 73.4% of patients, and 1912 patients received placebo. After a mean follow-up 2.1 years, the mean blood pressure while sitting was 15.0/6.1 mm Hg lower in the active-treatment group than in the placebo group. Active treatment was associated with a 30% reduction in the rate of fatal or nonfatal stroke ([95% CI −1-51%]; P=0.06). The active therapy shows a 39% reduction in the rate of death from stroke ([95% CI 1-62]; P=0.05) and in the rate of death from any cause reduced by 21% ([95% CI 4-35%]; P=0.02). The active therapy also shows that the rate of death from cardiovascular causes reduced by 23% ([95% CI −1-40%]; P=0.06), and a 64% reduction in the rate of heart failure ([95% CI 42-78%]; P<0.001.
3.4. Losartan/Hydrochlorothiazide vs. Atenolol/Hydrochlorothiazide (LIFE TRAIL)
The Losartan Intervention For Endpoint Reduction (LIFE) [154] randomized 9193 patients (mean age 67 years, male 46%, white origin 92%, black origin 6%, diabetes 13%, any vascular disease 25%, and current smoker 16%) with essential Hypertension and left ventricular hypertrophy, of whom 4605 patients received losartan(50-100mg/d) plus hydrochlorothiazide (12.5-25 mg/d) for 72% of the total follow up time and 4588 patients receive Atenolol plus hydrochlorothiazide (12.5-25 mg/d) for 70% of the total follow up time. After a mean follow-up 4.8 years, both group’s blood pressure was reduced by 30·2/16·6 mm Hg in the losartan-based group and 29·1/16·8 mm Hg in Atenolol based groups. The study shows the primary endpoint of cardiovascular mortality, myocardial infarction, and stroke for both groups was not significantly different (11% in losartan base vs. 13% atenolol base, HR 0·87, [95%CI 90·77-0·98]; P=0.021). The secondary endpoint of stoke occurred at 5% in patients with losartan base and 7% in patients with atenolol base (HR 0.75, [95% CI 0.63-0.89]; P=0.001). New onset of diabetes occurred at 6% in patients with losartan-based and 8% in patients with Atenolol-based (HR 0.75, [95% CI 0.63-0.88]; P=0.001). The atenolol-base-receiving patients died from cardiovascular causes more than losartan-based therapy (4.4% in losartan base vs. 5% in atenolol base, HR 0·89, [95% CI 0·73-1·07]; p=0·206).
3.5. A Calcium Antagonist vs. Non-Calcium Antagonist (INVEST TRAIL)
The International Verapamil-Trandolapril Study (INVEST) [155], randomized 22576 patients (mean age 66 years, male 48%, white race 48%, Hispanic race 36%, black race 13%, hypercholesterolemia 56%, diabetes 28%, peripheral vascular disease 12%, heart failure 6%, previous stoke 5%, renal impairment 2% and current smoker 12%) with Hypertension and coronary artery disease(CAD), of whom 11267 patients received sustain release verapamil (120-480 mg/d) plus trandolapril (0.5-8 mg/d) and/or hydrochlorothiazide(12.5-100 mg/d) and 11309 patients received Atenolol (25-200 mg/d) plus hydrochlorothiazide(12.5-100mg/day) and/or trandolapril(0.5-8 mg/d). After a mean follow-up 2.7 years, the calcium antagonist (CAS) and non-calcium antagonist strategies (NCAS) effectively reduced blood pressure by 18.7/10 mmHg and 19/10.2 mmHg, respectively. The study also shows that both CAS and NCAS were effective in reducing the occurrence of the primary outcome of death from any cause and nonfatal myocardial infarction and stroke (9.93% in CAS vs10.17% in NCAS, RR 0.98, [95% CI 0.9-1.06]; P=0.57). the study shows that the development of new-onset diabetes is more significant in NCAS than in CAS (7.03% in CAS vs. 8.23% in NCAS, RR 0.85, [95% CI 0.77-0.95]). Both groups show some adverse effects such as angina (2.32% in CAS vs. 2.02% in NCAS), coronary artery bypass graft/percutaneous coronary intervention (2.49% in CAS vs. 2.43% in NCAS), cough (1.78% in CAS vs. 1.34%in NCAS), constipation (1.73% in CAS vs 0.13% in NCAS), and symptomatic bradycardia (0.66% in CAS vs. 1.26% in NCAS).
3.6. Amlodipine-Based Regiment vs. Atenolol Based Regiment (ASCOT-BPLA TRAIL)
The Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA) [156], randomized 19257 patients (mean age 63 years, male 77%, white race 95%, diabetes 27%, previous stroke or transient ischemic attack 11%, Left-ventricular hypertrophy 22%, peripheral vascular disease 6% and current smoker 33%) with Hypertension, of whom 9639 patients received amlodipine based regiment (AMBR), amlodipine(5-10 mg/day) associated with perindopril (4-8 mg/d) for 50% of the total follow up time and 9618 patients received Atenolol based regiment(ABR), Atenolol (50-100 mg/d), plus Bendroflumethiazide (1.25-2.5 mg/d) for 55% of the total follow up time. After a median follow-up 5.5 years, both therapeutic regimens reduce blood pressure, obtaining an average of 26.6/16.6 mmHg throughout the trial. The study obtained the reduction of the primary endpoint of nonfatal myocardial infarction and fatal coronary heart disease (CHD) for both groups was similar (5% vs. 5%, HR 0.90, [95% CI 0·79-1·02]; p= 0·1052). The reduction of the secondary endpoint of nonfatal myocardial infarction (excluding silent) plus nonfatal coronary disease, cardiovascular mortality, and fatal and nonfatal stroke for both groups were not significantly different. The Development of diabetes mellitus was more in patients who received Atenolol based regimen than an amlodipine-based regimen (6% in AMBR vs. 8% in ABR, HR 0.7,[95% CI 0·63-0·78,] P<0.0001).some adverse effect was developed for both group including bradycardia(0.4% in AMBR vs. 6% in ABR, p<0.0001), cough(19% in AMBR vs. 8% in ABR, p<0.0001), dyspnea (6% in AMBR vs. 10% in ABR, p<0.0001), Dizziness (12% in AMBR vs. 16% in ABR, p<0.0001), joint swelling (14% in AMBR vs. 3% in ABR, p<0.0001), edema peripheral (23% in AMBR vs. 6% in ABR, p<0.0001).
3.7. Benazepril/Amlodipine vs. Benazepril/Hydrochlorothiazide (ACCOMPLISH TRAIL)
The Avoiding Cardiovascular Events through Combination Therapy in Patients Living with Systolic Hypertension (ACCOMPLISH) [157], randomized 11506 patients (mean age 68.4 years, male 60.5%, white race 83.5%, black race 12.3%, dyslipidemia 74.2%, diabetes mellitus 60.4 % previous coronary revascularization 35.8%, obesity 49.6%, left ventricular hypertrophy 13.2%, previous stoke 13%, renal disease 6.1% and current smoker 11.3%) with Hypertension, of whom 5744 patients received benazepril (20-40 mg/d) plus amlodipine(5-10 mg/d) and 5762 patients received benazepril (20-40 mg/d) plus hydrochlorothiazide (12.5-25 mg/d). After a mean follow-up 3 years, both groups had excellent blood pressure control. Mean blood pressures after dose adjustment were 131.6/73.3 mm Hg in the benazepril–amlodipine group and 132.5/74.4 mm Hg in the benazepril–hydrochlorothiazide group. The benazepril plus amlodipine receiving group was superior to the benazepril–hydrochlorothiazide receiving group in reducing primary endpoint of cardiovascular events(fatal and nonfatal stroke, fatal and nonfatal myocardial infarction, Hospitalization for unstable angina, Resuscitation after sudden cardiac arrest and Coronary revascularization procedure) and death from cardiovascular causes(9.6% in amlodipine based vs. 11.8% in hydrochlorothiazide based), representing an absolute risk reduction with benazepril–amlodipine therapy of 2.2% and a relative risk reduction of 19.6% (HR 0.8 [95% CI 0.72–0.90]; P < 0.001). the reduction of the secondary endpoint of death from cardiovascular causes, nonfatal myocardial infarction, and nonfatal stroke) was not significantly difference (HR 0.79,[95% CI 0.67-0.92]; P=0.002).Both groups show some adverse effects, including Dizziness (20.7% in amlodipine based vs. 25.4% in hydrochlorothiazide-based), Peripheral edema (31.2% in amlodipine based vs. 13.4%), Dry cough (20.5% in amlodipine based vs. 21.2% in hydrochlorothiazide based), and Hypotension (2.5% in amlodipine based vs. 3.6% in hydrochlorothiazide based).
3.8. Benazepril/Amlodipine vs. Benazepril/Hydrochlorothiazide (ACCOMPLISH TRAIL)
The Avoiding Cardiovascular Events through Combination Therapy in Patients Living with Systolic Hypertension (ACCOMPLISH) [158], randomized 11506 patients (male 60.5%, black origin 12.27%, white origin 83.39%, mean age 68 years, Serum creatinine 139·7 μ mol/L with chronic kidney disease, 81·33 μ mol/L without chronic kidney disease, urinary albumin-to-creatinine ratio 28·8 mg/mmol with chronic kidney disease and 8·7 mg/mmol without kidney disease) with Hypertension, of whom 5744 patients received benazepril(20-40mg/d) and amlodipine (5 mg/d) and 5762 patient receive benazepril (20-40mg/d) plus hydrochlorothiazide (12.5 mg/d). After a mean follow-up 2.9 years, both groups showed excellent blood pressure control; mean blood pressures after dose adjustment were 131.6/73.3 mm Hg in the benazepril–amlodipine group and 132.5/74.4 mm Hg in the benazepril–hydrochlorothiazide group. The benazepril plus amlodipine receiving group was superior to the benazepril–hydrochlorothiazide receiving group in reducing the primary endpoint of progression of chronic kidney disease (1.97% in amlodipine based vs. 3.62% in hydrochlorothiazide based, HR 0·52, [95% CI 0·41–0·65]; p<0·0001). the adverse event in patients with chronic kidney disease, peripheral edema was more frequent in benazepril plus amlodipine than in benazepril plus hydrochlorothiazide (33·7% in amlodipine-based vs. 16·0% in hydrochlorothiazide based, p< 0.0001. The study also shows that in patients with chronic kidney disease, angioedema was more frequent in the benazepril plus amlodipine group than in the benazepril plus hydrochlorothiazide group (1.6% in amlodipine based vs. 0.4% in hydrochlorothiazide based, p = 0.15). In patients without chronic kidney disease, Dizziness, hypokalemia, and Hypotension were more frequent in the benazepril plus hydrochlorothiazide group than in the benazepril plus amlodipine group.

Table 1.
Pharmacokinetic properties of the hypertensive drug.
| class | Cmax | F | T1/2 | VD | Proteinbinding | CLr | Refer-ence |
|---|---|---|---|---|---|---|---|
|
CCB Amlodipine |
6-8h |
64% |
40-50h |
21L/kg |
98% |
0.23-0.4L/h/kg |
[159] |
| Felodipine | 0.5-5h | 15% | 25 h | 10 L/kg | >99 | 1-1.5 L/min | [160] |
| Verapamil | 2.2h | 20% | 2.7-4.8h | 310-406 L | 90% | 875 ml/min | [161] |
|
ARB Olmesartan medoxomil/ Olmesartan |
1.7–2.5h |
26% |
15 h |
35L |
99.7% |
1.31 L/h |
[162, 163] |
| valsartan | 2h | 23% | 6.1h | 17L | 85-99% | 2.2 L/h | [164] |
| Telmisartan | 1h | 43% | 24h | 500L | >99% | >800ml/min | [165] |
| Candesartan cilexetil/candesartan | 3,5-6h | 40% | 3.5-11h | 0.13L/kg | 99.5% | 0.0222 L/h/kg | [166, 167] |
| Eprosartan | 1-2h | 13% | 5-9h | 13L | 98% | 130ml/min | [168] |
| Irbesartan | 1.3-3h | 60-80% | 11-18h | 53-93L | 90% | 167ml/min | [166] |
| Losartan | 1-2h | 33% | 1.7-2.1h | 34.4 ± 17.9L | 98.6–98.8% | 4.3-5.6L/h | [169] |
|
ACEI Benazepril/Benazeprilat |
1.5h |
37% |
22.3h |
8.7L |
95% |
1.4-1.7L/h |
[170] |
| Captopril | 0.75-1h | 65% | 2h | 0.8 L/kg | 23-31% | 0.7L/h/kg | [171] |
| Enalapril/ enalaprilat | 4h | 36-44% | 11h | 50% | 8-9.5L/h | [172,173] | |
| Fosinopril/Fosinoprilat | 2.8-3.1h | 25-29% | 11.5-12h | 9.8-10.6L | 95-99.8% | 1.55-2.35 L/h | [174] |
| Lisinopril | 8h | 20-28% | 12.6h | 24L | no | 6.36L/h | [175] |
| Quinapril/quinaprilat | 2.5h | 50-60% | 3.2h | 13.9L | 97% | 68 ml/min. | [176] |
| Diuretics hydrochlorothiazide |
1.5-4h |
60-70% |
5.6-14.8h |
275.3 L |
40-68% |
[177] |
Table 2.
Commonly prescribed combination drug for Hypertension.
| Combination type | Dose (mg) | Trade name | Cost |
|---|---|---|---|
|
CCB + ACEI Amlodipine-benazepril Hydrochloride |
2.5/10, 5/10, 5/20, 10/20 | Lotrel | $14 ($215)- $16 ($390) |
| Enalaprilmaleate-felodipine | 5/5 | Lexxel | |
| Trandolapril-verapamil | 2/180, 1/240, 2/240, 4/240 | Tarka | $47 ($185)- $65 ($185) |
|
CCB + ARB Amlodipine/Olmesartan medoxomil |
5/20,5/40,10/20,10/40 |
Azor |
$23 ($280)- $28 ($350) |
| Amlodipine/Valsartan |
5/160,320/5,10/160,10/320 | Exforge | $20 ($270)- $25 ($385) |
| Amlodipine/Telmisartan | 5/40,5/80,10/40,10/80 | Twynsta | $50 (NA)- $55 ($240) |
|
Diuretic + ACEI Benazepril-hydrochlorothiazide |
5/6.25, 10/12.5, 20/12.5, 20/25 | Lotensin HCT | $21 (NA)- $24 (NA) |
| Captopril-hydrochlorothiazide | 25/15, 25/25, 50/15, 50/25 | Capozide | |
| Enalapril-hydrochlorothiazide | 5/12.5, 10/25 | Vaseretic | $10 (NA)- $10 ($395) |
| Fosinopril-hydrochlorothiazide | 10/12.5, 20/12.5 | Monopril/HCT | |
| Lisinopril-hydrochlorothiazid | 10/12.5, 20/12.5, 20/25 | Prinzide, Zestoretic | $4 ($400)- $6 ($400) |
| Moexipril-hydrochlorothiazide | 7.5/12.5, 15/25 | Uniretic | |
| Quinapril-hydrochlorothiazide | 10/12.5, 20/12.5, 20/25 | Accuretic | $17 ($150) |
|
Diuretic + ARB Candesartan-hydrochlorothiazide |
16/12.5, 32/12.5,32/25 |
Atacand HCT | $48 ($150)- $50 ($165) |
| Eprosartan-hydrochlorothiazide | 600/12.5, 600/25 | Teveten-HCT | |
| Irbesartan-hydrochlorothiazide | 150/12.5, 300/12.5 | Avalide | $15 ($235)- $20 ($255) |
| Losartan-hydrochlorothiazide | 50/12.5, 100/12.5,100/25 | Hyzaar | $4 ($130)- $9 ($175) |
| Olmesartan medoxomil-hydrochlorothiazide | 20/12.5,40/12.5,40/25 | Benicar HCT | $14 ($225)- $16 ($310) |
| Telmisartan-hydrochlorothiazide | 40/12.5, 80/12.5 | Micardis-HCT | $47 ($220) |
| Valsartan-hydrochlorothiazide | 80/12.5, 160/12.5, 160/25,320/12.5,320/25 | Diovan-HCT | $14 ($270)- $18 ($420) |
4. Pharmacological Treatment Strategy
Current hypertension guidelines suggest treatment strategies for the proper management of Hypertension. The treatment strategy is shown in (Figure 5). on the left side British Hypertension Society guidelines recommendation [180], in the middle NICE guidelines [25], and on the right side the Japanese Society of Hypertension Guidelines recommendation [181].
5. Conclusions
Hypertension is the most common and significant risk factor for cardiovascular disease, and the prevalence rate is continuously increasing. Successful treatment of Hypertension most often requires a combination of different classes of antihypertensive drugs. The current guidelines suggest multiple antihypertensive agents are necessary to reach the target blood pressure goal. Although the combination of angiotensin system inhibitor with diuretics or calcium channel blocker is preferable, the ACCOMPLISH clinical trial shows angiotensin system inhibitor plus calcium channel blocker was superior to angiotensin system inhibitor plus diuretics in reducing the primary endpoint of a cardiovascular event, death from cardiovascular causes and progression of chronic kidney disease. Angiotensin-converting enzyme inhibitors inhibit the formation of bioactive angiotensin Ⅱ, and angiotensin receptor blockers block the AT1 receptor. Calcium channel blocker inhibits the influx of calcium ions into cells by blocking the L-type calcium channel. Diuretics show antihypertensive efficacy through the alteration of body fluid volume and electrolytes. Treatment of Hypertension initially started using a single agent, subsequently adding different classes of antihypertensive agents until reaching the target blood pressure goal. Four different classes of antihypertensive agents are often necessary to treat resistance hypertension.
Data Availability Statment
Restrictions apply to the availability of these data. Data can be made available upon reasonable request.
Funding
Self-funded.
Conflicts of Interest
The authors have no relevant financial or non-financial interests to disclose.
References
- Gaziano, T. , Reddy, K. S., Paccaud, F., Horton, S., & Chaturvedi, V. (2006). Cardiovascular disease. Disease Control Priorities in Developing Countries. 2nd edition.
- Nabel, E. G. (2003). Cardiovascular disease. New England Journal of Medicine, 349(1), 60-72.
- Kannel, W.B. Hypertension as a Risk Factor for Cardiac Events—Epidemiologic Results of Long-Term Studies. J. Cardiovasc. Pharmacol. 1993, 21 (Suppl. S2), S27–S37. [Google Scholar] [CrossRef] [PubMed]
- Snow, P.J.D.; Jones, A.M.; Daber, K.S. Coronary disease: a pathological study. Br. Heart J. 1955, 17, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A. Coronary heart disease: Overview. Lancet 1996, 348, S1–S4. [Google Scholar] [CrossRef]
- Libby, P.; Theroux, P. Pathophysiology of coronary artery disease. Circulation 2005, 111, 3481–3488. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, M., Oktay, A. A., Stewart, M. H., Milani, R. V., Ventura, H. O., & Lavie, C. J. (2020). Left ventricular hypertrophy and hypertension. Progress in cardiovascular diseases, 63(1), 10-21.
- Maganti, K.; Rigolin, V.H.; Sarano, M.E.; Bonow, R.O. Valvular Heart Disease: Diagnosis and Management. Mayo Clin. Proc. 2010, 85, 483–500. [Google Scholar] [CrossRef] [PubMed]
- Wijesurendra, R. S. , & Casadei, B. (2019). Mechanisms of atrial fibrillation. Heart.
- Wijesurendra, R.S.; Casadei, B. Atrial fibrillation: effects beyond the atrium? Cardiovasc. Res. 2015, 105, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Markus, H.S. Cerebral perfusion and stroke. J. Neurol. Neurosurg. Psychiatry 2004, 75, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Novak, V.; Chowdhary, A.; Farrar, B.; Nagaraja, H.; Braun, J.; Kanard, R.; Novak, P.; Slivka, A. Altered cerebral vasoregulation in hypertension and stroke. Neurology 2003, 60, 1657–1663. [Google Scholar] [CrossRef]
- Albright Jr, R. C. (2001, January). Acute renal failure: a practical update. In Mayo Clinic Proceedings (Vol. 76, No. 1, pp. 67-74). Elsevier.
- Bellomo, R.; Kellum, J.A.; Ronco, C. Defining acute renal failure: physiological principles. Intensiv. Care Med. 2003, 30, 33–37. [Google Scholar] [CrossRef]
- Luke, R. G. (1998). Chronic renal failure—a vasculopathic state. New England Journal of Medicine, 339(12), 841-843.
- Kjeldsen, S.E. Hypertension and cardiovascular risk: General aspects. Pharmacol. Res. 2018, 129, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.T.; Bundy, J.D.; Kelly, T.N.; Reed, J.; Kearney, P.M.; Reynolds, K.; Chen, J.; He, J. Abstract 16828: Global Disparities of Hypertension Prevalence and Control: A Systematic Analysis of Population-based Studies From 90 Countries. Circulation 2016, 134, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Fryar, C.D.; Ostchega, Y.; Hales, C.; Zhang, G.; Kruszon-Moran, D. Hypertension Prevalence and Control Among Adults: United States, 2015-2016. . 2017, 1–8. [Google Scholar]
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J. (2013). Epidemiology of hypertension. Clinical Queries: Nephrology, 2(2), 56-61.
- Giles, T.D.; Materson, B.J.; Cohn, J.N.; Kostis, J.B. Definition and Classification of Hypertension: An Update. J. Clin. Hypertens. 2009, 11, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Izzo, J. L. , Sica, D. A., & Black, H. R. (Eds.). (2008). Hypertension primer. Lippincott Williams & Wilkins.
- Beevers, G., Lip, G. Y., & O’Brien, E. (2001). The pathophysiology of hypertension. Bmj, 322(7291), 912-916.
- Messerli, F. H., Williams, B., & Ritz, E. (2007). Essential hypertension. The Lancet, 370(9587), 591-603.
- National Institute for Health and Care Excellence. (2019). Hypertension in adults: diagnosis and management. London: National Institute for Health and Care Excellence (UK).
- James, P. A. , Oparil, S., Carter, B. L., Cushman, W. C., Dennison-Himmelfarb, C., Handler, J.,... & Ortiz, E. (2014). 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). Jama, 311(5), 507-520.
- Watkins, P. J. , Drury, P. L., & Taylor, K. W. (1990). Diabetes and its management. Boston: Blackwell Scientific.
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K., Jafar, T. H., Nitsch, D., Neuen, B. L., & Perkovic, V. (2021). Chronic kidney disease. The lancet, 398(10302), 786-802.
- Webster, A. C., Nagler, E. V., Morton, R. L., & Masson, P. (2017). Chronic kidney disease. The lancet, 389(10075), 1238-1252.
- Levey, A. S., & Coresh, J. (2012). Chronic kidney disease. The lancet, 379(9811), 165-180.
- Cohn, J.N. Blood pressure and cardiac performance. Am. J. Med. 1973, 55, 351–361. [Google Scholar] [CrossRef]
- Loushin, M. K., Quill, J. L., & Iaizzo, P. A. (2015). Mechanical aspects of cardiac performance. Handbook of cardiac anatomy, physiology, and devices, 335-360.
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef]
- Michael, S.K.; Surks, H.K.; Wang, Y.; Zhu, Y.; Blanton, R.; Jamnongjit, M.; Aronovitz, M.; Baur, W.; Ohtani, K.; Wilkerson, M.K.; et al. High blood pressure arising from a defect in vascular function. Proc. Natl. Acad. Sci. 2008, 105, 6702–6707. [Google Scholar] [CrossRef]
- Beevers, G., Lip, G. Y., & O’Brien, E. (2001). The pathophysiology of hypertension. Bmj, 322(7291), 912-916.
- Hingorani, A.D.; Sharma, P.; Jia, H.; Hopper, R.; Brown, M.J. Blood Pressure and the M235T Polymorphism of the Angiotensinogen Gene. Hypertension 1996, 28, 907–911. [Google Scholar] [CrossRef]
- Canbakan, B. (2013). Rational approaches to the treatment of hypertension: drug therapy—monotherapy, combination, or fixed-dose combination?. Kidney international supplements, 3(4), 349-351.
- Guerrero-García, C., & Rubio-Guerra, A. F. (2018). Combination therapy in the treatment of hypertension. Drugs in context, 7.
- Chalmers, J. The Place of Combination Therapy in the Treatment of Hypertension in 1993. Clin. Exp. Hypertens. 1993, 15, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Gradman, A. H., Basile, J. N., Carter, B. L., Bakris, G. L., & American Society of Hypertension Writing Group. (2010). Combination therapy in hypertension. Journal of the American Society of Hypertension, 4(2), 90-98.
- Williams, B.; Mancia, G.; Spiering, W.; Rosei, E.A.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; De Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. The Task Force for the management of arterial hypertension of the European Society of Cardiology (ESC) and the European Society of Hypertension (ESH). Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef]
- Morris, L.S.; Schulz, R.M. Patient compliance—an overview. J. Clin. Pharm. Ther. 1992, 17, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, B. Patient compliance. New Engl. J. Med. 1973, 289, 249–252. [Google Scholar] [CrossRef]
- Salahuddin, A.; Mushtaq, M.; Materson, B.J. Combination therapy for hypertension 2013: An update. J. Am. Soc. Hypertens. 2013, 7, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Kalra, B.; Agrawal, N. Combination therapy in hypertension: An update. Diabetol. Metab. Syndr. 2010, 2, 44–11. [Google Scholar] [CrossRef] [PubMed]
- Gorostidi, M., & de la Sierra, A. (2013). Combination therapy in hypertension. Advances in therapy, 30, 320-336.
- Chaszczewska-Markowska, M.; Sagan, M.; Bogunia-Kubik, K. The renin-angiotensin-aldosterone system (RAAS) – physiology and molecular mechanisms of functioning. Postepy Hig. I Med. Doswiadczalnej 2016, 70, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Atlas, S.A. The Renin-Angiotensin Aldosterone System: Pathophysiological Role and Pharmacologic Inhibition. J. Manag. Care Pharm. 2007, 13, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, S. E. (2017). A brief introduction into the renin-angiotensin-aldosterone system: new and old techniques. The Renin-Angiotensin-Aldosterone System: Methods and Protocols, 1-19.
- Schweda, F. Salt feedback on the renin-angiotensin-aldosterone system. Pfl?gers Arch. Eur. J. Physiol. 2015, 467, 565–576. [Google Scholar] [CrossRef]
- Taugner, R., & Hackenthal, E. (1988). On the character of the secretory granules in juxtaglomerular epithelioid cells. International review of cytology, 110, 93-131.
- Barajas, L. (1979). Anatomy of the juxtaglomerular apparatus. American Journal of Physiology-Renal Physiology, 237(5), F333-F343.
- Schweda, F. Schweda, F., Friis, U., Wagner, C., Skott, O., & Kurtz, A. (2007). Renin release. Physiology, 22(5), 310-319.
- Trerattanavong, K. , & Chen, J. (2023). Biochemistry, Renin. In StatPearls. StatPearls Publishing.
- Morgan, L. , Pipkin, F. B., & Kalsheker, N. (1996). Angiotensinogen: molecular biology, biochemistry and physiology. The international journal of biochemistry & cell biology, 28(11), 1211-1222.
- Lu, H.; A Cassis, L.; Kooi, C.W.V.; Daugherty, A. Structure and functions of angiotensinogen. Hypertens. Res. 2016, 39, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Jeunemaitre, X.; Soubrier, F.; Kotelevtsev, Y.V.; Lifton, R.P.; Williams, C.S.; Charru, A.; Hunt, S.C.; Hopkins, P.N.; Williams, R.R.; Lalouel, J.-M.; et al. Molecular basis of human hypertension: Role of angiotensinogen. Cell 1992, 71, 169–180. [Google Scholar] [CrossRef]
- Wu, C.; Lu, H.; Cassis, L.A.; Daugherty, A. Molecular and Pathophysiological Features of Angiotensinogen: A Mini Review. Am. Chin. J. Med. Sci. 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M. E. , & Sigmund, C. D. (2006). Genetic basis of hypertension: revisiting angiotensinogen. Hypertension, 48(1), 14-20.
- Wu, C.; Lu, H.; Cassis, L.A.; Daugherty, A. Molecular and Pathophysiological Features of Angiotensinogen: A Mini Review. Am. Chin. J. Med. Sci. 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.; Murphy, T.J.; Alexander, R.W. Molecular biology of the renin-angiotensin system. Circulation 1993, 87, 1816–1828. [Google Scholar] [CrossRef] [PubMed]
- Sparks, M. A. , Crowley, S. D., Gurley, S. B., Mirotsou, M., & Coffman, T. M. (2014). Classical renin-angiotensin system in kidney physiology. Comprehensive Physiology, 4(3), 1201.
- Dorer, F.E.; Kahn, J.R.; Lentz, K.E.; Levine, M.; Skeggs, L.T. Hydrolysis of Bradykinin by Angiotensin-Converting Enzyme. Circ. Res. 1974, 34, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Pirahanchi, Y., & Sharma, S. (2019). Physiology, Bradykinin.
- Ng, K.K.F.; Vane, J.R. Conversion of Angiotensin I to Angiotensin II. Nature 1967, 216, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Erdös, E.G. Conversion of angiotensin I to angiotensin II. Am. J. Med. 1976, 60, 749–759. [Google Scholar] [CrossRef]
- Fountain, J. H. , Kaur, J., & Lappin, S. L. (2023). Physiology, renin angiotensin system. In StatPearls [Internet]. StatPearls Publishing.
- Peti-Peterdi, J.; Harris, R.C. Macula Densa Sensing and Signaling Mechanisms of Renin Release. J. Am. Soc. Nephrol. 2010, 21, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Unger, T. , Chung, O., Csikos, T., Culman, J., Gallinat, S., Gohlke, P.,... & Zhu, Y. Z. (1996). Angiotensin receptors. Journal of hypertension. Supplement: official journal of the International Society of Hypertension, 14(5), S95-103.
- Greindling, K.K.; Lassegue, B.; Alexander, R.W. Angiotensin Receptors and Their Therapeutic Implications. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Greindling, K.K.; Lassegue, B.; Alexander, R.W. Angiotensin Receptors and Their Therapeutic Implications. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 281–306. [Google Scholar] [CrossRef]
- Henrion, D. , Kubis, N., & Lévy, B. I. (2001). Physiological and pathophysiological functions of the AT2 subtype receptor of angiotensin II: from large arteries to the microcirculation. Hypertension, 38(5), 1150-1157.
- Padia, S.H.; Carey, R.M. AT2 receptors: beneficial counter-regulatory role in cardiovascular and renal function. Pfl?gers Arch. Eur. J. Physiol. 2013, 465, 99–110. [Google Scholar] [CrossRef]
- Volpe, M. , Musumeci, B., De Paolis, P., Savoia, C., & Morganti, A. (2003). Angiotensin II AT2 receptor subtype: an uprising frontier in cardiovascular disease?. Journal of hypertension, 21(8), 1429-1443.
- Kostenis, E.; Milligan, G.; Christopoulos, A.; Sanchez-Ferrer, C.F.; Heringer-Walther, S.; Sexton, P.M.; Gembardt, F.; Kellett, E.; Martini, L.; Vanderheyden, P.; et al. G-Protein–Coupled Receptor Mas Is a Physiological Antagonist of the Angiotensin II Type 1 Receptor. Circulation 2005, 111, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Oro, C.; Qian, H.; Thomas, W.G. Type 1 angiotensin receptor pharmacology: Signaling beyond G proteins. Pharmacol. Ther. 2007, 113, 210–226. [Google Scholar] [CrossRef]
- Thiriet, M. Signaling at the Cell Surface in the Circulatory and Ventilatory Systems; Springer Nature: Dordrecht, GX, Netherlands, 2012. [Google Scholar]
- Cat, A.N.D.; Touyz, R.M. Cell Signaling of Angiotensin II on Vascular Tone: Novel Mechanisms. Curr. Hypertens. Rep. 2011, 13, 122–128. [Google Scholar] [CrossRef]
- Abadir, P. M. , Periasamy, A., Carey, R. M., & Siragy, H. M. (2006). Angiotensin II type 2 receptor–bradykinin B2 receptor functional heterodimerization. Hypertension, 48(2), 316-322.
- Duka, A. , Duka, I., Gao, G., Shenouda, S., Gavras, I., & Gavras, H. (2006). Role of bradykinin B1 and B2 receptors in normal blood pressure regulation. American Journal of Physiology-Endocrinology and Metabolism, 291(2), E268-E274.
- Bernier, S.G.; Haldar, S.; Michel, T. Bradykinin-regulated Interactions of the Mitogen-activated Protein Kinase Pathway with the Endothelial Nitric-oxide Synthase. J. Biol. Chem. 2000, 275, 30707–30715. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Münzel, T. Organic Nitrate Therapy, Nitrate Tolerance, and Nitrate-Induced Endothelial Dysfunction: Emphasis on Redox Biology and Oxidative Stress. Antioxidants Redox Signal. 2015, 23, 899–942. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Michel, T. Subcellular Localization of Oxidants and Redox Modulation of Endothelial Nitric Oxide Synthase. Circ. J. 2012, 76, 2497–2512. [Google Scholar] [CrossRef] [PubMed]
- Stocco, D.M.; Clark, B.J. Regulation of the Acute Production of Steroids in Steroidogenic Cells*. Endocr. Rev. 1996, 17, 221–244. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Kim, J.; Soltys, S.; Koch, W.J. An adrenal β-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc. Natl. Acad. Sci. 2009, 106, 5825–5830. [Google Scholar] [CrossRef]
- Bollag, W. B. (2011). Regulation of aldosterone synthesis and secretion. Comprehensive physiology, 4(3), 1017-1055.
- Bollag, W. B. (2011). Regulation of aldosterone synthesis and secretion. Comprehensive physiology, 4(3), 1017-1055.
- Scott, J. H., Menouar, M. A., & Dunn, R. J. (2017). Physiology, aldosterone.
- Pearce, D.; Soundararajan, R.; Trimpert, C.; Kashlan, O.B.; Deen, P.M.; Kohan, D.E. Collecting Duct Principal Cell Transport Processes and Their Regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 135–146. [Google Scholar] [CrossRef]
- Sandgren, J. A. , Linggonegoro, D. W., Zhang, S. Y., Sapouckey, S. A., Claflin, K. E., Pearson, N. A.,... & Grobe, J. L. (2018). Fluid and Electrolyte Homeostasis: Angiotensin AT1A receptors expressed in vasopressin-producing cells of the supraoptic nucleus contribute to osmotic control of vasopressin. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 314(6), R770.
- Okuya, S.; Inenaga, K.; Kaneko, T.; Yamashita, H. Angiotensin II sensitive neurons in the supraoptic nucleus, subfornical organ and anteroventral third ventricle of rats in vitro. Brain Res. 1987, 402, 58–67. [Google Scholar] [CrossRef]
- Cuzzo, B. , Padala, S. A., & Lappin, S. L. (2023). Physiology, vasopressin. In StatPearls [Internet]. StatPearls Publishing.
- Hanoune, J. (2010). Vasopressin receptors, the signalling cascade and mechanisms of action. Perspectives on Vasopressin.
- Morla, L. , Edwards, A., & Crambert, G. (2016). New insights into sodium transport regulation in the distal nephron: Role of G-protein coupled receptors. World journal of biological chemistry, 7(1), 44.
- Severs, W.B.; Summy-Long, J. The role of angiotensin in thirst. Life Sci. 1975, 17, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimons, J. T. (1998). Angiotensin, thirst, and sodium appetite. Physiological reviews, 78(3), 583-686.
- Dibona, G. F. (2013). Sympathetic nervous system and hypertension. Hypertension, 61(3), 556-560.
- Dibona, G. F. (2004). The sympathetic nervous system and hypertension: recent developments. Hypertension, 43(2), 147-150.
- Esler, M. The sympathetic system and hypertension. Am. J. Hypertens. 2000, 13, 99S–105S. [Google Scholar] [CrossRef] [PubMed]
- Wnorowski, A. , & Jozwiak, K. (2014). Homo-and hetero-oligomerization of β2-adrenergic receptor in receptor trafficking, signaling pathways and receptor pharmacology. Cellular Signalling, 26(10), 2259-2265.
- Ecker, P. M. , Lin, C. C., Powers, J., Kobilka, B. K., Dubin, A. M., & Bernstein, D. (2006). Effect of targeted deletions of β1-and β2-adrenergic-receptor subtypes on heart rate variability. American Journal of Physiology-Heart and Circulatory Physiology, 290(1), H192-H199.
- Woo, A.Y.H.; Xiao, R.-P. β-Adrenergic receptor subtype signaling in heart: From bench to bedside. Acta Pharmacol. Sin. 2012, 33, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R. J. , Rockman, H. A., & Koch, W. J. (2000). Catecholamines, cardiac β-adrenergic receptors, and heart failure. Circulation, 101(14), 1634-1637.
- Stiles, G.L.; Caron, M.G.; Lefkowitz, R.J. Beta-adrenergic receptors: biochemical mechanisms of physiological regulation. Physiol. Rev. 1984, 64, 661–743. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D. Neural control of the circulation. Adv. Physiol. Educ. 2011, 35, 28–32. [Google Scholar] [CrossRef]
- Grassi, G. Renin–angiotensin–sympathetic crosstalks in hypertension: reappraising the relevance of peripheral interactions. J. Hypertens. 2001, 19, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Aldehni, F.; Tang, T.; Madsen, K.; Plattner, M.; Schreiber, A.; Friis, U.G.; Hammond, H.K.; Han, P.L.; Schweda, F. Stimulation of Renin Secretion by Catecholamines Is Dependent on Adenylyl Cyclases 5 and 6. Hypertension 2011, 57, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Messerli, F. H. , Bangalore, S., Bavishi, C., & Rimoldi, S. F. (2018). Angiotensin-converting enzyme inhibitors in hypertension: to use or not to use?. Journal of the American College of Cardiology, 71(13), 1474-1482.
- Uehara, Y.; Miura, S.-I.; Yahiro, E.; Saku, K. Non-ACE Pathway-induced Angiotensin II Production. Curr. Pharm. Des. 2013, 19, 3054–3059. [Google Scholar] [CrossRef]
- Hallberg, P.; Nagy, J.; Karawajczyk, M.; Nordang, L.; Islander, G.; Norling, P.; Johansson, H.-E.; Kämpe, M.; Hugosson, S.; Yue, Q.-Y.; et al. Comparison of Clinical Factors Between Patients With Angiotensin-Converting Enzyme Inhibitor–Induced Angioedema and Cough. Ann. Pharmacother. 2017, 51, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Robles, N. R. , Cerezo, I., & Hernandez-Gallego, R. (2014). Renin–angiotensin system blocking drugs. Journal of cardiovascular pharmacology and therapeutics, 19(1), 14-33.
- Hernández-Hernández, R.; Sosa-Canache, B.; Velasco, M.; Armas-Hernández, M.J.; Armas-Padilla, M.C.; Cammarata, R. Angiotensin II receptor antagonists role in arterial hypertension. J. Hum. Hypertens. 2002, 16, S93–S99. [Google Scholar] [CrossRef]
- Pantzaris, N.-D.; Karanikolas, E.; Tsiotsios, K.; Velissaris, D. Renin Inhibition with Aliskiren: A Decade of Clinical Experience. J. Clin. Med. 2017, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, H. Role of aliskiren in blood pressure control and renoprotection. Int. J. Nephrol. Renov. Dis. 2011, 4, 41–48. [Google Scholar] [CrossRef]
- Cooper, D. , & Dimri, M. (2020). Biochemistry, Calcium Channels.
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and Structure-Function Relationships of Voltage-Gated Calcium Channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yan, Z.; Li, Z.; Qian, X.; Lu, S.; Dong, M.; Zhou, Q.; Yan, N. Structure of the voltage-gated calcium channel Cav1.1 at 3.6 Å resolution. Nature 2016, 537, 191–196. [Google Scholar] [CrossRef]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of Voltage-Gated Calcium Channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef]
- Dolphin, A.C. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J. Physiol. 2016, 594, 5369–5390. [Google Scholar] [CrossRef]
- Shah, K.; Seeley, S.; Schulz, C.; Fisher, J.; Rao, S.G. Calcium Channels in the Heart: Disease States and Drugs. Cells 2022, 11, 943. [Google Scholar] [CrossRef]
- Hockerman, G.H.; Peterson, B.Z.; Johnson, B.D.; Catterall, W.A. Molecular determinants of drug binding and action on l-type calcium channels. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 361–396. [Google Scholar] [CrossRef] [PubMed]
- Neagoe, A. M. , Rexhaj, E., Grossman, E., & Messerli, F. H. (2019). Beta blockers and calcium channel blockers. Cardiovascular Hemodynamics: An Introductory Guide, 73-88.
- Weir, M.R. Calcium Channel Blockers: Their Pharmacologic and Therapeutic Role in Hypertension. Am. J. Cardiovasc. Drugs 2007, 7, 5–15. [Google Scholar] [CrossRef]
- Khalil, H. , & Zeltser, R. (2023). Antihypertensive medications.[Updated 2022 May 15]. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing.
- Elliott, W. J., & Ram, C. V. S. (2011). Calcium channel blockers. The Journal of Clinical Hypertension, 13(9), 687.
- Padilla, M.C.A.; Armas-Hernández, M.J.; Hernández, R.H.; Israili, Z.H.; Valasco, M. Update of Diuretics in the Treatment of Hypertension. Am. J. Ther. 2007, 14, 154–160. [Google Scholar] [CrossRef]
- Tamargo, J.; Segura, J.; Ruilope, L.M. Diuretics in the treatment of hypertension. Part 2: loop diuretics and potassium-sparing agents. Expert Opin. Pharmacother. 2014, 15, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Roush, G.C.; Sica, D.A. Diuretics for Hypertension: A Review and Update. Am. J. Hypertens. 2016, 29, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- McNally, R.J.; Morselli, F.; Farukh, B.; Chowienczyk, P.J.; Faconti, L. A review of the prescribing trend of thiazide-type and thiazide-like diuretics in hypertension: A UK perspective. Br. J. Clin. Pharmacol. 2019, 85, 2707–2713. [Google Scholar] [CrossRef] [PubMed]
- Liang, W. , Ma, H., Cao, L., Yan, W., & Yang, J. (2017). Comparison of thiazide-like diuretics versus thiazide-type diuretics: a meta-analysis. Journal of cellular and molecular medicine, 21(11), 2634-2642.
- Rapoport, R.M.; Soleimani, M. Mechanism of Thiazide Diuretic Arterial Pressure Reduction: The Search Continues. Front. Pharmacol. 2019, 10, 815. [Google Scholar] [CrossRef]
- Blowey, D. L. (2016). Diuretics in the treatment of hypertension. Pediatric nephrology, 31, 2223-2233.
- Duarte, J.D.; Cooper-DeHoff, R.M. Mechanisms for blood pressure lowering and metabolic effects of thiazide and thiazide-like diuretics. Expert Rev. Cardiovasc. Ther. 2010, 8, 793–802. [Google Scholar] [CrossRef]
- Carter, B. L. , Ernst, M. E., & Cohen, J. D. (2004). Hydrochlorothiazide versus chlorthalidone: evidence supporting their interchangeability. Hypertension, 43(1), 4-9.
- Wiggam, M. I. , Bell, P. M., Sheridan, B., Walmsley, A., & Atkinson, A. B. (1999). Low dose bendrofluazide (1.25 mg) effectively lowers blood pressure over 24 h: results of a randomized, double-blind, placebo-controlled crossover study. American journal of hypertension, 12(5), 528-531.
- Peterzan, M.A.; Hardy, R.; Chaturvedi, N.; Hughes, A.D. Meta-Analysis of Dose-Response Relationships for Hydrochlorothiazide, Chlorthalidone, and Bendroflumethiazide on Blood Pressure, Serum Potassium, and Urate. Hypertension 2012, 59, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Grossman, E., Verdecchia, P., Shamiss, A., Angeli, F., & Reboldi, G. (2011). Diuretic treatment of hypertension. Diabetes care, 34(Suppl 2), S313.
- Rockhold, R.W. Thiazide diuretics and male sexual dysfunction. Drug Dev. Res. 1992, 25, 85–95. [Google Scholar] [CrossRef]
- Krane, R. J., Goldstein, I., & de Tejada, I. S. (1989). Impotence. New England Journal of Medicine, 321(24), 1648-1659.
- Ravioli, S. , Bahmad, S., Funk, G. C., Schwarz, C., Exadaktylos, A., & Lindner, G. (2021). Risk of electrolyte disorders, syncope, and falls in patients taking thiazide diuretics: results of a cross-sectional study. The American Journal of Medicine, 134(9), 1148-1154.
- Akbari, P. , & Khorasani-Zadeh, A. (2023). Thiazide Diuretics. In StatPearls. StatPearls Publishing.
- Nochaiwong, S.; Chuamanochan, M.; Ruengorn, C.; Noppakun, K.; Awiphan, R.; Phosuya, C.; Tovanabutra, N.; Chiewchanvit, S.; Sood, M.M.; Hutton, B.; et al. Use of Thiazide Diuretics and Risk of All Types of Skin Cancers: An Updated Systematic Review and Meta-Analysis. Cancers 2022, 14, 2566. [Google Scholar] [CrossRef]
- Malha, L.; Mann, S.J. Loop Diuretics in the Treatment of Hypertension. Curr. Hypertens. Rep. 2016, 18, 1–10. [Google Scholar] [CrossRef]
- Pickkers, P.; Dormans, T.P.J.; Russel, F.G.M.; Hughes, A.D.; Thien, T.; Schaper, N.; Smits, P. Direct Vascular Effects of Furosemide in Humans. Circulation 1997, 96, 1847–1852. [Google Scholar] [CrossRef]
- Musini, V. M. , Rezapour, P., Wright, J. M., Bassett, K., & Jauca, C. D. (2015). Blood pressure-lowering efficacy of loop diuretics for primary hypertension. Cochrane Database of Systematic Reviews, (5).
- Blowey, D. L. (2016). Diuretics in the treatment of hypertension. Pediatric nephrology, 31, 2223-2233.
- Sica, D. A., Carter, B., Cushman, W., & Hamm, L. (2011). Thiazide and loop diuretics. The journal of clinical hypertension, 13(9), 639-643.
- Huxel, C. , Raja, A., & Ollivierre-Lawrence, M. D. (2023). Loop diuretics. In StatPearls [Internet]. StatPearls Publishing.
- Calhoun, D.A.; White, W.B. Effectiveness of the selective aldosterone blocker, eplerenone, in patients with resistant hypertension. J. Am. Soc. Hypertens. 2008, 2, 462–468. [Google Scholar] [CrossRef] [PubMed]
- PROGRESS Collaborative Group. (2001). Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6105 individuals with previous stroke or transient ischaemic attack. The Lancet, 358(9287), 1033-1041.
- Patel, A. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet 2007, 370, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Beckett, N.S.; Peters, R.; Fletcher, A.E.; Staessen, J.A.; Liu, L.; Dumitrascu, D.; Stoyanovsky, V.; Antikainen, R.L.; Nikitin, Y.; Anderson, C.; et al. Treatment of Hypertension in Patients 80 Years of Age or Older. New Engl. J. Med. 2008, 358, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Dahlöf, B.; Devereux, R.B.; E Kjeldsen, S.; Julius, S.; Beevers, G.; de Faire, U.; Fyhrquist, F.; Ibsen, H.; Kristiansson, K.; Lederballe-Pedersen, O.; et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002, 359, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Pepine, C. J. , Handberg, E. M., Cooper-DeHoff, R. M., Marks, R. G., Kowey, P., Messerli, F. H.,... & INVEST Investigators. (2003). A calcium antagonist vs a non–calcium antagonist hypertension treatment strategy for patients with coronary artery disease: the International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. Jama, 290(21), 2805-2816.
- Dahlöf, B.; Sever, P.S.; Poulter, N.R.; Wedel, H.; Beevers, D.G.; Caulfield, M.; Collins, R.; E Kjeldsen, S.; Kristinsson, A.; McInnes, G.T.; et al. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005, 366, 895–906. [Google Scholar] [CrossRef]
- Jamerson, K.; Weber, M.A.; Bakris, G.L.; Dahlöf, B.; Pitt, B.; Shi, V.; Hester, A.; Gupte, J.; Gatlin, M.; Velazquez, E.J. Benazepril plus Amlodipine or Hydrochlorothiazide for Hypertension in High-Risk Patients. New Engl. J. Med. 2008, 359, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; A Sarafidis, P.; Weir, M.R.; Dahlöf, B.; Pitt, B.; Jamerson, K.; Velazquez, E.J.; Staikos-Byrne, L.; Kelly, R.Y.; Shi, V.; et al. Renal outcomes with different fixed-dose combination therapies in patients with hypertension at high risk for cardiovascular events (ACCOMPLISH): a prespecified secondary analysis of a randomised controlled trial. Lancet 2010, 375, 1173–1181. [Google Scholar] [CrossRef]
- Meredith, P.A.; Elliott, H.L. Clinical Pharmacokinetics of Amlodipine. Clin. Pharmacokinet. 1992, 22, 22–31. [Google Scholar] [CrossRef]
- Edgar, B. , Lundborg, P., & Regårdh, C. G. (1987). Clinical pharmacokinetics of felodipine: a summary. Drugs, 34, 16-27.
- Hamann, S.R.; Blouin, R.A.; McAllister, R.G., Jr. Clinical Pharmacokinetics of Verapamil. Clin. Pharmacokinet. 1984, 9, 26–41. [Google Scholar] [CrossRef]
- Scott, L. J. , & McCormack, P. L. (2008). Olmesartan medoxomil: a review of its use in the management of hypertension. Drugs, 68, 1239-1272.
- Warner, G. T., & Jarvis, B. (2002). Olmesartan medoxomil. Drugs, 62, 1345-1353.
- Markham, A. , & Goa, K. L. (1997). Valsartan: a review of its pharmacology and therapeutic use in essential hypertension. Drugs, 54, 299-311.
- McClellan, K. J. , & Markham, A. (1998). Telmisartan. Drugs, 56(6).
- Israili, Z.H. Clinical pharmacokinetics of angiotensin II (AT1) receptor blockers in hypertension. J. Hum. Hypertens. 2000, 14 (Suppl. 1), S73–S86. [Google Scholar] [CrossRef]
- Gleiter, C.H.; Mörike, K.E. Clinical Pharmacokinetics of Candesartan. Clin. Pharmacokinet. 2002, 41, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Bottorff, M. B. , & Tenero, D. M. (1999). Pharmacokinetics of eprosartan in healthy subjects, patients with hypertension, and special populations. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy, 19(4P2), 73S-78S.
- A Sica, D.; Gehr, T.W.B.; Ghosh, S. Clinical Pharmacokinetics of Losartan. Clin. Pharmacokinet. 2005, 44, 797–814. [Google Scholar] [CrossRef] [PubMed]
- Balfour, J. A. , & Goa, K. L. (1991). Benazepril: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in hypertension and congestive heart failure. Drugs, 42, 511-539.
- Duchin, K.L.; McKinstry, D.N.; Cohen, A.I.; Migdalof, B.H. Pharmacokinetics of Captopril in Healthy Subjects and in Patients with Cardiovascular Diseases. Clin. Pharmacokinet. 1988, 14, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Gomez, H. J. , Cirillo, V. J., & Irvin, J. D. (1985). Enalapril: a review of human pharmacology. Drugs, 30, 13-24.
- Todd, P. A. , & Goa, K. L. (1992). Enalapril: a reappraisal of its pharmacology and therapeutic use in hypertension. Drugs, 43, 346-381.
- Murdoch, D. , & McTavish, D. (1992). Fosinopril: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in essential hypertension. Drugs, 43, 123-140.
- Goa, K. L. , Balfour, J. A., & Zuanetti, G. (1996). Lisinopril: a review of its pharmacology and clinical efficacy in the early management of acute myocardial infarction. Drugs, 52(4), 564-588.
- Kieback, A. G. , Felix, S. B., & Reffelmann, T. (2009). Quinaprilat: a review of its pharmacokinetics, pharmacodynamics, toxicological data and clinical application. Expert Opinion on Drug Metabolism & Toxicology, 5(10), 1337-1347.
- Commander, S.J.; Wu, H.; Boakye-Agyeman, F.; Melloni, C.; Hornik, C.D.; Zimmerman, K.; Al-Uzri, A.; Mendley, S.R.; Harper, B.; Cohen-Wolkowiez, M.; et al. Pharmacokinetics of Hydrochlorothiazide in Children: A Potential Surrogate for Renal Secretion Maturation. J. Clin. Pharmacol. 2020, 61, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Chobanian, A. V., Bakris, G. L., Black, H. R., Cushman, W. C., Green, L. A., Izzo Jr, J. L., ... & National High Blood Pressure Education Program Coordinating Committee. (2003). Seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. hypertension, 42(6), 1206-1252.
- Smith, D.K.; Lennon, R.P.; Carlsgaard, P.B. Managing Hypertension Using Combination Therapy. 2020, 101, 341–349.
- Williams, B.; Poulter, N.R.; Brown, M.J.; Davis, M.; McInnes, G.T.; Potter, J.F.; Sever, P.S.; Thom, S.M. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004, 328, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Umemura, S.; Arima, H.; Arima, S.; Asayama, K.; Dohi, Y.; Hirooka, Y.; Horio, T.; Hoshide, S.; Ikeda, S.; Ishimitsu, T.; et al. The Japanese Society of Hypertension Guidelines for the Management of Hypertension (JSH 2019). Hypertens. Res. 2019, 42, 1235–1481. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Drug action site of Angiotensin II synthesis pathway.
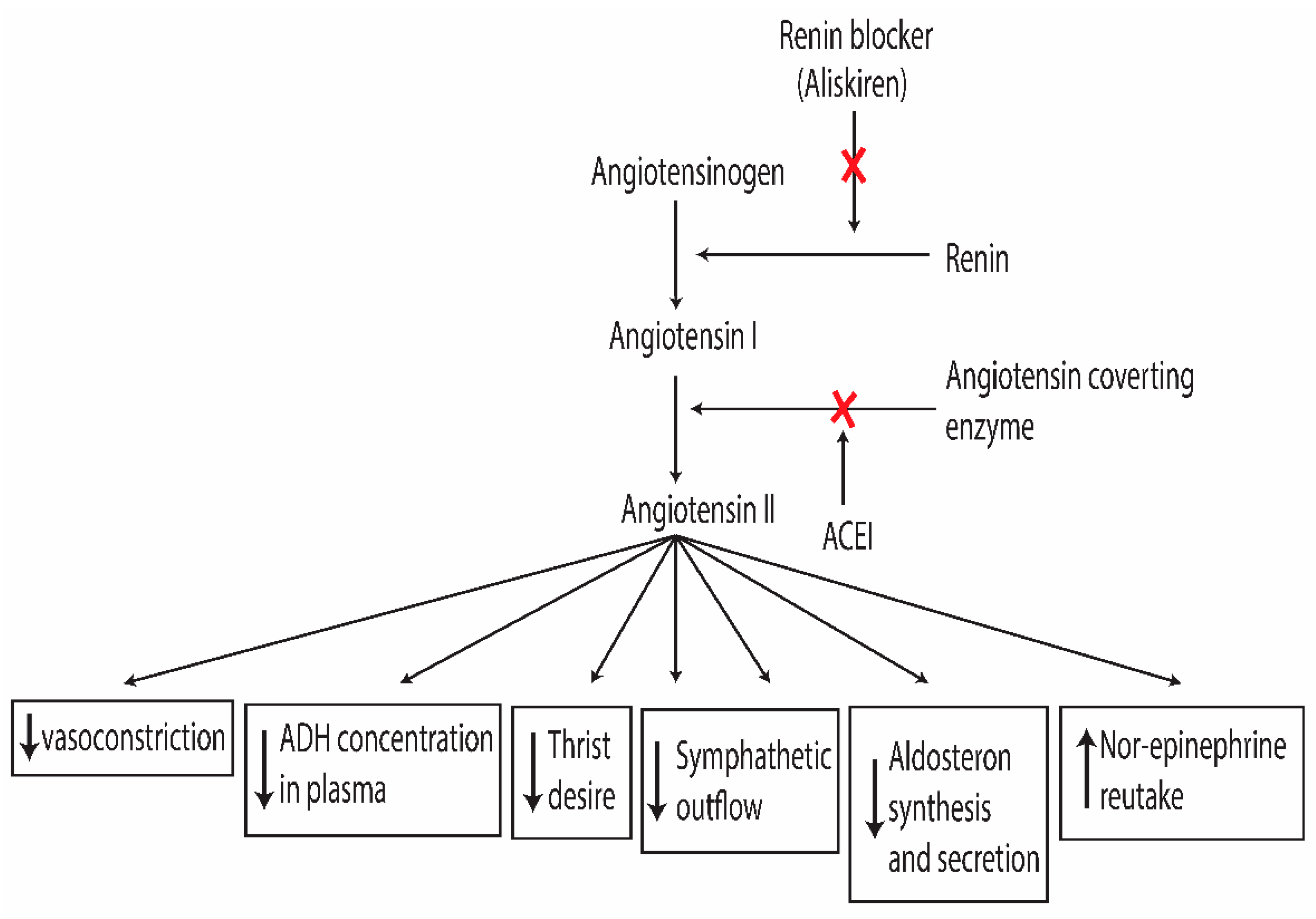
Figure 2.
Angiotensin system blocker reduces the synthesis of aldosterone synthesis and its effect on principle cell.
Figure 2.
Angiotensin system blocker reduces the synthesis of aldosterone synthesis and its effect on principle cell.
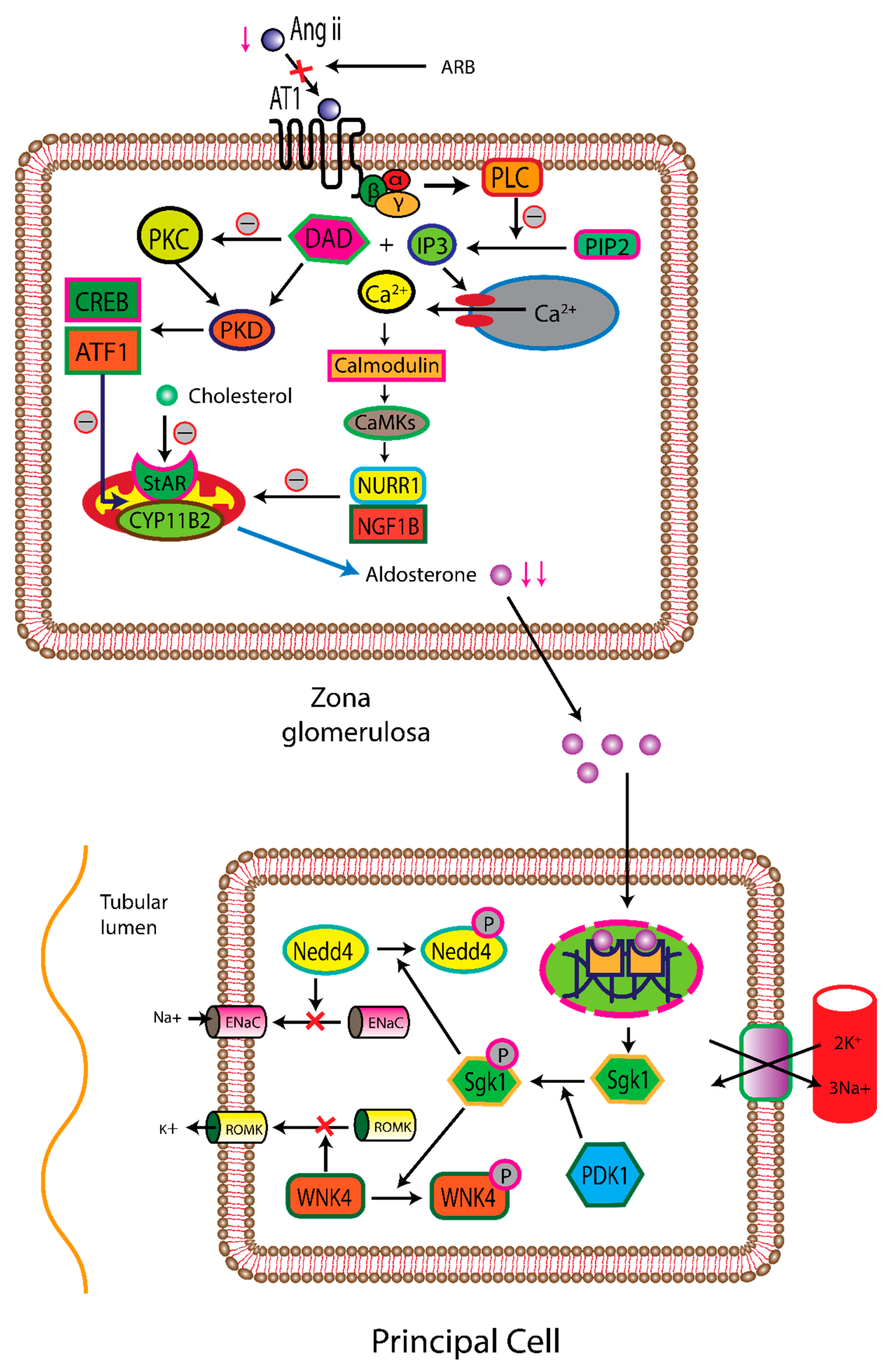
Figure 3.
By blocking of angiotensin system, reduce the effect of norepinephrine (NE), anti-diuretic hormone (ADH), and angiotensin Ⅱ on vascular smooth muscle, resulting in the reduction of vascular smooth muscle contraction. Muscle relaxation is facilitated by bradykinin.
Figure 3.
By blocking of angiotensin system, reduce the effect of norepinephrine (NE), anti-diuretic hormone (ADH), and angiotensin Ⅱ on vascular smooth muscle, resulting in the reduction of vascular smooth muscle contraction. Muscle relaxation is facilitated by bradykinin.
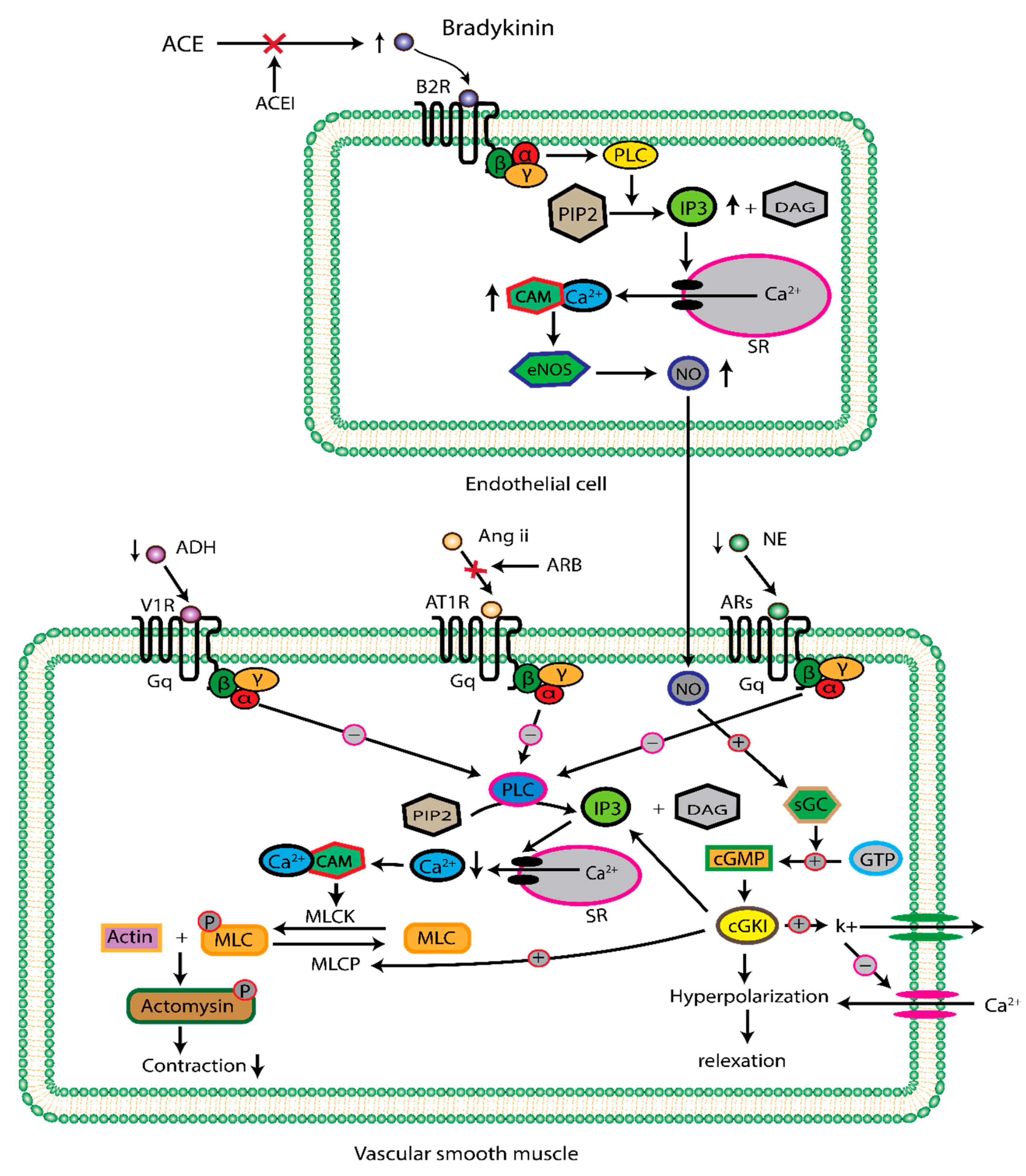
Figure 4.
The general mechanism of calcium channel blocker, resulting in vascular smooth muscle relaxation and reduced heart performance.
Figure 4.
The general mechanism of calcium channel blocker, resulting in vascular smooth muscle relaxation and reduced heart performance.
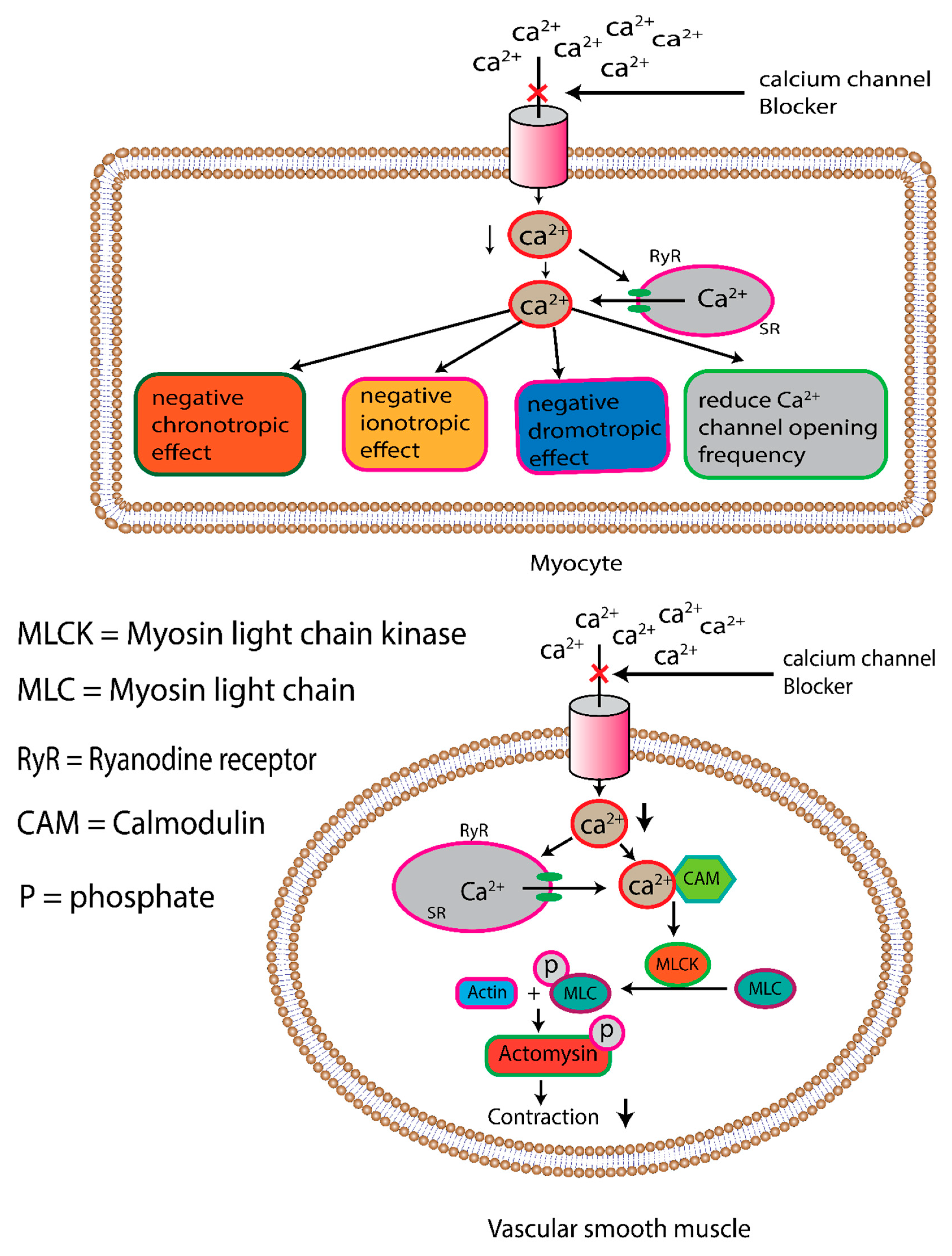
Figure 5.
treatment strategy for hypertension.
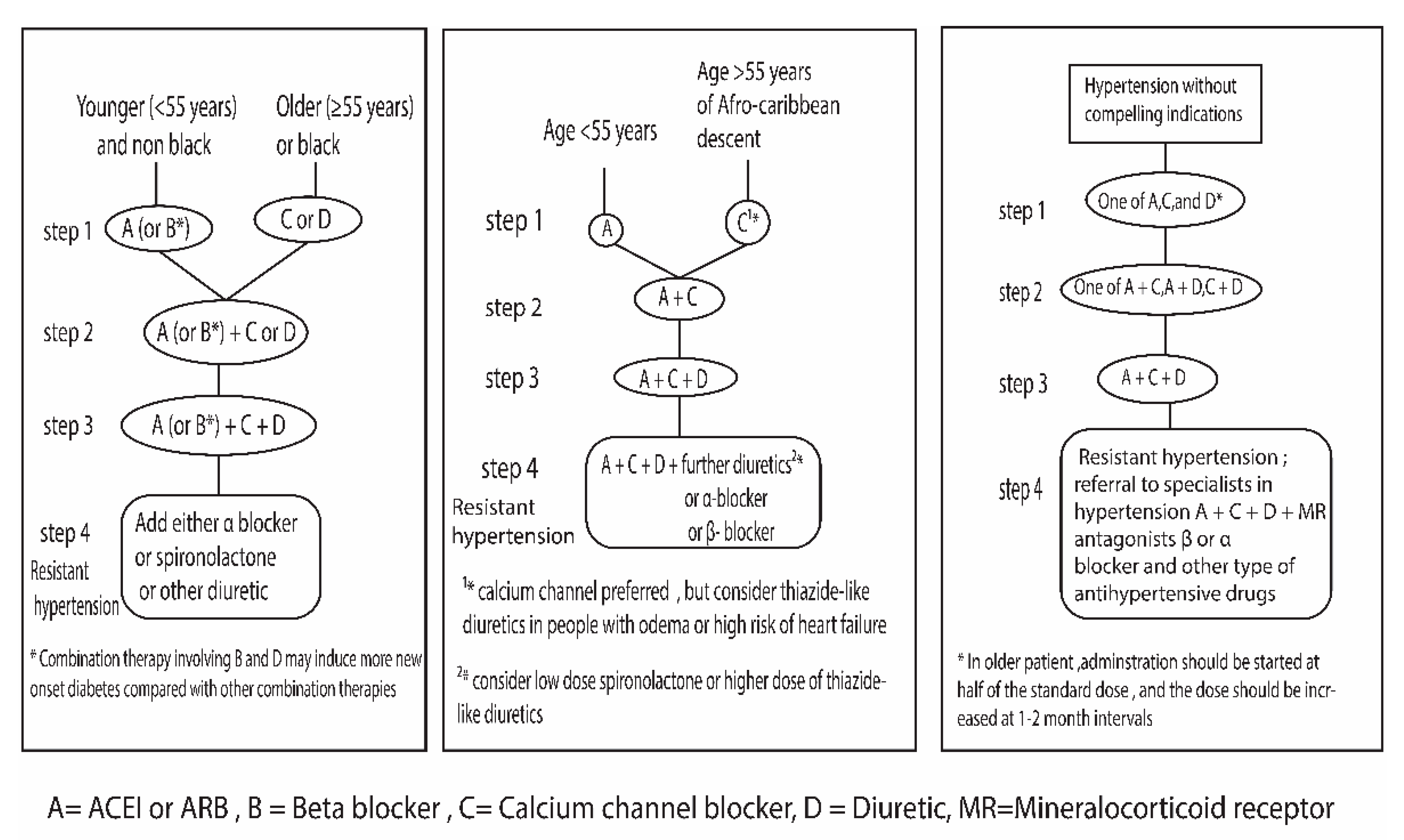
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



