Submitted:
16 February 2024
Posted:
19 February 2024
You are already at the latest version
Abstract
Post-ovulatory aging (POA) is an inevitable factor during some assisted reproduction technology procedures, and results in poor fertilization rates and impaired embryo development. This study used RNA sequencing analysis and experimental validation to study the similarities and differences between in vivo- and in vitro-matured porcine oocytes before and after POA.
Differentially expressed genes (DEGs) between fresh in vivo-matured oocyte (F_vivo) and aged in vivo-matured oocyte(A_vivo) and DEGs between fresh in vitro-matured oocyte (F_vitro) and aged in vitro-matured oocyte(A_vitro) were intersected to explore the co-effects of POA. It was found that "organelles", especially "mitochondria", were significantly enriched GO terms. The expression of genes related to the “electron transport chain” and “cell redox homeostasis” pathways related to mitochondrial function significantly showed low expression patterns in both A_vivo and A_vitro groups. WGCNA analysis to explore gene expression modules specific to A_vivo. Trait-modules association analysis showed that the red modules were most associated with in vivo aging. There are a total of 959 genes in the red module, which are mainly enriched in “RNA binding”, “mRNA metabolic process”, etc. GO terms, and “spliceosome” and “nucleotide excision repair” pathways. DNAJC7, IK, and DDX18 were at the hub of the gene regulatory network. And these genes were all expressed higher in A_vivo. In summary, POA affects the organelle function of oocytes. A_vivo oocytes have some unique gene expression patterns. These genes may be potential anti-aging targets. This study helps us understand the detailed mechanism of POA and provides strategies for researching oocyte aging.
Keywords:
post-ovulatory aging
; oocyte quality
; mitochondrial function
; post-transcriptional regulation
INTRODUCTION
Oocyte quality and maturation not only affect the embryo and fertilization success but also have long-term impacts on fetal growth and development. Notably, mature oocytes arrested at metaphase II (MII) can be successfully fertilized within a restricted time window after ovulation. When fertilization does not occur within an optimal time frame, MII oocytes experience a process of degradation known as “post-ovulatory aging”(POA) [1]. The POA refers to the oocytes released from the ovary. Over time, the cytoplasm ages and eventually dies, which is inevitable[1]. In mice, the quality of oocytes rapidly deteriorates six hours after ovulation if they are not fertilized, with them subsequently being lost after 12 hours [2]. For human oocytes, the time window for fertilization is 4–12 hours after ovulation [3]. However, women do not show natural visual signs of ovulation; fertilization can occur hours later and can involve aged oocytes and freshly ejaculated spermatozoa.
POA induces many abnormal effects on cell biology, including partial exocytosis of cortical granules [4, 5], hardening of the zona pellucida [5], decline in maturation-promoting factor and MAPK levels [6], abnormalities in the cytoskeleton, and condensation of the chromosome [7]. It can also induce mitochondrial dysfunction, leading to apoptosis [8-10]; perturbation of Ca2+ homeostasis; oxidative damage to lipids, proteins, and DNA components of the cell [11]; and epigenetic changes [12]. Furthermore, POA reduces the fertilization rate and embryo quality and increases the likelihood of abnormalities in the offspring. Therefore, the timely fertilization of oocytes is crucial to ensure embryonic development. Although oocytes fertilized via intracytoplasmic sperm injection (ICSI) can develop into viable embryos, the procedure takes 6–12 hours and, hence, induces POA [13]. Aging-induced defects such as oxidative stress, mitochondrial dysfunction, and chromosomal abnormalities have been detected in oocytes during assisted reproductive technology (ART) procedures, which can result in poor oocyte quality, lower fertilization rates, embryo development aberrancy, and even unhealthy offsprings [14-16]. Therefore, developing strategies to improve in-vitro fertilization (IVF) outcomes remains imperative.
Research comparing the anti-aging ability of in vivo- and vitro-matured porcine oocytes has not yet been reported. Compared with other species, porcine oocytes are more similar to human oocytes in many aspects, including the closed oocyte volume (120–125 μm in diameter), average time for oocyte maturation (40–44 h for pig, 40 h for human) [17], similar core transcriptional network required to maintain pluripotency [18], and similar developmental stage of embryonic genome activation [17, 19]. Moreover, they both contain a large quantity of endogenous lipids [20, 21]. Therefore, porcine oocytes are a good model for human reproductive research and clinical-assisted reproductive technology applications. Therefore, we collected in vivo- and in vitro-matured and aged porcine oocytes for RNA sequencing to explore the effects of POA on porcine oocytes and the key pathways and genes that provide in vivo-matured oocytes with stronger anti-aging ability.
In this study, we used transcriptomic analysis and experimental validation to study the similarities and differences between in vivo- and vitro-matured porcine oocytes during POA. Addressing this question will help us understand the detailed mechanisms of POA and provide strategies for researching oocyte aging.
METHODS
1. Collection of porcine cumulus–oocyte complexes (COCs) and in vitro maturation (IVM)
Pig ovaries were collected from a local slaughterhouse (Farm Story Dodarm B&F, Umsung, Chungbuk, Korea) and transported to the laboratory in a pre-warmed NaCl solution containing 75 mg/mL penicillin G and 50 mg/mL streptomycin sulfate. Porcine follicles approximately 3–6 mm in diameter were aspirated using a 10-mL disposable syringe. COCs with more than two layers of compact cumulus cells (CCs) were selected and washed three times with an IVM medium (TCM-199 [11150–059; Gibco, Grand Island, NY, USA] supplemented with 100 mg/L sodium pyruvate, 10 ng/mL EGF, 10 % (v/v) porcine follicular fluid, 10 IU/mL LH, and 10 IU/mL FSH). Finally, 50–100 COCs per well were cultured in four-well dishes covered with mineral oil for 44–48 h until maturation to the MII phase at 38.5 °C with 5 % CO2.
2. In vitro aging
CCs were removed with 1 mg/mL hyaluronidase by pipetting approximately 40 times. MII-stage oocytes were selected for further studies. To analyze oocyte POA, the selected MII-stage oocytes were continuously cultured in an IVM medium covered with mineral oil for an additional 48 hours.
3. In vivo- fresh and aged-oocyte collection
In vivo oocytes were collected as described previously [22]. All crossbred gilts (Duroc) were housed at the National Institute of Animal Science (NIAS, Cheonan, Republic of Korea). The gilts were synchronized using Altrenogest ReguMate or Virbagest for 15 d. Briefly, all animals were detected as being in estrus by back-pressure testing and were kept in separate pens without insemination for mature oocyte collection. On day 2 post-estrus, the gilts were stunned by electro-anesthesia, slaughtered by bleeding in a commercial slaughterhouse, and their reproductive organs were obtained immediately after slaughter. The mature oocytes were then flushed from the oviduct with 10 mL of pre-warmed PBS, after which the oocytes were carefully selected in a Petri dish under a stereomicroscope. For POA, the selected MⅡ-stage oocytes were continuously cultured in an IVM medium for an additional 48 h at 38.5 ℃ in a humidified atmosphere (5 % CO2). All samples were removed from the zona pellucidae using an acid Tyrode’s solution and collected in a tube, each containing five oocytes. The samples were then stored at -80 ℃ until library preparation.
4. Quantitative reverse transcription PCR (qRT-PCR)
mRNA was extracted from five MⅡ-stage oocyte using a Dyna Beads mRNA Direct Kit (61012; Thermo Fisher Scientific, Waltham, MA, USA), and cDNA was synthesized using the First Strand Synthesis Kit (cat# 6210; LeGene, San Diego, CA, USA) in accordance with the manufacturer’s instructions. qRT-PCR was performed using a WizPure qPCR Master (W1731-8; Wizbio Solutions, Seongnam, South Korea) according to the manufacturer’s instructions, on a Quant Studio™ six Flex Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). The PCR conditions were as follows: initial denaturation at 95 °C for 10 min, followed by 40 cycles of amplification at 95 °C for 15 s, 60 °C for 20 s, and 72 °C for 15 s, and a final extension at 95 °C for 15 s. Relative gene expression was calculated using the 2-∆∆CT method. The primers used in this study are listed in Table 1.
5. RNA sequencing and data analysis
Total RNA was extracted using a TRIzol reagent kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. RNA quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and verified by RNase-free agarose gel electrophoresis. After total RNA extraction, the eukaryotic mRNA was enriched with Oligo(dT) beads. The enriched mRNA was fragmented into short fragments using a fragmentation buffer and reverse transcribed into cDNA using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB #7530; New England Biolabs, Ipswich, MA, USA). The purified double-stranded cDNA fragments were end-repaired, a base was added and ligated to Illumina sequencing adapters. The ligation reaction was purified using AMPure XP Beads (1.0X) and PCR was performed to amplify the ligation products. The resulting cDNA library was sequenced using an Illumina Novaseq6000 (by Gene Denovo Biotechnology Co., Guangzhou, China). RNA differential expression analysis was performed using DESeq2 [23] software between two different groups. The genes/transcripts with the parameter of false discovery rate (FDR) <0.05 and absolute fold change ≥2 was considered differentially expressed genes/transcripts. The reference genome used in this study is ncbi_GCF_000003025.6.
RESULTS
1. Global transcriptome analysis of fresh and aged in vivo and vitro-matured porcine oocytes.
As shown in Figure 1, GV-stage oocytes were collected from pig ovaries and cultured for 44 and 92 h to obtain fresh in vitro-matured oocyte (F_vitro) and aged in vitro-matured oocyte(A_vitro). Fresh in vivo-matured oocyte (F_vivo) and aged in vivo-matured oocyte(A_vivo) were obtained by in vitro culturing of MII-stage oocytes, collected from pig fallopian tubes, for 0 and 48 h, respectively.
To investigate the differentiations in gene expression in response to POA, PE150 high-throughput RNA-seq analysis was performed on the four groups oocytes. The expression value for each gene was calculated as the expected number of fragments per kilobase of transcript sequence per million base pairs sequenced (FPKM). A total of 25,479 expressed genes in porcine oocytes were identified based on a cut-off of FPKM > 0.0. The Spearman correlation heatmap showed good intra-group reproducibility and the four groups were clearly distinguished from each other (Figure 2A). Genes with FPKM > 1 and an adjusted p-value of < 0.05 were considered significant differential expression genes (DEGs). As shown in Figure 2B, 3,989 genes (1,482 up and 2,507 down) were differentially expressed between the F_vivo and A_vivo, 5,880 genes (3,006 up and 2,874 down) were differentially expressed between the F_vivo and F_vitro, 1,130 genes (928 up and 202 down) were differentially expressed between the A_vivo and A_vitro, and 1,535 genes (642 up and 893 down) were differentially expressed between the fresh in vitro and aged in vitro oocytes. Heat map (Figure 2C) showing overall gene expression patterns across four groups. These results indicate that different culture environments had a profound influence on the quality and anti-aging abilities of porcine oocytes.
Then, we used qRT-PCR to detect the relative expression levels of randomly selected genes (BTF3, CEP95, CMC1, DBI, and NOUFB5), and the expression trends were consistent with the sequencing results (Figure 2D). The results indicated that the RNA sequencing data obtained in this study were reliable, reproducible, and of high quality.
2. POA interferes with the mitochondrial function in both in vivo and vitro-matured porcine oocytes
To exclude the great interference of the culture environment, DEGs between F_vivo and A_vivo and DEGs between F_vitro and A_vitro were intersected to explore the co-effects of POA (Figure 3A). To better understand the roles the DEGs played during the process of POA, the DEGs were enriched into Gene Ontology (GO) terminology. The top 15 enriched GO terms were shown in Figure 3B. The top enriched terms were mainly related to Intracellular, Organelle, and Mitochondrion. In addition, we found that the expression of electron transport chain-related genes POLG2, FDX1, GLRX2, COX8C, etc. and cell redox homeostasis-related genes TXNDC11, PRDX3, and GLRX2 decreased in both A_vivo and A_vitro groups (Figure 3C and 3D). In addition, the results of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis showed that the oxidative phosphorylation pathway was significantly enriched (Figure 4A). The expression of genes related to oxidative phosphorylation pathway COX6B1, NDUFB5, UQCRQ, etc. were downregulated in both A_vivo and A_vitro groups (Figure 4D). These results indicate that POA can cause damage to oocyte mitochondrial function and affect intracellular energy supply and redox status.
3. POA affects protein export and oocyte meiotic in both in vivo and vitro-matured porcine oocytes
The top 15 enriched KEGG pathways were shown in Figure 3B. Ribosome, protein export, spliceosome and thermogenesis were the most enriched pathways. In addition, we also found that the oocyte meiosis pathway which closely related to oocyte maturation was enriched. Heat map showing protein export and meiosis-related gene expression patterns. Genes related to protein export including SRP54, SPCS2, SEC11C, etc. were significantly downregulated in both A_vivo and A_vitro. The expression levels of CDK1, ANAPC10 and SPDYA were lower in both A_vivo and A_vitro groups. And the expression levels of PKMYT1 and PPP2R1A in aged group were lower than F_vitro but higher than F_vivo group. These results indicate that POA can lead to impaired oocyte protein export function and affect meiosis progression.
4. Selection of specific target modules for aged in vivo matured porcine oocytes
The sample clustering dendrograms was constructed based on the genes that FPKM > 2 (Total 11,804 genes). The applicable power value for this test was nine. Then, gene modules were detected based on the TOM matrix. A total of 22 modules were detected in the analysis (Figure 5A). And coded as brown, black, red, pink, green, grey60 module, etc., including 3,136, 1,705, 959, 773, 716, 655 genes, respectively (Figure 5B).
Next, we use module feature values and specific trait data to perform correlation analysis to find the modules most relevant to traits and phenotypes. The most significant module based on the correlation of the modules with the traits were red module (Figure 6A). The 959 target genes are included within the red module, and the expression level of genes within the red module is shown in Figure 6B. Most genes expressed higher in A_vivo group.
The gene significance (GS) and module membership (MM) values of genes were used to analyze the association between each trait and the module, and the module with high correlation played an important biological role in the trait. The intramodular connectivity (K.in) and GS values of genes within the module were used to analyze the association between the module and the gene and the trait. The result is shown in the Figure 6C,D. It shows that the red module has high correlation and connectivity with traits. Therefore, red module was selected for further analysis.
5. DNAJC7, DDX18 and IK were the hub genes in the gene regulation network in aged in vivo porcine oocytes.
We further analyzed the 959 candidate hub genes in the red module by GO/KEGG enrichment analysis. Ten significantly enriched GO terms were shown in Figure 7A. RNA binding, mRNA metabolic process, RNA splicing, and DNA repair, etc. were enriched. And ten significantly enriched KEGG pathways were shown in Figure 7B. Spliceosome, Nucleotide excision repair, DNA replication and RNA polymerase, etc. were enriched. These enrichment results are highly correlated, suggesting that oocytes matured in vivo may resist POA through RNA binding, modification, splicing and DNA repair, etc., which are related to post-transcriptional gene expression.
The gene regulatory relationship network diagram was used to obtain the core genes at the hub position in the regulatory relationship network within red module (Figure 8A). As the figure shows, DnaJ heat shock protein family (Hsp40) member C7 (DNAJC7), IK cytokine (IK), DEAD-Box Helicase 18 (DDX18) and transcription factors zinc finger protein 836 (ZNF836), ZNF667 were at the hub position. The expression patterns of these five genes are shown in the Figure 8B. All the five genes have higher expression levels in A_vivo than A_vitro group.
DISCUSSION
The exact mechanisms underlying ovarian aging are poorly understood. Oocyte senescence is thought to play a central role in ovarian aging. Previous studies have compared the differences in gene expression profiles in GV- or MII-stage oocytes from young and old individuals [24, 25], which were limited by the differences in in vivo and in vitro culture environments that are hard to ignore. Herein, we used RNA sequencing to determine the gene expression profiles of oocytes in the following four groups: fresh in vivo, fresh in vitro, aged in vivo, and aged in vitro. The similar and different effects of POA on in vivo- and in vitro-matured oocytes were explored.
Most DEGs appeared between fresh in vivo and in vitro oocytes, with a total of 5880 (Figure 2B). Different culture environments had a profound influence on the quality and anti-aging abilities of porcine oocytes. To exclude the influence of in vitro conditions, DEGs between F_vivo and A_vivo and DEGs between F_vitro and A_vitro were intersected to explore the co-effects of POA.
We performed enrichment analyses of common DEGs caused by POA to explore the effects of POA on both in vivo and in vitro-matured oocytes. The results revealed that many DEGs were related to important cellular function. These DEGs were enriched for intracellular, organelle, and membrane-bounded organelle. Among them, we are concerned that mitochondria-related terms are enriched (Figure 3B).
Our research showed that the dysfunction of mitochondrial function, including electron transport chain, cell redox homeostasis and oxidative phosphorylation, contributed to the aging of oocytes. As oocyte maturation requires a large amount of ATP for continuous transcription and translation, the availability of the right number of functional mitochondria is crucial[26]. It has been speculated that the higher ATP content in human and mouse MII oocytes was associated with better embryo potential for development and implantation[27]. The normal function of mitochondria is the guarantee of oocyte quality and embryo developmental potentiality, while oocyte aging is related to mitochondrial dysfunction and disturbance of energy metabolism[28]. The protein machinery to control the health of the mitochondria via unfolded protein response and mitophagy may be compromised in oocytes from aged females, which may result in inefficient clearance of dysfunctional mitochondria and reduced oocyte quality[29]. This highlights the importance of mitochondrial function in aging.
Abnormal distribution of mitochondria as well as mitochondrial dysfunction, resulting in severely impaired germinal vesicle breakdown (GVBD) of mouse oocytes. This conclusion was also confirmed by our data, as KEGG enrichment analysis showed that the meiosis pathway was enriched, and the expression of related genes was reduced in aging oocytes[30](Figure 4A, 4C).
Another enriched pathway in both in vivo and in vitro-matured oocytes was protein export (Figure 4A, 4B). This pathway contains DEGs named signal recognition particle (SRP), which was a ribonucleoprotein particle crucial for co-translational targeting of secretory and membrane proteins to the endoplasmic reticulum [31]. The eukaryotic SPC is composed of five subunits including two isoforms of catalytic subunits SEC11A and SEC11C and three regulatory subunits including signal peptidase complex subunit 1 (SPCS1), SPCS, and SPCS3 [32, 33]. Among the SPC subunits, SEC11 and SPCS3 are essential for signal peptidase activity and cell survival[34, 35]. SPCS2 interacts with the b subunit of SEC61 translocon complex likely facilitating co-translational signal peptidase processing[36] and, at high-temperature conditions, regulates the catalytic activity of the SPC and viability of yeast cells[37]. A previous study has shown that nuclear protein export is a common hallmark of pathological and physiological aging in the Hutchinson Gilford syndrome cellular phenotype of normal fibroblasts [38].
The co-effects of POA on in vivo and in vitro oocytes were observed through enrichment analysis, and then we performed WGCNA to explore the unique gene expression patterns of in vivo-matured oocytes during POA. 22 co-expression network modules were determined, and successfully mined the specific module related to A_vivo. WGCNA divides modules into soft thresholds, which reflect the effectiveness of biological networks more effectively than hard thresholds[39]. Functional enrichment analysis using GO/KEGG identified RNA binding and spliceosome as key pathways against POA in vivo (Figure 7A, 7B).
RNA-binding proteins (RBPs) achieve their biological function essentially by post-transcriptional gene regulation[40]. In eukaryotic cells, following RNA polymerase II-mediated transcript synthesis, RBPs dictate extensive pre-mRNA processing by interacting with the target RNA and partner proteins[41]. Alternative splicing generates diverse transcripts by removing introns and splicing exons. They are responsible for adding modifications to the transcript that affect stability and translation efficiency, including 5’-end capping and 3’-end polyadenylations. RBPs play a crucial role in the transport of transcripts from the nucleus to the cytoplasm, where protein synthesis occurs. Additionally, RBPs are involved in modulating the localization and half-life of mRNA within the cell by either promoting or delaying transcript degradation. Hence, RBPs have a significant impact on almost every aspect of RNA biology[42, 43].
Decades of work on aging have shed light into the fundamental role played within this context by a class of proteins termed RBPs[44, 45]. Loss of intracellular RBP AU-rich-element factor-1 will alter post-transcriptional regulation of targets particularly relevant for protection of genomic integrity and gene regulation, thus concurring to responses related to oxidative stress and accelerated aging[46]. RBP PUM1 overexpression protected MSCs against H2O2-induced cellular senescence by suppressing TLR4-mediated NF-κB activity[47].
Spliceosome induced by pharmacological and genetic inhibition of spliceosome genes, have been reported to trigger cell senescence, suggesting a key role of spliceosome genes as a gatekeeper [48]. Pre-mRNA splicing is fundamental for gene expression and regulation. Similar results were observed in transcriptome sequencing data from human mature defective oocytes, with spliceosomes being the most abundant pathway [49].
The hub genes DNAJC7, DDX18, and IK were screened out by constructing a gene regulatory relationship network. These hub genes are involved to a certain extent in maintaining genome stability, DNA damage repair and other life processes. DNAJC7 participates in p53/MDM2 negative feedback regulatory pathway, and thereby enhancing the stability and activity of tumor suppressor p53 which promotes apoptosis to eliminate cells with seriously damaged DNA to maintain genomic integrity[50]. DDX18 depletion leads to γH2AX accumulation and genome instability[51]. IK is a splicing factor that promotes spliceosome activation and contributes to pre-mRNA splicing[52]. And all the three genes had higher expression levels in A_vivo than A_vitro group.
These results demonstrate the impact of post-transcriptional regulation on cellular senescence. In vivo-matured oocytes have higher expression levels on these pathways than in vitro-matured oocytes. We predict that in vivo-matured oocytes can resist POA through post-transcriptional regulation of gene expression.
To summarize our findings, we have observed that POA has an impact on the quality of porcine oocytes. This is likely due to its effects on mitochondrial function and protein export. We have also noted that there are variations in the expression patterns of in vivo and in vitro oocytes during POA, particularly in pathways related to post-transcriptional regulation such as RNA-binding and spliceosome pathways (Figure 9). Our data serves as a foundation for understanding the mechanisms that underlie POA, but more research is needed to fully explore the relationship between these factors.
CONCLUSION
POA adversely affects the quality of porcine oocytes, both in vivo and in vitro. This is most likely due to the impairment of mitochondrial function and protein export. Further research has shown that during the POA process, RNA-binding and spliceosome pathways were the most differentially enriched pathways between oocytes that matured in vivo and those that matured in vitro. The hub genes that were identified in this study were DNAJC7, DDX18, and IK.
Author Contributions
XS Cui and D Zhou designed this experiment. XS Cui reviewed and edited this manuscript. CL Zhan analyzed the experimental data and drafted the manuscript. D Zhou completed culture and treatment of experimental samples. MH Sun, SH Lee, JD Kim and GH Lee participated in the collection and culture of in vivo samples. WJ Jiang, XH Li, QY Lu, and JM Sim participated in the data analysis and confirmation of sequencing data. HJ Chung, ES Cho and SJ Sa provided supports with experimental design and methodology for this experiment. CL Zhan and D Zhou contributed equally to this study. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea grant funded by the Korea government (MSIT) (No. 2022R1A2C300769), Republic of Korea.
Conflict of Interest Statement
No conflicts of interest, financial, or otherwise, are declared by the authors.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
References
- Lord, T.; Aitken, R.J. Oxidative stress and ageing of the post-ovulatory oocyte. Reproduction 2013, 146, R217–227. [Google Scholar] [CrossRef] [PubMed]
- Di Nisio, V.; Antonouli, S.; Damdimopoulou, P.; Salumets, A.; Cecconi, S. In vivo and in vitro postovulatory aging: when time works against oocyte quality? J Assist Reprod Genet 2022, 39, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; ShiYang, X.; Zhang, Y.; Miao, Y.; Chen, Y.; Cui, Z.; Xiong, B. Coenzyme Q10 ameliorates the quality of postovulatory aged oocytes by suppressing DNA damage and apoptosis. Free Radic Biol Med 2019, 143, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Ducibella, T.; Duffy, P.; Reindollar, R.; Su, B. Changes in the distribution of mouse oocyte cortical granules and ability to undergo the cortical reaction during gonadotropin-stimulated meiotic maturation and aging in vivo. Biol Reprod 1990, 43, 870–876. [Google Scholar] [CrossRef]
- Xu, Z.; Abbott, A.; Kopf, G.S.; Schultz, R.M.; Ducibella, T. Spontaneous activation of ovulated mouse eggs: time-dependent effects on M-phase exit, cortical granule exocytosis, maternal messenger ribonucleic acid recruitment, and inositol 1,4,5-trisphosphate sensitivity. Biol Reprod 1997, 57, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Naito, K.; Noguchi, J.; Kaneko, H.; Tojo, H. Maturation/M-phase promoting factor regulates aging of porcine oocytes matured in vitro. Cloning Stem Cells 2002, 4, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, S.; Thuan, N.V.; Kishigami, S.; Ohta, H.; Mizutani, E.; Hikichi, T.; Miyake, M.; Wakayama, T. Production of offspring from one-day-old oocytes stored at room temperature. J Reprod Dev 2004, 50, 627–637. [Google Scholar] [CrossRef]
- Lord, T.; Nixon, B.; Jones, K.T.; Aitken, R.J. Melatonin prevents postovulatory oocyte aging in the mouse and extends the window for optimal fertilization in vitro. Biol Reprod 2013, 88, 67. [Google Scholar] [CrossRef]
- Zhang, N.; Wakai, T.; Fissore, R.A. Caffeine alleviates the deterioration of Ca(2+) release mechanisms and fragmentation of in vitro-aged mouse eggs. Mol Reprod Dev 2011, 78, 684–701. [Google Scholar] [CrossRef]
- Tatone, C.; Di Emidio, G.; Barbaro, R.; Vento, M.; Ciriminna, R.; Artini, P.G. Effects of reproductive aging and postovulatory aging on the maintenance of biological competence after oocyte vitrification: insights from the mouse model. Theriogenology 2011, 76, 864–873. [Google Scholar] [CrossRef]
- Guérin, P.; El Mouatassim, S.; Ménézo, Y. Oxidative stress and protection against reactive oxygen species in the pre-implantation embryo and its surroundings. Hum Reprod Update 2001, 7, 175–189. [Google Scholar] [CrossRef]
- Liang, X.W.; Zhu, J.Q.; Miao, Y.L.; Liu, J.H.; Wei, L.; Lu, S.S.; Hou, Y.; Schatten, H.; Lu, K.H.; Sun, Q.Y. Loss of methylation imprint of Snrpn in postovulatory aging mouse oocyte. Biochem Biophys Res Commun 2008, 371, 16–21. [Google Scholar] [CrossRef]
- Nagy, Z.P.; Rienzi, L.F.; Ubaldi, F.M.; Greco, E.; Massey, J.B.; Kort, H.I. Effect of reduced oocyte aging on the outcome of rescue intracytoplasmic sperm injection. Fertil Steril 2006, 85, 901–906. [Google Scholar] [CrossRef]
- Gupta, S.; Sekhon, L.; Kim, Y.; Agarwal, A. The role of oxidative stress and antioxidants in assisted reproduction. Current Women's Health Reviews 2010, 6, 227–238. [Google Scholar] [CrossRef]
- Reis Soares, S.; Rubio, C.; Rodrigo, L.; Simón, C.; Remohí, J.; Pellicer, A. High frequency of chromosomal abnormalities in embryos obtained from oocyte donation cycles. Fertil Steril 2003, 80, 656–657. [Google Scholar] [CrossRef]
- Babayev, E.; Seli, E. Oocyte mitochondrial function and reproduction. Curr Opin Obstet Gynecol 2015, 27, 175–181. [Google Scholar] [CrossRef]
- Santos, R.R.; Schoevers, E.J.; Roelen, B.A. Usefulness of bovine and porcine IVM/IVF models for reproductive toxicology. Reprod Biol Endocrinol 2014, 12, 117. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, Y.; Yang, J.Y.; Cheng, D.; Liu, X.; Ma, X.; West, F.D.; Wang, H. Comparative gene expression signature of pig, human and mouse induced pluripotent stem cell lines reveals insight into pig pluripotency gene networks. Stem Cell Rev Rep 2014, 10, 162–176. [Google Scholar] [CrossRef]
- Jarrell, V.L.; Day, B.N.; Prather, R.S. The transition from maternal to zygotic control of development occurs during the 4-cell stage in the domestic pig, Sus scrofa: quantitative and qualitative aspects of protein synthesis. Biol Reprod 1991, 44, 62–68. [Google Scholar] [CrossRef]
- Haggarty, P.; Wood, M.; Ferguson, E.; Hoad, G.; Srikantharajah, A.; Milne, E.; Hamilton, M.; Bhattacharya, S. Fatty acid metabolism in human preimplantation embryos. Hum Reprod 2006, 21, 766–773. [Google Scholar] [CrossRef]
- McEvoy, T.G.; Coull, G.D.; Broadbent, P.J.; Hutchinson, J.S.; Speake, B.K. Fatty acid composition of lipids in immature cattle, pig and sheep oocytes with intact zona pellucida. J Reprod Fertil 2000, 118, 163–170. [Google Scholar] [CrossRef]
- Baek, S.Y.; Sa, S.J.; Jeong, Y.D.; Cho, E.S.; Hong, J.G.; YS, K.I.; Cho, K.H.; Park, S.H.; Kim, K.W.; Lee, H.C.; et al. Altrenogest affects expression of galectin-3 and fibroblast growth factor 9 in the reproductive tract of sows. Anim Biotechnol 2021, 32, 537–543. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef]
- Grøndahl, M.L.; Yding Andersen, C.; Bogstad, J.; Nielsen, F.C.; Meinertz, H.; Borup, R. Gene expression profiles of single human mature oocytes in relation to age. Hum Reprod 2010, 25, 957–968. [Google Scholar] [CrossRef]
- Reyes, J.M.; Silva, E.; Chitwood, J.L.; Schoolcraft, W.B.; Krisher, R.L.; Ross, P.J. Differing molecular response of young and advanced maternal age human oocytes to IVM. Hum Reprod 2017, 32, 2199–2208. [Google Scholar] [CrossRef]
- Kirillova, A.; Smitz, J.E.J.; Sukhikh, G.T.; Mazunin, I. The Role of Mitochondria in Oocyte Maturation. Cells 2021, 10. [Google Scholar] [CrossRef]
- Van Blerkom, J.; Davis, P.W.; Lee, J. ATP content of human oocytes and developmental potential and outcome after in-vitro fertilization and embryo transfer. Hum Reprod 1995, 10, 415–424. [Google Scholar] [CrossRef]
- Zhang, Q.; Hao, J.X.; Liu, B.W.; Ouyang, Y.C.; Guo, J.N.; Dong, M.Z.; Wang, Z.B.; Gao, F.; Yao, Y.Q. Supplementation of mitochondria from endometrial mesenchymal stem cells improves oocyte quality in aged mice. Cell Prolif 2023, 56, e13372. [Google Scholar] [CrossRef]
- Khan, S.A.; Reed, L.; Schoolcraft, W.B.; Yuan, Y.; Krisher, R.L. Control of mitochondrial integrity influences oocyte quality during reproductive aging. Mol Hum Reprod 2023, 29. [Google Scholar] [CrossRef]
- Xue, Y.; Meng, T.G.; Ouyang, Y.C.; Liu, S.L.; Guo, J.N.; Wang, Z.B.; Schatten, H.; Song, C.Y.; Guo, X.P.; Sun, Q.Y. Miro1 regulates mitochondrial homeostasis and meiotic resumption of mouse oocyte. J Cell Physiol 2022, 237, 4477–4486. [Google Scholar] [CrossRef]
- Piazzon, N.; Schlotter, F.; Lefebvre, S.; Dodré, M.; Méreau, A.; Soret, J.; Besse, A.; Barkats, M.; Bordonné, R.; Branlant, C.; et al. Implication of the SMN complex in the biogenesis and steady state level of the signal recognition particle. Nucleic Acids Res 2013, 41, 1255–1272. [Google Scholar] [CrossRef]
- Evans, E.A.; Gilmore, R.; Blobel, G. Purification of microsomal signal peptidase as a complex. Proc Natl Acad Sci U S A 1986, 83, 581–585. [Google Scholar] [CrossRef]
- Alzahrani, N.; Wu, M.J.; Sousa, C.F.; Kalinina, O.V.; Welsch, C.; Yi, M. SPCS1-Dependent E2-p7 processing determines HCV Assembly efficiency. PLoS Pathog 2022, 18, e1010310. [Google Scholar] [CrossRef]
- Fang, H.; Mullins, C.; Green, N. In addition to SEC11, a newly identified gene, SPC3, is essential for signal peptidase activity in the yeast endoplasmic reticulum. J Biol Chem 1997, 272, 13152–13158. [Google Scholar] [CrossRef]
- Böhni, P.C.; Deshaies, R.J.; Schekman, R.W. SEC11 is required for signal peptide processing and yeast cell growth. J Cell Biol 1988, 106, 1035–1042. [Google Scholar] [CrossRef]
- Antonin, W.; Meyer, H.A.; Hartmann, E. Interactions between Spc2p and other components of the endoplasmic reticulum translocation sites of the yeast Saccharomyces cerevisiae. J Biol Chem 2000, 275, 34068–34072. [Google Scholar] [CrossRef]
- Mullins, C.; Meyer, H.A.; Hartmann, E.; Green, N.; Fang, H. Structurally related Spc1p and Spc2p of yeast signal peptidase complex are functionally distinct. J Biol Chem 1996, 271, 29094–29099. [Google Scholar] [CrossRef]
- García-Aguirre, I.; Alamillo-Iniesta, A.; Rodríguez-Pérez, R.; Vélez-Aguilera, G.; Amaro-Encarnación, E.; Jiménez-Gutiérrez, E.; Vásquez-Limeta, A.; Samuel Laredo-Cisneros, M.; Morales-Lázaro, S.L.; Tiburcio-Félix, R.; et al. Enhanced nuclear protein export in premature aging and rescue of the progeria phenotype by modulation of CRM1 activity. Aging Cell 2019, 18, e13002. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef]
- Schieweck, R.; Ninkovic, J.; Kiebler, M.A. RNA-binding proteins balance brain function in health and disease. Physiol Rev 2021, 101, 1309–1370. [Google Scholar] [CrossRef]
- Lenzken, S.C.; Achsel, T.; Carrì, M.T.; Barabino, S.M. Neuronal RNA-binding proteins in health and disease. Wiley Interdiscip Rev RNA 2014, 5, 565–576. [Google Scholar] [CrossRef]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat Rev Genet 2021, 22, 185–198. [Google Scholar] [CrossRef]
- Varesi, A.; Campagnoli, L.I.M.; Barbieri, A.; Rossi, L.; Ricevuti, G.; Esposito, C.; Chirumbolo, S.; Marchesi, N.; Pascale, A. RNA binding proteins in senescence: A potential common linker for age-related diseases? Ageing Res Rev 2023, 88, 101958. [Google Scholar] [CrossRef]
- Conlon, E.G.; Manley, J.L. RNA-binding proteins in neurodegeneration: mechanisms in aggregate. Genes Dev 2017, 31, 1509–1528. [Google Scholar] [CrossRef]
- Cookson, M.R. RNA-binding proteins implicated in neurodegenerative diseases. Wiley Interdiscip Rev RNA 2017, 8. [Google Scholar] [CrossRef]
- Salvato, I.; Ricciardi, L.; Dal Col, J.; Nigro, A.; Giurato, G.; Memoli, D.; Sellitto, A.; Lamparelli, E.P.; Crescenzi, M.A.; Vitale, M.; et al. Expression of targets of the RNA-binding protein AUF-1 in human airway epithelium indicates its role in cellular senescence and inflammation. Front Immunol 2023, 14, 1192028. [Google Scholar] [CrossRef]
- Yoon, D.S.; Lee, K.M.; Choi, Y.; Ko, E.A.; Lee, N.H.; Cho, S.; Park, K.H.; Lee, J.H.; Kim, H.W.; Lee, J.W. TLR4 downregulation by the RNA-binding protein PUM1 alleviates cellular aging and osteoarthritis. Cell Death Differ 2022, 29, 1364–1378. [Google Scholar] [CrossRef]
- Kwon, S.M.; Min, S.; Jeoun, U.W.; Sim, M.S.; Jung, G.H.; Hong, S.M.; Jee, B.A.; Woo, H.G.; Lee, C.; Yoon, G. Global spliceosome activity regulates entry into cellular senescence. Faseb j 2021, 35, e21204. [Google Scholar] [CrossRef]
- Li, J.; Lu, M.; Zhang, P.; Hou, E.; Li, T.; Liu, X.; Xu, X.; Wang, Z.; Fan, Y.; Zhen, X.; et al. Aberrant spliceosome expression and altered alternative splicing events correlate with maturation deficiency in human oocytes. Cell Cycle 2020, 19, 2182–2194. [Google Scholar] [CrossRef]
- Kubo, N.; Wu, D.; Yoshihara, Y.; Sang, M.; Nakagawara, A.; Ozaki, T. Co-chaperon DnaJC7/TPR2 enhances p53 stability and activity through blocking the complex formation between p53 and MDM2. Biochem Biophys Res Commun 2013, 430, 1034–1039. [Google Scholar] [CrossRef]
- Lin, W.L.; Chen, J.K.; Wen, X.; He, W.; Zarceno, G.A.; Chen, Y.; Chen, S.; Paull, T.T.; Liu, H.W. DDX18 prevents R-loop-induced DNA damage and genome instability via PARP-1. Cell Rep 2022, 40, 111089. [Google Scholar] [CrossRef]
- Ka, H.I.; Seo, H.; Choi, Y.; Kim, J.; Cho, M.; Choi, S.Y.; Park, S.; Han, S.; An, J.; Chung, H.S.; et al. Loss of splicing factor IK impairs normal skeletal muscle development. BMC Biol 2021, 19, 44. [Google Scholar] [CrossRef]
Figure 1.
Experimental flowchart. In brief, GV-stage oocytes were collected from pig ovaries and cultured for 44 and 92 h to obtain two groups of oocytes, fresh in vitro and aged in vitro, respectively. Fresh in vivo and aged in vivo oocytes were obtained by in vitro culture of MII-stage oocytes collected from porcine fallopian tubes for 0 and 48 h, respectively.
Figure 1.
Experimental flowchart. In brief, GV-stage oocytes were collected from pig ovaries and cultured for 44 and 92 h to obtain two groups of oocytes, fresh in vitro and aged in vitro, respectively. Fresh in vivo and aged in vivo oocytes were obtained by in vitro culture of MII-stage oocytes collected from porcine fallopian tubes for 0 and 48 h, respectively.
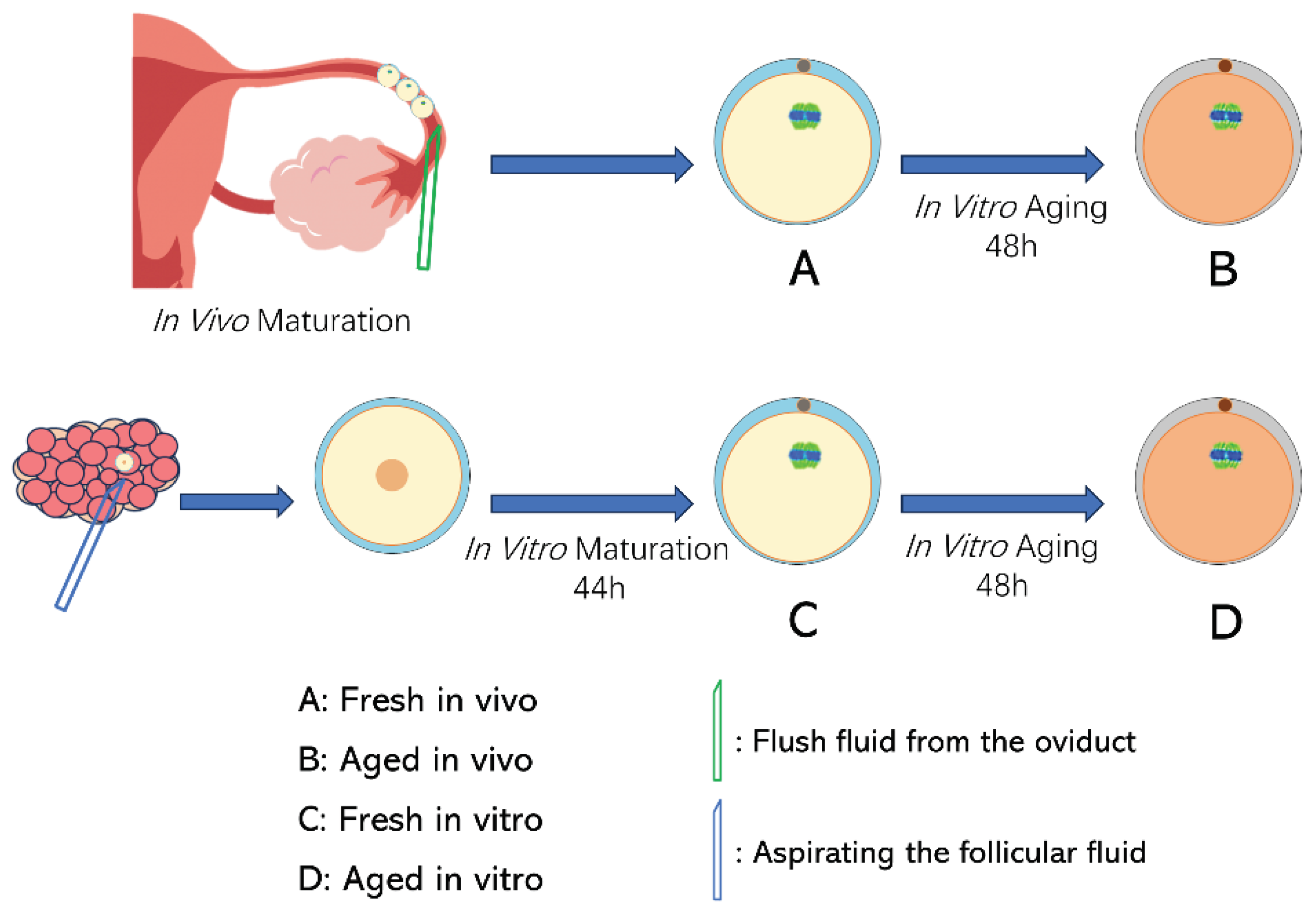
Figure 2.
Global transcriptome analysis of fresh and aged in vivo and vitro-matured porcine oocytes. (A) Spearman’s correlation heatmap shows the reproducibility between repetitive samples and the difference between groups. The color gradient indicates the magnitude of the correlation coefficient. (B) Scatterplot of differentially expressed genes between each two groups. The criteria for differentially expressed genes were FDR<0.05, and fold change>2. (C) Heatmap of mRNA expression profiles in fresh and aging oocytes, showing changes in a subset of genes in response to POA. The color key (from blue to red) of the Z-score value indicates low to high expression levels. (D) Relative mRNA expression levels of BTF3, CEP95, CMC1, DBI, and NOUFB5 in four groups.
Figure 2.
Global transcriptome analysis of fresh and aged in vivo and vitro-matured porcine oocytes. (A) Spearman’s correlation heatmap shows the reproducibility between repetitive samples and the difference between groups. The color gradient indicates the magnitude of the correlation coefficient. (B) Scatterplot of differentially expressed genes between each two groups. The criteria for differentially expressed genes were FDR<0.05, and fold change>2. (C) Heatmap of mRNA expression profiles in fresh and aging oocytes, showing changes in a subset of genes in response to POA. The color key (from blue to red) of the Z-score value indicates low to high expression levels. (D) Relative mRNA expression levels of BTF3, CEP95, CMC1, DBI, and NOUFB5 in four groups.
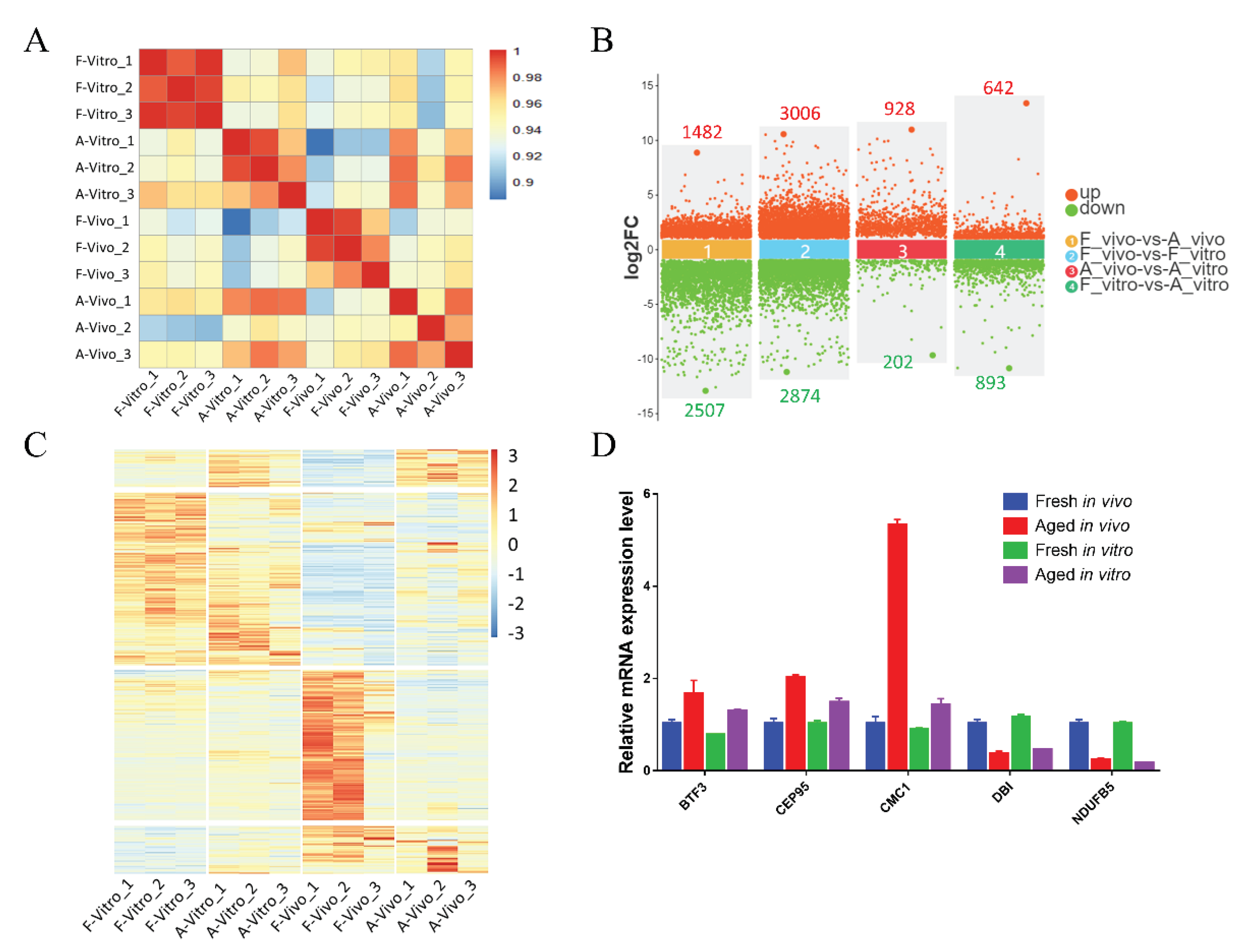
Figure 3.
GO enrichment analysis of common DEGs caused by POA in both in vivo and in vitro matured oocytes (A) Differential gene Venn diagram. The intersection of DEGs between F_vivo and A_vivo and DEGs between F_vitro and A_vitro were defined as common DEGs. (B) GO enrichment analysis on common DEGs showing the top 15 enriched GO items. (C) The heat map for gene expression levels associated with electron transport chain in enriched GO terms. (D) The heat map for gene expression levels associated with Cell REDOX homeostasis in enriched GO terms.
Figure 3.
GO enrichment analysis of common DEGs caused by POA in both in vivo and in vitro matured oocytes (A) Differential gene Venn diagram. The intersection of DEGs between F_vivo and A_vivo and DEGs between F_vitro and A_vitro were defined as common DEGs. (B) GO enrichment analysis on common DEGs showing the top 15 enriched GO items. (C) The heat map for gene expression levels associated with electron transport chain in enriched GO terms. (D) The heat map for gene expression levels associated with Cell REDOX homeostasis in enriched GO terms.
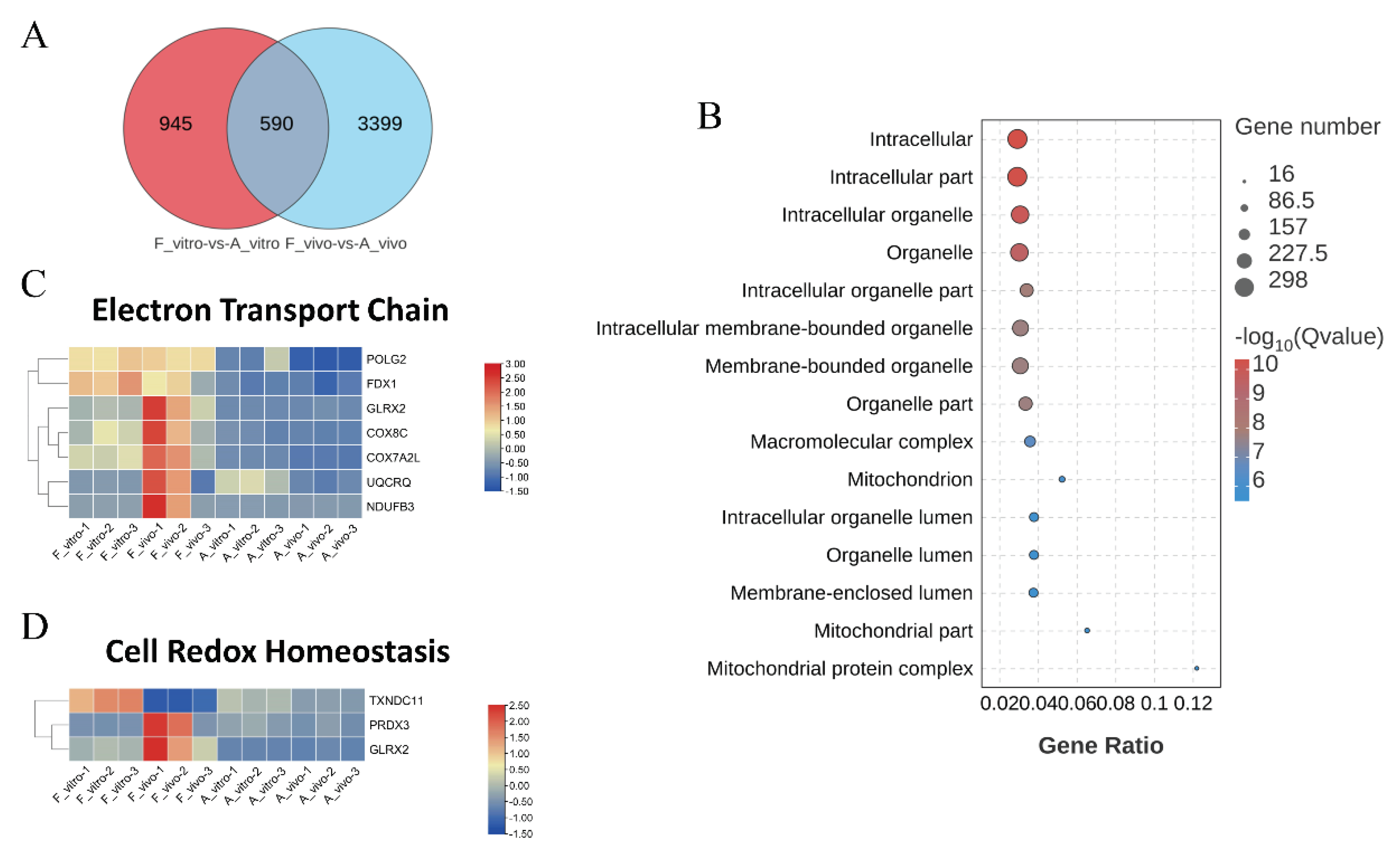
Figure 4.
KEGG enrichment analysis of common DEGs caused by POA in both in vivo and vitro-matured oocytes (A) KEGG enrichment analysis on common DEGs showing the top 15 enriched KEGG pathways. (B) The heat map for gene expression levels associated with protein export in enriched KEGG pathway. (C) The heat map for gene expression levels associated with oocyte meiosis in enriched KEGG pathway. (D) The heat map for gene expression levels associated with oxidative phosphorylation in enriched KEGG pathway.
Figure 4.
KEGG enrichment analysis of common DEGs caused by POA in both in vivo and vitro-matured oocytes (A) KEGG enrichment analysis on common DEGs showing the top 15 enriched KEGG pathways. (B) The heat map for gene expression levels associated with protein export in enriched KEGG pathway. (C) The heat map for gene expression levels associated with oocyte meiosis in enriched KEGG pathway. (D) The heat map for gene expression levels associated with oxidative phosphorylation in enriched KEGG pathway.
Figure 5.
Construction of weighted gene co-expression network analysis (WGCNA). (A) Module hierarchical clustering tree. Gene modules are divided according to the clustering relationship between genes. Genes with similar expression patterns will be classified into the same module. The branches of the cluster tree are cut and distinguished to generate different modules. Each color represents a module, gray Indicates genes that cannot be assigned to any module. After the preliminary module division, the preliminary divided “Dynamic Tree Cut” is obtained. Modules with similar expression patterns are then merged based on the similarity of module feature values to obtain the final dividend “Merged dynamic”. (B) The bar plot shows the number of genes in every module.
Figure 5.
Construction of weighted gene co-expression network analysis (WGCNA). (A) Module hierarchical clustering tree. Gene modules are divided according to the clustering relationship between genes. Genes with similar expression patterns will be classified into the same module. The branches of the cluster tree are cut and distinguished to generate different modules. Each color represents a module, gray Indicates genes that cannot be assigned to any module. After the preliminary module division, the preliminary divided “Dynamic Tree Cut” is obtained. Modules with similar expression patterns are then merged based on the similarity of module feature values to obtain the final dividend “Merged dynamic”. (B) The bar plot shows the number of genes in every module.
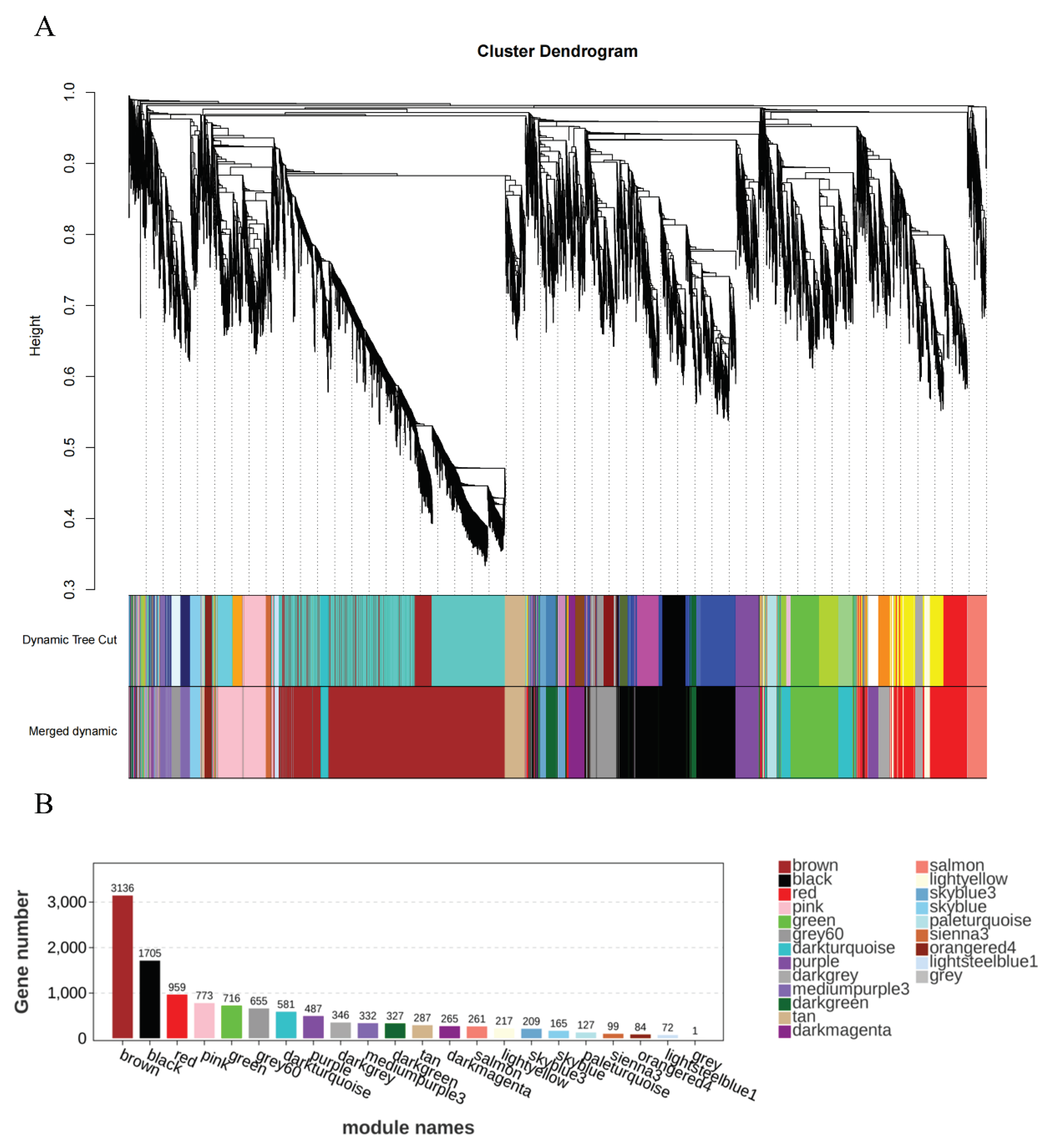
Figure 6.
(A)Module–trait relationships. Each row presents a module eigengene, each column presents a trait. Each cell contains the corresponding correlation and p-value (“*” means p-value < 0.05, “**” means p-value < 0.01). The table is color-coded by correlation according to the color legend. (B) The heat map for gene expression levels of genes in the red module among the four groups. (C) The (module membership) MM- gene significance (GS) correlation of genes in the red module. Use the GS and MM values of genes to analyze the correlation between each trait and the module. Modules with high correlation play an important biological role in the trait. (D) Intramodular connectivity (K.in) and GS correlation analysis of genes in the red module. Analyze the association between modules, genes, and traits using the connectivity of genes within the module (K.in value) and the correlation value between genes and traits (GS).
Figure 6.
(A)Module–trait relationships. Each row presents a module eigengene, each column presents a trait. Each cell contains the corresponding correlation and p-value (“*” means p-value < 0.05, “**” means p-value < 0.01). The table is color-coded by correlation according to the color legend. (B) The heat map for gene expression levels of genes in the red module among the four groups. (C) The (module membership) MM- gene significance (GS) correlation of genes in the red module. Use the GS and MM values of genes to analyze the correlation between each trait and the module. Modules with high correlation play an important biological role in the trait. (D) Intramodular connectivity (K.in) and GS correlation analysis of genes in the red module. Analyze the association between modules, genes, and traits using the connectivity of genes within the module (K.in value) and the correlation value between genes and traits (GS).
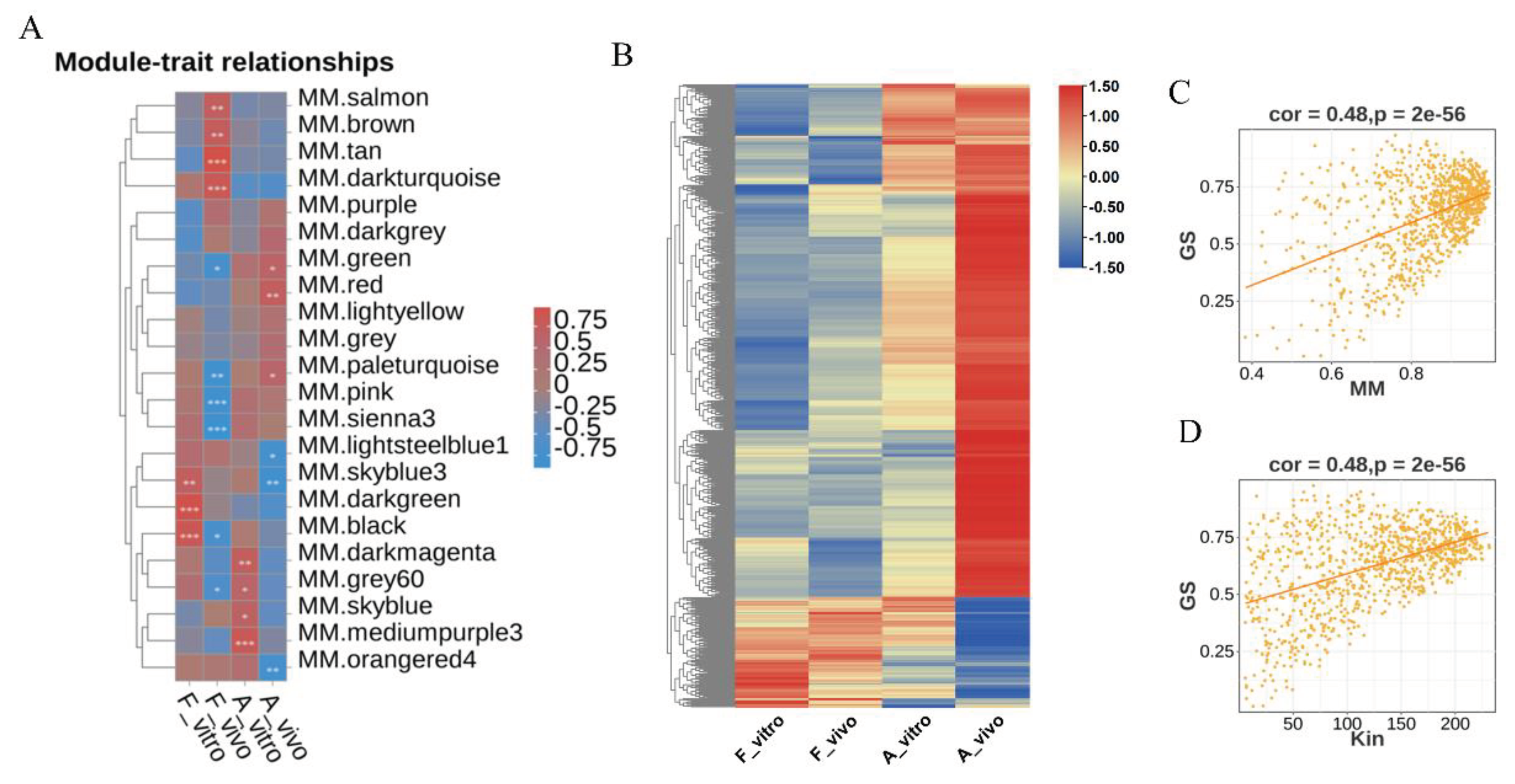
Figure 7.
Enrichment analysis of genes in the red module. (A) GO enrichment analysis of genes in the red module showing the top 15 enriched GO terms (B) KEGG enrichment analysis of genes in the red module showing the top 15 enriched KEGG pathways.
Figure 7.
Enrichment analysis of genes in the red module. (A) GO enrichment analysis of genes in the red module showing the top 15 enriched GO terms (B) KEGG enrichment analysis of genes in the red module showing the top 15 enriched KEGG pathways.
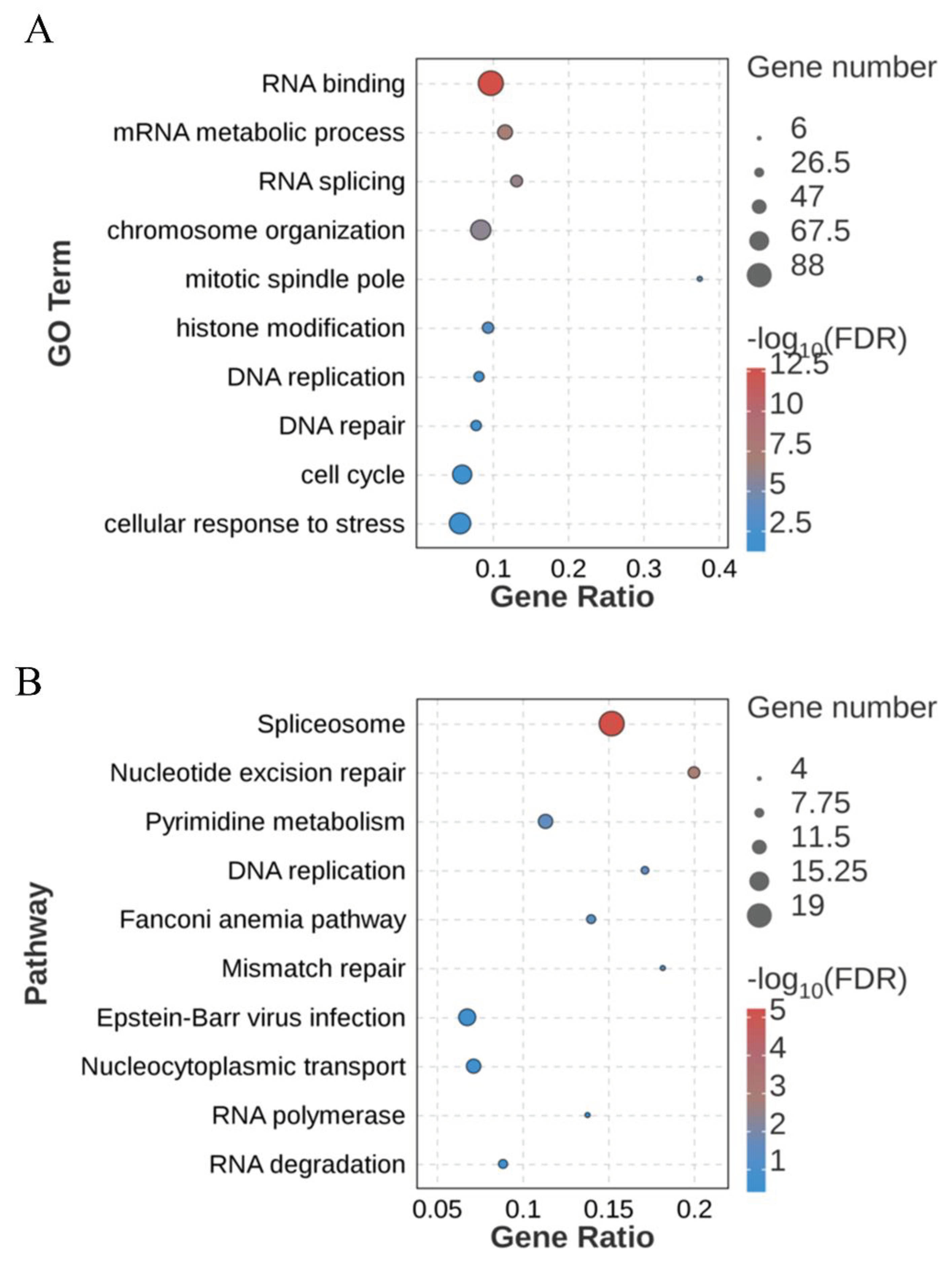
Figure 8.
Hub genes in the red module from the gene regulatory relationship network. (A) Construction of gene regulatory network. Each node in the figure is a gene, and each line represents the regulatory relationship between the nodes. The darker and larger the node color is, the higher the abundance and the stronger the connectivity. The weight value defines the color and thickness of the line. The darker the color and the thicker the line, the stronger the regulatory relationship between genes. (B) The boxplot shows the expression of hub genes in the A_vivo and A_vitro groups.
Figure 8.
Hub genes in the red module from the gene regulatory relationship network. (A) Construction of gene regulatory network. Each node in the figure is a gene, and each line represents the regulatory relationship between the nodes. The darker and larger the node color is, the higher the abundance and the stronger the connectivity. The weight value defines the color and thickness of the line. The darker the color and the thicker the line, the stronger the regulatory relationship between genes. (B) The boxplot shows the expression of hub genes in the A_vivo and A_vitro groups.
Figure 9.
Schematic representation depicting the same and different effects of POA on in vivo and in vitro-matured porcine oocytes. POA adversely affects the quality of porcine oocytes, both in vivo and in vitro. This is most likely due to the impairment of mitochondrial function and protein export. RNA-binding and spliceosome pathways were the most differentially enriched pathways between oocytes that matured in vivo and those that matured in vitro.
Figure 9.
Schematic representation depicting the same and different effects of POA on in vivo and in vitro-matured porcine oocytes. POA adversely affects the quality of porcine oocytes, both in vivo and in vitro. This is most likely due to the impairment of mitochondrial function and protein export. RNA-binding and spliceosome pathways were the most differentially enriched pathways between oocytes that matured in vivo and those that matured in vitro.
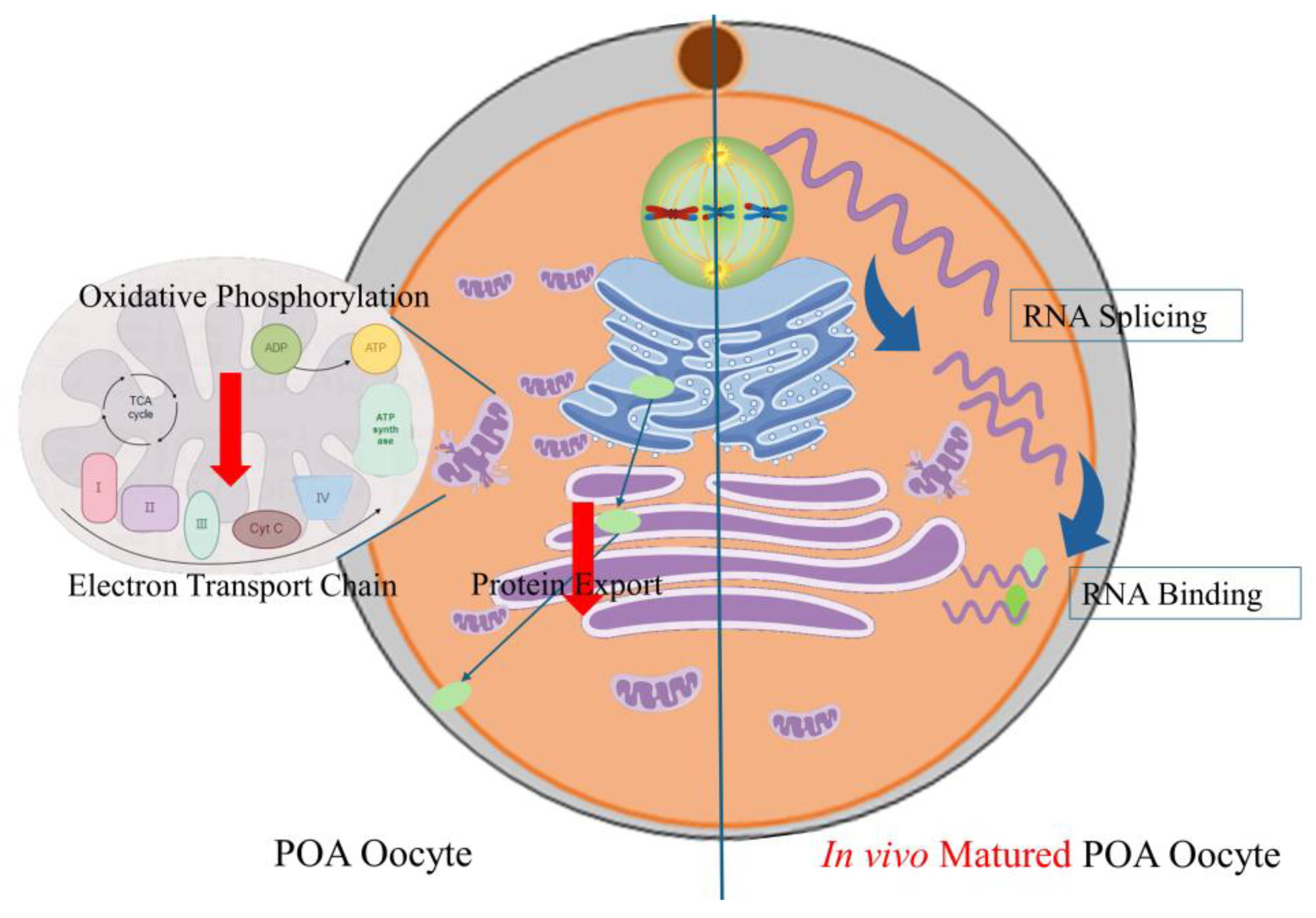

Table 1.
Information of primers used for qPCR.
| Genes | Primer sequences(5’-3’)F | Primer sequences(5’-3’)R |
|---|---|---|
| BTF3 | GTGTGTGCGCCTTATCTCAG | GTTTGGCGAGTTTCTCCTGG |
| CEP95 | AGAGGGCAGGAGAGAGGTTA | ACATCCTCCTCTTCACAGCC |
| CMC1 | CGCAGAACAGCATCTCAGAC | TCCAGAGTCCTTGCAGCATT |
| DBI | ACAGCCACTACAAACAAGCG | ACGCTTTCATGGCATCTTCC |
| NDUFB5 | GCTTTGCCCTCAGTCAACAT | CATGGCTACTATGGGCGAGA |
| 18s | CGCGGTTCTATTTTGTTGGT | AGTCGGCATCGTTTATGGTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



