
Submitted:
16 February 2024
Posted:
19 February 2024
You are already at the latest version
Abstract
Abstract: A. baumannii exhibits resilience in harsh environments and progressively develops resistance to diverse antibacterial agents over time. This adaptability poses a significant challenge in treating an infection caused by this bacterium within healthcare settings. The presence of peptidoglycan, a crucial precursor for cell wall synthesis, contributes to this challenge. Peptidoglycan serves not only as a structural support in bacterial cells but also provides protection against adverse environmental conditions. The synthesis of the peptidoglycan layer involves a group of substrates such as N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc), within a three-partitioned process occurring in the membrane, periplasmic space, and cytoplasm of each bacterium. Initiated in the cytoplasm and extending to the inner cell membrane, this synthesis starts with the production of nucleotide precursors. The first step involves the synthesis of UDP-GlcNAc, orchestrated by Glm (GlmS, GlmM, and GlmU) enzymes using fructose-6-phosphate. Subsequently, enzymes from the Mur family, namely MurB, MurA, MurD, MurC, MurI, MurF, and MurE, synthesize UDP-N-acetylmuramyl-pentapeptide (UDP-Mpp) from UDP-GlcNAc. Concurrently, on the cytosolic side, the production of membrane-surrounded undecaprenyl phosphate occurs, involving membrane-side proteins MraY and MurG in the biosynthesis of peptidoglycan. This review emphasizes updated information on Mur family proteins, encompassing physicochemical properties, nature of binding site, number and type of binding site residues, functional domain details, three-dimensional structural aspects, and their functions within bacterial cells. Special attention is directed towards their enzymatic role in peptidoglycan biosynthesis due to the limited availability of precursors in the market. Additionally, the review provides a comprehensive summary of existing inhibitors and ongoing efforts in identifying new targeted inhibitors, recognizing these enzymes as potential candidates for antibacterial drug design and discovery.
Keywords:
Antibiotic resistance
; Antibacterial drug design
; Peptidoglycan
; Mur
1. Introduction
A. baumannii possesses virulence factors contributing to various infections. These factors include the hydrophobic nature of the cell surface, enzymes within the outer membrane, toxic slime polysaccharides, and toxins like verotoxins. These elements potentially enhance the pathogenicity of the bacterium. Among these factors, the hydrophobic properties of the cell surface play a crucial role in bacterial cohesion, preventing phagocytosis [1,2]. Pathogenicity is further facilitated by extracellular enzymes present in A. baumannii, such as esterases, peptidases, phosphatases, and various toxins, produced in the organism’s cytoplasm. Additionally, secreted substances significantly impact the pathogenicity of A. baumannii in the host cell, particularly in the respiratory system. Due to its multidrug resistance (MDR), extensive drug resistance (XDR), and biofilm-forming nature, this organism is categorized as a critical and highly effective disease-causing bacterium, responsible for hospital-acquired or nosocomial infections in admitted patients, according to the World Health Organization (WHO) report [3]. Consequently, there is a pressing need to explore the physicochemical, structural and functional properties of all members of the Mur family present in this bacterium to showcase possible novel antibacterial candidate inhibitors [3].
In addition to Acinetobacter baumannii, Enterococcus faecium, Enterobacter, Pseudomonas aeruginosa, Staphylococcus aureus, and Klebsiella pneumoniae are classified as ESKAPE pathogenic microorganisms. These ESKAPE pathogens are commonly associated with hospital-acquired (or) nosocomial infections. Among them, A. baumannii stands out as the foremost and most challenging pathogen. Diseases attributed to Multidrug-Resistant A. baumannii (MDRAB) include bacteremia, pneumonia, urinary tract infections (UTI), meningitis, and septicemia [4,5,6]. According to reports from the World Health Organization (WHO), A. baumannii is designated as a critical and priority I pathogen due to its capability for multidrug resistance across various drug groups (Figure 1) [3]. A. baumannii accounts for 2-10% of infections caused by gram-negative bacteria in the USA and Europe. In India, Acinetobacter baumannii, Klebsiella pneumoniae, Escherichia coli, and Pseudomonas aeruginosa are classified as Indian priority pathogen developed by WHO country office for India in collaboration with Department of Biotechnology, Government of India [7]. Numerous studies have linked its MDR ability to an increase in infection and mortality rates [8,9]. Therefore, there is a compelling and imperative need to explore drug target proteins and novel groups of promising synthetic or phytochemicals that can effectively inhibit these targeted proteins.
The bacterial cell is encapsulated by a sturdy cell wall, comprised of a layered assembly called murein, resembling a threads like structure. This protective barrier shields the organism from internal and external factors, primarily safeguarding against cell turgidity and shrinkage. Among the Mur enzymes, MurA catalyzes the initial biochemical reaction in murein synthesis [10]. Similarly, MurB plays a pivotal role in synthesizing UDP-MurNAc in the murein biosynthetic pathway [10,11]. Subsequent involvement of Mur-ligase members (MurC, MurD, MurE and MurF) extends polypeptide subunits to UDP-MurNAc, which is a crucial step. MurC facilitates L-alanine addition, MurD participates in D-glutamate addition, and glutamate racemase switches L- to D-glutamate interconversion [12]. MurE adds meso-diaminopimelate or L-lysine, while MurF enzymes attach D-alanyl-D-alanine to UDP-N-acetylmuramic acid. Despite low sequence identities, all Mur ligase enzymes exhibit structural similarities [13,14].
MraY, a transferase protein, aids in the transfer of phosphate-linked peptides to a phosphate carrier lipid [15]. The peptidoglycan biosynthetic pathway’s final step is catalyzed by MurG, transferring lipid intermediate-I to form lipid intermediate-II [16]. In the sequential process of peptidoglycan synthesis, all Mur family enzymes contribute, constituting a distinctive pathway for bacteria. Disruption of this vital cell wall component synthesis results in bacterial cell death and eventual bursting. Currently effective antibiotics, such as β-lactams and glycopeptides, target the murein biosynthesis pathway. However, certain bacteria exhibit resistance to these drugs, necessitating the exploration of new antibacterial molecules. Thus, a comprehensive understanding of murein biosynthesis pathways, along with detailed insights into the sequential, structural and functional aspects of drug target proteins as well as showcase drug candidate of synthetic and phytochemical molecules, is crucial for addressing the challenge of multi-drug resistance A. baumannii.
2. Peptidoglycan Biosynthetic Pathways in A. baumannii
Typically, in the biochemical reactions of a specific metabolic pathway, the output of the initial reaction (product) serves as a reactant for the subsequent reaction. In bacteria, including A. baumannii, a multitude of diverse pathways are operational within the microorganism’s cellular environment. Among the numerous metabolic pathways present in bacteria, peptidoglycan biosynthetic pathways hold particular significance and uniqueness in cell wall synthesis. Numerous enzymes within this pathway play crucial roles (Figure 2) [17]. It has been considered as a target of antimicrobial drugs because it is essential for the survival of the organism, has high accessibility for drugs and is present in bacteria but absent in the animal metabolic pathways. A bacterial cell wall is majorly made up of murein (peptidoglycan). Murein is made up of large molecules, which are known as macromolecules. Macromolecules of murein comprise the complicated system of linear glycan chains interconnected with one another by moieties of small peptides [18]. Synthesizing of murein undergoes several steps, and these steps are different in one another.
This critical and very important pathway starts in the cytoplasm by synthesizing UDP-GlcNAc and UDP-MurNAc pentapeptide. The later substrate formed in 6 steps, MurA (UDP-N-acetylglucosamine 1-carboxyvinyltransferase) is used to perform the first step of the reaction, which is starting from an enolpyruvate (PEP) to UDP-GlcNAc. UDP-N-acetylmuramate is then obtained using UDP-N-acetylenolpyruvoylglucosamine reductase (MurB), by catalyzing PEP moiety to D-lactate. Following this, 4 amino-ligases (ATP-dependent) catalyze consecutive addition of pentapeptide chains to the D-lactate by using MurD, MurC, MurF and MurE enzymes [17,18]. The final stage of the peptidoglycan biosynthetic pathways involves via the catalytic activities of two crucial proteins, namely MraY and MurG, also known as membrane transferase enzymes. MraY transfers phospho-N-acetylmuramoyl with pentapeptide onto undecaprenyl phosphate [15], while MurG is utilized for the formation of lipid intermediate II [16]. All these proteins collaborate in a chronological manner for peptidoglycan biosynthesis, a process unique to prokaryotic cells.
2.1. UDP-N-Acetylglucosamine Biosynthesis
Initially, fructose-6-phosphate serves as the precursor for the synthesis of UDP-GlcNAc in the bacterial peptidoglycan biosynthetic pathway. This pathway involves four consecutive enzymatic actions, namely glucosamine-6-phosphate synthase (GlmS), phosphoglucosamine mutase (GlmM), N-acetylglucosamine-1-phosphate uridyltransferase (GlmU bifunctional enzyme), and glucosamine-1-phosphate acetyltransferase (GlmU bifunctional enzyme) (Figure 3). GlcNAc, a crucial component of major biological macromolecules such as chitin and glycoproteins in eukaryotes, becomes a potential antibacterial target pathways due to the dissimilarity between the prokaryotic and eukaryotic pathways in UDP-GlcNAc formation [17]. The hexosamine breakdown is initiated by GlmS, an amido-transferase enzyme, representing the first dedicated phase of this process. GlmM, the second enzyme in the pathway, catalyzes the isomers of glucosamine-6-phosphate and glucosamine-1-phosphate during UDP-GlcNAc biosynthesis. In the transfer of GlcN-1-phosphate to UDP-GlcNAc, GlmU (bifunctional enzyme) plays a crucial role by catalyzing both acetyl transfer and uridyl transfer processes [17].
2.2. UDP-N–Acetylmuramic Acid Biosynthesis
Now that UDP-GlcNAc has been synthesized, the polymerization of murein requires another precursor macromolecule known as UDP-MurNAc. The conversion of UDP-GlcNAc to UDP-MurNAc is facilitated by two enzymes, namely MurB and MurA. MurA initiates the primary step, followed by MurB catalyzing the second step. In the initial stage, the enolpyruvate moiety of phosphoenolpyruvate (PEP) is transferred to the 30-hydroxyl of UDP-GlcNAc, resulting in the formation of UDP-GlcNAc-enolpyruvate which is accompanied by the release of inorganic phosphate (Pi). Subsequently, with the assistance of the MurB enzyme (Figure 3), this compound is reduced to UDP-MurNAc. In the second reduction reaction, MurB undergoes the conversion using one equivalent of NADPH and a solvent-derived proton, making this step a NADPH-dependent reaction [17].
2.1.1. MurA
The specified enzyme exhibits two domains sharing a common folding pattern, complemented by six bending repetitions of a beta motif (Figure 3). With 418 amino acids, it has a molecular weight of 44.6 kDa. An isoaspartyl residue is situated near the hinge region, resulting from the post-translational modification of typical residues. Upon substrate binding, the reaction adheres to the induced fit model mechanism, involving significant conformational rearrangements within the two domains [19]. The reaction, facilitated by MurA, follows the principle of an addition and elimination mechanism. In theory, this reaction is unconventional because it involves the attack of phosphoenolpyruvate (PEP) at the C-2 location, leading to the cleavage of the C-O bond. However, PEP serves as a phosphoryl transfer agent in most PEP-dependent biochemical reactions utilizing biological catalysts known as enzymes [10].
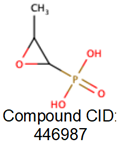
To disrupt this critical step, understanding the binding ability of drug like natural compound is very important. Previously, fosfomycin, a naturally occurring broad-spectrum antimicrobial compound was considered as a promising inhibitor of MurA. The mechanism of action involves the formation of a covalent adduct with a specific active site residue of the enzyme, rendering it an irreversible inhibitor of MurA [20]. This inhibitory effect is particularly potent in the presence of the substrate, UDP-GlcNAc, and displays time-dependent characteristics [21]. However, certain bacteria, such as Chlamydia trachomatis, Mycobacterium tuberculosis, and Borrelia burgdorferi, exhibit intrinsic resistance to these natural compounds through amino acid residue alterations. According to the World Health Organization [3], A. baumannii, classified as a crucial and priority-I pathogen, demonstrates resistance to currently available inhibitors. This resistance arises due to factors like (i) reduced permeability, (ii) modification of the active site, and (iii) enzymatic adjustments to fosfomycin itself. Consequently, there is an urgent need for an inhibitor with a distinct mechanism of action and chemical structure compared to existing inhibitors [20].
2.1.2. MurB
UDP-N-acetylenolpyruvoylglucosamine reductase (MurB) derived from E. coli consists of three distinct domains, encompassing 353 amino acids and a molecular weight of 39.7 kDa. The first and second domains are responsible for facilitating FAD binding, while the third domain mediates the binding of its substrate, namely UDP-GlcNAc-enolpyruvate (Figure 3). MurB activity has been enhanced by cations such as NH4+, Rb+, and K+ [22]. In the enzymatic reaction catalyzed by MurB for murein synthesis, two half-reaction steps occur. In the initial half-reaction, the enzyme-bound FAD undergoes oxidation-reduction transition, with FAD being reduced to FADH2 through the utilization of NADPH. The release of NADP+ is accompanied by the binding of EP-UDP-GlcNAc. The subsequent half of the reaction involves the formation of UDP-MurNAc through the reduction of EP-UDP-GlcNAc [10].
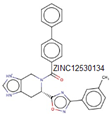
In the peptidoglycan biosynthesis process, the second step, known as the reduction reaction, occurs in the presence of MurB. This signifies that the products generated in the reaction catalyzed by MurA serve as the inputs for the MurB reaction. In this step, UDP-GlcNAc-enolpyruvate undergoes a reduction reaction catalyzed by MurB [17]. Like the preceding stage, this process is crucial for peptidoglycan synthesis, and disrupting it leads to the inhibition of the respective microorganism. According to literature surveys, certain synthetic and natural compounds have demonstrated inhibitory effects on MurB, halting this essential murein synthesis step. The initial report of a MurB inhibitor involves 4-thiazolidinone derivatives designed to mimic the diphosphate moiety of enolpyruvate-UDP-GlcNAc [23]. Additionally, an imidazolinone derivative has exhibited potent MurB inhibitory activity against Staphylococcus aureus, showing promising antibacterial effects. Despite some literature reports lacking sufficient information on the antibacterial activities of 4-thiazolidinones [20], there is an urgent need for new inhibitors, both from natural sources and synthetic compounds. These inhibitors should differ in their mechanism of action and chemical structure from the 4-thiazolidinone derivatives.
2.2. Biosynthesis of the UDP-MurNAc-Peptides Using the Mur Ligases
Mur ligase enzymes, MurC, MurD, MurE, MurI and MurF play a pivotal role in the stepwise reactions of the peptide portion during murein biosynthesis pathways. These enzymes are responsible for the sequential addition of D-glutamic acid, L-alanine, dipeptide D-Ala-D-Ala, and diamino acid onto the UDP-MurNAc D-lactoyl group, facilitated by MurD, MurC, MurF, and MurE, respectively. In these reactions, Mur ligase enzymes catalyze the concurrent formation of a peptide or amid bond, along with the generation of ADP and Pi from ATP. Essential for this reaction are cations such as Mg2+ or Mn2+. The functional and structural properties of these Mur ligase proteins have undergone thorough examination through extensive biochemical and amino acid sequence analyses (Figure 3). The common characteristics shared by these enzymes include a consistent mechanism of action, sequential similarity of six residues, and similar structures with three functional domains [17].
2.2.1. MurC
This enzyme falls outside the category of ribosomal peptide ligase and finds utility in murein synthesis, specifically in the addition of L-alanine to UDP-N-acetylmuramoyl. In this step, UMA (UDP-Nacetylmuramoyl-L-alanine), ATP and inorganic phosphate are required [10]. As per biological and chemical reports on this enzyme, ATP serves as the initial substrate, followed by UDP-MurNAc as the second substrate, and finally, L-Ala is utilized as the third substrate in this process [31]. A crucial interaction takes place at the interface of the third and second domains, particularly involving α and β-phosphates and the adenine ring, which constitutes the ATP-binding site [32]. The MurC enzyme exhibits various characteristics and forms a dimer interface through the interaction of loops, with residues Phe223 and Tyr224; however, these residues are not universally conserved in all bacteria [10,33]. Like other Mur ligase enzymes, MurC features three α/β domains. It has a total of 494 amino acid residues. The first functional domain occupies the N-terminal residue portion and consists of a central five-strand structure. Conversely, domain II, recognized as the ATP-binding domain and the largest among all MurC domains, encompasses seven central strands. The C-terminal portion houses the final domain, or domain III, comprising one antiparallel, five parallel, and six central β-sheet strands [10,32].
MurC’s most potent inhibitor, with an IC50 of 49 nM, appears to be a sequence of phosphinate transition-state analogs synthesized and tested on the MurC E. coli enzyme. According to the mechanism of action of this inhibitor, the UDP moiety plays a crucial role in inhibiting the specified enzyme [34]. In the three enzyme substrates states, such as UDP-MurNAc, L-Ala and ATP, the molecule act as a mixed type of inhibitor as per the compound biochemical characterization. This suggested that phosphinate inhibitor creates a good complex to several states of MurC enzymes [35]. The second inhibitor of MurC E. coli involves analogs of L-alanine, demonstrating competitive inhibition with L-alanine [31]. Another inhibitor, benzylidene rhodanines, exhibits significant IC50 inhibition against MurC. This compound displays specific whole-cell activities in methicillin-resistant S. aureus but not in E. coli, a gram-negative bacterium. While it shows a significant minimum inhibitory concentration against Methicillin-resistant Staphylococcus aureus (MRSA) (31 µM), a notable challenge is its antagonistic effects on animal cells. As a result, this compound is recognized for having a nonspecific mechanism of action [36].
2.2.2. MurD
This enzyme, like other ligase biological catalysts, is situated in the cytoplasm and plays a role in peptidoglycan synthesis within bacterial cells. MurD contributes to this pathway by adding D-glutamate to UDP-N-acetylmuramoyl-L-alanine, a nucleotide precursor (UMA) [37]. As usual, MurD also used as a target for different inhibitors to stop the synthesis of the cell wall and inhibit the growth of the bacteria. The biochemical characteristics of MurD include a molecular weight of 49.3 kDa, localization and expression in the cytoplasm, and a sequence of 496 amino acids [10]. The activity of E. coli MurD has been extensively studied, particularly in the catalytic process of forming the peptide bond between D-glutamate (D-Glu) and UDP-N-acetylmuramoyl L-alanine (UMA) [38,39]. The carboxylic acid chain of UMA undergoes catalysis in the presence of MurD, marking the initial step as an ATP-dependent reaction. Subsequently, the output is catalyzed to generate a tetrahedral intermediate through the incoming D-Glu, resulting in the production of inorganic phosphate and UMAG [10,39].
MurD is the first enzyme from the group of Mur ligase for which phosphinate inhibitors have been developed for inhibiting peptidoglycan synthesis. This compound represents the pioneering and paradigmatic instance of a mechanism-based inhibitor effectively disrupting UDP-N-acetylmuramic acid-pentapeptide biosynthesis. The biochemical attributes of this inhibitor suggest its close resemblance to the typical intermediate reaction and the preservation of the UDP moiety, which is presumably crucial for attachment [38]. In general, this protein has served as a target for a myriad of inhibitors, as documented in various studies. While a diverse array of inhibitors has been identified and examined to date [17,40], the focus of scientific investigations has primarily centered on both peptide and nonpeptide inhibitors, exploring their impact on E. coli’s MurD. Additionally, small molecules have been categorized into inhibitors with and without glutamic acid-based motifs [40].
Several MurD inhibitors, including potent macrocyclic, 9H-Xanthene derivatives, and polycyclic compounds, have been identified through computational approaches against E. coli MurD, demonstrating significant minimum inhibitory concentration levels [41,42]. However, the persisting challenge lies in the inhibitory effects of previously discovered inhibitors being easily overcome by bacteria, both gram-negative and gram-positive. Hence, the quest for new synthetic and natural inhibitors remains imperative to overcome the resistance exhibited by pathogenic bacteria.
2.2.3. MurI
The conversion of L and D-glutamate is facilitated by the cofactor-independent protein glutamate racemase (ordered locus name ABAYE0082) [29]. Given that the MurI gene encodes the Glutamate Racemase protein, playing a pivotal role in the initial phases of peptidoglycan synthesis, it emerges as a promising target for drug design and discovery processes. The transformation of L- to D-glutamate is a critical step in peptidoglycan synthesis, with glutamate racemase being a key contributor to this vital process. Another significant role of glutamate racemase (MurI) is the sequestration of the DNA gyrase enzyme. Naringenin and quercetin are currently recognized as potent molecules for inhibiting the MurI protein [12].
2.2.4. MurE
In the MurE enzyme, the reaction varies between gram-positive and gram-negative bacteria. It functions as an ATP-dependent ligase, incorporating amino acids like meso-diaminopimelic acid into the murein synthesis and catalyzes the process in a species-specific manner [43]. The third amino acid in the peptide during the reaction differs and can be L-lysine, L-ornithine, meso-A2pm, LLA2pm, meso-lanthionine, L-homoserine, or L-diaminobutyric acid [43]. MurE exhibits high specificity, and alterations in the third amino acid result in changes to MurE’s function. For instance, Bacillus sphaericus has two MurE enzymes, where one is active during bacterial vegetative growth, while the other becomes active during spore cortex formation [10]. The gene, located between nucleotides 303,112 and 304,611, is responsible for the synthesis of the Mur ligase enzyme in A. baumannii, specifically the MurE enzyme. This protein consists of 499 residues and has theoretical pI values of 9, with a molecular weight of 54.98 kDa [26]. MurE exhibits amino-terminal, central, and carboxyl-terminal domains [17,44].
The initial potent inhibitor identified for the MurE protein was a phosphinate molecule [14]. In many cases, ligase proteins share similar functions, and certain molecules can inhibit multiple proteins. For instance, sulfonamides serve as inhibitors for both MurD and MurE proteins, exhibiting an IC50 of 181 µM [45]. The MurE proteins in S. aureus and M. tuberculosis were inhibited using 3-Methoxynordomesticine molecules [46,47]. Additionally, a recently discovered potent inhibitor against M. tuberculosis demonstrated an IC50 of 75 µM [48].
2.2.5. MurF
Similar to other Mur enzymes, MurF is involved in catalyzing the biosynthetic pathways of peptidoglycan. In this process, MurF adds D-Ala-D-Ala to the UDP-MurNAc-tripeptide, a product of MurE enzymes [10,43]. All Mur ligase enzymes follow a comparable reaction mechanism, occurring in the presence of ATP. MurF, like other members of the Mur family, lacks a human complement, making it an attractive target for drug development. The predicated structure of MurF ligase, along with other Mur ligase enzymes, exhibits similarities. MurF consists of 510 residues organized into three domains. Interestingly, the three domains in gram-positive bacteria bear resemblance to those in gram-negative bacteria; for example, the MurF domain in E. coli shares similarity with M. tuberculosis and S. pneumonia [49].
As per Miller and colleagues’ findings, the initial inhibitors of MurF enzymes were tetra-peptide and tripeptide. These compounds share a common structure of XLys-PO2H-Gly-Ala and function as reversible competitive inhibitors in transition-state analogs, as determined through kinetic assays conducted on MurF from E. coli [50]. Through affinity selection screening technology and subsequent structure-based optimization, various compounds were identified against E. coli’s MurF [49,51]. However, even the most potent compound among these groups showed limited antimicrobial inhibition activity against E. coli [52].
Recently, a potent lead compound based on cyanothiophene was obtained through systematic modification of the original structure and tested on S. pneumoniae, S. aureus, and E. coli’s MurF. Interestingly, all the compounds exhibited activity against S. pneumoniae but not against E. coli and S. aureus. Presently, new MurF ligase inhibitors are being discovered through various in-silico techniques, including virtual screening based on target and ligand structure, leading to the identification of potent molecules from both natural and synthetic databases. For instance, the moderately potent molecule 1,3,5-triazine was obtained based on the structure of the target protein MurF [41]. Additionally, an inhibitor with active efficacy at the micro-molar level against E. coli and S. pneumoniae MurF was identified through a ligand-based approach [53].

Table 2.
Structural and functional properties of Mur family proteins.
| Protein/Gene Name | Structural and Functional Properties | |||||
|---|---|---|---|---|---|---|
| Conserved Region | AA position in the domain | Binding site residues | In-silico model/PDB ID | Inhibitor | References | |
| MurA | Substrate binding domain | Domain I (6-407) | Cys115, Lys22, and Asp305 |  |
 |
[25,54] |
| MurB | 2 conserved regions | Domain I (30-164) and domain II (218-342) | Leu57, Val58 Leu59, Ser60 Gly61, Gly62 Ser63, Asn64 Met65, Ala99 Ile124, Pro125 Gly126, Leu127 Gly129, Ala130 Val133, Gln134 Ile136, Ile185 Ile186, Val340 and Tyr342 |
 |
 |
[54,55] |
| MurC | Three domains | N-terminal domain (1-106), Central domain (107-314) and the C-terminal domain, (315-461) | LB site: (Arg314, Phe316, Asp334, Gly336, Hie337, Ile338, Glu341, Val342, Thr345, Gln447 and Ala348) |
 |
 |
[27,54] |
| MurD | Three domains | Domain 1 (1–93), domain 2 (94–298), domain 3 (299–437). | Leu15, Thr16 Thr36, Arg37 Gly73, Asn138 and His183 |
 |
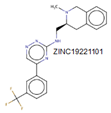 |
[28,54] |
| MurI | Two domains | - | Ser13, Gly14, Val15, Asn41, Gly42, Pro43, Tyr44, Gly45, Ile52, Ser77, Ala121, Cys185, His187, Val199 and etc. |
 |
 |
[12,29] |
| MurE | Three domains | Domain I (1-98), Domain II (99-332), Domain III (333-499) | Asp407, Arg383 Arg410, Glu463 and Asn408 |
 |
 |
[44,54] |
| MurF | Three domains | Domain I (37-84), Domain II (120-303), Domain III (325-405) | Thr42 and Asp43 |  |
ethyl pyridine substituted 3-cyanothiophene | [30,54] |
| MurG | 2 conserved regions | domain I (1-163 and 353-365) and domain II (164-352) | Arg170, Gly20 Ser201, Gly200 Phe254, Gln227 Thr276, Glu279 and Leu275 |
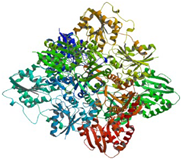 |
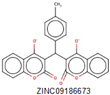 |
[54,56] |
| MraY | Substrate binding domain | Domain I (45-372) | - | 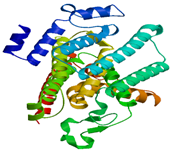 |
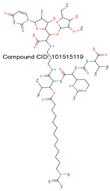 |
[15,54] |
UDP-N-acetylglucosamine 1-carboxyvinyltransferase = MurA; UDP-N-acetylenolpyruvoylglucosamine reductase = MurB; UDP-N-acetylmuramate--L-alanine ligase = MurC; UDP-N-acetylmuramoylalanine--D-glutamate ligase = MurD; Glutamate racemase = MurI; UDP-N-acetylmuramoyl-L-alanyl-D-glutamate--2,6-diaminopimelate ligase = MurE; UDP-N-acetylmuramoyl-tripeptide--D-alanyl-D-alanine ligase = MurF; Phospho-N-acetylmuramoyl-pentapeptide-transferase = MraY; UDP-N-acetylglucosamine--N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine =MurG.
2.3. Formation of Lipid Intermediates
Within the murein synthesis, the involvement of lipid intermediates constitutes a crucial stage achieved through a two-step mechanism employing the MraY and MurG enzymes. In this dual-phase process, MraY facilitates the transfer of the substrate derived from the prior step (phospho-MurNAc-pentapeptide) to lipid I. Subsequently, MurG playing a role (Figure 3), the next stage unfolds with the addition of N-acetylglucosamine subunits, ultimately resulting in the formation of lipid II [57,58].
2.3.1. MraY
MraY, a primary membrane protein, serves as a catalyst for the initial step in the membrane-related processes of the murein biosynthetic pathways. The MurNAc-pentapeptide, an outcome of the cytoplasmic reaction mediated by Mur ligase, undergoes transfer onto the undecaprenyl phosphate carrier (C55-P/bactoprenol) in the presence of MraY, occurring on the bacterial cytoplasm’s membrane side. This reaction involves a two-step process resulting in the formation of Lipid I. The first step involves the UDP-MurNAc-pentapeptide β-phosphate nucleophilic attack within the active site of the MraY protein, leading to the release of UMP and the formation of the enzyme-phospho-MurNAc-pentapeptide intermediate [59]. The subsequent phase entails the attack on the phosphate in the covalent intermediate by a C55-P oxyanion, resulting in the formation of Lipid I. This contributes to the creation of the intermediate known as Lipid I, accompanied by the release of UPM [15].
Regarding the MraY enzyme, sequence analysis and structure experiments conducted on both gram-negative and gram-positive bacteria reveal specific characteristics: five loops on the cytoplasmic side, ten transmembrane segments, and six loops in the periplasm. In this enzyme, the cytoplasmic side of the membrane contains high sequence conservation, and the active site is consistently oriented towards the cytoplasm [59,60]. To sum up, MraY plays a role in catalyzing a membrane-linked step involving the transition of the MurNAc-pentapeptide moiety to the undecaprenyl phosphate lipid carrier. It functions as a transmembrane protein, and its crystal structure has been recently resolved [59].
Similar to the aforementioned Mur proteins, MraY serves as a targeted enzyme for the development of promising molecules against it. However, despite ongoing efforts, only a few classes of MraY blockers have been considered thus far, and none have progressed to clinical studies due to challenges associated with delivering these molecules across membranes. The study of MraY inhibitors has historically employed two approaches: screening assays and the synthesis of molecules mimicking those previously identified as MraY inhibitors [61].
The Uridyl peptide antibiotics (UPAs) family stands out as the most recognized and effective group of small molecule inhibitors of MraY, featuring compounds such as muraymycin, tunicamycin, mureidomycin, capuramycin, and liposidomycin [62,63,64]. These inhibitors share a common amino-ribosyl-O-uridine skeleton crucial for their inhibitory activities, indicating their attachment to the binding sites of MraY [62,63,64]. Another strategy in the development of novel MraY inhibitors involves the structural modification of existing inhibitors [63].
2.3.2. MurG
MurG represents the final stage in the cytoplasmic segment of the murein biosynthetic pathways. This enzyme primarily facilitates the transformation of lipids, specifically converting lipid I to lipid II through the provision of UDP-activated N-acetyl D-glucosamine [65]. Following MraY, MurG catalyzes an essentially irreversible reaction as a glycosyltransferase enzyme within the membrane. The enzyme binds to GlcNAc derived from UDP-GlcNAc, leading to the transformation into Lipid I, ultimately resulting in the production of Lipid II [63].
The exploration of MurG inhibitors has traditionally centered around two main strategies: (i) the synthesis of molecules mimicking UDP-GlcNAc and (ii) the utilization of natural inhibitors to craft analogous compounds. Another effective avenue for identifying new inhibitors involves screening compounds from targeted libraries [63,66,67]. Notably, previous reports have highlighted a MurG inhibitor, specifically a vancomycin derivative and the known antibiotic moenomycin. While effective in inhibiting MurG, its in-vivo activities has been less optimal [63,68].
More recently, the discovery of murgocil, exhibiting narrow-spectrum properties, has emerged as a MurG protein inhibitor [69]. Interestingly, the in-vitro activity of murgocil towards the isolated and purified MurG of S. aureus is notably less than its bioactivity effect towards S. aureus. However, further research is imperative to inhibit MurG activities across various gram-negative and gram-positive bacteria [63,70].
2.4. Broad Spectrum Inhibitors of Mur Enzymes
In the bacterial cell, various stages unfold during murein synthesis, making studies on Mur enzymes crucial and pertinent [57,71]. Given the multi-target hypothesis addressing multiple Mur enzymes, the strategic design of a single effective inhibitor becomes vital, offering advantages in terms of time, cost, and mitigating drug resistance concerns. Drawing on existing literature, pan or broad-spectrum inhibitors within the Mur family encompass Naphthyl Tetronic Acid Derivatives, Furan-based Benzene Mono and Dicarboxylic Acid Derivatives, N-acylhydrazones Derivatives, 5-amido-1-(2, 4-dinitrophenyl)-1H-4-pyrazolecarbonitriles Derivatives, Thiazolidinone Derivatives, and 5-amido-1-(2, 4-dinitrophenyl)-1H-4-pyrazolecarbonitriles Derivatives [57]. Among these multi-target inhibitors, Mansour and colleagues specifically prepared and evaluated Naphthyl Tetronic Acid family, identifying 11 derivatives within this family as potential inhibitors against multiple Mur enzymes [72].
3. Conclusion
As per this review, A. baumannii stands out as a formidable pathogen exhibiting resistance to inhibitors from various groups. Among the diverse pathways in bacteria, including A. baumannii, the biosynthetic pathway of peptidoglycan emerges as the most crucial for bacterial survival. Recognizing the significance of enzymes in this biosynthetic pathway as potential drug targets, efforts are focused on identifying compounds that disrupt their proper activity. Hence, comprehensive structural and functional studies of Mur members (MurB, MurA, MurD, MurC, MurF, MurE, MurG, and MraY) are of paramount importance to pinpoint promising lead molecules towards potential drug targets proteins of multidrug-resistant A. baumannii (MDRAB). The escalating levels of multidrug resistance in A. baumannii strains contribute to severe mortality rates. Consequently, a thorough structural and functional analysis of essential drug targets from this menacing pathogen is imperative for uncovering novel antibacterial agents. The presented review serves as a gateway to identifying diverse therapeutic molecules against MDRAB, employing both computational and experimental approaches.
Author Contributions
Gizachew Muluneh Amera and Amit Kumar Singh conceived the concept of the study. Gizachew Muluneh Amera, Jayaraman Muthukumaran and Silvano Piazza wrote the manuscript as well as prepared the figures and tables. All authors collected and analyzed the literature survey from various resources, edited manuscript and approved the contents of manuscript.
Funding
we would like to thank for the contributions the Italian Agency for Development Cooperation (AICS) within the framework of the BIOTECHNET initiative (AID n. 12098) and the International Centre for Genetic Engineering and Biotechnology (ICGEB) for supports with internal funds. GC is a PhD student of the Molecular Biomedicine program of the University of Trieste.
Data availability
According to the requirements of the journal, the corresponding author will provide the data examined during the current study upon reasonable request.
Acknowledgements
Dr. Amit Kumar Singh thanks Indian Council of Medical Research (ICMR) and Indian National Science Academy (INSA), New Delhi, India. Dr. Gizachew Muluneh Amera thanks Bahir Dar, Wollo and Sharda University for the support. Silvano Piazza thanks the International Centre for Genetic Engineering and Biotechnology (ICGEB) for the support.
Competing Interest
None
References
- Almasaudi SB. Acinetobacter spp. as nosocomial pathogens: Epidemiology and resistance features. Saudi J Biol Sci2018;25:586–96. [CrossRef]
- Peleg AY, Seifert H, Paterson DL. Acinetobacter baumannii: Emergence of a successful pathogen. Clin Microbiol Rev2008;21:538–82. [CrossRef]
- Shrivastava SR, Shrivastava PS, Ramasamy J. World health organization releases global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. JMS - Journal of Medical Society2018;32:76–7. [CrossRef]
- Bravo Z, Orruño M, Parada C, Kaberdin VR, Barcina I, Arana I. The long-term survival of Acinetobacter baumannii ATCC 19606T under nutrient-deprived conditions does not require the entry into the viable but non-culturable state. Arch Microbiol [Internet] 2016;198:399–407. [CrossRef]
- Lee CR, Lee JH, Park M, Park KS, Bae IK, Kim YB, et al. Biology of Acinetobacter baumannii: Pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front Cell Infect Microbiol 2017;7. [CrossRef]
- Haeili M, Kafshdouz M, Feizabadi MM. Molecular mechanisms of colistin resistance among pandrug-resistant isolates of acinetobacter baumannii with high case-fatality rate in intensive care unit patients. Microbial Drug Resistance 2018;24:1271–6. [CrossRef]
- Sharma, Anuj. Indian Priority Pathogen List TO GUIDE RESEARCH, DISCOVERY AND DEVELOPMENT OF NEW ANTIBIOTICS IN INDIA.
- Apisarnthanarak A, Chaiyakunapruk N, Goh BH, Kongpakwattana K, Kulpokin D, Lee LH, et al. A Systematic Review of the Burden of Multidrug-Resistant Healthcare-Associated Infections Among Intensive Care Unit Patients in Southeast Asia: The Rise of Multidrug-Resistant Acinetobacter baumannii. Infect Control Hosp Epidemiol [Internet] 2018;39:525–33. Available from: https://www.cambridge.org/core/product/2CEF98E0840839B6E25B248A00899504. [CrossRef]
- Blevins A, Chiang HY, Chorazy ML, Khader K, Nelson RE, Nelson SD, et al. Costs and Mortality Associated With Multidrug-Resistant Healthcare-Associated Acinetobacter Infections. Infect Control Hosp Epidemiol [Internet] 2016;37:1212–8. Available from: https://www.cambridge.org/core/product/6BB596E6DC6729B102C1519E3E6B8E2C. [CrossRef]
- Moraes GL, Gomes GC, Monteiro De Sousa PR, Alves CN, Govender T, Kruger HG, et al. Structural and functional features of enzymes of Mycobacterium tuberculosis peptidoglycan biosynthesis as targets for drug development. Tuberculosis2015;95:95–111. [CrossRef]
- Benson TE, Walsh CT, Hogle JM. The structure of the substrate-free form of MurB, an essential enzyme for the synthesis of bacterial cell walls. Structure [Internet] 1996;4:47–54. Available from: https://www.sciencedirect.com/science/article/pii/S0969212696000081. [CrossRef]
- Pawar A, Jha P, Chopra M, Chaudhry U, Saluja D. Screening of natural compounds that targets glutamate racemase of Mycobacterium tuberculosis reveals the anti-tubercular potential of flavonoids. Sci Rep 2020;10. [CrossRef]
- Gordon E, Flouret B, Chantalat L, Van Heijenoort J, Mengin-Lecreulx D, Dideberg O. Crystal Structure of UDP-N-acetylmuramoyl-L-alanyl-D-glutamate: meso-Diaminopimelate Ligase from Escherichia Coli. Journal of Biological Chemistry 2001;276:10999–1006. [CrossRef]
- Ziegler K, Diener A, Herpin C, Richter R, Deutzmann R, Lockau W. Molecular characterization of cyanophycin synthetase, the enzyme catalyzing the biosynthesis of the cyanobacterial reserve material multi-L- arginyl-poly-L-aspartate (cyanophycin). Eur J Biochem 1998;254:154–9. [CrossRef]
- Al-Dabbagh B, Henry X, Ghachi M El, Auger G, Blanot D, Parquet C, et al. Active Site Mapping of MraY, a Member of the Polyprenyl-phosphate N-Acetylhexosamine 1-Phosphate Transferase Superfamily, Catalyzing the First Membrane Step of Peptidoglycan Biosynthesis. Biochemistry [Internet] 2008;47:8919–28. [CrossRef]
- Fakhar Z, Naiker S, Alves CN, Govender T, Maguire GEM, Lameira J, et al. A comparative modeling and molecular docking study on Mycobacterium tuberculosis targets involved in peptidoglycan biosynthesis. J Biomol Struct Dyn [Internet] 2016;34:2399–417. [CrossRef]
- Barreteau H, Kovač A, Boniface A, Sova M, Gobec S, Blanot D. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol Rev [Internet] 2008;32:168–207. [CrossRef]
- van Dam V, Olrichs N, Breukink E. Specific Labeling of Peptidoglycan Precursors as a Tool for Bacterial Cell Wall Studies. ChemBioChem [Internet] 2009;10:617–24. [CrossRef]
- Schönbrunn E, Svergun DI, Amrhein N, Koch MHJ. Studies on the conformational changes in the bacterial cell wall biosynthetic enzyme UDP-N-acetylglucosamine enolpyruvyltransferase (MurA). Eur J Biochem [Internet] 1998;253:406–12. [CrossRef]
- Hrast M, Sosič I, Šink R, Gobec S. Inhibitors of the peptidoglycan biosynthesis enzymes MurA-F. Bioorg Chem [Internet] 2014;55:2–15. Available from: https://www.sciencedirect.com/science/article/pii/S0045206814000224. [CrossRef]
- Marquardt JL, Brown ED, Lane WS, Haley TM, Ichikawa Y, Wong CH, et al. Kinetics, Stoichiometry, and Identification of the Reactive Thiolate in the Inactivation of UDP-GlcNAc Enolpyruvoyl Transferase by the Antibiotic Fosfomycin. Biochemistry [Internet] 1994;33:10646–51. [CrossRef]
- Dhalla AM, Yanchunas JJr, Ho HT, Falk P, Villafranca JJ, Robertson JG. Steady-State Kinetic Mechanism of Escherichia coli UDP-N-Acetylenolpyruvylglucosamine Reductase. Biochemistry [Internet] 1995;34:5390–402. [CrossRef]
- Bronson JJ, DenBleyker KL, Falk PJ, Mate RA, Ho HT, Pucci MJ, et al. Discovery of the first antibacterial small molecule inhibitors of MurB. Bioorg Med Chem Lett [Internet] 2003;13:873–5. Available from: https://www.sciencedirect.com/science/article/pii/S0960894X02010764. [CrossRef]
- Amera GM, Khan RJ, Jha RK, Pathak A, Muthukumaran J, Singh AK. Prioritization of Mur family drug targets against A. baumannii and identification of their homologous proteins through molecular phylogeny, primary sequence, and structural analysis. Journal of Genetic Engineering and Biotechnology 2020;18. [CrossRef]
- Sonkar A, Shukla H, Shukla R, Kalita J, Pandey T, Tripathi T. UDP-N-Acetylglucosamine enolpyruvyl transferase (MurA) of Acinetobacter baumannii (AbMurA): Structural and functional properties. Int J Biol Macromol [Internet] 2017;97:106–14. Available from: https://www.sciencedirect.com/science/article/pii/S0141813016324904. [CrossRef]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein Identification and Analysis Tools on the ExPASy Server [Internet]. In: Walker JM, editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005. page 571–607. [CrossRef]
- Ahmad S, Murtaza UA, Raza S, Azam SS. Blocking the catalytic mechanism of MurC ligase enzyme from Acinetobacter baumannii: An in Silico guided study towards the discovery of natural antibiotics. J Mol Liq [Internet] 2019;281:117–33. Available from: https://www.sciencedirect.com/science/article/pii/S0167732218337401. [CrossRef]
- Jha RK, Khan RJ, Amera GM, Singh E, Pathak A, Jain M, et al. Identification of promising molecules against MurD ligase from Acinetobacter baumannii: insights from comparative protein modelling, virtual screening, molecular dynamics simulations and MM/PBSA analysis. J Mol Model [Internet] 2020;26:304. [CrossRef]
- Poen S, Nakatani Y, Opel-Reading HK, Lassé M, Dobson RCJ, Krause KL. Exploring the structure of glutamate racemase from Mycobacterium tuberculosis as a template for anti-mycobacterial drug discovery. Biochemical Journal [Internet] 2016;473:1267–80. [CrossRef]
- Ahmad S, Raza S, Uddin R, Azam SS. Binding mode analysis, dynamic simulation and binding free energy calculations of the MurF ligase from Acinetobacter baumannii. J Mol Graph Model [Internet] 2017;77:72–85. Available from: https://www.sciencedirect.com/science/article/pii/S1093326317303388. [CrossRef]
- Emanuele JJ, Jin H, Yanchunas J, Villafranca JJ. Evaluation of the Kinetic Mechanism of Escherichia coli Uridine Diphosphate-N-acetylmuramate:l-Alanine Ligase. Biochemistry [Internet] 1997;36:7264–71. [CrossRef]
- Mol CD, Brooun A, Dougan DR, Hilgers MT, Tari LW, Wijnands RA, et al. Crystal structures of active fully assembled substrate- and product-bound complexes of UDP-N-acetylmuramic acid:L-alanine ligase (MurC) from Haemophilus influenzae. J Bacteriol 2003;185:4152–62. [CrossRef]
- Jin H, Emanuele John J., Fairman R, Robertson JG, Hail ME, Ho HT, et al. Structural Studies of Escherichia coli UDP-N-Acetylmuramate:l-Alanine Ligase. Biochemistry [Internet] 1996;35:1423–31. [CrossRef]
- Reck F, Marmor S, Fisher S, Wuonola MA. Inhibitors of the bacterial cell wall biosynthesis enzyme MurC. Bioorg Med Chem Lett [Internet] 2001;11:1451–4. Available from: https://www.sciencedirect.com/science/article/pii/S0960894X01002517. [CrossRef]
- Marmor S, Petersen CP, Reck F, Yang W, Gao N, Fisher SL. Biochemical Characterization of a Phosphinate Inhibitor of Escherichia coli MurC. Biochemistry [Internet] 2001;40:12207–14. [CrossRef]
- Sim MM, Ng SB, Buss AD, Crasta SC, Goh KL, Lee SK. Benzylidene Rhodanines as Novel Inhibitors of UDP-N-Acetylmuramate/l-Alanine Ligase. Bioorg Med Chem Lett [Internet] 2002;12:697–9. Available from: https://www.sciencedirect.com/science/article/pii/S0960894X01008320. [CrossRef]
- Bertrand JA, Fanchon E, Martin L, Chantalat L, Auger G, Blanot D, et al. “Open” structures of MurD: domain movements and structural similarities with folylpolyglutamate synthetase11Edited by R. Huber. J Mol Biol [Internet] 2000;301:1257–66. Available from: https://www.sciencedirect.com/science/article/pii/S0022283600939949. [CrossRef]
- El Zoeiby A, Sanschagrin F, Levesque RC. Structure and function of the Mur enzymes: development of novel inhibitors. Mol Microbiol [Internet] 2003;47:1–12. [CrossRef]
- Arvind A, Kumar V, Saravanan P, Mohan CG. Homology modeling, molecular dynamics and inhibitor binding study on MurD ligase of Mycobacterium tuberculosis. Interdiscip Sci [Internet] 2012;4:223–38. [CrossRef]
- Šink R, Barreteau H, Patin D, Mengin-Lecreulx D, Gobec S, Blanot D. MurD enzymes: some recent developments. 2013;4:539–56. [CrossRef]
- Turk S, Kovač A, Boniface A, Bostock JM, Chopra I, Blanot D, et al. Discovery of new inhibitors of the bacterial peptidoglycan biosynthesis enzymes MurD and MurF by structure-based virtual screening. Bioorg Med Chem [Internet] 2009;17:1884–9. Available from: https://www.sciencedirect.com/science/article/pii/S0968089609000935. [CrossRef]
- Perdih A, Kovač A, Wolber G, Blanot D, Gobec S, Solmajer T. Discovery of novel benzene 1,3-dicarboxylic acid inhibitors of bacterial MurD and MurE ligases by structure-based virtual screening approach. Bioorg Med Chem Lett [Internet] 2009;19:2668–73. Available from: https://www.sciencedirect.com/science/article/pii/S0960894X09004636. [CrossRef]
- Basavannacharya C, Robertson G, Munshi T, Keep NH, Bhakta S. ATP-dependent MurE ligase in Mycobacterium tuberculosis: Biochemical and structural characterisation. Tuberculosis [Internet] 2010;90:16–24. Available from: https://www.sciencedirect.com/science/article/pii/S1472979209001152. [CrossRef]
- Amera GM, Khan RJ, Pathak A, Kumar A, Singh AK. Structure based in-silico study on UDP-N-acetylmuramoyl-L-alanyl-D-glutamate-2,6-diaminopimelate ligase (MurE) from Acinetobacter baumannii as a drug target against nosocomial infections. Inform Med Unlocked 2019;16. [CrossRef]
- Humljan J, Kotnik M, Boniface A, Šolmajer T, Urleb U, Blanot D, et al. A new approach towards peptidosulfonamides: synthesis of potential inhibitors of bacterial peptidoglycan biosynthesis enzymes MurD and MurE. Tetrahedron [Internet] 2006;62:10980–8. Available from: https://www.sciencedirect.com/science/article/pii/S0040402006013007. [CrossRef]
- Guzman JD, Gupta A, Evangelopoulos D, Basavannacharya C, Pabon LC, Plazas EA, et al. Anti-tubercular screening of natural products from Colombian plants: 3-methoxynordomesticine, an inhibitor of MurE ligase of Mycobacterium tuberculosis. Journal of Antimicrobial Chemotherapy [Internet] 2010;65:2101–7. [CrossRef]
- Paradis-Bleau C, Lloyd A, Sanschagrin F, Maaroufi H, Clarke T, Blewett A, et al. Pseudomonas aeruginosa MurE amide ligase: enzyme kinetics and peptide inhibitor. Biochemical Journal [Internet] 2009;421:263–72. [CrossRef]
- Shiu WKP, Malkinson JP, Rahman MM, Curry J, Stapleton P, Gunaratnam M, et al. A new plant-derived antibacterial is an inhibitor of efflux pumps in Staphylococcus aureus. Int J Antimicrob Agents [Internet] 2013;42:513–8. Available from: https://www.sciencedirect.com/science/article/pii/S0924857913002975. [CrossRef]
- Longenecker KL, Stamper GF, Hajduk PJ, Fry EH, Jakob CG, Harlan JE, et al. Structure of MurF from Streptococcus pneumoniae co-crystallized with a small molecule inhibitor exhibits interdomain closure. Protein Science [Internet] 2005;14:3039–47. [CrossRef]
- Miller D, HAMMOND S, Anderluzzi D, BUGG T. ChemInform Abstract: Aminoalkylphosphinate Inhibitors of D-Ala-D-Ala Adding Enzyme. Cheminform 2010;29. [CrossRef]
- Comess KM, Schurdak ME, Voorbach MJ, Coen M, Trumbull JD, Yang H, et al. An Ultraefficient Affinity-Based High-Throughout Screening Process: Application to Bacterial Cell Wall Biosynthesis Enzyme MurF. SLAS Discovery [Internet] 2006;11:743–54. Available from: https://www.sciencedirect.com/science/article/pii/S2472555222085173. [CrossRef]
- Gu YG, Florjancic AS, Clark RF, Zhang T, Cooper CS, Anderson DD, et al. Structure–activity relationships of novel potent MurF inhibitors. Bioorg Med Chem Lett [Internet] 2004;14:267–70. Available from: https://www.sciencedirect.com/science/article/pii/S0960894X03010655. [CrossRef]
- Stefane B, Gobec S, Sosič I, Kovač A, Turk S, Blanot D. The Synthesis of Novel 2,4,6-Trisubstituted 1,3,5-Triazines: A Search for Potential MurF Enzyme Inhibitors. Heterocycles 2010;81. [CrossRef]
- Studer G, Rempfer C, Waterhouse AM, Gumienny R, Haas J, Schwede T. QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 2020;36:1765–71. [CrossRef]
- Amera GM, Khan RJ, Pathak A, Jha RK, Jain M, Muthukumaran J, et al. Structure based drug designing and discovery of promising lead molecules against UDP-N-acetylenolpyruvoylglucosamine reductase (MurB): A potential drug target in multi-drug resistant Acinetobacter baumannii. J Mol Graph Model 2020;100. [CrossRef]
- Amera GM, Khan RJ, Pathak A, Jha RK, Muthukumaran J, Singh AK. Screening of promising molecules against MurG as drug target in multi-drug-resistant-Acinetobacter baumannii - insights from comparative protein modeling, molecular docking and molecular dynamics simulation. J Biomol Struct Dyn 2020;38:5230–52. [CrossRef]
- Sangshetti J, Joshi S, Patil R, Shinde D. Mur Ligase Inhibitors as Anti-bacterials: A Comprehensive Review. Curr Pharm Des 2017;23. [CrossRef]
- Heijenoort J van. Recent advances in the formation of the bacterial peptidoglycan monomer unit. Nat Prod Rep [Internet] 2001;18:503–19. [CrossRef]
- Lovering AL, Safadi SS, Strynadka NCJ. Structural Perspective of Peptidoglycan Biosynthesis and Assembly. Annu Rev Biochem [Internet] 2012;81:451–78. [CrossRef]
- Bouhss A, Crouvoisier M, Blanot D, Mengin-Lecreulx D. Purification and Characterization of the Bacterial MraY Translocase Catalyzing the First Membrane Step of Peptidoglycan Biosynthesis*. Journal of Biological Chemistry [Internet] 2004;279:29974–80. Available from: https://www.sciencedirect.com/science/article/pii/S0021925819710059. [CrossRef]
- Mihalyi A, Jamshidi S, Slikas J, Bugg TDH. Identification of novel inhibitors of phospho-MurNAc-pentapeptide translocase MraY from library screening: Isoquinoline alkaloid michellamine B and xanthene dye phloxine B. Bioorg Med Chem [Internet] 2014;22:4566–71. Available from: https://www.sciencedirect.com/science/article/pii/S0968089614005513. [CrossRef]
- Rodolis MT, Mihalyi A, O’Reilly A, Slikas J, Roper DI, Hancock REW, et al. Identification of a Novel Inhibition Site in Translocase MraY Based upon the Site of Interaction with Lysis Protein E from Bacteriophage ϕX174. ChemBioChem [Internet] 2014;15:1300–8. [CrossRef]
- Liu Y, Breukink E. The membrane steps of bacterial cell wall synthesis as antibiotic targets. Antibiotics2016;5. [CrossRef]
- Rodolis MT, Mihalyi A, Ducho C, Eitel K, Gust B, Goss RJM, et al. Mechanism of action of the uridyl peptide antibiotics: an unexpected link to a protein–protein interaction site in translocase MraY. Chemical Communications [Internet] 2014;50:13023–5. Available from: http://dx.doi.org/10.1039/C4CC06516F. [CrossRef]
- Lemieux MJ, Huang Y, Wang DN. Crystal structure and mechanism of GlpT, the glycerol-3-phosphate transporter from E. coli. J Electron Microsc (Tokyo) [Internet] 2005;54:i43–6. [CrossRef]
- Hu Y, Helm JS, Chen L, Ginsberg C, Gross B, Kraybill B, et al. Identification of Selective Inhibitors for the Glycosyltransferase MurG via High-Throughput Screening. Chem Biol [Internet] 2004;11:703–11. Available from: https://www.sciencedirect.com/science/article/pii/S1074552104001103. [CrossRef]
- Helm JS, Hu Y, Chen L, Gross B, Walker S. Identification of Active-Site Inhibitors of MurG Using a Generalizable, High-Throughput Glycosyltransferase Screen. J Am Chem Soc [Internet] 2003;125:11168–9. [CrossRef]
- Liu H, Ritter TK, Sadamoto R, Sears PS, Wu M, Wong CH. Acceptor Specificity and Inhibition of the Bacterial Cell-Wall Glycosyltransferase MurG. ChemBioChem [Internet] 2003;4:603–9. [CrossRef]
- Mann PA, Müller A, Xiao L, Pereira PM, Yang C, Ho Lee S, et al. Murgocil is a Highly Bioactive Staphylococcal-Specific Inhibitor of the Peptidoglycan Glycosyltransferase Enzyme MurG. ACS Chem Biol [Internet] 2013;8:2442–51. [CrossRef]
- Mitachi K, Siricilla S, Klaić L, Clemons WM, Kurosu M. Chemoenzymatic syntheses of water-soluble lipid I fluorescent probes. Tetrahedron Lett [Internet] 2015;56:3441–6. Available from: https://www.sciencedirect.com/science/article/pii/S0040403915000593. [CrossRef]
- Von Döhren H. Antibiotics: Actions, origins, resistance, by C. Walsh. 2003. Washington, DC: ASM Press. 345 pp. $99.95 (hardcover). Protein Science [Internet] 2004;13:3059–60. [CrossRef]
- Mansour TS, Caufield CE, Rasmussen B, Chopra R, Krishnamurthy G, Morris KM, et al. Naphthyl Tetronic Acids as Multi-Target Inhibitors of Bacterial Peptidoglycan Biosynthesis. ChemMedChem [Internet] 2007;2:1414–7. [CrossRef]
Figure 1.
WHO priority pathogen list and A. baumannii as critical pathogenic bacteria.
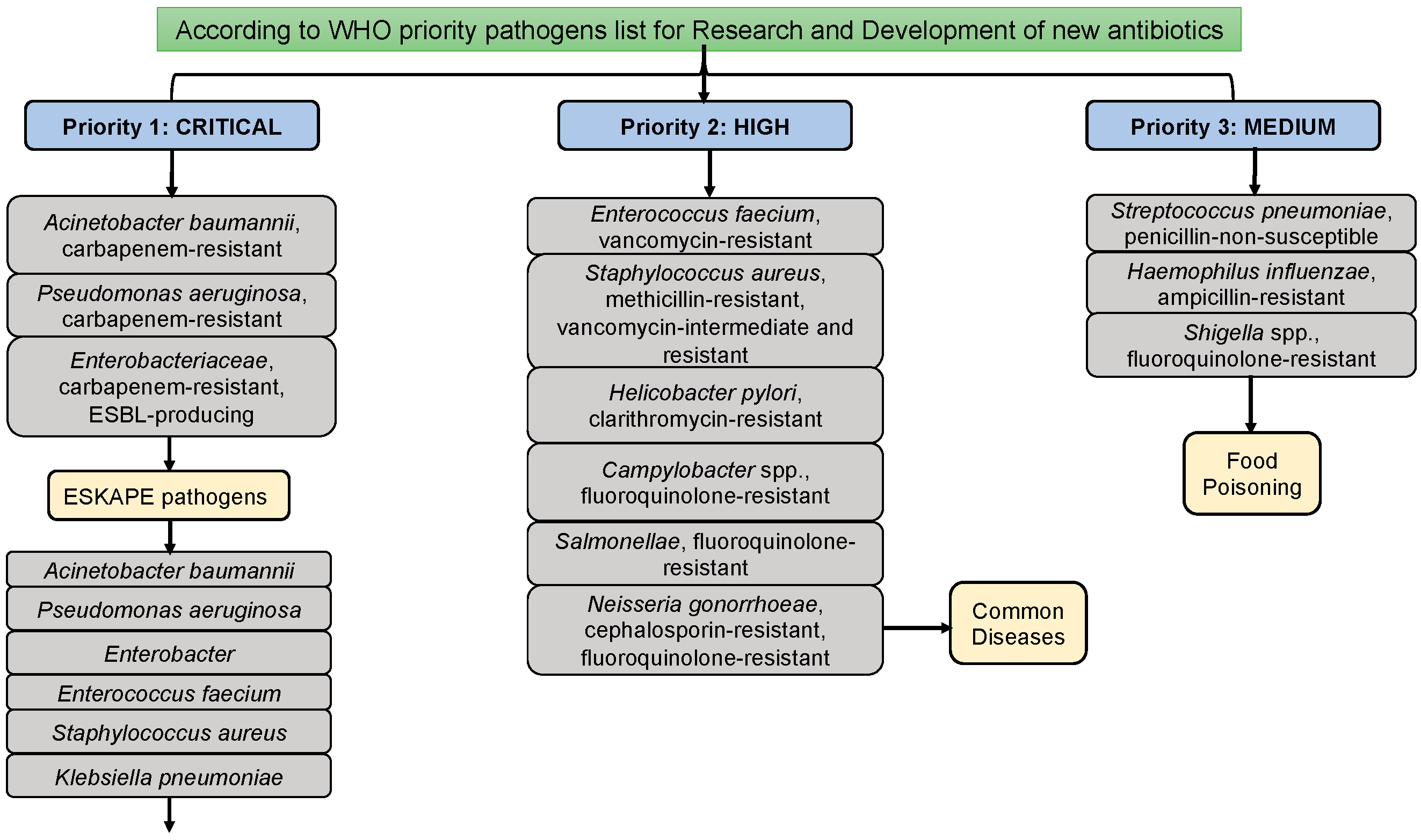
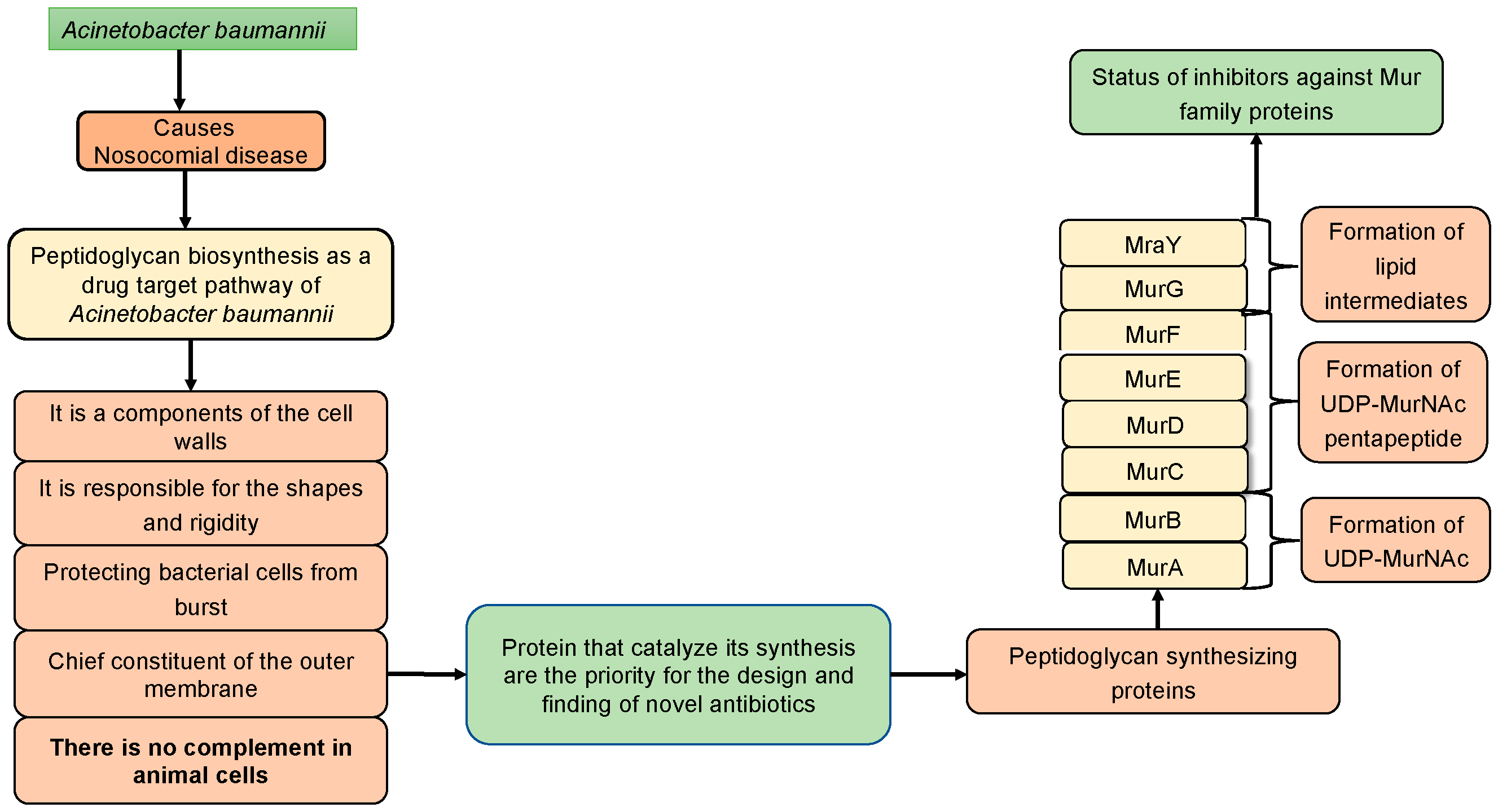
Figure 2.
Stepwise Peptidoglycan biosynthetic pathway.
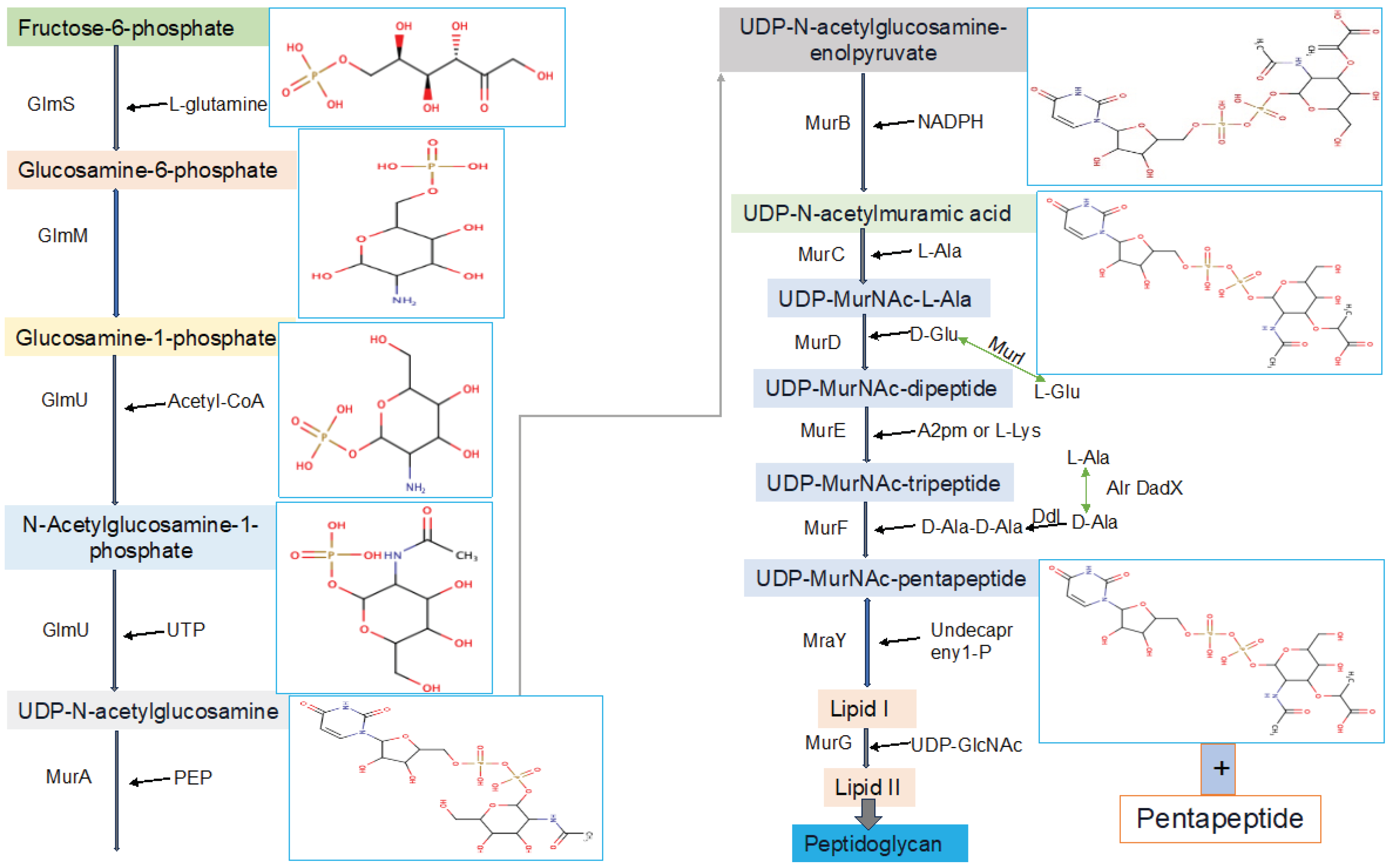
Figure 3.
The summery of functional features of enzymes involved in the assembly of the peptidoglycan monomer unit.
Figure 3.
The summery of functional features of enzymes involved in the assembly of the peptidoglycan monomer unit.
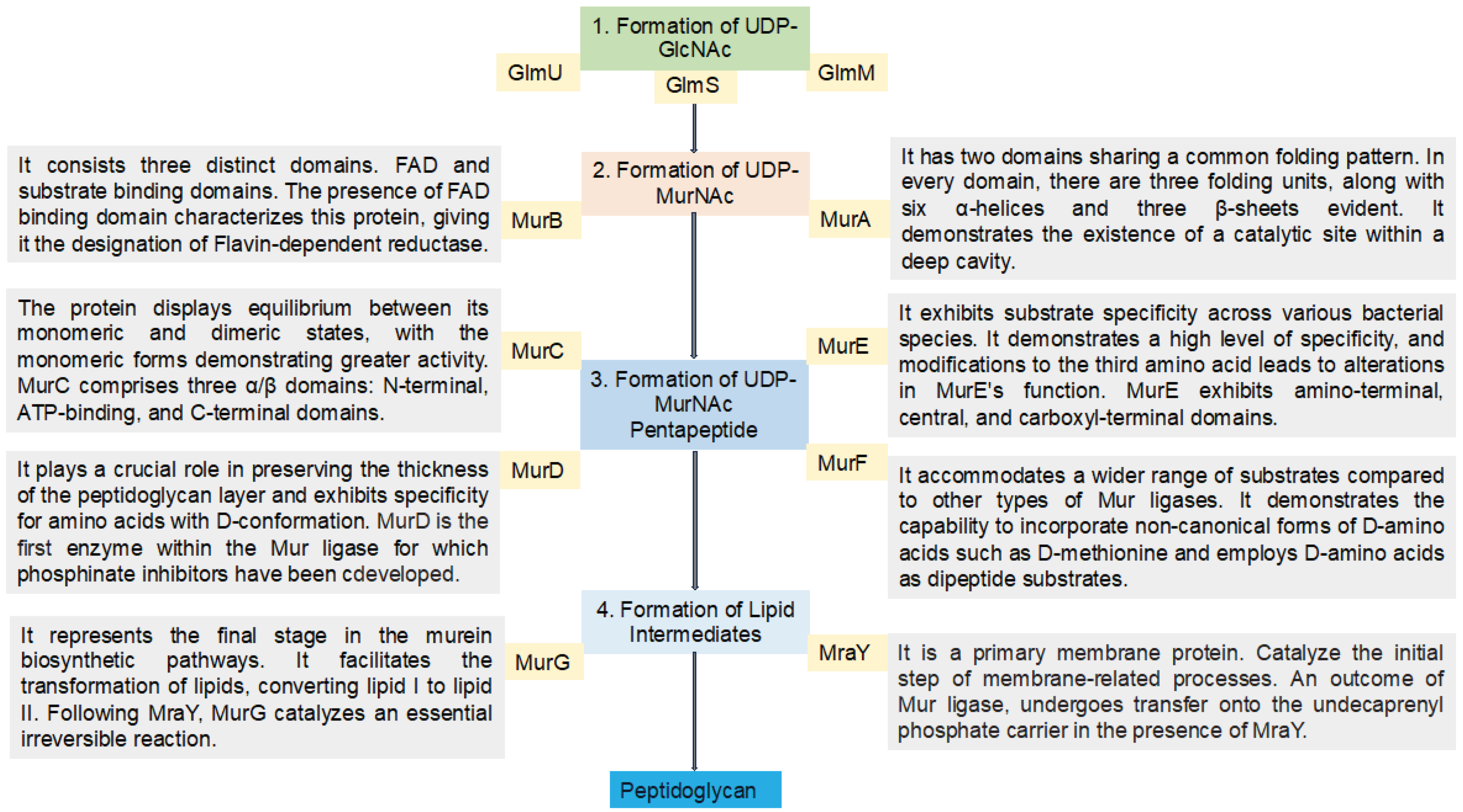
Table 1.
Sequential properties of Mur family proteins.
| Gene Name | Sequential Properties |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Length | MW (kDa) | pI | Hydropathicity | NAS | EGA (% of identity) | VFA (hit score) | SSC | References | |
| MurA | 418 | 44.6 | 5.12 | Hydrophobic | 20 | 100 | No hits found. | Alpha-Beta | [24,25,26] |
| MurB | 353 | 39.7 | 6.18 | Hydrophobic | 16 | 99.65 | 54 | Alpha-Beta | [24,26] |
| MurC | 482 | 52.8 | 5.64 | Hydrophobic | 25 | 100 | No hits found. | Alpha-helix and random coil | [24,26,27] |
| MurD | 455 | 48.65 | 6.14 | Hydrophobic | 21 | 99.31 | No hits found. | Alpha-Beta | [24,26,28] |
| MurI | 266 | 29.03 | 5.40 | Hydrophobic | - | 41.47 | No hits found. | α/β structure | [26,29] |
| MurE | 499 | 54.98 | 5.48 | Hydrophobic | 22 | 99.52 | No hits found | Alpha-Beta | [24,26] |
| MurF | 466 | 50.79 | 5.87 | Hydrophobic | 19 | 98.74 | No hits found | Alpha-helix and random coil | [24,26,30] |
| MurG | 365 | 39.35 | 9.02 | Hydrophobic | 14 | 86.03 | No hits found. | mixed αβ class | [24,26] |
| MraY | 372 | 40.9 | 9.54 | Hydrophobic | 13 | 92.47 | 53 | Alpha-helix and random coil | [15,24,26] |
Number of antigenic sites = NAS; Secondary structural class = SSC; Virulence Factors Alignment = VFA; Essential Gene Alignment=EGA.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



