
Submitted:
19 February 2024
Posted:
19 February 2024
You are already at the latest version
Abstract
(S)-Atenolol ((S)-2-(4-(2-Hydroxy-3-(isopropylamino)propoxy)phenyl)acetamide) has been synthesized in >99% ee with use of Candida antarctica lipase B from Syncozymes (Shanghai, China), in a kinetic resolution of the corresponding racemic chlorohydrin. Deprotonation of the starting phenol building block was base catalyzed. The enantiopurity of the chlorohydrin building block remained unchanged upon subsequent amination. All four steps in the synthesis protocol have been optimized compared to previously reported methods, which makes this new protocol more sustainable and in accordance with green chemistry principles compared to previously reported protocols.
Keywords:
Enantiopure (S)-atenolol
; Candida antarctica lipase B
; base catalysis
; Chiralcel OD-H column
1. Introduction
The American Heart Association reported in March 2019 their update on heart diseases and stroke statistics. The report states that high blood pressure concerned 46% of the total population at ages 20 years and older in the United States between 2013-2016. It was the cause of death for 82,735 Americans in 2016 and costed the American society approximately $ 55.9 billion in the period 2014-2015 [1]. In 2018, cardiovascular treatment made up 4.9% of the total pharmaceutical marked in Norway, which corresponded to 1,17 million Norwegian kroner [2]. A class of drugs that have been used in the treatment of both cardiovascular and non-cardiovascular diseases are the β-adrenergic blocking agents (beta-blockers). Approximately 300,000 patients in Norway use beta-blockers [3]. Worldwide, the use of beta-blockers increase year by year and the sales are estimated to account for 13,684 million USD by 2030 [4].
The highly polar cardio selective β1-antagonist atenolol is selective towards β1-receptors found in the heart. This drug is used in the treatment of hypertension, angina pectoris and arrythmia. Atenolol is one of the most widely used beta-blockers clinically and is often used as a reference drug for comparisons with other antihypertensives. The drug might however be even more effective in preventing myocardial infarction [5].
Atenolol is manufactured with enantiomerically pure active pharmaceutical ingredient (API) as Atpure® by Emcure Pharmaceuticals, (Pune, India) and with racemic API under the names Tenormin®, Mylan® and others. The eudismic ratio is 46 in favour of (S)- to (R)-atenolol [6], and studies in rats show that the R-enantiomer has no effect. While the racemic drug causes a lowering of the heart rate, this side effect is not observed with enantiopure (S)-atenolol [7].
Several synthesis protocols for producing enantiopure (S)-atenolol have been published. Emcure Pharmaceuticals uses enantiopure epichlorohydrin to produce enantiopure (S)-atenolol from deprotonated 2-(4-hydroxyphenyl)acetamide. The specific rotation of the final drug (S)-atenolol of = −17.1 (1.0, 1N HCl) is reported [8]. Dwivedee et al. also started with deprotonation of 2-(4-hydroxyphenyl)acetamide gaining only the epoxide 2-(4-(2-oxiran-2-ylmethoxy)phenyl)benzeneacetamide. Reaction of this epoxide with acetyl chloride in methanol gave the racemic 4-(3-chloro-2 hydroxypropoxy)benzeneacetamide, which was resolved by several lipase preparations and vinyl acetate as the acyl donor. The authors claim to have formed (S)-atenolol from (S)-4-(3-chloro-2-hydroxypropoxy)benzeneacetamide, which is not possible according to their reaction conditions [9]. To our knowledge, addition of isopropylamine in water to (S)-4-(3-chloro-2-hydroxypropoxy)benzeneacetamide leads to (R)-atenolol. The authors do not report any specific rotation of their (claimed) (S)-atenolol product, nor of their enantiopure building blocks. Agustian et al. present a similar study of (S)-atenolol synthesis using several lipase preparations to resolve the racemic 4-(3-chloro-2 hydroxypropoxy)benzeneacetamide [10]. The authors give no evidence of the absolute configuration of the product, nor any yields or evidence of the enantiomeric excess (ee) of the product. Sikora et al. reported in 2020 the kinetic resolution of racemic atenolol catalyzed by lipase from Candida rugosa with isopropenyl acetate as the acyl donor, giving the acetate of (S)-atenolol in 94% ee. The authors give no evidence of the absolute configuration of this acetate, nor of the unreacted (R)-atenolol [11,12]. The authors have previously published several articles of lipase catalyzed kinetic resolution of racemic atenolol with the amide of (S)-atenolol presented as the acetate [13-15]. (S)-Atenolol has been synthesized in 98% ee in a seven step method using Jacobsen’s catalyst ((R,R)-salen Co(III)OAc) [16], and in 94% ee through kinetic resolution of racemic atenolol using lipase from Pseudomonas cepacia [17]. We have produced the enantiopure building block (R)-4-(3-chloro-2 hydroxypropoxy)benzeneacetamide starting with a deprotonation of 2-(4-hydroxyphenyl)acetamide with sodium hydroxide, and by using lipase B from Candida antarctica (CALB) in the kinetic resolution of the racemic chlorohydrin 4-(3-chloro-2 hydroxypropoxy)benzeneacetamide the enantiopure chlorohydrin was obtained with 99% ee [18]. We have now improved the yield of the building block and reduced the amounts of reactants used. The enantiopure drug (S)-atenolol has been synthezised from the enantiopure chlorohydrin.
2. Results and discussion
2.1. Deprotection of the phenol proton
The first step in the synthesis of (S)-atenolol ((S)-4) is the formation of 4-(3-chloro-2-hydroxypropoxy)benzeneacetamide (2a) and 2-(4-(2-oxiran-2-ylmethoxy)phenyl)benzeneacetamide (2b) from phenol 2-(4-hydroxyphenyl)acetamide (1), sodium hydroxide and epichlorohydrin. The impact of the amount of sodium hydroxide, epichlorohydrin and acetic acid used in the reaction was investigated, in order to improve the overall yield of the product compared to earlier reported data (Scheme 1).
2.2. Dimer Formation With Use of Stoichiometric Amount of Base
When stoichiometric amounts of sodium hydroxide was used to deprotonate the phenolic proton of 1 a small peak at tR = 13.37 min is seen in the HPLC chromatogram together with the wanted product chlorohydrin 2a (tR = 12.06 min) and the epoxide 2b (tR = 9.58 min), see Figure 1. LC-MS analysis of the reaction mixture on an AQUITY UPLC BEH C18-column with isocratic mobile phase composition of water and acetonitrile and (30:70) with 1% formic acid and a flow of 0.2 mL/min showed a peak with the molecular mass of 382.04 g/mol, which corresponds to the mass of C19H21N2O5Na (Figure 2). The compound has then a molecular mass of 358.39 g/mol, which corresponds to the dimer 2c. We have previously predicted the mechanism of dimer formation in syntheses of similar beta-blockers [19].
2.3. Base Catalyzed Deprotection of Phenol 1
When two equivalents of epichlorohydrin were added to a solution containing phenol 1 and catalytical amounts of sodium hydroxide, the by-product 2c was not observed after 4 hours of reaction. A proposed mechanism for the reaction of 1 to 2a and 2b with the regeneration of the base is shown in Scheme 2. Phenol 1 is deprotonated forming alkoxide 1’ which can attack epichlorohydrin at carbon 1 and 3 (reaction mechanism a and b, respectively) giving alkoxide 2a’ and epoxide 2b. Alkoxide 2a’ reacts in two ways; by protonation from water forming chlorohydrin 2a, thus regenerating the base (reaction mechanism c), and by an intramolecular SN2-like reaction forming epoxide 2b (reaction mechanism d).
Use of catalytic amounts of base is possible due to the regeneration of the base during the deprotonation of water during the formation of 2a from 2a‘. For the same reason it is also possible to use less acetic acid than previously reported in the syntheses of other beta-blockers. We have previously used between 5 and 10 equivalents of acetic acid [20]. By the addition of one equivalent of lithium chloride and five equivalents of acetic acid to the mixture of 2a and 2b, a 52% yield of 2a was achieved.
2.4. Lipase Catalyzed Kinetic Resolution of Chlorohydrin 2a
A CALB catalyzed kinetic resolution of 2a with vinyl butanoate as the acyl donor produced (R)-2a in 99% ee with 32% yield (Scheme 1). Figur X shows the chromatogram of the reaction after 24 hours of a total of 27 hours reaction time. AS shown, the product ester S-2b was not separated on this column, however, it was hydrolyzed and analyzed as the S-2a as shown in outr previous published work on this topic. We have previously obtained a yield of 16% of (R)-2a, and the E-value of the kinetic resolution of 2a was >200. In our previous article we also present data of the (S)-enantiomer of the chlorohydrin, (S)-2a [18].
2.5. Synthesis of racemic atenolol (4) and attempt to resolve 4 with CALA
Racemic atenolol (4) was produced directly from the reaction mixture of 2a and 2b (without the ring opening of 2b with lithium chloride) with addition of isopropylamine in water giving 4 in 42% yield (Scheme 3).
Attempts to resolve 4 in n-hexane with lipase A from Candida antarctica (CALA) as catalyst and vinyl butanoate as acyl donor was performed with an E-value of 1.8 (Scheme 4). We have previously had success with using CALA as catalyst in kinetic resolution of secondary alcohols with two (quite) large groups connected to the stereocenter, which atenolol also has [21].
The ee of the unreacted (S)-atenolol was 4% (ees ((S)-atenolol, (S)-4)) and ee for the product ester (R)-4b was 27% (eep). The retention times were assigned due to the known stereo preference of CALA [21], tR (S)-4 = 18.36 min and tR (R)-4 = 28.97 min, RS = 2.72, tR (S)-4b = 25.04 min and tR (R)-4b = 33.87 min, Chiralcel OD-H column with a gradient mobile phase i-PrOH/n-hexane; 9:95 (0 min) -10:90 (10 min) - 60:40 (80 min), flow 0.5 mL/min (Figure 4).
These results show that CALA exhibits low selectivity for the enantiomers of atenolol (4). The racemic compound with tR = 14.15 min and tR = 16.94 min is anticipated to be the amide product 4d (Scheme 4). As mentioned, Chalupka (Sikora) et al. have succeeded in resolving racemic atenolol with Candida rugosa lipase with vinyl acetate as the acyl donor giving 94% ee of the (S)-atenolol acetate [11].
3. Materials and methods
3.1. Chemicals
All chemicals are commercially available and of analytic grade. The chemicals were bought from Sigma-Aldrich Norway (Oslo, Norway). HPLC grade solvents were used for HPLC analyses. Dry solvents (tetrahydrofuran and acetonitrile) were prepared with a solvent purifier, MBraun MDSPS800. (München, Germany). n-Hexane was dried manually by adding molecular sieves (4Å) to the solvent 24 h before use. Molecular sieves (1/8 pellets, pore diameter 4Å) were placed in a porcelain dish and dried at 1000°C for 24 h and kept in a desiccator thereafter.
3.2. Enzymes
Candida antarctica Lipase B (CALB) (activity ≥ 10,000 PLU/g, 1 unit corresponds to the synthesis of 1 mmol per minute propyl laureate from lauric acid and 1-propanol at 60℃, lot no. 20170315), immobilized at high hydrophobic macroporous resin, produced in fermentation with genetically modified Pichia pastoris was gifted from Syncozymes Co, Ltd. (Shanghai, China). Candida antarctica lipase A (activity = 725 U/g, lot no. VZ1030-12, batch no 080116) immobilized on microporous beads was a gift from Viazym BV (Delft, The Netherlands).
The enzymatic reactions were performed in a New Brunswick G24 Environmental Incubator Shaker from New Brunswick Co. Inc. (Edison, NJ, USA) or in an Infors Minitron (Infors AG, Bottmingen, Switzerland).
3.3. General Analyses
TLC was performed on Merck silica 60 F254 and detected by UV at λ = 254 nm. Flash chromatography was performed on silica gel from Sigma-Aldrich (Oslo, Norway). Pore size 60 Å, 230-400 mesh particle size, 40-63 μm particle size.
3.4. High-Performance Liquid Chromatography (HPLC)
Achiral HPLC analyses were performed on an Agilent 1290 system equipped with an auto injector (4 μL), detection was done by a diode array detector (DAD, l = 254 nm). All separations of 1, 2a, and 2b were performed on an Agilent Zorbax Eclipse XBD-C18 column (150 mm L × 4.6 mm i.d., 5 μm particle size) from Matriks (Oslo, Norway) with an isocratic eluent (H2O:MeCN, 75:25) over 5 min with a flow of 1.0 mL/min, which gave tR 1b = 1.86 min, tR 2b = 3.04 min and tR 2a = 3.32 min. When stoichiometric amount of sodium hydroxide was used also the dimer 2c was seen, then a linear gradient mobile phase composition of H2O and MeCN (75:25) - (65:35) over 20 min with 0.5 mL/min flow was the method on the Zorbax Eclipse XDB C18-column, giving tR 1 = 3.03 min, tR 2a = 12.06 min, tR 2b = 9.59 min and the by-product dimer tR 2c = 13.38 min.
LC-MS analysis of 2c was performed on a ACQUITY UPLC BEH C18 column (100 mm L x 2.1 mm i.d., 1.7 μm particle size) from WatersTM (Waters Norway, Oslo, Norway) with isocratic mobile phase (H2O:MeCN, 30:70) with 1% formic acid and a flow of 0.2 mL/min giving a mass of 382.04 g/mol which is which corresponds to C19H21N2O5Na. The calculated mass of 2c is 358.39 g/mol, the formula for is C19H22N2O5 .
Chiral HPLC analyses were performed on an Agilent HPLC 1100 with a manual injector (Rheodyne 77245i/Agilent 10 μl loop). A Chiralcel OD-H column from Daicel, Chiral Technologies Europe (Gonthier d´Andernach, Illkirch, France) was used (250 mm L, x 4.6 mm i.d., 5 μm particle size) in addition to the reverse phase corresponding column chiralcel OD-RH (150 x 4.6 mm i.d., 5 μm particle size). The method used for all analyses was i-PrOH:n-hexane, 17:83, flow 1 mL min-1, UV 254 nm. The enantiomers of 2a eluted with tR (S)-2a = 41.52 min, tR (R)-2a = 46.32 min, RS = 1.74. The purified (R)-2a was analyzed by the same method as racemic 2a: tR (R)-2a = 46.32 min, with no presence of (S)-2a in the chromatogram, resulting in an enantiomeric excess of > 99% ee. For determination of the E-value of the enzyme catalyzed kinetic reesolution of 2a, see Lund et al. [18]. With the use of Chiralcel OD-RH (reverse phase) the retention times were tR (R)-2a = 6.32 min and tR (S)-2a = 7.17 min. Retention times of 4 on Chiralcel OD-H: tR (S)-4 = 18.37 min and tR (R)-4 = 28.98 min, RS = 2.72. Purified (S)-atenolol: tR (S)-4 = 18.39 min.
3.5. NMR Analyses
NMR-analyses were recorded on a Bruker 400 MHz Avance III HD instrument equipped with a 5 mm SmartProbe Z-gradient probe operating at 400 MHz for 1H and 100 MHz for 13C, respectively, or on a Bruker 600 MHz Avance III HD instrument equipped with a 5 mm cryogenic CP-TCI Z-gradient probe operating at 600 MHz for 1H and 150 MHz for 13C (Bruker, Rheinstetten, Germany). Chemical shifts are in ppm relative to TMS and coupling constants are in hertz (Hz).
3.6. Mass Spectroscopy Analyses
Accurate mass determination in positive and negative mode was performed on a ”Synapt G2-S” Q-TOF instrument from Waters™ (Waters Norway, Oslo, Norway). Samples were ionized by the use of ASAP probe (APCI). Calculated exact mass and spectra processing was done by Waters™ Software (Masslynxs V4.1 SCN871).
3.7. Infrared Spectroscopy Analyses
Infrared spectroscopy was performed on a NEXUS FT-IR model 470 instrument from Thermo Nicolet Corporation (Madison, WI, USA).
3.8. Specific Rotation Analyses
Specific rotation was determined on a PerkinElmer Model 341 Polarimeter (Waltham, MA, USA), with a cell of 10 cm length, λ 589 nm.
3.9. Assignment of Absolute Configurations
Absolute configuration of the faster reacting enantiomer in lipase catalyzed resolution was determined by the known enantioselectivity of CALA [21] and CALB [22] and by comparing the elution orders of the atenolol building block enantiomers and drug enantiomer with previous analyses of similar α-halogenated 1-(4-benzyloxy)phenyl)ethanols on the Daicel Chiralcel OD-H column. In general, the R-enantiomers are the most retained [23].
3.10. Synthesis Protocols
3.10.1. Synthesis of chlorohydrin 2a and epoxide 2b
To an aqueous solution of NaOH (0.2M, 8.71 mL, 1.98 mmol) and 2-(4-hydroxyphenyl)-acetamide (1) (1.00 g, 6.62 mmol), epichlorohydrin (1.04 mL, 13.26 mmol) were added and set to stir at rt. for 4 h. The reaction was monitored by TLC (MeOH:CH2Cl2, 1:5): Rf 2a = 0.56, Rf 2b = 0.63. The reaction mixture was washed with H2O in vacuo giving a 1.44 g mixture of chlorohydrin 2a and epoxide 2b as a white solid, analyzed on the Zorbax Eclipse XBD-C18 HPLC column with an isocratic mobile phase (MeCN:H2O:, 25:75) over 5 min, flow = 1.0 mL/min, tR (1) = 1.86 min, tR 2a =3.32 min and tR 2b = 3.04 min.
3.10.2. Synthesis of chlorohydrin 2a by ring-opening of epoxide 2b
To a mixture of chlorohydrin 2a and epoxide 2b (1.44 g), MeCN (20 mL), LiCl (0.28 g, 6.6 mmol) and AcOH (1.98 mL, 34.62 mmol) was added and stirred at rt. for 24 h. The reaction was monitored by TLC (CH2Cl2:MeOH, 1:5), Rf 2a = 0.56. The reaction mixture was quenched with Na2CO3 (pH 12) to a neutral pH followed by extraction with CH2Cl2 (3 x 50 mL). The organic layer was dried over MgSO4 and the solvent was removed under reduced pressure and further in vacuo giving chlorohydrin 2a in 52% yield (0.85 g, 3.49 mmol) as a white solid; mp. 143-145°C. The conversion of epoxide 2b was analyzed on the Zorbax Eclipse XBD-C18 HPLC column with an isocratic mobile phase (MeCN:H2O:, 25:75) over 5 min, flow = 1.0 mL/min, tR 2a =3.32 min. 1H NMR (600 MHz, DMSO-d6) δ ppm: 7.38 (s, 1H, -NH-H), 7.17-7.16 (m, 2H aromatic), 6.88-6.86 (m, 2H, aromatic), 6.82 (s, 1H, -NH-H), 5.54-5.53 (d, 1H, 3JHH = 5.13 Hz, -OH), 4.04-4.00 (sextet, 1H, 3JHH = 5.13 Hz, -CH-OH), 3.97-3.92 (m, 2H, -O-CH2-), 3.68-3.65 (2H, m, 2H, CH2-Cl), 3.28 (s, 2H, -CH2-CONH2); 13C NMR (600 MHz, DMSO-d6) δ ppm: 172.9, 157.5, 130.5 (2C), 129.2, 114.7 (2C), 69.5, 69.1, 47.2, 41.8. MS (TOF-ASAP): [M+H]+ 244.0739, (calcd. C11H14NO3Cl, 243.654). IR (cm-1): 3349, 1633, 1241, 706.
3.10.3. Synthesis of Racemic Atenolol (4) Directly from Phenol 1
2-(4-Hydroxyphenyl)acetamide (1) (2.52 g, 16.67 mmol) was stirred in 2-(chloromethyl)oxirane (epichlorohydrin) (13 mL, 165.80 mmol) at rt, and was added a solution of NaOH (0.33 g, 8.25 mmol) in H2O (5 mL). After 48 h, full conversion was observed by TLC (MeOH: CH2Cl2: 1:4), which showed the presence of both 2a and 2b. The mixture was filtered, and the solids were dried under reduced pressure, before the crude product was dissolved in MeOH (25 mL) and added i-PrNH2 (10 mL, 116.39 mmol). After stirring for 24 h, full conversion of 1 to 4 was observed by TLC. The solvent was removed under reduced pressure. This afforded 5.49 g crude product of which 1.03 g was recrystallized from MeCN, afforded atenolol (4) in 42% total yield (0.35 g, 1.31 mmol) and > 98% purity (NMR). 1H NMR (400 MHz, CD3OD) δ ppm: 7.26-7.24 (d, 2H, aromatic, 3JHH = 7.5 Hz), 6.95-6.93 (d, 2H, aromatic, 3JHH = 7.5 Hz), 4.24-4.22 (m, 1H, CH-OH, 3JHH = 4.4 Hz), 4.08-3.99 (m, 2H, -O-CH2-, 3JHH = 5.0 Hz, 9.5 Hz), 3.49-3.43 (m, 3H, CO-CH2-, -NH-CH-, 3JHH = 6.6 Hz), 3.30-3.27 (m, 1H, -CH2-NH-, 3JHH = 13.2 Hz), 3.18-3.12 (t, 1H, -CH2-NH-, 3JHH = 10.5 Hz), 1.39-1.38 (m, 6H, 3JHH = 3.5 Hz). 13C NMR (400 MHz, CD3OD) δ ppm: 175.8, 157.5, 129.9, 128.4, 114.3, 69.6, 65.6, 50.6, 47.1, 41.0, 18.0, 17.4.
3.10.4. Synthesis of (R)-2-(4-(3-chloro-2 hydroxypropoxy)phenyl)acetamide, (R)-2a
Chlorohydrin 2a (0.56 g, 2.3 mmol) and vinyl butanoate (1.43 g, 12.5 mmol) were added to a flask with dry MeCN (40 mL) and molecular sieve. CALB (0.71 g) was added, and the reaction was incubated at 30°C and 200 rpm for 27 h in an incubator shaker. The enzyme and the molecular sieves were filtered off and the solvent was removed under reduced pressure. The ester (S)-3 and the chlorohydrin (R)-2a were separated on a silica column with EtOAc as eluent. This yielded (R)-2a (0.090 g, 0.37 mmol, 32% yield), ee > 99%. = −3.0 (1.0, MeOH), which is in accordance with our previous reported data of (R)-2a [18]. The E-values and Keq were calculated by the software program E&K Calculator 2.1b0 PPC [24].
3.10.5. Synthesis of (S)-Atenolol, (S)-4
To (R)-2a (0.090 g, 0.37 mmol) i-PrNH2 (3 mL, 34.9 mmol), and dist. H2O (1.0 mL) was added. The reaction was stirred at room temp. for 48 h until TLC (MeOH:CH2Cl2, 1:5) showed full conversion. This gave (S)-4 as a white powder (0.054 g, 0.22 mmol, 60% yield), 99% purity (NMR), ee > 99%. = −17.0 (1.0, 1N HCl). NMR spectra as for racemic 4.
3.10.6. Kinetic resolution of 2-(4-(2-hydroxy-3 (isopropylamino)propoxy)phenyl)acetamide (4)
To a solution of 2-(4-(2-hydroxy-3-(isopropylamino)propoxy)phenyl)acetamide (4) (35.1 mg, 0.13 mmol) and vinyl butanoate (81.3 mg, 0.71 mmol) in MeCN (3 mL) placed in an incubator shaker at 30°C and 200 rpm, CALA (20.4 mg) was added. Samples were collected over four days, which were analyzed by chiral HPLC (n-hexane:i-PrNH2:Et2NH: 80:20:0.1) which showed low enantioselectivity (E = 1.8) and the reaction was therefore not further analyzed.
4. Conclusions
A four-step synthesis of (S)-atenolol ((S)-4) in >99% ee has been performed, starting from 2-(4-hydroxyphenyl)-acetamide (1). Base catalyzed deprotonation of the starting material avoided formation of the dimer by-product (2c), thus increasing the overall yield of the racemic chlorohydrin 2a to 52%, an improvement from our previous reported yield of 22%. CALB catalyzed kinetic resolution of chlorohydrin 2a gave the enantiopure (R)-2a in >99% ee with 32% yield. CALB catalyzed kinetic resolution of chlorohydrin 2a is an efficient method to obtain enantiopure building blocks for beta-blockers, however, the yield of this enzymatic step is limited to 50%. By use of f. inst. dynamic kinetic resolution inverting the “wrong” enantiomer to the right one, the yield could be increased further [25]. Our new protocol to achieve enantiopure (S)-atenolol has fewer steps and uses reduced amounts of all reagents in the synthesis compared to previous published protocols.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Chromatograms.
Author Contributions
Conceptualization, E.E.J. and F.H.B.; methodology, E.E.J., M.B.H. and F.H.B.; validation, F.H.B., L.H.Y.B. and E.E.J.; formal analysis, M.B.H and A.L.T.; investigation, M.B.H. and A.L.T.; resources, E.E.J.; data curation, M.B.H. and A.L.T.; writing—original draft preparation, E.E.J. and M.B.H.; writing—review and editing, E.E.J. and L.H.Y.B.; supervision, E.E.J.; project administration, E.E.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Acknowledgments
EEA project 18-COP-0041 GreenCAM is thanked for support. Syncozymes Co. LTD, Shanghai, China, is thanked for gift of CALB, Viazym BV (Delft, The Netherlands) is thanked for gift of CALA.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Benjamin, E.J., Muntner, P., Alonso, A., Bittencourt, M.S., Callaway, C.W., Carson, A.P., Chamberlain, A.M., Chang, A.R., Cheng, S., Das, S.R., Delling, F.N., Djousse, L., Elkind, M.S.V., Ferguson, J.F., Fornage, M., Jordan, L.C., Khan, S.S., Kissela, B.M., Knutson, K.L., Kwan, T.W., Lackland, D.T., Lewis, T.T., Lichtman, J.H., Longenecker, C.T., Loop, M.S., Lutsey, P.L., Martin, S.S, Matsushita, K., Moran, A.E., Mussolino, M.E., O’Flaherty, M., Pandey, A., Perak, A.M., Rosamond, W.D., Roth, G.A., Sampson, U.K.A., Satou, G.M., Schroeder, E.B., Shah, S.H., Spartano, N.L., Stokes, A., Tirschwell, D.L., Tsao, C.W., Turakhia, M.P., VanWagner, L.B., Wilkins, J.T., Wong, S.S., Virani, S.S. Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation 2019, 139 (10), e56-e528. [CrossRef]
- Dansie, L.S., Bakken, G.V., Berg, C.L., Blix, H.S., Ilic, M., Litleskare, I., Sharikabad, M.N., Skoufa, I.I. , Torheim S. and Granum. T. Drug Consumption in Norway 2017-2021. Norwegian Institute of Public Health, Sept. 2022, pp 172, ISBN electronic ed.: 978-82-8406-313-3.
- Norwegian Health Information. www.nhi.no Accessed Feb. 11, 2024.
- https://www.databridgemarketresearch.com/reports/global-beta-blockers-market. Accessed Feb. 11, 2024.
- Carlberg, B., Samuelsson, O., Lindholm. L.J. Atenolol in hypertension: is it a wise choice? Lancet 2004, 364, 1684-1689. [CrossRef]
- Stoschitzky K, Egginger G, Zernig G, Klein W, Lindner W. Stereoselective features of (R)- and (S)-atenolol. Stereoselective Features of (R)- and (S)-Atenolol: Clinical, Pharmacological, Pharmacokinetic, and Radioligand Binding Studies. Chirality 1993, 5, 15-24. [CrossRef]
- Stoschitzky, K.; Lindner, W.; Zernig, G. Racemic beta-blockers – fixed combinations of different drugs. J. Clin. Bas. Cardiol. 1998, 1, 15–19. [Google Scholar]
- Mehta, S.R., Bhawal, B.M., Deshpande, V.H., Gurjar, M.K. PROCESS FOR PRODUCING ATENOLOL OF HIGH OPTICAL PURITY. Emcure Pharmaceuticals Limited, Pune, India, US patent 6,982,349 B1, Jan. 2006.
- Dwivedee, B.P., Gosh, S., Bhaumik, J., Banoth, L. Banerjee, U.C. Lipase-catalyzed green synthesis of enantiopure atenolol. RSC Adv. 2015, 5 (21), 15850-15860. [CrossRef]
- Agustian, J.; Kamaruddin, A.H.; Aboul-Enein, H.Y. Enantio-conversion and -selectivity of racemic atenolol kinetic resolution using free Pseudomonas fluorescens lipase (Amano) conducted via ransesterification reaction. Rsc. Adv. 2016, 6, 26077–26085. [Google Scholar] [CrossRef]
- Sikora, A.; Chałupka, J.; Marszałł, M.P. The Use of Ion Liquids as a Trojan Horse Strategy in Enzyme-Catalyzed Biotransformation of (R,S)-Atenolol. Catalysts 2020, 10((7)), 787–798. [Google Scholar] [CrossRef]
- Chałupka, J., Sikora, A., Marszałł, M. P. The Utilization of Two-Phase Catalytic System in Enantioselective Biotransformation of Racemic Atenolol. Catalysts 2022, 12, 1068-1080. [CrossRef]
- Sikora, A., Chelminiak-Dudkiewicz, D., Ziegler-Borowska, M., Marszałł, M.P. Enantioseparation of (RS)-atenolol with the use of lipases immobilized onto new-synthesized magnetic nanoparticles. Tetrahedron-Asymmetry 2017, 28, 374-380. [CrossRef]
- Sikora, A., Chełminiak-Dudkiewicz, D., Siódmiak, T., Tarczykowska, A., Sroka, W.D., Ziegler-Borowska, M., Marszałł, M.P. Enantioselective acetylation of (R,S)-atenolol: The use of Candida rugosa lipases immobilized onto magnetic chitosan nanoparticles in enzyme-catalyzed biotransformation. J. Mol. Catal. B Enzym. 2016, 134, 43-50. [CrossRef]
- Sikora, A., Sroka, W.D., Siodmiak, T., Marszałł, M.P. Kinetic Resolution of (R, S)-atenolol with the Use of Lipases in Various Organic Solvents. Curr. Org. Synth. 2017, 14, 747-754. [CrossRef]
- Subhas Bose, D., Venkat Narsaiah, A. An efficient asymmetric synthesis of (S)-atenolol: using hydrolytic kinetic resolution. Bioorg. & Med. Chem. 2005, 13 (3), 627-630. [CrossRef]
- Darnle, S.V., Patil, P.N., Salunkhe, M.M. Chemoenzymatic Synthesis of (R) - and (S) -Atenolol and Propranolol employing Lipase Catalyzed Enantioselective Esterification and Hydrolysis. Synth. Comm. 1999, 29 (22), 3855-3862. [CrossRef]
- Lund, I.T., Bøckmann, P.L., Jacobsen, E.E. Highly enantioselective CALB-catalyzed kinetic resolution of building blocks for b-blocker atenolol. Tetrahedron, 2016, 72, (46), 7288-7292. [CrossRef]
- Troøyen, S.H., Tennfjord, A.L., Klungseth, K., Bocquin, L.H.Y., Jacobsen, E.E. Green Chemo-Enzymatic Protocols for the Synthesis of Enantiopure b-Blockers (S)-Esmolol and (S)-Penbutolol. Catalysts 2022, 12, 980-992. [CrossRef]
- Bøckmann, P.L., Jacobsen, E.E. Chemo-Enzymatic Synthesis of Enantiopure β-Blocker (S)-Metoprolol and Derivatives. Top. Catal. 2023, 1-9. [CrossRef]
- Tjosaas, F., Anthonsen, T., Jacobsen, E.E. Biocatalytic resolution of saphenic acid. Substrate preferences for lipases A and B from Candida antarctica. ARKIVOC 2008, vi, 81-90. DOI: http://hdl.handle.net/11250/2430662.
- Hoff, B.H. Anthonsen, T. Gas chromatographic enantiomer separation of C-3 and C-4 synthons: Prediction of absolute configuration from elution order and enzymatic resolution. Chirality 1999, 11, 760-767. [CrossRef]
- Lystvet, S., Hoff, B. H., Anthonsen, T., Jacobsen, E. E., Chemoenzymatic synthesis of enantiopure 1-phenyl-2-haloethanols and their esters. Biocatal. Biotransform., 2010, 28 (4), 272-278. [CrossRef]
- Anthonsen, H.W, Hoff, B.H., Anthonsen, T. Calculation of Enantiomer Ratio and Equilibrium Constants in Biocatalytic Ping-Pong Bi-Bi Resolutions. Tetrahedron: Asymmetry 1996, 7, 2633-2639. [CrossRef]
- Verho, O., Bäckvall, J.-E. Chemoenzymatic Dynamic Kinetic Resolution: A Powerful Tool for the Preparation of Enantiomerically Pure Alcohols and Amines. J. Am. Chem. Soc. 2015 137, 3996-4009. [CrossRef]
Scheme 1.
Synthesis of (S)-atenolol ((S)-4), starting from a sodium hydroxide catalyzed deprotonation of 2-(4-hydroxyphenyl)acetamide (1) with addition of epichlorohydrin giving a mixture of 4-(3-chloro-2-hydroxypropoxy)benzeneacetamide (2a) and 2-(4-(2-oxiran-2-ylmethoxy)phenyl)benzeneacetamide (2b). Subsequent addition of lithium chloride and acetic acid in tetrahydrofuran to the mixture gave pure 2a, which was resolved in acetonitrile with vinyl butanoate and with lipase B from Candida antarctica (CALB). Addition of isopropylamine in water to (R)-2a (>99% ee) gave (S)-atenolol ((S)-4) in >99% ee, 60% yield and 99% purity.
Scheme 1.
Synthesis of (S)-atenolol ((S)-4), starting from a sodium hydroxide catalyzed deprotonation of 2-(4-hydroxyphenyl)acetamide (1) with addition of epichlorohydrin giving a mixture of 4-(3-chloro-2-hydroxypropoxy)benzeneacetamide (2a) and 2-(4-(2-oxiran-2-ylmethoxy)phenyl)benzeneacetamide (2b). Subsequent addition of lithium chloride and acetic acid in tetrahydrofuran to the mixture gave pure 2a, which was resolved in acetonitrile with vinyl butanoate and with lipase B from Candida antarctica (CALB). Addition of isopropylamine in water to (R)-2a (>99% ee) gave (S)-atenolol ((S)-4) in >99% ee, 60% yield and 99% purity.
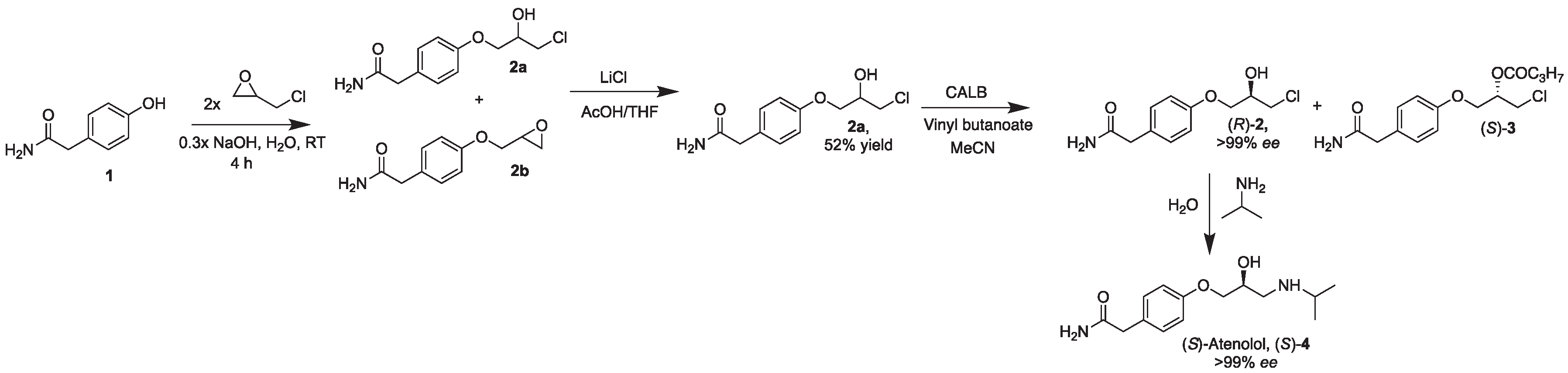
Figure 1.
HPLC-chromatogram of the reaction starting with phenol 1 (tR = 3.03 min), with stoichiometric amount of sodium hydroxide producing halohydrin 2a (tR = 12.06 min) and epoxide 2b (tR = 9.58 min) and the by-product dimer 2c (tR = 13.38 min) analyzed on an (achiral ) Zorbax Eclipse XDB C18-column. A linear gradient mobile phase composition of water and acetonitrile (75:25) - (65:35) over 20 minutes with 0.5 mL/min flow was used.
Figure 1.
HPLC-chromatogram of the reaction starting with phenol 1 (tR = 3.03 min), with stoichiometric amount of sodium hydroxide producing halohydrin 2a (tR = 12.06 min) and epoxide 2b (tR = 9.58 min) and the by-product dimer 2c (tR = 13.38 min) analyzed on an (achiral ) Zorbax Eclipse XDB C18-column. A linear gradient mobile phase composition of water and acetonitrile (75:25) - (65:35) over 20 minutes with 0.5 mL/min flow was used.
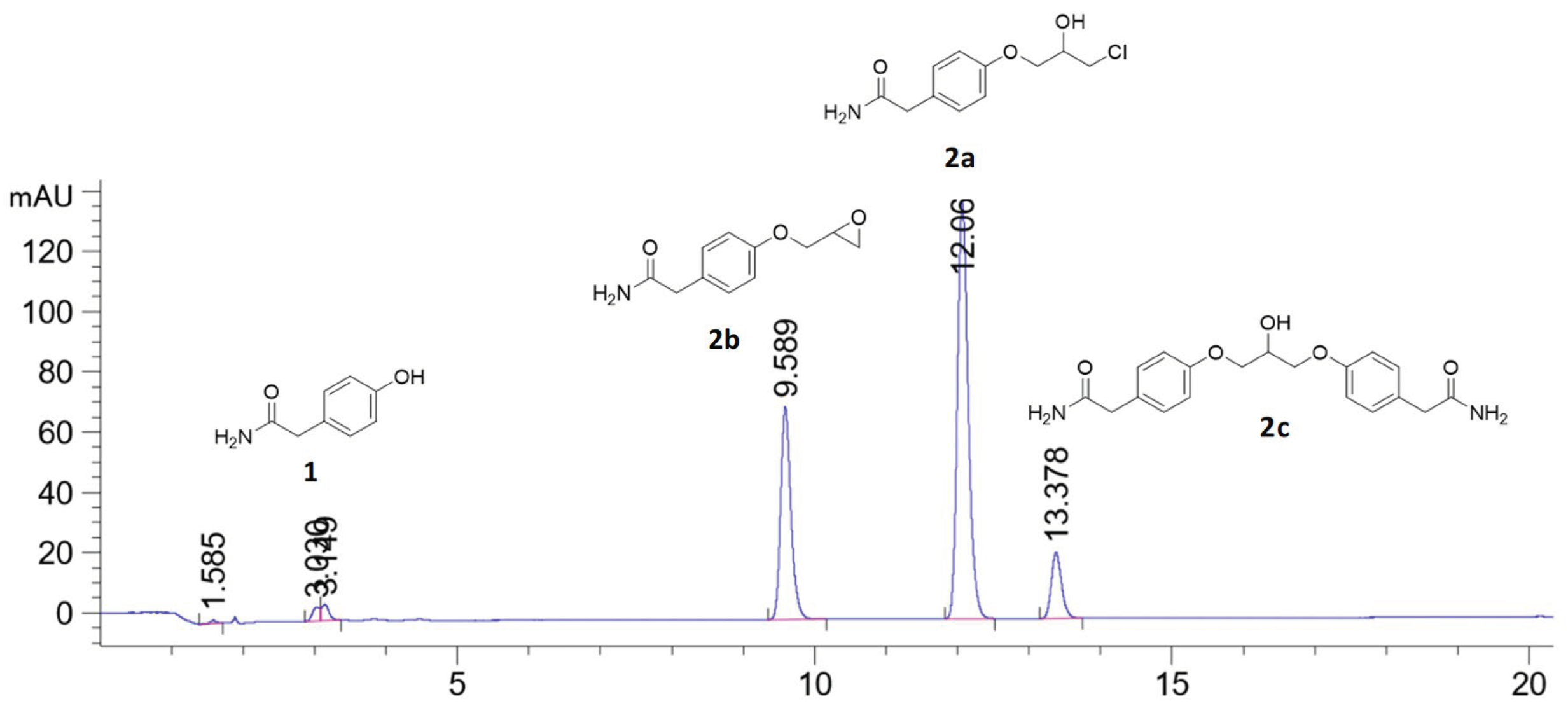
Figure 2.
LC-MS analysis of the by-product 2c showed a molecular mass of 382.04 g/mol, which corresponds to C19H21N2O5Na. The calculated mass of 2c is 358.39 g/mol, C19H22N2O5, which is the evidence of this dimeric structure. The reaction mixture showed in the chromatogram in Figure 1 was analyzed on an AQUITY UPLC BEH C18-column with isocratic mobile phase composition of water and acetonitrile (30:70) with 1% formic acid and a flow of 0.2 mL/min.
Figure 2.
LC-MS analysis of the by-product 2c showed a molecular mass of 382.04 g/mol, which corresponds to C19H21N2O5Na. The calculated mass of 2c is 358.39 g/mol, C19H22N2O5, which is the evidence of this dimeric structure. The reaction mixture showed in the chromatogram in Figure 1 was analyzed on an AQUITY UPLC BEH C18-column with isocratic mobile phase composition of water and acetonitrile (30:70) with 1% formic acid and a flow of 0.2 mL/min.
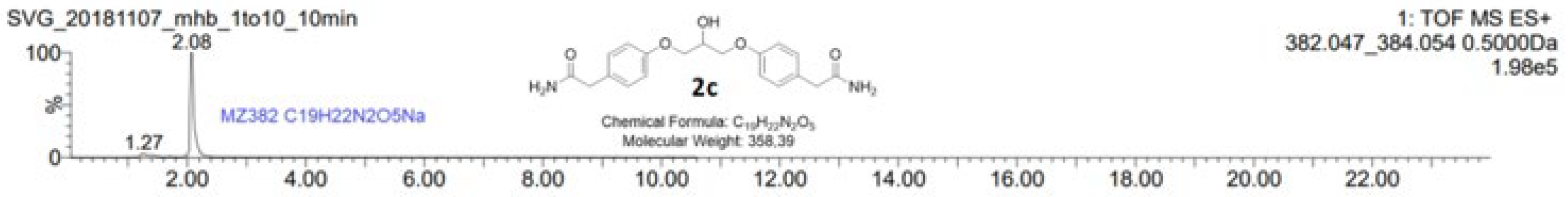
Scheme 2.
Reaction mechanism for the sodium hydroxide catalyzed reaction of phenol 1 forming chlorohydrin 2a and epoxide 2b. Phenol 1 is deprotonated forming alkoxide 1’ which can attack epichlorohydrin at carbon 1 and 3 (reaction mechanism a and b, respectively) giving alkoxide 2a’ and epoxide 2b. Alkoxide 2a’ reacts in two ways; by protonation from water forming chlorohydrin 2a, thus regenerating the base (reaction mechanism c), and by an intramolecular SN2-like reaction forming epoxide 2b (reaction mechanism d).
Scheme 2.
Reaction mechanism for the sodium hydroxide catalyzed reaction of phenol 1 forming chlorohydrin 2a and epoxide 2b. Phenol 1 is deprotonated forming alkoxide 1’ which can attack epichlorohydrin at carbon 1 and 3 (reaction mechanism a and b, respectively) giving alkoxide 2a’ and epoxide 2b. Alkoxide 2a’ reacts in two ways; by protonation from water forming chlorohydrin 2a, thus regenerating the base (reaction mechanism c), and by an intramolecular SN2-like reaction forming epoxide 2b (reaction mechanism d).
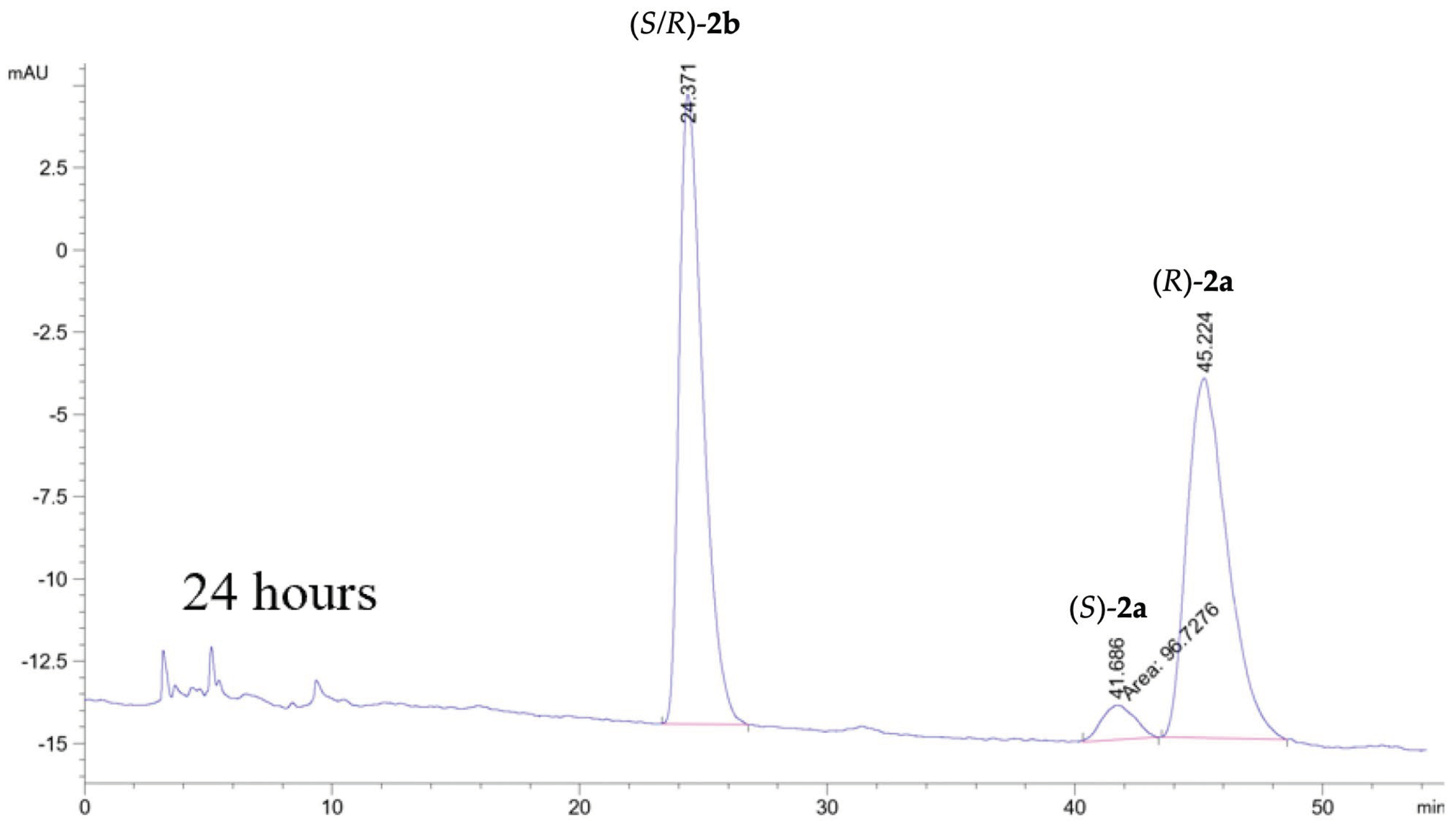
Figure 3.
Kinetic resolution of 2a catalyzed by CALB after 24 hours of reaction time. HPLC analyses were performed on a Chiralcel OD-H column (250 mm L x 4.6 mm i.d, 5 μm particle size) with isocratic eluent i-PrOH:n-hexane, 17:83, flow 1 mL/min. This gave unseparated ester enantiomers tR (R/S)-2b = 25.37 min, tR (S)-2a = 41.66 min and tR (R)-2a = 45.22 min with RS = 1.4. After 27 hours no (S)-2a was seen in ht echromatogram and the ee of (R)-2a was >99.0%.
Figure 3.
Kinetic resolution of 2a catalyzed by CALB after 24 hours of reaction time. HPLC analyses were performed on a Chiralcel OD-H column (250 mm L x 4.6 mm i.d, 5 μm particle size) with isocratic eluent i-PrOH:n-hexane, 17:83, flow 1 mL/min. This gave unseparated ester enantiomers tR (R/S)-2b = 25.37 min, tR (S)-2a = 41.66 min and tR (R)-2a = 45.22 min with RS = 1.4. After 27 hours no (S)-2a was seen in ht echromatogram and the ee of (R)-2a was >99.0%.
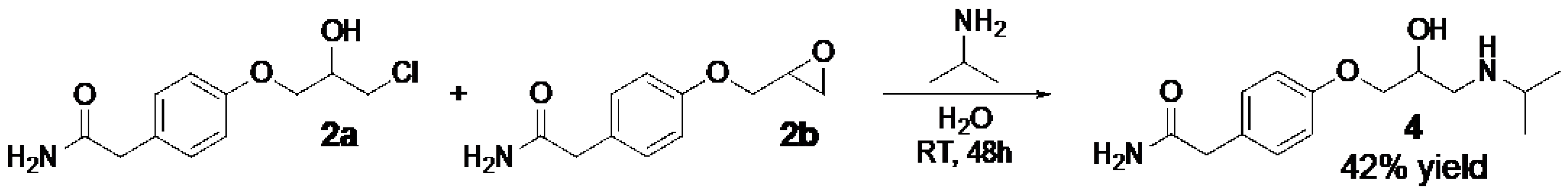
Scheme 3.
Atenolol (4) was prepared directly from the reaction mixture of 2a and 2b with addition of isopropylamine in water giving 4 in 42% yield.
Scheme 3.
Atenolol (4) was prepared directly from the reaction mixture of 2a and 2b with addition of isopropylamine in water giving 4 in 42% yield.
Scheme 4.
Kinetic resolution of 4 in dry n-hexane with lipase A from Candida antarctica (CALA) as the catalyst and vinyl butanoate as acyl donor at 40oC and 200 rpm showing low selectivity and several acetylation products.
Scheme 4.
Kinetic resolution of 4 in dry n-hexane with lipase A from Candida antarctica (CALA) as the catalyst and vinyl butanoate as acyl donor at 40oC and 200 rpm showing low selectivity and several acetylation products.

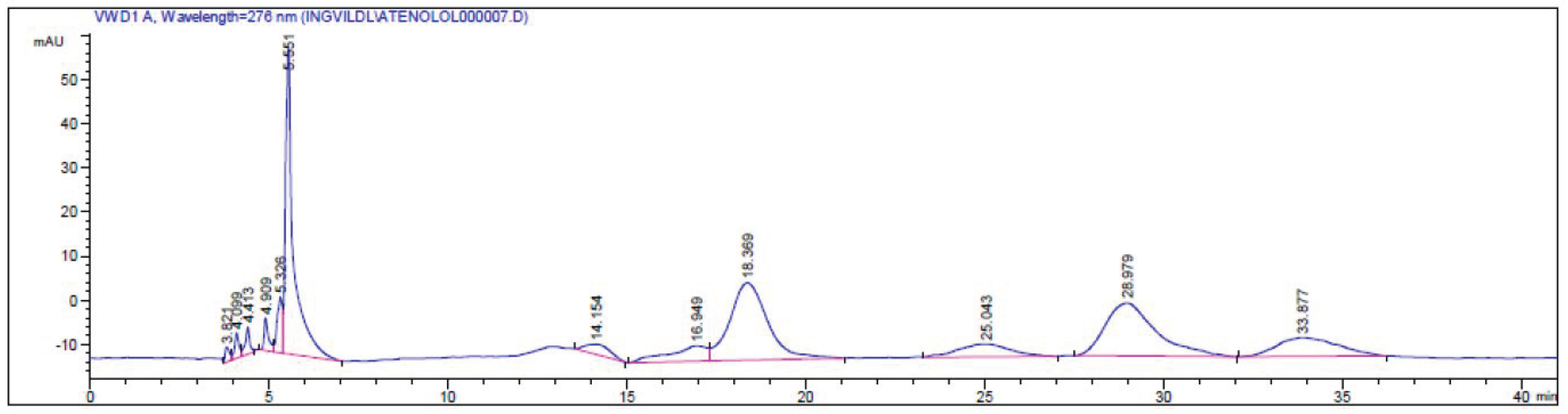
Figure 4.
HPLC chromatogram of the kinetic resolution of atenolol (4) with CALA as the catalyst, showing retention times tR (S)-4 = 18.36 min and tR-(R)-4 = 28.97 min, the esters 4b tR (S)-4b = 25.04 min and tR (R)-4b = 33.87 min and a by-product (tR = 14.15 min and tR = 16.94 min) analyzed on a Chiralcel OD-H column (250 mm L × 4.6 mm i.d., 5 μm particle size) with a gradient mobile phase i-PrOH/n-hexane; 9:95 (0 min) – 10:90 (10 min)- 60:40 (80 min), at 0.5 mL/min.
Figure 4.
HPLC chromatogram of the kinetic resolution of atenolol (4) with CALA as the catalyst, showing retention times tR (S)-4 = 18.36 min and tR-(R)-4 = 28.97 min, the esters 4b tR (S)-4b = 25.04 min and tR (R)-4b = 33.87 min and a by-product (tR = 14.15 min and tR = 16.94 min) analyzed on a Chiralcel OD-H column (250 mm L × 4.6 mm i.d., 5 μm particle size) with a gradient mobile phase i-PrOH/n-hexane; 9:95 (0 min) – 10:90 (10 min)- 60:40 (80 min), at 0.5 mL/min.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



