Submitted:
21 February 2024
Posted:
22 February 2024
You are already at the latest version
Abstract
During the last few years, advancement in the area of biochemistry, science of the material world, engineering and computer-aided testing has directed towards advancement of high-throughput tools for the sake of profiling information encoded in a gene. Single-cell RNA-sequencing (scRNA-seq) tools capable to examine the sequence data from each individual cells and it shows within population variety and permit exploring of cell conditions and transformation by extreme resolution, possibly give out cell sub-types or gene expression fluctuations that are shaded in mass sequencing processes, which shows population-averaged evaluations. Yet, the major disadvantage for this tool is the lack of success to pick out location related details of the RNA transcriptome, as this needs tissue detachment and cell isolation. Location based transcript determination is among the advancements in the area of medical biotechnology as this can find out the molecules like RNA dataset in their intact physical placement in tissue segment with spatial context at the scale of a single-cell, which is very advantageous as compare to single-cell sequencing techniques. These approaches give key observation in the areas of biomedical field sub-disciplines like neurology, embryology, carcinoma study, immune cell investigation and histological activities. Generally, this review mainly focused towards single-cell sequencing methods, technology development, its challenges observed, different expression data analysis mechanisms and their applications in different areas, such as in cancer research, microbes, central nervous system, sex organs and immune-biology, intensifying the essentiality of sequencing tools at single-cell level for the characterization of extremely dynamic individual cells.
Keywords:
Single cell RNA sequencing
; Transcriptome
; high-throughput
; spatial transcriptomics
1. Introduction
During the last few years, advancement in the area of biochemistry, science of the material world, engineering and computer-aided testing has directed towards advancement of high-throughput tools for the sake of profiling information encoded in a gene and sequence figures from different biological specimens. Technologies are now capable of sequencing many fragments of DNA in parallel, such as RNA sequencing; let intense and full understanding of complex events involved in an organism including whole organism development process, damaged tissue re-establishment into normal structure and function and carcinogenesis [1]. The genetic Testing and Sequencing technology advancement recently encouraged the progression of applying a sequencing-supported tool for exploring genomics heterogeneity and fluctuations within a body system [2]. Currently, there are a lot of techniques used for gene expression profiling, from these techniques, RNA-sequencing mechanism (RNA-seq) permit transcript investigation along extraordinary precision and broadness, directing towards uncovering of newly discovered RNA species and boost our perception of transcriptomic changing [3,4].
Just recently, low-input RNA-sequencing techniques came to be used to study in single-cell characterizations [5]. Single-cell RNA-sequencing (scRNA-seq) tools capable to examine the sequence data from each individual cells and it shows within population variety and permit exploring of cell conditions and transformation by extreme resolution, possibly give out cell sub-types or gene expression fluctuations that are shaded in mass sequencing processes, which shows population-averaged evaluations [6,7].
The investigation of every individual cells by global mechanisms has the capability to transform persons looking of full organisms from that point on cell ancestry can be drawn and variability within a tissue be summarized with extraordinary resolution [8]. Exploring cells from a tissue at the individual level create special scope to assess the interaction between inherent cellular activities and outside signals like the surrounding circumstances or adjacent cellular interplays in cell destiny determination. On the other hand single-cell exploration are furthermore of highly helpful in the medical centers, assisting to investigate how an ‘outlier cell’ may decide the consequence of a disease progression [9], medication or anti-microbial resistance [10,11]and tumor reversion [12]. Moreover, since most of cells from the life forms are unable to be cultured in the outside environment (usually we call it ‘microbial dark matter’); examination at the single-cell level hold the opportunity of finding unrecognized species or governing mechanisms of biotechnological or therapeutic importance [13].
High-efficiency single-cell RNA profiling has allowed extraordinary looking into cellular heterogeneity of tissues everywhere in different life forms. Gene expression analysis using mass tissue considers and characterizes all the cells in the way that identical body, because of this there is taking no notice of the statistical probability of gene expression [14,15]. To solve problems related to the Probabilistic sort of gene expression; single-cell sequencing techniques are incredibly appropriate.
Single-cell sequencing techniques means it is the process of sequencing of every isolated cells hereditary material to get genomic, transcriptomic, or multi-omics data figures at single-cell scale to indicate cell population heterogeneity and cellular developmental associations. Ordinary sequencing techniques can hardly dissect the median of numerous cells, cannot explore a small amount of cells and it masks important cellular variability information outcomes. In comparison with conventional sequencing tools, single-cell sequencing tools have the importance of dissecting gene expression variability’s information among each individual cells [16], differentiate a limited number of cells in amount, and tracing out cell maps. In the mean time of 2013 single-cell sequencing techniques taken as , named “Nature Methods” as the yearly innovation [17]. Nonetheless, previous single-cell sequencing protocols diminished its extensive usage for the reason that it’s expensive price to apply the techniques without much effort.
The global analysis of single cells came to be accomplished by an enormous progress in the sensitivity of scientific instrument and a constantly increasing robotization of every single steps from specimen preparation to information analysis, so as it enables to detect gene expression dynamics at tiny level in the cell population. High throughput technologies nowadays allow rapid sequencing of the genomes of a lot of single cells side by side fashion to produce large amounts of information with in short time periods [18], or figure out expressed proteins by utilizing techniques like fluorescence and mass cytometry [19]. Messenger RNA exploration of single-cells has been started by a host of probe-based techniques together with reporter merging to fluorescent proteins, fluorescence in situ hybridization (FISH), quantitative real-time PCR (qRT-PCR), and microarrays [20], quite a lot of which can give a statement of expression variability of many genetic materials in side to side fashion.
Generally, this write up mainly focused towards single-cell sequencing methods, technology development, its challenges and their applications in different areas, such as in cancer research, microbes, central nervous system, sex organs and immune-biology, intensifying the essentiality of sequencing tools at single-cell level for the characterization of extremely dynamic individual cells. Nonetheless, single-cell RNA sequencing tools up to this time have difficulty related with skilled man power and high profiling price, which diminishes the usage of sequencing technologies in transcriptomic studies.
2. From bulk to single-cell transcriptomic dissection
Transcriptomic examination at individual cell resolution level were first started a period of 20 years before by Norman Iscove, with Exponentially amplification techniques of single-cell cDNAs by Polymerase chain reaction and by James Eberwine applying straight-forward amplification of cDNAs via T7 RNA polymerase based outside the living body transcription [21,22,23]. The above mentioned mechanisms have speeded-up looking into deeper molecular systems of developmental process and working mechanisms of the vertebrate’s nervous system, particularly for the reason that these cells are very likely the most varied class of cells. Under these circumstances, transcriptomes come single-cell or lower level determination in a prolonged axon can be descriptive [24,25]. Afterward, the implementing of mass-produced accessible high-density DNA microarray chips accelerates the advancement of individual cell microarray [26].
Latest findings have demonstrated that there exists big cell to cell transcriptomic dissimilarity [27], even amongst ancestral invariable cell cluster. As a result, mass assessments can hide principal cellular variability [28] and generate averaging biases, because of this the scientific investigation done in the experiment did not give us the reality of gene expression dynamics among cell population. The main importance of doing single cell RNA sequence is it enables to characterize such cell-to-cell variability and take advantages to come across population structure and cell changes masked at the mass scale.
So far, almost all the transcriptomic analysis is conducted at a ‘population level’ consistently results come out as mean to the transcriptomes of a lot of cells. Although, with in certain conditions like stem cells, malignant tumor cells and other scarce populations, there is a difficulty to get sufficient material to execute determination on such degree. Moreover, mass techniques unable to investigate the ultra-fine but definitely biologically significant variability between apparently similar cells. Despite each vertebrate animal cells are approximate to accommodate 10⁵-10⁶ mRNA expressed genes [29], the relative percentage of various transcriptomic groups in a population are exceedingly dissimilar [30]: a quantifiable characterization in yeast [31] has proven that most of messenger-RNAs are available in a small number of (͔͔less than 5 transcriptome) duplicates within each cell, and the majority long non-coding RNAs surprisingly in less than 0.5 duplicates per each cell. Also in certain bacteria, the mean copy number of messenger-RNA in Escherichia coli is 0.4 per each cell [32]. Figure 1 below shows workflow for mass and single- cell RNA sequencing process in relation to data generated between them.
Single-cell RNA sequencing method has been powerful in characterizing multiplex, varied cell populations, allowing freely knowing of population composition and the finding out of new sub-types and cell types that are infrequent nature[33,34]. In the situation of changing process, cell path reorganized from single-cell profiling data have afforded looking into short term in-between cell states and have assisted to examine main controller genes [35].
Additionally, single cell RNA sequencing method also display great possibilities for understanding probabilistic transcriptional bouncing and reasoning gene controller systems. Nevertheless, system hypothesis from single cell RNA sequencing information is computationally difficult and problematic to confirm; deduced system models should thus be seriously characterized and practically approved where viable. Moreover, a particular mRNA will be expressed at numerous scales in-between a cell population either by the cause of no randomness grounds as a result it is fraction of an operated cellular process or as a result of arbitrary varied scales of expression in between cells, a condition also known as transcriptional trouble that fail to be acknowledge unimportant because it has great significance in cell destiny determination [36].
RNA-seq of single cells enable researchers to find the special nature of biological cells at individual levels in complex tissues and organ systems and it also helps to understand cellular sub-population response to environmental conditions. RNA-seq has allowed the first determination of the scope of transcriptomic variability’s of the two coding and non-coding RNAs covering a genome-vast level. Besides to divergent gene expression scales, other layers of transcript varieties become visible within individual cells. It is known that splicing mechanisms [37] and allelic casual expression [38] are extensively different within cells. The techniques of single-cell RNA sequencing will similarly assist to redevelop time related transcriptional networks in time of transitional mechanisms [7] or when cells are faced to external environment [37], every one of which can be hidden on a population scale. This scale of throughput determination capable scientists to find out at the single-cell scale just which genes are expressed and what genes are off at a particular situation, in what amount, and how they varied entire cells between a heterogeneous cell population.
3. Progresses in single-cell RNA profiling techniques and innovation
Progress in the area of life science investigation continues to look things deepen, because of these working with single-cell RNA sequencing tools go on with to become greater and move forward towards minimal dissecting prices, make progress researchers investigation on the molecular workings deeply into every individual cell level. As proposed by another findings [39] a single-cell integrative label sequencing tools (SCI-seq) that can at the same time build many of single-cell information libraries and dissect heterogeneity in body cell duplicated number. This method maximizes the amount of tissue cells characterized and minimizes the price of library preparation, and has significant advantage for the analysis of body cell heterogeneity.
According to an investigation [40] they innovated a new single-cell entire-genome amplification technique that enable to examine CNV at kilobase level and further efficiently characterize mutations in a number of diseases. Another tools developed by a studies [41] mainly based upon sequencing single-cells by parallel method (scCOOL-seq) which enables concurrent detection of individual cell genomic condition or nuclear micro-environment organization, copy number heterogeneity, repetition of chromosome sets and analysis of the methylated DNA, which capable to show various activities and forms of genomic condition and DNA a heritable epigenetic methylation (a methyl group attaches to a specific spot on a gene). Another studies [42] Designed a mechanisms of Topographic Single-Cell RNA Sequencing (TSCS) that enables precise spatial positional data for every individual cells. Again this method has high degree of sensitivity to investigate and express the particular nature of every single cancer cells positional information’s and allows studying the occupation and malignant nature of cancer cells. Figure 2: below describes events arranged based on the time of single cell RNA sequencing techniques progression as well as advancements.
Another mechanism described a very high-efficiency and minimal-divergence single-cell RNA sequencing techniques which utilizes droplet based micro-fluidics to isolate, expand, and barcode the genetic material of an individual cell. The methodology allows wider genetic material characterizations for a number of cell masses [43]. Additionally Microwell-seq created by another investigator [44] is an advanced and minimal-price single-cell RNA sequencing methodologies. But also, it enhance the investigation plenty of single-cell RNA tools, apart from this, again it minimize the price of examination by a sequence of dimensions analyzed to single-cell RNA sequencing methods covered with oil droplets. The SPLit-seq tool from developed, depend on the idea of a minimal-price combined barcode; capable to decrease the price of single-cell RNA transcriptomic sequencing to one cent level. After more broke the price threshold for single-cell characterization, this makes the technology more accessible to researchers [45].
Currently, there are no standardized single-cell sequencing techniques and this means that researchers choose the method they follow based on their objective of study and other range of options. On the basis of transcript reportage, single-cell transciptome sequencing tools currently utilized can be grouped into different types [46]: (i) entire-length transcriptome sequencing mechanism [one example- MATQ-seq [47], SMART-seq2 [48], ICELL8 [49] and SUPeR-seq [50]), (ii) 5′ -end transcriptome sequencing techniques (for instance- STRT-seq ([51])), (iii)3′ -end transcriptome sequencing technique (one prototype- Chromium ([52]) 10X Genomics, Fluidigm C1 ([53]), Drop-seq ([54]), inDrop ([55])).
Following an entire-length transcriptomic sequencing method, there is a problem of strength of character, rate, and sequencing price. On the contrary, a main problem of sequencing cDNA is emphasizing either 5′ or 3′ - end transcriptome of the DNA gives lacking the ability of exploring allele-related gene expression or alternative splice fashion. Various techniques depend on flow-activated cell sorting (FACS) based arranging, including MARS-seq that prepares them dependent on a huge starting volume [56] and is discouraging when there is minimal starting volume, with respect to fine-needle suck out. The other issue of utilizing flow-activated cell sorting (FACS) method is the demands of applying antibody beads that prey particular proteins for categorizing; this creates challenges while categorizing rare cell sub-types [57]. Thus, every rules has its own nature of pros and cons that decide the “depth” of a stated data collection, and it may greatly influence the numerical and biological perception [58].
Single cell RNA sequencing is not a “one-size-fits all” method the same as the mass sequencing mechanism since the deepness can differ with the rules being followed, types of cell being characterized, isolation techniques, sequencing mechanism, and association strictness during library preparation [59]. Single-cell RNA sequencing technique first started by separation of individual cells among biological samples, although the seizing capability is a main problem for single-cell RNA sequencing work flow. It needs to choose highly sensitive isolation methods and skilled man to increase the accuracy of detecting transcriptome expression dynamics. Now a day, many diverse techniques are accessible for separating single cells from biological samples, consists of limiting dilution techniques, flow-activated cell sorting (FACS), micromanipulation, laser capture micro-dissection (LCM) and micro-fluidics [60,61,62].
Limiting dilution method utilizes pipettes to separate targeted cells from heterogynous population by process of dilution; the major drawback of using this technique is not achieving a maximum productivity, as compare to other methods. Micro-dissection or microinjection is typical mechanisms applied to recover cells out of specimens with a minimal quantity of cells, thus early developing embryos or uncultured micro-organism, during the time that using this method is time taking process and small throughput. Fluorescence-activated cell sorting has been mostly utilized for select out targeted individual cells, that needs huge proceeding volumes (almost >10,000 cells) in suspension solution. However there are some drawbacks related with using FACS from these speed of processing the sample is slow. Laser capture micro-dissection is an improved technique applied for select out the individual cells mostly from a specimen of solid tissues by utilizing a laser mechanism assisted by computer. The main advantage of LCM is its speed of processing cell samples. The main weakness of this technique is that optical microscopic investigation is required to pick out single cells in a sophisticated tissue. This needs an expert mastered in cell characterization. Micro-fluidics is progressively become well known techniques because of its character of minimal specimen using up, accurate fluid regulator and minimal investigation price. These single cells pick out procedures have its own importances and manifest typical achievement with regard to picking out effectiveness and clarity of the prey cells [56,62].
Different scRNA-seq procedures may possess its own advantage and disadvantages, and numerous published analysis have correlated a part of them from top to bottom [63]. A previous analysis illustrated that Smart-seq2 method can identify a huge quantity of expressed genes than other forms of scRNA-seq tools along with CEL-seq2 [64], MARS-seq [65] , Smart-seq [66] , and Drop-seq mechanisms[67]. Currently, Sheng et al. displayed that an additional full-length transcriptome sequencing mechanisms MATQ-seq could overcome Smart-seq2 in examining minimal-occurrence genes [47]. Correlated to 3′-end or 5′-end figure up procedures, full-length single-cell RNA sequencing techniques have extraordinary importance in isoform usage investigation, allelic expression characterization, and RNA editing examination owing to their dominance of transcriptome scope. Furthermore, for investigating inescapable low ranking expressed transcripts, full-length single-cell RNA methods could be greater than 3′ sequencing approaches [67].
particularly, droplet-centered tools [example, Drop-seq[54], InDrop [55] , and Chromium[52] can predominantly give a huge parallel sequencing of single cells and a minimal sequencing price each cell in comparison with the entire-transcriptome single-cell RNA sequencing. Thus, droplet-based mechanisms are more comfortable for producing numerous quantities of cells to detect the cell sub-populations of sophisticated tissues or cancer specimens. Notably, many single-cell RNA sequencing tools can catch the two polyA+ and polyA− RNAs, like SUPeR-seq [50] and MATQ-seq [47]. These mechanisms are severely important for sequencing long non-coding RNAs and circular RNAs.
In comparison with traditional mass RNA-seq tools, single-cell transciptomic sequencing protocols face a problem of huge technical heterogeneities. In order to guesstimate the technical differences between various type of cells and spike-ins like External RNA Control Consortium (ERCC) controls [68] and UMIs became mostly applied in related single-cell RNA sequencing techniques.
RNA spike-ins are RNA transcripts (with recognized sequences and amount) that are used to grading the determinations of RNA hybridization diagnosis, like RNA sequencing, and UMIs allow to hypothetically empower the consideration of total molecular scores. It is value noting that ERCC and UMIs are not relevant to each one of the single-cell RNA sequencing tools due to the innate rule variations. Spike-ins is utilized in mechanisms such as Smart-seq2 and SUPeR-seq yet is not comparable with droplet-based techniques, although UMIs are mainly used to 3′-end sequencing mechanisms [like Drop-seq [54], InDrop [55] and MARS-seq [69]. Consequently, experimenters can select the suitable single-cell RNA sequencing approach on the basis of practical possessions and importances, the cell quantity to be sequenced and price thought.
3.1. Current progresses in single-cell RNA sequencing tools
Cells from organisms were investigated for the actual beginning time in the 16 ͭ ͪ centuries, and then after a number of developments and progress of novel techniques, mechanisms emerged from simple to powerful way. whereas the introductory microscope designed by Zacharias Janssen and Hans Lippershey in the early 16 ͭ ͪ century allowed Robert Hooke and Anton van Leeuwenhoek to look the initial living cell in the 17 ͭ ͪ century, the process took nearly two centuries to reevaluate cells apart from being as of the structural as well as functional unit of life [70]. preceding in time, a number of investigation and techniques were performed for the desire of high-grade sense of perception and investigating cells in heterogeneous multi-cellular networks [71].
The initial visionary and practical advancement of the single-cell RNA sequencing techniques was done [5], and that sequenced the transcripts of single blastomeric and oocytes. The idea and tools carried by this analysis unlocked a novel mechanism of looming a difficulty to upgrade the quantity of the cells and generate agreeable improved RNA sequencing mechanisms achievable for the beginning period. Considering that, a growing number of reformed and upgraded single-cell RNA sequencing mechanisms were enhanced to set up necessary alterations and advancements in specimen collection, single-cell pick out, bar-coded reverse transcription, cDNA amplification, library construction, sequencing and streamlined bioinformatics assessment. Above all, price has been surprisingly minimized, while robotization and advancement have been remarkably accelerated. All this procedures split into more developed scRNA-seq mechanisms, but the idea of utilizing single-cell RNA sequencing rest not changed [72].
A number of single-cell RNA sequencing techniques are this day widely used in fundamental research and clinically rephrasing background, any way, they demand specially-designed instruments and experts in isolation of specimen, manipulation and processing, sequencing library construction and information analysis. Because of this, single-cell sequencing analysis has turn into one of the quickly-developing areas in life science, generating very interesting new looking’s into tissue architecture and changing biological activity [72].
Now a day, the carrying capacity of single-cell RNA sequencing progresses from a small quantity of cells per test to many cells, where the price has been impressively diminished from time to time. remarkably, research work publications utilizing the single-cell RNA sequencing technology method each progresses annually because of quick and precise single-cell RNA sequencing tools like micro-fluidic, microwell, droplet based, in situ bar-coding and spatial transcript study [73].
In case of humans, single-cell investigation has upgraded detail awareness of of developmental and biological activity in our body systems [74], aging [75] and a number of sickness characterizations like tumor development [76]. However, there is an issue to produce universal blueprints for single-cell RNA transcript investigation in view of the fact that each method needs the exploiter to generate notified judgment in order to get explainable outcomes. These comprise the choice of specimen categories, cell amount and readying mechanisms; the selection of single-cell RNA sequencing methods and sequencing principles; and the outline of computational investigation grand design to create observation from single-cell database. Principally, favorable single-cell RNA transcriptomic analysis with definable data file and relevant scientific result can be realized uniquely through the use of conformed experimental outline.
3.2. Single- cell isolation process and library preparation
The variations of one cell from another cell in RNA transcriptomic and gene expression can be an important way to solving problem in tumor, neuroscience, stem cell differentiation, immune-biology, and evolutionary biology. For this purpose, first individual cell need to be isolated from the tissues, which is the way of catching high-grade single cells from a given body sample, because of this taking out accurate genomic and biochemical processes that occur in cellular activity and enabling the analysis of particular genomic and molecular operations [77]. Conventional transcriptomic, epigenomic or proteomic from mass RNA or DNA specimens can solely find out the sum total of information’s from tissues or organs, which unable to detect every individual cells heterogeneity. To deeply understand variations from in between cells, researchers desire to utilize the approach of single cell RNA sequencing analysis that can give highly organized dataset for treatment choice production in accurateness medicament. Isolation of single-cell and capturing techniques are to a great degree diverse relying on the life forms, tissues or cell natures [78].
Before executing a single cell investigation, researchers are supposed to isolate individual cells from a tissue. Cell separation can be executed by picking out entire cells, cell particular nucleus or cell distinct organelles, and still by isolating the preferred cells expressing particular marker proteins [79]. The main purpose isolating individual cells, and specifically in parallel mechanisms, is that every individual cell is isolated in a separated reaction combination, in particular all transcriptomes from each individual single-cell will be specifically bar-coded following changed into complementary DNAs (cDNA) [72].
Yet, the single-cell RNA sequencing methods has progressively uncovered several intrinsic procedural problems, like ‘artificial transcriptional stress responses’. This thing clearly tells us the separation mechanisms could activate the expression of stress related genes that cause synthetically converts in cell transcription profiles. This has been approved by numerous experimental analyses. An investigation [80] Confirmed that the mechanisms of protease separation at 37 °C could cause the expression of stress related genes, offer practical fallacy and lead to imprecise cell form recognition.
Following the procedure of changing mRNA directly into the first strand cDNA, the generated cDNA are amplified by the process of polymerase chain reaction (PCR) techniques or test-tube transcription [61]. The cDNA is built from entire length mRNA transcriptomes utilizing a reverse transcriptase that has terminal transferase workout. This, when integrated with a second “template-switch” primer, enables for cDNAs to be bullied that have two universal priming sequences. After the individual cells bar-coded cDNAs are produced from each separated cells or single nucleus, the cDNA can be sequenced utilizing a numerous detail sequencing techniques [72].
3.3. Single-cell transcriptomic sequence data analysis
Due to the growth of scope for advanced single-cell RNA sequencing (scRNA-seq) techniques, including clinical specimens, the investigation of these immense volumes of dataset has developed into an intimidating hope for scientists to get involved in this field of study. The investigation of single-cell RNA sequencing (scRNA-seq) dataset is an additional important key element, and this day the main requirement, to widen the implementation of this advanced mechanisms in life science and clinical studies. To safeguard the utilization of single-cell RNA sequencing analysis methods, a number of designers have made great achievements. Almost 1000 various bioinformatic techniques have been evolved and made accessible by May 28 ͭ ͪ, 2021 [81].
In the time of preparing of single-cell mixture, intact cell may encounter dying, injury to cell membrane or multi-cellular adherence because of inescapable natural conditions, investigational activities and practical problems. To remove the gene expression intervention from poor quality cells, it is mandatory to execute another step of quality regulator measures with satisfactory techniques, like Seurat, [82] scran [83] and scanpy [84].
Different studies show that, Seurat is the highest well known techniques with integrated usage to hold poor quality cell purification. Mainly, the preceding quality control (QC) signals should be utilized to determine in case a cell should be maintained: the amount of genes, the amount of UMI (transcripts), the proportion of mitochondrial genes and the proportion in relation to ribosomal protein genes in every individual cell. There is no perfect means for the configuring of strainer borderline, which mostly relies on the groups of cell and tissue being analyzed. [85] Purified cells with less or equal 100 or great or equal 6000 expressed genes, less or equal 200 UMIs and great or equal 10% mitochondrial genes as stated in their investigation. According to another view [86] maintained excellent standard cells utilizing the following framework: (1) 200 less than overall amount of expressed genes per a cell (nGenes) less than 2500; (2) 300 less than full amount of UMIs per a cell (nUMIs) less than 15000; and (3) proportion of UMIs tracked to mitochondrial genes (MT%) less than 10% .
Indistinguishable to the determination of conventional mass RNA sequencing dataset, every cell is counted as a separate specimen in time of analyzing single-cell RNA sequencing dataset. The beginning expression model incapable to directly utilized for downstream analysis for the reason of expression scale within cells are not equivalent because of systemic biases or practical noises, like variations in sequencing intensity and transcript pick out degree for every cell. Normalization is considered to prevent practical noise and to secure comparability with in every single-cell. An investigation [87] assessed the validity of seven normalization techniques, such as BASiCS, GRM, Linnorm, SAMstrt, SCnorm, scran and Simple Norm.
The scRNA-seq dataset has huge number of features, with numerous thousands of cells in a specimen and millions of genes expressed in every single-cell. The majority of genes in every cell included in to housekeeping genes, which are detected by there is no important variations in the expression scale within each cell, and their existence leads to mask the real biological information’s. The sub-sets of characters that show great cell to cell differences in the dataset are also known as highly variable genes (HVGs). In fact HVGs not only identify biological stimuli’s but also highly hasten the downstream investigation of individual cell RNA sequencing dataset because of the important minimization in the computation volume. A good-quality HVGs should incorporate genes that can characterize various cell forms, and the good quality of HVGs has a huge outcome on the accuracy of grouping. Another study [88] assessed seven techniques for characterizing HVGs, such as BASiCS, Brennecke, scLVM, scran, scVEGs, and Seurat, recognized huge heterogeneities in the grouping of outcomes furthermore in the operational times of the various mechanisms. Correlation with another tool, scran can identify a reliable number of HVGs with very good operating time and independence from the mean. Brennecke was verified to have secure and persistent achievement with a huge scope of datasets. scran and Seurat were illuminated to execute in the best mechanism with portion of datasets. BASiCS and scLVM_LogVar were observed to be much steady than other techniques.
3.3.1. Data preprocessing
Fundamental configuration of unprocessed sequencing dataset for single-cell transcript comprises FASTQ and BCL layout that rely on the dataset origin and sequencing program. Afterward solely FASTQ data’s can be straightly used for standard control, if the unprocessed dataset are not in FASTQ layout, the initial procedure is to change it in to FASTQ setup with the relevant techniques. FASTQ data’s can be produced from the BCL data’s utilizing cellranger mkfastq, a pipeline that has banded bcl2fastq program. Mainly, a recognizable CSV matrix data along with minima of 3 columns (lane, sample and index) need to be accommodated additionally to the route of BCL extracts. After that Fast QC can be executed to determine the standard of raw single-cell RNA sequencing dataset [72].
3.3.2. Exploratory analysis
To vigorously disclose functional error and biological importance of a given cell folk, it is crucial to carry out functional enrichment analyses on a focused divergently expressed gene dataset. Global investigation approaches for activity enrichment are also great for single-cell dataset, like gene ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway. A numerous ways of functional methods have been evolved for functional enrichment determination. A study done by Huang et al. comprehensively differentiate 68 enrichment analysis techniques in 2009 after assessing corresponding robustness and drawbacks [89].
Additionally, GSVA is again mostly applicable in functional enrichment analysis process and extra control investigations in a pathway-approached method. GSVA can measure enrichment outcomes for various signaling pathways in every specimen to determine the origins of morphological type variations, which can be applied as an additional to the KEGG pathway to generate the output better physiologically explanative [90].
To detect the transcription factors enriched in every one of cell group from single-cell RNA sequencing dataset, a study [91] developed SCENIC techniques, which allowed extrapolating transcription factors for the reason that it primarily obtains the enrichment of TF motifs by finding the supposed regulatory sections of focused genes. Then after TF motif enrichment can find out the relationship of candidate TF regulatory factors with candidate desire genes [92]. Though SCENIC can be practiced in both R and Python, pySCENIC is greatly suggested for processing huge datasets because of its quicker application of the SCENIC outlets. Note that the most recent model of SCENIC helps Homo sapiens, Musmusculus and Drosophila melanogaster, with the opportunity of handcrafted composing a custom repository for another species [93]. Though SCENIC was largely utilized for the reason that it’s outstanding adjustability and robustness for a broad degree of repository, it disregarded the dynamic shift in gene regulation systems in various cell forms. IRIS3 developed by [94] , an affiliated cell-class-specific regulon reasoning server from single-cell RNA sequencing [94]. In practicable implications, IRIS3 was highly appropriated for the scientists devoid of substantial programming Expertise with its adaptable web server. Nevertheless, uninterrupted advancement is needed by IRIS3 in truthfulness and competence.
4. Spatial Single-Cell RNA sequencing
Every cell within a multi-cellular life interchanges with the neighborhood circumstances. For instance, stem cells divergence in the course of development fundamentally by way of cell-to-cell interactivity and consequential signaling, which is regulated by the comparative locality of cells inside the embryo [95]. The spatial localization of tissues controls the expression of transcription factors linked to divergence and at the very end of the process give rise to a robust association of cellular organization linked to their functions [96]. Location based Transcriptomics is the capability to pick out the positional information of transcriptional movement inside undisturbed tissue, either for sections or only one cells. It explains an array of advanced tools allowing scientists to track down transcripts, regularly towards the bottom to the sub-cellular level, delivering an unbiased map of RNA molecules in every part of tissue sections [97].
The scRNA-seq has been a strong enough to knock down into single-cell level genomic investigation technique. It enables RNA examination and quantifiable inquiry extending far down to the principal basic unit of life, the cell. Yet, the major disadvantage for this tool is the lack of success to pick out location related details of the RNA transcriptome, as this needs tissue detachment and cell isolation, the necessity to capture alive cells from entire tissue in the absence of inducement of pressure, death of cell or cell assemblage [98].
The spatial context is crucial to explaining basic problems about the heterogeneousness of a tissue what is expressed in it, which cell expressed it, and where exactly expressed. Location based transcript determination is among the advancements in the area of medical biotechnology as this can find out the molecules like RNA dataset in their intact physical placement in tissue segment with spatial context at the scale of a single-cell, which is very advantageous as compare to single-cell sequencing techniques. This technique can also be utilized to detect sub-cellular placement of mRNA unit [99]. This method was adopted by and practiced for the determination of mRNA data’s with great resoluteness and bigger sensitiveness [100]. following the undertaking of Stahl et al. in spatial aligning of RNA, molecules are carried out with a number of methods like in situ sequencing mechanism, fluorescent in situ hybridization techniques, in situ capture approaches, and in silico ways[101]. Even so, spatial transcripts of RNA molecules is classified into two main types[102,103] , (1) next-generation sequencing (NGS), consisting of positional determination of RNA transcriptome prior to next-generation sequencing and (2) imaging-based methods together with in situ sequencing-based mechanism, which concern amplification of RNA and their sequencing in a tissue specimen and in situ hybridization-based procedures. The investigation of transcriptome is executed by utilizing imaging probes that mixture progressively in the tissue target [104,105].
Elucidating the spatial localization of mRNA transcriptome enables for the investigator to reveal cellular diversity in tissues, tumors, immune system cells along with detect the sub-cellular dissemination of transcripts in several situations. This output gives a special chance to interpret both at cellular and sub-cellular framework in both tissues and every individual cells. The spatial transcriptomics slogan is “any target, any region, any sample.” The placement of whatever designated cell, comparative to its neighbors and other structures out of the cell, can lay out fundamental details for explaining cellular characteristics, body cell condition, and basically cell and tissue purpose in biological networks. Position can detect the ligands to which cells are prone. Although endocrine hormones perform at observable levels, a great number of other kinds of signals take action upon neighboring cells by a means of cell to cell intercommunication or through soluble ligands performing in the surrounding area. One can design this substances which uses cell outer surface bound sensory receptors and ligand pairs, the mRNA for which can be investigated by transcriptomic mechanisms [106].
These approaches give key observation in the areas of biomedical field sub-disciplines like neurology, embryology, carcinoma study, immune cell investigation and histological activities. The operating of each and every individual cell in multi-cellular life forms can solely be fully described in the circumstance of pick out their partikecular position in the body system. Location based transcript dissection tools find out to illuminate cells’ characteristics this process[101]. Ideal example that shows the significance of spatial organization is tumor tissue, in which cells strongly interplay with the neighboring cancer microenvironment to advance into suppressive situations that prevent the activity of immune cell performance, thereby bypassing immune cell defense systems and fastening multiplication into another level [107].
To comprehend the sophistication of biological networks covering from several physiological circumstances to the pathological concepts of infection progression, it is very important to examine the importance of each and every single cell and their interplay to integrate complicated purpose of tissues and organs in the body organization. Approaches for determining these natural processes of living things working approaches consists of finding out cells that present in the tissue and their spatial locations and interconnections with one another [108].
5. Single-cell sequencing Applications in the field of biomedical research
Now days, single-cell RNA transcriptomic investigation is speedily advancing and develop into an outstanding techniques for a number of scientific investigations consisting humans, animals and plants allowing highly precise, fast determination of infrequent and discovery of new cells in body like never previously. Furthermore, with the findings related to gene expression at messenger RNA molecule and protein amounts, biotransformation, cell to cell interaction and positional organization, it develop into achievable to investigate the mystery of cell configuration and their implication in well-being and illness conditions. Whereas the initial discovery and usage of single-cell RNA sequencing were predominantly performed on animal and following human cells, the sequencing in the case of plant science is even now in its premature level and has numerous interesting issues preserved to be overcome [109].
Currently, the implication of single-cell transcriptomic sequencing mechanisms remains restricted to solely a small number of plants, because of practical issues or extremely restricted details on the cell forms and findings in evolutionary biology. Numerous plant investigation assemblage applied the majority utilized model plant in molecular genomics, Arabidopsis thaliana root for advanced single-cell transcriptomic sequencing and spatial transcriptomics investigation as a result of the comparatively few amount of cells, recognized gene markers and simple techniques to pick out every single cells through enzymatic cell wall degeneration mechanism [110-112].
Analyzing cells at the individual cell level provides special opening to characterize the interaction with in inherent cellular processes and external signal like the regional information or adjacent cells in cell fate investigation. Dissection at single-cell level are also of good insight in the clinical diagnosis, assisting to realize in what way an ‘outlier cell’ may influence the result of disease process [9], antimicrobial drug adaptation [10,11]and tumor reversion [12]. The latest technological breakthrough have brought single-cell RNA sequencing a progressively influential techniques for elucidating biology and cellular activity, disease investigation, treatment feedback prognostication, and medication choices [72].
Every single tissue/organs organized exceedingly structurally and functionally different groups of cells in diverse conditions, physiological transformations, divergence routes and spatial location. This sophisticated but well-organized microenvironment maintains equilibrium up to the time of extreme situations happened that might transform the ordinary cell framework into another one, for instance, tumors. To elucidate the development of tumors, developmental origin of cells, tumor proliferation, malignant and treatment feedback, it is significant to increase our perception of tumor microenvironments with key immunological and stromal penetrates [113].
Single-cell transcriptomic sequencing investigation can examine working healthy cells from tumor cells at different growing levels of tumors. This enables to get accurate outcome and examinations amongst the distinguishing and determination of accuracy to various medicines and develops the majority efficient therapeutic mechanisms for tumors. From the beginning, single-cell RNA sequencing (scRNA-seq) tools were concentrated on analyzing every individual region of the tissue, its diversity and cell forms concerned resulting in generating extensive dataset [114].
Most of challenges arise from the truth that tumor cells are variably located in the tissue organization; as a result their microenvironmental organization consists heterogeneity of tumor and other non-tumor cells in various levels and conditions. Furthermore, the cells specimen combination and distribution even between the uniform sections of a tumor cell could be highly dissimilar if the autopsy was drawn in the process of variable point of times and situations. Additionally, single-cell gene expression level dataset usually comprise a number of noises, and thus cells of the similar form could become eventually in various groups, and cells of variable forms can be in the similar group because of batch effects [115].
In spite of the fact that single-cell transcriptomic sequencing is highly important, RNA expression profiling do not all the time deliver findings regarding protein level or post-translational transformations. Currently, single-cell RNA sequencing investigations are encouraged with another methods like mass cytometry (cytometry by time-of-flight, CyTOF) where for instance both approaches proved that regulatory T lymphocyte cells in the tumor express elevated degrees of tumor necrosis factor receptor super family member 9 (TNFRSF9; encoding 4-1BB), such as T cell co-stimulator (ICOS) and cytotoxic T lymphocyte-related antigen 4 (CTLA4) than T regulatory cells in blood or closest healthy tissue, hopefully insightful of an activated condition [116]. Additionally, by attaching spatial information to single-cell RNA sequencing dataset, we are capable to examine molecular, cellular and spatial tissue networkings and one cell with another cell inter-connection in situ [117-119].
Single-cell RNA sequencing is also regularly applied to investigate cellular transformation through various conditions and to trace cell path across steps such as differentiation. Numerous analytical platforms have been recommended for understanding such path: Monocle proposed the idea of “pseudo-time” as a quantifiable weigh of “progress through a biological process” and utilizes methods from mathematical geometry to organize cells in pseudo-time on the ground of their transcriptome outline. Right away cells have been organized across a path; gene expression model over the route of the deep-seated developmental path can be evaluated to find principal regulators and genes with “switch-like” actions [7].
The intrinsic gene expression difference within cells in single-cell transcriptomic sequencing information’s can be performed to deduce gene regulatory networks (GRNs) [120]. Typically, genes are clustered into co-regulated “modules” on the grounds of expression model resemblance [121].
5.1. Applications in cancer research
The single-cell RNA sequencing (scRNA-seq) has influenced numerous fields of cancer research and reformed our level of understanding of intra-cancer heterogeneity, the tumor microenvironment, malignancy, and treatment resistance. Investigations have demonstrated that hereditary or genomic difference can direct to cells with variable genetic and physical composition traits between the cancerous tissue, causing the cancer cells extremely diversified in nature [122,123]. This elevated level of variability may be connected to the approaches of onceogenesis [124] and malignancy [125-127], so investigators require executing more precise examination of cancer cells.
Conventional sequencing mechanisms can solely determine cell masses, generate the midpoint of the signals in a cluster of cells, but the variability in cancer cells could be covered. As a result, conventional sequencing techniques absolutely not detect cancer cells very carefully. scRNA-seq tools can surely make good for the imperfection of conventional sequencing mechanisms. The cell tracing of cancer cells and its microenvironment was outlined by determining the variability of cancer cells. Furthermore, make clear the cell type within the cancer tissue and search particular markers, additionally define a chain of issues like onceogenesis and its proliferation. Consequently, scRNA-seq tools were mainly applied in the investigation of numerous cancers, and were of huge importance for the creation of novel diagnostic and anti-cancer therapeutic techniques [128] tracked the T cell immune-receptor of colo-rectal tumor by single-cell RNA sequencing tool, and uncovered the subclass grouping, tissue organization traits, cancer variability and drug focused gene expression of colo-rectal tumor T cells.
A possible circumstance transformation association between T cell classes and subclasses disseminated throughout tissues was distinguished. By that year, another study [129] utilized this method to evaluate the traits and correlation of genomic copy number heterogeneity, DNA methylation irregularity and gene expression alteration in the time of the appearance and proliferation of human colo-rectal tumor from single-cell resolution and multi-class degree.
scRNA-seq tools can detect alterations in gene expression in time of cancer proliferation. Coordinating individual cell transcriptomic sequencing of neighboring healthy tissues and adenomas at various levels of progression in patients efficiently exposed genomic variation, clonal configuration and metabolic instability in the process of tumorigenesis, giving deep information’s into the restraint of cancer development level [130]. For instance, the transition of familial adenomatous polyposis to adenocarcinoma, Chen et al. introduced a moderate epithelial mesenchymal status, demonstrating that malignant cells keep epithelial traits at the time that performing fast relocation in breast tumor [131].
Another illustration is a detection executed on treatment-refractory bladder tumor victims. In this investigation, single-cell transcriptomic sequencing was performed to elucidate the cancer microenvironment, which incorporate immune cells, the extracellular pattern, blood vessels and another cells, such as fibroblasts [132]. Likewise, single-cell evaluation was carried out in renal cell carcinomas contrasted to benign kidney tissues. This task furnished perception into the biology behind the way to renal cell carcinoma progress and the way it react to treatment [133].
Additionally, individual cell investigation is being run to understand molecular control point or stimulation focuses inside cancers and to detect how a sick person reacts to focusing a definite protein or pathway. For instance, genome broad investigations of DNA became tracked to detect mutations that can adapt the design one is behaved towards [134]. These mechanisms are essential in situations where victims are not reactive to normal therapeutics. Conditions like this regularly happen because of the sophistication and variability of infected tissues within persons. Because of this, there show to be a flashing future for the applying of single-cell transcriptomic sequencing technologies for personalized treatments [135].
5.1. Implications in the area of immunology
Single-cell RNA sequencing (scRNA-seq) tools can examine every single immune cells, as a result differentiating distinct classes of immune cells, along with uncovering novel immune cell masses and their association this aids to puzzle out the sophisticated immune network and suggest new mechanisms for disease therapy. One study determined sub-populations of the spleen and blood Natural killer cells in mouse and human by scRNA-seq, exposing two different characters that differentiate blood and spleen Natural killer cells [136] and using the correlation of transcripts, the resemblance within two main sub-classes NK1 and NK2 in organs and species is identified. This investigation gives comprehensively figuring out the biological nature of Natural killer cells and provides to the transformation of animal exploration into human-associated studies. Another study determined several subclasses of Dendritic cells and monocytes in human bloodstream by scRNA-seq and uncovered a novel subdivision of DC cells that have the characteristics of plasmacytoid dendritic cells but are successful in triggering T lymphocyte cells [137].
In order to investigate immune cells and cytokines in time of continual infection period, using single-cell RNA sequencing method is very informative. One study establish that the variability of IL-10 revealing CD4 T cells and the formation of IL-10 by a subset of helper cells in the course of distinct diseases show a great involvement in advancing humeral immunity [138].
Single-cell RNA sequencing protocols can investigate immunological cells with elevated variability arising by disease causing agents, precisely figure out the genetic substance of every single immune cells, and assist to make clear the sophisticated immunological working process of the body [139]. Furthermore to disease causing agents, age increment can also cause to increased cellular variability among each individual cells. A study executed scRNA-seq on CD4+ T cells in various conditions of young and aged mice, and established that aging influences cell transcriptomic dynamics, causing in increment of variability of gene expression level within immune cells [140].
5.2. Implications in the gastro-intestinal system and urinary tract system
scRNA-seq has developed into an influential technique for investigator of several research fields because of its capacity to understand the diversity and complex cell-class organizations of varied tissues and cell populations. Studies done by Haber et al. establish numerous new gut-epithelial cell sub-classes by scRNA-seq techniques and tracked the expression profile of gastro-intestinal epithelial cells [141]. From this transcriptomics expression profile, the nature of gut cells keeping homeostasis and reacting to disease causing microbes is described. Another investigator Gao et al. utilized top level accuracy single-cell transcriptomic sequencing to examine the four digestive organs of human embryonic phase and several cells of the adult large intestine, exposing the related operation of gene regulation in the progress of four human digestive organs [142].
5.3. Implications in the neurology
In the nervous system organization, there is dissimilarity among each single neuron for the reason that there is certain specific copy number variance in brain cells [143]. The variability in-between these neurons create a difficulty to investigate that in what manner the brain circuits are organized and to work out neuronal reconnections. Nevertheless, single-cell RNA sequencing methods can detect numerous various stages of nerve cells, and generate in depth single-cell trace to elucidate and point out divergent classes of neurons and their attaching molecules in the brain [144]. Studies done by [145] differentiated sub-classes of mouse and human frontal cortical neuronal cells by advanced single-cell methylation sequencing mechanisms. They found a novel organization of neurons in the human frontal cortex and re-describe the neuron class on the basis of the methylation type of neurons. On the other hand Lake et al. utilized a new single-cell nuclear sequencing mechanism to track the second generation single-cell of an adult brain [146]. It is effective to investigate the importance of principal cell classes and to trace normal human brain cells. Another researcher [147] determined a main subset of cerebellar cells and subclasses that is advantageous to cerebellar progress in a mouse cerebellar progression trace designed by single-cell sequencing. These investigations will make easy futurity for research execution on cerebellar progress, neuro-biology and diseases.
5.4. Implications in the area of reproductive and embryonic medicine
Single cell RNA sequencing tools capable to investigate the importance of a limited quantity of cells that could be utilized to prenatal identification and facilitated reproduction. Characterization of women egg cell polar cells or embryonic cells by scRNA-seq to pick out safe and healthy embryo transfer capable to minimize the birth rate of infants with hereditary genetic disorder and assist to stop hereditary diseases [148,149].
Embryonic growth could be taken as the differentiation transformation from the early zygote to the fully developed stage. Investigating the premature levels of embryonic growth needs techniques that are harmonious with small amounts of cells. Single-cell transcriptomic sequencing examinations have allowed global investigations of premature mammalian development [150] assisting to replace hypothesis with uncovering-driven science. In case of animals, single-cell RNA sequencing method is adopted to track the cell growth of zebra fish and an African cockroach embryos, giving useful suggestions for deep elucidating of transformational biology [151]. Another study[152] traced the genome-wide map of human embryos before to implantation by single-cell multi-sequence sequencing techniques. This analysis has applications for the examination of sophisticated and extremely integrated epigenetic courses in the pre-implantation growth of human embryos. A study by Vento-Tormo et al. [153] executed a transcriptome determination of placental cells in prior to gestation by single-cell RNA sequencing tool and tracked placental cell maps. Through the cell map, three sub-populations of perivascular and stromal cells existed in various decidual sheets and dNK (decidual natural killer) were established. Additionally it has also detected regulatory reactions that may reduce the immune feedback of harmful mothers, besides uncovering interplays that provide to the favorable outcome of placenta and reproduction.
Detecting is useful for elucidating early gestation course and enhancing the examination and therapy of gestation-linked diseases. Another study [154] uncovered the fluctuation activity and molecular mechanisms of gene expression in the course of generation of spermatozoa in mice and the particular model of alternative splicing by single-cell RNA sequencing tool, also the finding of principal regulators for specific phases of male germ cell growth. By that year, the group executed scRNA-seq investigation of human ordinary testicular cells and unhealthy testicular cells. On the basis of the outcomes, a hierarchical pattern of spermato-gonial subclasses, spermatocyte sub-types and sperm cell subclasses was founded, and particular markers for human germ cells were more distantly established. Additionally, alterations of expression profiles in testicular somatic cells in a non-obstructive azoospermia (NOA) victims were recognized, which may be the pathogenesis of non-obstructive azoospermia NOA [155].
6. Challenges in single-cell RNA sequencing technologies
A broad detection of the transcriptomic situations of every isolated cell capable us to obtain complete understanding into the interaction of transcripts inside single cells. Yet, today single-cell transcriptomic sequencing tools still face a multitude of obstacles. Generally, current single-cell RNA sequencing (scRNA-seq) techniques have minimal capture efficiency. For the reason that, solely a few fragment of each cell’s transcriptome complement (nearly 10% for numerous procedures [29] is illustrated in the last sequencing libraries, single-cell RNA sequencing (scRNA-seq) has diminished sensitivity and is incapable to accurately determine low-abundance transcripts [38,57]. The minimal quantity of input material for single-cell RNA sequencing libraries also directs to elevated degrees of technical noise that sophisticates information investigation and can cover underlying biological process heterogeneity [61,156].
Techniques for mapping technical divergence in single-cell transcriptomic sequencing dataset have been suggested [157-159]; though, the majority of techniques utilize the specimen-to-specimen heterogeneity in ERCC read counts to pattern and regulate for technical noise in the individual cell dataset and thus could be utilized solely with trials integrating spike-in controls. Furthermore, these methods supposed that the spike-in transcript is considered invariable as cellular RNA in time of library preparation. Though, naked spike-in RNA does not proceed across cellular lysis and is not in sophisticated with ribosome’s or RNA-binding proteins. Thus, though spike-in operations function as important signs of transcriptome occurrence and accuracy in an investigation, there are numerous origins of heterogeneity that stay problems to regulate in single-cell transcriptomic sequencing protocols.
The other possible origin of influence arises from protocols to pick out and capture every individual cell from the specimen tissue. Whereas techniques like micro-manipulation or laser dissection mechanisms capable to pick out individual cells from known positions between a cells mass or tissue forms, these techniques are work-intensive or need extensive instrument. Major single-cell RNA sequencing mechanisms and all of the presenting advanced high-throughput techniques initially separate tissues to form an individual cell suspension early catching every single-cell. This cell separation procedure is regularly nontrivial, and enzymatic treatments applied to disintegrate tissues may affect cell aliveness, possibly influencing cells’ transcriptional status [61]. To prevent biases coming from like enzymatic treatments, one examination [160] have established methods for executing RNA-sequencing straightly on individual nuclei without the use of harsh protease enzymatic treatments. Single-cell RNA sequencing (scRNA-seq) tools have progressed remarkably after their foundation, enhancing with regard to both transcriptome quantization and investigational throughput. However low capture capability and high degrees of technical noise reduce the accuracy and precision of single-cell transcriptomic sequencing techniques, further advanced investigational configuration are appearing to make easy the elucidation of single-cell RNA sequencing dataset [158].
As another option, coming out in situ sequencing techniques are capable to catch and amplify RNA inside the primary tissue circumstance, though today techniques can quantify up to solely a few hundred genes per cell. The indicated techniques sequence RNA straightly within intact cells: cDNA amplicons are produced and circularized, amplified by means of rolling circle amplification mechanism, and after that sequenced by ligation in situ by utilizing the SOLiD platform [161].
Lastly, the mass of single-cell RNA sequencing (scRNA-seq) written works has concentrated merely on polyadenylated messenger RNAs; nearly all published single-cell transcriptomic sequencing protocols pick out cellular RNA transcript by utilizing poly-T priming techniques that catches solely polyadenylated transcriptomes. As a result, present-day techniques are poorly compatible to examine non-polyadenylated transcriptomic categories, like regulatory non-coding RNA or bacterial RNA [162]. Random hexamer priming has been proposed as a procedure to concurrently fishing both polyadenylated and non-polyadenylated transcripts in individual cells and computationally preferred “not-so-random” primers could possibly be applied to fishing poly(A)+ and poly(A)– species while reducing for ribosomal RNA [163].
7. Future perspectives and Concluding Remarks
Analysis of single-cell is a promising and speedily growing area that maintains enormous possibilities to upgrade our elucidating of basic biological complications in life science and support us to greater elucidate the condition and complexity of human disease process for the purpose to find out extra efficient treatments. The single-cell transcriptomic sequencing techniques has confirmed as one of the progressing tools in the field of life sciences during the past 10 years. The progression of advanced scRNA-seq tools and the computational devices form the tool capable of being reached and usable in nearly all operations in life sciences areas. The other usable ground knowledge to revising by utilizing the scRNA-seq tool is the creation of single-cell atlas in tissues, bodies and organism levels. Because of advancement of the new techniques and implementations, a huge scRNA-seq dataset are predicted to be produced and incorporated in a openly accessible database to smooth the path of the elucidating gene and cell implications in well-being and sickness conditions [164].
In the upcoming future, great resolution maps will enable utilizers to magnify on the present dataset, bypassing high-cost and time-taking specimen reprocessing. Micro-fluidics techniques possess now driven a radical change in investigational outlines, and theoretically many possible techniques like combinatorial bar-coding, might drive the obstacle back even more distant. For the reason that, they do not need physical detachment of every single cells, these methods enable for less-costly side by side processing of cells, which will make it able to be done for cell numbers to be increased even to a greater extent [165].
Integrating single-cell RNA sequencing techniques and other huge scale genetic testing technologies would be advance amplifying the implication of the instruments. For instance an important combinational tool is integrating single-cell RNA sequencing (scRNA-seq) and CRISPR-established genome-level genetic testing, like Perturb-seq that allows the investigation of transcriptional factors of deleting numerous genes with CRISPR, [166] LinTIMaT, which combines individual cell transcript dataset and mutation dataset for ancestry track downing [167]. With the continual progress of single-cell RNA sequencing and CRISPR gene editing process, like prime editing, [168] more of this kind integrational advancements and implications are supposed to be reached and offered to the deeper elucidating of gene and cell purposes in body system working. A huge amount of multi-omics investigations and analyses are proposed to be performed to completely assess the gene regulatory mechanism, importance’s, molecules and interconnections for cell classes in normal tissues or organs and in sickness states[169].
Another hopeful implication in the upcoming time is union of the single-cell RNA sequencing (scRNA-seq) technology with regular clinical examination strategies and personalized treatment mechanisms. Nevertheless, right now the majority of single-cell transcriptomic sequencing-based clinical trials are until now at their investigative stages, largely concentrating on reexamining and excellent elucidating the infection pathogenesis process and determination of diagnosis and curative markers. Despite the fact that the price per cell has been minimized remarkably, the price per each specimen (together with the library construction and sequencing process) is until now extensively high. This becomes a continual drawback to apply the single-cell transcriptomic sequencing methods as a common diagnostic protocol. Another problem includes the single-cell RNA sequencing dataset operation, analysis, display and interpretation. Automated single-cell RNA sequencing (scRNA-seq) data investigation pipelines with easily adoptable interphase, and very significantly that could be applied by labor force in the absence of any bioinformatic expertise’s and background, are required to further broaden the single-cell transcriptomic sequencing based clinical applications.
Generally single-cell RNA-sequencing techniques became improved in the past few years by an advancement of novel tools and computational tools allowing the characterization of the transcript distribution of millions of individual cells in sophisticated multi-cellular living things. Currently, highly accurate, automated and low cost of sequencing tools is being constantly evolved and vows to give excellent data quality and greater output with minimal hands-on time. The magnificent quantity of intelligence that is turning to be generated from current and future investigations will have an intense effect in several features of life science research areas, from the establishment of absolutely tailored tumor therapies, to an excellent elucidating of antimicrobial resistance and host-disease causing agent interplays; from the finding out of the mechanisms controlling stem cell divergence to the elaboration of the premature activity of human embryogenesis and deeply understanding of fundamental biological problems.
Funding
Not applicable
Authors’ Contributions
GETNET MOLLA DESTA prepared the whole manuscript text and figures with close supervision and support of the review by ALEMAYEHU GODANA BIRHANU
Availability of Data and Materials
Not applicable
Acknowledgements
Not applicable
Competing Interests
The authors report no conflicts of interest in this work.
Ethics Approval and Consent to Participate
Not applicable
Consent for Publication
Not applicable
Authors’ Information
Not applicable
Notations
| cDNA | Complementary Deoxyribonucleic Acid |
| CNV | Copy number variation |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| DNA | Deoxyribonucleic Acid |
| GSVA | Gene set variation analysis |
| NK | Natural killer cell |
| NOA | Non-obstructive azoospermia |
| PCR | Polymerase chain reaction |
| RNA | Ribonucleic acid |
| scRNA-seq | Single-cell RNA sequencing |
| UMIs | Unique molecular identifiers |
References
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Soon, W.W.; Hariharan, M.; Snyder, M.P. High-throughput sequencing for biology and medicine. Molecular systems biology 2013, 9, 640. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Ozsolak, F.; Milos, P.M. RNA sequencing: advances, challenges and opportunities. Nature reviews. Genetics 2011, 12, 87–98. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nature methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Wills, Q.F.; Livak, K.J.; Tipping, A.J.; Enver, T.; Goldson, A.J.; Sexton, D.W.; Holmes, C. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nature biotechnology 2013, 31, 748–752. [Google Scholar] [CrossRef]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature biotechnology 2014, 32, 381–386. [Google Scholar] [CrossRef]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nature reviews. Genetics 2013, 14, 618–630. [Google Scholar] [CrossRef]
- Snijder, B.; Sacher, R.; Rämö, P.; Damm, E.M.; Liberali, P.; Pelkmans, L. Population context determines cell-to-cell variability in endocytosis and virus infection. Nature 2009, 461, 520–523. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Helaine, S.; Cheverton, A.M.; Watson, K.G.; Faure, L.M.; Matthews, S.A.; Holden, D.W. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science (New York, N.Y.) 2014, 343, 204–208. [Google Scholar] [CrossRef]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nature biotechnology 2013, 31, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Munsky, B.; Neuert, G.; van Oudenaarden, A. Using gene expression noise to understand gene regulation. Science (New York, N.Y.) 2012, 336, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Tang, F. Boosting the power of single-cell analysis. 2018, 36, 408–409. [Google Scholar] [CrossRef]
- The biology of genomes. Single-cell sequencing tackles basic and biomedical questions. Science (New York, N.Y.) 2012, 336, 976–977. [CrossRef]
- Metzker, M.L. Sequencing technologies - the next generation. Nature reviews. Genetics 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Bendall, S.C.; Nolan, G.P. From single cells to deep phenotypes in cancer. Nature biotechnology 2012, 30, 639–647. [Google Scholar] [CrossRef]
- Kalisky, T.; Blainey, P.; Quake, S.R. Genomic analysis at the single-cell level. Annual review of genetics 2011, 45, 431–445. [Google Scholar] [CrossRef]
- Brady, G.; Barbara, M.A.M.; Iscove, N.N. Representative in Vitro cDNA Amplification From Individual Hemopoietic Cells and Colonies. Corpus ID: 39940906.
- 1999 Sheet 2 of 3 5, 958, 688 xxx xx : rir rrrrr.
- Conlin, D.G.; Piffat, K.A. SYNTHESIS OF NUCLEC ACDS USINGA SOLID SUPPORT. 2017.
- Dulac, C.; Axel, R. A novel family of genes encoding putative pheromone receptors in mammals. Cell 1995, 83, 195–206. [Google Scholar] [CrossRef]
- Shumyatsky, G.P.; Tsvetkov, E.; Malleret, G.; Vronskaya, S.; Hatton, M.; Hampton, L.; Battey, J.F.; Dulac, C.; Kandel, E.R.; Bolshakov, V.Y. Identification of a signaling network in lateral nucleus of amygdala important for inhibiting memory specifically related to learned fear. Cell 2002, 111, 905–918. [Google Scholar] [CrossRef]
- Sul, J.Y.; Wu, C.W.; Zeng, F.; Jochems, J.; Lee, M.T.; Kim, T.K.; Peritz, T.; Buckley, P.; Cappelleri, D.J.; Maronski, M.; et al. Transcriptome transfer produces a predictable cellular phenotype. Proceedings of the National Academy of Sciences of the United States of America 2009, 106, 7624–7629. [Google Scholar] [CrossRef]
- Kumar, R.M.; Cahan, P.; Shalek, A.K.; Satija, R.; DaleyKeyser, A.; Li, H.; Zhang, J.; Pardee, K.; Gennert, D.; Trombetta, J.J.; et al. Deconstructing transcriptional heterogeneity in pluripotent stem cells. Nature 2014, 516, 56–61. [Google Scholar] [CrossRef] [PubMed]
- de Vargas Roditi, L.; Claassen, M. Computational and experimental single cell biology techniques for the definition of cell type heterogeneity, interplay and intracellular dynamics. Current opinion in biotechnology 2015, 34, 9–15. [Google Scholar] [CrossRef]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lönnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nature methods 2014, 11, 163–166. [Google Scholar] [CrossRef]
- Sanchez, A.; Golding, I. Genetic determinants and cellular constraints in noisy gene expression. Science (New York, N.Y.) 2013, 342, 1188–1193. [Google Scholar] [CrossRef]
- Marguerat, S.; Schmidt, A.; Codlin, S.; Chen, W.; Aebersold, R.; Bähler, J. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 2012, 151, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Choi, P.J.; Li, G.W.; Chen, H.; Babu, M.; Hearn, J.; Emili, A.; Xie, X.S. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science (New York, N.Y.) 2010, 329, 533–538. [Google Scholar] [CrossRef]
- Luo, Y.; Coskun, V.; Liang, A.; Yu, J.; Cheng, L.; Ge, W.; Shi, Z.; Zhang, K.; Li, C.; Cui, Y.; et al. Single-cell transcriptome analyses reveal signals to activate dormant neural stem cells. Cell 2015, 161, 1175–1186. [Google Scholar] [CrossRef]
- Mahata, B.; Zhang, X.; Kolodziejczyk, A.A.; Proserpio, V.; Haim-Vilmovsky, L.; Taylor, A.E.; Hebenstreit, D.; Dingler, F.A.; Moignard, V.; Göttgens, B.; et al. Single-cell RNA sequencing reveals T helper cells synthesizing steroids de novo to contribute to immune homeostasis. Cell reports 2014, 7, 1130–1142. [Google Scholar] [CrossRef]
- Bendall, S.C.; Davis, K.L.; Amir el, A.D.; Tadmor, M.D.; Simonds, E.F.; Chen, T.J.; Shenfeld, D.K.; Nolan, G.P.; Pe'er, D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 2014, 157, 714–725. [Google Scholar] [CrossRef]
- Chang, H.H.; Hemberg, M.; Barahona, M.; Ingber, D.E.; Huang, S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 2008, 453, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D.; et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef]
- Deng, Q.; Ramsköld, D.; Reinius, B.; Sandberg, R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science (New York, N.Y.) 2014, 343, 193–196. [Google Scholar] [CrossRef]
- Vitak, S.A.; Torkenczy, K.A. Sequencing thousands of single-cell genomes with combinatorial indexing. 2017, 14, 302–308. [Google Scholar] [CrossRef]
- Chen, C.; Xing, D. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). 2017, 356, 189–194. [Google Scholar] [CrossRef]
- Guo, F.; Li, L.; Li, J.; Wu, X.; Hu, B.; Zhu, P.; Wen, L.; Tang, F. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell research 2017, 27, 967–988. [Google Scholar] [CrossRef] [PubMed]
- Casasent, A.K.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, N.E. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018, 172, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Demaree, B.; Weisgerber, D.; Lan, F.; Abate, A.R. An Ultrahigh-throughput Microfluidic Platform for Single-cell Genome Sequencing. Journal of visualized experiments : JoVE 2018. [Google Scholar] [CrossRef]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 172, 1091–1107. [Google Scholar] [CrossRef]
- Rosenberg, A.B.; Roco, C.M. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. 2018, 360, 176–182. [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Frontiers in genetics 2019, 10, 317. [Google Scholar] [CrossRef]
- Sheng, K.; Cao, W.; Niu, Y.; Deng, Q.; Zong, C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nature methods 2017, 14, 267–270. [Google Scholar] [CrossRef]
- Picelli, S.; Björklund, Å.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Goldstein, L.D.; Chen, Y.J.; Dunne, J.; Mir, A.; Hubschle, H.; Guillory, J.; Yuan, W.; Zhang, J.; Stinson, J.; Jaiswal, B.; et al. Massively parallel nanowell-based single-cell gene expression profiling. BMC genomics 2017, 18, 519. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, X.; Wu, X.; Guo, H.; Hu, Y.; Tang, F.; Huang, Y. Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome biology 2015, 16, 148. [Google Scholar] [CrossRef]
- Islam, S.; Kjällquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lönnerberg, P.; Linnarsson, S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome research 2011, 21, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. 2017, 8, 14049. [CrossRef]
- DeLaughter, D.M. The Use of the Fluidigm C1 for RNA Expression Analyses of Single Cells. Current protocols in molecular biology 2018, 122, e55. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Zhang, W.; Xin, H.; Deng, G. Single Cell Isolation and Analysis. Frontiers in cell and developmental biology 2016, 4, 116. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: advances and future challenges. Nucleic acids research 2014, 42, 8845–8860. [Google Scholar] [CrossRef]
- Menon, V. Clustering single cells: a review of approaches on high-and low-depth single-cell RNA-seq data. Briefings in functional genomics 2018, 17, 240–245. [Google Scholar] [CrossRef]
- Cembrowski, M.S. Single-cell transcriptomics as a framework and roadmap for understanding the brain. Journal of neuroscience methods 2019, 326, 108353. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Experimental & molecular medicine 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Molecular cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Gross, A.; Schoendube, J.; Zimmermann, S.; Steeb, M.; Zengerle, R.; Koltay, P. Technologies for Single-Cell Isolation. International journal of molecular sciences 2015, 16, 16897–16919. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Kovacs, D.; Konijnenberg, A.; Timmerman, E.; Volkov, A.; Guharoy, M.; De Decker, M.; Jaspers, T.; Ryan, V.H. Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Molecular cell 2017, 65, 1044–1055. [Google Scholar] [CrossRef]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; De Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome biology 2016, 17, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science (New York, N.Y.) 2014, 343, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Ramsköld, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature biotechnology 2012, 30, 777–782. [Google Scholar] [CrossRef]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular cell 2017, 65, 631–643. [Google Scholar] [CrossRef]
- Proposed methods for testing and selecting the ERCC external RNA controls. BMC genomics 2005, 6, 150. [CrossRef]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science (New York, N.Y.) 2014, 343, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Arendt, D.; Musser, J.M.; Baker, C.V.H.; Bergman, A.; Cepko, C.; Erwin, D.H.; Pavlicev, M.; Schlosser, G.; Widder, S.; Laubichler, M.D.; et al. The origin and evolution of cell types. Nature reviews. Genetics 2016, 17, 744–757. [Google Scholar] [CrossRef]
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education 2002, 26, 256–270. [Google Scholar] [CrossRef]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. 2022, 12, e694. [CrossRef]
- Rodriques, S.G.; Stickels, R.R. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. 2019, 363, 1463–1467. [CrossRef]
- Shahbazi, M.N.; Scialdone, A.; Skorupska, N.; Weberling, A.; Recher, G.; Zhu, M.; Jedrusik, A.; Devito, L.G.; Noli, L.; Macaulay, I.C.; et al. Pluripotent state transitions coordinate morphogenesis in mouse and human embryos. Nature 2017, 552, 239–243. [Google Scholar] [CrossRef]
- Enge, M.; Arda, H.E.; Mignardi, M.; Beausang, J.; Bottino, R.; Kim, S.K.; Quake, S.R. Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell 2017, 171, 321–330. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Voet, T. Single cell genomics: advances and future perspectives. PLoS genetics 2014, 10, e1004126. [Google Scholar] [CrossRef]
- Sanz, E.; Yang, L.; Su, T.; Morris, D.R.; McKnight, G.S.; Amieux, P.S. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proceedings of the National Academy of Sciences of the United States of America 2009, 106, 13939–13944. [Google Scholar] [CrossRef]
- Zeb, Q.; Wang, C.; Shafiq, S.; Liu, L. An overview of single-cell isolation techniques. Single-cell omics 2019, 101-135.
- van den Brink, S.C.; Sage, F.; Vértesy, Á. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. 2017, 14, 935–936. [CrossRef]
- Zappia, L.; Phipson, B. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. 2018, 14, e1006245. [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Lun, A.T.; McCarthy, D.J.; Marioni, J.C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research 2016, 5, 2122. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome biology 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S. Phenotype molding of stromal cells in the lung tumor microenvironment. 2018, 24, 1277–1289. [CrossRef]
- Fan, X.; Bialecka, M.; Moustakas, I. Single-cell reconstruction of follicular remodeling in the human adult ovary. 2019, 10, 3164. [CrossRef]
- Lytal, N.; Ran, D.; An, L. Normalization Methods on Single-Cell RNA-seq Data: An Empirical Survey. Frontiers in genetics 2020, 11, 41. [Google Scholar] [CrossRef]
- Yip, S.H.; Sham, P.C.; Wang, J. Evaluation of tools for highly variable gene discovery from single-cell RNA-seq data. Briefings in bioinformatics 2019, 20, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics 2013, 14, 7. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: single-cell regulatory network inference and clustering. Nature methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B. SCENIC: single-cell regulatory network inference and clustering. 2017, 14, 1083–1086. [CrossRef]
- Van de Sande, B.; Flerin, C. A scalable SCENIC workflow for single-cell gene regulatory network analysis. 2020, 15, 2247–2276. [CrossRef]
- Ma, A.; Wang, C.; Chang, Y.; Brennan, F.H.; McDermaid, A.; Liu, B.; Zhang, C.; Popovich, P.G.; Ma, Q. IRIS3: integrated cell-type-specific regulon inference server from single-cell RNA-Seq. Nucleic acids research 2020, 48, W275–w286. [Google Scholar] [CrossRef]
- Lewis, J. From signals to patterns: space, time, and mathematics in developmental biology. Science (New York, N.Y.) 2008, 322, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Cremisi, F.; Philpott, A.; Ohnuma, S. Cell cycle and cell fate interactions in neural development. Current opinion in neurobiology 2003, 13, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome medicine 2022, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. 2017, 9, 75. [CrossRef]
- Terabayashi, T.; Germino, G.G.; Menezes, L.F. Pathway identification through transcriptome analysis. Cellular signalling 2020, 74, 109701. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science (New York, N.Y.) 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration. 2020, 42, e1900221. [CrossRef]
- Zhuang, X. Spatially resolved single-cell genomics and transcriptomics by imaging. Nature methods 2021, 18, 18–22. [Google Scholar] [CrossRef]
- Larsson, L.; Frisén, J.; Lundeberg, J. Spatially resolved transcriptomics adds a new dimension to genomics. 2021, 18, 15–18. [CrossRef]
- Crosetto, N.; Bienko, M.; van Oudenaarden, A. Spatially resolved transcriptomics and beyond. Nature reviews. Genetics 2015, 16, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Moor, A.E.; Itzkovitz, S. Spatial transcriptomics: paving the way for tissue-level systems biology. Current opinion in biotechnology 2017, 46, 126–133. [Google Scholar] [CrossRef]
- Armingol, E.; Officer, A. Deciphering cell-cell interactions and communication from gene expression. 2021, 22, 71–88. [CrossRef]
- Rompolas, P.; Mesa, K.R.; Greco, V. Spatial organization within a niche as a determinant of stem-cell fate. Nature 2013, 502, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-E.; Jo, S.H.; Lee, R.H.; Macks, C.P.; Ku, T.; Park, J.; Lee, C.W.; Hur, J.K.; Sohn, C.H. Spatial Transcriptomics: Technical Aspects of Recent Developments and Their Applications in Neuroscience and Cancer Research. Advanced Science n/a, 2206939. [CrossRef]
- Shaw, R.; Tian, X.; Xu, J. Single-Cell Transcriptome Analysis in Plants: Advances and Challenges. Molecular plant 2021, 14, 115–126. [Google Scholar] [CrossRef]
- Denyer, T.; Ma, X.; Klesen, S.; Scacchi, E.; Nieselt, K.; Timmermans, M.C.P. Spatiotemporal Developmental Trajectories in the Arabidopsis Root Revealed Using High-Throughput Single-Cell RNA Sequencing. Developmental cell 2019, 48, 840–852. [Google Scholar] [CrossRef]
- Shulse, C.N.; Cole, B.J.; Ciobanu, D.; Lin, J.; Yoshinaga, Y.; Gouran, M.; Turco, G.M.; Zhu, Y.; O'Malley, R.C.; Brady, S.M.; et al. High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types. Cell reports 2019, 27, 2241–2247. [Google Scholar] [CrossRef]
- Zhang, T.Q.; Xu, Z.G.; Shang, G.D.; Wang, J.W. A Single-Cell RNA Sequencing Profiles the Developmental Landscape of Arabidopsis Root. Molecular plant 2019, 12, 648–660. [Google Scholar] [CrossRef]
- Levitin, H.M.; Yuan, J.; Sims, P.A. Single-Cell Transcriptomic Analysis of Tumor Heterogeneity. Trends in cancer 2018, 4, 264–268. [Google Scholar] [CrossRef]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Haghverdi, L.; Lun, A.T.L. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. 2018, 36, 421–427. [CrossRef]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 1661–1662. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer's Disease. Cell 2020, 182, 976–991. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.; Belcastro, V.; Ambesi-Impiombato, A.; di Bernardo, D. How to infer gene networks from expression profiles. Molecular systems biology 2007, 3, 78. [Google Scholar] [CrossRef]
- Xue, Z.; Huang, K.; Cai, C.; Cai, L.; Jiang, C.Y.; Feng, Y.; Liu, Z.; Zeng, Q.; Cheng, L.; Sun, Y.E.; et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature 2013, 500, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell reports 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhou, X. Applications of Single-Cell Sequencing for Multiomics. Methods in molecular biology (Clifton, N.J.) 2018, 1754, 327–374. [Google Scholar] [CrossRef]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef]
- Lawson, D.A.; Kessenbrock, K. Tumour heterogeneity and metastasis at single-cell resolution. 2018, 20, 1349–1360. [CrossRef]
- Caswell, D.R.; Swanton, C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC medicine 2017, 15, 133. [Google Scholar] [CrossRef]
- Alderton, G.K. Tumour evolution: Epigenetic and genetic heterogeneity in metastasis. Nature reviews. Cancer 2017, 17, 141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, X.; Zheng, L.; Zhang, Y.; Li, Y.; Fang, Q.; Gao, R.; Kang, B.; Zhang, Q.; Huang, J.Y.; et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018, 564, 268–272. [Google Scholar] [CrossRef]
- Bian, S.; Hou, Y. Single-cell multiomics sequencing and analyses of human colorectal cancer. 2018, 362, 1060–1063. [CrossRef]
- Li, J.; Wang, R.; Zhou, X.; Wang, W. Genomic and transcriptomic profiling of carcinogenesis in patients with familial adenomatous polyposis. 2020, 69, 1283–1293. [CrossRef]
- Chen, Y.C.; Sahoo, S.; Brien, R.; Jung, S.; Humphries, B.; Lee, W.; Cheng, Y.H.; Zhang, Z.; Luker, K.E.; Wicha, M.S.; et al. Single-cell RNA-sequencing of migratory breast cancer cells: discovering genes associated with cancer metastasis. The Analyst 2019, 144, 7296–7309. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Chung, W.; Lee, H.-O.; Jeong, D.E.; Jo, A.; Lim, J.E.; Hong, J.H.; Nam, D.-H.; Jeong, B.C.; Park, S.H. Single-cell RNA sequencing reveals the tumor microenvironment and facilitates strategic choices to circumvent treatment failure in a chemorefractory bladder cancer patient. Genome medicine 2020, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Narayanan, S.P.; Mannan, R.; Raskind, G.; Wang, X.; Vats, P.; Su, F.; Hosseini, N.; Cao, X.; Kumar-Sinha, C. Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proceedings of the National Academy of Sciences 2021, 118, e2103240118. [Google Scholar] [CrossRef]
- Shalek, A.K.; Benson, M. Single-cell analyses to tailor treatments. 2017, 9. [CrossRef]
- Mustachio, L.M.; Roszik, J. Single-Cell Sequencing: Current Applications in Precision Onco-Genomics and Cancer Therapeutics. Cancers 2022, 14, 657. [Google Scholar] [CrossRef]
- Crinier, A.; Milpied, P.; Escalière, B.; Piperoglou, C.; Galluso, J.; Balsamo, A.; Spinelli, L.; Cervera-Marzal, I.; Ebbo, M.; Girard-Madoux, M.; et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity 2018, 49, 971–986. [Google Scholar] [CrossRef]
- Villani, A.C.; Satija, R. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. 2017, 356. [CrossRef]
- Xin, G.; Zander, R.; Schauder, D.M.; Chen, Y.; Weinstein, J.S.; Drobyski, W.R.; Tarakanova, V.; Craft, J. Single-cell RNA sequencing unveils an IL-10-producing helper subset that sustains humoral immunity during persistent infection. 2018, 9, 5037. [CrossRef]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nature reviews. Immunology 2018, 18, 35–45. [Google Scholar] [CrossRef]
- Martinez-Jimenez, C.P.; Eling, N.; Chen, H.C.; Vallejos, C.A.; Kolodziejczyk, A.A.; Connor, F.; Stojic, L.; Rayner, T.F.; Stubbington, M.J.T.; Teichmann, S.A.; et al. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science (New York, N.Y.) 2017, 355, 1433–1436. [Google Scholar] [CrossRef]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef]
- Gao, S.; Yan, L. Tracing the temporal-spatial transcriptome landscapes of the human fetal digestive tract using single-cell RNA-sequencing. 2018, 20, 721–734. [CrossRef]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science (New York, N.Y.) 2013, 342, 632–637. [Google Scholar] [CrossRef]
- Li, H.; Horns, F.; Wu, B.; Xie, Q.; Li, J.; Li, T.; Luginbuhl, D.J.; Quake, S.R.; Luo, L. Classifying Drosophila Olfactory Projection Neuron Subtypes by Single-Cell RNA Sequencing. Cell 2017, 171, 1206–1220. [Google Scholar] [CrossRef]
- Luo, C.; Keown, C.L. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. 2017, 357, 600–604. [CrossRef]
- Lake, B.B.; Ai, R.; Kaeser, G.E.; Salathia, N.S.; Yung, Y.C.; Liu, R.; Wildberg, A.; Gao, D.; Fung, H.L.; Chen, S.; et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science (New York, N.Y.) 2016, 352, 1586–1590. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.A.; Bihannic, L.; Rosencrance, C.; Hadley, J.L.; Tong, Y.; Phoenix, T.N.; Natarajan, S.; Easton, J.; Northcott, P.A.; Gawad, C. A Single-Cell Transcriptional Atlas of the Developing Murine Cerebellum. Current biology : CB 2018, 28, 2910–2920. [Google Scholar] [CrossRef]
- Hou, Y.; Fan, W.; Yan, L.; Li, R.; Lian, Y.; Huang, J.; Li, J.; Xu, L.; Tang, F.; Xie, X.S.; et al. Genome analyses of single human oocytes. Cell 2013, 155, 1492–1506. [Google Scholar] [CrossRef]
- Li, W.; Ma, Y.; Yu, S.; Sun, N.; Wang, L.; Chen, D.; Yang, G.; Lu, S.; Li, Y.; Yang, B.; et al. The mutation-free embryo for in vitro fertilization selected by MALBAC-PGD resulted in a healthy live birth from a family carrying PKD 1 mutation. Journal of assisted reproduction and genetics 2017, 34, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nature structural & molecular biology 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Briggs, J.A.; Weinreb, C.; Wagner, D.E. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. 2018, 360. [CrossRef]
- Li, L.; Guo, F. Publisher Correction: Single-cell multi-omics sequencing of human early embryos. 2018, 20, 1227. [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, Y.; Gao, Y.; Lin, Z.; Yang, S.; Wang, T.; Wang, Q.; Xie, N.; Hua, R.; Liu, M.; et al. Single-cell RNA-seq uncovers dynamic processes and critical regulators in mouse spermatogenesis. 2018, 28, 879–896. [CrossRef]
- Wang, M.; Liu, X.; Chang, G.; Chen, Y.; An, G.; Yan, L.; Gao, S.; Xu, Y.; Cui, Y.; Dong, J.; et al. Single-Cell RNA Sequencing Analysis Reveals Sequential Cell Fate Transition during Human Spermatogenesis. Cell stem cell 2018, 23, 599–614. [Google Scholar] [CrossRef]
- Marinov, G.K.; Williams, B.A.; McCue, K.; Schroth, G.P.; Gertz, J.; Myers, R.M.; Wold, B.J. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome research 2014, 24, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, P.; Anders, S.; Kim, J.K.; Kołodziejczyk, A.A.; Zhang, X.; Proserpio, V.; Baying, B.; Benes, V.; Teichmann, S.A.; Marioni, J.C.; et al. Accounting for technical noise in single-cell RNA-seq experiments. Nature methods 2013, 10, 1093–1095. [Google Scholar] [CrossRef]
- Ding, B.; Zheng, L.; Zhu, Y.; Li, N.; Jia, H.; Ai, R.; Wildberg, A.; Wang, W. Normalization and noise reduction for single cell RNA-seq experiments. Bioinformatics (Oxford, England) 2015, 31, 2225–2227. [Google Scholar] [CrossRef]
- Grün, D.; Kester, L.; van Oudenaarden, A. Validation of noise models for single-cell transcriptomics. Nature methods 2014, 11, 637–640. [Google Scholar] [CrossRef]
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O'Shaughnessy, A.L.; Lambert, G.M.; Araúzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-sequencing from single nuclei. Proceedings of the National Academy of Sciences of the United States of America 2013, 110, 19802–19807. [Google Scholar] [CrossRef]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. 2015, 10, 442–458. [CrossRef]
- Kang, Y.; Norris, M.H.; Zarzycki-Siek, J.; Nierman, W.C.; Donachie, S.P.; Hoang, T.T. Transcript amplification from single bacterium for transcriptome analysis. Genome research 2011, 21, 925–935. [Google Scholar] [CrossRef]
- Armour, C.D.; Castle, J.C.; Chen, R.; Babak, T.; Loerch, P.; Jackson, S.; Shah, J.K.; Dey, J.; Rohl, C.A.; Johnson, J.M.; et al. Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nature methods 2009, 6, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Fincher, C.T.; Wurtzel, O.; de Hoog, T.; Kravarik, K.M.; Reddien, P.W. Cell type transcriptome atlas for the planarian Schmidtea mediterranea. 2018, 360. 360. [CrossRef]
- Cao, J.; Packer, J.S. Comprehensive single-cell transcriptional profiling of a multicellular organism. 2017, 357, 661–667. [CrossRef]
- Replogle, J.M.; Norman, T.M.; Xu, A. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. 2020, 38, 954–961. [CrossRef]
- Zafar, H.; Lin, C.; Bar-Joseph, Z. Single-cell lineage tracing by integrating CRISPR-Cas9 mutations with transcriptomic data. 2020, 11, 3055. [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Perkel, J.M. Single-cell proteomics takes centre stage. Nature 2021, 597, 580–582. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mass and single cell RNA sequencing technique procedures.
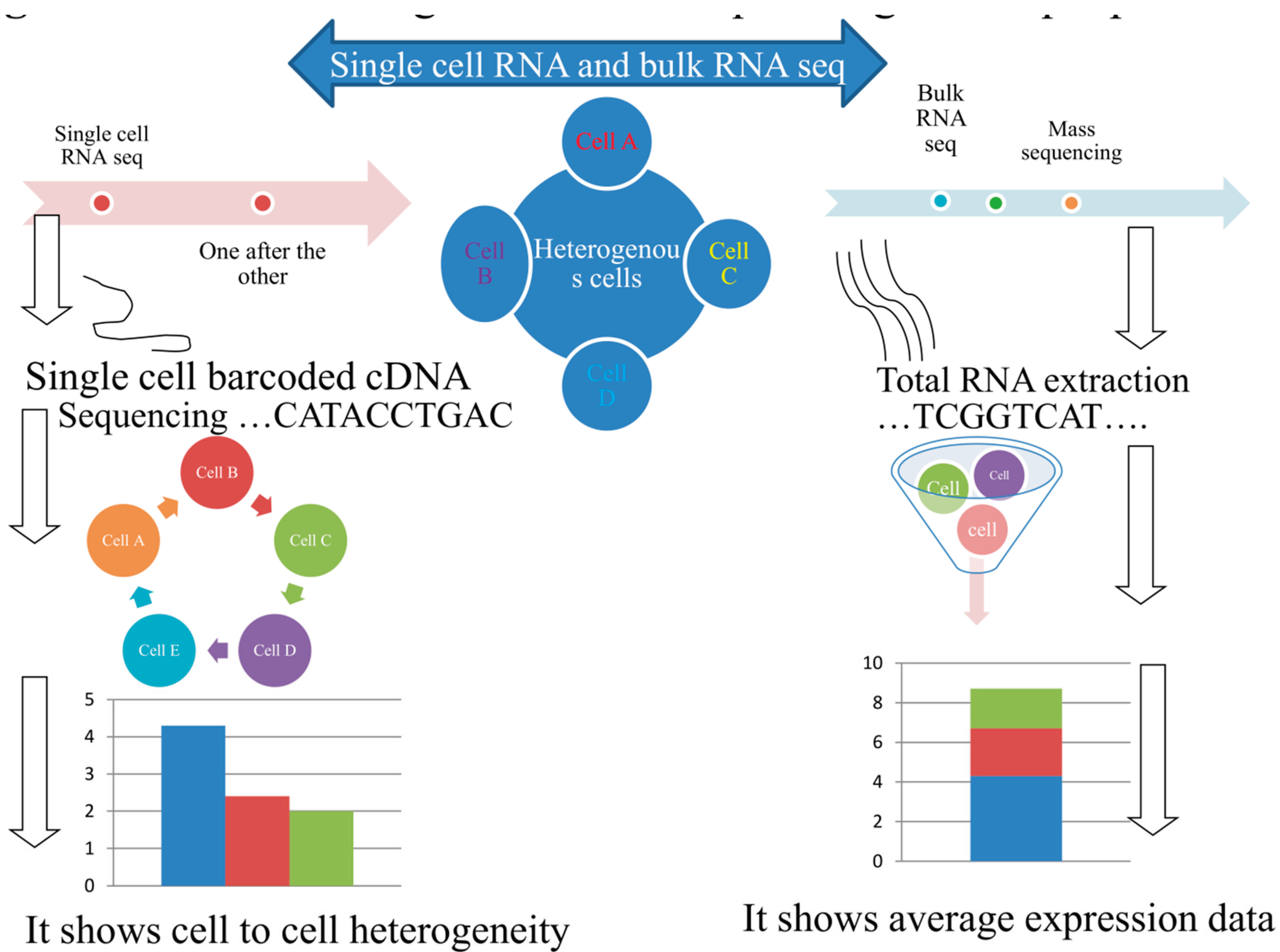
Figure 2.
Timeline of single cell sequencing methods milestones.
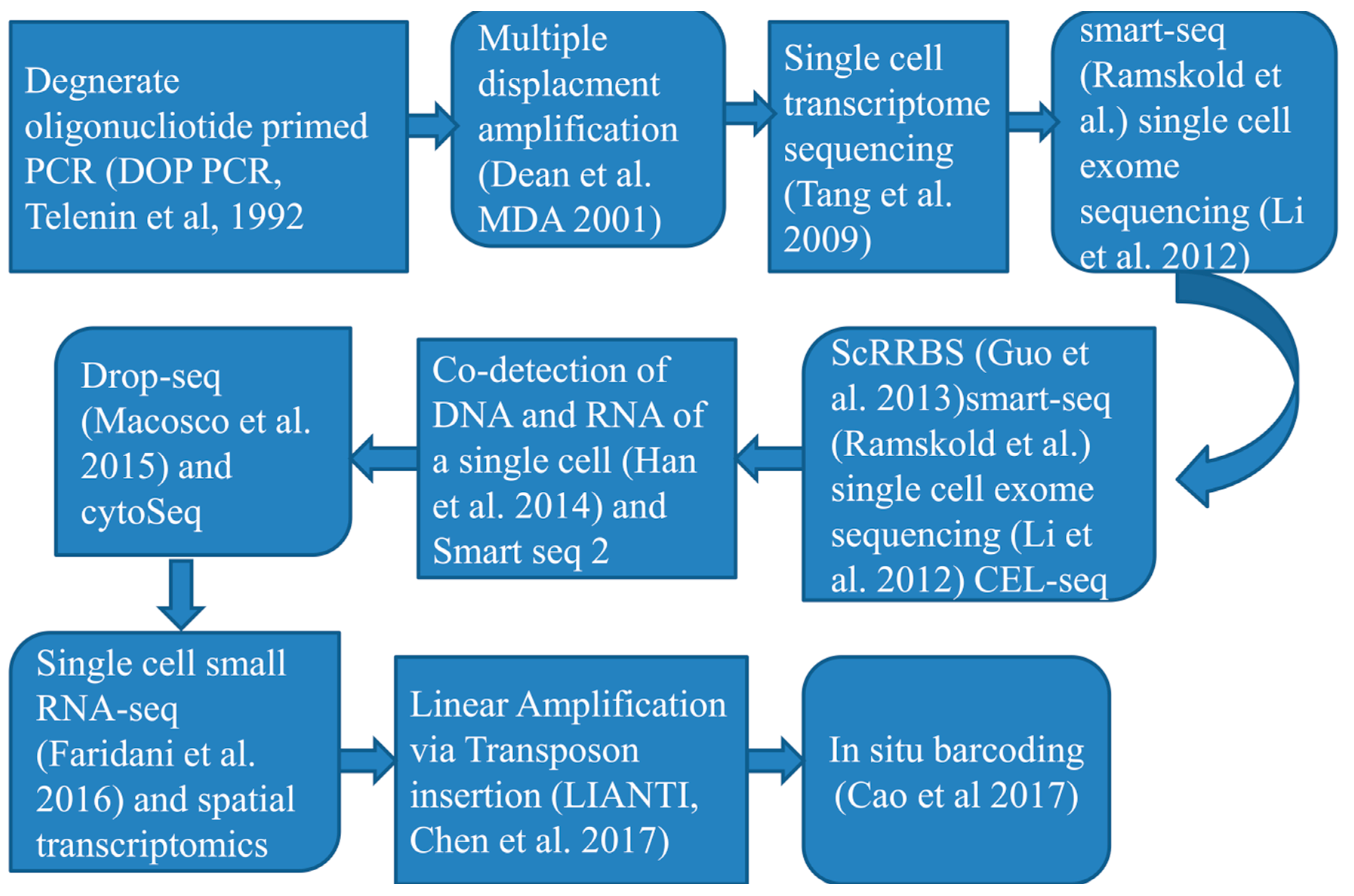
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



