
Submitted:
22 February 2024
Posted:
23 February 2024
You are already at the latest version
Abstract
This study investigates the metabolic parallels between stimulated pancreatic beta cells and cancer cells, emphasizing glucose and glutamine metabolism. Addressing the significant public health challenges of Type 2 Diabetes Mellitus (T2DM) and cancer, we aim to deepen understanding of the mechanisms driving insulin secretion and cellular proliferation. Our analysis of anaplerotic cycles and NADPH's role in biosynthesis elucidates their vital functions in both processes. Additionally, we find that both cell types share an antioxidative response mediated by the Nrf2 signaling pathway, glutathione synthesis, and UCP2 upregulation. Notably, UCP2 facilitates the transfer of C4 metabolites, enhancing reductive TCA cycle metabolism. We also uncover that hypoxic responses are transient in beta cells post-stimulation but persistent in cancer cells. By synthesizing these insights, our research suggests novel therapeutic targets for T2DM, highlighting the shared metabolic strategies of stimulated beta cells and cancer cells. This comparative analysis not only illuminates the metabolic complexity of these conditions but also emphasizes the crucial role of metabolic pathways in cell function and survival, offering fresh perspectives for tackling T2DM and cancer challenges.
Keywords:
Diabetes
; Cancer
; Reductive metabolism
; NADPH
; GSH
; Nrf2 pathway
; UCP2
; Succinate
; HIF
1. Introduction
Cancer and Type 2 Diabetes Mellitus (T2DM) are significant public health challenges in contemporary society, each with substantial impacts on global health and economies. Cancer, with its rapid and often lethal progression, captures considerable attention in medical research and public concern. In contrast, T2DM, characterized by more gradual health decline, garners less immediate fear and urgency despite its profound societal and individual burdens. The global prevalence of T2DM is staggering, with an estimated 463 million individuals affected as of 2019, and projections suggest this number could rise to 700 million by 2045 (Saeedi et al., 2019). The economic implications are equally daunting; for instance, the global healthcare expenditure on diabetes was estimated to be at least 760 billion USD in 2019, a figure expected to surge as the prevalence of T2DM increases (International Diabetes Federation, 2019). These statistics underscore the critical need for enhanced research, prevention strategies, and therapeutic interventions for T2DM, paralleling the urgency accorded to cancer research.
Building upon extensive research in cancer metabolism, we can derive valuable insights for a deeper understanding of T2DM, particularly in the context of pancreatic beta-cell metabolism. The metabolic response of pancreatic beta cells to glucose, crucial for insulin secretion, remains a vibrant area of scientific inquiry. Despite numerous studies, the mechanisms by which beta cells detect and respond to glucose and other metabolites to regulate insulin release are not fully understood. Traditionally, glucose-stimulated insulin secretion (GSIS) has been attributed to the increase in mitochondrial ATP production leading to the closure of KATP channels. Our research group has focused on this ATP-centric model, examining its role as the primary signal following glucose uptake by beta cells (Grubelnik et al., 2020a,b). In our more recent papers (Grubelnik et al., 2022, 2024), we have expanded our investigation through pathways involving pyruvate carboxylase (PC) and phosphoenolpyruvate (PEP) formation, which locally elevates ATP concentrations near KATP channels, providing a nuanced understanding of GSIS (Abulizi et al., 2020; Corkey, 2020; Foster et al., 2022; Lewandowski et al., 2020; Merrins et al., 2022). While the elevation in ATP levels and subsequent insulin secretion in response to glucose stimulation remains undisputed, a revised conceptual framework acknowledges beta cells' ability to sense the overall abundance of postprandial metabolites, particularly glucose and glutamine, which are closely associated with the need for energy storage through anabolic processes. This recognition of the anabolic state appears to hold greater significance for insulin secretion in beta cells than mere ATP production via oxidative phosphorylation (OxPhos) within mitochondria (Lu et al., 2002; Jesinkey et al., 2019; Lewandowski et al., 2020; Grubelnik et al., 2024). Such insights suggest that the metabolic pathways engaged by beta cells in response to glucose are multifaceted, implicating not only traditional ATP production but also the integration of signals from various metabolic intermediates that reflect the cell's broader nutritional and energetic status.
Importantly, while mitochondrial ATP may not be the sole determinant of glucose-stimulated insulin secretion (GSIS), particularly during the amplification phase, the indispensable role of mitochondria in insulin release is evident. Experimental evidence supports the involvement of mitochondria in the amplification pathway of insulin secretion, observed when beta cells are stimulated by glucose and other mitochondrial substrates, with cytosolic Ca2+ maintained at levels conducive to secretion (Maechler, 2013). The amplification phase is influenced by mitochondrial coupling factors, derived from tricarboxylic acid (TCA) cycle metabolites, necessitating replenishment through anaplerotic processes (Rustenbeck et al., 2021). Thus, the integration of mitochondrial anaplerotic pathways with cytosolic processes is essential for a thorough understanding of the cellular dynamics underpinning insulin secretion. In the postprandial state, glucose, glutamine, amino acids, and fatty acids significantly influence insulin secretion. This period is characterized by an anabolic state, conducive to metabolite synthesis, with insulin functioning as a key anabolic hormone. For effective regulation of body-wide anabolic processes, beta cells are tasked with detecting this metabolic abundance, particularly sensing elevations in glucose and glutamine levels. Analogously, cancer cells capitalize on the anabolic state, exploiting it to furnish the necessary building blocks for their proliferation and invasion. This suggests that cancer cells not only thrive in but also potentially simulate an anabolic environment to facilitate their unchecked growth and division. In contrast, beta cells are finely tuned to recognize and respond to the body’s anabolic cues, secreting insulin to modulate anabolic activity. This distinction highlights a fundamental divergence in how cancer cells and beta cells interact with and respond to the body’s metabolic state, with the former hijacking these processes for malignancy and the latter orchestrating physiological responses to maintain metabolic homeostasis.
We propose that metabolic pathways exploited by cancer cells for proliferation and survival could similarly be operational in beta cells for identifying the anabolic state and facilitating insulin secretion. These mechanisms, essential for the uncontrolled growth and division of cancer cells, are hypothesized to play a pivotal role in beta cells for the initiation and enhancement of insulin secretion. This paper focuses on the metabolism of glucose and glutamine, highlighting the shared aspects between cancer cells and beta cells. We underscore the common cellular mechanisms in both cell types, particularly concerning the role of anaplerotic cycles that generate citrate and malate, and especially NADPH, which is vital for biosynthesis. NADPH is acknowledged as one of the key enhancers of insulin secretion and is critical in cancer pathogenesis.
Furthermore, we explore the production of H2O2 and ROS and the antioxidant defenses in both beta and cancer cells. Our study delves into the Nrf2 signaling pathway and glutathione (GSH) synthesis, highlighting their significance in the antioxidant capacity and mechanisms within these cells. We also consider the role of UCP2, not primarily as a potential decoupler but more for its antioxidative significance in conditions of elevated ROS levels. UCP2 is additionally recognized for its role in the transport of C4 metabolites, particularly malate, oxaloacetate, and aspartate, from the mitochondria to the cytosol. Lastly, we examine the pathophysiology of T2DM and cancer within the realm of hypoxic conditions, focusing on the mechanisms behind succinate accumulation and HIF stabilization. Specifically, we discuss the distinct impacts of temporal versus more sustained, permanent responses to hypoxic conditions.
Investigating these shared pathways and mechanisms between pancreatic beta cells and cancer cells offers a unique opportunity to bridge existing research gaps and potentially unveil novel therapeutic avenues for T2DM. This approach leverages the rich research foundation in cancer research to foster a new understanding and tackle the complexities of T2DM that remains a significant challenge to global health.
2. Reductive metabolism and NADPH production
Cancer cells and stimulated beta cells, characterized by their high metabolic activity, typically necessitate substantial energy, predominantly in the form of ATP, to support their rapid growth and functional demands. However, the comprehensive metabolic needs of proliferating cancer cells extend beyond mere ATP production, as elucidated by Vander Heiden et al. (2009). For a cell to successfully divide and produce two viable daughter cells during mitosis, it must duplicate its entire cellular content, imposing significant demands for nucleotides, amino acids, and lipids. In this process, glucose not only serves as a source of ATP but also as a precursor for biomass generation. While ATP hydrolysis releases the free energy needed for some biochemical replication reactions, the synthesis of complex molecules like palmitate – a crucial component of cellular membranes – demands additional inputs: 7 molecules of ATP, 16 carbons from 8 acetyl-CoA molecules, and 28 electrons from 14 NADPH molecules. Similarly, the synthesis of amino acids and nucleotides requires more carbon and NADPH equivalents than ATP. A single glucose molecule can yield up to 36 ATPs, or alternatively, 30 ATPs and 2 NADPHs if channeled into the pentose phosphate pathway (PPP), besides providing 6 carbons for macromolecular biosynthesis. Consequently, synthesizing a 16-carbon fatty acyl chain would require the ATP from one glucose molecule fivefold, whereas seven glucose molecules are necessary to furnish the needed NADPH. This 35-fold asymmetry underscores that proliferating cells prioritize NADPH generation and carbon for biosynthesis over ATP production from carbon catabolism. Lane (2022) vividly articulates this preference: “If you were a cancer cell, the last thing you’d want to do is burn glucose through cell respiration, as it will produce a pile-up of ATP.” Notably, glucose and glutamine are the primary sources of carbon, nitrogen, free energy, and reducing equivalents for cell growth and division. Thus, this paper will focus on the metabolism of glucose and glutamine, pivotal for understanding the metabolic intricacies of both cancer and stimulated beta cells.
In stimulated beta cells, research has predominantly focused on ATP production following glucose stimulation. However, the longstanding notion that the primary trigger for insulin secretion is the increase in ATP levels, which leads to the closure of KATP channels, an increase in Ca2+ concentration, and the subsequent signal transmission to insulin granules, is being reevaluated. Recent insights suggest that, beyond ATP's role, other metabolic signals are critically important. Notably, metabolites from reductive metabolism in mitochondria, once released into the cytosol, play a significant role. The significance of mitochondrial anaplerotic products in insulin secretion has been a subject of intense research over the past two decades (MacDonald et al., 2005; Affourtit and Brand, 2008; Maechler, 2013; Rustenbeck et al., 2021). Significant strides have been made in acknowledging the role of anabolic signals in beta cells, particularly the redirection of glucose-derived pyruvate into reductive anaplerotic pathways via pyruvate carboxylase (PC), as opposed to the cataplerotic pathway via pyruvate dehydrogenase (PDH), as highlighted by recent studies (Abulizi et al., 2020; Foster et al., 2022; Lewandowski et al., 2020; Merrins et al., 2022). While the rise in ATP levels and its role in insulin secretion following glucose stimulation is well-established, a revised conceptual framework now recognizes the beta cells' capacity to detect the overall abundance of postprandial metabolites, especially glucose and glutamine. This sensitivity to the anabolic state is deemed more critical for insulin secretion than mere ATP generation through oxidative phosphorylation (OxPhos) within mitochondria (Lu et al., 2002; Jesinkey et al., 2019; Lewandowski et al., 2020; Grubelnik et al., 2024). Importantly, although mitochondrial ATP is not the sole determinant of glucose-stimulated insulin secretion (GSIS), particularly during the amplification phase, the role of mitochondria in this process is incontrovertible. Therefore, it is essential to consider mitochondrial anaplerotic pathways together with cytosolic processes, alongside OxPhos, to achieve a comprehensive understanding of the complex cellular mechanisms driving insulin secretion.
A common trait shared by both cancer and beta cells is the upregulation of reductive anaplerotic pathways at the expense of the catabolic conversion of pyruvate to acetyl-CoA via pyruvate dehydrogenase (PDH), which typically leads to NADH and subsequent ATP production. Specifically, in cancer cells, as Luengo et al. (2021) demonstrated, when there's an increased demand for NAD+ relative to ATP turnover, cells shift their metabolism towards aerobic glycolysis (the Warburg effect), converting pyruvate into lactate. This conversion necessitates NADH, which is then transformed into the needed NAD+. Consequently, lactate production becomes a hallmark of most cancer cells. In contrast, beta cells express very low levels of lactate dehydrogenase (LDH), indicating a minimal capacity for converting pyruvate into lactate, thus not exhibiting the Warburg effect (Sekine et al., 1994). Instead, cellular mechanisms in beta cells preferentially direct pyruvate towards oxaloacetate (OAA) via pyruvate carboxylase (PC), and subsequently to a reductive metabolism producing phosphoenolpyruvate (PEP) and malate. This reductive pathway of PEP production is vital for ATP generation near KATP channels, triggering insulin secretion (Abulizi et al., 2020; Foster et al., 2022; Lewandowski et al., 2020; Merrins et al., 2022). Thus, while the reductive metabolism plays a pivotal role in both cell types, there are nuanced differences. Cancer cells address the imbalance of a high NADH/NAD+ ratio relative to ATP turnover via the Warburg effect, whereas beta cells channel pyruvate through PC into PEP and malate production. Both cell types rely on anaplerotic cycles, especially the pyruvate-malate and pyruvate-citrate cycles, to support their metabolic needs.
Furthermore, the conversion of NADH to NADPH is critically important, with Nicotinamide Nucleotide Transhydrogenase (NNT) playing a key role in both cell types. In beta cells, NNT mediates the interconversion between NADH and NADPH in both forward and reverse modes, depending on glucose concentration (Santos et al., 2017). Similarly, in cancer cells, NNT is instrumental in converting NADH to NADPH, a process that, when disrupted, can affect reductive carboxylation (Gameiro et al., 2013). Overexpression of NNT has been shown to enhance glutamine oxidation and reductive carboxylation while inhibiting glucose catabolism in the TCA cycle (Gameiro et al., 2013). Recent findings also indicate that that the NNT-induced tumor cell “slimming” reverses the pro-carcinogenesis effect of HIF2a in tumors (Xiong et al., 2021). When NADPH is generated within mitochondria, its transport across the inner mitochondrial membrane is primarily facilitated by the isocitrate/α-KG shuttle (Ferdaoussi et al., 2015; Zhang et al., 2021). Figure 1 schematically presents the principal processes involved in producing and transporting metabolites that signal for cancer cell division and the amplification of insulin secretion.
In Figure 1, we illustrate a comprehensive overview of the processes involved in producing and transporting metabolites essential for cancer cell division and the amplification of insulin secretion. Within this intricate network, the role of NADPH is particularly notable, emphasizing its critical significance in two fundamental aspects shared by both cancer and stimulated beta cells. First, NADPH emerges as a key anaplerotically derived building block indispensable for biosynthesis. Second, it plays a crucial role in the cellular oxidative defense mechanism, acting in concert with other pathways. This role becomes especially vital in highly active cells, where every process, including oxidative phosphorylation (OxPhos), must function optimally, necessitating rigorous control of reactive oxygen species (ROS) to preserve cellular integrity and function. The forthcoming section will explore the role of NADPH more thoroughly, especially focusing on its involvement in the synthesis of glutathione (GSH), the foremost antioxidant in biological cells. This discussion aims to highlight the paramount importance of NADPH in maintaining cellular health and stability.
3. Antioxidative defense by GSH and the Nrf2 pathway
In all highly active cells, ROS production is notably high, primarily generated by mitochondria through the electron transport chain (ETC), as well as by H2O2 and subsequent ROS production via the pentose phosphate pathway and membrane-associated NADPH oxidases (NOX). Both stimulated beta cells and cancer cells, with their heightened activity, require stringent regulation of ROS levels. A critical mechanism for this regulation is the ROS-induced activation of the Nrf2 pathway (Blandino et al., 2023). The production of H2O2, whether by NOX in the cytoplasm or mitochondria, triggers the activation of Nrf2, setting off a cascade of downstream events that bolster the cell's antioxidant machinery and support the maintenance of mitochondrial function and integrity (Blandino et al., 2023). Among these events is the increased production of GSH within the cell. The Nrf2-mediated antioxidant response is a major defense mechanism against oxidative stress. The pathway's complexity is highlighted by Wu et al. (2019), showing that it regulates the import of cysteine into the cell and activates glutamate-cysteine ligase (GCL), the enzyme catalyzing the rate-limiting step in GSH biosynthesis (see Figure 2). Additionally, Nrf2 controls four NADPH-producing enzymes: glucose-6-phosphate dehydrogenase (G6PD), 6-phosphogluconate dehydrogenase (PGD), isocitrate dehydrogenase-1 (IDH1), and malic enzyme-1 (ME1), essential for providing NADPH required by many antioxidant enzymes and for GSH production (Wu et al., 2019). This aspect is especially crucial for cancer cells, which often face high oxidative stress levels due to their rapid metabolism and proliferation. Elevated GSH levels enhance antioxidant capacity and resistance to oxidative stress in many cancer cells, aiding their survival and proliferation (Traverso et al., 2013). Furthermore, GSH plays a vital role in detoxifying xenobiotics, including chemotherapy drugs, thereby offering cancer cells a protective advantage against the cytotoxic effects of anticancer agents. The association of increased GSH levels in cancer cells with enhanced resistance to various drugs and radiation therapy underscores this protective mechanism (Estrela et al., 2006).
Interestingly, unstimulated beta cells exhibit very limited antioxidant capacity (Robertson and Harmon, 2007; Lenzen et al., 2008). However, upon stimulation, particularly with glucose, these cells develop a robust antioxidative defense akin to that observed in cancer cells, characterized by activated Nrf2 signaling pathway and elevated GSH production. The relatively low antioxidant capacity of unstimulated beta cells can be attributed to the role of ROS, primarily its precursor H2O2, in signaling. The inherent weak antioxidant capacity in the resting state renders beta cells as efficient redox systems, with ROS serving as a critical signal for regulating insulin secretion (Ježek et al., 2021). Upon glucose stimulation, locally produced H2O2 by NOX4, in conjunction with an increase in ATP near KATP channels, contributes to the closure of these channels, thereby triggering insulin secretion (Plecitá-Hlavatá et al., 2020; Ježek et al., 2021). Thus, ROS plays a pivotal role in the initial phase of insulin secretion. It has been suggested that excessive use of antioxidants in prediabetes or early stages of T2DM could be counterproductive (Pi et al., 2010), a notion recently echoed by findings on the long-term antioxidative treatments in schizophrenia (Blandino et al., 2023). Conversely, chronic and sustained high levels of ROS are detrimental. In T2DM, the first phase of insulin secretion may be significantly impaired, potentially due to the diminished H2O2 signaling; the beta cell loses its ability to sense transient increases in H2O2 following glucose stimulation. Chronic exposure to hyperlipidemic or lipotoxic conditions could overwhelm beta cells, leading to excessive ROS production beyond the cell's capacity to mitigate. This scenario compromises the beta cells' function as effective redox sensors for glucose-induced ROS production, thereby impairing the mechanism that triggers the closure of KATP channels and subsequent insulin secretion.
Building on the understanding of ROS's role in beta cells as discussed, it is important to acknowledge the nuanced function of ROS in stimulated beta cells. On one hand, ROS acts as an essential signaling molecule for triggering insulin secretion, a process vital for physiological regulation. On the other hand, its accumulation can become detrimental, necessitating efficient scavenging during the extended second phase of insulin secretion (Newsholme et al., 2019). The neutralization of ROS through glutathione (GSH) activation of SENP1 highlights this delicate balance, promoting enhanced insulin secretion (Ferdaoussi et al., 2015; Jensen et al., 2017; Lin et al., 2023). This dualistic nature underscores the critical need to distinguish between the immediate and prolonged effects of nutrients on beta cell redox changes, particularly at the subcellular level. Research indicates that the interplay between nutrient regulation of beta cell redox signaling, ROS toxicity, and the effectiveness of antioxidant mechanisms in the context of gluco-lipotoxic conditions associated with T2DM is complex (Roma and Jonas, 2020). Therefore, in light of this complexity and the dual role of ROS, the indiscriminate use of antioxidants in the early stages of T2DM could potentially be counterproductive (Pi et al., 2010), emphasizing the importance of a nuanced approach to managing redox states for optimal beta cell function.
4. UCP2 upregulation and C4 transport
Uncoupling protein 2 (UCP2) activation can occur in response to superoxide and ROS elevations (Krauss et al., 2003), as well as through mechanisms not directly linked to superoxide production (Produit-Zengaffinen et al., 2007; Li et al., 2017). UCP2's function extends beyond that of classical uncoupling proteins, playing a significant role in cell-stress regulation, especially when contrasted with UCP1. In the context of increased ROS levels observed in cancer cells and beta cells postprandial stimulation, UCP2 assumes a crucial role. Notably, an upregulation in UCP2 expression characterizes both cancer cells and stimulated beta cells, with UCP2 exhibiting a markedly short half-life of 30 minutes, compared to the 30 days of UCP1 (Rousset et al., 2007). This rapid turnover is particularly vital for beta cells following stimulation, enabling swift adaptation to oxidative environments in the postprandial state.
In the realm of ROS signaling, UCP2's function is pivotal in both beta and cancer cells. Its expression is markedly elevated in most cancer cells, where UCP2 primarily serves to shield these cells from ROS, thereby averting cell death and fostering proliferation (Valle et al., 2010). Post-stimulation upregulation of UCP2 expression in beta cells underscores its importance, with its half-life ranging from approximately 30 minutes (Rousset et al., 2007) to 60 minutes in INS-1E insulinoma cells, a model for pancreatic beta cells (Azzu et al., 2008). This nutrient-induced expression of UCP2 in INS-1E cells has been scrutinized to understand the dynamics of UCP2 expression in response to nutrient availability. The findings reveal that UCP2 levels decrease in the absence of glutamine and increase with 20 mM glucose, indicating UCP2's variable expression in light of its short protein half-life, around one hour (Azzu et al., 2008), demonstrating its responsive nature to the cellular metabolic state.
Environmental factors significantly modulate Ucp2 transcription; its message levels rise in beta cells exposed to cold, high glucose, non-esterified fatty acids, and hydrogen peroxide (Affourtit & Brand, 2008). Postprandially, UCP2 expression escalates to manage the increased ROS production. Glucose, amino acids, and free fatty acids induce UCP2 expression (Patané et al., 2002), and UCP2 is activated by superoxide (Krauss et al., 2003), generated during metabolite oxidation (Affourtit & Brand, 2008). Despite UCP2's short half-life, nutrient-induced expression and subsequent activation are comparatively slower than the initial phase of glucose-stimulated insulin secretion (GSIS). Postprandially, the insulin release must significantly increase, requiring additional signals for GSIS amplification. These signals might include NADPH, acetyl-CoA, malonyl-CoA, and α-ketoglutarate, whose production is contingent on TCA cycle activity, a process facilitated by UCP2. This illustrates UCP2's role in connecting metabolic amplifiers of insulin secretion, previously discussed in the section "Reductive Metabolism and NADPH Production."
To comprehend how UCP2 aids GSIS amplifiers' production, its function as a transporter of carbon metabolites, especially C4 metabolites, across the mitochondrial membrane, must be considered. The anaplerotic export of carbon metabolites from mitochondria is crucial for beta and cancer cells, supplying metabolites during the body's anaplerotic state. This process is vital for providing building blocks for rapidly dividing cancer cells and amplifying insulin secretion. Vozza et al. (2014) demonstrated that UCP2 facilitates the exchange of malate, oxaloacetate, and aspartate for phosphate, thereby exporting C4 metabolites from mitochondria. This activity limits the oxidation of acetyl-CoA-producing substrates like glucose, enhances glutaminolysis, and prevents the mitochondrial accumulation of glutamine-derived C4 metabolites. The phosphate influx through UCP2 positively affects glutaminase, enhancing the conversion of glutamine to glutamate and then to oxaloacetate in the TCA cycle (Han et al., 2021). UCP2's regulation of C4 metabolite export and mitochondrial oxidation underscores its pivotal role in cellular bioenergetics and metabolic reprogramming under various conditions (Vozza et al., 2014; Ježek et al., 2018; Segalés et al., 2023). Further, Raho et al. (2020) highlighted UCP2's crucial role in transferring aspartate from mitochondria to the cytosol, essential for protein and nucleotide synthesis in fast-dividing cancer cells. Citrate and malate mainly contribute to NADPH production, vital for GSH biosynthesis and other metabolites (Prentki et al., 2013; Ju et al., 2020). These insights reveal UCP2's unique regulatory mechanism in cell bioenergetics, highlighting its importance in maintaining cellular redox states and NADPH production for anabolic processes and antioxidant defenses, pivotal in both cancer and nutrient-stimulated beta cells. This function of UCP2 elucidates a mechanistic link between enhanced glutamine oxidation and the Warburg effect, mitigating redox pressure on the respiratory chain and ROS production (Vozza et al., 2014). Figure 3 schematically presents UCP2's main regulatory mechanisms, emphasizing its role in metabolic flexibility and the anaplerotic export of C4 metabolites from mitochondria to the cytosol, crucial for both beta and cancer cells.
The observation that UCP2 facilitates a metabolic shift from glucose oxidation towards enhanced glutamine and lipid utilization (Pecqueur et al., 2008; Vozza et al., 2014; Ježek et al., 2018; Raho et al., 2020) has underscored its role in optimizing substrate use. This shift may allow pyruvate to be conserved for biosynthetic processes rather than being fully oxidized in mitochondria (Bouillaud, 2009). Such a metabolic adaptation is crucial for vigorous biosynthesis, necessitating enhanced anaplerotic flux to replenish TCA cycle intermediates. This process, involving pyruvate carboxylase and glutaminolysis, highlights the glutamine dependency seen in cancer cells (Baffy, 2017), attributed to glutamine's role in supporting rapid cell proliferation. Similarly, in stimulated beta cells, a comparable reliance on glutamine becomes evident in the amplification of GSIS. Glutamine, being the most abundant amino acid in the bloodstream, acts as a significant amplifier of GSIS. This effect is mediated through its metabolites, glutamate and NADPH, and further supported by the production of GSH, which are key products of glutaminolysis (Han et al., 2021). This intricate interplay underscores the critical function of UCP2 in modulating cellular energy metabolism and biosynthetic pathways, particularly in the context of the metabolic demands of both cancer and beta cells.
5. Hypoxia, succinate accumulation, and HIF activation
Hypoxic conditions, characterized by reduced oxygen availability, are a critical characteristic of tumor microenvironments, significantly influencing cancerogenesis and tumor progression. Hypoxia triggers a variety of biological responses, including the activation of pathways that govern proliferation, angiogenesis, and resistance to apoptosis. Tumors adeptly adjust to flourish in hypoxic conditions, correlating strongly with a poorer prognosis and increased resistance to traditional treatments like radiation therapy (Harris, 2002). The hypoxia-inducible factor (HIF), especially HIF-1α, serves as the primary molecular mediator for cellular adaptation to hypoxia, regulating numerous genes that equip tumor cells to endure, expand, and metastasize in oxygen-deprived settings (Vaupel, 2004; Brahimi-Horn, 2007). HIF activation promotes angiogenesis, shifts metabolism (notably, the Warburg effect), and facilitates invasion and metastasis via processes such as epithelial-to-mesenchymal transition (EMT). Consequently, targeting the hypoxia and HIF signaling pathways emerges as a promising approach in cancer therapy, seeking to thwart the adaptive mechanisms enabling cancer cell proliferation in hypoxic environments. Delving into the complex roles of hypoxia within tumor biology is essential for devising efficacious cancer treatments.
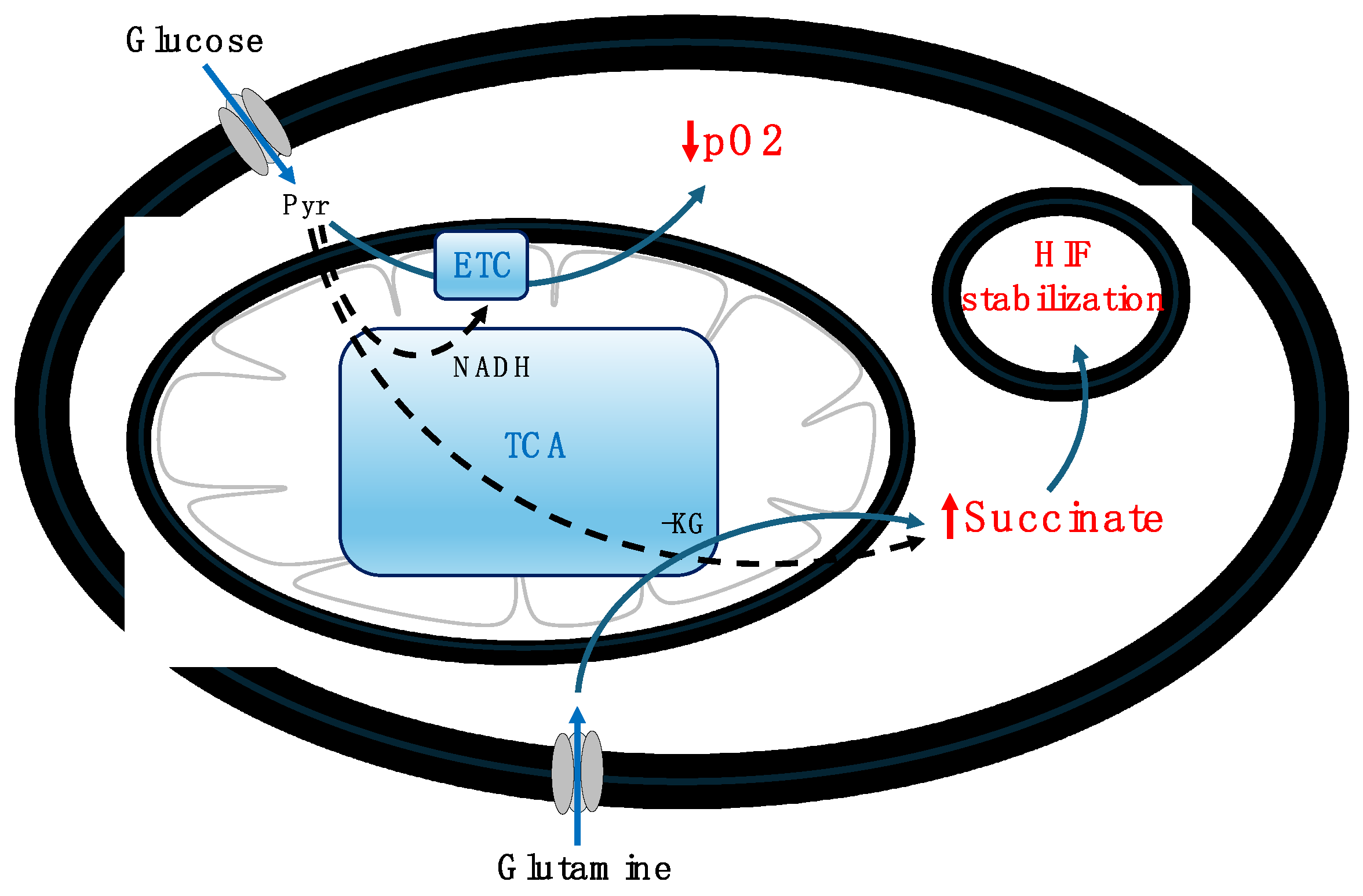
Similarly, hypoxic conditions manifest in beta cells following glucose stimulation under normal physiological scenarios, transitioning from an initial normoxic state. High glucose levels are known to induce hypoxia in pancreatic beta cells, leading to a decrease in oxygen partial pressure (pO2) due to glucose oxidation (Bensellam et al., 2012; Gerber & Rutter, 2017). These transient hypoxic states are significant for insulin secretion (Bensellam et al., 2012). Studies have shown that hypoxia enhances the cellular uptake and mitochondrial utilization of glutamine, a process detailed in studies by Jiang et al. (2017) and Yoo et al. (2020). Further investigations, indicate that glutamine, upon conversion to glutamate and succinate, serves as a primary source for succinate accumulation under hypoxic conditions (Slaughter et al., 2016; Martínez-Reyes & Chandel, 2020). This accumulation of succinate notably activates HIF. In rat beta cells, glucose-induced oxygen consumption has been shown to create intracellular hypoxia that activates both HIF1 and HIF2 (Bensellam et al., 2012). Furthermore, van Diepen et al. (2017) demonstrated that hypoxia and hyperglycemia independently promote succinate release from mouse adipose tissue (17-fold and up to 18-fold, respectively), with higher plasma levels of succinate observed in individuals with type 2 diabetes compared to non-diabetic counterparts. These studies underscore succinate's pivotal role as a metabolic signal in the stabilization and activation of HIF under hypoxic conditions, illustrating the intricate link between cellular metabolism and adaptive responses to hypoxia. This interplay, particularly the glucose-induced hypoxia leading to succinate accumulation and subsequent HIF stabilization, is depicted in Figure 4.
The cellular mechanism through which succinate activates HIF involves the inhibition of prolyl hydroxylases (PHDs), enzymes tasked with HIF degradation under normoxic conditions. Koivunen et al. (2007) elucidated that succinate directly inhibits HIF prolyl 4-hydroxylases (HIF-P4Hs) in vitro, thereby stabilizing HIF-1α and establishing a direct connection between succinate accumulation and the activation of the cellular hypoxic response through HIF hydroxylases. Moreover, cancer cells with deficiencies in fumarate hydratase (FH) and succinate dehydrogenase (SDH) exhibit succinate and fumarate accumulation, leading to HIF-1α stabilization. This phenomenon suggests that mutations in metabolic enzymes can drive cancer progression by disrupting hypoxia signaling pathways (Pollard et al., 2005), highlighting the intricate link between cellular metabolism and hypoxic adaptation.
In the context of beta cells, Edalat et al. (2015) highlighted SDH as a critical regulator of insulin secretion and ROS production, underscoring its essential role in glucose homeostasis. The inhibition of SDH, either through 3-nitropropionic acid (3-NPA) or monoethyl fumarate (MEF), was shown to diminish glucose-stimulated insulin secretion, pointing to SDH's significance in metabolic regulation (Edalat et al., 2015). Complementarily, Lee & Annes (2020) discovered that a reduction in SDH activity in diabetic beta cells, evidenced by decreased succinate dehydrogenase subunit B (SDHB) expression, led to compromised insulin secretion and the onset of diabetes in a beta cell-specific SDHB knockout (SDHBβKO) mouse model, indicating the importance of SDH in beta cell functionality (Lee & Annes, 2020). Further, Lee et al. (2022) demonstrated that SDH deficiency in beta cells impairs both glucose-stimulated insulin secretion and beta cell proliferation, presenting SDH deficiency as a key factor in the progressive failure of beta cells in diabetes. Metabolomic and transcriptomic analyses revealed that SDH loss results in excessive succinate accumulation, which aberrantly activates mTOR complex I–regulated metabolic anabolism, including the upregulation of lipid biosynthesis. These changes, reflective of beta cell dysfunction in diabetes, were shown to be partially mitigated by acute mTOR inhibition using rapamycin (Lee et al., 2022), offering insights into potential therapeutic avenues to counteract beta cell dysfunction by modulating metabolic pathways.
Building on the insights into the role of succinate dehydrogenase (SDH) deficiency and chronic hypoxia in activating hypoxia-inducible factor 1-alpha (HIF1α), such conditions have been identified as critical factors in the progression of pathologies associated with Type 2 Diabetes Mellitus (T2DM), including beta cell dedifferentiation (Liu et al., 2020). This chronic activation of HIF underscores the potential of HIF inhibitors as a novel therapeutic strategy for T2DM. Ilegems et al. (2022) provided evidence that pancreatic beta cells, under the strain of prolonged metabolic overload characteristic of type 2 diabetes progression, develop a hypoxic phenotype mediated by HIF1α. Their findings reveal that administration of the HIF-1α inhibitor PX-478 can significantly ameliorate beta cell function. Islets isolated from treated mice exhibited signs of enhanced beta cell functionality, such as increased insulin content, upregulation of genes associated with beta cell function and maturation, reduction in dedifferentiation markers, and the formation of mature insulin granules (Ilegems et al., 2022).
Transitioning from the discussion on chronic hypoxia, it's imperative to distinguish between transient and prolonged or chronic hypoxic conditions. While the previous sections have delved into the detrimental effects of chronic hypoxia, temporary hypoxia, such as that experienced by beta cells post-glucose stimulation, presents physiological benefits crucial for health and normal functioning. Intriguingly, recent research by Rogers et al. (2023) suggests that temporary hypoxia might also play a role in lifespan extension. The mechanism behind this phenomenon is complex, yet hypoxia has been documented to prolong lifespan in various models, including yeast (Pan et al., 2011), C. elegans (Lee et al., 2010; Schieber et al., 2014), and to delay replicative senescence in primary human lung fibroblasts (Bell et al., 2007), potentially through an increase in ROS production that activates life-extending pathways, a concept known as hormesis (Cox et al., 2018). Future investigations are essential to thoroughly assess the impact of hypoxia on ROS levels and to determine whether changes in ROS have beneficial or deleterious effects on health and longevity.
6. Discussion
Pancreatic beta cells, upon stimulation, share several signaling and metabolic pathways with cancer cells, responding to the abundance of key metabolites, particularly glucose and glutamine. Beta cells act as sensors of the postprandial metabolic state, recognizing and responding to the surge in nutrient availability by promoting insulin secretion. Conversely, cancer cells exploit these nutrient-rich conditions to support their uncontrolled proliferation, often enhancing their metabolic supply through mechanisms like forced angiogenesis. This parallel suggests that while beta cells modulate the body's anabolic state by regulating insulin release, cancer cells selfishly harness these conditions for rampant growth. The similarity in cellular response to this anabolic environment underscores the adaptability of beta cells, which, under certain conditions such as the early stages of T2DM, can proliferate rapidly, a characteristic commonly associated with cancer cells (Sachdeva & Stoffers, 2009; Xu et al., 2009; Sharma & Alonso, 2014).
This manuscript has delineated the shared metabolic responses between beta and cancer cells, both of which exhibit intense metabolic demands. Beta cells require an efficient metabolism to produce insulin in response to elevated levels of glucose and glutamine, whereas cancer cells adjust their metabolism to support their rapid proliferation. Key common metabolic pathways have been highlighted, particularly those favoring reductive metabolism that facilitates the anaplerotic export of critical metabolites such as malate, citrate, aspartate, succinate, and notably NADPH. The role of NADPH is pivotal in GSH production, essential for regulating ROS, a factor crucial for both cancer cell division and insulin secretion from beta cells. The upregulation of UCP2 in both stimulated beta cells and cancer cells underlines its importance in managing the high anaplerotic flux from the reductive TCA cycle. Special attention has been given to succinate, a metabolite of paramount importance in HIF regulation, underscoring the intertwined metabolic pathways of stimulated beta cells and rapidly dividing cancer cells. While this exploration covers significant ground, numerous additional aspects warrant further investigation, opening avenues for future research into the complex metabolic interplay between these cell types.
Building on the discussion of anaplerotic metabolites' significance, aspartate emerges as a noteworthy component, intricately linked through the urea cycle to arginine utilization (Fu et al., 2020). This linkage underscores the multifaceted roles of arginine, not only as a direct enhancer of insulin secretion from beta cells but also as a key player in various cellular processes, including inflammation (Henquin et al., 2000; Krausse et al., 2011; Newsholme et al., 2017; Fu et al., 2020). The critical importance of arginine in cancer cells has led to the exploration of arginine deprivation as a potential therapeutic approach to combat cancer cell metastasis (Al-Koussa et al., 2020). The conversion of aspartate to argininosuccinate, and its subsequent transformation into fumarate and succinate within the urea cycle, highlights not only arginine's significance but also points to the pivotal role of succinate. As previously discussed, succinate's accumulation is instrumental in HIF activation, a phenomenon observed both in cancer cells and transiently in stimulated beta cells.
Delving further into the relationship between succinate, HIF activation, and the function of UCP2 invites a deeper understanding of their interconnected roles. Given UCP2's essential role in modulating mitochondrial function and ROS production, it is plausible that UCP2 indirectly influences the cellular response to hypoxia and the stabilization of HIF. By potentially altering the mitochondrial ROS landscape, UCP2 could modify the cellular redox state, impacting the stability and activity of HIF-1α. This intricate balance between oxygen sensing, HIF stabilization, and subsequent effects reveals a broader regulatory context in which UCP2 may exert its influence. For example, the review by Sakashita et al. (2019) on the role of PHD inhibitors in chronic kidney disease provides insight into HIF stabilization under hypoxic conditions and its broader biological implications, suggesting a framework for UCP2's potential indirect effects on HIF activity. Further exploration by McGettrick and O'Neill (2020) into HIF's role in immunity and inflammation, as well as Lee et al. (2019)'s discussion on the dual nature of HIF signaling, underscore the significance of mitochondrial and redox regulation. Additionally, research by Kuan et al. (2021) on the protective effects of HIF stabilization in models of neonatal brain injury indirectly highlights the critical nature of cellular adaptations to hypoxia, where UCP2's regulation of mitochondrial function may play a key role. These studies pave the way for future inquiries into UCP2's impact on hypoxia signaling pathways, further integrating the roles of anaplerotic metabolites within this complex network.
These investigations could significantly enhance our understanding of beta cell pathologies. Drawing insights from cancer research might unveil new avenues for exploring the pathophysiology of beta cells. Particularly, the chronic elevations of ROS and persistent hypoxic conditions are potentially hazardous for beta cells. While excessive ROS production can be detrimental, a balanced generation of ROS within beta cells is essential for normal cellular signaling, including the finely tuned regulation of insulin secretion. ROS, including those produced by NOX4, serve as signaling molecules that influence cellular responses to metabolic signals, illustrating the dual role of ROS in both the physiological and pathological aspects of beta-cell function (Kowluru, 2020). Chronic hypoxia presents another challenge. This manuscript has highlighted the critical physiological role of transient, time-limited hypoxia in insulin secretion from beta cells following glucose stimulation. However, sustained hypoxia, especially when associated with succinate accumulation, could contribute to the development of Type 2 Diabetes Mellitus (T2DM), as evidenced by research showing that beta cell succinate dehydrogenase (SDH) deficiency leads to metabolic dysfunction and insulinopenic diabetes (Lee et al., 2022). Furthermore, this study revealed that SDH deficiency, resulting in excess succinate accumulation, activates mTOR complex I–regulated metabolic anabolism, a process that can be partially mitigated by acute mTOR inhibition with rapamycin. These findings draw parallels with cancer cell behavior, suggesting that the extensive knowledge base from cancer research could be instrumental in addressing T2DM, which continues to pose a significant challenge to modern societies. Leveraging the wealth of cancer research insights could thus provide valuable strategies for the treatment and management of T2DM, underscoring the importance of interdisciplinary research in uncovering novel therapeutic approaches.
Acknowledgments
The author acknowledges the support from the Slovenian Research and Innovation Agency (research core funding no. P1-0055, and research project no. J3-3077).
Conflicts of Interest
The author declares no conflicts of interest.
References
- Abulizi, A.; Cardone, R.L.; Stark, R.; Lewandowski, S.L.; Zhao, X.; Hillion, J.; Ma, L.; Sehgal, R.; Alves, T.C.; Thomas, C.; Kung, C.; Wang, B.; Siebel, S.; Andrews, Z.B.; Mason, G.F.; Rinehart, J.; Merrins, M.J.; Kibbey, R.G. Multi-Tissue Acceleration of the Mitochondrial Phosphoenolpyruvate Cycle Improves Whole-Body Metabolic Health. Cell Metabolism 2020, 32, 751-766.e11. [CrossRef]
- Affourtit, C., Brand, M.D. (2008). On the role of uncoupling protein-2 in pancreatic beta cells. Biochim. Biophys. Acta – Bioenergetics 2008, 1777, 973–979. [CrossRef]
- Al-Koussa, H.; El Mais, N.; Maalouf, H.; Abi-Habib, R.; El-Sibai, M. Arginine deprivation: a potential therapeutic for cancer cell metastasis? A review. Cancer Cell Int. 2020, 20, 150. [CrossRef]
- Azzu, V.; Affourtit, C.; Breen, E.P.; Parker, N.; Brand, M.D. Dynamic regulation of uncoupling protein 2 content in INS-1E insulinoma cells. Biochim. Biophys. Acta 2008, 1777, 1378–1383. [CrossRef]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Schumacker, P.T.; Chandel, N.S. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol Cell Biol. 2007, 27, 5737–45. [CrossRef]
- Bensellam, M.; Duvillié, B.; Rybachuk, G.; Laybutt, D.R.; Magnan, C.; Guiot, Y.; Pouysségur, J.; Jonas, J.-C. Glucose-Induced O2 Consumption Activates Hypoxia Inducible Factors 1 and 2 in Rat Insulin-Secreting Pancreatic Beta-Cells. PLoS ONE 2012, 7, e29807. [CrossRef]
- Blandino, G.; Fiorani, M.; Canonico, B.; De Matteis, R.; Guidarelli, A.; Montanari, M.; Buffi, G.; Coppo, L.; Arnér, E.S.J.; Cantoni, O. Clozapine suppresses NADPH oxidase activation, counteracts cytosolic H2O2, and triggers early onset mitochondrial dysfunction during adipogenesis of human liposarcoma SW872 cells. Redox Biology 2023, 67, 102915. [CrossRef]
- Bouillaud, F. UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim. Biophys. Acta 2009, 1787, 377–383. [CrossRef]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [CrossRef]
- Baffy, G. Mitochondrial uncoupling in cancer cells: Liabilities and opportunities. Biochim. Biophys. Acta 2017, 1858, 655–664. [CrossRef]
- Corkey, B.E. Targeting Pyruvate Kinase PEPs Up Insulin Secretion and Improves Glucose Homeostasis. Cell Metabolism 2020, 32, 693–694. [CrossRef]
- Cox, C.S.; McKay, S.E.; Holmbeck, M.A.; Christian, B.E.; Scortea, A.C.; Tsay, A.J.; Newman, L.E.; Shadelal, G.S. Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell Metab. 2018, 28, 776–86e5. [CrossRef]
- Edalat, A.; Schulte-Mecklenbeck, P.; Bauer, C.; Undank, S.; Krippeit-Drews, P.; Drews, G.; Düfer, M. Mitochondrial succinate dehydrogenase is involved in stimulus-secretion coupling and endogenous ROS formation in murine beta cells. Diabetologia 2015, 58, 1532–1541. [CrossRef]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in Cancer Biology and Therapy. Critical Reviews in Clinical Laboratory Sciences 2006, 43, 143–181. [CrossRef]
- Ferdaoussi, M.; Dai, X.; Jensen, M.V.; Wang, R.; Peterson, B.S.; Huang, C.; Ilkayeva, O.; Smith, N.; Miller, N.; Hajmrle, C.; Spigelman, A.F.; Wright, R.C.; Plummer, G.; Suzuki, K.; Mackay, J.P.; van de Bunt, M.; Gloyn, A.L.; Ryan, T.E.; Norquay, L.D.; … MacDonald, P.E. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J. Clin. Invest. 2015, 125, 3847–3860. [CrossRef]
- Foster, H.R.; Ho, T.; Potapenko, E.; Sdao, S.M.; Huang, S.M.; Lewandowski, S.L.; VanDeusen, H.R.; Davidson, S.M.; Cardone, R.L.; Prentki, M.; Kibbey, R.G.; Merrins, M.J. β-cell deletion of the PKm1 and PKm2 isoforms of pyruvate kinase in mice reveals their essential role as nutrient sensors for the KATP channel. eLife 2022, 11. [CrossRef]
- Fu, A.; Alvarez-Perez, J.C.; Avizonis, D.; Kin, T.; Ficarro, S.B.; Choi, D.W.; Karakose, E.; Badur, M.G.; Evans, L.; …. Danial, N.N. Glucose-dependent partitioning of arginine to the urea cycle protects β-cells from inflammation. Nat. Metab. 2020, 2, 432–446. [CrossRef]
- Gameiro, P.A.; Laviolette, L.A.; Kelleher, J.K.; Iliopoulos, O.; Stephanopoulos, G. Cofactor balance by nicotinamide nucleotide transhydrogenase (NNT) coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid (TCA) cycle. J. Biol. Chem. 2013, 288, 12967-12977. [CrossRef]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [CrossRef]
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Mitochondrial Dysfunction in Pancreatic Alpha and Beta Cells Associated with Type 2 Diabetes Mellitus. Life 2020a, 10, 348. https://www.mdpi.com/2075-1729/10/12/348. [CrossRef]
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Modelling of energy-driven switch for glucagon and insulin secretion. J. Theor. Biol. 2020b, 493, 110213. [CrossRef]
- Grubelnik, V.; Zmazek, J.; Završnik, M.; Marhl, M. Lipotoxicity in a Vicious Cycle of Pancreatic Beta Cell Exhaustion. Biomedicines 2022, 10, 1627. [CrossRef]
- Grubelnik, V.; Zmazek, J.; Gosak, M.; Marhl, M. The Role of Anaplerotic Metabolism of Glucose and Glutamine in Insulin Secretion: A Model Approach. Available at SSRN 2024. https://ssrn.com/abstract=4716886. [CrossRef]
- Han, G.; Takahashi, H.; Murao, N.; Gheni, G.; Yokoi, N.; Hamamoto, Y.; Asahara, S.; Seino, Y.; Kido, Y.; Seino, S. Glutamate is an essential mediator in glutamine-amplified insulin secretion. J. Diabetes Investig. 2021, 12, 920–930. [CrossRef]
- Harris, A.L. Hypoxia – a key regulatory factor in tumour growth. Nat. Rev. Cancer. 2002, 2, 38–47. https://www.nature.com/articles/nrc704.
- Henquin, J.C. Triggering and Amplifying Pathways of Regulation of Insulin Secretion by Glucose. Diabetes 2000, 49, 1751–60. [CrossRef]
- Ilegems, E.; Bryzgalova, G.; Correia, J.; Yesildag, B.; Berra, E.; Ruas, J.L.; 2, Pereira, T.S.; Berggren, P.-O. HIF-1α inhibitor PX-478 preserves pancreatic β cell function in diabetes. Sci. Transl. Med. 2022, 14, eaba9112. [CrossRef]
- IDF Diabetes Atlas, 9th edn. 2019, Brussels, Belgium: International Diabetes Federation. https://diabetesatlas.org.
- Jesinkey, S.R.; Madiraju, A.K.; Alves, T.C.; Yarborough, O.H.; Cardone, R.L.; Zhao, X.; Parsaei, Y.; Nasiri, A.R.; Butrico, G.; Liu, X.; Molina, A.J.; Rountree, A.M.; Neal, A.S.; Wolf, D.M.; Sterpka, J.; Philbrick, W.M.; Sweet, I.R.; Shirihai, O.H.; Kibbey, R.G. Mitochondrial GTP Links Nutrient Sensing to β Cell Health, Mitochondrial Morphology, and Insulin Secretion Independent of OxPhos. Cell Reports 2019, 28, 759-772.e10. [CrossRef]
- Ježek, P.; Holendová, B.; Garlid, K.D.; Jabůrek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid Redox Signal. 2018, 29, 667–714. [CrossRef]
- Ježek, P.; Holendová, B.; Jabůrek, M.; Tauber, J.; Dlasková, A.; Plecitá-Hlavatá, L. The Pancreatic β-Cell: The Perfect Redox System. Antioxidants 2021, 10, 197. [CrossRef]
- Jensen, M.V.; Gooding, J.R.; Ferdaoussi, M.; Dai, X.; Peterson, B.S.; MacDonald, P.E.; Newgard, C.B. Metabolomics applied to islet nutrient sensing mechanisms. Diabetes, Obesity and Metabolism 2017, 19(S1), 90–94. [CrossRef]
- Jiang, Z.-F.; Wang, M.; Xu, J.-L.; Ning, Y.-J. Hypoxia promotes mitochondrial glutamine metabolism through HIF1α-GDH pathway in human lung cancer cells. Biochem. Biophys. Res. Commun. 2017, 483, 32e38. [CrossRef]
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduction and Targeted Therapy 2020, 5, 231. [CrossRef]
- Koivunen, P.; Hirsilä, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of Hypoxia-inducible Factor (HIF) Hydroxylases by Citric Acid Cycle Intermediates: Possible Links between Cell Metabolism and Stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [CrossRef]
- Kowluru, A. Oxidative Stress in Cytokine-Induced Dysfunction of the Pancreatic Beta Cell: Known Knowns and Known Unknowns. Metabolites 2020, 10, 480. [CrossRef]
- Krauss, S.; Zhang, C.-Y.; Scorrano, L.; Dalgaard, L.T.; St-Pierre, J.; Grey, S.T.; Lowell, B.B. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic β-cell dysfunction. J. Clin. Invest. 2003, 112, 1831–1842. [CrossRef]
- Krause, M.S.; McClenaghan, N.H.; 3, Flatt, P.R.; Homem de Bittencourt, P.I.; Murphy, C.; Newsholme, P. L-Arginine is essential for pancreatic b-cell functional integrity, metabolism and defense from inflammatory challenge. J. Endocrinol. 2011, 211, 87–97. [CrossRef]
- Kuan, C.-Y.; Chen, H.-R.; Gao, N.; Kuo, Y.-M.; Chen, C.-W.; Yang, D.; Kinkaid, M.M.; Hu, E.; Sun, Y.-Y. Brain-targeted hypoxia-inducible factor stabilization reduces neonatal hypoxic-ischemic brain injury. Neurobiology of Disease 2021, 148, 105200. [CrossRef]
- Lane, N. Transformer. The deep chemistry of life and death. Profile Books Ltd. 2022, London. ISBN 978 1 78816 054 4. eISBN 978 1 78283 450 2.
- Lee, S.-J.; Hwang, A.B.; Kenyon, C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 2010, 20, 2131–6. [CrossRef]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51, 1–13. https://www.nature.com/articles/s12276-019-0235-1.
- Lee, S.; Annes, J.P. Mitochondrial Dysfunction Promotes Diabetes via A Previously Unrecognized Mechanism: Protein Succinylation. FASEB J. 2020, 34, Supplement: Experimental Biology 2020 Meeting Abstracts. [CrossRef]
- Lee, S.; Xu, H.; Van Vleck, A.; Mawla, A.M.; Li, A.M.; Ye, J.; Huising, M.O.; Annes, J.P. β-Cell Succinate Dehydrogenase Deficiency Triggers Metabolic Dysfunction and Insulinopenic Diabetes. Diabetes 2022, 71, 1439–1453. [CrossRef]
- Lenzen, S. Oxidative stress: the vulnerable β-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [CrossRef]
- Lewandowski, S.L.; Cardone, R.L.; Foster, H.R.; Ho, T.; Potapenko, E.; Poudel, C.; VanDeusen, H.R.; Sdao, S.M.; Alves, T.C.; Zhao, X.; Capozzi, M.E.; de Souza, A.H.; Jahan, I.; Thomas, C.J.; Nunemaker, C.S.; Davis, D.B.; Campbell, J.E.; Kibbey, R.G.; Merrins, M.J. Pyruvate Kinase Controls Signal Strength in the Insulin Secretory Pathway. Cell Metabolism 2020, 32, 736-750.e5. [CrossRef]
- Li, N.; Karaca, M.; Maechler, P. Upregulation of UCP2 in beta-cells confers partial protection against both oxidative stress and glucotoxicity. Redox Biology 2017, 13, 541–549. [CrossRef]
- Lin, H.; Suzuki, K.; Smith, N.; Li, X.; Nalbach, L.; Fuentes, S.; Spigelman, A.F.; Dai, X.; Bautista, A.; Ferdaoussi, M.; Aggarwal, S.; Pepper, A.R.; Roma, L.P.; Ampofo, E.; Li, W.; MacDonald, P.E. β-cell responses to high fat feeding: A role and mechanism for redox sensing by SENP1. BioRxiv 2023. [CrossRef]
- Liu, N.; Cai, X.; Liu, T.; Zou, J.; Wang, L.; Wang, G.; Liu, Y.; Ding, X.; Zhang, B.; Sun, P.; Liang, R.; Wang, S. Hypoxia-inducible factor-1α mediates the expression of mature β cell- disallowed genes in hypoxia-induced β cell dedifferentiation. Biochem. Biophys. Res. Commun. 2020, 523, 382e388. [CrossRef]
- Lu, D.; Mulder, H.; Zhao, P.; Burgess, S.C.; Jensen, M.V.; Kamzolova, S.; Newgard, C.B.; Sherry, A.D. 13C NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS). Proc. Natl. Acad. Sci. 2002, 99, 2708–2713. [CrossRef]
- Luengo,A.; Li,Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; Thomas, C.J.; Spranger, S.; Matheson, N.J.; Vander Heiden, M.G. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Molecular Cell 2021, 81, 691–707. [CrossRef]
- MacDonald, M.J.; Fahien, L.A.; Brown, L.J.; Hasan, N.M.; Buss, J.D.; Kendrick, M.A. Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1–E15. [CrossRef]
- McGettrick, A.F., O’Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metabolism. 2020, 32, 524–536. [CrossRef]
- Maechler, P. Mitochondrial function and insulin secretion. Molecular and Cellular Endocrinology 2013, 379, 12–18. [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nature Communications 2020, 11, 102. [CrossRef]
- Merrins, M.J.; Corkey, B.E.; Kibbey, R.G.; Prentki, M. Metabolic cycles and signals for insulin secretion. Cell Metabolism 2022, 34, 947–968. [CrossRef]
- Newsholme, P.; Keane, K.N.; Elahy, M.; Cruzat, V.F. L-Arginine, Pancreatic Beta Cell Function, and Diabetes: Mechanisms of Stimulated Insulin Release and Pathways of Metabolism. In: Patel, V., Preedy, V., Rajendram, R. (eds) L-Arginine in Clinical Nutrition. Nutrition and Health. Humana Press 2017. [CrossRef]
- Newsholme, P., Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Cell. Physiol. 2019, 317, C420–C433. [CrossRef]
- Pan, Y.; Schroeder, E.A.; Ocampo, A.; Barrientos, A.; Shadel, G.S. Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 2011, 13, 668–78. [CrossRef]
- Patané, G.; Anello, M.; Piro, S.; Vigneri, R.; Purrello, F.; Rabuazzo, A.M. Role of ATP production and uncoupling protein-2 in the insulin secretory defect induced by chronic exposure to high glucose or free fatty acids and effects of peroxisome proliferator-activated receptor-γ inhibition. Diabetes 2002, 51, 2749–2756. [CrossRef]
- Pecqueur, C.; Bui, T.; Gelly, C.; Hauchard, J.; Barbot, C.; Bouillaud, F.; Ricquier, D.; Miroux, B.; Thompson, C.B. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J. 2008, 22, 9 –18. https://faseb.onlinelibrary.wiley.com/doi/epdf/10.1096/fj.07-8945com.
- Pi, J.; Zhang, Q.; Fu, J.; Woods, C.G.; Hou, Y.; Corkey, B.E.; Collins, S.; Melvin, E.; Andersen, M.E. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicology and Applied Pharmacology 2010, 244, 77–83. [CrossRef]
- Plecitá-Hlavatá, L.; Jabůrek, M.; Holendová, B.; Tauber, J., Pavluch, V.; Berková, Z.; Cahová, M.; Schröder, K.; Brandes, R.P.; Siemen, D.; Ježek, P. Glucose-Stimulated Insulin Secretion Fundamentally Requires H2O2 Signaling by NADPH Oxidase 4. Diabetes 2020, 69, 1341–1354. [CrossRef]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; … Tomlinson, I.P.M. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Human Molecular Genetics 2005, 14, 2231–2239. [CrossRef]
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.R.M. Metabolic Signaling in Fuel-Induced Insulin Secretion. Cell Metabolism 2013, 18, 162–185. [CrossRef]
- Produit-Zengaffinen, N.; N. Davis-Lameloise, N.; Perreten, H.; Bécard, D.; Gjinovci, A.; & Keller, P.A.; Wollheim, C.B.; Herrera, P.; Muzzin, P.; Assimacopoulos-Jeannet, F. Increasing uncoupling protein-2 in pancreatic beta cells does not alter glucose-induced insulin secretion but decreases production of reactive oxygen species. Diabetologia 2007, 50, 84–93. [CrossRef]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, V.; Piazzolla, C.; De Leonardis, F.; Gorgoglione, R.; Scarcia, P.; …. & Fiermonte, G. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat. Metab. 2020, 2, 1373–1381. [CrossRef]
- Robertson, R.P. and Harmon, J.S. Pancreatic islet β-cell and oxidative stress: The importance of glutathione peroxidase. FEBS Letters 2007, 581, 3743–3748. [CrossRef]
- Rogers, R.S.; Wang, H.; Durham, T.J.; Stefely, J.A.; Owiti, N.A.; Markhard, A.L.; Sandler, L.; To, T.-L.; Mootha, V.K. Hypoxia extends lifespan and neurological function in a mouse model of aging. PLoS Biol. 2023, 21, e3002117. [CrossRef]
- Roma, L.P. and Jonas, J.-C. Nutrient Metabolism, Subcellular Redox State, and Oxidative Stress in Pancreatic Islets and β-Cells. J. Mol. Biol. 2020, 432, 1461–1493. [CrossRef]
- Rousset, S.; Mozo, J.; Dujardin, G.; Emre, Y.; Masscheleyn, S.; Ricquier, D.; Cassard-Doulcier, A.-M. UCP2 is a mitochondrial transporter with an unusual very short half-life. FEBS Letters 2007, 581, 479–482. [CrossRef]
- Rustenbeck, I.; Schulze, T.; Morsi, M.; Alshafei, M.; Panten, U. What Is the Metabolic Amplification of Insulin Secretion and Is It (Still) Relevant? Metabolites 2021, 11, 355. [CrossRef] [PubMed]
- Sachdeva, M.M.; Stoffers, D.A. Minireview: Meeting the Demand for Insulin: Molecular Mechanisms of Adaptive Postnatal ß-Cell Mass Expansion. Molecular Endocrinology 2009, 23, 747–758. [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.;... & IDF Diabetes Atlas Committee. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Research and Clinical Practice 2019, 157, 107843. [CrossRef]
- Sakashita, M.; Tanaka, T.; Nangaku, M. (2019). Hypoxia-Inducible Factor-Prolyl Hydroxylase Domain Inhibitors to Treat Anemia in Chronic Kidney Disease. Contributions to Nephrology 2019, 198, 112–123. [CrossRef]
- Santos, L.R.B.; Muller, C.; de Souza, A.H.; Takahashi, H.K.; Spégel, P.; Sweet, I.R.; Chae, H.; Mulder, H.; Jonas, J.-C. NNT reverse mode of operation mediates glucose control of mitochondrial NADPH and glutathione redox state in mouse pancreatic β-cells. Molecular Metabolism 2017, 6, 535–547. [CrossRef]
- Schieber, M.; Chandel, N.S. TOR signaling couples oxygen sensing to lifespan in C. elegans. Cell Rep. 2014, 9, 9–15. [CrossRef]
- Segalés, J.; Sánchez-Martín, C.; Pujol-Morcillo, A.; Martín-Ruiz, M.; de los Santos, P.; Lobato-Alonso, D.; Oliver, E.; Rial, E. Role of UCP2 in the Energy Metabolism of the Cancer Cell Line A549. Int. J. Mol. Sci. 2023, 24, 8123. [CrossRef]
- Sekine, N.; CirulliS, V.; Regazzi, R.; Brown, L.J.; Ginen, E.; Tamarit-Rodrigued, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; Rutter, G.A. Low Lactate Dehydrogenase and High Mitochondrial Glycerol Phosphate Dehydrogenase in Pancreatic β-Cells. J Biol Chem. 1994; 269, 4895–902. [Google Scholar] [CrossRef]
- Sharma, R.B.; Alonso, L.C. Lipotoxicity in the Pancreatic Beta Cell: Not Just Survival and Function, but Proliferation as Well? Curr. Diabetes Rep. 2014, 14, 492. [CrossRef]
- Slaughter, A.L.; D’Alessandro, A.; Moore, E.E.; Banerjee, A.; Silliman, C.C.; Hansen, K.C.; Reisz, J.A.; … Peltz, E.D. Glutamine metabolism drives succinate accumulation in plasma and the lung during hemorrhagic shock. J. Trauma Acute Care Surg. 2016, 81, 1012–1019. [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of Glutathione in Cancer Progression and Chemoresistance. Oxid. Med. Cell. Longev. 2013, 972913. [CrossRef]
- Valle, A.; Oliver, J.; Roca, P. Role of Uncoupling Proteins in Cancer. Cancers 2010, 2, 567–591. [CrossRef]
- van Diepen, J.A.; Robben, J.H.; Hooiveld, G.J.; Carmone, C.; Alsady, M.; Boutens, L.; Bekkenkamp-Grovenstein, M.; Hijmans, A.; Engelke, U.F.H.; Wevers, R.A.; Netea, M.G.; Tack, C.J.; Stienstra, R.; Deen, P.M.T. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia 2017, 60, 1304–1313. [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.G. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029. [CrossRef]
- Vaupel, P. The role of hypoxia-induced factors in tumor progression. The Oncologist 2004, 9, 10–7. [CrossRef]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; Paradies, E.; Scarcia, P.; Palmieri, F.; Bouillaud, F.; Fiermonte, G. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. 2014, 111, 960–965. [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Medicine 2019, 8, 2252–2267. [CrossRef]
- Xiong, Z.; Xiong, W.; Xiao, W.; Yuan, C.; Shi, J.; Huang, Y.; Wang, C.; Meng, X.; Chen, Z.; Yang, H.; Chen, K.; Zhang, X. NNT-induced tumor cell “slimming” reverses the pro-carcinogenesis effect of HIF2a in tumors. Clin. Transl. Med. 2021, 11, e264. [CrossRef]
- Xu, J.; Long, Y.S.; Gozal, D.; Epstein, P.N. Beta-cell death and proliferation after intermittent hypoxia: role of oxidative stress. Free Radical Biology & Medicine, 2009, 46, 783–790. [CrossRef]
- Yoo, H.C.; Yu, Y., C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [CrossRef]
- Zhang, G.-F.; Jensen, M.V.; Gray, S.M.; El, K.; Wang, Y.; Lu, D.; Becker, T.C.; Campbell, J.E.; Newgard, C.B. Reductive TCA cycle metabolism fuels glutamine- and glucose-stimulated insulin secretion. Cell Metabolism 2021, 33, 804-817.e5. [CrossRef]
Figure 1.
Schematic representation of the key processes involved in the production and transport of metabolites that signal for cancer cell division and the amplification of insulin secretion. This figure illustrates how both cancer and stimulated beta cells utilize common metabolic pathways for their proliferation and function. Abbreviations: α-KG – alpha-ketoglutarate, Asp – aspartate, Cit – citrate, Gln – glutamine, Glu – glutamate, IsoCit – isocitrate, Mal – malate, Mal-CoA – Malonyl-CoA, NNT – nicotinamide nucleotide transhydrogenase, OAA – oxaloacetate, Pyr – pyruvate, Succ – succinate, TCA – tricarboxylic acid cycle.
Figure 1.
Schematic representation of the key processes involved in the production and transport of metabolites that signal for cancer cell division and the amplification of insulin secretion. This figure illustrates how both cancer and stimulated beta cells utilize common metabolic pathways for their proliferation and function. Abbreviations: α-KG – alpha-ketoglutarate, Asp – aspartate, Cit – citrate, Gln – glutamine, Glu – glutamate, IsoCit – isocitrate, Mal – malate, Mal-CoA – Malonyl-CoA, NNT – nicotinamide nucleotide transhydrogenase, OAA – oxaloacetate, Pyr – pyruvate, Succ – succinate, TCA – tricarboxylic acid cycle.
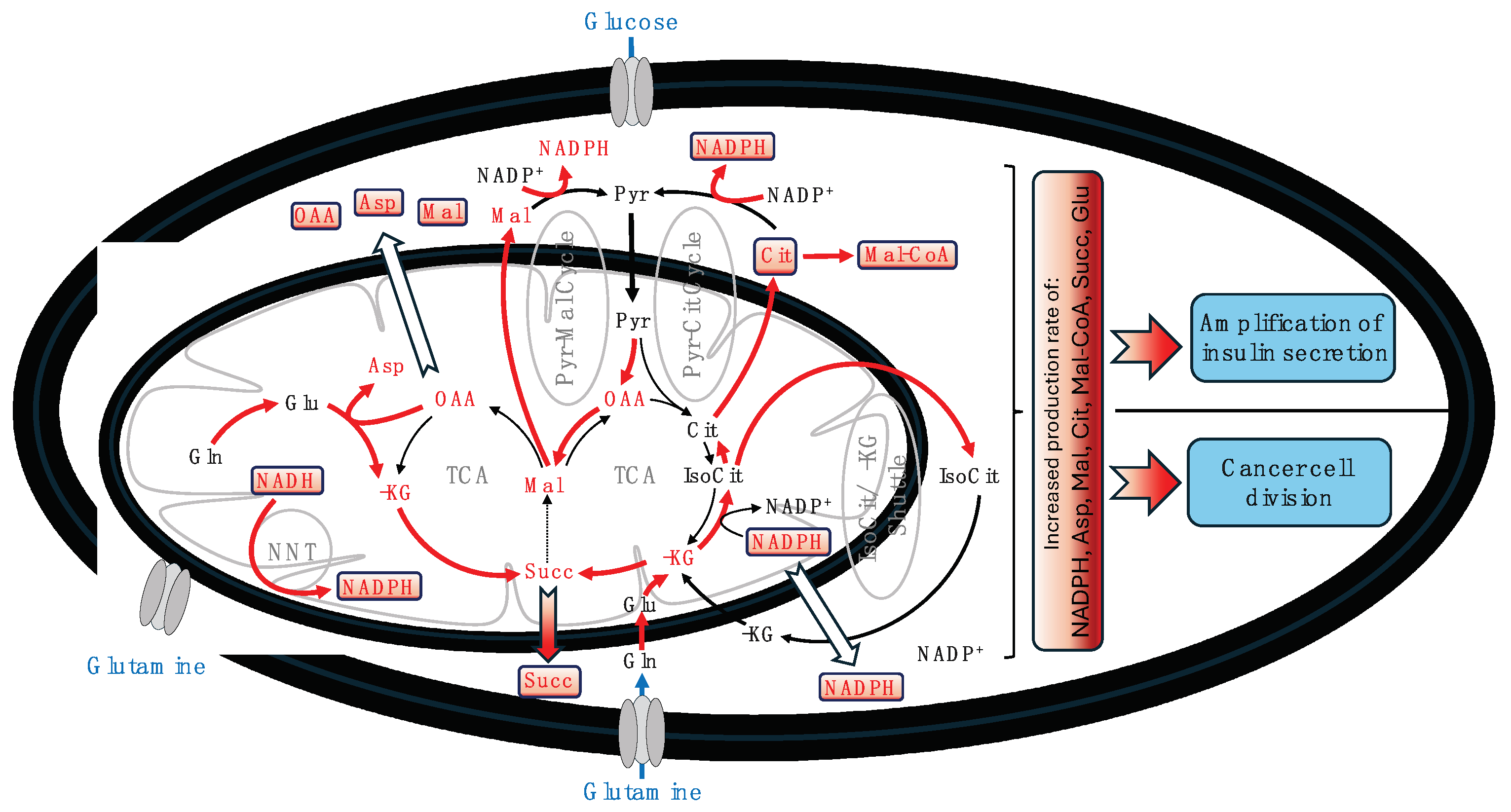
Figure 2.
Schematic depicting the activation of the Nrf2 pathway by H2O2, leading to the enhanced influx of cysteine, activation of glutamate-cysteine ligase (GCL), and the upregulation of four key NADPH-generating enzymes: glucose-6-phosphate dehydrogenase (G6PD), 6-phosphogluconate dehydrogenase (PGD), isocitrate dehydrogenase-1 (IDH1), and malic enzyme-1 (ME1). This coordinated upregulation results in increased NADPH production, which, in conjunction with cysteine and GCL, facilitates glutathione (GSH) biosynthesis, bolstering the cell's antioxidant defense mechanism. Abbreviations: Cyst – cysteine, NOX4 – NADPH Oxidase 4, PPP – pentose phosphate pathway, TCA – tricarboxylic acid cycle.
Figure 2.
Schematic depicting the activation of the Nrf2 pathway by H2O2, leading to the enhanced influx of cysteine, activation of glutamate-cysteine ligase (GCL), and the upregulation of four key NADPH-generating enzymes: glucose-6-phosphate dehydrogenase (G6PD), 6-phosphogluconate dehydrogenase (PGD), isocitrate dehydrogenase-1 (IDH1), and malic enzyme-1 (ME1). This coordinated upregulation results in increased NADPH production, which, in conjunction with cysteine and GCL, facilitates glutathione (GSH) biosynthesis, bolstering the cell's antioxidant defense mechanism. Abbreviations: Cyst – cysteine, NOX4 – NADPH Oxidase 4, PPP – pentose phosphate pathway, TCA – tricarboxylic acid cycle.
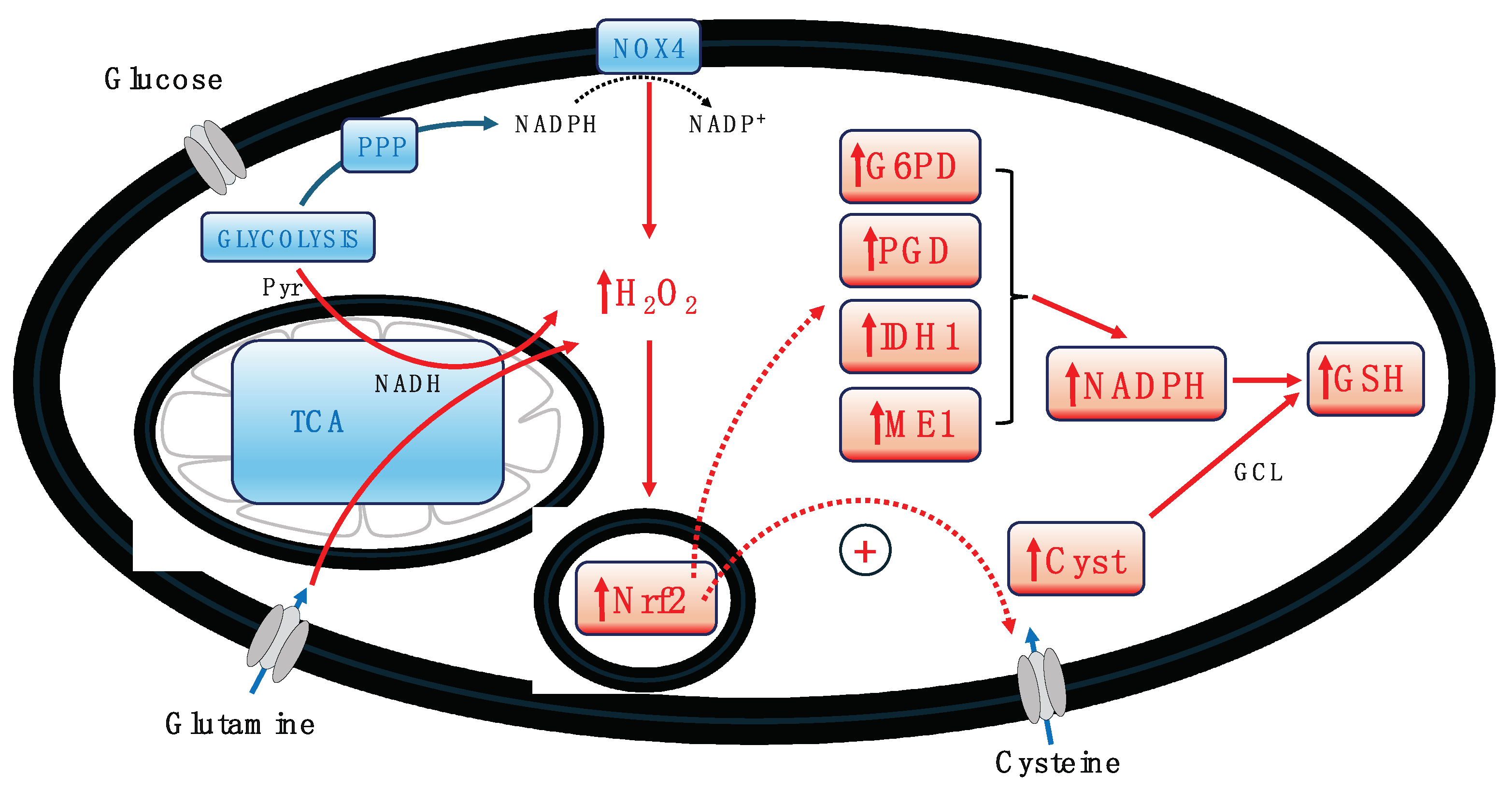
Figure 3.
Schematic representation of UCP2's regulatory mechanisms, emphasizing its role in metabolic flexibility, particularly by enhancing glutamine utilization and facilitating the anaplerotic export of C4 metabolites across the mitochondrial membrane. This process is crucial for biosynthetic activities and the amplification of GSIS. Abbreviations: α-KG – alpha-ketoglutarate, Asp – aspartate, GLS – glutaminase, Gln – glutamine, Glu – glutamate, Mal – malate, ME1 – malic enzyme-1, Pyr – pyruvate, OAA – oxaloacetate, Pi – phosphate, TCA – tricarboxylic acid cycle.
Figure 3.
Schematic representation of UCP2's regulatory mechanisms, emphasizing its role in metabolic flexibility, particularly by enhancing glutamine utilization and facilitating the anaplerotic export of C4 metabolites across the mitochondrial membrane. This process is crucial for biosynthetic activities and the amplification of GSIS. Abbreviations: α-KG – alpha-ketoglutarate, Asp – aspartate, GLS – glutaminase, Gln – glutamine, Glu – glutamate, Mal – malate, ME1 – malic enzyme-1, Pyr – pyruvate, OAA – oxaloacetate, Pi – phosphate, TCA – tricarboxylic acid cycle.
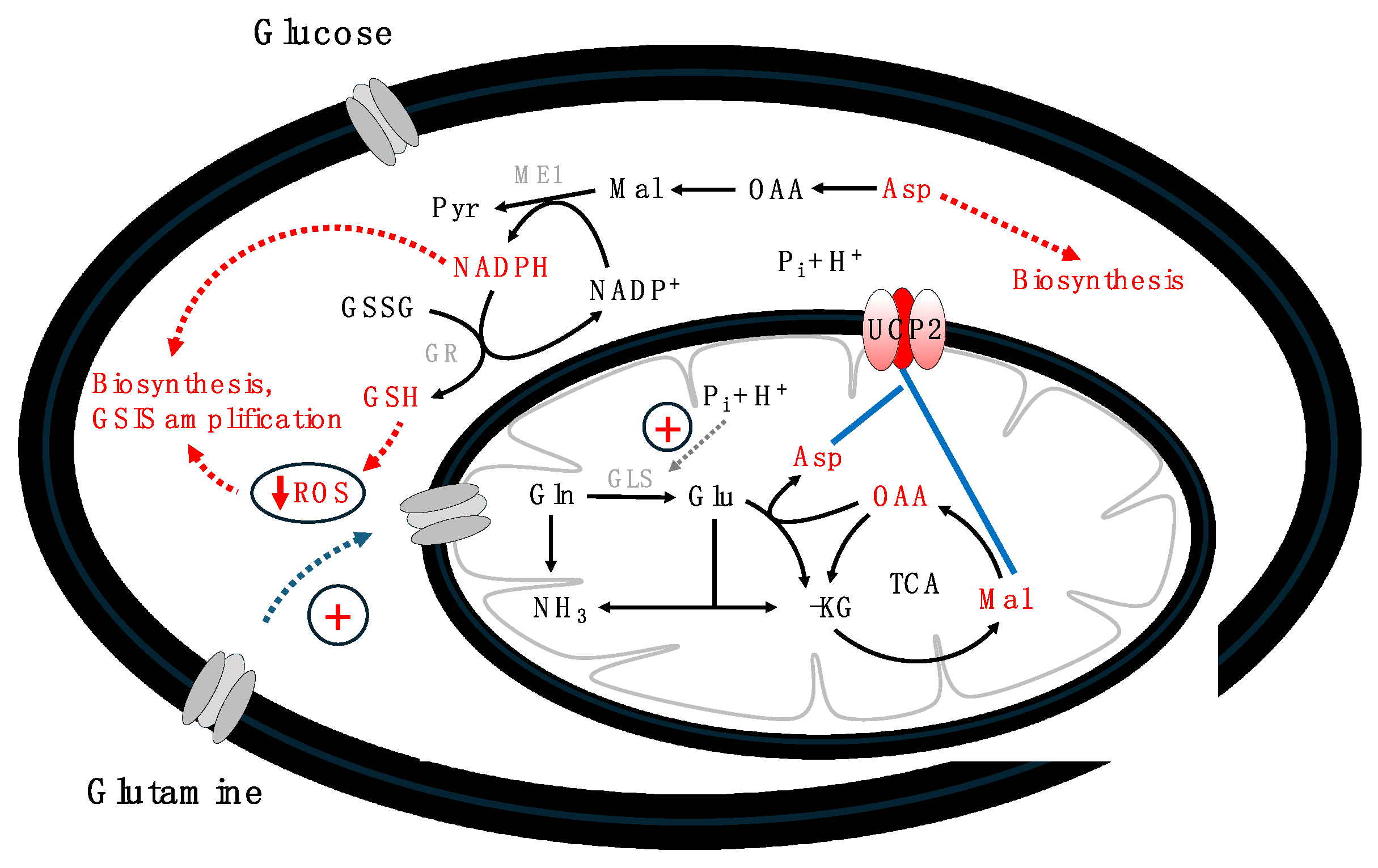
Figure 4.
A schematic representation of a temporal hypoxia in beta cells after glucose stimulation, glutamine-induced succinate accumulation, and HIF stabilization. Abbreviations: α-KG – alpha-ketoglutarate, ETC – electron transport chain, pO2 – oxygen partial pressure, Pyr – pyruvate, TCA – tricarboxylic acid cycle.
Figure 4.
A schematic representation of a temporal hypoxia in beta cells after glucose stimulation, glutamine-induced succinate accumulation, and HIF stabilization. Abbreviations: α-KG – alpha-ketoglutarate, ETC – electron transport chain, pO2 – oxygen partial pressure, Pyr – pyruvate, TCA – tricarboxylic acid cycle.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



