
Submitted:
26 February 2024
Posted:
27 February 2024
You are already at the latest version
Abstract
Transcription factors (TFs) regulate gene expression by recognizing specific target enhancers in the genome. The DNA-binding and regulatory activity of TFs depend on the presence of additional protein partners, leading to the formation of versatile and dynamic multimeric protein complexes. Visualizing these protein-protein interactions (PPIs) in the nucleus is key for decrypting the molecular cues underlying TF specificity in vivo. Over the last years, Bimolecular Fluorescence Complementation (BiFC) has been developed in several model systems and applied for the analysis of different types of PPIs. In particular, BiFC has been applied for analyzing PPIs with hundreds of TFs in the nucleus of live Drosophila embryos. However, the visualization of PPIs at the level of specific target enhancers or genomic regions of interest awaits the advent of DNA-labelling methods that could be coupled with BiFC. Here, we present a novel experimental strategy that we called BiFOR and that is based on the coupling of BiFC with the bacterial ANCHOR DNA-labelling system. We demonstrate that BiFOR enables to precisely quantify the enrichment of specific dimeric protein complexes on target enhancers in Drosophila salivary gland nuclei. Given its versatility and sensitivity, BiFOR could be applied more largely in other tissues during Drosophila development. Our work set up the experimental basis for these future applications.
Keywords:
protein-protein interaction
; BiFC
; ANCHOR
; ParB-INT
; Hox
1. Introduction
Cell fate is determined by specific gene expression programs that are primarily occurring at the level of transcription. This key regulatory process leads to the production of precise doses of mRNA molecules that will subsequently be translated into proteins. Among the different cis-regulatory elements that control mRNA production are the so-called “enhancers”, which are DNA elements of relatively small size (less than 1 kilobases) and located at various distances from the transcription start site [1]. Enhancers contain specific DNA sequences that are recognized by transcription factors (TFs). Importantly, the full activity of an enhancer relies on the combinatorial binding and activity of several TFs with their cofactors [2]. Therefore, understanding the molecular rules that govern enhancer activity requires the visualization of protein-protein interactions (PPIs) that are specifically occurring on it in the nucleus.
Several techniques have been developed to visualize the genomic regions of interest (ROIs). These techniques rely on the use of either fluorescent oligonucleotide probes for doing fluorescent in situ hybridization (FISH), or DNA-binding proteins fused to fluorescent proteins (FPs). FISH provides kilobase resolution but requires stringent experimental procedures, such as DNA denaturation, that may affect PPIs ([3], see also results). Alternatively, different DNA-binding proteins have been fused to different FPs to target and label a genomic ROI in the nucleus. These strategies depend on multiplexing for getting enough FPs bound to the target locus for subsequent analysis by microscopy. For example, fluorescent dCas9 (catalytically dead Cas9) has been used to label repeated sequences in the genome [4,5,6]. Fluorescent dCas9 can also be used to label non-repetitive cis-regulatory elements by multiplexing guide RNAs (gRNAs) oligonucleotides [7]. However, these Cas9-based strategies have only been established in culture cells and await further development for applications in a multicellular organism. Other strategies exist to label genomic ROI in the nucleus. For example, the Lactose Operon Repressor/Operator (LacI/LacO) and Tetracycline Operon Repressor/Operator (TetR/TetO) systems have been used to visualize chromosome translocations in human live cells [8]. Still, this system requires intensive engineering since it is based on the insertion of a high number of LacO and TetO DNA sequences for exploiting fluorescent signals emitted by the FP-LacI and FP-TetR fusion proteins. Another more promising approach is based on the Partition Protein B/Binding Sequence (ParB-INT or ANCHOR) system. In this system, multimerization of the fluorescent ParB proteins is achieved through PPIs on the INT sequence. First applied in yeast [9] and human cell culture [10,11], this system has also been developed to visualize enhancer-promoter interactions [12] or a DNA locus in Drosophila [13]. Moreover, a recent analysis showed that the ANCHOR system did not locally perturb transcription in Drosophila (in contrast to the LacI/lacO system: [14]).
Here, we present a unique experimental approach coupling the ANCHOR and Bimolecular Fluorescence Complementation (BiFC) systems to visualize and measure the specific enrichment of dimeric protein complexes on a target DNA sequence in the nucleus of Drosophila larval salivary gland cells. BiFC relies on the property of monomeric FPs to be reconstituted from two separate sub-fragments upon spatial proximity [15]. In particular, BiFC has been extensively used to reveal and analyze PPIs between various classes of TFs in live Drosophila embryos [16,17,18]. As a proof-of-concept, we used the well-characterized enhancer of the salivary gland selector gene forkhead (enhancer fkh250) as a regulatory model system. This enhancer is specifically regulated by the Hox protein Sex combs reduced (Scr) in association with the Extradenticle (Exd) cofactor [19]. This regulation occurs through the recognition of a unique Scr/Exd DNA-binding site. Mutations affecting this binding site have been generated to change the specificity of the regulation in vitro and in vivo [19]. These variants of the fkh250 enhancer constitute ideal tools for testing the specificity of the ANCHOR and BiFC systems. Altogether, our work demonstrates that BiFC and ANCHOR coupling (hereafter called “BiFOR”) is a powerful approach for the sensitive quantification of PPI enrichment on specific target enhancers in Drosophila salivary gland nuclei.
2. Material and methods
2.1. Fly lines
The salivary gland sgs3-Gal4 driver was from the Bloomington stock center (line 6870). ParB1-mCherry fly lines were given by François Payre (CBI, Toulouse, France).
2.2. Generation of the INT1-fkh enhancer fly lines
The different enhancers fkh250, fkh250MUT and fkh250CONS were synthesized in ten copies (GenScript) and cloned between the EcoRI and XhoI sites, downstream of the three INT1 sequences of the pattB-INT1-hslacZ vector (provided by F. Payre). All constructs were verified by sequencing before sending for injection (Bestgene Inc. company). INT1-fkh constructs were all inserted in the same landing on the third chromosome through the ФC-31 integrase system [21].
2.3. Salivary gland preparation for imaging.
Dissected salivary glands were fixed at room temperature for 20 min in phosphate-buffered saline (PBS) containing 3.7% formaldehyde (formaldehyde methanol free, Thermo Fisher Scientific) and washed 2 times for 30 min in 1x PBS. Salivary glands were either mounted in Vectashield with DAPI (Vector labs) between slide and coverslip for directly visualizing BiFC and ParB1-mCherry, or prepared for standard immunostaining before mounting for revealing the VC-Ubx construct (using rabbit anti-GFP (A11122, Molecular Probes, 1:500) recognizing the VC fragment [16], coupled to secondary Alexa Fluor 488 (A11039, Molecular Probes)).
2.4. FISH probes synthesis for the forkhead gene
The protocol for synthesizing DNA probes is taken from [22]. Briefly, a 12 kb long sequence centered on the fkh gene locus was chosen. Twelve pairs of 21 bp oligonucleotides were determined via the Primer3 website to cover the entire region (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). Each fragment was then amplified by PCR from genomic DNA (Figure S4). Each fragment is approximately 1 kb long. PCR products are then digested and labelled with the FISH Tag DNA orange kit (Alexa Fluor 555 dye, Thermofisher), following provider instructions.
2.5. Acquisition of confocal “lightning” images
Images were acquired on a confocal Leica SP8 microscope with the x63 oil objective (HC PL APO 63x/1.40). The Lightning option increases confocal resolution thanks to optimal acquisition parameters for stack thickness, objective used, mounting medium and laser wavelength. These parameters enable maximum details to be extracted from each image voxel. The images acquired are oversampled (a large number of pixels) to enable the processing. The image is scanned by a "lightning decision mask", and various parameters are measured to determine the image quality of each pixel and adapt the processing according to the result. This differs from traditional methods, which apply reconstruction schemes with global rather than local efficiency. This mask removes much of the background noise and detects signals with a limited number of photons, making the image more resolute (theoretical resolution achieved: 120 nm). (https://www.leica-microsystems.com/fr/produits/microscopes-confocaux/informations-detaillees/product/show/Products/lightning/).
Acquisition parameters for each experimental condition were set (2656x2656 pixels; laser scan speed: 574 Hz; x1.5 zoom). A 405 laser is used to image DAPI. A 488 nm laser excites Venus for BiFC signals and a 552 nm laser excites mCherry to localize the fkh250 enhancer (with Leica HyD detector).
3. Results
3.1. Visualizing Hox/Exd interactions by doing BiFC in salivary gland nuclei
To validate that we could get exploitable BiFC signals in third instar larva (L3) salivary gland nuclei, we used previously published fly lines compatible with the UAS/Gal4 system. More precisely, Hox (Scr and Ultrabithorax, Ubx) and Exd constructs are fused to the C-terminal (UAS-VCScr and UAS-VCUbx) or N-terminal (UAS-VNExd) fragment of Venus [16,20]. Interaction between Hox and Exd fusion proteins should lead to the reconstitution of an excitable Venus fluorescent protein upon complementation between the VN and VC fragments (Figure 1A,B). The UAS-VC-Hox constructs are inserted on the same landing site on the second chromosome and expressed at comparable levels [16,20]. VC-Hox and VN-Exd fusion proteins were expressed in L3 salivary glands with the sgs3-Gal4 driver (see materials and methods). It should be noticed that L3 salivary gland nuclei do not contain endogenous Exd given that its nuclear transporter Homothorax (Hth) is repressed by Scr (by stage 11, [13]; see also https://flybase.org/reports/FBgn0001235 for RNA-seq data in L3 salivary glands).
As expected, the two Hox proteins led to BiFC signals when co-expressed with Exd in salivary gland nuclei (Figure 1A’,B’). However, the overall quantification of BiFC showed that signals were three times higher on average with Ubx when compared to Scr (see materials and methods and Figure S1). This differential level of BiFC might be explained by the presence of endogenous Scr (although expressed at very low levels: https://flybase.org/reports/FBgn0003339), which could compete with VC-Scr for complex formation with VN-Exd. Not exclusively, the fusion topology may also affect the interaction between Scr and Exd, and therefore the resulting BiFC signals. In order to get a better resolution of signals, we applied an adaptative and automated deconvolution method on confocal acquisitions (see materials and methods). This manipulation revealed loci with higher fluorescence intensity and with a pattern following the polytene chromosome organization for both Scr/Exd and Ubx/Exd complexes (Figure 1A’’,B’’). Moreover, BiFC signals are not systematically co-localized with DAPI (which depicts AT-rich and more condensed chromatin), underlining that Hox/Exd complexes are bound on different types of chromatin region in salivary gland nuclei (enlargements in Figure 1A’–A”,B’–B’’). Overall, these observations confirm that our fusion constructs and expression system are compatible for doing BiFC in L3 salivary gland nuclei.
3.2. Experimental conditions for DNA-FISH are detrimental for BiFC
We next wanted to assess whether we could quantify BiFC signals specifically on the fkh locus by doing DNA-FISH (Figure 2A,B). To this end, we designed fluorescent oligonucleotides spanning 12kb upstream and downstream the fkh gene (see material and methods) and performed FISH staining on BiFC-positive salivary gland nuclei. Although FISH signals were clearly observable, we could not detect BiFC signals for Scr/Exd complexes (neither in the whole nucleus, nor on the fkh locus: Figures 2A’ and S2). BiFC signals were also extremely weak with Ubx/Exd complexes, making any quantification difficult. A more defined pattern of BiFC signals (Figure S2), especially after deconvolution (Figure 2B’), could be observed in a few nuclei. In these rare nuclei, the fkh FISH signal was systematically localized in a zone with low or no BiFC (Figure 2B’), which in accordance with previous work showing that Ubx/Exd complexes can neither bind the fkh250 enhancer in vitro nor regulate its expression in vivo [19]. Altogether, these experiments reveal that experimental conditions for FISH are detrimental for BiFC, underlining the need of using alternative approaches for doing colocalization studies with a genomic ROI.
3.3. BiFOR allows to reveal the enrichment of specific dimeric protein complexes on a target enhancer in salivary gland nuclei
Previous work established ANCHOR as a specific and sensitive method for labelling target ROIs in the genome without affecting transcriptional regulation in vivo [13,14].
We therefore decided to couple INT1 sequences to different versions of the fkh250 enhancer: wild type, mutant (with mutations abolishing the DNA-binding of Scr and Exd: fkh250MUT) and consensus (with mutations transforming the specific Scr/Exd into a generic Hox/Exd binding site and allowing the recognition by different Hox/Exd complexes: fkhCONS). These mutations were based on previous work on the fkh250 regulation by Hox/Exd complexes [13]. INT1-fkh250 fusion constructs were all inserted at the same landing site in the genome ([21] and material and methods). ParB1-mCherry fusion proteins were expressed in the salivary glands with the UAS/Gal4 system to label the different INT1-fkh250 fusion constructs ([13] and material and methods). All INT1-fkh250 fusion constructs were specifically recognized by ParB1-mCherry, leading to a clear red fluorescent signal in the nucleus. This signal can be of variable size and shape depending on the orientation of the nucleus, and was also usually less intense in the case of the INT1-fkh250MUT enhancer construct (compare Figure 3 and Figure 4).
We first performed BiFC by expressing VC-Scr and VN-Exd in ParB1-mCherry/INT1-fkh250 positive salivary gland nuclei (Figure 3A). In contrast to the previous observation under DNA-FISH condition, VC-Scr/VN-Exd BiFC signals were not affected. Quantification (see also material and methods) showed that BiFC signals were significantly enriched with ParB1-mCherry, demonstrating a preferential localization on the fkh250 enhancer when compared to other genomic loci in the salivary gland nuclei (Figures 3A’–A”, S3 and S4). This observation is in accordance with previous work showing the specific recognition and regulation of fkh250 by Scr/Exd in vitro and in vivo [13]. As negative control experiments, we repeated the analysis by expressing VC-Ubx either in combination with VN-Exd or alone. As expected, VC-Ubx/VN-Exd signals were globally stronger than the ones obtained with VC-Scr/VN-Exd in salivary gland nuclei, but the quantification showed a significant diminution of enrichment on the fkh250 enhancer (Figures 3B’–B” and S4). Expression of VC-Ubx alone also led to numerous strong signals along the polytene chromosomes, but quantification showed the absence of any specific enrichment on INT1-fkh250 enhancers (Figures 3C’–C” and S4). This observation underlines the absence of any preferential binding of either Ubx/Exd complexes or Ubx on the fkh250 enhancer, which is in accordance with previous work [13]. Together with the specific enrichment of VC-Scr/VN-Exd BiFC, our results indicate that BiFOR can recapitulate the specific recognition of target enhancers by a dimeric protein complex in salivary gland nuclei.
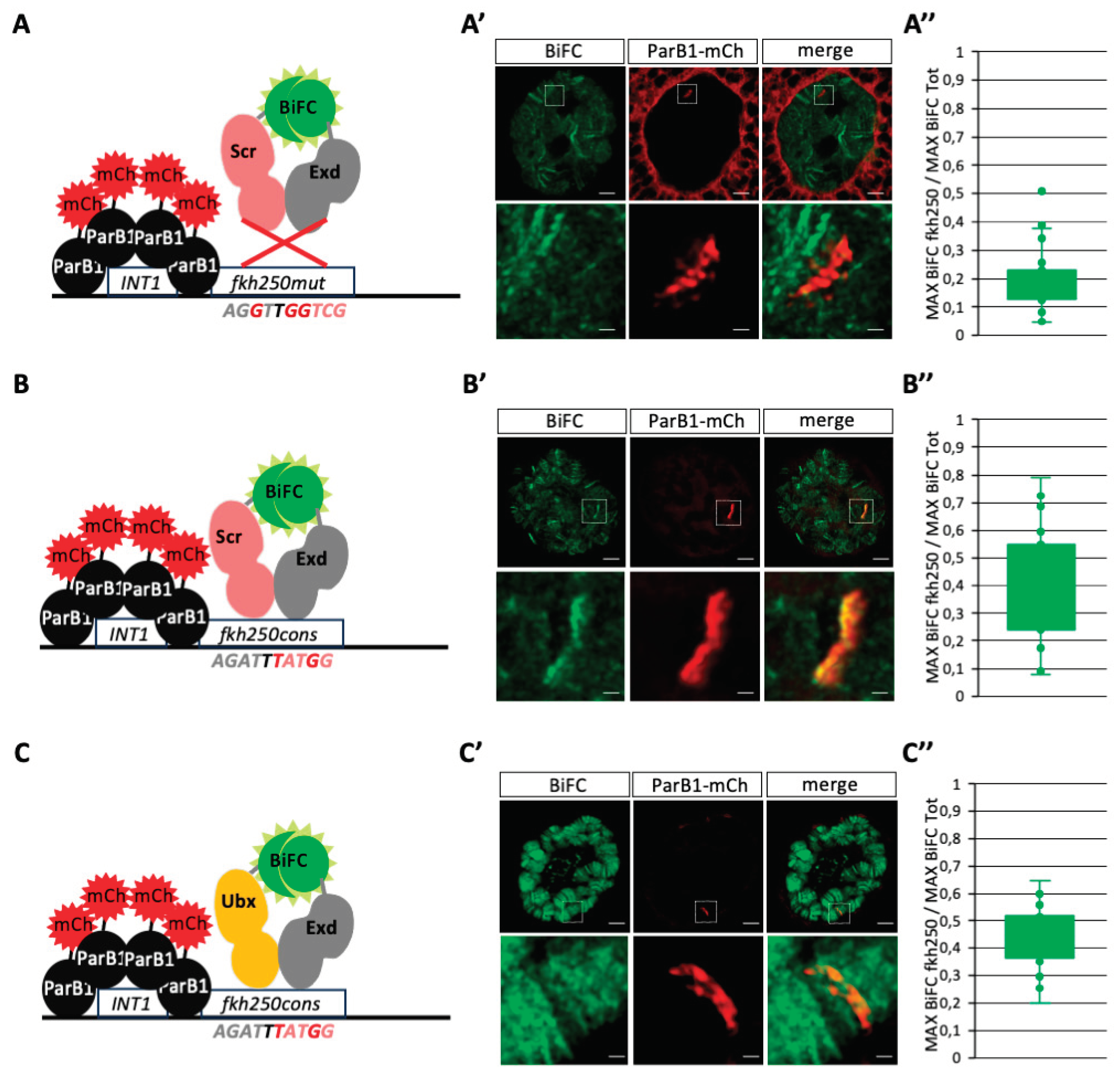
3.4. BiFOR allows to reveal dimeric complex enrichment on specific target enhancers in salivary gland nuclei
To further confirm the specificity of BiFOR, two additional variants of the fkh250 enhancer have been used: fkh250MUT and fkh250CONS. As previously mentioned, mutations in the flh250MUT enhancer abolish any Hox/Exd binding in vitro [13]. Not surprisingly, the analysis in salivary gland nuclei showed the absence of any significant enrichment of BiFC resulting from Scr/Exd complex assembly at the level of the fkh250MUT-containing genomic locus (Figures 4A–A” and S2). In contrast, Scr/Exd and Ubx/Exd BiFC signals were similarly and significantly enriched on fkh250CONS (Figures 4B–B”,C–C” and S2), confirming previous in vitro observations related to the gained ability of this modified enhancer to be recognized by different Hox/Exd complexes [13].
In conclusion, both the use of different Hox/Exd complexes and variants of the fkh250 enhancer confirmed the specificity and sensitivity of BiFOR for visualizing and quantifying PPI enrichment on a target enhancer of interest in salivary gland nuclei.
4. Discussion
4.1. Advantages of ANCHOR over other DNA-labelling systems for BiFC coupling
Several technics exist to visualize genomic ROIs in the nucleus. Here, we first tried to perform FISH for analyzing BiFC on the genomic locus of fkh. We found that the experimental conditions of FISH were detrimental for BiFC, forbidding any quantification measurements and underlining the need of using alternative and softer DNA-labelling approaches.
Several alternative methods have been developed to label genomic ROIs, the main challenge being to have enough and specific fluorescent signals for proper quantification in the nucleus. Among them are Crispr/Cas9-based approaches, which are long and labor intensive for the cloning and expression of multiple copies of the specific gRNAs. These issues might explain that CrispR/Cas9-based DNA-labeling approaches have not yet been applied in a multicellular organism [4,5,6,7]. The LacO/LacI system has been used in Drosophila, but it also requires hundred copies of the LacO cassette to exploit fluorescent signals emitted by the DNA-bound LacI-FP fusion protein [8]. In addition, the binding of LacI has been shown to interfere with gene transcription and is therefore not best appropriate for quantifying DNA-bound TFs in the genomic vicinity [14]. In contrast, the ANCHOR system is more neutral on gene transcription [14]. Moreover, the property of ParB proteins to oligomerize on ParS binding sites through protein-protein interactions allows seeing the ParB-FP without multiplying the number of ParS cassettes (usually three copies are used, [9]). The existence of different ParB proteins recognizing different ParS sequences (ParB1/ParS1 and ParB2/ParS2 for instance) also allows visualizing two different loci simultaneously when each ParB protein is fused to a different FP [14]. Thus, ANCHOR appears as a quite straightforward and neutral DNA-labelling system for visualizing genomic ROIs in the nucleus. Here, we demonstrated that BiFC could be coupled with ANCHOR to visualize PPIs on a specific target enhancer in salivary gland nuclei. Moreover, since BiFC is stabilizing the dimeric complex upon complementation (with the formation of covalent bonds between the two sub-fragments of the FP), it allows analyzing not only strong and stable but also weak and transient PPIs on a ROI.
4.2. Futures perspectives for BiFOR
Our system was based on transgenic constructs, with ten copies of wild type or modified fkh enhancers to ensure the visualization of BiFC signal enrichment in salivary gland nuclei. Salivary gland nuclei are characterized by multi-replicated polytene chromosomes, which also favors the visualization of TF binding events. Our analysis revealed clear and strong signals for both ParB1-mCherry and BiFC, suggesting that a much weaker number of INT1 sequences and fkh250 copies could have been used. The intensity of fluorescent signals also suggest that BiFOR could be exploited in diploid tissues, although it remains to be demonstrated. Along this line, ANCHOR has previously been shown to efficiently label a genomic ROI in Drosophila imaginal discs [14]. The exploitation of BiFC might depend on the number of DNA-binding sites in the target enhancer. For example, fkh250 contains only one Hox/Exd binding site, and a minimum number of copies is probably required for getting exploitable BiFC signals in diploid nuclei under conventional confocal microscope resolution. An alternative strategy to further increase the intensity and/or resolution of BiFC signals in this context could be the use of a BiFC-specific nanobody [23]. Indeed, the reconstitution of the FP upon complementation provides only 20% of the brightness of the original FP used for BiFC. This issue could be circumvented by using a BiFC-specific nanobody fused to a bright FP (such as mNeonGreen, [24]) or other fluorescent-emitting proteins (such as HaloTag, [25]). These imaging tools are compatible for STED microscopy and could therefore also increase the resolution scale of PPI analysis on the target enhancer. The same rational could apply for the ANCHOR labelling system. Here we used ParB1-mCherry fusions proteins, but other ParB proteins fused to brighter fluorescent proteins or systems are available [14].
In conclusion, our work established the experimental foundation for future technological developments of BiFOR in Drosophila and other model organisms.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
S.M. and S.V. designed the study. S.V. performed the experiments and acquired the data. S.M. and S.V. were responsible for data analysis. S.M. wrote the paper, and S.V. amended it.
Funding
This research was funded by FRM (grant 160896), CNRS and ENSL (recurrent funding).
Data Availability Statement
Dataset available on request from the authors.
Acknowledgments
We thank Christophe Place for advices on the quantification analysis, Marilyne Duffraisse for the discussions, and Marilyne Duffraisse and Christelle Forcet for comments on the manuscript. We thank the Bloomington stock center, Philippe Valenti and F. Payre for the fly lines, the IGFL imaging facility and the Arthrotools platform of the UAR3444/US8. This work was supported by Fondation pour la Recherche Médicale (Team FRM 160896 and 4th year PhD fellow to S.V.), CNRS and ENS-Lyon.
Conflicts of Interest
The authors declare no conflict of interest
References
- Kim S, W.J. Deciphering the multi-scale, quantitative cis-regulatory code. Mol Cell. 2022, 83, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E.M. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–26. [Google Scholar] [CrossRef] [PubMed]
- JG., G. The origin of in situ hybridization - A personal history. Methods 2016, 98, 4–9. [CrossRef] [PubMed]
- Fu Y, Rocha PP, Luo VM, Raviram R, Deng Y, Mazzoni EO, S.J. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat. Commun. 2016, 7. [CrossRef]
- Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, P.T. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol. 2016, 34, 528–530. [CrossRef] [PubMed]
- Chen B, Hu J, Almeida R, Liu H, Balakrishnan S, Covill-Cooke C, Lim WA, H.B. Expanding the CRISPR imaging toolset with Staphylococcus aureus Cas9 for simultaneous imaging of multiple genomic loci. Nucleic Acids Res. 2016, 44, e75. [CrossRef]
- Gu B, Swigut T, Spencley A, Bauer MR, Chung M, Meyer T, W.J. Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 2018, 359, 1050–1055. [CrossRef]
- Roukos V, Voss TC, Schmidt CK, Lee S, Wangsa D, M.T. Spatial dynamics of chromosome translocations in living cells. Science 2013, 341, 660–664. [CrossRef]
- Saad H, Gallardo F, Dalvai M, Tanguy-le-Gac N, Lane D, B.K. DNA dynamics during early double-strand break processing revealed by non-intrusive imaging of living cells. PLoS Genet. 2014, 10, e1004187. [CrossRef]
- Germier T, Audibert S, Kocanova S, Lane D, B.K. Real-time imaging of specific genomic loci in eukaryotic cells using the ANCHOR DNA labelling system.le. Methods 2018, 1, 16–23. [CrossRef]
- Mariamé B, Kappler-Gratias S, Kappler M, Balor S, Gallardo F, B.K. Real-Time Visualization and Quantification of Human Cytomegalovirus Replication in Living Cells Using the ANCHOR DNA Labeling Technology. J Virol. 2018, 92, e00571-18. [CrossRef] [PubMed]
- Chen H, Levo M, Barinov L, Fujioka M, Jaynes JB, G.T. Dynamic interplay between enhancer-promoter topology and gene activity. Nat. Genet. 2018, 50, 1296–1303. [CrossRef]
- Gomez-Lamarca, M.J.; Falo-Sanjuan, J.; Stojnic, R.; Abdul Rehman, S.; Muresan, L.; Jones, M.L.; Pillidge, Z.; Cerda-Moya, G.; Yuan, Z.; Baloul, S.; et al. Activation of the Notch Signaling Pathway In Vivo Elicits Changes in CSL Nuclear Dynamics. Dev. Cell 2018, 44, 611–623. [Google Scholar] [CrossRef]
- Rebecca K. Delker, R.H.M.; Michelle Hu, R.S.M. Fluorescent labeling of genomic loci in Drosophila imaginal discs with heterologous DNA-binding proteins. Cell Rep. 2022, 2, 100175. [CrossRef]
- Miller, K.E.; Kim, Y.; Huh, W.K.; Park, H.O. Bimolecular fluorescence complementation (BiFC) analysis: Advances and recent applications for Genome-Wide interaction studies. J. Mol. Biol. 2015, 427, 2039–2055. [Google Scholar] [CrossRef]
- Hudry, B.; Viala, S.; Graba, Y.; Merabet, S. Visualization of protein interactions in living Drosophila embryos by the bimolecular fluorescence complementation assay. BMC Biol. 2011, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Baeëza, M.; Viala, S.; Heim, M.; Dard, A.; Hudry, B.; Duffraisse, M.; Rogulja-Ortmann, A.; Brun, C.; Merabet, S. Inhibitory activities of short linear motifs underlie hox interactome specificity in vivo. Elife 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.; Duffraisse, M.; Furger, E.; Ajuria, L.; Giraud, G.; Vanderperre, S.; Paul, R.; Björklund, M.; Ahr, D.; Ahmed, A.W.; et al. Generation of a versatile bifc orfeome library for analyzing protein–protein interactions in live drosophila. Elife 2018, 7. [Google Scholar] [CrossRef]
- Ryoo, H.D.; Mann, R.S. The control of trunk Hox specificity and activity by Extradenticle. Genes Dev. 1999, 13, 1704–1716. [Google Scholar] [CrossRef]
- Hudry, B.; Remacle, S.; Delfini, M.-C.; Rezsohazy, R.; Graba, Y.; Merabet, S. Hox proteins display a common and ancestral ability to diversify their interaction mode with the PBC class cofactors. PLoS Biol. 2012, 10, e1001351. [Google Scholar] [CrossRef]
- Bischof, J.; Maeda, R.K.; Hediger, M.; Karch, F.; Basler, K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 3312–7. [Google Scholar] [CrossRef]
- Bantignies, F.; Cavalli, G. Topological organization of drosophila hox genes using DNA fluorescent in situ hybridization. Methods Mol. Biol. 2014, 1196, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Ariotti, N.; Rae, J.; Giles, N.; Martel, N.; Sierecki, E.; Gambin, Y.; Hall, T.E.; Parton, R.G. Ultrastructural localisation of protein interactions using conditionally stable nanobodies. PLoS Biol. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, W.J. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods 2013, 10, 407–409. [CrossRef]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, W.K. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008, 3, 373–382. [CrossRef]
Figure 1.
Visualization of BiFC signals in L3 salivary gland nuclei. A-B. Principle of the BiFC obtained with the VC-Scr and VN-Exd (A) or VC-Ubx and VN-Exd (B) constructs. A’-B’. Illustrative L3 salivary gland nucleus with BiFC (green) between Scr and Exd (A’) or Ubx and Exd (B’), with dapi staining of polytene chromosomes (cyan). Enlargement of a particular zone is shown to highlight both overlapping and non-overlapping staining between BiFC and dapi. Images are confocal acquisition without lightning deconvolution. A”-B”. Same illustrative salivary gland nuclei as in A’ and B’ but with applied lightning deconvolution to improve the resolution and signal-to-noise ratio of raw acquisitions. VN: N-terminal fragment of Venus (1-172). VC: C-terminal fragment of Venus (155-235). Scale bars=5μm (upper panels) or 1μm (enlargements).
Figure 1.
Visualization of BiFC signals in L3 salivary gland nuclei. A-B. Principle of the BiFC obtained with the VC-Scr and VN-Exd (A) or VC-Ubx and VN-Exd (B) constructs. A’-B’. Illustrative L3 salivary gland nucleus with BiFC (green) between Scr and Exd (A’) or Ubx and Exd (B’), with dapi staining of polytene chromosomes (cyan). Enlargement of a particular zone is shown to highlight both overlapping and non-overlapping staining between BiFC and dapi. Images are confocal acquisition without lightning deconvolution. A”-B”. Same illustrative salivary gland nuclei as in A’ and B’ but with applied lightning deconvolution to improve the resolution and signal-to-noise ratio of raw acquisitions. VN: N-terminal fragment of Venus (1-172). VC: C-terminal fragment of Venus (155-235). Scale bars=5μm (upper panels) or 1μm (enlargements).
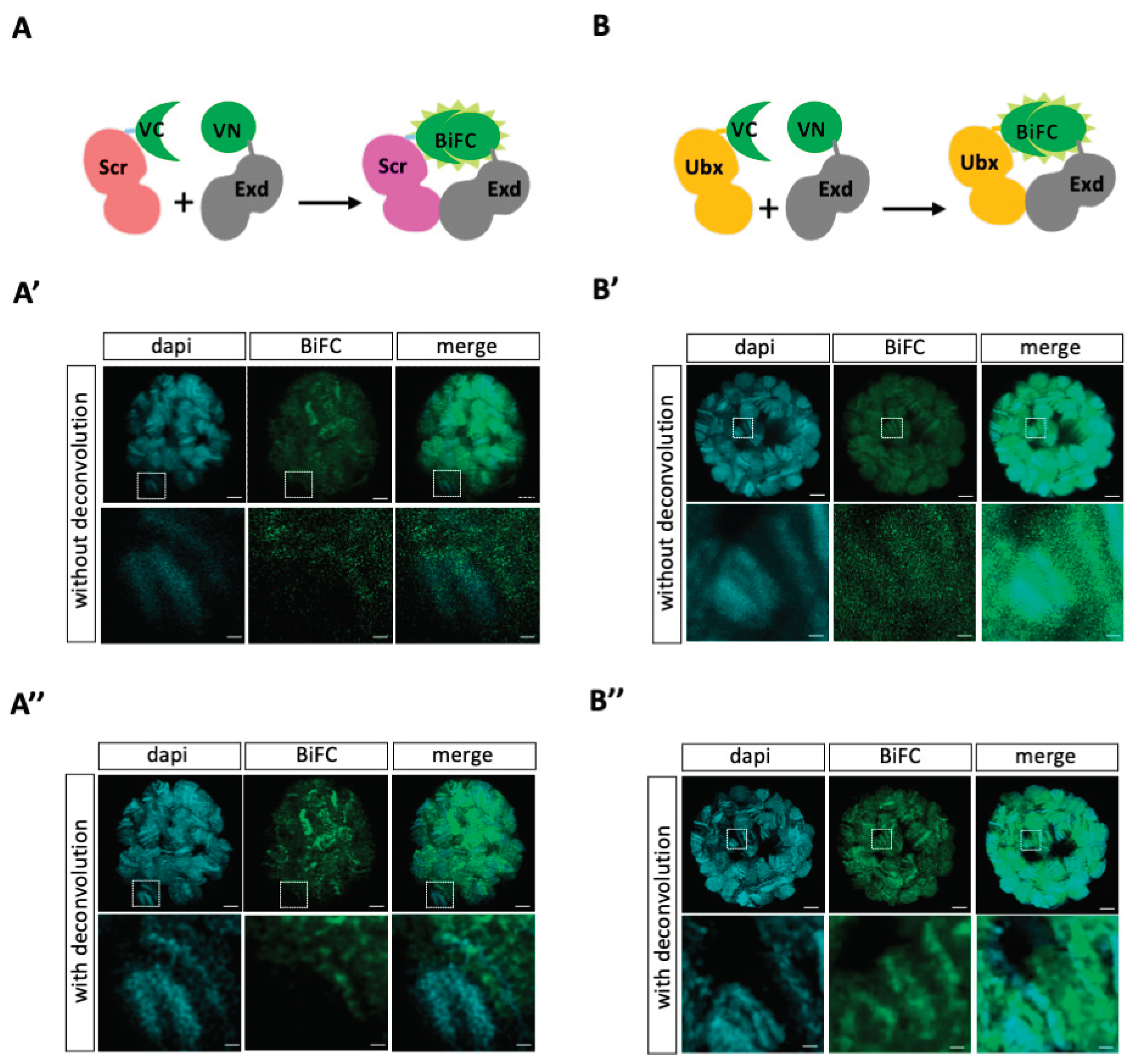
Figure 2.
Experimental conditions for DNA-FISH are detrimental to BiFC. A-B. Principle of the coupling between BiFC resulting from VC-Scr/VN-Exd (A) or VC-Ubx/VN-Exd (B) complex assembly and FISH staining of the forkhead (fkh) genomic locus. The nucleotide sequence of the DNA-binding site that is specifically recognized by Scr/Exd complexes in the fkh250 enhancer is indicated. Gray and light-red nucleotides are bound by Exd or the Hox protein, respectively. The two central nucleotides (black) are recognized by the two proteins (based on [13]). A’-B’. Illustrative L3 salivary gland nucleus with BiFC (green) resulting from VC-Scr/VN-Exd (A’) or VC-Ubx/VN-Exd (B’) complex assembly, FISH of the fkh locus (magenta) and dapi staining of polytene chromosomes (cyan). No exploitable BiFC signals is obtained under FISH experimental conditions for VC-Scr/VN-Exd complexes. Very few nuclei showed a weak BiFC pattern with VC-Ubx/VN-Exd. Enlargement in B’ highlights the non-overlapping pattern between BiFC and FISH signals. Images are confocal acquisition with lightning deconvolution (see also Figure S2). Scale bars=5μm (upper panels) or 1μm (enlargements).
Figure 2.
Experimental conditions for DNA-FISH are detrimental to BiFC. A-B. Principle of the coupling between BiFC resulting from VC-Scr/VN-Exd (A) or VC-Ubx/VN-Exd (B) complex assembly and FISH staining of the forkhead (fkh) genomic locus. The nucleotide sequence of the DNA-binding site that is specifically recognized by Scr/Exd complexes in the fkh250 enhancer is indicated. Gray and light-red nucleotides are bound by Exd or the Hox protein, respectively. The two central nucleotides (black) are recognized by the two proteins (based on [13]). A’-B’. Illustrative L3 salivary gland nucleus with BiFC (green) resulting from VC-Scr/VN-Exd (A’) or VC-Ubx/VN-Exd (B’) complex assembly, FISH of the fkh locus (magenta) and dapi staining of polytene chromosomes (cyan). No exploitable BiFC signals is obtained under FISH experimental conditions for VC-Scr/VN-Exd complexes. Very few nuclei showed a weak BiFC pattern with VC-Ubx/VN-Exd. Enlargement in B’ highlights the non-overlapping pattern between BiFC and FISH signals. Images are confocal acquisition with lightning deconvolution (see also Figure S2). Scale bars=5μm (upper panels) or 1μm (enlargements).
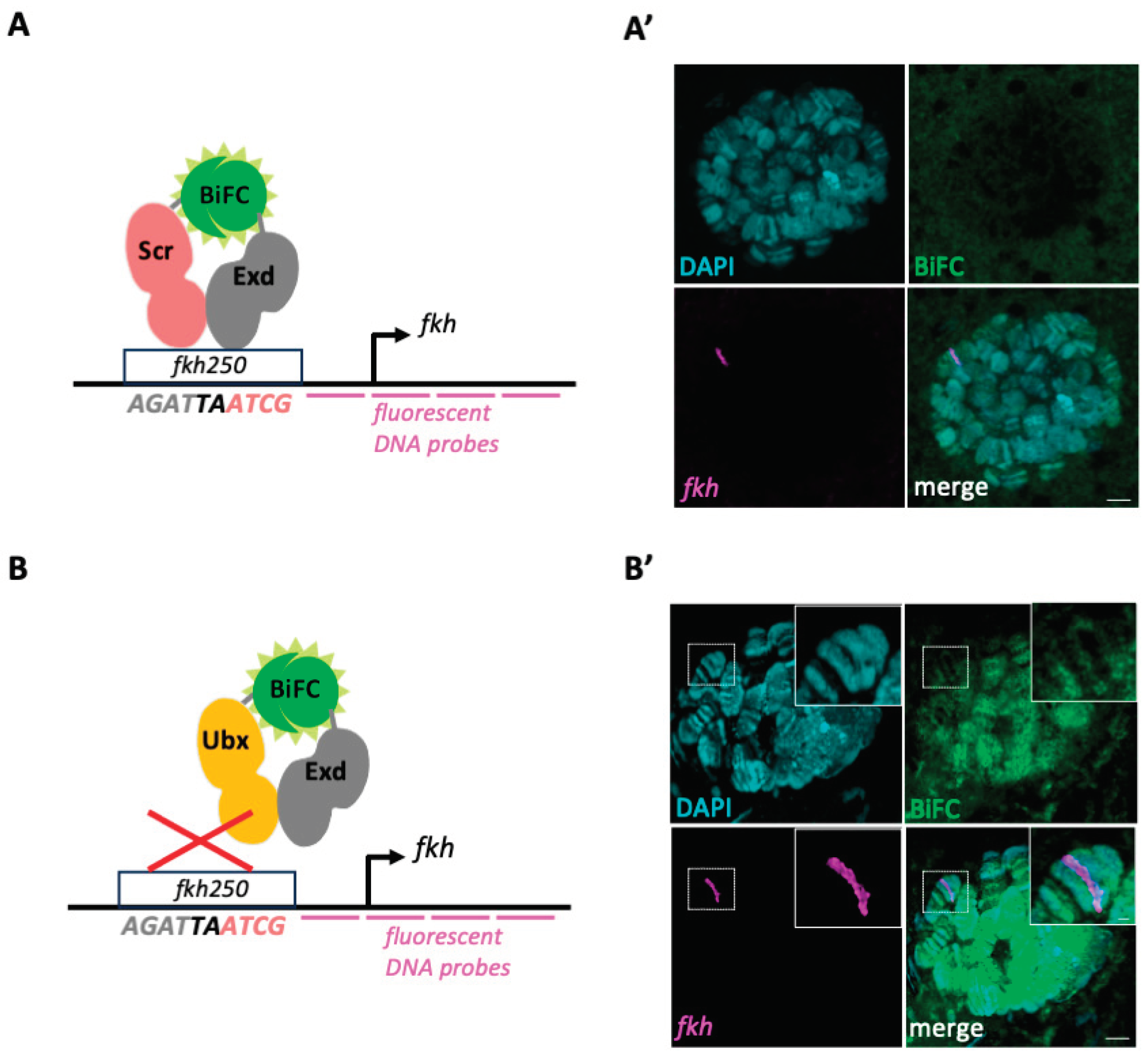
Figure 3.
Assessing the specificity of BiFOR with the wild type fkh250 enhancer in salivary gland nuclei. A-C. Schematic representation of the different conditions. The ParB1-mCherry (mCh) fusion proteins oligomerize on the target INT1 cassettes that are inserted closed to the fkh250 enhancer (see material and methods). DNA-Binding or not (red cross) is based on published work in vitro [13]. A’-C’. Illustrative confocal acquisition with lightning deconvolution of salivary gland nuclei expressing the different constructs, as indicated. Protein enrichment (green) was analyzed at the level of the inserted fkh250 enhancers (red) and compared to the overall maximum green fluorescence intensity in the nucleus (see material and methods). Enlargements are shown at the level of the ParB1-mCherry signal to better highlight the presence or absence of colocalization with BiFC (A’-B’) or VC-Ubx (C’). A”-C”. Statistical quantification of BiFC (A”-B”) or immunostaining (C”) at the level of the ParB1-mCherry signal. This quantification is obtained from three independent biological replicates. Scale bars=5μm (upper panels) or 1μm (enlargements). See also Figures S3 and S4.
Figure 3.
Assessing the specificity of BiFOR with the wild type fkh250 enhancer in salivary gland nuclei. A-C. Schematic representation of the different conditions. The ParB1-mCherry (mCh) fusion proteins oligomerize on the target INT1 cassettes that are inserted closed to the fkh250 enhancer (see material and methods). DNA-Binding or not (red cross) is based on published work in vitro [13]. A’-C’. Illustrative confocal acquisition with lightning deconvolution of salivary gland nuclei expressing the different constructs, as indicated. Protein enrichment (green) was analyzed at the level of the inserted fkh250 enhancers (red) and compared to the overall maximum green fluorescence intensity in the nucleus (see material and methods). Enlargements are shown at the level of the ParB1-mCherry signal to better highlight the presence or absence of colocalization with BiFC (A’-B’) or VC-Ubx (C’). A”-C”. Statistical quantification of BiFC (A”-B”) or immunostaining (C”) at the level of the ParB1-mCherry signal. This quantification is obtained from three independent biological replicates. Scale bars=5μm (upper panels) or 1μm (enlargements). See also Figures S3 and S4.
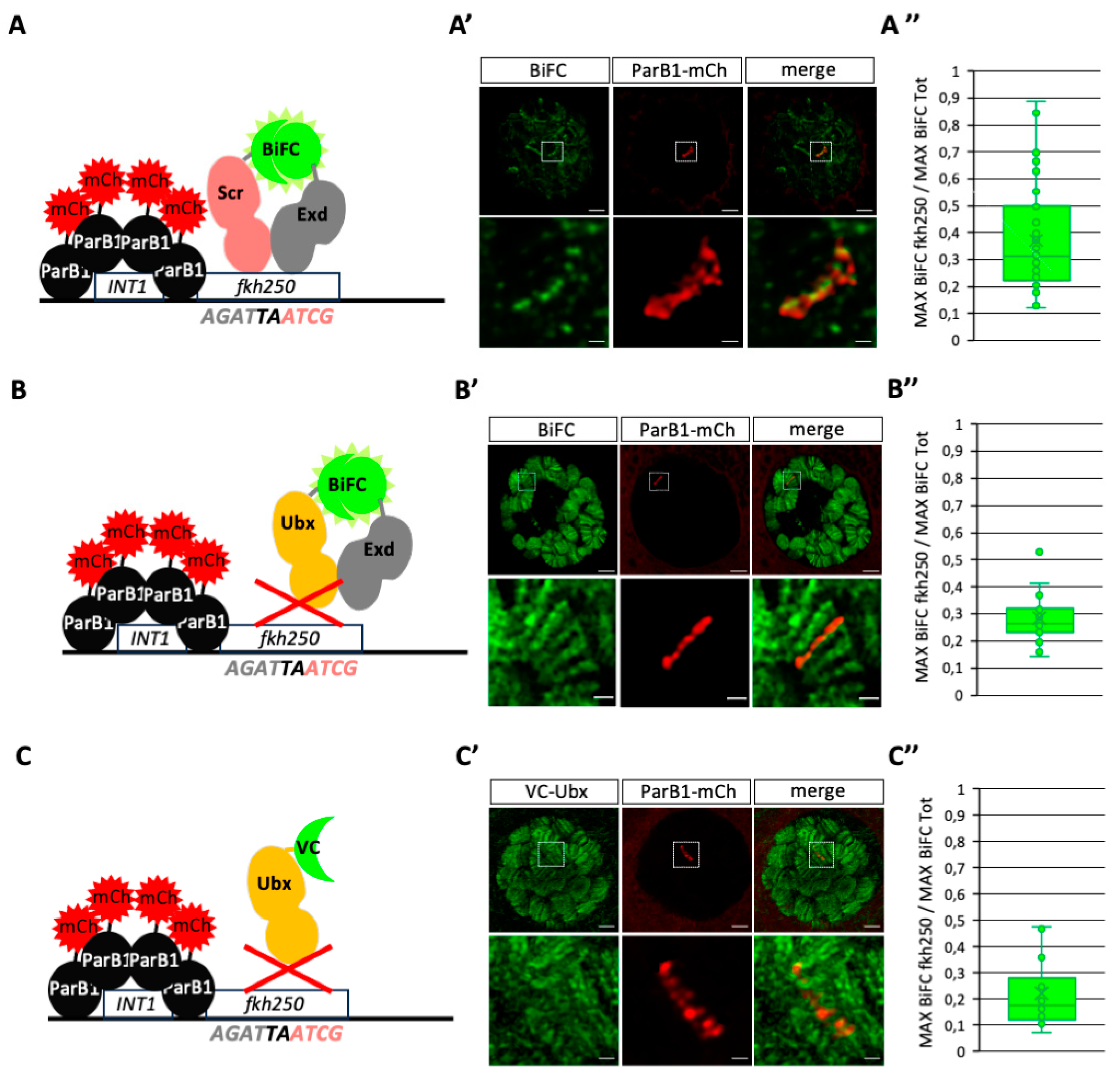
Figure 4.
Assessing the specificity of BiFOR on two different variants of the fkh250 enhancer in salivary gland nuclei. A-C. Schematic representation of the fkh250MUT (A) and fkh250CONS (B-C) enhancer variants. Mutated residues in the Hox and/or Exd DNA-binding site are highlighted in bold-red. Their negative (red cross) or positive effect on Hox/Exd binding is based on previous in vitro and in vivo studies [13]. A’-B’. Illustrative confocal acquisition with lightning deconvolution of salivary gland nuclei expressing the different constructs, as indicated. BiFC enrichment (green) was analyzed at the level of the fkh250MUT (A’) or fkh250CONS (B’-C’) enhancers (labelled with ParB1-mCherry, red) and compared to the overall maximum green fluorescent intensity in the nucleus (see material and methods). Enlargements are shown at the level of the ParB1-mCherry signal to better highlight the absence (A’) or presence (B’-C’) of colocalized green and red signals. A”-C”. Statistical quantification of BiFC at the level of the ParB1-mCherry signal. This quantification is obtained from three independent biological replicates. Scale bars=5μm (upper panels) or 1μm (enlargements). See also Figure S4.
Figure 4.
Assessing the specificity of BiFOR on two different variants of the fkh250 enhancer in salivary gland nuclei. A-C. Schematic representation of the fkh250MUT (A) and fkh250CONS (B-C) enhancer variants. Mutated residues in the Hox and/or Exd DNA-binding site are highlighted in bold-red. Their negative (red cross) or positive effect on Hox/Exd binding is based on previous in vitro and in vivo studies [13]. A’-B’. Illustrative confocal acquisition with lightning deconvolution of salivary gland nuclei expressing the different constructs, as indicated. BiFC enrichment (green) was analyzed at the level of the fkh250MUT (A’) or fkh250CONS (B’-C’) enhancers (labelled with ParB1-mCherry, red) and compared to the overall maximum green fluorescent intensity in the nucleus (see material and methods). Enlargements are shown at the level of the ParB1-mCherry signal to better highlight the absence (A’) or presence (B’-C’) of colocalized green and red signals. A”-C”. Statistical quantification of BiFC at the level of the ParB1-mCherry signal. This quantification is obtained from three independent biological replicates. Scale bars=5μm (upper panels) or 1μm (enlargements). See also Figure S4.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



