
Submitted:
27 February 2024
Posted:
28 February 2024
You are already at the latest version
Abstract
Erastin (ER) induces cell death by utilizing the process of ferroptosis via generation of reactive oxygen species (ROS). Ferroptosis is characterized by accumulation of ROS within the cell, leading to an iron-dependent oxidative damage-mediated cell death. Here, we have examined ferroptosis-dependent mechanism(s) of cytotoxicity of ER in K-RAS mutant human ovarian tumor OVCAR-8 and NCI/ADR-RES cell lines. We used these cell lines to decipher the mechanisms of ER as NCI/ADR-RES cells express higher activities of SOD, glutathione peroxidase and transferases than OVCAR-8 cells. We found that ER was equally cytotoxic to both cells; ER formed more ROS in OVCAR-8 cells than in NIH/ADR-RES cells. Ferrostatin-1, an inhibitor of ferroptosis, inhibited ER-induced cell death in both cell lines. In contrast, RSL3 (RAS-selective lethal 3), an inducer of ferroptosis, significantly enhanced cell death in both cell lines. ER induced significant lipid peroxidation only in OVCAR-8 cells. We found that RSL3 was more cytotoxic to NCI/ADR-RES cells and significantly enhanced ER-induced lipid peroxidation in both cells, indicating GPX4 was involved in the process of ER-mediated ferroptosis. ER treatment also differentially modulated several ferroptosis related genes (e.g., HMOX1/OH1, and CHAC1) in these cells, indicating ER-induced cell death may be mediated differently in OVCAR-8 and NCI/ADR-RES ovarian cells. Our studies suggest that combinations of ER and RSL3 may contribute significantly to the treatment of ovarian cancers, including those that are resistant to other therapeutic agents due to the presence of P-gp or tumors with Ras mutation in the clinic. ER induced ferroptosis may also be an important treatment modality for resistant tumors expressing MDR1 in the clinic.
Keywords:
Erastin
; Ferroptosis
; Ovarian Tumor calls
; RSL3
; Reactive Oxygen Species
1. Introduction
Ovarian cancer affects a significant number of women worldwide and is a leading cause of death among gynecological cancers. It is also considered one of the most lethal malignancies due to its high recurrence rate and inadequate early detection methods, placing a significant burden on patients. Ovarian cancers are highly heterogeneous and have distinct etiology, morphology, molecular biology and prognosis, but unfortunately, they are treated as a single cancer [1,2]. The standard treatment consists of cytoreductive surgery and combination chemotherapy containing cis-platin, taxanes and/or adriamycin [2]. While the response rate to chemotherapy (taxanes and/or adriamycin) is reported to be high, patients develop chemotherapy resistance and relapse. Taxanes and adriamycin are substrates of P-glycoprotein 170 (P-gp), a family of ABC transporter proteins that are highly expressed in various clinical tumor samples. Emergence of chemotherapy resistance and failure to respond to chemotherapy poses significant challenges in the treatment of ovarian cancers, requiring innovative approaches for therapy development for more effective anti-cancer agents or combinations of drugs.
Recently, drugs that induce tumor cell death through ferroptosis have been suggested to represent a promising avenue for cancer therapy, especially in cancers that are resistant to conventional treatments [3,4]. Ferroptosis is a regulated cell death characterized by the accumulation of lipid peroxides and the dependence on iron and is different from necrosis, autophagy, and apoptosis [5,6]. The cellular death resulting from ferroptosis has been reported to arise from the inhibition of glutathione peroxidase 4 (GPX4) and the accumulation of intracellular lipid hydroperoxides (LOOH), resulting in damage to cellular membranes (lipid peroxidation) in the presence of iron [7,8]. The damaging species is the reactive ●OH, formed from the reaction of H2O2 with Fe2+ (the Fenton reaction). Erastin (ER) is a small molecule that induces cell death in various tumor cells by ferroptosis. Erastin-induced killing of tumor cells primarily involves disrupting the cellular antioxidant defense system and triggering lipid peroxidation. However, several mechanisms of ER-dependent tumor cell death have been proposed, including the inhibition of the system XC– (glutamate/cystine antiporter), the inhibition of the mitochondria-bound voltage-dependent anion channel (VDAC) and the modulation of the tumor suppressor p53 gene [4,9,10,11]. Inhibition of the cystine/glutamate antiporter system Xc- by ER leads to a decrease in intracellular cystine and, subsequently, a decrease in GSH levels. Xc- transporter plays a crucial role in maintaining intracellular levels of cystine, a precursor for the synthesis of the antioxidant glutathione (GSH). This decrease in cellular GSH results in an increase in ROS/RNS formation (and oxidative stress), causing cellular damage and death. In addition, ER has been reported to inhibit VDAC which plays an important role in the induction of ferroptosis. The VDAC, an ion channel located in the outer mitochondrial membrane, mediates and controls molecular and ion exchange between the mitochondria and the cytoplasm. The permeability of the VDAC can be altered by drugs, causing mitochondrial metabolic dysfunction, ROS production, oxidative damage and cell death [12].
Erastin and its analogs are promising anticancer agents and ER also sensitizes resistant tumor cells to chemotherapy agents [13,14]. Furthermore, ER has shown activity in ovarian cancers in vitro. Therefore, understanding the mechanisms of ER cytotoxicity is crucial for developing strategies to design better drug combinations for the treatment of human cancers. Here, we have utilized both OVCAR-8 and OVCAR-8-derived adriamycin (ADR) resistant (NCI/ADR-RES) cell lines to understand and decipher the mechanisms of ER cytotoxicity. There are several reasons to use these ovarian cells as model cell lines to evaluate the mechanisms of ER cytotoxicity:- first, NCI/ADR-RES cells are extremely resistant to free radical generating drugs due to the presence of higher expressions of SOD, and catalase compared to OVCAR-8 cells [15,16]. Furthermore, NCI/ADR-RES cells have higher expression of both glutathione peroxidase (GPx1) glutathione transferase (GST) compared to OVCAR-8 cells. These antioxidant enzymes and GSH/GST systems protect cells from oxidative damage, resulting from the formation of ROS as well as other reactive species generated in tumor cells [17,18]. Therefore, we reasoned that if the mechanism of ER cytotoxicity is dependent upon the formation of ROS, then ER would be less cytotoxic to NCI/ADR-RES cells compared to OVCAR-8 cells as these reactive species would be detoxified due to the presence of glutathione peroxidase as well as by SOD and catalase in NCI/ADR-RES cells.
2. Materials and Methods
Materials: Adriamycin, the cystine analog, Seleno-L-cystine, the fluorescent molecule fluorescein O,O’-diacrylate, and the system XC- inhibitor sulfasalazine were purchased from Sigma Aldrich, (St. Louis, MO). Erastin, (2-[1-[4-[2-(4-Chlorophenoxy)acetyl]-1-piperazinyl]ethyl]-3-(2-ethoxyphenyl)-4(3H)-quinazolinone), RSL3, Ferrostatin-1 and N-acetyl cysteine (NAC) were purchased from Cayman Chemicals (Ann Arbor, MI) and were dissolved in DMSO. Stock solutions were stored at -80C. Fresh drug solutions, prepared from the stock solutions, were used in all experiments.
2.1. Cell Culture
Authenticated human ovarian OVCAR-8 and ADR-selected OVCAR-8 cells (NCI/ADR-RES) cells were obtained from the Division of Cancer Treatment and Diagnosis Tumor Repository, National Cancer Institute the NCI-Frederick Cancer Center (Frederick, MD, USA). Cells were grown in Phenol Red-free RPMI 1640 media supplemented with 10% fetal bovine serum and antibiotics. Cells were routinely used for 20-25 passages, after which the cells were discarded, and a new cell culture was started from the frozen stock.
2.2. Cytotoxicity Studies
The cytotoxicity studies were carried out with both TiterGLO (Promega) and Trypan Exclusion methods. For TiterGlo assay, roughly 2500-3000 cells/well were seeded in opaque white, 96-well plates and allowed to attach overnight. Cells were then treated with various concentrations of ER or ADR and incubated for 72h. Following 72h incubations, cytotoxicity of drugs were determined according to the manufacturer’s instructions.
For the Trypan Blue assay, about 25,000-50,000 cells/well were seeded onto a 6-well plate (in duplicate) and allowed to attach for 18h. Various concentrations of drugs (ER or combinations of ER, and minimally cytotoxic concentrations of ferrostatin-1, (FeS), or RSL3, or NAC (100 µM) were added to cells in fresh complete media (2 ML) and were incubated for either 24, 48 or 72h. When used, NAC, FeS or RSL3 were preincubated with cells for 1-2 h before the addition of ER. DMSO (0.01-0.1%) was included as the vehicle control when used. Following trypsinization, surviving cells were collected and 15 µL of cell mixtures were combined with 15 µL of trypan blue and counted in T20 automatic cell counter (Bio-Rad, Hercules, CA).
2.3. Cystine Uptake Assay via XC- Antiporter
50,00-75,000/well (OVCAR-8 and NCI-ADR/RES) were plated into a Greiner Fluotrac flat-bottomed 96-well black plate and allowed to attach for 18h. The media was removed, and 200 µL of pre-warmed cystine analog solution (200 µM selenocysteine) was added to other wells except to controls. Various concentrations of ER (1-20 µM) and 250 and 500 µM sulfasalazine were added. To the controls, 200 µL of cystine-free, serum-free media was added. Cells were then incubated at 37C for 60 minutes. Supernatant was removed and cells were washed three times with 200 µL of ice-cold PBS. Supernatant was removed and 60 µL of 100% methanol was added to each well. A working solution was prepared immediately before use that consisted of 100 mM MES buffer (pH 6.0), 10µM fluorescein O,O’-diacrylate, and 200µM tris-(2-carboxyethyl)phosphine hydrochloride. 200 µL of working solution was added to each well, mixed by pipetting, and incubated for 30 minutes at 37C. Following incubation, fluorescence was read using a Bi-Tek Synergy microplate reader. Fluorescence intensity was measured at Ex/Em = 490/535 and samples were read in triplicate.
2.4. Flow Cytometric Analysis of Mitochondrial ROS
The formation of ROS was determined as described previously [19]. Briefly, the cells were loaded with MitoSox Red (5 uM final concentration; Life Technologies, Carlsbad, CA) for 30 minutes at 37C, 7% CO2 atmosphere before the addition of ER. Cells were examined at 2h intervals with the addition of Sytox Blue as a vital dye by flow cytometry. A LSRFortessa flow cytometer (Benton Dickinson, San Jose, CA), equipped with FACSDiVa software, was used to analyze all samples. MitoSox and Sytox Blue were excited using a 561 nm and 405 nm laser and detected using a 610/20 nm and 450/50 nm filter, respectively. For each sample, 10, 000 cells were analyzed using FACSDiVa software.
2.5. Lipid Peroxidation Assay
Assay for the peroxidation of cellular lipids was carried out by measuring the formation of malondialdehyde (MDA) using 2-thiobarbituric acid as previously published [20,21]. Briefly, about 3-4.0 x 106 cells were incubated with various concentrations of ER or ADR (10 µM) for 4h at 37C. Following incubations, the reactions were stopped by adding 10% trichloroacetic acid (2 ml) and the mixtures were centrifuged (5 min at 1000g). Aliquots (1.5 ml) of the supernatant fractions were then reacted with 1.5 ml of 2% 2-thiobarbituric acid and the chromophore was developed at 90.0C for 10 min. After the samples were cooled, the absorbance at 532 nm was determined.
2.6. Flow Cytometric Analysis of Annexin-V Binding
Changes in membrane phosphatidylserine symmetry was determined using Annexin-5 V binding assay kit (Trevigen, Gaithersburg, MD) according to the manufacturer’s instructions. Briefly, cells were washed in 1X PBS, then incubated with 1 ul Annexin-V FITC and propidium iodide (PI) in Annexin-V binding buffer for 15 minutes at room temperature. After this time, the samples were diluted with 1X binding buffer and examined immediately by flow cytometry. Cells were analyzed using a LSRFortessa flow cytometer (Benton Dickinson, San Jose, CA) equipped with FACSDiVa software. Annexin-V FITC and PI was excited using a 488 nm and 561 nm laser and detected using a 530/30 nm and 582/15 nm filter, respectively. For each sample, 10, 000 cell were analyzed using FACSDiVa software.
2.7. Real Time RT-PCR
The expression levels of selected transcripts were examined by real-time polymerase chain reaction (RT-PCR) using absolute SYBR green ROX Mix (ThermoFisher Scientific, Rochester, NY) as previously described[19]. Cells (OVCAR-8 or NCI/ADR-RES) were treated with ER (2.5 µM) for 0h, 4h or 24h. Total RNA was isolated using Trizol following treatment with NCX4040 (5µM) for 4 and 24h and purified. Data were analyzed using ΔΔCt method of relative quantification in which cycle times were normalized to β-actin (or GADPH) from the same sample. Primers for the selected genes were purchased from Origene (Gaithersburg, MD). All real-time fluorescence detection was carried out on an iCycler (Bio-Rad, Hercules, CA). Experiments were carried out three different times and results are expressed as the mean ± SEM. Analyses were performed using unpaired Student’s t-test and considered significant when p ≤0.05.
2.8. Western Blot Assay
Cell pellets following treatment with ER (2.5µM) for 4 and 24h were collected, washed (ice-cold PBS) and homogenized in 200µl PierceTM RIPA (Thermo Fisher, 89900) supplemented with 25x Complete Protease Inhibitor, EDTA-free (Roche, 11836170001). Samples were centrifuged for 15 min at 14,000g at 4C, and supernatant aliquoted and stored at -80C. The concentration of protein in the samples was determined using the PierceTM BCA Protein Assay (Thermo Fisher, 23225). 15µg protein was combined with NuPage LDS Sample Buffer (Invitrogen, NP0007) and NuPage Sample Reducing Agent (Invitrogen, NP 0009) and incubated at 70C for 10min. Samples were loaded onto a NuPAGE™ 4 to 12%, Bis-Tris gel (Invitrogen, NP0336BOX), and run for 35 min at 200V in 1x NuPage MES Run Buffer (Invitrogen, NP0002) to which NuPage Antioxidant (Invitrogen, NP0005) had been added. Spectra™ Multicolor Broad Range Protein Ladder (ThermoFisher) was used as a reference for protein size. Proteins were transferred from the gel to nitrocellulose membranes using the iBlot™ Gel Transfer Device (ThermoFisher). Ponceau S solution (Sigma, P7170) was used to visualize total protein transfer. EveryBlot Blocking Reagent (Bio-Rad, 120110020) was used to block membranes. Membranes were incubated with primary antibody overnight at 4C diluted in EveryBlot Blocking Reagent: rabbit anti-vinculin (Novus, NBP2-20859), [add in ordering information for additional antibodies: GPX4, NRF2, NOX4]. Membranes were washed 3x5 min in 1x tris-buffered saline (TBS; Bio-Rad, 1706435) plus 0.1% tween-20 (Sigma, P7949) (TBST). Membranes were incubated for 45 min at room temperature with secondary antibody diluted 1:5000 in EveryBlot Blocking Reagent (Bio-Rad): goat-anti-rabbit HRP (Novus, NB7160) or goat-anti-mouse HRP (Invitrogen, 32230). Excess secondary antibody was removed by washing 3x5 min in TBS. HRP was visualized using SuperSignal™ West Pico Plus Chemiluminescent Substrate (Thermo Scientific, 34580).
2.9. Statistical Analysis
The results are expressed as mean ± SEM of minimum of 3 independent experiments (n = 3). One-way analysis of variance (ANOVA) was used for statistical analysis using Graph Pad Prism (GraphPad Software, Inc, La Jolla, CA). For multiple comparisons, the Tukey Multiple comparison’s test was utilized and were considered statistically significant when p < 0.05.
3. Results
3.1. Cytotoxicity Studies with Erastin
Previous studies have found that ER is cytotoxic to human ovarian tumor cells [19]. In this study, we found ER to be highly cytotoxic to both human OVCAR-8 and its P-gp expressing variant NCI/ADR-RES cells (Figure 1) and there were small differences in cytotoxicity, as found both with TiterGlo (A) and Trypan Blue (B) assay. Most interestingly, we found that the resistant variant to be slightly more sensitive to ER. This is in contrast to previous finding in other P-gp-expressing ovarian tumor cells which found Taxol resistant ovarian cells resistant to ER [22].
3.2. Effects of Ferrostatin-1 and RSL3 on Erastin Cytotoxicity
Since cytotoxicity of ER is mediated by ferroptosis, inhibitors of ferroptosis were used to confirm this in OVCAR-8 and NCI/ADR-RES cells. We used both Ferrostatin-1 (FES), a known inhibitor of ferroptosis [23,24,25], and RSL3, an inducer of ferroptosis [26,27], on ER cytotoxicity. As shown in Figure 2, FES significantly attenuated ER cytotoxicity while RSL3 markedly enhanced ER cytotoxicity in both cells, strongly suggesting that ER killed these ovarian cells by inducing ferroptosis. ER is also known to induce apoptosis in certain tumor cell types; however, neither Annexin binding assay [28] nor CaspaTag assay [29] showed any apoptosis induced by ER in either OVCAR-8 or NCI/ADR-RES cells (data not shown).
3.3. Cytotoxicity of RSL3 in OVCAR-8 and NIH/ADR-RES Cells
3.4. Effects of ER on Xc- Transporter
ER is known to inhibit Xc- transporter in tumor cells which then leads to decreases in cellular glutathione and oxidative damage [4]. We, therefore, examined effects of ER on Xc- transporter in both OVCAR-8 and NCI/ADR-RES cells. Data depicted in Figure 4 clearly indicate that ER is effective in inhibiting Xc- transporter in both OVCAR-8 and NIH/ADR-RES tumor cells and as little as 1.0 µM ER was sufficient to inhibit Xc- transporter in both cells.
3.5. ROS Formation by ER in OVCAR-8 and NIH/ADR-RES Cells
Studies have shown that ER generates ROS in tumor cells, leading to oxidative damage and cell death; Mitosox was utilized to detect formation of ER-dependent ROS species in both OVCAR-8 and NCI/ADR-RES cells. While use of Mitosox has been questioned for the selective detection of superoxide anion radical, we as well as others have successfully used this compound to detect ROS. Figure 5 shows that significant amounts of ROS were detected in OVCAR-8 cells; while little or no ROS could be detected in NCI/ADR-RES cells. Lower levels of ROS formation and/or detection most likely results from a rapid detoxification of active species formed due to presence of SOD, catalase, and GPX1 expressed in NCI/ADR-RES cells. We have reported similar findings with NCX4040 previously in these cell lines [19].
3.6. ER Induces Lipid Peroxidation in OVCAR-8 and NCI/ADR-RES Ovarian Cells
Formation of ER-dependent lipid peroxides was examined in OVCAR-8 and NIH/ADR-RES cells. As shown in Figure 6, small but significant amounts of peroxidation, measured as MDA, was detected in WT cells compared to the resistant variant. Adriamycin, a known inducer of peroxidation, was included as the positive control. MDA formation was dose-dependent in OVCAR-8 cells (WT). In contrast, neither ADR nor ER significantly induced peroxidation of lipids in the resistant NCI/ADR-RES cells.
3.7. RSL3 Enhances ER-Induced Lipid Peroxidation in OVCAR-8 and NIH/ADR-RES Cells
Because RSL3 enhanced ER cytotoxicity in these ovarian cells and that we found that RSL3 is more cytotoxic to NCI/ADR-RES cells, we investigated if RSL3 would also selectively enhance lipid peroxide formation in the resistant NCI/ADR-RES cells. Our results (Figure 6B) indicated that RSL3 enhanced the lipid peroxidation in NCI/ADR-RES cells such that there was no significant difference between OVCAR-8 and NCI/ADR-RES cells in MDA formation. RSL3 also increased MDA formation in OVCAR-8 cells (Figure 6B).
3.8. N-Acetyl Cysteine (NAC) Attenuates ER Cytotoxicity
Since ER-mediated cytotoxicity in tumor cells results from ROS formation, cytotoxicity of ER in OVCAR-8 and NCI/ADR-RES cells were further evaluated in the presence of NAC, a known scavenger of ROS and ferroptosis [30,31]. Our results (Figure 7) show that NAC significantly attenuated the cytotoxicity of ER in both cell lines, suggesting that ER cytotoxicity was mediated by ROS generation in these cells.
3.9. RT-PCR Studies in OVCAR-8 and NCI/ADR-RES Cells
RT-PCR was utilized to examine the expression level of genes related to oxidative damage-, ferroptosis- and oxidative DNA damage repair DNA Glycosyse1 (OGG1) following treatment of OVCAR-8 and NCI/ADR-RES cells with ER (2.5 µM) for 4h and 24h. We found that OGG1 was rapidly induced by ER in OVCAR-8 cells (4h) and decreased at 24h. OGG1 was significantly induced only at 24h in NCI/ADR-RES cells. These observations suggest that ER caused oxidative DNA damage in both cells. The anti-apoptotic gene BCl2 expression was unchanged in both OVCAR-8 and NCI/ADR-RES cells while the pro-apoptotic BAX gene expression was marginally decreased in both cells (Not Shown).
ER treatment also resulted in significant modulations of various gene expressions that are related to ferroptosis and oxidative stress in both ovarian cell lines (Figure 8). Heme oxygenase (HMOX1/OX1), a biomarker for the oxidative stress in cells [32,33], was significantly induced by ER treatment in both OVCAR-8 and NCI/ADR-RES cells. In contrast, NOX4, gene responsible for generating superoxide anion radical (O2.-) it was not affected in either OVCAR-8 or NCI/ADR-RES cells (Figure 8). CHAC1, a biomarker for ferroptosis [34,35,36], was highly induced in NCI/ADR-RES ovarian tumor cells. GPX4, the key ferroptosis indicator in cells and responsible for suppressing the process of ferroptosis [26,37], was decreased in OVCAR-8 cells at 4h and 24h while it was not significantly affected in NCI/ADR-RES cells. Glutathione reductase, responsible for maintaining GSH hemostasis was decreased only in NCI/ADR-RES cells. Nuclear erythroid factor 2 (NRF2), the master regulator of antioxidant pathways [38,39], was unchanged in both cells.
3.10. Western Blot in OVCAR-8 and NCI/ADR-RES Cells
Western blots were used to confirm our findings with RT-PCR and our studies show that GPX4 protein expression was unchanged in these ovarian cells following treatment with ER, suggesting that the enzyme activity was suppressed by ER and not its expression (Figure 8). Similarly, ER did not modulate the protein expressions of NRF2 or NOX4 as observed with RNA transcripts levels (Figure 8).
4. Discussion
Ferroptosis has been shown to be a novel type of iron-dependent, oxidative damage induced cell death that is different than other forms of cell death based on its genetic, morphological, and biochemical characteristics. It is dependent upon the formation and accumulation of lipid peroxides and iron, resulting in cellular damage via lipid peroxidation. It has been suggested that lipid peroxidation arises from the decrease in cellular GSH via the inhibition of Xc-transporter by ER resulting in inhibition of transport of cystine in exchange for glutamate into cells. Cystine is then reduced to cysteine by NADPH ultimately generating cellular GSH, which is utilized by GPX4 to reduce hydrogen peroxide, organic hydroperoxides and lipid hydroperoxides. Thus, the inhibition of cystine import by ER decreases GSH synthesis, inhibiting GPX4 activity, producing lipid ROS, and inducing oxidative damage and ferroptosis. RSL3, and other related compounds/agents have been shown to be direct inhibitors of GPX4, enhancing the process of ferroptosis.
We have shown that NCX4040, a non-steroidal nitric oxide donor, induces ferroptosis in human colon cancer cells [40]. We found that ER and RSL3 significantly enhances cytotoxicity of NCX4040, suggesting that these combinations could be highly effective for the treatment of both Ras-mutated and non-Ras tumor mutated cells. It is clear that understanding of molecular mechanisms that regulate ferroptosis will help scientists in designing better combinations of drugs to treat human cancers. We believe that elucidation and understanding the mechanisms of ER that underlie the sensitivity of a given cell type to ferroptosis is critical to understanding the process of ferroptosis and therefore, that it can be better utilized for the treatment of cancers in the clinic. Utilizing ER (and its recently synthesized analogs) as a novel combinational strategy for cancer therapy requires elucidation of its mechanism of action in different cancer types.
In this study, we investigated and compared the sensitivity to ER in human ovarian cancer OVCAR-8 and its ADR-selected NCI/ADR-RES cells. These ovarian tumors have P121H Ras mutation [41,42] and contain mutated p53 [43,44] and thus are markedly resistant to standard chemotherapy. The utilization of these cells to elucidate mechanisms of ER were several fold:- ADR-selected NCI/ADR-RES cells contain higher expressions of various enzymes (e.g. SOD, Catalase, and glutathione-dependent peroxidase (GPX1) and transferase) that are involved in detoxifications of ROS and one of the mechanisms of ER has suggested to depend upon the formation of ROS, inducing lipid peroxidation and ferroptosis. If this scenario is correct, then NCI/ADR-RES cells would show resistance to ER-induce ferroptosis and cell death. Secondly, NCI/ADR-RES cells also overexpress P-gp. A recent article suggests that ER [22] and some of its analogs are substrates for P-gp [22,45], which would also contribute to ability of NCI/ADR-RES cells to resist ER-dependent cell death. Finally, due to difficulties in curing clinical resistance and lack of suitable combinational chemotherapy agents for ovarian cancers, ferroptosis inducing agents are considered one of the novel approaches for a successful therapy.
Our studies show that ER inhibits Xc- cystine/glutamate transporters in both cell lines at similar ER concentrations and that ER induces cell death which was significantly enhanced by RSL3 in both cell lines. In addition, we found that ferrostatin-1, an inhibitor of ferroptosis, attenuated ER-mediated cell death in both cells. These observations are consistent with the process of ferroptosis-mediated cell death. Studies also show that ER is equally cytotoxic to both OVCAR-8 and its resistant variant NCI/ADR-RES cells lines. In fact, NCI/ADR-RES cell are slightly more sensitive to ER than OVCAR-8 cells. Our findings are similar to those recently reported by Frye et al. in these cell lines [45]. However, our results are in contrast to those reported by Zhou et al., showing that Taxol-selected P-gp-overexpressing ovarian cell line were significantly resistance to ER [22]. The reason for this discrepancy is not clear but it may result from a different selecting agent as cell line was selected for resistance to Taxol (different mechanisms of resistance in addition to P-gP expression) whereas NCI/ADR-RES cells were selected for resistance with adriamycin.
We found that ER-induced ROS formation only in OVCAR-8 cells and ROS could not be detected in NCI/ADR-RES cells. This was not unexpected as the resistant NCI/ADR-RES ovarian cells express SOD, Catalase and GSH-dependent peroxidases and transferase [15,16]. Furthermore, OVCAR-8 cells generated significant amounts of lipid peroxides (and MDA) compared to NCI/ADR-RES cells both with ER and adriamycin, used as the positive control. However, ER was equally cytotoxic to both cell lines and our studies showed that ER cytotoxicity is ROS-mediated as NAC completely attenuated ER cytotoxicity in both cells. These observations suggest that ER-mediated ferroptotic-cell death in NCI/ADR-RES cells may involve different pathways than OVCAR-8 cells. While it is clear that ER-mediated cell death is ROS-dependent, the pathway (s) to generate and induce oxidative damage in NCI/ADR-RES cells must be different in addition to decreases in cellular GSH from inhibition of Xc transporter as ER was equally effective in inhibiting cysteine transporter in both cells (Figure 4). Our studies also showed that ER inhibits GSR activity in NCI/ADR-RES cells, further reducing cellular GSH in NCI/ADR-RES cells and thus, enhancing oxidative damage by ER. It is also clear from our studies that GPX4 is involved in the detoxification of lipid peroxides formed in both cells as direct inhibition of GPX4 by RSL3 significantly increased lipid peroxide formation in both cells which resulted in enhanced cytotoxicity of ER in both cells, more in NCI/ADR-RES cells.
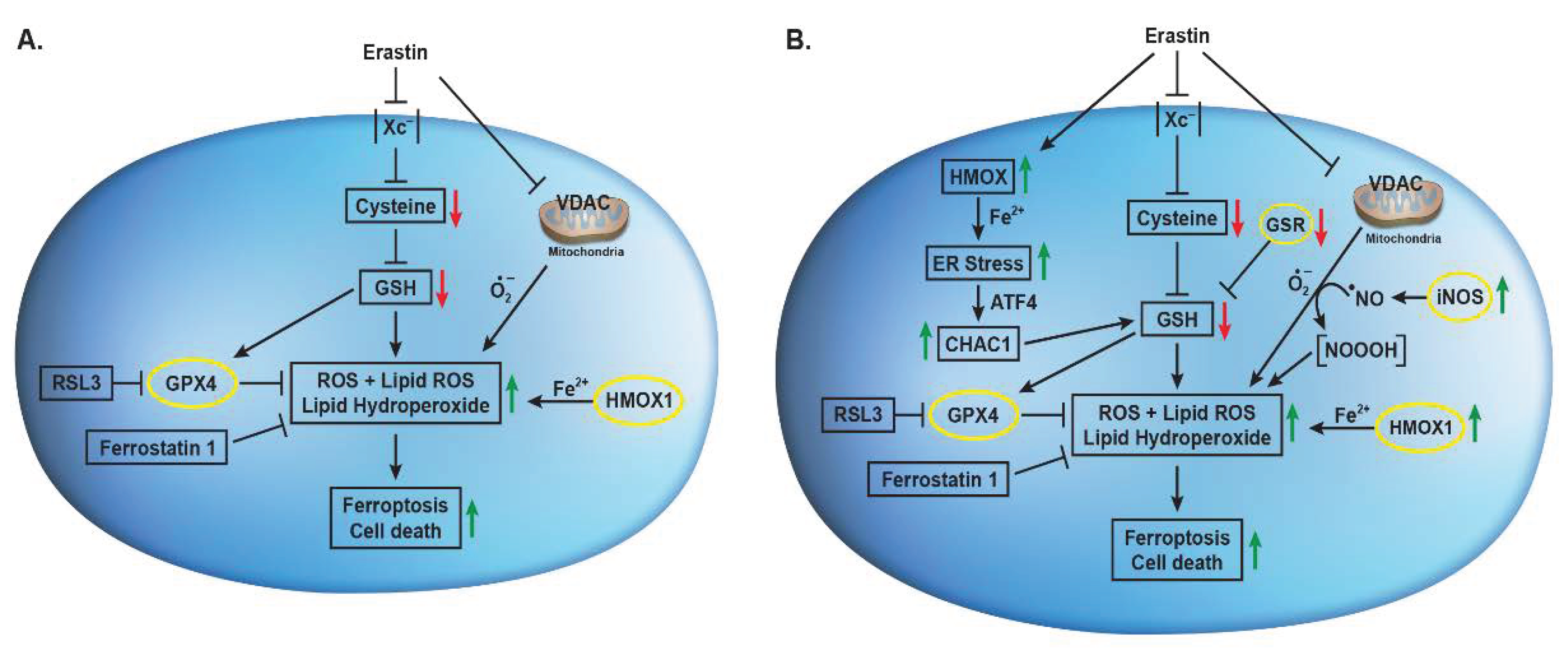
Erastin has been shown to induce nitric oxide (●NO) formation in MDA-MB-231 breast cancer cells and has been implicated in the induction of ferroptosis [46]. It has been reported that ER-induced GSH depletion activated disulfide isomerase (PDI) which induced iNOS, resulting in accumulation of cellular lipid ROS. Hou et al., have also shown that ER-induced GSH depletion leads to activation of PDI, which then mediates ferroptosis by catalyzing nNOS dimerization, resulting in the accumulation of cellular ●NO, ROS and lipid ROS which ultimately causes ferroptotic cell death in immortalized HT22 mouse hippocampal neuronal cells [47]. We have also previously reported that ●NO-generating drug, NCX4040, induces ferroptosis in colon cancer cells which was further enhanced in the presence of ER [40]. These observations suggest that ER may induce/activate iNOS in NCI/ADR-RES cells to generate ●NO which then reacts with O2●- to generates NOOOH which participate in ER-induced ferroptotic cell death. These observations are further supported by our recent findings that ER reverses ADR resistance in NCI/ADR-RES cells without significantly modulating ADR cytotoxicity in OVCAR-8 cells (manuscript in submission). Furthermore, increase in ADR cytotoxicity in NCI/ADR-RES cells was inhibited by 1400W, a specific inhibitor of iNOS suggesting formation of ●NO. We have shown that ●NO inhibits ATPase activity [48], including those of the ABC transporter [49,50]. 1400W had no effects on ADR cytotoxicity in OVCAR-8 cells suggest that ER may not induce/activate iNOS in OVCAR-8 cells and thus the contributions of ●NO would be minimal in this cell line. However, this needs to be confirmed in future studies. Figure 9 summarizes our findings.
Additionally, we found that CHAC1 was differentially induced by ER in NCI/ADR-RES cells. CHAC1 has been shown to hydrolyze cellular GSH, depleting GSH [51]. This would indicate that the cellular GSH is further decreased in NCI/ADR-RES cells in addition to that resulting from the inhibition of XC- transporter by ER and GSR. While we did not observe any depletion of GSH in these cells at 4h and cells were significantly killed at 24h by higher concentrations of ER (10, 20µM) and GSH could not be measured by using flow cytometeric analysis [19]. However, our recent metabolomic analysis (ER at 2.5µM) showed significant decreases in cellular GSH in both cells at 24h but not at 4h (data not shown). Taken together, our studies suggest that ER induces oxidative stress in both cell lines, however, ER-induced cellular oxidative damages are mediated by different pathways in NCI/ADR-RES cells, causing ferroptotic cell death. Additionally, our studies show that ER is not a substrate for P-gp and that it can be utilized to treat ovarian tumors both that either harbor RAS mutation or show MDR phenotypes in the clinic. Our studies also suggest that combinations of ER and RSL3 provide excellent venue to consider for future treatment of ovarian cancers. While we have not presented our studies here, combinations of ER with other anticancer drugs, e.g., adriamycin may also be given considerations for treating ABC transporter-expressing tumors in the clinic.
5. Conclusions
In conclusion, studies presented show that Erastin, a ferroptosis inducer, is not a substrate for P-gp and that it induces ferroptotic cell death in both OVCAR-8 and its P-gp-expressing NCI/ADR-RES tumor cells. While significant amounts of ROS was detected in OVCAR-8 cells, little or no ROS was detected in NCI/ADR-RES cells, most likely due to presence of various GSH-based detoxifying enzymes. However, significant oxidative damages were evident in both cell types as several oxidative damage markers were observed, e.g. induction of HMOX1/OX1, OGG1 and CHAC1. In addition, we found that the cytotoxicity of ER was completely attenuated by NAC in both cells, indicating free radical based cell death. Our studies suggest that GPX4 may be central to ER mediated cell death as the direct inhibition of GPX4 by RSL3 treatment enhanced ER-mediated lipid peroxidation in both cells which significantly enhanced ER-dependent cell death. Our studies further indicate that ER and RSL3 are important modulators of ovarian cell death and may provide excellent combinatorial therapy for difficult to treat cancers, including those show K-RAS mutations and/or express MDR phenotype.
Author Contributions
Conceptualization Birandra K. Sinha, Writing—BKS, Methods---BKS, CM, SMB, BS, EJT, and CDB.
Funding
This research was supported by the intramural research program (grant numbers ZIA E505013922, CARCI-HEI #, and ZIA ES103383-01) of the National Institute of Environmental Health Sciences, NIH. Statements contained herein do not necessarily represent the statements, opinions, or conclusions of NIEHS, NIH, or the US Government.
Acknowledgments
Authors would like to thank Drs. B. Alex Merrick and Ronald C. Cannon for critical review of the manuscript.
Conflicts of Interest
The authors declare no actual or potential conflicts of interest.
References
- Kossai, M.; Leary, A.; Scoazec, J.Y.; Genestie, C. Ovarian Cancer: A Heterogeneous Disease. Pathobiology 2018, 85, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Parmar, M.K.; Ledermann, J.A.; Colombo, N.; du Bois, A.; Delaloye, J.F.; Kristensen, G.B.; Wheeler, S.; Swart, A.M.; Qian, W.; Torri, V.; et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet 2003, 361, 2099–2106. [Google Scholar] [CrossRef]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dancik, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 2019, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem Biophys Res Commun 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med 2019, 133, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front Oncol 2017, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol 2015, 2, e1054549. [Google Scholar] [CrossRef]
- Cheng, Q.; Bao, L.; Li, M.; Chang, K.; Yi, X. Erastin synergizes with cisplatin via ferroptosis to inhibit ovarian cancer growth in vitro and in vivo. J Obstet Gynaecol Res 2021, 47, 2481–2491. [Google Scholar] [CrossRef]
- Batist, G.; Tulpule, A.; Sinha, B.K.; Katki, A.G.; Myers, C.E.; Cowan, K.H. Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells. J Biol Chem 1986, 261, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Cowan, K.H.; Batist, G.; Tulpule, A.; Sinha, B.K.; Myers, C.E. Similar biochemical changes associated with multidrug resistance in human breast cancer cells and carcinogen-induced resistance to xenobiotics in rats. Proc Natl Acad Sci U S A 1986, 83, 9328–9332. [Google Scholar] [CrossRef]
- Sinha, B.K. Free radicals in anticancer drug pharmacology. Chemico-biological interactions 1989, 69, 293–317. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.K.; Mimnaugh, E.G.; Rajagopalan, S.; Myers, C.E. Adriamycin activation and oxygen free radical formation in human breast tumor cells: protective role of glutathione peroxidase in adriamycin resistance. Cancer research 1989, 49, 3844–3848. [Google Scholar]
- Sinha, B.K.; Tokar, E.J.; Bortner, C.D. Molecular Mechanisms of Cytotoxicity of NCX4040, the Non-Steroidal Anti-Inflammatory NO-Donor, in Human Ovarian Cancer Cells. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Trush, M.A.; Gram, T.E. Stimulation by adriamycin of rat heart and liver microsomal NADPH-dependent lipid peroxidation. Biochemical pharmacology 1981, 30, 2797–2804. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Kennedy, K.A.; Trush, M.A.; Sinha, B.K. Adriamycin-enhanced membrane lipid peroxidation in isolated rat nuclei. Cancer research 1985, 45, 3296–3304. [Google Scholar]
- Zhou, H.H.; Chen, X.; Cai, L.Y.; Nan, X.W.; Chen, J.H.; Chen, X.X.; Yang, Y.; Xing, Z.H.; Wei, M.N.; Li, Y.; et al. Erastin Reverses ABCB1-Mediated Docetaxel Resistance in Ovarian Cancer. Front Oncol 2019, 9, 1398. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vuckovic, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol 2020, 28, 101328. [Google Scholar] [CrossRef]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett 2020, 25, 10. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Liu, C.X.; Song, R.; Li, Q.L. Ferrostatin-1 protects HT-22 cells from oxidative toxicity. Neural Regen Res 2020, 15, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol 2018, 9, 1371. [Google Scholar] [CrossRef] [PubMed]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci 2017, 108, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, M.; Funahashi, H.; Tsukamura, K.; Imai, T.; Hayakawa, A.; Kiuchi, T.; Nakao, A. Intracellular signaling in the induction of apoptosis in a human breast cancer cell line by water extract of Mekabu. Int J Clin Oncol 2005, 10, 122–126. [Google Scholar] [CrossRef]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. The Biochemical journal 2013, 454, 201–208. [Google Scholar] [CrossRef]
- Zhang, Q.; Yi, H.; Yao, H.; Lu, L.; He, G.; Wu, M.; Zheng, C.; Li, Y.; Chen, S.; Li, L.; et al. Artemisinin Derivatives Inhibit Non-small Cell Lung Cancer Cells Through Induction of ROS-dependent Apoptosis/Ferroptosis. J Cancer 2021, 12, 4075–4085. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, Y.; Zheng, T.; Yang, G.; Zhang, X.; Sun, Z.; Shi, C.; Zhao, S. Inhibition of heme oxygenase-1 enhances anti-cancer effects of arsenic trioxide on glioma cells. J Neurooncol 2011, 104, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; He, X.; Fang, H.; Liao, S.; Liu, Y.; Tian, L.; Niu, J. Identification of heme oxygenase-1 from golden pompano (Trachinotus ovatus) and response of Nrf2/HO-1 signaling pathway to copper-induced oxidative stress. Chemosphere 2020, 253, 126654. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Wang, S.F.; Hsu, C.Y.; Yin, P.H.; Yeh, T.S.; Lee, H.C.; Tseng, L.M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zeng, G.Z.; Yin, J.L.; Bian, Z.X. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt's Lymphoma. Biochem Biophys Res Commun 2019, 519, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Wang, S.; Guo, J.; Liu, S.; Ding, A.; Wang, G.; Li, W.; Zhang, Y.; Bian, X.; Zhao, S.; et al. Ferroptosis-related gene NOX4, CHAC1 and HIF1A are valid biomarkers for stomach adenocarcinoma. J Cell Mol Med 2022, 26, 1183–1193. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid Redox Signal 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Sinha, B.K.; Bortner, C.D.; Jarmusch, A.K.; Tokar, E.J.; Murphy, C.; Wu, X.; Winter, H.; Cannon, R.E. Ferroptosis-Mediated Cell Death Induced by NCX4040, The Non-Steroidal Nitric Oxide Donor, in Human Colorectal Cancer Cells: Implications in Therapy. Cells 2023, 12. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Stewart, M.L.; Tamayo, P.; Wilson, A.J.; Wang, S.; Chang, Y.M.; Kim, J.W.; Khabele, D.; Shamji, A.F.; Schreiber, S.L. KRAS Genomic Status Predicts the Sensitivity of Ovarian Cancer Cells to Decitabine. Cancer research 2015, 75, 2897–2906. [Google Scholar] [CrossRef]
- De Feudis, P.; Debernardis, D.; Beccaglia, P.; Valenti, M.; Graniela Sire, E.; Arzani, D.; Stanzione, S.; Parodi, S.; D'Incalci, M.; Russo, P.; et al. DDP-induced cytotoxicity is not influenced by p53 in nine human ovarian cancer cell lines with different p53 status. Br J Cancer 1997, 76, 474–479. [Google Scholar] [CrossRef]
- Ogretmen, B.; Safa, A.R. Expression of the mutated p53 tumor suppressor protein and its molecular and biochemical characterization in multidrug resistant MCF-7/Adr human breast cancer cells. Oncogene 1997, 14, 499–506. [Google Scholar] [CrossRef]
- Frye, W.J.E.; Huff, L.M.; Gonzalez Dalmasy, J.M.; Salazar, P.; Carter, R.M.; Gensler, R.T.; Esposito, D.; Robey, R.W.; Ambudkar, S.V.; Gottesman, M.M. The multidrug resistance transporter P-glycoprotein confers resistance to ferroptosis inducers. Cancer Drug Resist 2023, 6, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, P.; Zhu, B.T. Mechanism of Erastin-Induced Ferroptosis in MDA-MB-231 Human Breast Cancer Cells: Evidence for a Critical Role of Protein Disulfide Isomerase. Mol Cell Biol 2022, 42, e0052221. [Google Scholar] [CrossRef]
- Hou, M.J.; Wang, P.; Zhu, B.T. Biochemical mechanism of erastin-induced ferroptotic cell death in neuronal cells. Acta Biochim Biophys Sin (Shanghai) 2023, 55, 853–865. [Google Scholar] [CrossRef]
- Kumar, A.; Ehrenshaft, M.; Tokar, E.J.; Mason, R.P.; Sinha, B.K. Nitric oxide inhibits topoisomerase II activity and induces resistance to topoisomerase II-poisons in human tumor cells. Biochimica et biophysica acta 2016, 1860, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.K.; Bortner, C.D.; Mason, R.P.; Cannon, R.E. Nitric oxide reverses drug resistance by inhibiting ATPase activity of p-glycoprotein in human multi-drug resistant cancer cells. Biochimica et biophysica acta. General subjects 2018, 1862, 2806–2814. [Google Scholar] [CrossRef]
- Sinha, B.K.; Perera, L.; Cannon, R.E. NCX-4040, a Unique Nitric Oxide Donor, Induces Reversal of Drug-Resistance in Both ABCB1- and ABCG2-Expressing Multidrug Human Cancer Cells. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J Biol Chem 2015, 290, 15878–15891. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cytotoxicity of Erastin in OVCAR-8 and NCI/ADR-RES cells following 72h of treatment using TiterGlo (A) and Trypan Blue (B) cytotoxicity assays.
Figure 1.
Cytotoxicity of Erastin in OVCAR-8 and NCI/ADR-RES cells following 72h of treatment using TiterGlo (A) and Trypan Blue (B) cytotoxicity assays.
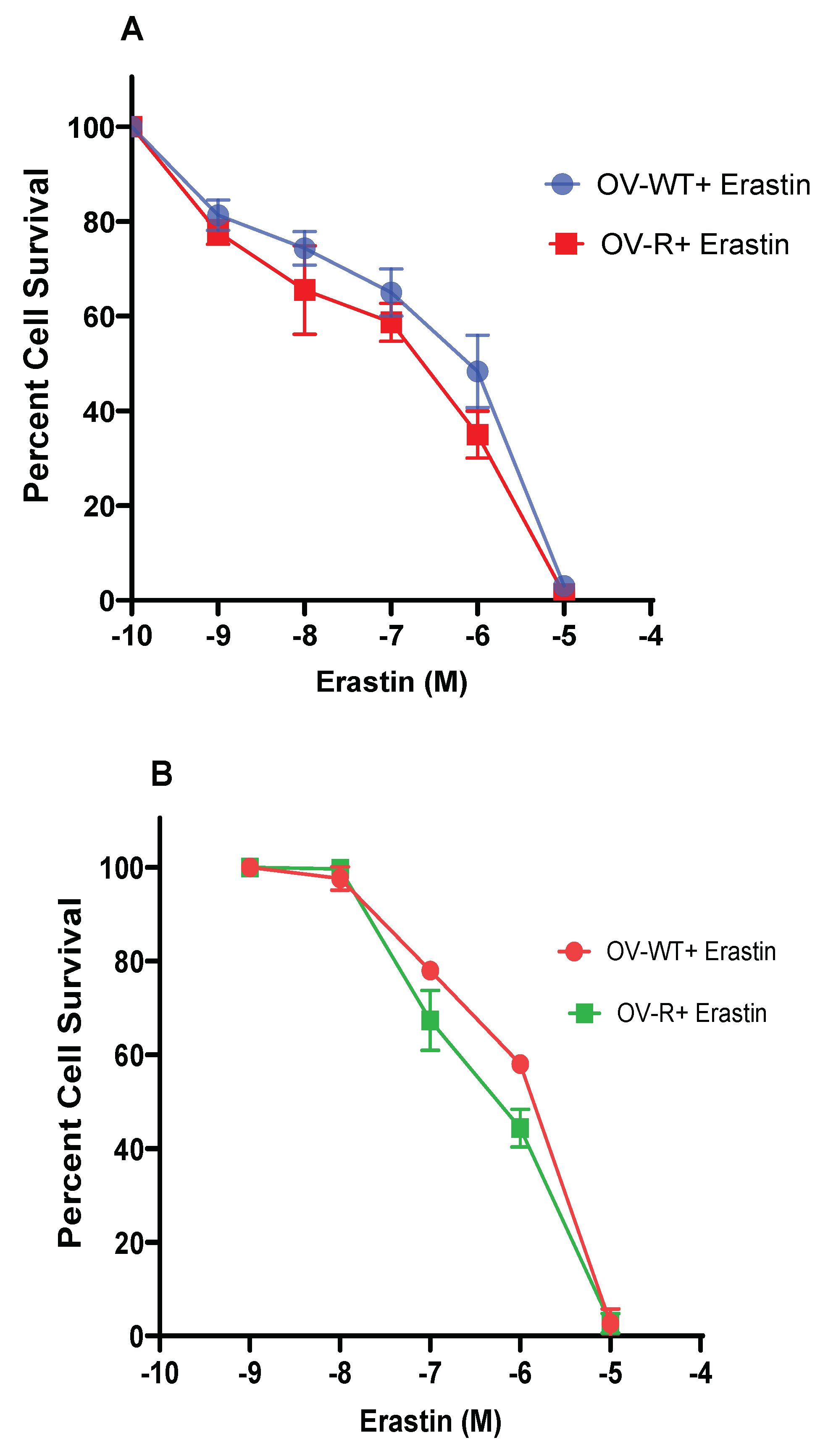
Figure 2.
Effects of Ferrostatin-1 (A, B) and RSL3 (C, D) on the cytotoxicity of Erastin in ovarian cells following 48h incubations. (A) OVCAR-8 and (B) NCI/ADR-RES cells, respectively. Effects of RSL3 on cytotoxicity ER following 24h of incubations (C), OVCAR-8 and (D) NCI/ADR-RES cells. *, ** and *** p values) >0.05, 0.005 and 0.001 respectively compared to untreated control. ##, and ### p values >0.005 and 0.0001, respectively compared to treated FES or RSL3 alone to treated ER + FES or ER + RSL3. $$$ p values respectively compared to treated ER alone.
Figure 2.
Effects of Ferrostatin-1 (A, B) and RSL3 (C, D) on the cytotoxicity of Erastin in ovarian cells following 48h incubations. (A) OVCAR-8 and (B) NCI/ADR-RES cells, respectively. Effects of RSL3 on cytotoxicity ER following 24h of incubations (C), OVCAR-8 and (D) NCI/ADR-RES cells. *, ** and *** p values) >0.05, 0.005 and 0.001 respectively compared to untreated control. ##, and ### p values >0.005 and 0.0001, respectively compared to treated FES or RSL3 alone to treated ER + FES or ER + RSL3. $$$ p values respectively compared to treated ER alone.
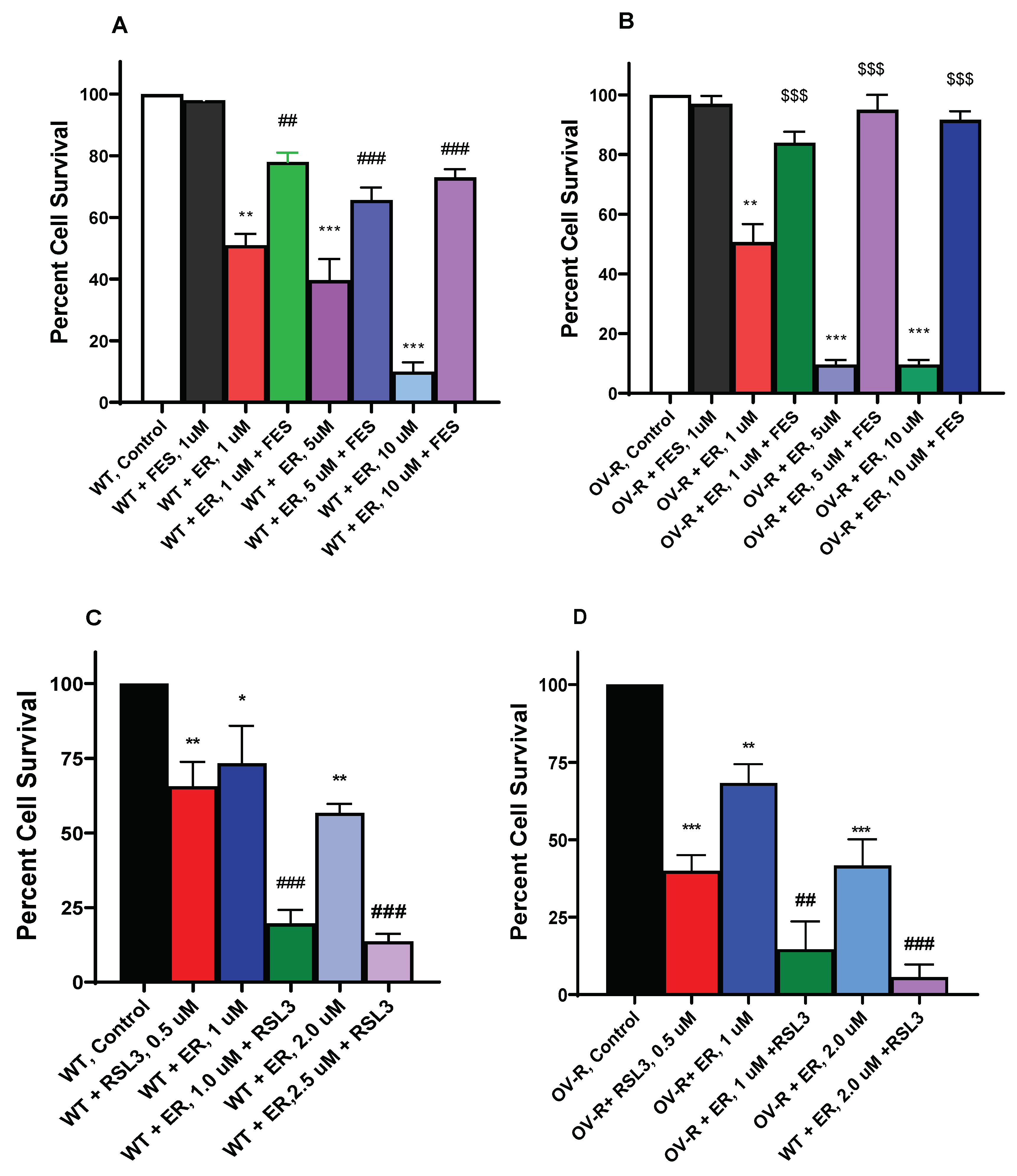
Figure 3.
Dose-dependence of RSL3 cytotoxicity in OVCAR-8 (A) and NCI/ADR-RES cells (B). The cells were incubated with different concentrations of RSL3 for 24h. ##, and ### and ** and *** p values <0.005 and 0.001 respectively compared to untreated control.
Figure 3.
Dose-dependence of RSL3 cytotoxicity in OVCAR-8 (A) and NCI/ADR-RES cells (B). The cells were incubated with different concentrations of RSL3 for 24h. ##, and ### and ** and *** p values <0.005 and 0.001 respectively compared to untreated control.
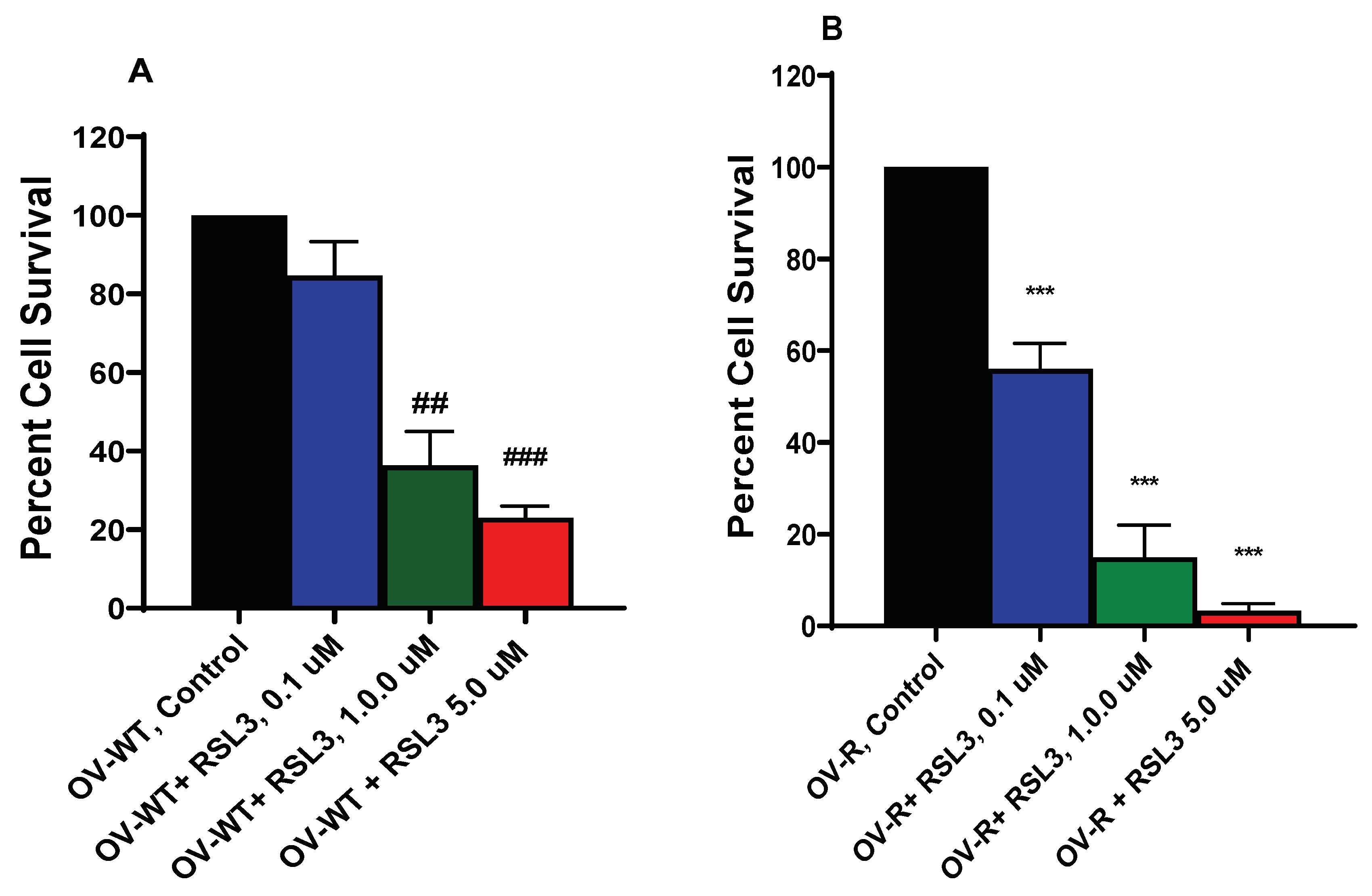
Figure 4.
Effects of ER on the Xc- transporter in OVCAR-8 (A) and NCI/ADR-RES (B) cells. *** p values < 0.001 compared to control untreated cells; ### and $$$ p values > 0.0001, respectively compared to Se-Cysteine treated cells.
Figure 4.
Effects of ER on the Xc- transporter in OVCAR-8 (A) and NCI/ADR-RES (B) cells. *** p values < 0.001 compared to control untreated cells; ### and $$$ p values > 0.0001, respectively compared to Se-Cysteine treated cells.
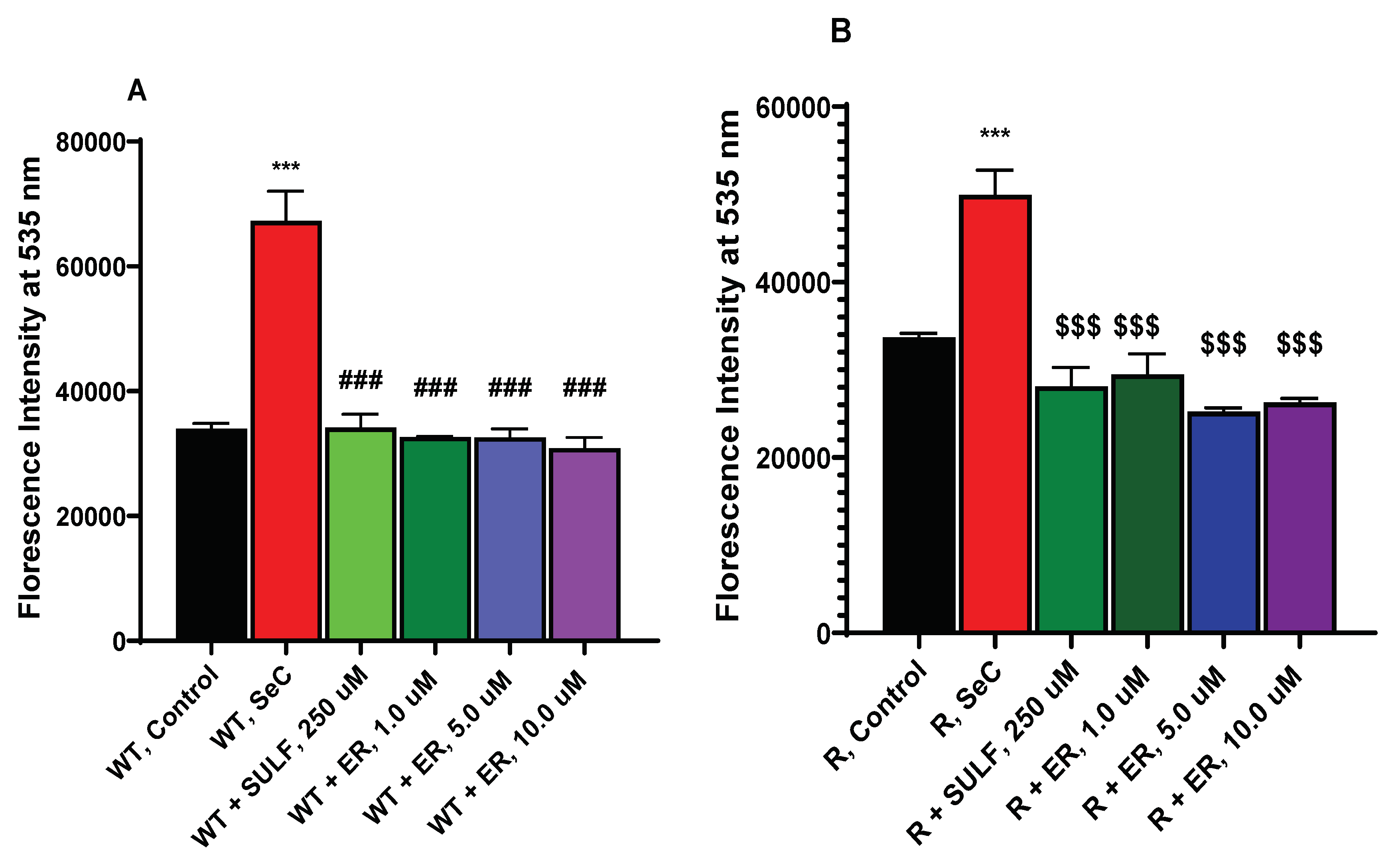
Figure 5.
Formation and detection of Erastin-dependent oxygen reactive species (ROS) in OVCAR-8 and NCI/ADR-RES cells following 4h incubations. ** and *** p values 0.005 and 0.001 respectively compared to untreated control.
Figure 5.
Formation and detection of Erastin-dependent oxygen reactive species (ROS) in OVCAR-8 and NCI/ADR-RES cells following 4h incubations. ** and *** p values 0.005 and 0.001 respectively compared to untreated control.
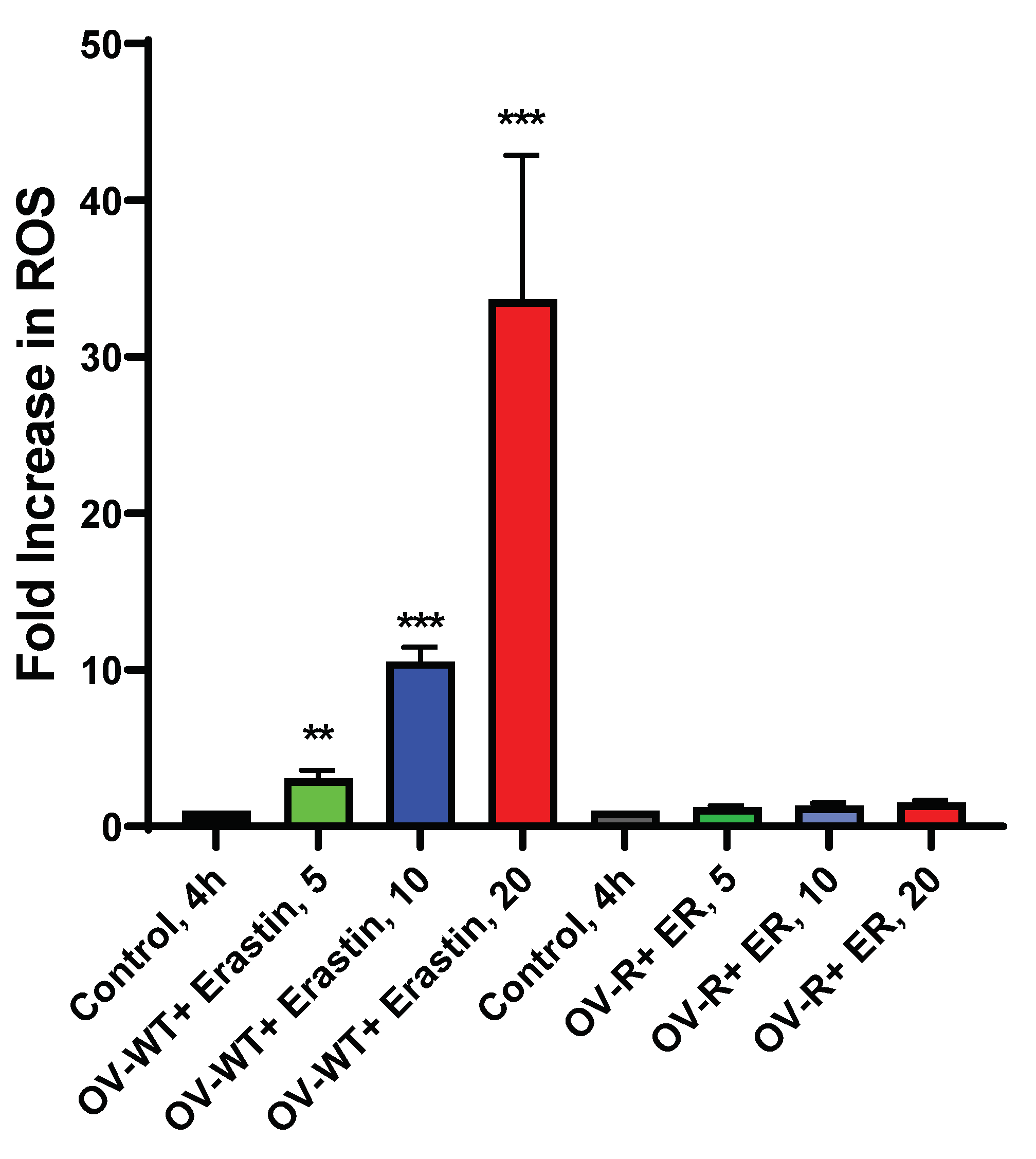
Figure 6.
Dose-dependence of ER-induced lipid peroxidation in OVCAR-8 and NCI/ADR-RES cells (A) and effects of RSL3 on MDA formation (B). The MDA formation was measured at 532mM. ** and *** p values 0.005 and 0.001 respectively compared to untreated control.
Figure 6.
Dose-dependence of ER-induced lipid peroxidation in OVCAR-8 and NCI/ADR-RES cells (A) and effects of RSL3 on MDA formation (B). The MDA formation was measured at 532mM. ** and *** p values 0.005 and 0.001 respectively compared to untreated control.
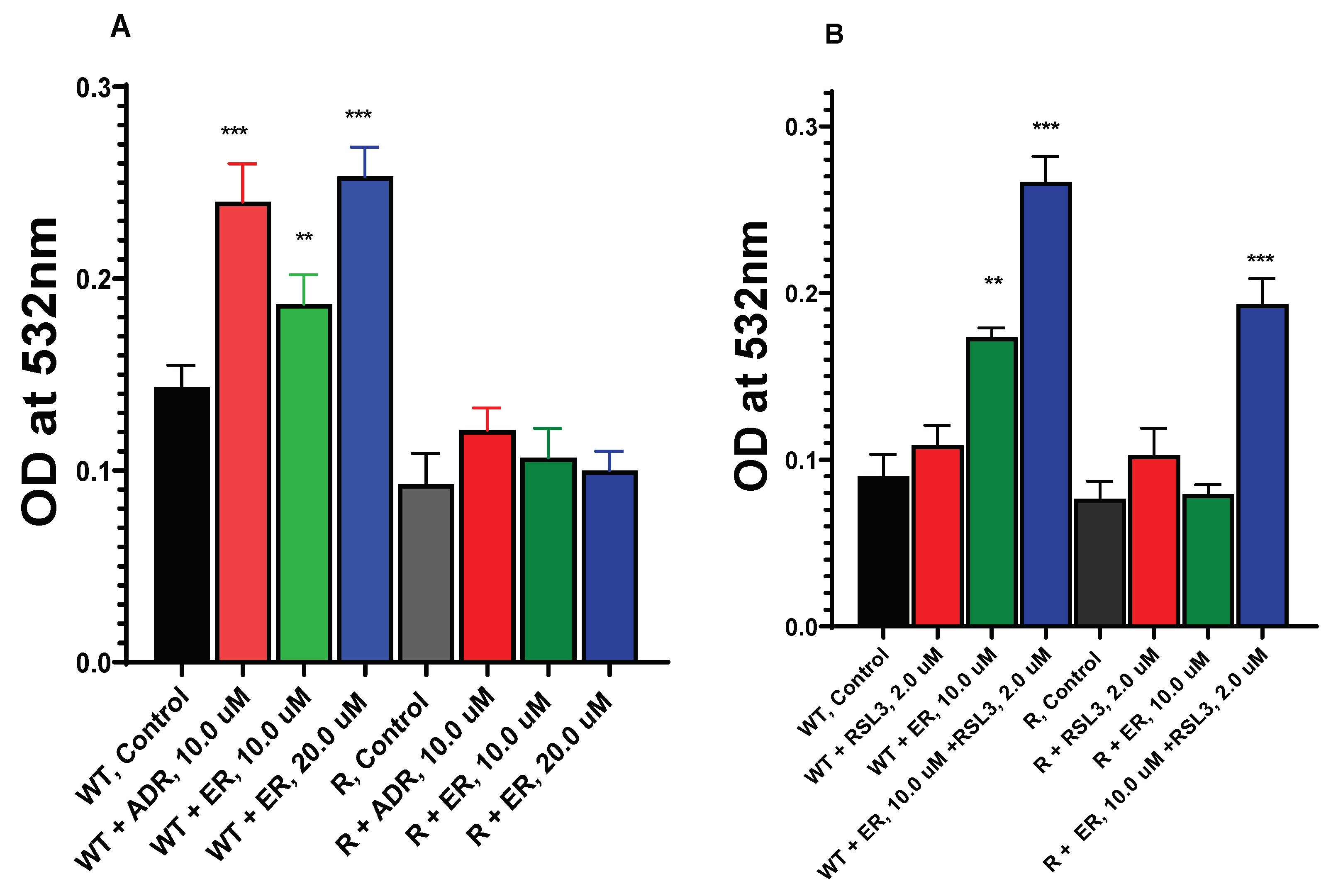
Figure 7.
Effects of N-acetyl Cysteine (NAC) on ER cytotoxicity in OVCAR-8 (A) and NCI/ADR-RES cells (B). The cells were incubated with 100 µM NAC for 30 min before adding ER for 24h. ** and *** p values 0.005 and 0.001 compared to untreated control. $$ and ###, p values 0.005 and 0.001, respectively compared to ER values alone.
Figure 7.
Effects of N-acetyl Cysteine (NAC) on ER cytotoxicity in OVCAR-8 (A) and NCI/ADR-RES cells (B). The cells were incubated with 100 µM NAC for 30 min before adding ER for 24h. ** and *** p values 0.005 and 0.001 compared to untreated control. $$ and ###, p values 0.005 and 0.001, respectively compared to ER values alone.
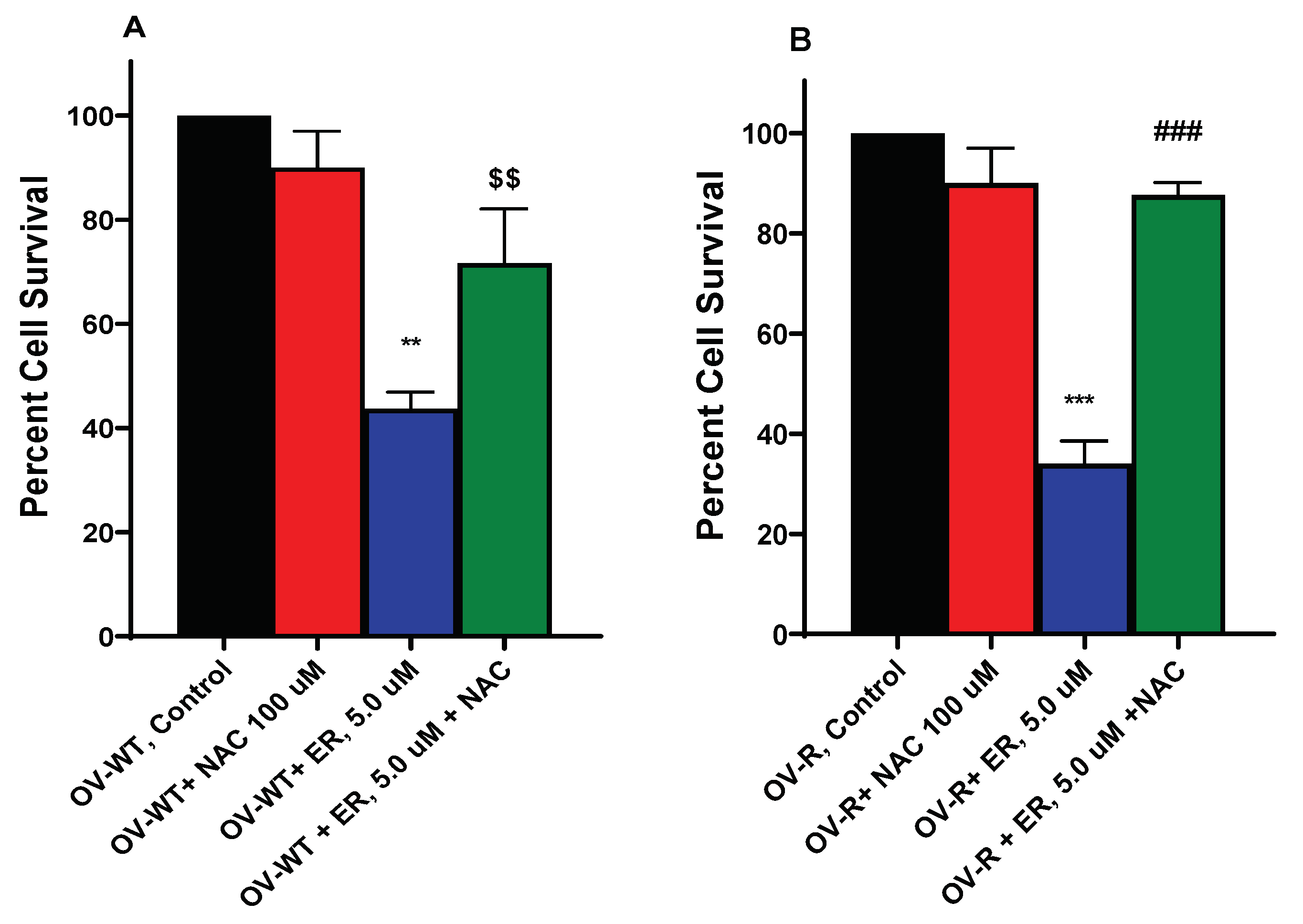
Figure 8.
Effects of Erastin (2.5µM) on various oxidative and ferroptosis-related genes in OVCAR-8 (A) and NCI/ADR-RES cells (B) cells following treatment with Erastin for 4h and 24h. Protein levels for GPX4 NrF2, and NOX4 following treatment with 2.5µM for 4 and 24h in OVCAR-8 and NCI/ADR-RES cells (C). Vinculin was used as the loading control. ** and *** p values < 0.005 and 0.001 respectively compared to control (β-Actin at 4h and 24h, respectively).
Figure 8.
Effects of Erastin (2.5µM) on various oxidative and ferroptosis-related genes in OVCAR-8 (A) and NCI/ADR-RES cells (B) cells following treatment with Erastin for 4h and 24h. Protein levels for GPX4 NrF2, and NOX4 following treatment with 2.5µM for 4 and 24h in OVCAR-8 and NCI/ADR-RES cells (C). Vinculin was used as the loading control. ** and *** p values < 0.005 and 0.001 respectively compared to control (β-Actin at 4h and 24h, respectively).
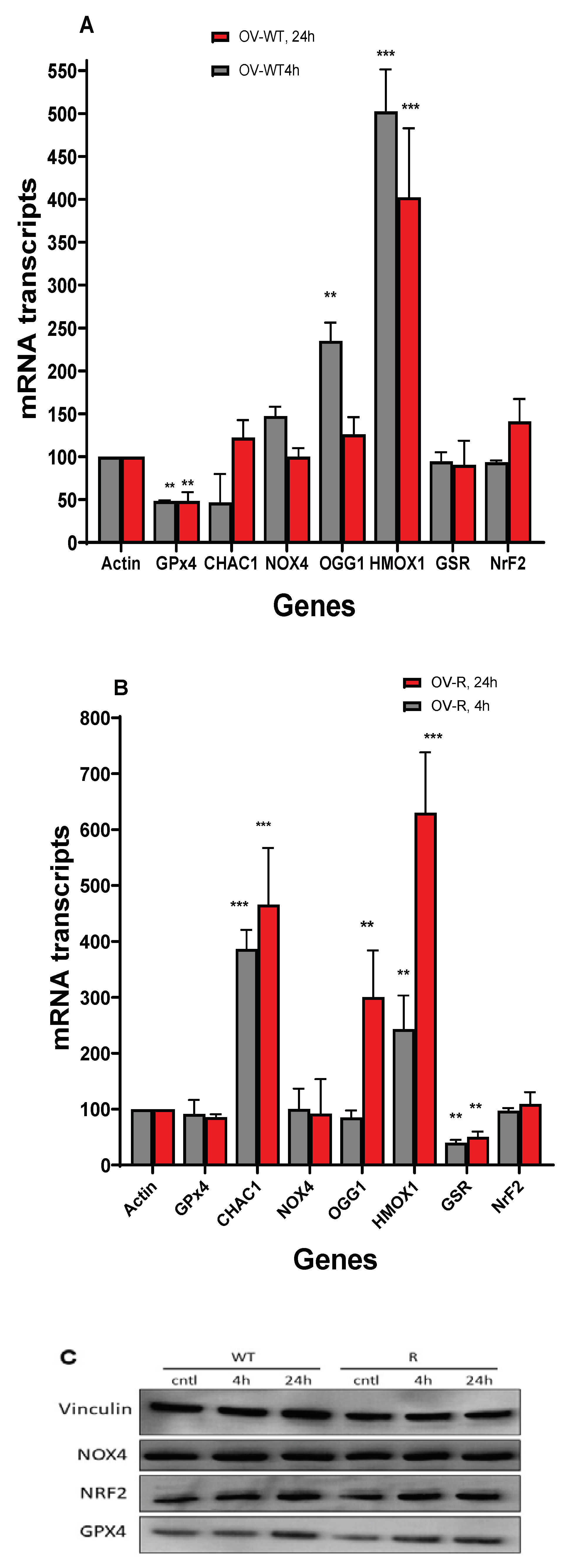
Figure 9.
Effects of Erastin on Xc- transporter, VDAC, CHAC1, HMOX1, iNOS and their implications in Erastin-induced lipid peroxidation and ferroptosis in (A) OVCAR-8 and (B) NCI/ADR-RES cells.
Figure 9.
Effects of Erastin on Xc- transporter, VDAC, CHAC1, HMOX1, iNOS and their implications in Erastin-induced lipid peroxidation and ferroptosis in (A) OVCAR-8 and (B) NCI/ADR-RES cells.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



