
Submitted:
28 February 2024
Posted:
28 February 2024
You are already at the latest version
Abstract
Coxsackievirus A6 (CV-A6) has emerged as the predominant causative agent of hand, foot, and mouth disease (HFMD) in young children. Since the declaration of coronavirus disease 2019 (COVID-19) as a global pandemic, the incidence of infectious diseases, including HFMD, has decreased markedly. When social mitigation was relaxed during the COVID-19 pandemic in 2022, re-emergence of HFMD was observed in Gwangju, South Korea, and seasonal characteristics of the disease appeared to have changed. To investigate the molecular characteristics of enterovirus (EV) associated with HFMD during 2022, 277 specimens were collected. Children aged under 5 years accounted for the majority of affected individuals. EV detection and genotyping were performed using real-time RT-PCR and nested RT-PCR followed by sequence analysis. EV detection rate was found to be 82.3%, and the main genotype identified was CV-A6. Sixteen CV-A6 samples were selected for whole genome sequencing. According to phylogenetic analysis, all CV-A6 strains from this study belonged to the sub-genotype D3 clade based on VP1 sequences. Analysis of 3D polymerase phylogeny showed that only the recombinant RF-A group was identified. In conclusion, circulating EV types should be continuously monitored to understand pathogen emergence and evolution during the post-pandemic era.
Keywords:
HFMD
; coxsackievirus A6
; molecular epidemiology
; phylogenetic tree
1. Introduction
Hand, foot, and mouth disease (HFMD) is an acute viral infectious disease that mostly affects in early childhood (children younger than 5 years of age). The typical symptoms of HFMD include fever, rashes, or blisters on the hands and feet, and mouth sores. HFMD is primarily caused by a group of human enteroviruses (HEVs), in particular HEV-A, including coxsackievirus (CV) A16 (CV-A16) and enterovirus A71 (EV-A71) [1]. Although CV-A16 and EV-A71 have historically been considered the leading pathogens causing HFMD across the Asia-Pacific region [2,3,4], CV-A6–related HFMD outbreaks have occurred frequently worldwide in recent years. Since the initial outbreak in Finland in 2008 [5], numerous CV-A6–related HFMD outbreaks have occurred in European countries [6,7,8], America [9], Singapore [10], Hong Kong [11], Japan [12], Thailand [13,14], and China [15,16,17]. Therefore, rapidly spreading HFMD cases caused by CV-A6 infections have markedly increased the public health burden. CV-A6 has gradually played a key role in the outbreak of HFMD, thereby replacing CV-A16 and EV-A71 as the major causative agent of HFMD; thus, CV-A6–associated HFMD has become an important part of enterovirus surveillance.
Since the announcement of coronavirus disease 2019 (COVID-19) as a global pandemic in early 2020, the incidence of viral infectious diseases, including HFMD, had decreased considerably. However, when social mitigation was relaxed in 2022, HFMD epidemics caused by CV-A6 infections were first detected in Gwangju during the COVID-19 pandemic. This study aimed to elucidate the molecular and epidemiological characteristics of CV-A6–associated HFMD cases diagnosed in surveillance hospitals across Gwangju in 2022. Toward this end, we analyzed CV-A6–positive samples using next-generation sequencing and described their genetic characteristics of CV-A6 in Gwangju.
2. Materials and Methods
2.1. Specimen Collection
Annual reports on HFMD are accessible to the public on the website of the Korea Disease Control and Prevention Agency (KDCA). Weekly national surveillance data on HFMD from 2019 to 2022 were obtained from the KDCA Infectious Disease Web Portal (https://www.kdca.go.kr/npt). This surveillance was conducted via a network of pediatric hospitals that reported all cases of EV infection, including HFMD. The Korea Enterovirus Surveillance System has been implemented by regional Health and Environment Research Institutes and medical institutions. We participated in a national surveillance program to monitor EV circulation in Gwangju, South Korea. All clinical specimens from patients presenting with HFMD were collected by the hospitals in the Gwangju area and sent weekly to the Institute of Health and Environment Research for detecting HEV.
2.2. Detection and Identification of CV-A6
HEV RNA was extracted from the specimens using the QIAamp viral RNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. To directly detect HEVs, real-time reverse transcription-PCR (RT-PCR) was performed firstly by using the human EV/EV71 multiplex real-time PCR kit (Kogenebiotech, Seoul, South Korea). Real-time RT-PCR amplifications were run on the 7500 Fast Real-Time PCR system (Applied Biosystems, CA, USA), and PCR was conducted according to the manufacturer’s instructions. Samples positive for HEV were subjected to nested RT-PCR for identification of the HEV genotypes. VP1 gene sequences were amplified using VP1 1st RT-PCR kit and VP1 2nd PCR kit (iNtRON Biotechnology, Gyeonggi, South Korea). All PCR products were subsequently sequenced in both forward and reverse directions. Obtained sequences were assembled using BioEdit software and then compared sequence homology with other enterovirus sequences in GenBank.
2.3. Complete Genome Sequencing
Six HEV samples underwent whole genome sequencing. A customized Ion AmpliSeq panel (Thermo Fisher Scientific, MA, USA) with two primer pools was designed following the manufacturer’s protocol to cover the entire human enterovirus genome. The HEV Ion AmpliSeq Custom Panel was designed with 567 amplicons, split into Pool 1 (284 amplicons) and Pool 2 (283 amplicons). The amplicon range is 125 to 375 bp, providing 99.94% coverage according to the reference genome.
cDNA synthesis, library preparation, templating, and sequencing were executed using the HEV Ion AmpliSeq Custom Panel and the Ion Torrent Genexus Integrated Sequencer, following the manufacturer’s instructions (Thermo Fisher Scientific, MA, USA). 6 HEV samples were sequenced per lane using Ion Torrent GX5 chip. Sequencing reads were processed, and their quality was assessed by using Genexus software version 6.6.2 Revision F.0 (Publication Number MAN0017910, Thermo Fisher Scientific, Waltham, MA, USA). A custom assay definition file was employed for initial QC, covering chip loading density, median read length, and the number of mapped reads. Notably, the Genexus system automates all QC and data analysis steps post-sequencing, including mapping, variant calling, and optional report generation in a single day. The sequencing reads were mapped on the reference genome of HEV using the Torrent Mapping Alignment Program (TMAP), and the variant calling was performed using the CLC Genome workbench. (Qiagen, Germany) The complete genomes were named CVA6/strain number/GJ/KOR/2022, and the genome sequences were deposited in NCBI GenBank (accession No PP191111-PP191126).
2.4. Phylogenetic Analyses
For the global strains, 67 VP1 sequences and 59 3D polymerase (3Dpol) sequences from Finland, France, Spain, Japan, and China were obtained from the NCBI GenBank database. Multiple genome sequences were aligned using Cluster-W algorithm in MEGA X software. A phylogenetic tree was constructed by the maximum likelihood method in MEGA X. The Kimura 2-parameter model with a variation rate among sites given by gamma distributed with invariant sites (G + I) was selected as a nucleotide substitution model. During phylogenetic tree construction, statistical support for tree nodes was assessed using 1,000 bootstrap replicates.
2.5. Mutation Analyses
A reference CV-A6 genome (KM114057), 7,423 nucleotides in length, was obtained from the NCBI GenBank and used to identify mutations in the genomes of Gwangju strains.
3. Results
According to the EV laboratory surveillance system in Gwangju in 2022, the number of manifestations suggestive of HFMD infection began to increase rapidly at week 32 and continued until week 38 (Figure 1). In total, 277 clinical specimens were collected from patients with HFMD. Of these, 148 were male (53.4%; 148/277) and 129 were female (46.6%; 129/277), with a male-to-female sex ratio of 1.15:1. Patient age ranged from three months to 15 years, although most patients were less than five years old. Of these, 251 (90.6%; 251/277) were under 5 years of age, of which 224 (80.9%; 224/277) were 1−4 years old and the other 27 (9.7%; 27/277) were less than one year old (Table 1). Among the 277 cases of HFMD with laboratory results, 93.5% (259/277) had fever. Respiratory symptoms, including pharyngodynia and digestive symptoms, were also reported in 5.4% (15/277) and 1.4% (4/277) patients, respectively (Table 1). Of the 277 specimens, 82.3% (228/277) were positive for HEV. The main genotype detected in the HEV-positive samples was CV-A6, which accounted for 95.6% cases (218/228), whereas the remaining 10 samples were not typed (4.4%; 10/228).
In total, 16 complete genomes of CV-A6 were obtained in this study. Recombination events were not observed. Phylogenetic analysis of CV-A6 was performed by aligning its genome sequences. The 16 CV-A6 strains in this study and other 67 global CV-A6 strains from GenBank were analyzed using the sequences of their VP1 capsid region. A recent study has showed that CV-A6 variants could be categorized into four major groups, namely, A–D genotypes, and that genotype D could be further divided into three sub-genotypes (D1–D3) using VP1 gene analysis [18]. Following the above classification, phylogenetic analysis revealed that CV-A6 strains from this study were all grouped under sub-genotype D3 (Figure 2). All Gwangju strains displayed close genetic relationships with the strains collected in China between 2020 and 2021.
To elucidate mutations at the amino acid level of the VP1 gene, we compared 16 sequences from this study with that of Finland strain (KM114057), which was reported as the sub-genotype D3 of CV-A6. Nineteen amino acid substitution positions were detected in the VP1 gene (A5T, S27N, T28I, V30A/D, T32S/N, S34Y, L35M, A45V, E47V/D, S97N/I, L98F, D99N, W104R, G110D, F111L, V112G, S137N, L215V, and T283A). All 16 strains harbored four mutations (A5T, S27N, S137N, and T283A).
CV-A6 strains were assigned recombinant forms (RFs) based on the 3Dpol coding region [11,19,20]. Accordingly, all 16 CV-A6 strains prevalent in Gwangju clustered within the previously assigned recombinant RF-A group (Figure 3). RF-A was first described in Finland in 2008 and variants within the RF-A group were subsequently detected in Asia. Based on the 3Dpol sequences, the recombinant variant RF-A was determined to be the predominant form in Gwangju. Taken together, we found that sub-genotype D3 and RF-A was the predominant genotype of CV-A6 circulating in Gwangju during the late summer and early autumn of 2022.
4. Discussion
HFMD outbreaks and epidemics have become a critical public health threat to younger children. In this study, the major population susceptible to HFMD in Gwangju includes children under five years of age. This is similar to the results of previous epidemiological studies on HFMD [21,22]. Our analysis of weekly reported number of HFMD cases revealed that the HFMD incidence commonly showed seasonal peaks, with a large peak from late spring to early summer, followed by a smaller peak in autumn. After the outbreak of COVID-19, the incidence of HFMD declined sharply and occurred rarely in South Korea in 2020 and 2021. However, in 2022, a resurgence of the HFMD epidemic was observed in Gwangju, and the seasonal characteristics changed. Compared to that observed in the previous year of 2019, the peak period shifted from May−June to August−October. The HFMD epidemic started in week 27 of 2022, whereas it started between weeks 19 and 20 of 2019, thereby revealing a delayed start of at least 8 weeks. In late summer and early fall of 2022, we identified a sharp increase in the number of HFMD cases in Gwangju. This was the first HFMD epidemic to occur since the COVID-19 pandemic.
Our laboratory surveillance revealed that this atypical epidemic was primarily caused by CV-A6 infection and that other types of EV were not present. The degree of sequence similarity in VP1 region at both the nucleotide and amino acid levels was used as a criterion for the identification and assignment of distinct EV subtypes. CV-A6 strains have been assigned to four genotypes, A- D, and sub-classified into seven sub-genotypes (B1-B2, C1-C2 and D1-D3) based on VP1 sequences. CV-A6 prototype strain Gdula (AY421764) from the United States formed a single branch that was denoted as genotype A. Genotypes B and C comprised few isolates from China and India, respectively. Sub-genotype D1 consisted of strains from Japan, Australia, Taiwan, Spain, and France. Sub-genotype D2 was primarily assigned to isolates from China, with the exception of one strain isolated from Japan. Sub-genotype D3 included strains from Finland, Taiwan, Spain, France, Germany, Australia, Japan, and China. In this study, molecular and phylogenetic analyses of VP1 sequences showed that all strains from Gwangju clearly belonged to the sub-genotype D3. Since 2008, the D3 strains have become predominant in many countries throughout the world [8,18]. Therefore, the transmission of the sub-genotype D3 may have been responsible for persistent global outbreaks of CV-A6–associated HFMD.
In addition, CV-A6 strains from Gwangju had 19 amino acid alterations including A5T, V30A, S137N, and T283A in the VP1 gene compared to Finland strain (KM114057). CV-A6 strains circulating in Guangxi from 2010–2017 also had six amino acid switches, namely, A5T, V30A, S137N, V174I, I242V, and T283A, which might be associated with CV-A6 related HFMD pandemic [23].
Nevertheless, only phylogenetic and molecular analyses based on the VP1 sequences have some limitations because of recombination. Recombination events are frequently observed within EV genomes and commonly occur in non-structural protein-coding regions of EVs. Among the non-structural protein domains, the 3Dpol coding region encodes RNA-dependent RNA polymerase and is essential for virus replication, which implies that recombination of the 3Dpol region may lead genetic diversity of EVs. According to the phylogenetic analysis of 3D polymerase sequences, CV-A6 strains have been assigned into RF-A to X [14]. Although recombination events were absent in the genomes obtained, RF-A was the prevalent recombinant variant in this study, thereby suggesting that D3/RF-A was the predominant form in Gwangju.
The detection of a single CV-A6 subclade may have been affected by the health regulations implemented during the COVID-19 period. Actually, extremely low levels of EVs were detected owing to strict public health interventions, such as school/kindergarten closure, social distancing, mandatory use of face masks, and maintenance of hand hygiene, in 2020 and 2021. Therefore, non-pharmaceutical interventions over the past 2 years may have reduced EV transmission and resulted in lack of immune stimulation. Reduction in the number of infected or immunized persons may lead to higher susceptibility to viral infections, including EV infections. The emergence of HFMD outbreaks during the COVID-19 period has been reported in other countries, including in France and Brazil in 2021 [21,24].
This study highlights the re-emergence of CV-A6–associated HFMD after the relaxation of social mitigation. Therefore, CV-A6 infections should be monitored continuously to understand the increased risk during the COVID-19 pandemic. Moreover, the genomic characteristics of CV-A6 should be shared and analyzed to enable the interpretation of the alterations in its transmissibility, infectivity, and pathogenicity. Therefore, continued surveillance of circulating EV types is required to monitor the pathogen spectrum of HFMD and epidemiological trend. Surveillance for HFMD may provide valuable information for the prevention and control of the disease. Further laboratory and clinical investigations are essential for managing HFMD. Public health interventions and control measures against COVID-19 have suppressed the occurrence of HFMD. Based on these findings, we concluded that the prevention of EV infections by following personal protective measures is important for reducing the impact of HFMD on health.
Author Contributions
Conceptualization, J.-E.L. and Y.-S.C.; Methodology, J.-E.L.; Formal Analysis, J.-E.L.; Investigation, M.K., M.L., S.H. J.K. S.K. and E.H.; Writing—original draft, J.-E.L.; Writing—review and editing, J.-E.L. and Y.-S.C.; Project administration, J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study was approved by the Institutional Review Board of the Korea Disease Control and Prevention Agency (approval number: 2018-08-02-2C-A).
Informed Consent Statement
Patient consent was waived in accordance with institutional IRB policies, based on national surveillance and diagnostic data.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We thank the cooperative hospitals that helped us monitor enterovirus cases in Gwangju.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Oberste, M.S.; Peñaranda, S.; Maher, K.; Pallansch, M.A. Complete genome sequences of all members of the species Human enterovirus A. Journal of General Virology 2004, 85, 1597-1607. [CrossRef]
- Zhang, Y.; Tan, X.-J.; Wang, H.-Y.; Yan, D.-M.; Zhu, S.-L.; Wang, D.-Y.; Ji, F.; Wang, X.-J.; Gao, Y.-J.; Chen, L. An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. Journal of Clinical Virology 2009, 44, 262-267. [CrossRef]
- Xing, W.; Liao, Q.; Viboud, C.; Zhang, J.; Sun, J.; Wu, J.T.; Chang, Z.; Liu, F.; Fang, V.J.; Zheng, Y. Hand, foot, and mouth disease in China, 2008–12: an epidemiological study. The Lancet infectious diseases 2014, 14, 308-318. [CrossRef]
- Zhang, Y.; Wang, D.; Yan, D.; Zhu, S.; Liu, J.; Wang, H.; Zhao, S.; Yu, D.; Nan, L.; An, J. Molecular evidence of persistent epidemic and evolution of subgenotype B1 coxsackievirus A16-associated hand, foot, and mouth disease in China. Journal of clinical microbiology 2010, 48, 619-622. [CrossRef]
- Österback, R.; Vuorinen, T.; Linna, M.; Susi, P.; Hyypiä, T.; Waris, M. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerging infectious diseases 2009, 15, 1485. [CrossRef]
- Montes, M.; Artieda, J.; Pineiro, L.D.; Gastesi, M.; Diez-Nieves, I.; Cilla, G. Hand, foot, and mouth disease outbreak and coxsackievirus A6, northern Spain, 2011. Emerging infectious diseases 2013, 19, 676. [CrossRef]
- Sinclair, C.; Gaunt, E.; Simmonds, P.; Broomfield, D.; Nwafor, N.; Wellington, L.; Templeton, K.; Willocks, L.; Schofield, O.; Harvala, H. Atypical hand, foot, and mouth disease associated with coxsackievirus A6 infection, Edinburgh, United Kingdom, January to February 2014. Eurosurveillance 2014, 19, 20745. [CrossRef]
- Tomba Ngangas, S.; Bisseux, M.; Jugie, G.; Lambert, C.; Cohen, R.; Werner, A.; Archimbaud, C.; Henquell, C.; Mirand, A.; Bailly, J.-L. Coxsackievirus A6 recombinant subclades D3/A and D3/H were predominant in hand-foot-and-mouth disease outbreaks in the paediatric population, France, 2010–2018. Viruses 2022, 14, 1078. [CrossRef]
- Control, C.f.D.; Prevention. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6-Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR. Morbidity and mortality weekly report 2012, 61, 213-214.
- Wu, Y.; Yeo, A.; Phoon, M.; Tan, E.; Poh, C.; Quak, S.; Chow, V.T. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. International Journal of Infectious Diseases 2010, 14, e1076-e1081. [CrossRef]
- Lau, S.K.; Zhao, P.S.; Sridhar, S.; Yip, C.C.; Aw-Yong, K.L.; Chow, E.Y.; Cheung, K.C.; Hui, R.W.; Leung, R.Y.; Lai, Y.S. Molecular epidemiology of coxsackievirus A6 circulating in Hong Kong reveals common neurological manifestations and emergence of novel recombinant groups. Journal of Clinical Virology 2018, 108, 43-49. [CrossRef]
- Fujimoto, T.; Iizuka, S.; Enomoto, M.; Abe, K.; Yamashita, K.; Hanaoka, N.; Okabe, N.; Yoshida, H.; Yasui, Y.; Kobayashi, M. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerging infectious diseases 2012, 18, 337. [CrossRef]
- Puenpa, J.; Chieochansin, T.; Linsuwanon, P.; Korkong, S.; Thongkomplew, S.; Vichaiwattana, P.; Theamboonlers, A.; Poovorawan, Y. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerging infectious diseases 2013, 19, 641. [CrossRef]
- Puenpa, J.; Saengdao, N.; Khanarat, N.; Korkong, S.; Chansaenroj, J.; Yorsaeng, R.; Wanlapakorn, N.; Poovorawan, Y. Evolutionary and Genetic Recombination Analyses of Coxsackievirus A6 Variants Associated with Hand, Foot, and Mouth Disease Outbreaks in Thailand between 2019 and 2022. Viruses 2022, 15, 73. [CrossRef]
- Han, J.-F.; Xu, S.; Zhang, Y.; Zhu, S.-Y.; Wu, D.-L.; Yang, X.-D.; Liu, H.; Sun, B.-X.; Wu, X.-Y.; Qin, C.-F. Hand, foot, and mouth disease outbreak caused by coxsackievirus A6, China, 2013. Journal of Infection 2014, 69, 303-305. [CrossRef]
- Zeng, H.; Lu, J.; Zheng, H.; Yi, L.; Guo, X.; Liu, L.; Rutherford, S.; Sun, L.; Tan, X.; Li, H. The epidemiological study of coxsackievirus A6 revealing hand, foot and mouth disease epidemic patterns in Guangdong, China. Scientific reports 2015, 5, 10550. [CrossRef]
- Zhang, M.; Chen, X.; Wang, W.; Li, Q.; Xie, Z. Genetic characteristics of Coxsackievirus A6 from children with hand, foot and mouth disease in Beijing, China, 2017–2019. Infection, Genetics and Evolution 2022, 106, 105378. [CrossRef]
- Song, Y.; Zhang, Y.; Ji, T.; Gu, X.; Yang, Q.; Zhu, S.; Xu, W.; Xu, Y.; Shi, Y.; Huang, X. Persistent circulation of Coxsackievirus A6 of genotype D3 in mainland of China between 2008 and 2015. Scientific reports 2017, 7, 5491. [CrossRef]
- Gaunt, E.; Harvala, H.; Österback, R.; Sreenu, V.B.; Thomson, E.; Waris, M.; Simmonds, P. Genetic characterization of human coxsackievirus A6 variants associated with atypical hand, foot and mouth disease: a potential role of recombination in emergence and pathogenicity. The Journal of general virology 2015, 96, 1067. [CrossRef]
- Puenpa, J.; Vongpunsawad, S.; Österback, R.; Waris, M.; Eriksson, E.; Albert, J.; Midgley, S.; Fischer, T.K.; Eis-Hübinger, A.M.; Cabrerizo, M. Molecular epidemiology and the evolution of human coxsackievirus A6. Journal of General Virology 2016, 97, 3225-3231. [CrossRef]
- Carmona, R.C.; Machado, B.C.; Reis, F.C.; Jorge, A.M.; Cilli, A.; Dias, A.M.; Morais, D.R.; Leme, L.; Ana, L.; Silva, M.R. Hand, foot, and mouth disease outbreak by Coxsackievirus A6 during COVID-19 pandemic in 2021, São Paulo, Brazil. Journal of Clinical Virology 2022, 154, 105245. [CrossRef]
- Ji, T.; Han, T.; Tan, X.; Zhu, S.; Yan, D.; Yang, Q.; Song, Y.; Cui, A.; Zhang, Y.; Mao, N. Surveillance, epidemiology, and pathogen spectrum of hand, foot, and mouth disease in mainland of China from 2008 to 2017. Biosafety and Health 2019, 1, 32-40. [CrossRef]
- Chen, M.; Zuo, X.; Tan, Y.; Ju, Y.; Bi, F.; Wang, H.; Chen, M. Six amino acids of VP1 switch along with pandemic of CV-A6-associated HFMD in Guangxi, southern China, 2010–2017. Journal of Infection 2019, 78, 323-337. [CrossRef]
- Mirand, A.; Cohen, R.; Bisseux, M.; Tomba, S.; Sellem, F.C.; Gelbert, N.; Béchet, S.; Frandji, B.; Archimbaud, C.; Brebion, A. A large-scale outbreak of hand, foot and mouth disease, France, as at 28 September 2021. Eurosurveillance 2021, 26, 2100978. [CrossRef]
Figure 1.
Weekly number of HFMD cases reported by the sentinel surveillance from the Korea Disease Control and Prevention Agency (KDCA), 2019−2022 (n = 3,479), and collected clinical samples with HFMD from enterovirus laboratory surveillance system in Gwangju, 2022 (n = 277).
Figure 1.
Weekly number of HFMD cases reported by the sentinel surveillance from the Korea Disease Control and Prevention Agency (KDCA), 2019−2022 (n = 3,479), and collected clinical samples with HFMD from enterovirus laboratory surveillance system in Gwangju, 2022 (n = 277).
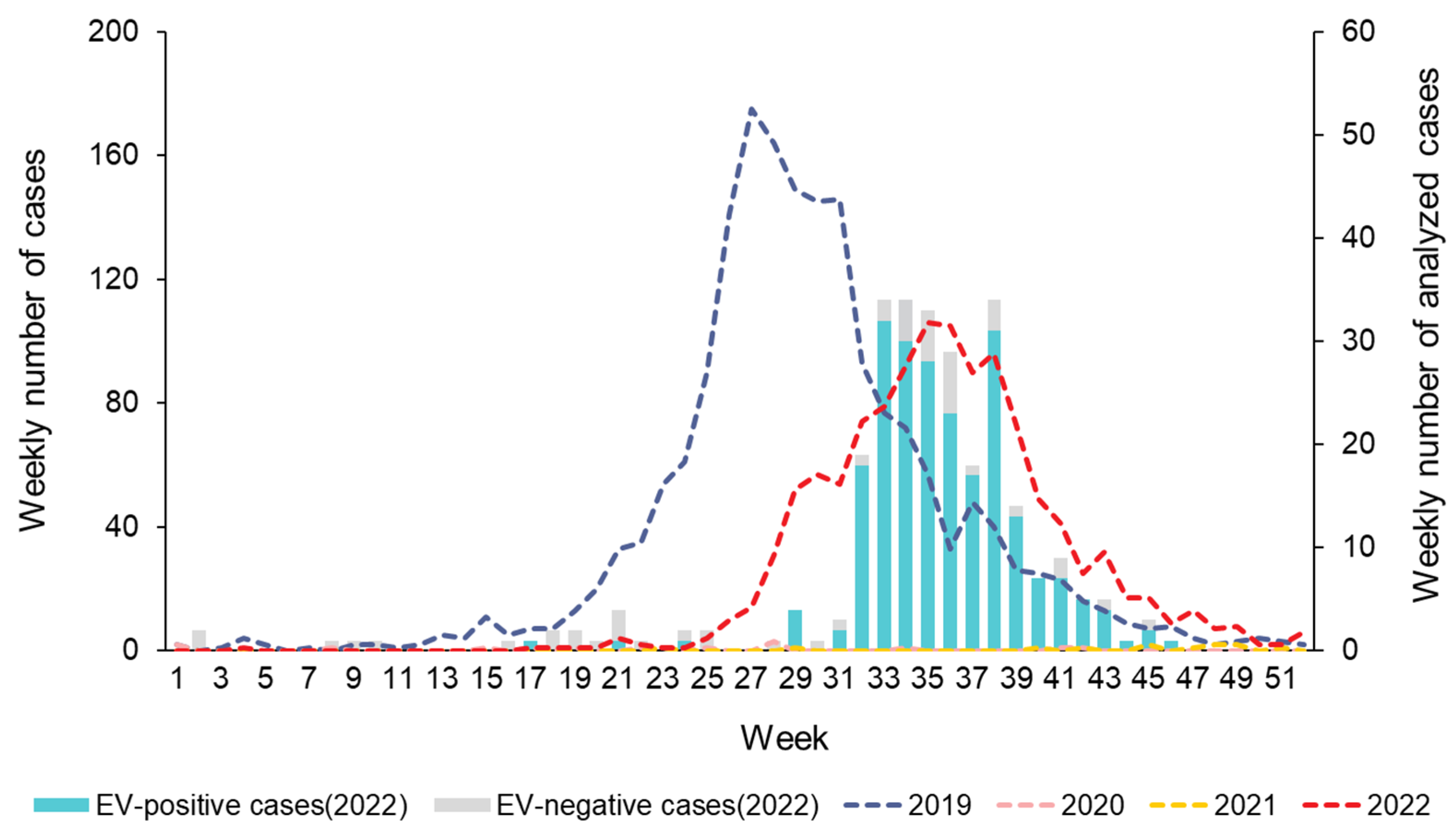
Figure 2.
Phylogenetic tree of partial VP1 region with CV-A6 sequences. Tree was constructed using the maximum likelihood method with the Kimura-2 parameter model of MEGA X. Bar denotes the evolutionary distance according to the number of nucleotide substitutions per site. Bootstrap was performed with 1,000 replicates. Bootstrap values lower than 70% are not shown. For clarity, the 16 CV-A6 sequences obtained in this study are highlighted with red dots.
Figure 2.
Phylogenetic tree of partial VP1 region with CV-A6 sequences. Tree was constructed using the maximum likelihood method with the Kimura-2 parameter model of MEGA X. Bar denotes the evolutionary distance according to the number of nucleotide substitutions per site. Bootstrap was performed with 1,000 replicates. Bootstrap values lower than 70% are not shown. For clarity, the 16 CV-A6 sequences obtained in this study are highlighted with red dots.

Figure 3.
Phylogenetic tree of partial 3Dpol region with CV-A6 sequences. Tree was constructed using the maximum likelihood method with the Kimura-2 parameter model of MEGA X. Bar denotes the evolutionary distance according to the number of nucleotide substitutions per site. Bootstrap was performed with 1,000 replicates. Bootstrap values lower than 70% are not shown. For clarity, the 16 CV-A6 sequences obtained in this study are highlighted with red dots.
Figure 3.
Phylogenetic tree of partial 3Dpol region with CV-A6 sequences. Tree was constructed using the maximum likelihood method with the Kimura-2 parameter model of MEGA X. Bar denotes the evolutionary distance according to the number of nucleotide substitutions per site. Bootstrap was performed with 1,000 replicates. Bootstrap values lower than 70% are not shown. For clarity, the 16 CV-A6 sequences obtained in this study are highlighted with red dots.
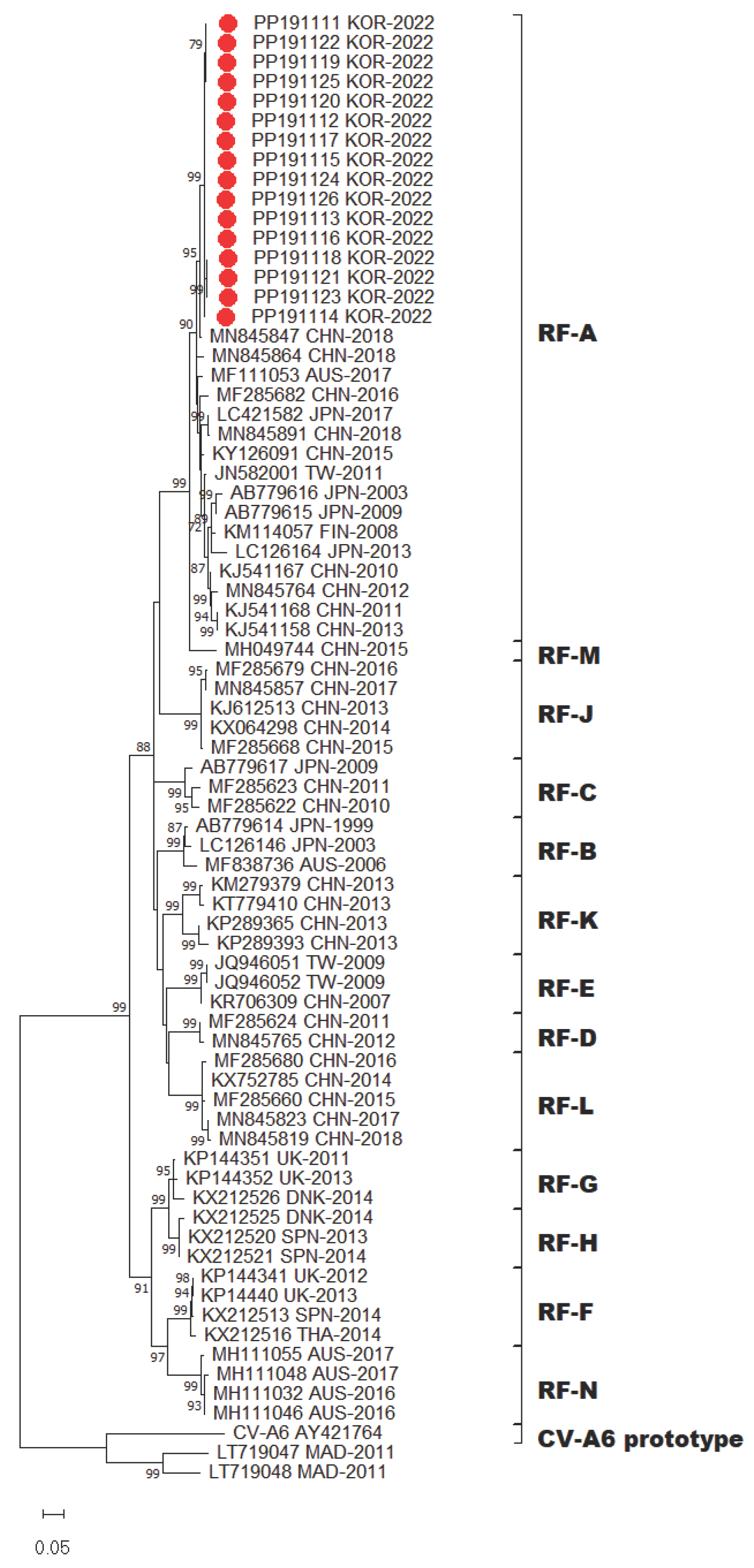

Table 1.
Demographic characteristics of hand, foot, and mouth disease cases, Gwangju, 2022.
| Total (n = 277) | % | ||
|---|---|---|---|
| Gender | Male | 148 | 53.4 |
| Female | 129 | 46.6 | |
| Age group (years) |
<1 | 27 | 9.7 |
| 1 to 4 | 224 | 80.9 | |
| 5 to 9 | 21 | 7.6 | |
| >9 | 5 | 1.8 | |
| Symptoms | Fever | 259 | 93.5 |
| Respiratory | 15 | 5.4 | |
| Digestive | 4 | 1.4 | |
| Unknown | 10 | 4.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



