
Submitted:
28 February 2024
Posted:
28 February 2024
You are already at the latest version
Abstract
Recently, the composite materials consisting of ionic liquids (ILs) and metal-organic frameworks (MOFs) have attracted a great of attention due to their fantastic properties. Many theoretical studies have been performed towards its special structures and applications. Yet, the mechanism for the diffusion of ILs inside MOFs channels still remain unclear. Here, the DFT calculations together with frontier orbital analysis, natural charge analysis, and energy decomposition analysis were performed to investigate the diffusion behavior of a typical IL, [C4mim][PF6], into the ZIF-8 SOD cage. The potential energy surface (PES) profiles indicate that it is quite difficult for the cation [C4min]+ to diffuse into the cage of ZIF-8 through the pristine pores because of its large imidazole steric hindrance. Moreover, the PES reveals that a success diffusion could be obtained by the thermal contributions, by which the pore size is enlarged through the swing effects at a higher temperature. Subsequently, electronic structure analyses reveal that the main interactions between [PF6]-or [C4mim]+ and ZIF-8 is the steric repulsion interactions. Finally, the effects of amounts of [C4mim][PF6]on the ZIF-8 were investigated, and the results show that two pairs of [C4mim][PF6] per SOD cage is more reasonable in terms of interaction energies and structural changes.
Keywords:
ionic liquids
; ZIF-8
; density functional theory
1. Introduction
Recently, the composite materials consisting of ionic liquids (ILs) and metal-organic frameworks (MOFs) have attracted considerable attention because of their exceptional properties1, 2, therefore, they have been used in many important fields, e.g. the gas adsorption and separation, catalysis, and sensors3-10. In particular, the large number of adsorption sites of MOFs with ILs play an important role in the mixture gas separation (e.g. CO2/CH4) 11-14. For example, Ban et al15 loaded [C4mim][TF2N] into the cages of ZIF-8, and the results showed the adsorption selectivity for CO2/CH4 significantly increased from 7.5 to 41. Following this work, Kinik et al.16 examined the [C4mim][PF6]/ZIF-8 system via the density-functional theory (DFT) and Monte Carlo (MC) simulations. The authors found that the interactions between [C4mim][PF6] and ZIF-8 created certain new adsorption sites, which increases the ideal selectivity for CO2/N2 from 7.82 to 24.21. Burak et al.17 obtained a similar result by incorporating [C4mim][BF4] into ZIF-8, of which the selectivity of 13.3 has been obtained for CO2/N2. Afterwards, a large number of systems have been reported with different combination of ILs and MOFs18.
In general, the reasons for ILs@MOFs composites materials with high gas separation or selectivity have been attributed into the two main aspects. On the one hand, the rich hydrogen bonds network of ILs19, 20 could effectively dissolve CO2 molecules against other gases, e.g. CH4 or N2,21 hence, it improves the separation performance compared to the pure MOF materials22-24. For example, Thomas et al25 performed DFT and Giant Canonical Monte Carlo (GCMC) simulations to investigate the selectivity for a series of IL@ZIF-8 systems. Their findings reveal that the hydrophobic fluorinated anion (e.g. [BF4]-, [Tf2N]-, and [PF6]-) exhibit higher selectivity compared to hydrophilic non-fluorinated anions (e.g. [NO2]-, [NO3]-, [SCN]-). Kavak et al.26 investigated the CO2 separation efficiency of five different ILs encapsulated MIL-53 systems, and they found that the CO2/CH4 separation selectivity of [C4mim][PF6]/MIL-53 was 2.8 times higher compared to the pure MIL-53. Zhang et al.27 conducted an investigation on ILs with three different cation functional groups, revealing that ILs containing amino functional groups exhibited the highest CO2 molecule affinity and CO2/CH4 adsorption selectivity.
On the other hand, it is believed that the introduction of ILs could also modify the structures of the MOFs, and increase the gas selectivity, subsequently. Based on the experimental techniques (e.g. high pressure X-ray diffraction, XRD) with molecular simulations, Fairen et al.28 demonstrated that the imidazolate linker of the ZIF-8 changed because of the swing effects, and change the gas selectivity, subsequently.29 Uzun and colleagues30 coated 1-(2-hydroxyethyl)-3-methylimidazolium dicyanamide ([Hemim][DCA]) on the ZIF-8 surface to eliminate the nonselective voids, and authors found that the CO2 selectivity was increased by 45 times compared to pure ZIF-8. Chang et al.31 successfully constructed an zwitterionic MOF material (MOF UiO-66-SO3--NH3+), and the adsorption capacity for CO2 increased by 32-48%, because the positive and negative charges on the modified MOF material interacts with the ILs of [Emim][SCN]. Our previous theoretical studies 32, 33 revealed that the ZIF-8 aperture configurations have significant impact on the separation of CO2/CH4. Specific speaking, the DFT calculations showed that the pristine ZIF-8 aperture (with a pore size of 3.4 Å) exhibits the best separation performance, which has the largest energy barrier difference between CO2 and CH4, and the MD simulations revealed that ILs (e.g. [C4mim][PF6]) could effectively maintain pristine aperture configurations, and remain the high separation properties.
Yet, there is still an open question following the reported mechanistic studies, that is, how does the ILs diffusion into the cage of MOFs (e.g. ZIF-8), and the how many ILs could be accommodated per cage. For this vein, we performed DFT calculations to investigate the diffusion behavior of ILs into MOFs cages, by taking [C4mim][PF6] and ZIF-8 as an example. Firstly, the potential energy surface (PES) scan were performed to study the passing of [C4mim]+ and [PF6]- through different ZIF-8 apertures. Secondly, several electronic analysis method, including frontier orbital energies, natural charge analysis, and energy decomposition analysis, were used to investigate the interactions between [C4mim]+/[PF6]- and ZIF-8. Lastly, a series of ILs encapsulated ZIF-8 with different loading amount were investigated to obtain stabilities of ionic pairs in the SOD cage of ZIF-8.
2. Computational Details
The crystal structure of the ZIF-8 was obtained from the Cambridge Crystallographic Data Centre (CCDC) Crystal Library21, and the simplified SOD cage presentation is depicted in Figure 1a. We choose the five typical pore apertures of the ZIF-8 structure as the result of the rotation of the zinc-imidazole-zinc and methyl functional groups, as described by our previous work, 32, including "Pristine (3.4 Å)", "Closed" (4.1 Å), "Semi-open (4.2 Å)", "Closed (4.7 Å)", "Open" (6.9 Å), respectively, which are shown in Figure 1b – Figure 1f.
As demonstrated in Figure 2a, the [PF6]- and [C4mim]+ were firstly placed at 10 Å away from the aperture of the ZIF-8, then gradually (e.g. with a step size of 0.1 Å), penetrate passed through the center of the aperture (in other words, the center of mass, COM). In principle, [PF6]- and [C4mim]+ might have many orientations when pass through the five pore structures as shown in Figure 1. Here, we considered only the orientations which have possible low energy barriers. Summarizing in Figure 2 (b), three different orientations of [PF6]- have been studied, denoted as case I, II and III, with one, two, three fluorine atoms (F) towards the aperture, respectively. For the [C4mim]+, one case was chosen towards the aperture, of which the imidazole ring is almost vertical to the aperture.
With above-described models, rigid PES scans with single point (SP) energy calculations were performed, where all atoms in the aperture of ZIF-8 and [PF6]-/[C4mim]+ were kept frozen. Afterward, we performed the relaxation PES scan within the regions nearby the highest energy barrier positions from rigid PES scans (e.g. ca. -2 Å to 2 Å). In these cases, the atoms in the aperture of ZIF-8 were kept frozen, while [PF6]-/[C4mim]+ were fully optimized. Both PES scans were carried out with the Gaussian16 software package41. Subsequently, the orbital energies and charge transfer were computed by natural population analysis (NPA)34 using the Multiwfn35 software package, and visualized using the VESTA36 software package. To quantitatively study the interactions between [PF6]-/[C4mim]+ and ZIF-8, the energy decomposition analysis were performed based on the generalized Kohn-Sham energy density analysis (GKS-EDA) method37, which is implemented in the GAMESS program package 38. All above-mentioned DFT calculations were performed at the B3LYP level of theory, and a basis set of 6-311+G*39, together with the damping scheme of Becke and Johnson, which denoted as DFT-D3 (BJ)40, 41.
Lastly, to study how many pairs are stable inside the SOD cage of ZIF-8, the periodic structures of ILs@ZIF-8 were examined by the BLYP functional with the dispersion correction scheme (DFT-D3). The geometry optimizations were performed to account for both changes in atomic positions and lattice dimensions, which were updated with the efficient L-BFGS algorithm. Within the DFT calculations, the DZVP-MOLOPT-SR-GTH basis set was adopt for Zn atoms, while the other atoms used the TZVP-MOLOPT -GTH basis set. The energy cutoff set to 400 Ry, and the energy convergence for the self-consistent field (SCF) calculation was set to 1 × 10-5 Hartree. All those periodic calculations were performed by employing the CP2K’s Quickstep module42.
3. Results and Discussion
The PES scan of the [C4mim]+ and [PF6]- passing through the pristine ZIF-8 aperture (with a pore size of 3.4 Å) were shown in Figure 3a. It is shown that both [C4mim]+ and [PF6]- almost cannot pass through the pore, because of their large energy barriers. For [C4mim]+, an energy barrier of 135. 5 kcal mol-1 were obtained, and the corresponding structure is the one when the COM of imidazole is almost overlap with COM of the pristine ZIF-8 aperture. For [PF6]-, three cases have been considered, as shown in Figure 2b. The lowest energy barrier, which is 69.17 kcal mol-1, was found for the case III, in which three F atoms pointed to the plane of the ZIF-8 apertures. This finding could be explained by their molecular diameters, in which case I has a diameter of 3.29 Å, case II has a diameter of 3.20 Å, and case III has a diameter of 2.94 Å (Figure S1). Meanwhile, as shown in Figure S2, the results of the energy decomposition analysis show that the repulsive interactions between the [PF6]- and ZIF-8 is the smallest when [PF6]- adopted the case III. After obtaining the configurations with the highest energy barriers, we then refined the PES with the relax scan (see Figure 3b), and we found that both [C4mim]+ and [PF6]- still have relative high energy barriers, being 39.87 kcal mol-1 and 16.61 kcal mol-1, respectively, which still prohibited [C4mim][PF6] diffusing into the SOD cage of ZIF-8 at finite temperatures, e.g. the room temperature. In details, only one energy barrier was found when [PF6]- moved from the left to the right side of the pore, and the highest energy point is corresponding to the structure of which the P atom is locating at the plane of the pristine ZIF-8, with three F atom locating at each side (see Figure S3a). The average distance between the H atoms of ZIF-8 and the F atom of the [PF6]- is computed to be 2.52 Å. While for the [C4mim]+, two energy barriers were found when [C4mim]+ moved from left to the right of the pristine ZIF-8 aperture. The first one, 14.60 kcal·mol-1, is corresponding to passing of alkyl chain, in which the average distance between alkyl chain and ZIF-8 aperture is 2.61 Å. The second one, 39.87 kcal mol-1, is corresponding to the passing of imidazole ring, in which the average distance between imidazole ring and ZIF-8 aperture is only 1.66 Å (Figure S3b), hence, such small distances result in large steric hindrances or strong repulsion interactions.
Afterwards, the frontier orbitals, including the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were plotted in Figure 4. In general, the results indicate that there is no significant orbital interactions between the [C4mim]+/[PF6]- and the pristine ZIF-8 aperture within the frontier orbital scheme. For example, both LUMOs and HOMOs of [C4mim]+-ZIF-8 and [PF6]--ZIF-8 are almost the same compare to that of the free pristine ZIF-8 aperture, in other words, the frontier orbitals are mainly consisted by the contributions from ZIF-8 aperture. The HOMO-LUMO gaps of [C4mim]+-ZIF-8 and [PF6]--ZIF-8 are also identical to that of the free pristine ZIF-8 aperture, e.g. 0.64 eV versus 0.65 eV. Moreover, the natural population analysis (NPA) was performed and the natural charges as summarized in Table S1. The values show that certain amount of charges (ca. 0.08 e) have been transferred between [C4mim]+ and [PF6]- and the pristine ZIF-8 apertures.
To have a deep understanding of the interactions between [C4mim]+/[PF6]- and the pristine ZIF-8 aperture, the GKS-EDA energy decomposition analysis has been performed based on the [PF6]--ZIF-8(3.4 Å) and [C4mim]+-ZIF-8(3.4 Å) structures and the results are plotted in Figure 5. Generally, the interaction energies are decomposed according to following equations:
where , , , , , and represent exchange, repulsion, correlation, electrostatic, polarization, and dispersion components, respectively. As shown in Figure 5, the interaction energies are dominant by three terms, including the repulsion, exchange and electrostatic energies, of which the repulsions are the main reasons for the high energy barriers shown in the Figure 3. The rest components are relatively small (e.g. less than 28 kcal mol-1 in absolute values, and with a total ratio being less than 6.6%). Here, we take [C4mim]+-ZIF-8(3.4 Å) as an example, and discuss those three terms individually: 1) The repulsion component. This term is responsible for steric repulsion, and it consists of the destabilizing interactions between occupied orbitals of the fragments. Usually, this term is repulsive (with a positive computed value). Here, a value of 304.27 kcal mol-1 was obtained, which can be attributed to large steric repulsion between imidazole ring of [C4mim]+ and the pristine ZIF-8 aperture. As discussed in the PES scan section, the pristine ZIF-8 aperture has a pore size of only 3.4 Å, hence, leaving the imidazole ring few spaces to pass through; 2) The exchange component. This term essentially is related to electrons with the same spin exchange their positions in degenerate orbitals, to increase stability of electronic structure states. Here, we found an value of -162.2 kcal mol-1, in the case of [C4mim]+-ZIF-8; 3) The electrostatic component. This term is the energy between the unperturbed charge distributions of the prepared fragments, which is usually attractive. As shown in Figure 5, the computed electrostatic interaction is -50 kcal mol-1, which is much small compare to the repulsion and exchange interactions. This finding is somehow consistent with the NPA charge analysis, in which the pristine ZIF-8 is slightly charged (ca. 0.08 e) via the charge transfer from [C4mim]+.
Our previous MD simulations showed that the pristine ZIF-8 aperture could be distorted because of the thermal oscillations32. When the simulations were performed at a temperature of 300 K, an average of 6.8 Å movements was obtained for the atoms defined the size and shape of pores, which are highlighted in Figure 1a. Following that study, here, we computed the PES scan of [C4mim]+ and [PF6]- passing through the ZIF-8 aperture with the pore size of 4.1 Å, 4.2 Å, 4.7 Å, 6.9 Å, respectively, (the pore structures are depicted in Figure 1b ~ Figure 1e), and the results are summarized in Figure 6. Generally, the results show that the [PF6]- is able to freely pass through these pores with energies barriers under zero. However, [C4mim]+ can only pass through the pore with a diameter of 6.9 Å. Together with our previous room temperature MD simulations, we conclude that a higher temperature might be needed to obtain larger swing effect and to encapsulate [C4mim][PF6] into the ZIF-8 SOD cage. These findings are rather qualitatively consistent with a reported experiment study, in which the authors employed a two-step adsorption/infiltration method to incorporate [C4mim][PF6] into ZIF-843. In that work, [C4mim][PF6] were firstly adsorbed on the outer-surface of ZIF-8, then through heat treatment (e.g. 105 ℃), [C4mim][PF6] molecules infiltrated into the SOD cage. Afterwards, the temperature was down to the room temperature, and then the [C4mim][PF6] encapsulated ZIF-8 composite materials were obtained.
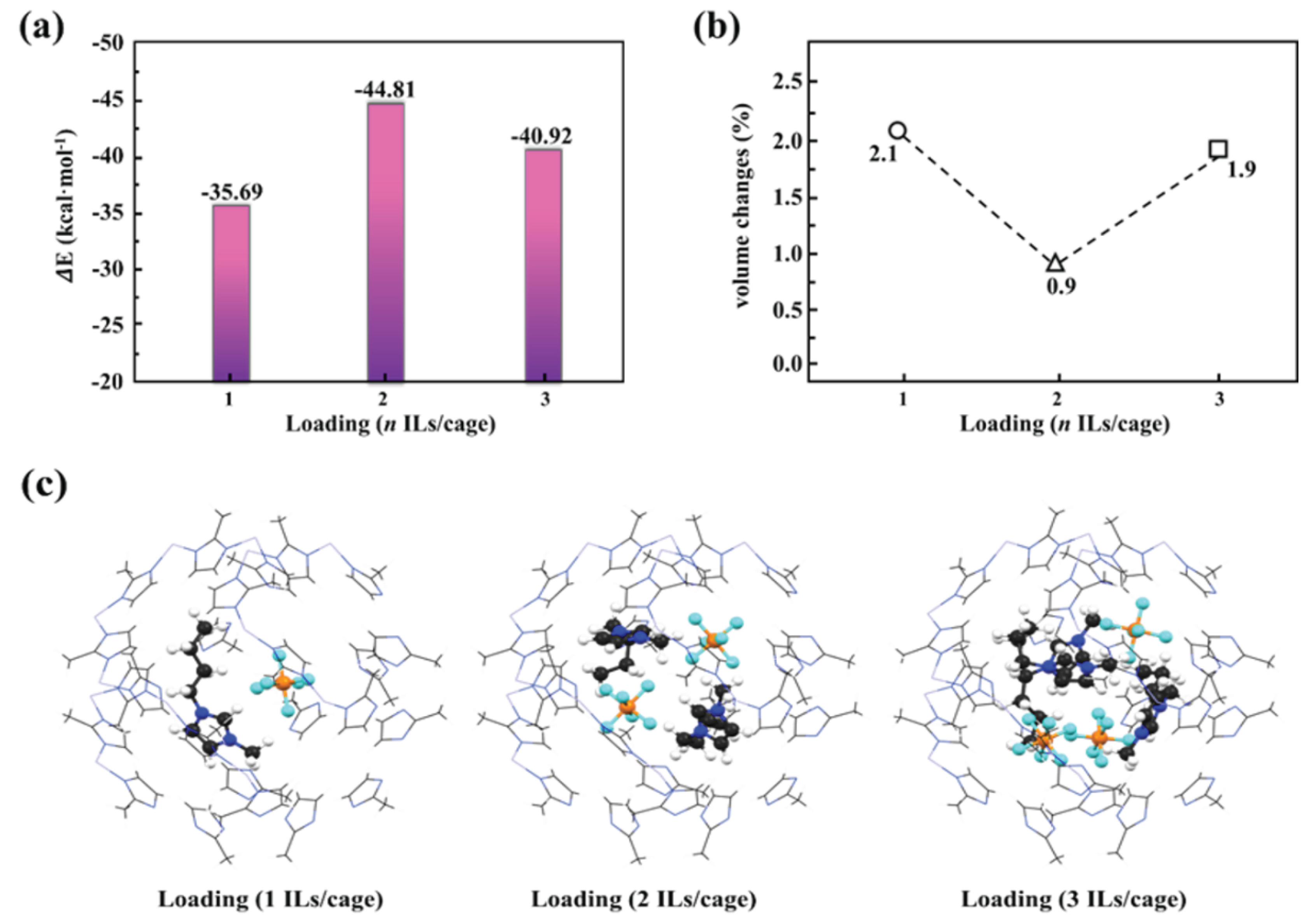
Lastly, to study how many pairs of [C4mim][PF6] could be stable inside the ZIF-8 SOD cage, and to examine their impact of contents on the structures of the ZIF-8 apertures, the structural optimizations were performed by adding one, two, and three pairs of [PF6][C4mim] in the pristine ZIF-8 SOD cage. As shown in Figure 7a, it is shown that the interaction energies between [C4mim][PF6] and ZIF-8 was strongest when two pairs [C4mim][PF6] were added, with a computed value of -44.81 kcal·mol-1, while the interactions energies of one and three pairs were computed to be -35.69 kcal·mol-1 and -40.92 kcal mol-1. As such, we conclude that most of the ZIF-8 SOD cage could accommodate two pairs of [C4mim][PF6], and some of them are even able to accommodate three pairs. Overall, a value slightly larger than 2 would be obtained for the pairs of [C4mim][PF6] per SOD cage. These finding are comparable to the experimental results obtained by Ban et al.15, in which average 1.4 pairs of [C4mim][TF2N] have been identified per single ZIF-8 SOD cage. The smaller value of 1.4 versus 2.0 could be attributed to the fact that the anion [TF2N]- is relatively larger than [PF6]- in the space size. Compared to the experimental values44, we found that the volume of ZIF-8 SOD cage has been increased when [C4mim][PF6] was encapsulated, of which the smallest value was obtained (0.9%) for the case with two pairs. Moreover, we also observed certain confinement effects from the results shown in Figure 7c, e.g. the structure of one pair of [C4mim][PF6] is very similar compared to the free pair (e.g. gas phase calculations), while the two and three pairs show structures closer to the condense phase45.
4. Conclusions
In summary, the DFT calculations and several electronic structure analysis have been performed to study the diffusion behavior of the ILs, [C4mim][PF6], through the different aperture configurations of ZIF-8, and their stability inside the ZIF-8 cage. The results indicate that the original aperture configuration (with a 3.4 Å pore size) eventually prohibit the diffusion of [C4mim][PF6] into the SOD cage of ZIF-8, as a minimum energy barrier of 39.87 kcal mol-1 has been identified, which is mainly due to imidazole ring of the [C4mim]+. The energy decomposition analysis based on the GKS-EDA method revealed that the large repulsion component are the main reason for the high energy barriers when [C4mim]+ and [PF6]- passing through the pristine ZIF-8 aperture. Nevertheless, we found that the diffusion properties could be largely enhanced by modified the ZIF-8 apertures, e.g. the pore size, and the it is proved that this could be achieved via thermal contributions (e.g. employing a higher temperature during the synthesis procedure). Moreover, we found that two pairs of [C4mim][PF6] are the stable state to accommodate inside the ZIF-8 cage, according to interaction energies and volume changes. Certain confinement effects were obtained as well, in which the structure of one pair of [C4mim][PF6] is similar compared to the free pair, while the two and three pairs show structures closer to the condense phase.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
L.L. is thankful for the financial support in the form of start-up funding from Wuhan Textile University (No. 20220321).
Conflicts of Interest
The authors declare that they have no known competing financial interests.
References
- Li, X.; Chen, K.; Guo, R.; Wei, Z. Ionic Liquids Functionalized MOFs for Adsorption. Chemical Reviews 2023, 123, 10432–10467. [Google Scholar] [CrossRef] [PubMed]
- Durak, O.; Zeeshan, M.; Habib, N.; Gulbalkan, H.C.; Alsuhile, A.A.A.M.; Caglayan, H.P.; Kurtoğlu-Öztulum, S.F.; Zhao, Y.; Haslak, Z.P.; Uzun, A.; Keskin, S. Composites of porous materials with ionic liquids: Synthesis, characterization, applications, and beyond. Microporous and Mesoporous Materials 2022, 332. [Google Scholar] [CrossRef]
- Wu, K.; Miao, X.; Zhao, H.; Liu, S.; Fei, T.; Zhang, T. Selective Encapsulation of Ionic Liquids in UiO-66-NH2 Nanopores for Enhanced Humidity Sensing. ACS Applied Nano Materials 2023, 6, 9050–9058. [Google Scholar] [CrossRef]
- Qian, Y.; Zhang, F.; Pang, H. A Review of MOFs and Their Composites-Based Photocatalysts: Synthesis and Applications. Advanced Functional Materials 2021, 31. [Google Scholar] [CrossRef]
- Friess, K.; Izák, P.; Kárászová, M.; Pasichnyk, M.; Lanč, M.; Nikolaeva, D.; Luis, P.; Jansen, J.C. A Review on Ionic Liquid Gas Separation Membranes. Membranes 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.; G. Saiz, P.; Peřinka, N.; Wuttke, S.; Fernández de Luis, R. Printed Capacitive Sensors Based on Ionic Liquid/Metal-Organic Framework Composites for Volatile Organic Compounds Detection. Advanced Functional Materials 2021, 31. [Google Scholar] [CrossRef]
- Tuffnell, J.M.; Morzy, J.K.; Kelly, N.D.; Tan, R.; Song, Q.; Ducati, C.; Bennett, T.D.; Dutton, S.E. Comparison of the ionic conductivity properties of microporous and mesoporous MOFs infiltrated with a Na-ion containing IL mixture. Dalton Transactions 2020, 49, 15914–15924. [Google Scholar] [CrossRef]
- Zeeshan, M.; Nozari, V.; Keskin, S.; Uzun, A. Structural Factors Determining Thermal Stability Limits of Ionic Liquid/MOF Composites: Imidazolium Ionic Liquids Combined with CuBTC and ZIF-8. Industrial & Engineering Chemistry Research 2019, 58, 14124–14138. [Google Scholar]
- Yoshida, Y.; Fujie, K.; Lim, D.W.; Ikeda, R.; Kitagawa, H. Superionic Conduction over a Wide Temperature Range in a Metal–Organic Framework Impregnated with Ionic Liquids. Angewandte Chemie International Edition 2019, 58, 10909–10913. [Google Scholar] [CrossRef]
- Li, H.; Tuo, L.; Yang, K.; Jeong, H.-K.; Dai, Y.; He, G.; Zhao, W. Simultaneous enhancement of mechanical properties and CO2 selectivity of ZIF-8 mixed matrix membranes: Interfacial toughening effect of ionic liquid. Journal of Membrane Science 2016, 511, 130–142. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Z.; Yang, C.; Liu, J.; Shen, H.; Yang, K.; Wang, Z. PIM-based mixed-matrix membranes containing MOF-801/ionic liquid nanocomposites for enhanced CO2 separation performance. Journal of Membrane Science 2021, 636. [Google Scholar] [CrossRef]
- Zeeshan, M.; Keskin, S.; Uzun, A. Enhancing CO2/CH4 and CO2/N2 separation performances of ZIF-8 by post-synthesis modification with [BMIM][SCN]. Polyhedron 2018, 155, 485–492. [Google Scholar] [CrossRef]
- Krokidas, P.; Moncho, S.; Brothers, E.N.; Castier, M.; Economou, I.G. Tailoring the gas separation efficiency of metal organic framework ZIF-8 through metal substitution: a computational study. Physical Chemistry Chemical Physics 2018, 20, 4879–4892. [Google Scholar] [CrossRef] [PubMed]
- Gulbalkan, H.C.; Haslak, Z.P.; Altintas, C.; Uzun, A.; Keskin, S. Assessing CH4/N2 separation potential of MOFs, COFs, IL/MOF, MOF/Polymer, and COF/Polymer composites. Chemical Engineering Journal 2022, 428. [Google Scholar] [CrossRef]
- Ban, Y.; Li, Z.; Li, Y.; Peng, Y.; Jin, H.; Jiao, W.; Guo, A.; Wang, P.; Yang, Q.; Zhong, C.; Yang, W. Confinement of Ionic Liquids in Nanocages: Tailoring the Molecular Sieving Properties of ZIF-8 for Membrane-Based CO2 Capture. Angew Chem Int Ed Engl 2015, 54, 15483–7. [Google Scholar] [CrossRef]
- Kinik, F.P.; Altintas, C.; Balci, V.; Koyuturk, B.; Uzun, A.; Keskin, S. [BMIM][PF6] Incorporation Doubles CO2 Selectivity of ZIF-8: Elucidation of Interactions and Their Consequences on Performance. ACS Applied Materials & Interfaces 2016, 8, 30992–31005. [Google Scholar]
- Koyuturk, B.; Altintas, C.; Kinik, F.P.; Keskin, S.; Uzun, A. Improving Gas Separation Performance of ZIF-8 by [BMIM][BF4] Incorporation: Interactions and Their Consequences on Performance. The Journal of Physical Chemistry C 2017, 121, 10370–10381. [Google Scholar] [CrossRef]
- Ali, S.A.; Khan, A.U.; Mulk, W.U.; Khan, H.; Nasir Shah, S.; Zahid, A.; Habib, K.; Shah, M.U.H.; Othman, M.H.D.; Rahman, S. An Ongoing Futuristic Career of Metal–Organic Frameworks and Ionic Liquids, A Magical Gateway to Capture CO2; A Critical Review. Energy & Fuels 2023, 37, 15394–15428. [Google Scholar] [CrossRef]
- Dong, K.; Liu, X.; Dong, H.; Zhang, X.; Zhang, S. Multiscale Studies on Ionic Liquids. Chemical Reviews 2017, 117, 6636–6695. [Google Scholar] [CrossRef]
- Chen, J.; Dong, K.; Liu, L.; Zhang, X.; Zhang, S. Anti-electrostatic hydrogen bonding between anions of ionic liquids: a density functional theory study. Physical Chemistry Chemical Physics 2021, 23, 7426–7433. [Google Scholar] [CrossRef]
- Kwon, H.T.; Jeong, H.-K.; Lee, A.S.; An, H.S.; Lee, J.S. Heteroepitaxially Grown Zeolitic Imidazolate Framework Membranes with Unprecedented Propylene/Propane Separation Performances. Journal of the American Chemical Society 2015, 137, 12304–12311. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Prakash, M. Tuning the CO2 adsorption by the selection of suitable ionic liquids at ZIF-8 confinement: A DFT study. Applied Surface Science 2019, 491, 633–639. [Google Scholar] [CrossRef]
- Mohamed, A.M.O.; Moncho, S.; Krokidas, P.; Kakosimos, K.; Brothers, E.N.; Economou, I.G. Computational investigation of the performance of ZIF-8 with encapsulated ionic liquids towards CO2 capture. Molecular Physics 2019, 117, 3791–3805. [Google Scholar] [CrossRef]
- Gao, W.; Zheng, W.; Sun, W.; Zhao, L. Understanding the Effective Capture of H2S/CO2 from Natural Gas Using Ionic Liquid@MOF Composites. The Journal of Physical Chemistry C 2022, 126, 19872–19882. [Google Scholar] [CrossRef]
- Thomas, A.; Ahamed, R.; Prakash, M. Selection of a suitable ZIF-8/ionic liquid (IL) based composite for selective CO(2) capture: the role of anions at the interface. RSC Adv 2020, 10, 39160–39170. [Google Scholar] [CrossRef] [PubMed]
- Kavak, S.; Polat, H.M.; Kulak, H.; Keskin, S.; Uzun, A. MIL-53(Al) as a Versatile Platform for Ionic-Liquid/MOF Composites to Enhance CO2 Selectivity over CH4 and N2. Chemistry – An Asian Journal 2019, 14, 3655–3667. [Google Scholar] [CrossRef]
- Zhang, Z.; Jia, X.; Sun, Y.; Guo, X.; Huang, H.; Zhong, C. Pore engineering of ZIF-8 with ionic liquids for membrane-based CO2 separation: bearing functional group effect. Green Chemical Engineering 2021, 2, 104–110. [Google Scholar] [CrossRef]
- Fairen-Jimenez, D.; Moggach, S.A.; Wharmby, M.T.; Wright, P.A.; Parsons, S.; Düren, T. Opening the Gate: Framework Flexibility in ZIF-8 Explored by Experiments and Simulations. Journal of the American Chemical Society 2011, 133, 8900–8902. [Google Scholar] [CrossRef]
- Zhang, C.; Lively, R.P.; Zhang, K.; Johnson, J.R.; Karvan, O.; Koros, W.J. Unexpected Molecular Sieving Properties of Zeolitic Imidazolate Framework-8. The Journal of Physical Chemistry Letters 2012, 3, 2130–2134. [Google Scholar] [CrossRef]
- Zeeshan, M.; Nozari, V.; Yagci, M.B.; Isık, T.; Unal, U.; Ortalan, V.; Keskin, S.; Uzun, A. Core–Shell Type Ionic Liquid/Metal Organic Framework Composite: An Exceptionally High CO2/CH4 Selectivity. Journal of the American Chemical Society 2018, 140, 10113–10116. [Google Scholar] [CrossRef]
- Chang, Y.; Wang, L.; Jiang, Z.; Zhang, R.; Zhu, H.; Zhang, D.; Zhu, J.; Kong, X.; Huang, H. Zwitterionic metal–organic framework with highly dispersed ionic liquid for enhancing CO2 capture. Separation and Purification Technology 2023, 326. [Google Scholar] [CrossRef]
- Yu, T.; Cai, Q.; Lian, G.; Bai, Y.; Zhang, X.; Zhang, X.; Liu, L.; Zhang, S. Mechanisms behind high CO2/CH4 selectivity using ZIF-8 metal organic frameworks with encapsulated ionic liquids: A computational study. Chemical Engineering Journal 2021, 419. [Google Scholar] [CrossRef]
- Yu, T.; Cai, Q.; Lian, G.; Liu, L. Molecular dynamics studies on separation of CO2/CH4 by the ionic liquids encapsulated ZIF-8. Journal of Membrane Science 2022, 644. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.; Glendening, E.J.I. r. i. p. c., What is NBO analysis and how is it useful? 2016, 35, 399–440. [CrossRef]
- Lu, T.; Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 2012, 33, 580–92. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: a three-dimensional visualization system for electronic and structural analysis. Journal of Applied Crystallography 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Su, P.; Jiang, Z.; Chen, Z.; Wu, W. Energy decomposition scheme based on the generalized Kohn-Sham scheme. J Phys Chem A 2014, 118, 2531–42. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; Windus, T.L.; Dupuis, M.; Montgomery Jr, J.A. General atomic and molecular electronic structure system. 1993, 14, 1347–1363. [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J.J.T.J. o. p. c., Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. 1994, 98, 11623–11627. [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 2011, 32, 1456–65. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; Golze, D.; Wilhelm, J.; Chulkov, S.; Bani-Hashemian, M.H.; Weber, V.; Borštnik, U.; Taillefumier, M.; Jakobovits, A.S.; Lazzaro, A.; Pabst, H.; Müller, T.; Schade, R.; Guidon, M.; Andermatt, S.; Holmberg, N.; Schenter, G.K.; Hehn, A.; Bussy, A.; Belleflamme, F.; Tabacchi, G.; Glöß, A.; Lass, M.; Bethune, I.; Mundy, C.J.; Plessl, C.; Watkins, M.; VandeVondele, J.; Krack, M.; Hutter, J. CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations. The Journal of Chemical Physics 2020, 152. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zheng, W.; Yan, X.; Dai, Y.; Ruan, X.; Yang, X.; Li, X.; Zhang, N.; He, G. Ionic liquid tuning nanocage size of MOFs through a two-step adsorption/infiltration strategy for enhanced gas screening of mixed-matrix membranes. Journal of Membrane Science 2020, 605. [Google Scholar] [CrossRef]
- Park, K.S.; Ni, Z.; Côté, A.P.; Choi, J.Y.; Huang, R.; Uribe-Romo, F.J.; Chae, H.K.; O’Keeffe, M.; Yaghi, O.M. Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. 2006, 103, 10186–10191. [Google Scholar] [CrossRef] [PubMed]
- Bühl, M.; Wipff, G. Insights into Uranyl Chemistry from Molecular Dynamics Simulations. ChemPhysChem 2011, 12, 3095–3105. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) The SOD cage presentation of the ZIF-8, and its typical (pristine) pore structure is highlighted in red color; (b-f) five different pore structures of ZIF-8 with their diameters (ref. 33). Color legend: C black, N blue, Zn light grey, H white.
Figure 1.
(a) The SOD cage presentation of the ZIF-8, and its typical (pristine) pore structure is highlighted in red color; (b-f) five different pore structures of ZIF-8 with their diameters (ref. 33). Color legend: C black, N blue, Zn light grey, H white.
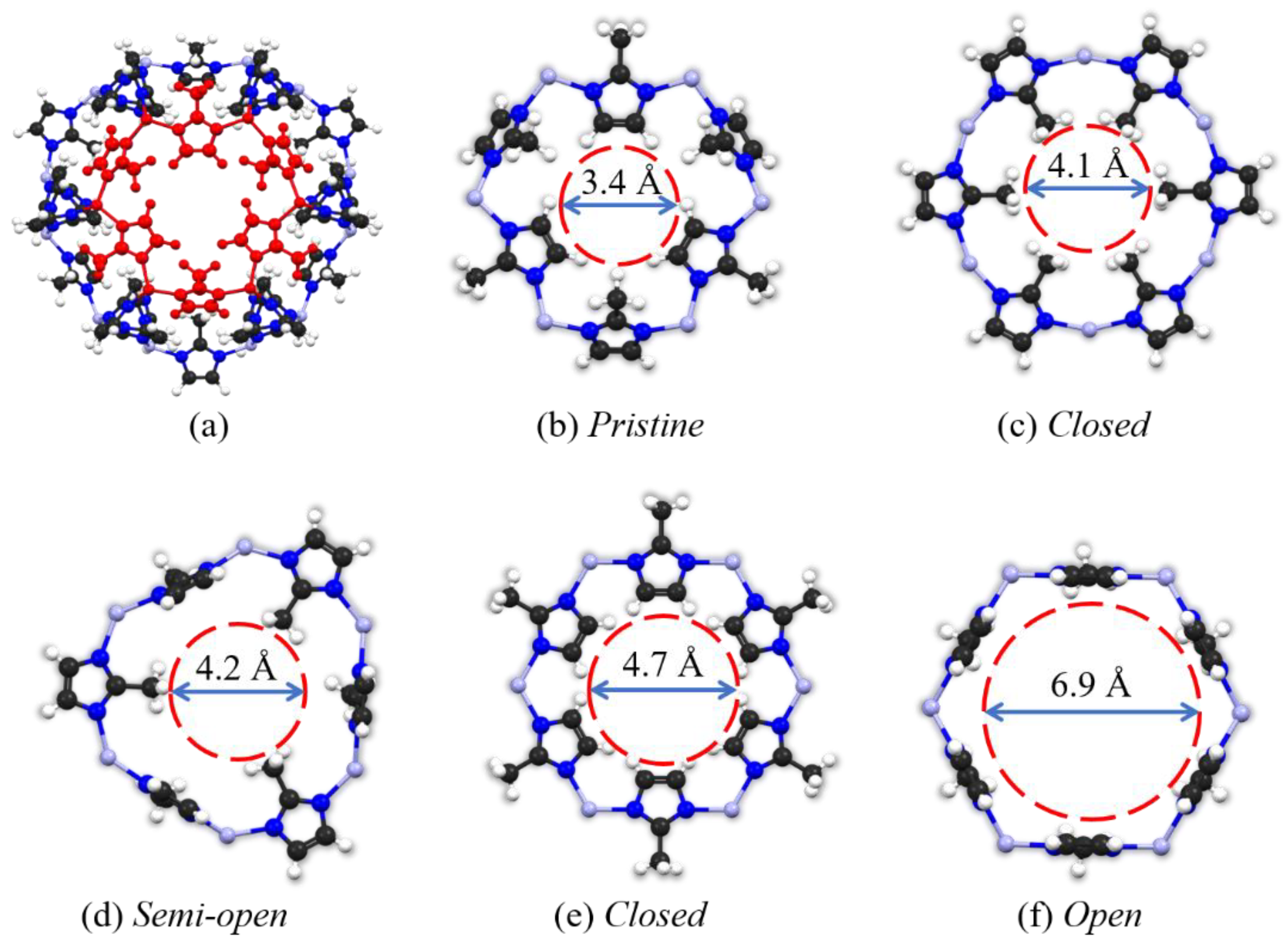
Figure 2.
(a) Schematic representation of [PF6]-/[C4mim]+ passing through ZIF-8 apertures. (b) The geometric structures of [PF6]- /[C4mim]+, and their orientations when passing through the ZIF-8 aperture.
Figure 2.
(a) Schematic representation of [PF6]-/[C4mim]+ passing through ZIF-8 apertures. (b) The geometric structures of [PF6]- /[C4mim]+, and their orientations when passing through the ZIF-8 aperture.
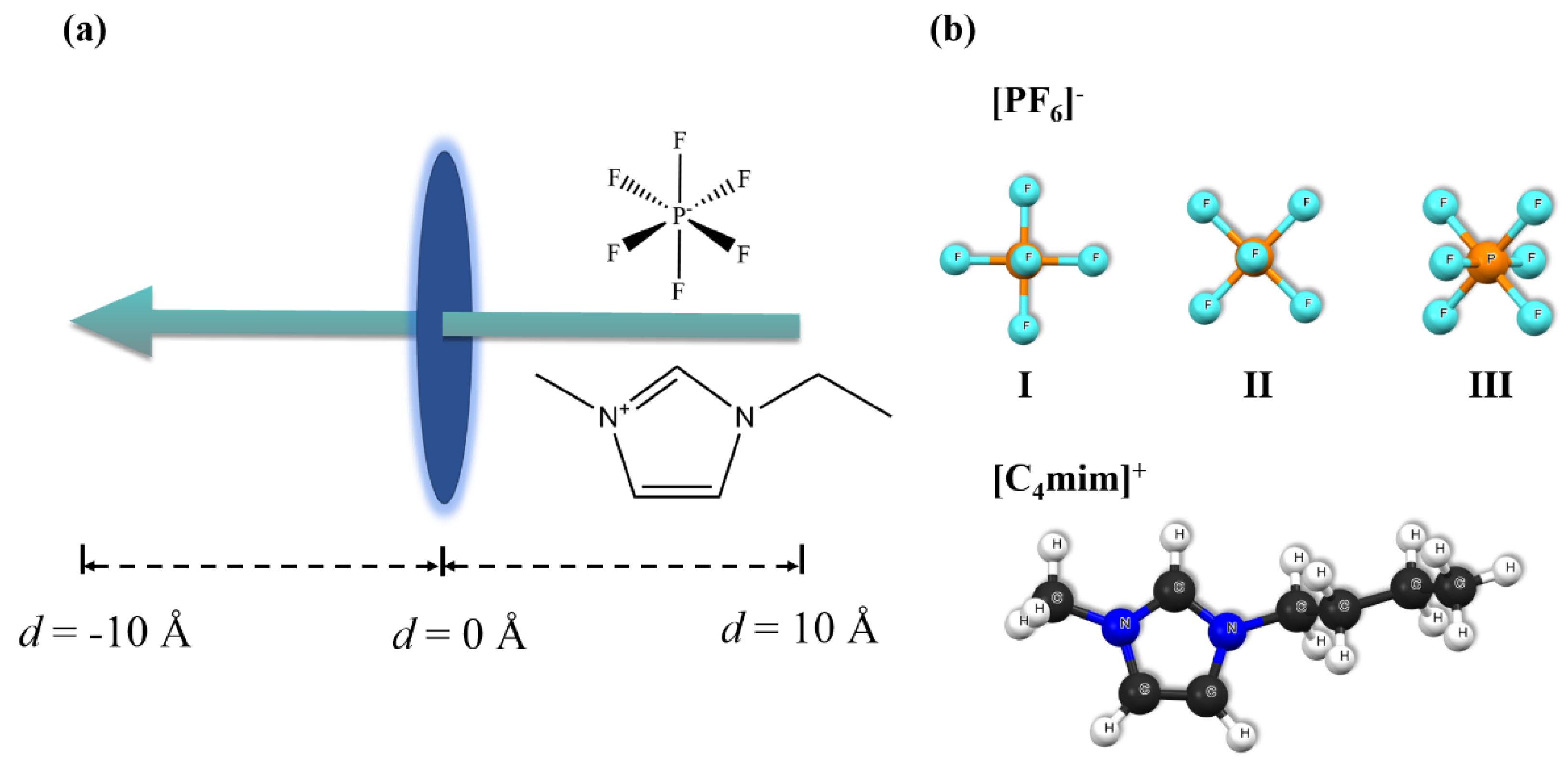
Figure 3.
The potential energy surface curves for the anion [C4mim]+ and [PF6]- passing through ZIF-8 structures: (a) the rigid PES scan, and (b) the relax PES scan.
Figure 3.
The potential energy surface curves for the anion [C4mim]+ and [PF6]- passing through ZIF-8 structures: (a) the rigid PES scan, and (b) the relax PES scan.
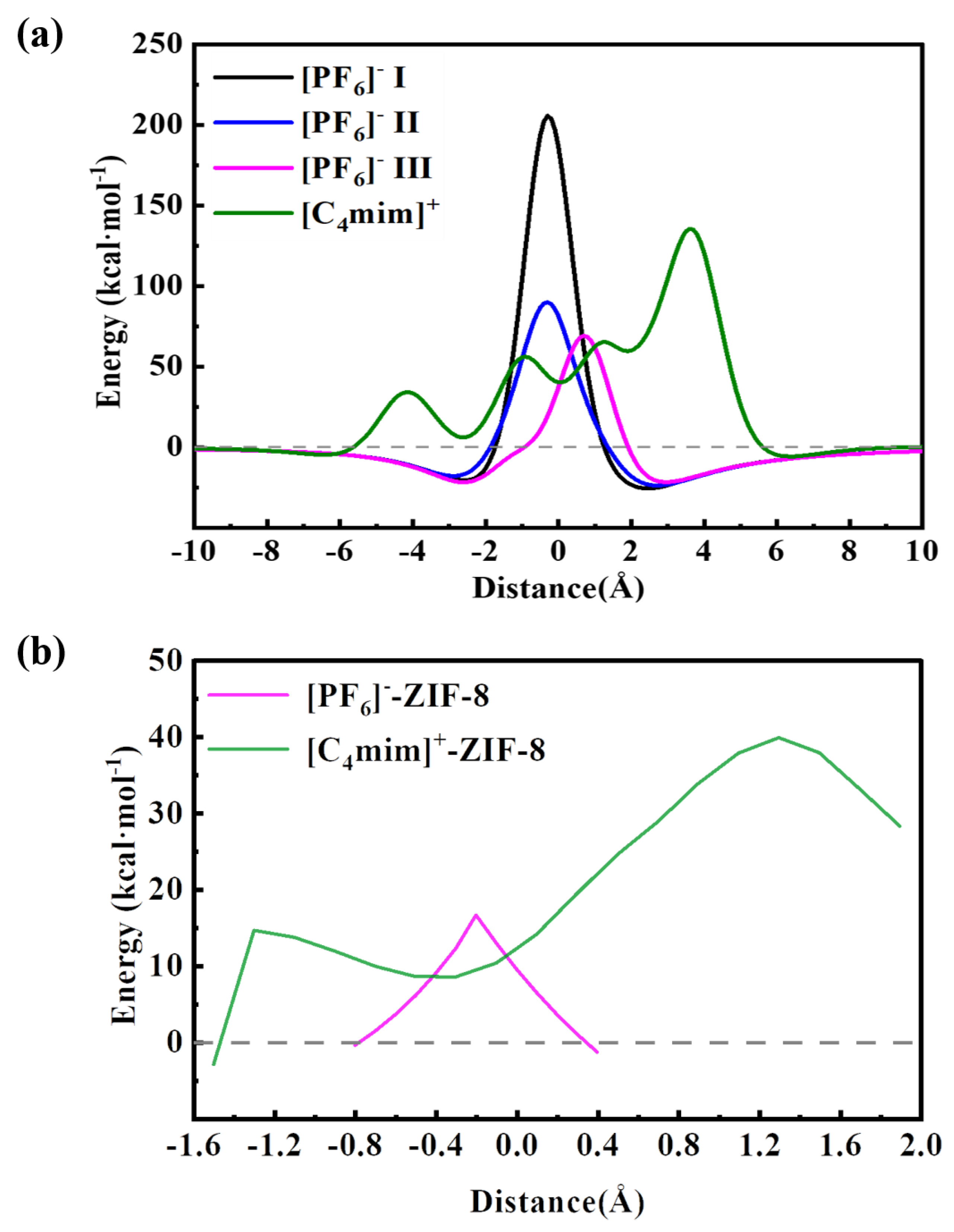
Figure 4.
Frontier orbitals and their energies, with an isosurface value of 0.05 e/ų.
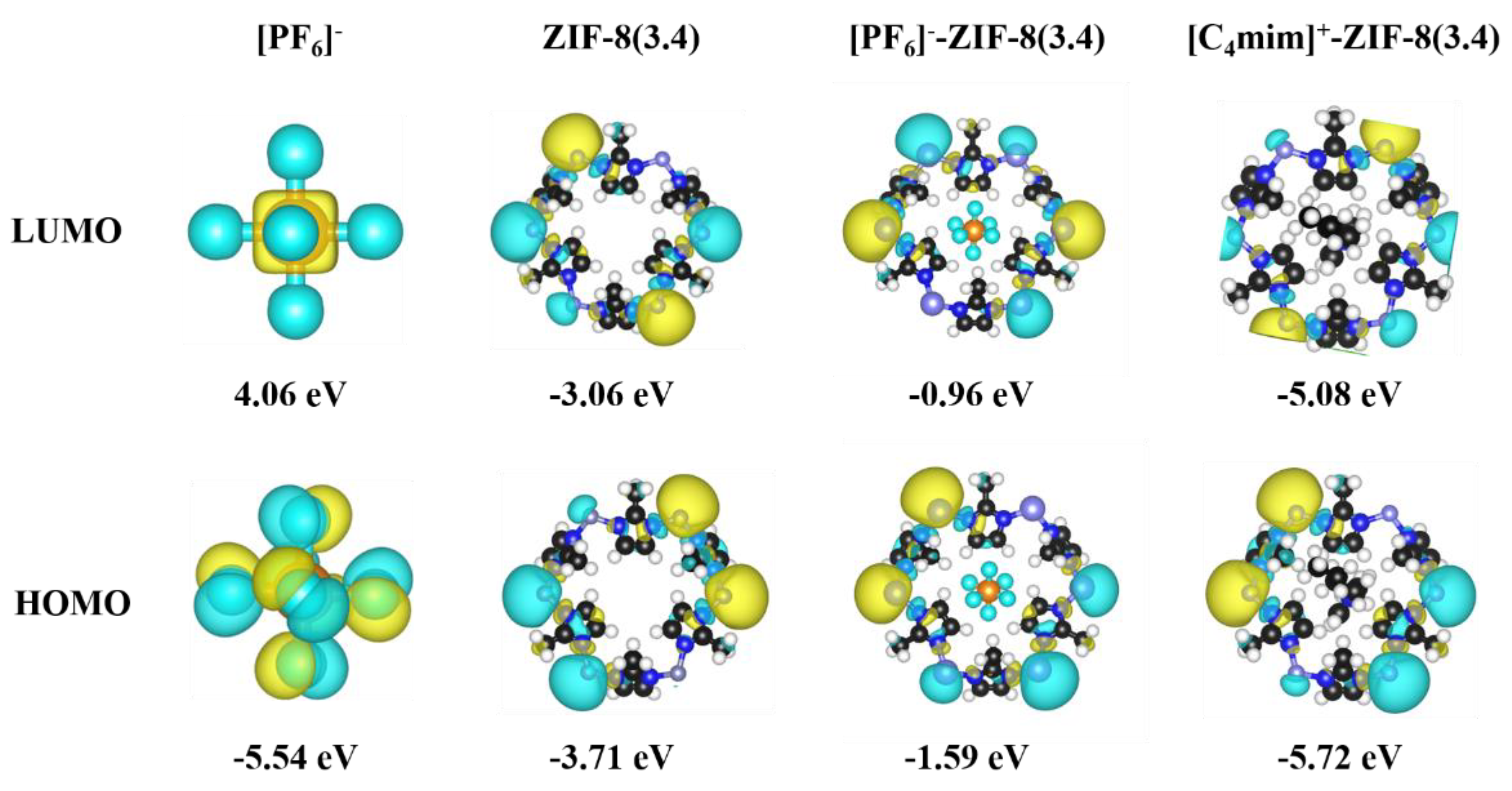
Figure 5.
Energy decomposition analysis calculated with the GKS-EDA method at the B3LYP-D3(BJ)/6-311+G* level of theory for the [PF6]-/[C4mim]+-ZIF-8 (3.4) systems.
Figure 5.
Energy decomposition analysis calculated with the GKS-EDA method at the B3LYP-D3(BJ)/6-311+G* level of theory for the [PF6]-/[C4mim]+-ZIF-8 (3.4) systems.
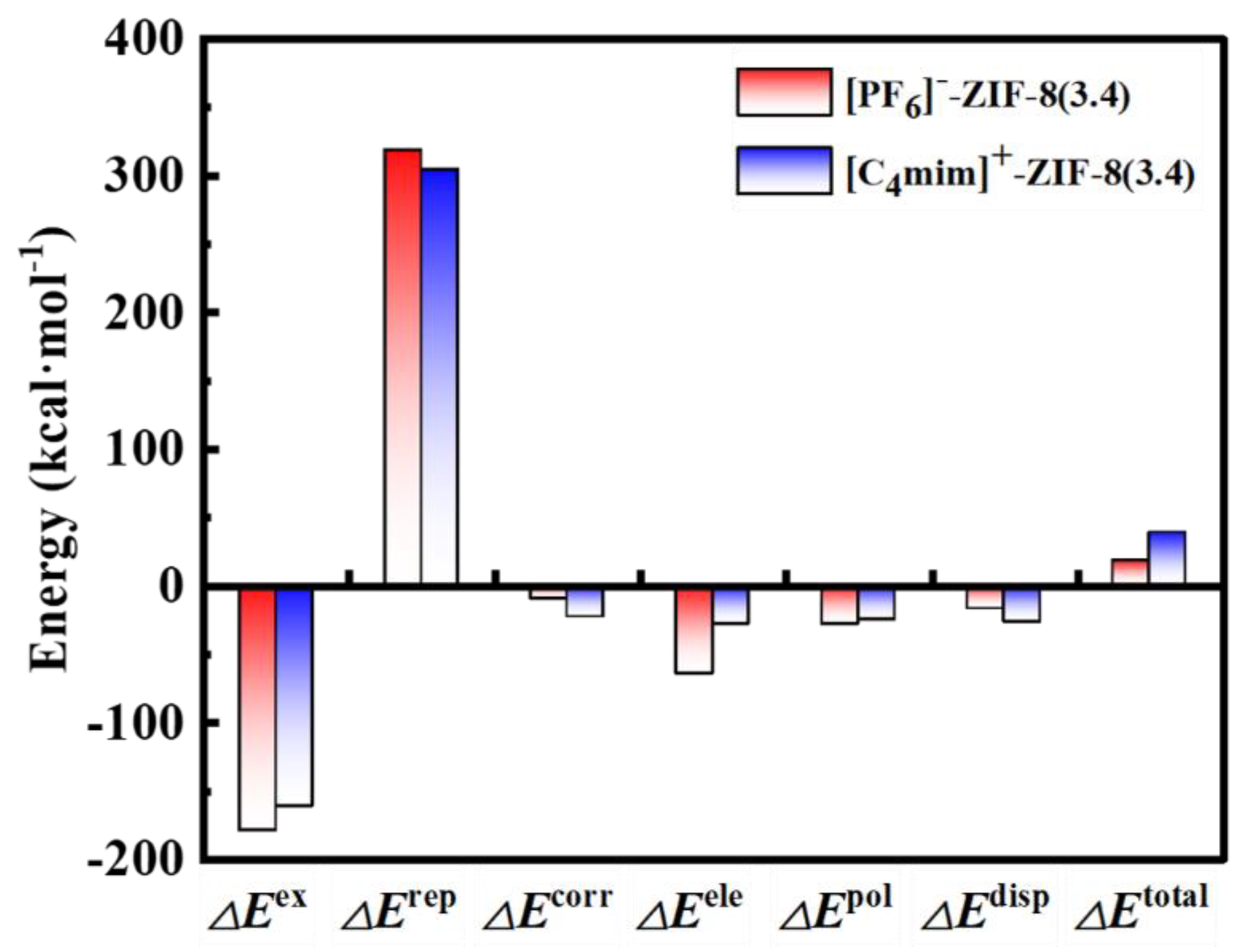
Figure 6.
Potential energy surface curves for [PF6]- (a) and [C4mim]+ (b) passing through different ZIF-8 apertures.
Figure 6.
Potential energy surface curves for [PF6]- (a) and [C4mim]+ (b) passing through different ZIF-8 apertures.
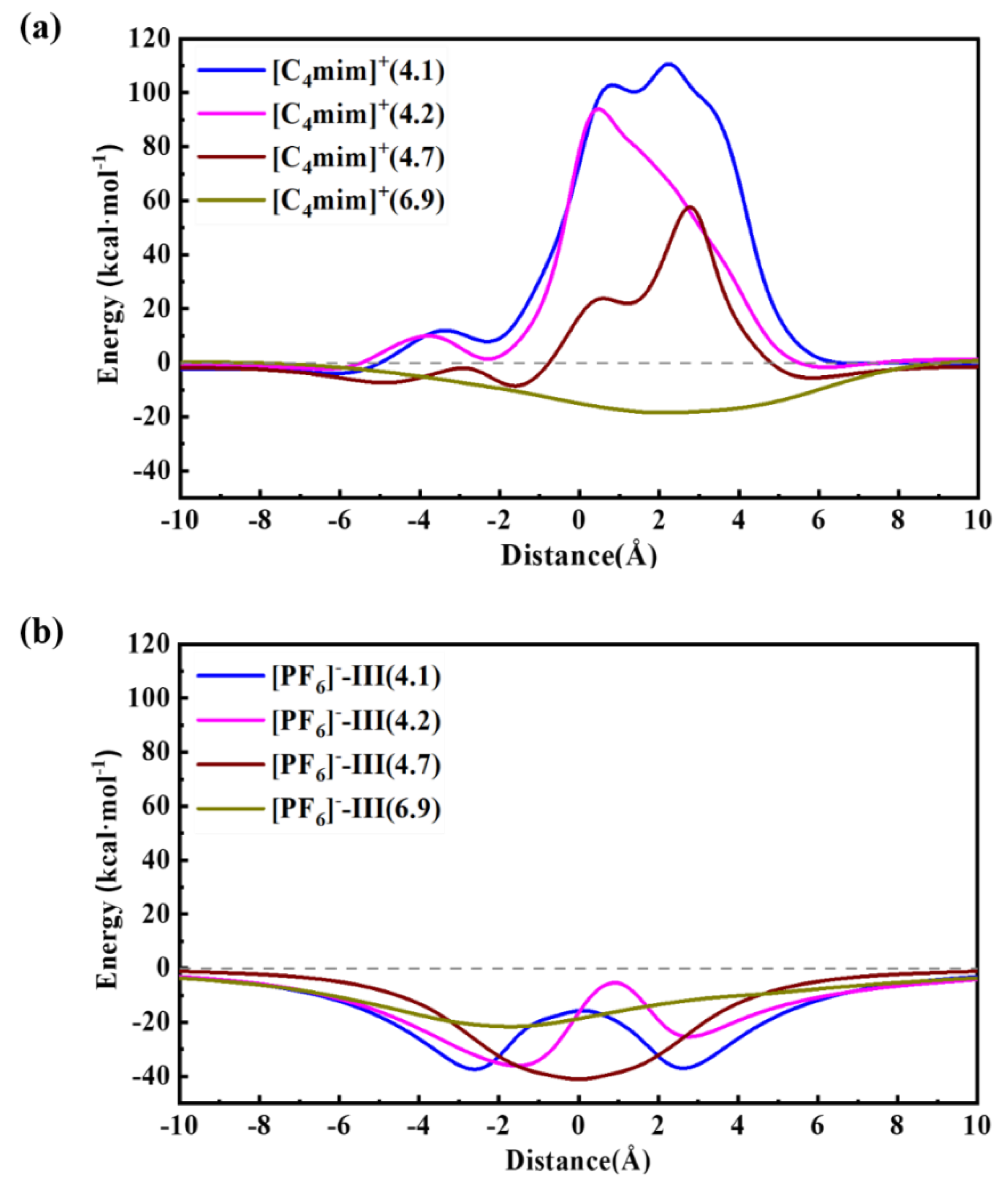
Figure 7.
Interaction energies (a), volume changes (b), and geometric structures (c) the ZIF-8 SOD cage containing one, two, and three pairs of [PF6][C4mim], respectively.
Figure 7.
Interaction energies (a), volume changes (b), and geometric structures (c) the ZIF-8 SOD cage containing one, two, and three pairs of [PF6][C4mim], respectively.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



