
Submitted:
29 February 2024
Posted:
29 February 2024
You are already at the latest version
Abstract
Pancreatic cancer is an intractable disease with the worst prognosis of all cancers. The currently used treatment regimens do not significantly impact patient survival, and therefore, effective treatment strategies are urgently needed. Drug repurposing, which identifies new indications for existing and approved drugs, has proven to be a promising approach to anti-cancer drug discovery. Indeed the antihelmintic drug, pyrvinium pamoate has shown promise as an anti-pancreatic cancer drug, but to date, the inhibition of mitochondrial function is the only mechanism of action described in pancreatic cancer. This study showed, using pancreatic cancer 2D cell cultures and 3D spheroids, that pyrvinium pamoate exhibited short- and long-term cytotoxicity, inhibited epithelial-to-mesenchymal transition and cell invasion and migration. Mechanistically, pyrvinium pamoate induced DNA damage, inhibited the PI3K/AKT cell survival pathway, and triggered cell cycle arrests, as well as apoptotic and autophagic cell death. Importantly, pyrvinium pamoate acted synergistically with the first-line drug, gemcitabine in 2D and 3D pancreatic cancer cell culture models. This study provided evidence that pyrvinium pamoate is effective as a single agent and in combination with gemcitabine for the treatment of pancreatic cancer.
Keywords:
pyrvinium pamoate
; pancreatic cancer
; drug repurposing
; drug combination
; gemcitabine
Introduction
Pancreatic cancer (PC) has the worst prognosis of all cancers and its worldwide incidence is continuously rising [1]. Pancreatic ductal adenocarcinoma (PDAC) and pancreatic neuroendocrine tumours (PNET) are the two major PC subtypes with PDAC being the more aggressive subtype and accounting for 90% of all PC cases [2,3]. PDAC has no specific symptoms and is consequently diagnosed late and only 24% of PDAC patients survive for one year and 9% for 5 years after diagnosis [4]. At present, surgery is the only curative option for PDAC, however, only 15−20% of patients present with resectable disease, and of these a large proportion are not eligible for immediate surgical resection due to immense vascular involvement [5]. The remaining 80% of patients present with either locally advanced or metastatic disease [1]. Clinical care for these patients is through systemic chemotherapy with the first lines of therapy being Gemcitabine (GEM) alone, GEM administered with a nanoparticle-bound paclitaxel (nab-paclitaxel), or a combination of 5-fluorouracil (5-FU), leucovorin, irinotecan and oxaliplatin (FOLFIRINOX) [6]. However, these treatment modalities do not significantly impact patient survival due to drug resistance, tumour recurrence, and their high toxicity profiles which lead to debilitating side effects. Over the past decades, efforts to address these drawbacks have been marginally successful [7] and limited by the drug development pipeline being time-consuming, risky, and expensive [8]. Drug repurposing, which identifies new indications for existing and approved drugs, has therefore gained traction as an alternative approach to anti-cancer drug discovery [9]. The rationale for this is that the candidate drugs have well-documented and established pharmacokinetic, pharmacodynamic, dosing, and toxicity profiles which accelerate their clinical development [8].
Pyrvinium pamoate (PP) is a small, fluorescent, and lipophilic compound that was first described in the 1940s for use as a dye, fluorescent probe, and antihelminthic drug [10]. Although the development of more effective anti-parasitic agents halted its use as an anthelminthic, PP remained clinically relevant due to its potential to target other disease-causing organisms as well as its ability to reduce toxicity, promote wound repair, and inhibit fibrotic tissue development [11]. Of late, PP has gained significant attention in cancer research. Indeed, several studies have reported on its anti-cancer activities which involve its ability to inhibit the Wnt signalling pathway and mitochondrial function [10]. In the context of PC, Esumi et al. (2004) showed that PP is highly toxic to serum-starved PANC-1 cells and can clear PANC-1 xenografts in vivo [12]. Tomitsuka et al. (2012) compared its effects under hypoxic-hypoglycemic conditions, which mimics the tumour microenvironment (TME), and normoxia-normoglycemic and found that in PDAC, PP inhibits mitochondrial energy metabolism through inhibition of the NADH-fumarate reductase (NADH-FR) system [13]. More recently, a study by Schultz et al. (2021) showed that PP does not only function by specific inhibition of NADH-FR but through a more global inhibition of mitochondrial function in PDAC [14]. Collectively, these studies revealed the potential of PP as a TME-specific anti-cancer drug. Since the efficacy of the drugs currently used to treat PDAC, including GEM, is negatively affected by the dense and desmoplastic PDAC TME [15], combining PP and GEM may be an effective option for treating PDAC.
This study showed that under normal serum conditions, PP inhibited the viability and survival of PDAC 2D cell cultures and 3D spheroids through the inhibition of the PI3K/AKT cell survival pathway and induction of apoptotic and autophagic cell death. PP also inhibited epithelial-to-mesenchymal transition (EMT), cell migration, and invasion in 2D and 3D PDAC cell culture models. Importantly, low concentrations of PP and GEM acted synergistically and their combination was more effective at exhibiting anti-pancreatic cancer activities than when used as single agents. Overall, this study extends our understanding of the anti-cancer activities of PP and provides additional evidence that PP is effective as a single agent and in combination with GEM for the treatment of pancreatic cancer.
Method and Materials
Cell Culture
The human PANC-1 and CFPAC-1 pancreatic ductal adenocarcinoma (PDAC) cell lines were purchased from the American Type Culture Collections (ATCC) and maintained in Dulbecco’s Modified Eagle Medium (DMEM) and Iscove's Modified Dulbecco's Medium (IMDM) (Gibco, Life Technologies/Thermo Fisher Scientific, USA), respectively. Media were supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin and streptomycin (pen/strep) (Gibco, USA). Cells were maintained at 37°C in a 95% air and 5% CO2 humidified incubator. Media were replaced every 2 – 3 days and the cells were routinely tested for mycoplasma infections to ensure that only mycoplasma-negative cells were used.
Treatments
The experimental drug PP (P0027) and a positive control drug GEM were purchased from Sigma Aldrich (Missouri, USA), dissolved in dimethyl sulfoxide (DMSO) (Sigma Aldrich, USA) to a final concentration of 5 mM, and stored at -20˚ C with limited freeze/thaw cycles. For cell culture treatments, the drugs were diluted in supplemented media to achieve the desired final concentration(s) and the percentage of DMSO in the highest drug concentration was used as a vehicle control. Unless stated, cells were treated at a confluency of 60%. To inhibit autophagy, cells were treated with 10 nM Bafilomycin A1 (B1793; Sigma Aldrich, USA) for 1h before treatment with PP or GEM. For cell migration experiments, cells were treated with 10 µM Mitomycin C (M4287; Sigma Aldrich, USA) to inhibit cell proliferation.
Cell Viability Assay
The effects of PP and GEM on the viability of PANC-1 and CFPAC-1 cells were assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltrazolium bromide (MTT) assay (11465007001; Sigma Aldrich, USA) following the manufacturer’s instructions. Briefly, cells were seeded in 96-well plates at a density of 8 x 103 cells/well overnight then treated with vehicle (0.1% DMSO) or increasing concentrations (2 – 10 µM) of PP or GEM. After 72h, cells were incubated with 10 μL of 5 mg/mL MTT for 4h in the dark, after which the wells were emptied followed by the addition of 100 μL DMSO to dissolve the formazan crystals. Absorbance was read at 600 nm using the GloMax® plate reader (Promega, USA), and mean cell viability was calculated and expressed as a percentage of the vehicle control. Half maximum inhibitory concentrations (IC50) were determined using the GraphPad Prism version 8.0 software (GraphPad Prism software, USA) from sigmoidal plots of data obtained from three independent experiments performed in quadruplicate.
Clonogenic Assays
Cells were plated in 6-well plates at a density of 3 x 105 cells/well overnight then treated with vehicle or ½IC50 or IC50 of PP or IC50 of GEM. After 48h, cells were harvested, counted, and replated in 35mm dishes at a low density (500 cells/dish) in duplicate and incubated in drug-free media for 14 days to allow for colony formation. The colonies were then fixed for 10 mins with 3:1 methanol: acetic acid, stained for 15 mins with 0.5% (w/v) crystal violet (Sigma Aldrich, USA) in 100% methanol, imaged and quantified using the ColonyArea plugin of the ImageJ software. Colony areas were determined for each treatment condition and expressed as a percentage of vehicle-treated control.
Cell Cycle Analyses
Cells treated with vehicle, or ½IC50 or IC50 of PP or IC50 GEM for 72h were collected, washed with 1x PBS, counted, diluted to 1 x 106 cells/mL, and permeabilised with 70% ethanol at -20°C overnight. Cells were then pelleted, incubated for 15 mins with 50 µg/mL RNase (Fermentas, Massachusetts, USA) in 1 X PBS at 37°C, transferred into FACS tubes, and stained with propidium iodide. A minimum of 5 x 104 cells/sample were analysed using a Becton Dickinson FACS Calibur flow cytometer (Becton Dickinson, New Jersey, USA) with a 488nm coherent laser. Data were acquired using CellQuest Pro version 5.2.1. software (Becton Dickinson, New Jersey, USA) and analysed using ModFit version 2.0. software (Verity Software House Inc, Maine, USA).
Immunofluorescence
Cells were plated on coverslips in a 12-well plate at a density of 1.5 x 105 cells/well overnight, treated with vehicle or ½IC50 or IC50 of PP or IC50 of GEM for 72h and immunofluorescence was performed as previously described [16]. Briefly, the treated cells were washed three times with 1x PBS, fixed for 5 mins with 100% methanol at -20˚ C, permeabilised for 10 mins with 0.2% (v/v) Triton X-100 in PBS, blocked for 1h with 1% (w/v) BSA in PBS/Tween at RT and incubated overnight with rabbit monoclonal anti- Phospho-Histone H2A.X (#2577; 1:100 dilution) or LC3 (#2775; 1:200 dilution) from Cell Signaling Technology (Massachusetts, USA) at 4˚ C. After incubation, the cells were washed as above and incubated with a donkey anti-rabbit Alexa 488 conjugated secondary antibody (1:500 dilution; Jackson Immuno Research Laboratories Inc., Pennsylvania, USA) for 2 hours in the dark. Both primary and secondary antibodies were diluted in 1% (w/v) BSA in PBS/Tween. Cells were subsequently stained for 10 mins with 1 µg/mL DAPI in PBS in the dark before being visualised using a Zeiss immunofluorescence microscope at 400× magnification or an LSM510 Meta confocal microscope (Zeiss, Oberkochen, Germany).
Western Blotting
Total proteins were harvested from cells treated for 72h with vehicle or ½ IC50 or IC50 PP or IC50 GEM and western blotting was performed as previously described [17]. Briefly, the proteins were resolved on 8 – 15% gels using sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS/PAGE) until a desired separation was obtained and transferred to Hybond Enhanced chemiluminescence (ECL) membranes (Amersham Biosciences, UK) for 1 – 2h. The ECL membranes were blocked with 5% fat-free milk in PBS/TBS containing 1% (v/v) Tween 20 for 1h and incubated with primary antibodies in blocking buffer (PBS/TBS-Tween 20/Milk) overnight at 4˚ C. Unless stated, the following primary antibodies were used at a 1:1000 dilution: rabbit polyclonal antibodies to Phospho-Histone H2A.X (Ser139) (#2577), PARP (#9542), Caspase-9 (#9502), LC3 (#2775), Vimentin (R28) (#3932); rabbit monoclonal antibody to Cleaved Caspase-7 (Asp198) (D6H1) (#8438) and β-catenin (D10A8) (#8480); mouse monoclonal antibodies to Cyclin B1 (V152) (#4135), Caspase-8 (1C12) (#9746), SQSTM1/p62 (D5L7G) (#88588), E-cadherin (4A2) (#14472), N-cadherin (13A9) (#14215) from Cell Signaling Technology (Massachusetts, USA). Mouse monoclonal antibodies to Cyclin A (H-432) (sc-751) and β-actin (1:3000; sc-47778) were purchased from Santa Cruz Biotechnology (California, USA). Secondary antibodies used were: horseradish peroxidase (HRP)-conjugated goat anti-rabbit (Biorad, USA), and goat anti-mouse (Biorad, USA) antibodies at 1:3000 dilution for 1h in a blocking buffer. β-actin was used as a loading control and the band intensities were measured using the ImageJ software. Densitometric readings were calculated as a ratio of the protein of interest/β-actin and normalised to either the vehicle control or first lane with a band.
Apoptosis Assay
Cells were plated on coverslips in a 12-well plate at a density of 1.5 x 105 cells/well overnight and treated with vehicle or ½IC50 or IC50 of PP or IC50 of GEM. After 72h, the cells were washed three times with 1X PBS, fixed for 10 mins with 3:1 methanol: acetic acid, and stained with 1 µg/ml of ethidium bromide and 1 µg/ml of acridine orange for 10 mins in the dark. After washing as above, the cells were mounted onto microscope slides, dried, and imaged under a fluorescence microscope (EVOS M5000 Imaging System, Thermo Fisher Scientific, USA).
Transwell Invasion Assay
Cells were plated in 6-well plates at a density of 3 x 105 cells/well overnight and then treated with vehicle or IC50 of PP or IC50 of GEM for 24h. The cells were then harvested and counted and 1 x 105 cells were resuspended in 200 µL of media with 1% FBS and replated in the upper chambers of the transwell inserts that were coated with 0.3 µg/ml of Matrigel (CLS354234; Merk, USA). The inserts were then placed in 12-well plates with media supplemented with 10% FBS as a chemoattractant and incubated for 20 h for cell invasion to occur. The non-invaded cells were removed using cotton swaps and invaded cells were fixed for 10 mins with 3.7% (w/v) paraformaldehyde and stained with 0.5% (w/v) crystal violet (Sigma Aldrich, USA). After 15 mins, the inserts were washed three times with 1x PBS to remove excess crystal violet, air-dried, and imaged under an inverted light microscope (EVOS M5000 Imaging System, Thermo Fisher Scientific, USA). The invasive cells were quantified by ImageJ and expressed as a percentage of the vehicle control.
In Vitro 2D Scratch Motility Assays
Cells were plated in 24-well plates at a density of 1.5 x 105 cells/well and incubated for 2 days to form a 100% confluent cell monolayer. A vertical wound was created on the cell monolayers using a 20 µL sterile pipette tip. The wounded cells were then treated with vehicle or ½IC50 or IC50 of PP or IC50 of GEM and 10 µM mitomycin C (M4287; Sigma Aldrich, USA) to inhibit cell proliferation and imaged at 0, 3, 6, 9, 12, and 24h. Marks were made on the plates to ensure that the cells were imaged in the same area at all time points. The wound areas were measured using ImageJ and the total migrated areas were calculated by subtracting the area for each treatment condition at a specific time point from the area measured at 0h.
Spheroid Growth Assays
Cells were plated at a density of 5000 cells/well in 96-well plates coated with 70 µL of 1.2% (w/v) agarose (SeaKem LE Agarose 50004, Lonza, USA) to prevent cell adhesion and incubated for 6 days for compact spheroid formation. Once formed, the spheroids were imaged and treated with vehicle or IC50 or 2x IC50 of PP or IC50 of GEM for 6 days (on day 0 and day 3). To avoid disrupting the spheroids during treatment, 50 µL of the media was removed and replaced with 50 µL of a doubled concentration of vehicle or drugs. After treatment, the spheroids were imaged and their areas were measured using ImageJ. A minimum of four spheroids per treatment condition were analyzed and the spheroid growth rates were calculated by dividing the areas of the spheroids on day 6 by their respective areas on day 0 and expressed relative to the vehicle-treated spheroids.
Spheroid Viability Assay
The effects of PP and GEM on spheroid viability were assessed by Calcein-AM staining. Briefly, a 2x staining solution was prepared by mixing 2 μM Calcein-AM (C1430; Invitrogen, USA) which stains viable cells, and 20 μg/mL of 4,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, USA) as a counterstain. To stain the spheroids, 50 µL of the media with/without treatment was removed and replaced with 50 µL of the 2x staining solution (which was diluted to 1x, i.e., 1 μM Calcein-AM and 10 μg/mL DAPI by the remaining 50 µL in each well) and incubated at 37°C and 5% CO2 for 1h in the dark. The spheroids were imaged under a fluorescence microscope (EVOS M5000 Imaging System, Thermo Fisher Scientific, USA) and the calcein-AM fluorescence intensity was measured using ImageJ, normalized to the spheroid areas, and expressed relative to the vehicle-treated spheroids.
Spheroid Invasion Assay
The spheroids generated as above were transferred to a new 96-well plate where they were embedded in 70 µL of 1.5 mg/ml collagen I rat tail matrix (Gibco, A1048301, Thermo Fisher Scientific, USA), imaged under a light microscope (EVOS M5000 Imaging System, Thermo Fisher Scientific, USA), and treated with 100 µL of vehicle or IC50 or 2xIC50 of PP or IC50 of GEM for 72h. A minimum of four spheroids per treatment condition were analysed per treatment condition.
Analyses of Drug Combination
Cells were treated for 72h with vehicle or ⅛ IC50, ⅟4 IC50, or ½ IC50 of PP or GEM as single agents and in combination and subjected to MTT cell viability assays as described above. The MTT assay data were analysed using the highest single agent (HSA) model synergy and antagonism model by the Combenefit software (Cancer Research, Cambridge, UK) and the CompuSyn version 1.0 software (ComboSyn, Inc., Paramus, USA) according to instructions [18,19].
Statistical Analysis
Unless stated, data were obtained from at least three independent experimental repeats and analysed by a parametric unpaired t-test using the GraphPad Prism version 8.0 Software (GraphPad Prism software, USA). Error bars represent the standard error of the mean (SEM) and significance was accepted at *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
PP Exerts Short- and Long-Term Cytotoxicity in PDAC Cells
The short-term cytotoxic effects of PP (Figure 1A) were investigated by treating the PDAC cell lines, PANC-1 (derived from a primary pancreatic tumour), and CFPAC-1 (derived from a liver metastasis) for 72 h with the drug, followed by MTT assays. The gold-standard drug gemcitabine (GEM) was included as a positive control drug in all experiments. A concentration-dependent reduction in cell viability was noted for PP with IC50 values of 3.4 µM and 4.4 µM obtained in the PANC-1 and CFPAC-1 cell lines respectively (Figure 1B). The IC50 values obtained for GEM were 5.7 µM and 1.5 µM in the same cell lines respectively suggesting that whereas the PANC-1 cells were more sensitive to PP, the CFPAC-1 cells were more sensitive to GEM. Tumour recurrence is common in PDAC, and therefore, the long-term cytotoxic effects of PP were next investigated using clonogenic assays. Briefly, PANC-1, and CFPAC-1 cells were treated with PP or GEM at the indicated concentrations for 48 h after which they were replated in a drug-free medium for up to 14 days. As shown in Figure 1C, there was a complete absence of colonies in cells treated with IC50 PP, and cells treated with ½ IC50 PP formed comparable numbers of colonies to those treated with IC50 GEM. These results showed that in both PDAC cell lines, PP was more potent than GEM in long-term cytotoxicity assays.
PP Induces DNA Damage and Triggers Cell Cycle Arrest in PDAC Cells
To begin to explore the mechanism(s) by which PP exerts its cytotoxicity in PDAC cells, we determined whether it induced DNA damage. Briefly PANC-1 and CPFAC-1 cells were treated with PP and we assessed the levels of γH2AX, a phosphorylated form of histone H2AX that is recruited to damaged DNA following cytotoxic stress [20]. Western blotting results showed that PP induced a concentration-dependent increase in the levels of γH2AX (Figure 2A) and immunocytochemistry showed a significant increase in γH2AX foci in the nuclei of the PP-treated PDAC cells (Figure 2B). To determine whether the PP-induced DNA damage led to cell cycle arrest(s), flow cytometry and western blotting were performed. The results showed that, in both PDAC cell lines, PP treatment led to an S-phase cell cycle arrest (Figure 2C) and this correlated with a decrease in levels of the cell cycle regulators, cyclin A and cyclin B1 (Figure 2D). Consistent with previous reports, GEM induced an S-phase arrest in both cell lines as well as a G1 arrest in PANC-1 cells [21,22] and this was accompanied by increased cyclin A and cyclin B1 levels. Importantly, there was a significant increase in the sub-G1 peak in PP-treated cells which is indicative of DNA fragmentation and cell death [23] (Figure 2C).
PP Induces Apoptosis in PDAC Cells
To explore if PP induced cell death by apoptosis, we examined the effects of the drug on morphological, biochemical, and molecular markers of apoptosis. Light microscopy images showed that PP-treated cells exhibited morphological features of apoptosis, including cell shrinkage and membrane blebbing (Figure 3A). The pro-apoptotic effects of PP were confirmed by the acridine orange/ethidium bromide (AO/EB) staining assay, which distinguishes between viable and apoptotic cells based on changes in their membrane permeability (Figure 3B). AO is a green fluorescent dye which is membrane permeable and thus stains viable and dead cells and EB is a membrane-impermeable red fluorescent dye and therefore only stains dead cells which have a compromised membrane [24]. As expected, compared to vehicle-treated cells, there were a combination of PP-treated cells that stained yellow/green (early apoptosis) and reddish/orange (late apoptosis) (Figure 3B). Furthermore, treatment with PP led to a concentration-dependent increase in cleaved caspase-8 and cleaved caspase-9 which are molecular markers of the extrinsic and intrinsic pathways of apoptosis respectively, and this correlated with increased levels of the cleaved forms of executioner caspase-7 and its substrate, PARP (Figure 3C). Similar results were obtained for GEM-treated cells (Figure 3A-C) which is in line with previous studies that showed that it induces apoptosis in PDAC cells [21,22,25].
PP Induces Autophagic Cell Death in PDAC Cells
To determine whether PP induced autophagy as an additional mode of cell death, we measured the levels of LC3II, a marker of autophagosome formation, in drug-treated PANC-1 and CFPAC-1 cells. Western blotting and immunocytochemistry show that treatment with PP led to a concentration-dependent increase in LC3II levels (Figure 4A) and LC3 puncta in the cytoplasm of PDAC cells (Figure 4B) respectively. Interestingly, the levels of p62, a cargo protein that gets incorporated and degraded with the autophagosome, decreased in PP-treated PANC-1 cells but increased in PP-treated CFPAC-1 cells (Figure 4A). These results suggest that PP induced autophagic flux in PANC-1 cells but not in CFPAC-1 cells. Treatment with GEM induced LC3II levels and puncta and decreased p62 levels confirming that it induced autophagic flux in both cell lines (Figure 4A and B). To determine whether the PP-induced autophagic flux in PANC-1 cells leads to cell death, the levels of cleaved PARP were measured in the presence and absence of Bafilomycin A1, a chemical inhibitor of autophagy. Figure 4C shows that inhibiting autophagy reduced PARP cleavage in PP- and GEM-treated cells which confirms that PP and GEM induced autophagic cell death in PANC-1 cells.
PP Inhibits the AKT Signalling Pathway in PDAC Cells
The AKT signalling pathway is constitutively active in PDAC and it is well known to inhibit cell death in order to promote cancer cell survival [26]. Figure 4D confirmed that the AKT pathway was constitutively active in the PDAC cells used in this study because vehicle-treated cells expressed high levels of total AKT, AKT1 (the predominant AKT isoform in PDAC), and pAKT (the activated form of AKT). Importantly, levels of total AKT, AKT1, and pAKT decreased in PP-treated cells which indicated that PP inhibited this survival pathway in PDAC cells (Figure 4D). It is worth noting that while GEM also inhibited the levels of these AKT signalling molecules in CFPAC-1 cells, it had no effect on these proteins in PANC-1 cells. This suggests that PP may be more effective than GEM in preventing the survival of primary PDAC cells.
PP Inhibits PDAC Cell Invasion and Migration
Metastasis is the main cause of death in PDAC patients and therefore, the effects of PP on cell invasion and migration were next assessed. Results from transwell invasion assays showed that PP significantly reduced the number of PANC-1 and CFPAC-1 cells that invaded the lower chamber of the transwell plate and that it was more effective than GEM (Figure 5A). In vitro 2D scratch motility assay results revealed that PP also significantly decreased the migratory abilities of PANC-1 and CFPAC-1 cells (Figure 5B). In these assays, GEM was found to have no effect on PANC-1 migration and was less effective than PP at inhibiting CFPAC-1 migration. Cell invasiveness and migration are preceded by epithelial-to-mesenchymal transition (EMT) and consistent with the results shown in Figure 5A and B, treatment with PP led to an increase in the epithelial marker, E-cadherin, and a decrease in the mesenchymal markers, N-cadherin, β-catenin, and vimentin (Figure 5C). The results shown in Figure 5C also indicated that PP was more effective at inhibiting EMT than GEM. Taken together, the data showed that PP inhibited PDAC cell invasiveness and migration probably through the inhibition of EMT and that it was more effective in doing so than GEM.
PP Inhibits PDAC Spheroid Growth, Viability, and Invasiveness
Compared to 2D cell culture models, 3D spheroids more accurately recapitulate the architecture of tumours in vivo [27], and therefore, the cytotoxic and anti-invasive effects of PP were next validated and compared to GEM in PDAC spheroids. For cytotoxic effects, the spheroids were treated with two doses of PP or GEM on day 0 and day 3, and their sizes were monitored by light microscopy. The results showed that compared to vehicle-treated spheroids, treatment with PP or GEM disintegrated and reduced the size of PDAC spheroids and there were no significant differences in their effects (Figure 6A and B). Analysis of the effects of the drugs on PDAC spheroid viability using Calcein-AM, a fluorescent dye that stains viable cells only, showed that PP and GEM significantly inhibited PDAC spheroid viability (Figure 6C). To determine the effect of PP on spheroid invasion, the PDAC spheroids were embedded in collagen I, a component of the extracellular matrix, and treated with the drug. The results showed that while vehicle-treated spheroids displayed spindles that protruded from their periphery into the collagen I matrix, PP-treated spheroids showed no spindles (Figure 6D). Notably, GEM-treated spheroids showed spindles that protruded into the collagen I matrix, however, these spindles were shorter compared to those observed in vehicle-treated spheroids (Figure 6D). Together, the results showed that PP inhibited PDAC spheroid growth and invasion, and that it was more effective at inhibiting PDAC invasiveness than GEM in our 3D spheroid model.
PP Enhances the Effects of Gemcitabine on DNA Damage and PDAC Spheroid Viability
Drug combinations are a cornerstone for cancer therapy [28] because combining two drugs with different mechanisms of action and at lower doses may overcome drug resistance and lead to fewer side effects [29]. Our data suggest that PP and GEM may have different modus opperandi and we speculated that PP may improve the anti-cancer activity of GEM in PDAC patients. To test this, ⅛ IC50, ⅟4 IC50, and ½ IC50 concentrations of each drug and their combinations were assessed for their effect on PANC-1 and CFPAC-1 cell viability using the MTT assay. These concentrations were tested so as to determine the lowest concentrations at which PP and GEM acted synergistically. The results showed that all combinations significantly inhibited cell viability more than the drugs on their own in PANC-1 cells (Figure 7A). In drug cocktails, concentrations that give a response greater than what is expected when used together (i.e. synergy) are desired over those that give an expected response (i.e. additive) [30]. To determine the concentrations that result in a synergistic effect, the MTT assay results were analysed using publicly available Combenefit software, and the highest single agent (HSA) model synergy and antagonism surface maps were generated. In the surface maps, the blue colour shows synergy, the green colour shows an additive effect and the red colour shows antagonism. As shown in Figure 7B, PP and GEM showed a synergistic effect in PANC-1 cells but an additive effect in CFPAC-1 cells. To determine the lowest concentrations of the drugs that gave the highest synergy in PANC-1 cells, the MTT results were further analysed using publicly available Compusyn software to generate the combination index (CI). In this analysis, CI<1 shows synergy, CI=1 shows an additive effect, and CI>1 shows antagonism [31]. The combination of ⅛ IC50 PP + ⅛ IC50 GEM (hereafter referred to as PP+GEM) showed the strongest synergy with a CI of 0.38 and only the ½ IC50 PP + ½ IC50 GEM combination showed an antagonistic effect with a CI of 1.14 (Figure 7C). We therefore next compared the ability of PP+GEM to PP (⅛ IC50) and GEM (⅛ IC50) on their own to induce anti-cancer activity in PANC-1 cells. Western blotting showed that all treatments resulted in increased levels of the DNA damage marker γH2AX but the band for γH2AX was almost twice as intense in the cells treated with PP+GEM (Figure 7D). Light microscopy images show that cells treated with ⅛ IC50 PP or ⅛ IC50 GEM on their own had a flattened morphology reminiscent of cells undergoing senescence and PP+GEM led to cell detachment and cell shrinkage indicative of cell death (Figure 7E). Furthermore, ⅛ IC50 GEM had no significant effect on spheroid growth and viability but ⅛ IC50 PP on its own significantly inhibited spheroid growth and viability and this was significantly enhanced when it was combined with GEM (PP+GEM) (Figure 7F and G). Taken together, the data showed that PP+GEM was more effective than low concentrations (i.e., ⅛ IC50) of PP and GEM as single agents in 2D and 3D PDAC culture models.
Discussion
Pancreatic cancer, commonly referred to as PDAC, is a fatal disease with an incredibly poor prognosis and a 5-year survival rate of less than 10% [32]. Drug repurposing has shown great promise in the rapid identification of effective anti-PDAC drugs and several FDA-approved non-cancer drugs are currently being explored for the treatment of this lethal neoplasm [8]. The antihelminthic drug pyrvinium pamoate (PP) was of interest to this study because it was shown to inhibit mitochondrial function under conditions that mimic the PDAC TME, but details of its anti-PDAC activites and the mechanism(s) involved are not known [12,13,14]. Here we showed using 2D and 3D PDAC cell culture models that PP induces double-strand DNA breaks, cell cycle arrests, apoptotic and autophagic cell deaths and inhibits the PI3K/AKT survival pathway. Furthermore, we provided important data that show that PP can enhance the anti-cancer effects of GEM in these cell culture models.
Systemic chemotherapies used to treat PDAC do not significantly improve patient survival due to their high toxicity profiles, which lead to adverse side effects, as well as tumour drug resistance and their failure to inhibit metastatic spread [6]. Here we provide several lines of evidence that PP may be an effective drug that can be repurposed to treat PDAC patients because it exerts cytoxocity in PDAC cells while exerting minimal cytotoxicity to normal cells, inhibits PDAC long-term survival and recurrence as well as metastasis. Indeed, we showed, using 2D and 3D cell culture models, that PP was cytotoxic in PDAC cells. Our findings together with that of others who showed that PP induced cytoxicity in PDAC cells while sparing nonmalignant human pancreatic epithelial cells suggest that it is selective for PDAC cells and may therefore be associated with limited side effects [33,34,35]. Using clonogenic assays we showed that PP prevented the colony forming ability of PDAC cells and hence inhibited their long-term survival and recurrence. These results are significant because post-treatment, tumour cells can retain their clonogenicity which ultimately leads to tumour recurrence and relapse and the clonogenic assay is therefore an effective tool for predicting a patient's long-term sensitivity to anti-cancer drugs [36]. Furthermore, our findings that PP inhibited PDAC spheroid growth, viability and invasion are promising, because compared to 2D cell cultures, 3D spheroids give a more accurate drug response because they closely resemble PDAC tumours in terms of their structure and their increased chemoresistance [37]. Furthermore, 3 in 4 PDAC patients develop recurrence within 2 years after resection which suggests that even the small proportion of patients that undergo surgical resection harbor micro-metastasis [6]. Indeed, PDAC cells acquire pro-metastatic traits which enable them to disseminate and spread before primary tumour formation, and as a result, PDAC patients present with liver (76–80%), peritoneum (48%), or lung (45%) metastasis at diagnosis [38,39]. Among the key regulators of PDAC metastasis is the canonical Wnt/β-catenin pathway which, upon activation, leads to the translocation of β-catenin into the nucleus where it facilitates the transcription of genes that induce EMT, invasion, and migration to distant sites [40,41]. PP is a well-known inhibitor of the Wnt/β-catenin pathway and it is therefore tempting to speculate that it may have in part exerted its anti-metastatic effects in our PDAC cells by inhibiting this pathway. Future studies should further explore this.
An understanding of the mechanism(s) by which anti-cancer drugs function may provide insight into the potential mechanisms of tumour drug resistance [42]. This study provided evidence that the anti-cancer effects of PP occur through mechanisms that may enable it to escape resistance by PDAC cells. Indeed, we show for the first time in PDAC that PP induced double-strand DNA breaks, an S-phase cell cycle arrest, inhibited the PI3K/AKT cell survival pathway, and induced intrinsic and extrinsic apoptosis as well as autophagic cell death. Our findings that PP triggers apoptotic and autophagic cell death is important because anti-cancer agents that induce more than one form of programmed cell death are more efficacious and less prone to resistance [43]. Furthermore, its ability to activate intrinsic and extrinsic apoptotic pathways is significant because the inactivation of the intrinsic apopotic pathway is one of the mechanisms by which PDAC cells become resistant to GEM [43,44]. In addition, our observations that PP induced autophagic cell death is of interest because autophagy is elevated in the late stages of PDAC where it fuels tumour metabolism to promote tumour growth and drug resistance [45]. Consistent with our data, another antihelminthic drug niclosamide inhibited PARP cleavage in the presence of a late inhibitor of autophagy, Bafilomycin A1, in COLO 357 and SW1990 PDAC cells [46]. Finally, the PI3K/AKT pathway contributes to the mechanisms by which PDAC cells develop drug resistance and here we showed that PP inhibited this pathway [47]. Interestingly, inhibiting the PI3K/AKT pathway with wortmannin and LY294002 sensitised PDAC cells to GEM [48] and inhibition of phosphatidylinositide 3-kinase-protein kinase B (PKB)/AKT by wortmannin was shown to promote the anti-cancer activities of GEM in PDAC mouse models [49]. Together, these studies suggests that when combined with GEM, inhibitors of the AKT pathway such as PP may be effective in the treatment of PDAC.
Combination therapies have proven to be an effective strategy for the management of PDAC. Indeed combining low doses of drugs with dinstict mechanisms of action may overcome drug resistance and lead to an increased efficacy without or with minimal side effects [29]. Most anti-PDAC combination regimens are centered on GEM because it is the only FDA-approved monotherapy and several of these including the FDA-approved GEM plus nab-paclitaxel have improved patient survival [50]. However, these combinations fail to achieve complete remission and they are associated with high toxicities and a high degree of intrinsic or acquired resistance due to the PDAC TME [51]. Indeed, the PDAC TME limits delivery of GEM to tumours by downregulating the concentrative and equilibrative nucleoside transporters and enzymes that are involved in GEM transport and metabolism, respectively [52]. Therefore, less cytotoxic combinations that target both PDAC cells and the TME are urgently needed to combat PDAC. PP was previously shown to be effective under conditions that mimic the PDAC TME [12,13,14] and we therefore investigated whether GEM and PP acted synergistically in PDAC. Using 2D cell cultures and 3D spheroids, we showed for the first time that PP can improve the anti-cancer activities of GEM in PDAC. It is therefore tempting to speculate that combining PP with GEM may lead to significant improvements in PDAC treatment. In this regard, it is worth noting that there is an ongoing Phase I clinical trial (NCT05055323) which is investigating the safety and tolerability of pyrvinium pamoate in early-stage PDAC patients [53] and based on our findings we recommend that this be extended to include its combination with gemcitabine.
Conclusions
This study our study provided compelling evidence that pyrvinium pamoate may be an effective drug that can be repurposed to treat PDAC patients on its own or together with gemcitibine and we revealed novel mechanisms by which pyrvinium pamoate achieved anti-PDAC activities. Additional experiments are required to fully characterise and investigate the mechanisms of action of the PP+GEM combination and their efficacy needs to be validated in vivo, in for example PDAC patient-derived xenografts models.
Author Contributions
K.S: Formal analysis, Investigation, Validation, Data curation, Writing - original draft. J.B: Writing - review & editing. S.P: Conceptualization, Formal analysis, Resources, Writing an original draft, Writing - review & editing, Supervision, Project administration, Funding acquisition.
Funding
Prof Prince gratefully acknowledges and thank the University of Cape Town under the UCT Vision 2030 Grand Challenges Programme, the National Research Foundation of South Africa under a Competitive Programme for Rated Researchers, the International Centre for Genetic Engineering and Biotechnology (ICGEB) under a Collaborative Research Programme, the South African Medical Research Council (SAMRC) under a Self-Initiated Research Grant for financial support as well as through the SAMRC Gynaecological Cancer Research Centre (GCRC). The views and opinions expressed are those of the author(s) and do not necessarily represent the official views of the SAMRC.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this article are available.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, J.; Zhang, H.; Xiu, C.; Gao, C.; Wu, S.; Bai, J.; Shen, Q.; Yin, T. METTL3 Promotes Pancreatic Cancer Proliferation and Stemness by Increasing Stability of ID2 MRNA in a M6A-Dependent Manner. Cancer Lett. 2023, 565, 216222. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.; Kumar, S.; Sandeep, K.; Patel, S.K.S. Therapeutic Approaches in Pancreatic Cancer: Recent Updates. Biomedicines 2023, 11, 1611. [Google Scholar] [CrossRef] [PubMed]
- Pourhoseingholi, M.A.; Ashtari, S.; Hajizadeh, N.; Fazeli, Z.; Zali, M.R. Systematic Review of Pancreatic Cancer Epidemiology in Asia-Pacific Region: Major Patterns in GLOBACON 2012. Gastroenterol. Hepatol. from bed to bench 2017, 10, 245–257. [Google Scholar]
- de Scordilli, M.; Michelotti, A.; Zara, D.; Palmero, L.; Alberti, M.; Noto, C.; Totaro, F.; Foltran, L.; Guardascione, M.; Iacono, D.; et al. Preoperative Treatments in Borderline Resectable and Locally Advanced Pancreatic Cancer: Current Evidence and New Perspectives. Crit. Rev. Oncol. Hematol. 2023, 186, 104013. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic Cancer: Advances and Challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef]
- Miller, A.L.; Garcia, P.L.; Yoon, K.J. Developing Effective Combination Therapy for Pancreatic Cancer: An Overview. Pharmacol. Res. 2020, 155, 104740. [Google Scholar] [CrossRef] [PubMed]
- De Lellis, L.; Veschi, S.; Tinari, N.; Mokini, Z.; Carradori, S.; Brocco, D.; Florio, R.; Grassadonia, A.; Cama, A. Drug Repurposing, an Attractive Strategy in Pancreatic Cancer Treatment: Preclinical and Clinical Updates. Cancers (Basel). 2021, 13, 3946. [Google Scholar] [CrossRef]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug Repurposing in Cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Momtazi-borojeni, A.A.; Abdollahi, E.; Ghasemi, F.; Caraglia, M.; Sahebkar, A. The Novel Role of Pyrvinium in Cancer Therapy. J. Cell. Physiol. 2018, 233, 2871–2881. [Google Scholar] [CrossRef]
- Schultz, C.W.; Nevler, A. Pyrvinium Pamoate: Past, Present, and Future as an Anti-Cancer Drug. Biomedicines 2022, 10, 3249. [Google Scholar] [CrossRef]
- Esumi, H.; Lu, J.; Kurashima, Y.; Hanaoka, T. Antitumor Activity of Pyrvinium Pamoate, 6-(Dimethylamino)-2-[2-(2,5-Dimethyl-1-Phenyl-1H-Pyrrol-3-Yl)Ethenyl]-1-Methyl-Quinolinium Pamoate Salt, Showing Preferential Cytotoxicity during Glucose Starvation. Cancer Sci. 2004, 95, 685–690. [Google Scholar] [CrossRef]
- Tomitsuka, E.; Kita, K.; Esumi, H. An Anticancer Agent, Pyrvinium Pamoate Inhibits the NADH–Fumarate Reductase System—a Unique Mitochondrial Energy Metabolism in Tumour Microenvironments. J. Biochem. 2012, 152, 171–183. [Google Scholar] [CrossRef]
- Schultz, C.W.; McCarthy, G.A.; Nerwal, T.; Nevler, A.; DuHadaway, J.B.; McCoy, M.D.; Jiang, W.; Brown, S.Z.; Goetz, A.; Jain, A.; et al. The FDA-Approved Anthelmintic Pyrvinium Pamoate Inhibits Pancreatic Cancer Cells in Nutrient-Depleted Conditions by Targeting the Mitochondria. Mol. Cancer Ther. 2021, 20, 2166–2176. [Google Scholar] [CrossRef]
- Liu, Q.; Liao, Q.; Zhao, Y. Chemotherapy and Tumor Microenvironment of Pancreatic Cancer. Cancer Cell Int. 2017, 17, 68. [Google Scholar] [CrossRef]
- Willmer, T.; Peres, J.; Mowla, S.; Abrahams, A.; Prince, S. The T-Box Factor TBX3 Is Important in S-Phase and Is Regulated by c-Myc and Cyclin A-CDK2. Cell Cycle 2015, 14, 3173–3183. [Google Scholar] [CrossRef]
- Prince, S.; Wiggins, T.; Hulley, P.A.; Kidson, S.H. Stimulation of Melanogenesis by Tetradecanoylphorbol 13-Acetate (TPA) in Mouse Melanocytes and Neural Crest Cells. Pigment Cell Res. 2003, 16, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An Interactive Platform for the Analysis and Visualization of Drug Combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef]
- Plesca, D.; Mazumder, S.; Almasan, A. Chapter 6 DNA Damage Response and Apoptosis. In; 2008; pp. 107–122.
- Li, D.; Jia, Y.-M.; Cao, P.-K.; Wang, W.; Liu, B.; Li, Y.-L. Combined Effect of 125 I and Gemcitabine on PANC-1 Cells: Cellular Apoptosis and Cell Cycle Arrest. J. Cancer Res. Ther. 2018, 14, 1476. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Trivieri, N.; Villani, A.; Terracciano, F.; Latiano, T.P.; Potenza, A.; Perri, F.; Binda, E.; Pazienza, V. Improving Gemcitabine Sensitivity in Pancreatic Cancer Cells by Restoring MiRNA-217 Levels. Biomolecules 2021, 11, 639. [Google Scholar] [CrossRef]
- Riccardi, C.; Nicoletti, I. Analysis of Apoptosis by Propidium Iodide Staining and Flow Cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef]
- Chi, C.-F.; Hu, F.-Y.; Wang, B.; Li, T.; Ding, G.-F. Antioxidant and Anticancer Peptides from the Protein Hydrolysate of Blood Clam (Tegillarca Granosa) Muscle. J. Funct. Foods 2015, 15, 301–313. [Google Scholar] [CrossRef]
- Park, S.H.; Sung, J.H.; Kim, E.J.; Chung, N. Berberine Induces Apoptosis via ROS Generation in PANC-1 and MIA-PaCa2 Pancreatic Cell Lines. Brazilian J. Med. Biol. Res. 2015, 48, 111–119. [Google Scholar] [CrossRef]
- Meng, Y.; Wang, W.; Kang, J.; Wang, X.; Sun, L. Role of the PI3K/AKT Signalling Pathway in Apoptotic Cell Death in the Cerebral Cortex of Streptozotocin-Induced Diabetic Rats. Exp. Ther. Med. 2017, 13, 2417–2422. [Google Scholar] [CrossRef]
- Damerell, V.; Ambele, M.A.; Salisbury, S.; Neumann-Mufweba, A.; Durandt, C.; Pepper, M.S.; Prince, S. The C-Myc/TBX3 Axis Promotes Cellular Transformation of Sarcoma-Initiating Cells. Front. Oncol. 2022, 11. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- KalantarMotamedi, Y.; Choi, R.J.; Koh, S.-B.; Bramhall, J.L.; Fan, T.-P.; Bender, A. Prediction and Identification of Synergistic Compound Combinations against Pancreatic Cancer Cells. iScience 2021, 24, 103080. [Google Scholar] [CrossRef] [PubMed]
- Roell, K.R.; Reif, D.M.; Motsinger-Reif, A.A. An Introduction to Terminology and Methodology of Chemical Synergy—Perspectives from Across Disciplines. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. The Combination Index (CI < 1) as the Definition of Synergism and of Synergy Claims. Synergy 2018, 7, 49–50. [Google Scholar] [CrossRef]
- Chitty, J.L.; Yam, M.; Perryman, L.; Parker, A.L.; Skhinas, J.N.; Setargew, Y.F.I.; Mok, E.T.Y.; Tran, E.; Grant, R.D.; Latham, S.L.; et al. A First-in-Class Pan-Lysyl Oxidase Inhibitor Impairs Stromal Remodeling and Enhances Gemcitabine Response and Survival in Pancreatic Cancer. Nat. Cancer 2023. [Google Scholar] [CrossRef]
- Zheng, W.; Hu, J.; Lv, Y.; Bai, B.; Shan, L.; Chen, K.; Dai, S.; Zhu, H. Pyrvinium Pamoate Inhibits Cell Proliferation through ROS-Mediated AKT-Dependent Signaling Pathway in Colorectal Cancer. Med. Oncol. 2021, 38, 21. [Google Scholar] [CrossRef]
- Wiegering, A.; Uthe, F.-W.; Hüttenrauch, M.; Mühling, B.; Linnebacher, M.; Krummenast, F.; Germer, C.-T.; Thalheimer, A.; Otto, C. The Impact of Pyrvinium Pamoate on Colon Cancer Cell Viability. Int. J. Colorectal Dis. 2014, 29, 1189–1198. [Google Scholar] [CrossRef]
- Feng, J.; Jiang, W.; Liu, Y.; Huang, W.; Hu, K.; Li, K.; Chen, J.; Ma, C.; Sun, Z.; Pang, X. Blocking STAT3 by Pyrvinium Pamoate Causes Metabolic Lethality in KRAS-Mutant Lung Cancer. Biochem. Pharmacol. 2020, 177, 113960. [Google Scholar] [CrossRef]
- Fiebig, H. .; Maier, A.; Burger, A.. Clonogenic Assay with Established Human Tumour Xenografts. Eur. J. Cancer 2004, 40, 802–820. [Google Scholar] [CrossRef]
- Liu, X.; Gündel, B.; Li, X.; Liu, J.; Wright, A.; Löhr, M.; Arvidsson, G.; Heuchel, R. 3D Heterospecies Spheroids of Pancreatic Stroma and Cancer Cells Demonstrate Key Phenotypes of Pancreatic Ductal Adenocarcinoma. Transl. Oncol. 2021, 14, 101107. [Google Scholar] [CrossRef]
- Joshi, V.B.; Gutierrez Ruiz, O.L.; Razidlo, G.L. The Cell Biology of Metastatic Invasion in Pancreatic Cancer: Updates and Mechanistic Insights. Cancers (Basel). 2023, 15, 2169. [Google Scholar] [CrossRef]
- Miquel, M.; Zhang, S.; Pilarsky, C. Pre-Clinical Models of Metastasis in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, S.; Jin, R.; Xu, H.; Li, Y.; Chen, Y.; Zhao, G. Pyrvinium Pamoate Regulates MGMT Expression through Suppressing the Wnt/β-Catenin Signaling Pathway to Enhance the Glioblastoma Sensitivity to Temozolomide. Cell Death Discov. 2021, 7, 288. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Li, Y.; Li, B.; Zhang, M.; Liu, Y.; Yao, Y.; Li, D. NMIIA Promotes Tumor Growth and Metastasis by Activating the Wnt/β-Catenin Signaling Pathway and EMT in Pancreatic Cancer. Oncogene 2019, 38, 5500–5515. [Google Scholar] [CrossRef] [PubMed]
- Cattley, R.C.; Radinsky, B.R. Cancer Therapeutics: Understanding the Mechanism of Action. Toxicol. Pathol. 2004, 32, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Omoruyi, S.I.; Ekpo, O.E.; Semenya, D.M.; Jardine, A.; Prince, S. Exploitation of a Novel Phenothiazine Derivative for Its Anti-Cancer Activities in Malignant Glioblastoma. Apoptosis 2020, 25, 261–274. [Google Scholar] [CrossRef]
- Jia, Y.; Xie, J. Promising Molecular Mechanisms Responsible for Gemcitabine Resistance in Cancer. Genes Dis. 2015, 2, 299–306. [Google Scholar] [CrossRef]
- Yamamoto, K.; Iwadate, D.; Kato, H.; Nakai, Y.; Tateishi, K.; Fujishiro, M. Targeting Autophagy as a Therapeutic Strategy against Pancreatic Cancer. J. Gastroenterol. 2022, 57, 603–618. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, J.B.; Bhatia, R.; Kanchan, R.K.; Raut, P.; Mallapragada, S.; Ly, Q.P.; Batra, S.K.; Rachagani, S. Repurposing Niclosamide for Targeting Pancreatic Cancer by Inhibiting Hh/Gli Non-Canonical Axis of Gsk3β. Cancers (Basel). 2021, 13. [Google Scholar] [CrossRef]
- Mehra, S.; Deshpande, N.; Nagathihalli, N. Targeting PI3K Pathway in Pancreatic Ductal Adenocarcinoma: Rationale and Progress. Cancers (Basel). 2021, 13, 4434. [Google Scholar] [CrossRef]
- Ng SSW; Tsao, M. S.; Chow, S.; Hedley, D.W. Inhibition of Phosphatidylinositide 3-Kinase Enhances Gemcitabine-Induced Apoptosis in Human Pancreatic Cancer Cells. Cancer Res. 2000, 60, 5451–5455. [Google Scholar]
- Ng, S.S.; Tsao, M.S.; Nicklee, T.; Hedley, D.W. Wortmannin Inhibits Pkb/Akt Phosphorylation and Promotes Gemcitabine Antitumor Activity in Orthotopic Human Pancreatic Cancer Xenografts in Immunodeficient Mice. Clin. Cancer Res. 2001, 7, 3269–3275. [Google Scholar]
- Lei, F.; Xi, X.; Batra, S.K.; Bronich, T.K. Combination Therapies and Drug Delivery Platforms in Combating Pancreatic Cancer. J. Pharmacol. Exp. Ther. 2019, 370, 682–694. [Google Scholar] [CrossRef]
- Beutel, A.K.; Halbrook, C.J. Barriers and Opportunities for Gemcitabine in Pancreatic Cancer Therapy. Am. J. Physiol. Physiol. 2023, 324, C540–C552. [Google Scholar] [CrossRef]
- Natu, J.; Nagaraju, G.P. Gemcitabine Effects on Tumor Microenvironment of Pancreatic Cancer: Special Focus on Resistance Mechanisms and Metronomic Therapies. Cancer Lett. 2023, 216382. [Google Scholar] [CrossRef] [PubMed]
- Ponzini, F.M.; Schultz, C.W.; Leiby, B.E.; Cannaday, S.; Yeo, T.; Posey, J.; Bowne, W.B.; Yeo, C.; Brody, J.R.; Lavu, H.; et al. Repurposing the FDA-Approved Anthelmintic Pyrvinium Pamoate for Pancreatic Cancer Treatment: Study Protocol for a Phase I Clinical Trial in Early-Stage Pancreatic Ductal Adenocarcinoma. BMJ Open 2023, 13, e073839. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pyrvinium pamoate exerts short- and long-term cytotoxicity in PDAC cells. (A) Chemical structure of pyrvinium pamoate (PP). (B) MTT cell viability assays of PANC-1 and CFPAC-1 cells post 72h treatment with PP or gemcitabine (GEM). The IC50 values were determined using GraphPad Prism version 8.0 software (GraphPad Prism software, USA) from sigmoidal plots of data obtained from three independent experiments performed in quadruplicate. (C) Representative images and quantification of clonogenic assays of PANC-1 and CFPAC-1 cells treated for 48h with ½ IC50 or IC50 PP or IC50 GEM and replated at low densities and left for 14 days in drug-free medium to form colonies. Colonies were stained with crystal violet and images from three independent experiments were quantified using the ImageJ Colony Area plugin. The graph represents the mean colony area ± SEM of each treatment condition expressed as a percentage of the vehicle control. Data are represented as mean ± SEM and ***p < 0.001 and **** < 0.0001.
Figure 1.
Pyrvinium pamoate exerts short- and long-term cytotoxicity in PDAC cells. (A) Chemical structure of pyrvinium pamoate (PP). (B) MTT cell viability assays of PANC-1 and CFPAC-1 cells post 72h treatment with PP or gemcitabine (GEM). The IC50 values were determined using GraphPad Prism version 8.0 software (GraphPad Prism software, USA) from sigmoidal plots of data obtained from three independent experiments performed in quadruplicate. (C) Representative images and quantification of clonogenic assays of PANC-1 and CFPAC-1 cells treated for 48h with ½ IC50 or IC50 PP or IC50 GEM and replated at low densities and left for 14 days in drug-free medium to form colonies. Colonies were stained with crystal violet and images from three independent experiments were quantified using the ImageJ Colony Area plugin. The graph represents the mean colony area ± SEM of each treatment condition expressed as a percentage of the vehicle control. Data are represented as mean ± SEM and ***p < 0.001 and **** < 0.0001.
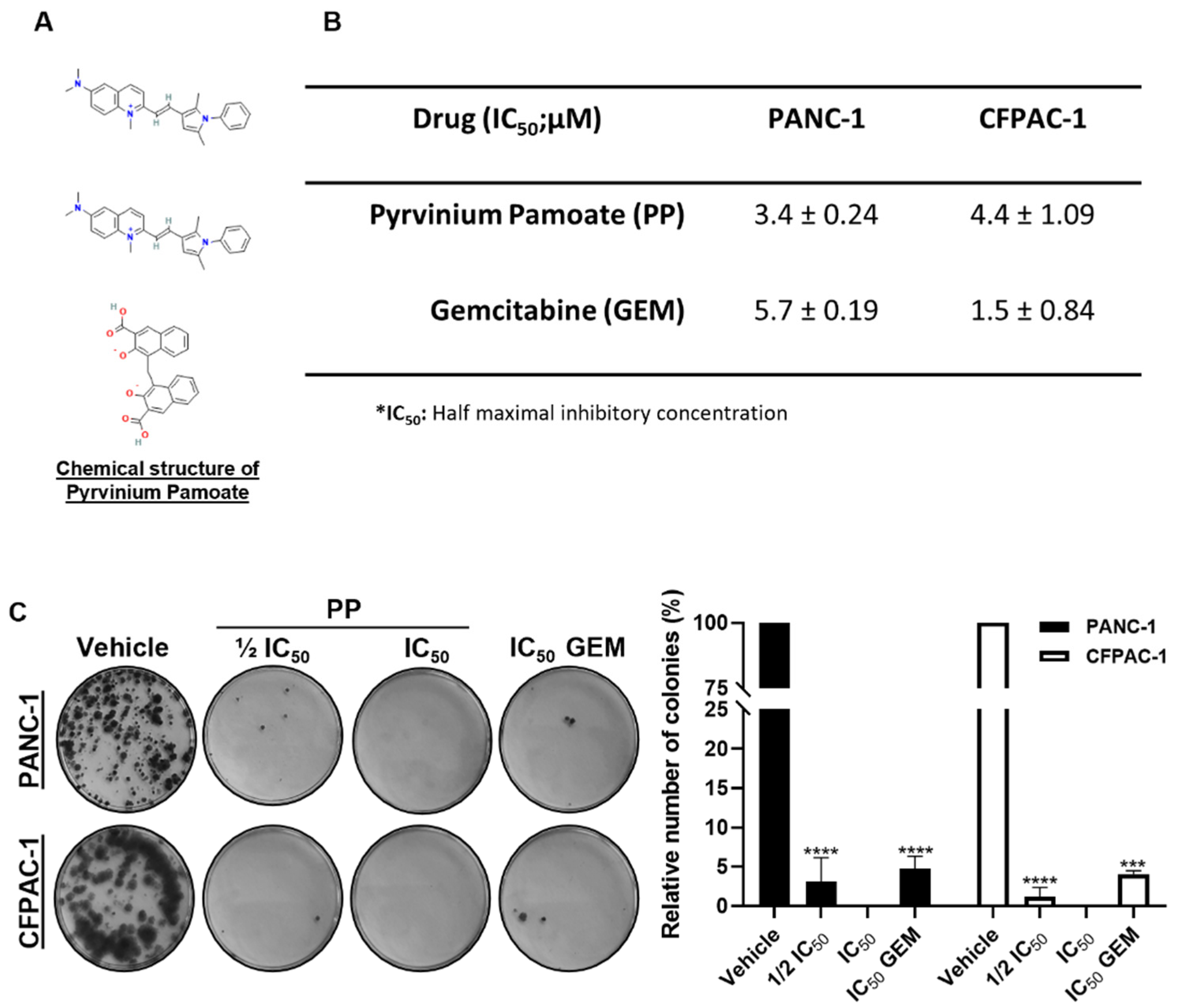
Figure 2.
: DNA damage induction and cell cycle arrest by pyrvinium pamoate in PDAC cells. (A) Western blot showing the levels of γH2AX in PANC-1 and CFPAC-1 cells post 72h treatment with indicated concentrations of PP or GEM. (B) Representative confocal microscope images (×630; Carl Zeiss LSM 510; Scale bar = 20 μM) and quantitation of γH2AX foci in PANC-1 and CFPAC-1 cells treated for 72h with IC50 PP or GEM. γH2AX was detected using a fluorophore-conjugated Alexa-488 secondary antibody and DAPI was used to stain the nuclei. (C) Flow cytometry analysis of cells treated for 72h with IC50 PP or GEM and stained with propidium iodide for cell cycle analysis. Graphs represent the mean proportion of cells in each phase of the cell cycle. (D) Western blot analyses of protein harvested from cells treated as indicated and incubated with antibodies against cell cycle markers cyclin A and cyclin B1. The blots shown are representative of three independent experiments where β-actin was used as a loading control. Band intensities used for densitometry readings were obtained using ImageJ and protein levels are represented as a ratio of protein of interest/β-actin normalized to the vehicle control (or first lane with a band). Data are represented as mean ± SEM and *, p < 0.05; **, p < 0.01.
Figure 2.
: DNA damage induction and cell cycle arrest by pyrvinium pamoate in PDAC cells. (A) Western blot showing the levels of γH2AX in PANC-1 and CFPAC-1 cells post 72h treatment with indicated concentrations of PP or GEM. (B) Representative confocal microscope images (×630; Carl Zeiss LSM 510; Scale bar = 20 μM) and quantitation of γH2AX foci in PANC-1 and CFPAC-1 cells treated for 72h with IC50 PP or GEM. γH2AX was detected using a fluorophore-conjugated Alexa-488 secondary antibody and DAPI was used to stain the nuclei. (C) Flow cytometry analysis of cells treated for 72h with IC50 PP or GEM and stained with propidium iodide for cell cycle analysis. Graphs represent the mean proportion of cells in each phase of the cell cycle. (D) Western blot analyses of protein harvested from cells treated as indicated and incubated with antibodies against cell cycle markers cyclin A and cyclin B1. The blots shown are representative of three independent experiments where β-actin was used as a loading control. Band intensities used for densitometry readings were obtained using ImageJ and protein levels are represented as a ratio of protein of interest/β-actin normalized to the vehicle control (or first lane with a band). Data are represented as mean ± SEM and *, p < 0.05; **, p < 0.01.
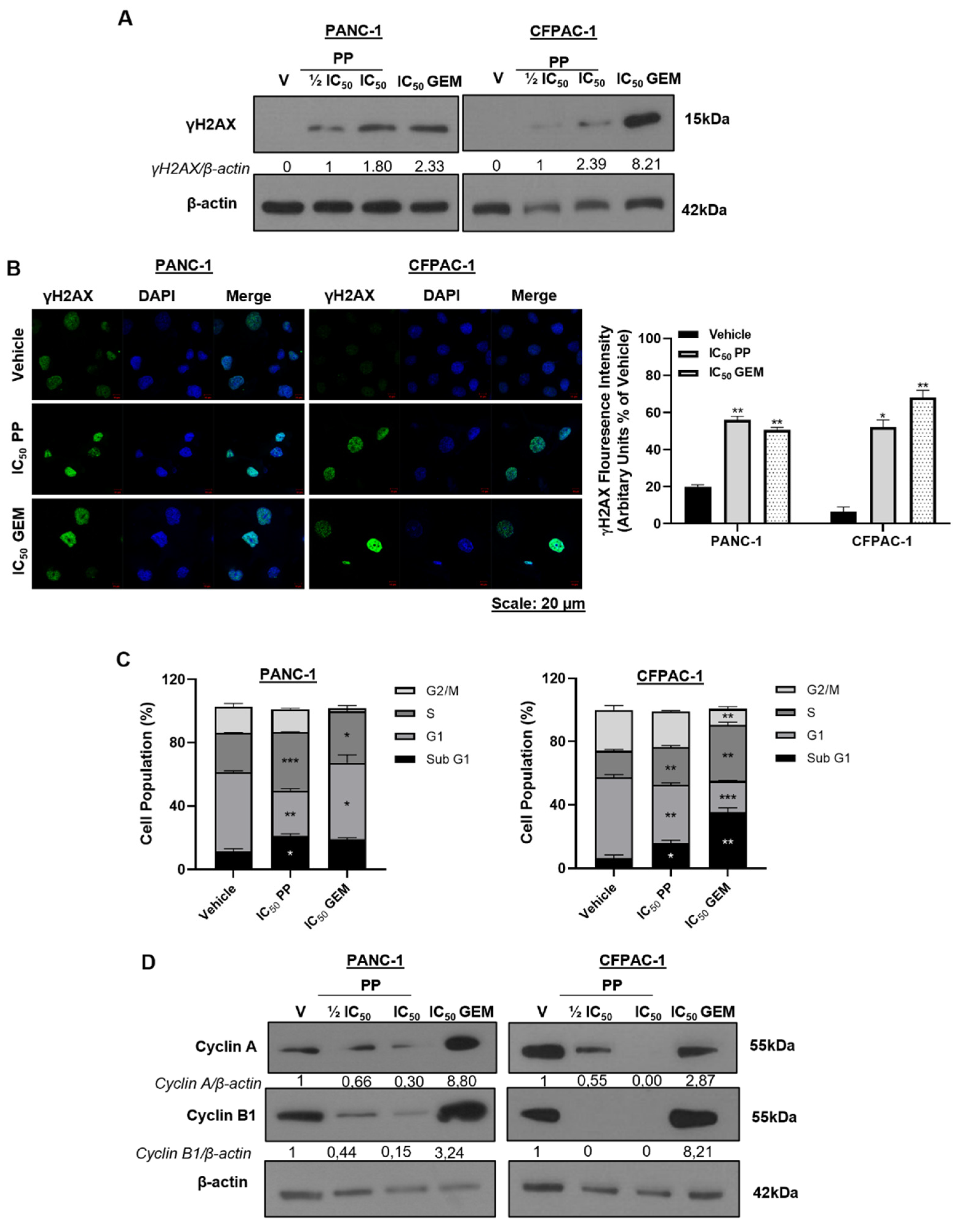
Figure 3.
Apoptosis induction by pyrvinium pamoate in PDAC cells. (A) Representative light microscopy images (×200; EVOS XL AMEX1000 Core Imaging System) showing the impact of PP or GEM on the morphology of PANC-1 and CFPAC-1 cells. The coloured circles correspond to the magnified images on the right and indicate morphological features of apoptosis (Red: cell shrinking; yellow: membrane blebbing; and black: apoptotic bodies). (B) Representative fluorescence microscope images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) showing the effects of PPP or GEM on biochemical features of apoptosis post 72h of treatment PP or IC50 of GEM as indicated. Cells were stained with acridine orange and ethidium bromide, and the arrows indicate cells in different stages of apoptosis (white arrows: viable cells; red arrows: early apoptotic cells, and green arrows: late apoptotic cells). (C) Western blot analysis of molecular markers of apoptosis caspase-8, caspase-9, cleaved caspase-7, and PARP in cells treated as indicated. The blots shown are representative of three independent experiments where β-actin was used as a loading control. Band intensities used for densitometry readings were obtained using ImageJ and protein levels are represented as a ratio of protein of interest/β-actin normalized to the vehicle control (or first lane with a band).
Figure 3.
Apoptosis induction by pyrvinium pamoate in PDAC cells. (A) Representative light microscopy images (×200; EVOS XL AMEX1000 Core Imaging System) showing the impact of PP or GEM on the morphology of PANC-1 and CFPAC-1 cells. The coloured circles correspond to the magnified images on the right and indicate morphological features of apoptosis (Red: cell shrinking; yellow: membrane blebbing; and black: apoptotic bodies). (B) Representative fluorescence microscope images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) showing the effects of PPP or GEM on biochemical features of apoptosis post 72h of treatment PP or IC50 of GEM as indicated. Cells were stained with acridine orange and ethidium bromide, and the arrows indicate cells in different stages of apoptosis (white arrows: viable cells; red arrows: early apoptotic cells, and green arrows: late apoptotic cells). (C) Western blot analysis of molecular markers of apoptosis caspase-8, caspase-9, cleaved caspase-7, and PARP in cells treated as indicated. The blots shown are representative of three independent experiments where β-actin was used as a loading control. Band intensities used for densitometry readings were obtained using ImageJ and protein levels are represented as a ratio of protein of interest/β-actin normalized to the vehicle control (or first lane with a band).
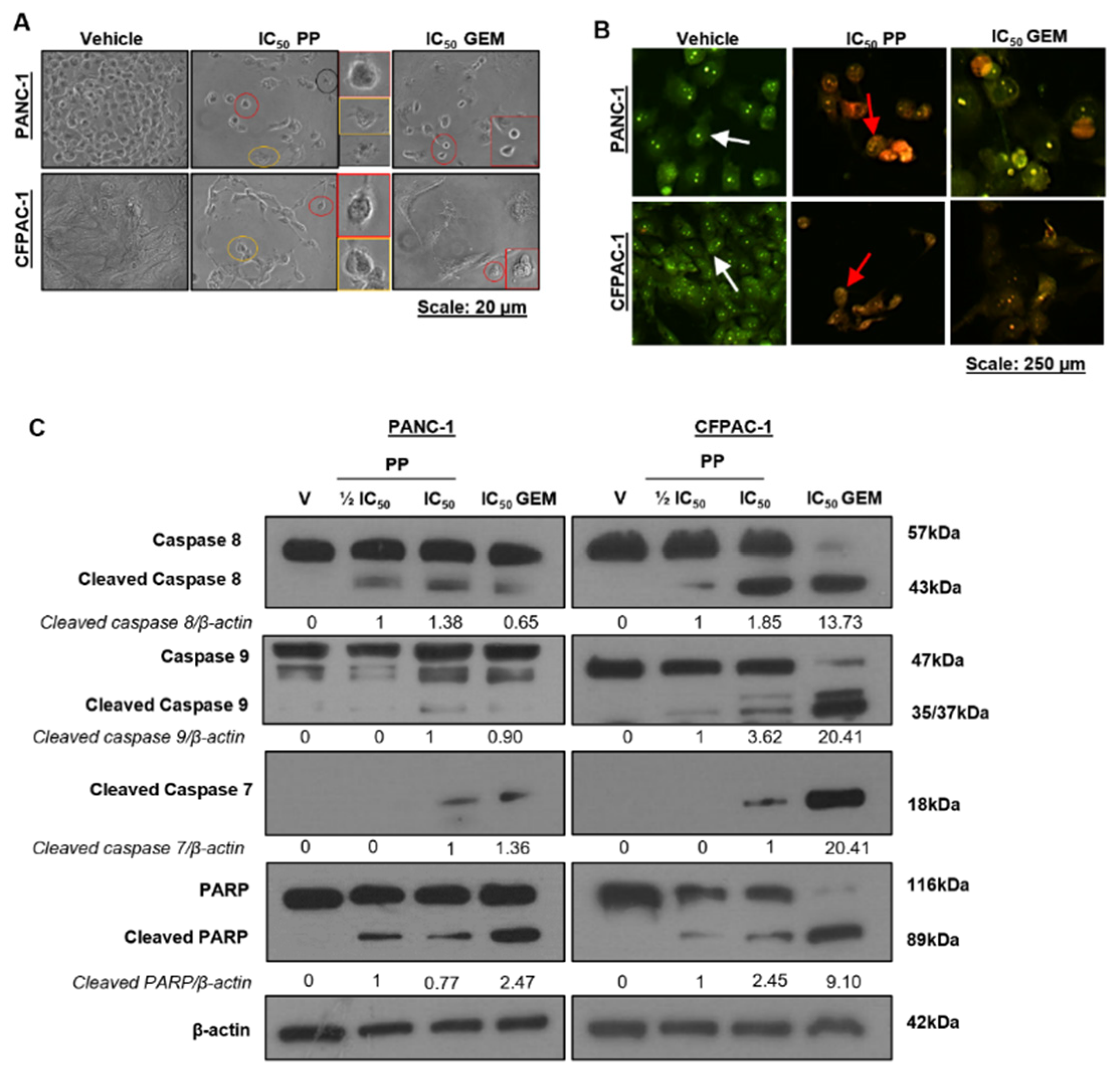
Figure 4.
Pyrvinium pamoate induces autophagic cell death and inhibits the PI3K/AKT pathway in PDAC cells. (A) Western analysis of molecular markers of autophagy LC3I/II and p62/SQSTM1 in PANC-1 and CFPAC-1 cells post 72h treatment as indicated. (B) Representative confocal microscope images (×630; Carl Zeiss LSM 510; Scale bar = 20 μM) showing LC3 puncta in cells treated as in (A). LC3 was detected using a fluorophore-conjugated Alexa-488 secondary antibody and DAPI was used to stain the nuclei. (C) Western blot analysis of LC3I/II and PARP in PANC-1 cells treated for 2h with 10 nM Bafilomycin A1 followed by 72h treatment with IC50 PP or GEM. (D) Western blots show levels of AKT, AKT1, and pAKT in PANC-1 and CFPAC-1 cells treated for 72h as indicated. The blots shown represent three independent experiments where β-actin was used as a loading control and the band intensities used for densitometry readings were obtained using ImageJ. Protein levels are represented as a ratio of protein of interest/β-actin normalized to the vehicle control (or first lane with a band).
Figure 4.
Pyrvinium pamoate induces autophagic cell death and inhibits the PI3K/AKT pathway in PDAC cells. (A) Western analysis of molecular markers of autophagy LC3I/II and p62/SQSTM1 in PANC-1 and CFPAC-1 cells post 72h treatment as indicated. (B) Representative confocal microscope images (×630; Carl Zeiss LSM 510; Scale bar = 20 μM) showing LC3 puncta in cells treated as in (A). LC3 was detected using a fluorophore-conjugated Alexa-488 secondary antibody and DAPI was used to stain the nuclei. (C) Western blot analysis of LC3I/II and PARP in PANC-1 cells treated for 2h with 10 nM Bafilomycin A1 followed by 72h treatment with IC50 PP or GEM. (D) Western blots show levels of AKT, AKT1, and pAKT in PANC-1 and CFPAC-1 cells treated for 72h as indicated. The blots shown represent three independent experiments where β-actin was used as a loading control and the band intensities used for densitometry readings were obtained using ImageJ. Protein levels are represented as a ratio of protein of interest/β-actin normalized to the vehicle control (or first lane with a band).
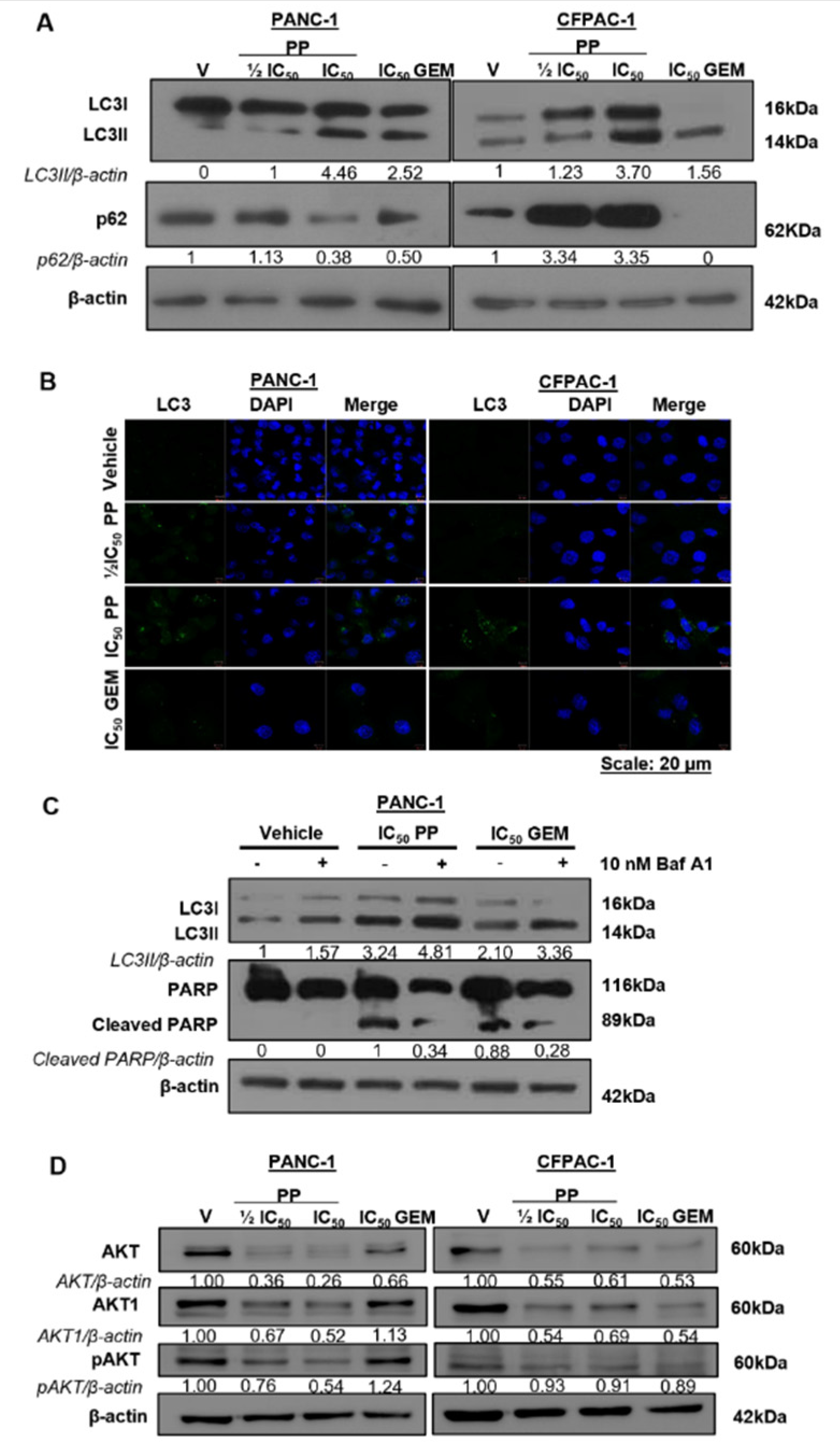
Figure 5.
Inhibition of PDAC cell invasion and migration by pyrvinium pamoate. (A) Representative light microscope images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) and quantification of Transwell invasion assays where PANC-1 and CFPAC-1 cells were treated for 24h with IC50 PP or GEM and replated in the upper chamber of the Matrigel-coated Transwell insert with medium + 1% FBS for 20 h. Invasive cells were stained with crystal violet and images from three independent experiments were quantified using the ImageJ software. The graph represents the mean number of invasive cells ± SEM of each treatment condition expressed as a percentage of the vehicle control. (B) Representative light microscope images (4X; EVOS M5000 Imaging System; scale bars = 600 μm) of scratch motility assays where PANC-1 and CFPAC-1 cells were grown to 100% confluency, wounded using a sterile 20 µL pipette tip and treated as indicated in the presence of 10 µM Mitomycin C. The wound areas were measured using the ImageJ software and cell migration was calculated by subtracting the wound area at each time point from the initial wound area at 0 h and expressed as a percentage of the vehicle control. Accompanying graphs represent the mean percentage of cell migration ± SEM pooled from three independent experiments. (C) Western blot analysis of proteins harvested from cells in (B) showing the effect of PP or GEM on EMT markers, E-cadherin, N-cadherin, β-catenin, and vimentin. The blots shown are representative of three independent experiments where β-actin was used as a loading control. Band intensities used for densitometry readings were obtained using ImageJ and protein levels are represented as a ratio of protein of interest/β-actin normalized to the vehicle control (or first lane with a band). Data are represented as mean ± SEM and p < 0.05, **p < 0.01 ***p < 0.001, and **** < 0.0001. .
Figure 5.
Inhibition of PDAC cell invasion and migration by pyrvinium pamoate. (A) Representative light microscope images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) and quantification of Transwell invasion assays where PANC-1 and CFPAC-1 cells were treated for 24h with IC50 PP or GEM and replated in the upper chamber of the Matrigel-coated Transwell insert with medium + 1% FBS for 20 h. Invasive cells were stained with crystal violet and images from three independent experiments were quantified using the ImageJ software. The graph represents the mean number of invasive cells ± SEM of each treatment condition expressed as a percentage of the vehicle control. (B) Representative light microscope images (4X; EVOS M5000 Imaging System; scale bars = 600 μm) of scratch motility assays where PANC-1 and CFPAC-1 cells were grown to 100% confluency, wounded using a sterile 20 µL pipette tip and treated as indicated in the presence of 10 µM Mitomycin C. The wound areas were measured using the ImageJ software and cell migration was calculated by subtracting the wound area at each time point from the initial wound area at 0 h and expressed as a percentage of the vehicle control. Accompanying graphs represent the mean percentage of cell migration ± SEM pooled from three independent experiments. (C) Western blot analysis of proteins harvested from cells in (B) showing the effect of PP or GEM on EMT markers, E-cadherin, N-cadherin, β-catenin, and vimentin. The blots shown are representative of three independent experiments where β-actin was used as a loading control. Band intensities used for densitometry readings were obtained using ImageJ and protein levels are represented as a ratio of protein of interest/β-actin normalized to the vehicle control (or first lane with a band). Data are represented as mean ± SEM and p < 0.05, **p < 0.01 ***p < 0.001, and **** < 0.0001. .
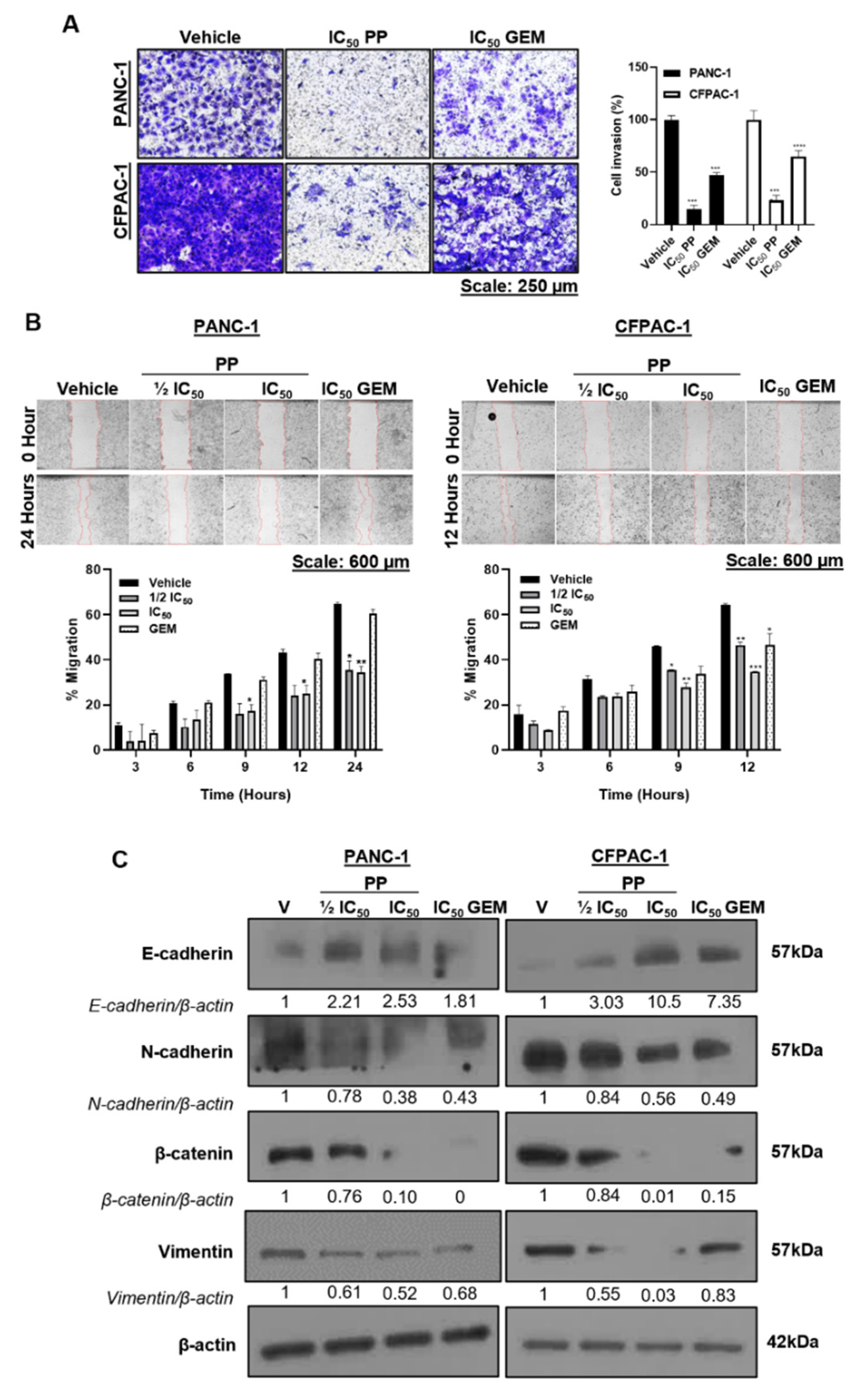
Figure 6.
Pyrvinium Pamoate inhibits PDAC spheroid growth, viability, and invasiveness. (A) Representative light microscope images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) of 3D spheroids that were established by plating PANC-1 cells on agarose-coated plates to prevent cell adhesion and treated for 6 days with PP or GEM. (B) Quantitation of spheroid growth rate calculated by dividing each spheroid area on day 6 by its respective area on day 0. The graph represents the mean spheroid growth rate ± SEM pooled from three independent experiments performed in hexaplicate. (C) Representative fluorescence microscope images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) of the spheroids in (A) stained with Calcein-AM on day 6. The accompanying graph show the mean calcein-AM fluorescence intensity measured using the ImageJ software and expressed as the percentage of the vehicle control. (D) Representative light microscope images (4X; EVOS M5000 Imaging System; scale bars = 600 μm) of PANC-1 spheroids that were established as in (A), embedded in collagen I, and treated for 72h as indicated. The red circles indicate invasion into the collagen matrix and corresponds to the magnified views on the bottom conners. Data are represented as mean ± SEM and **p < 0.01 ***p < 0.001, and **** < 0.0001.
Figure 6.
Pyrvinium Pamoate inhibits PDAC spheroid growth, viability, and invasiveness. (A) Representative light microscope images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) of 3D spheroids that were established by plating PANC-1 cells on agarose-coated plates to prevent cell adhesion and treated for 6 days with PP or GEM. (B) Quantitation of spheroid growth rate calculated by dividing each spheroid area on day 6 by its respective area on day 0. The graph represents the mean spheroid growth rate ± SEM pooled from three independent experiments performed in hexaplicate. (C) Representative fluorescence microscope images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) of the spheroids in (A) stained with Calcein-AM on day 6. The accompanying graph show the mean calcein-AM fluorescence intensity measured using the ImageJ software and expressed as the percentage of the vehicle control. (D) Representative light microscope images (4X; EVOS M5000 Imaging System; scale bars = 600 μm) of PANC-1 spheroids that were established as in (A), embedded in collagen I, and treated for 72h as indicated. The red circles indicate invasion into the collagen matrix and corresponds to the magnified views on the bottom conners. Data are represented as mean ± SEM and **p < 0.01 ***p < 0.001, and **** < 0.0001.
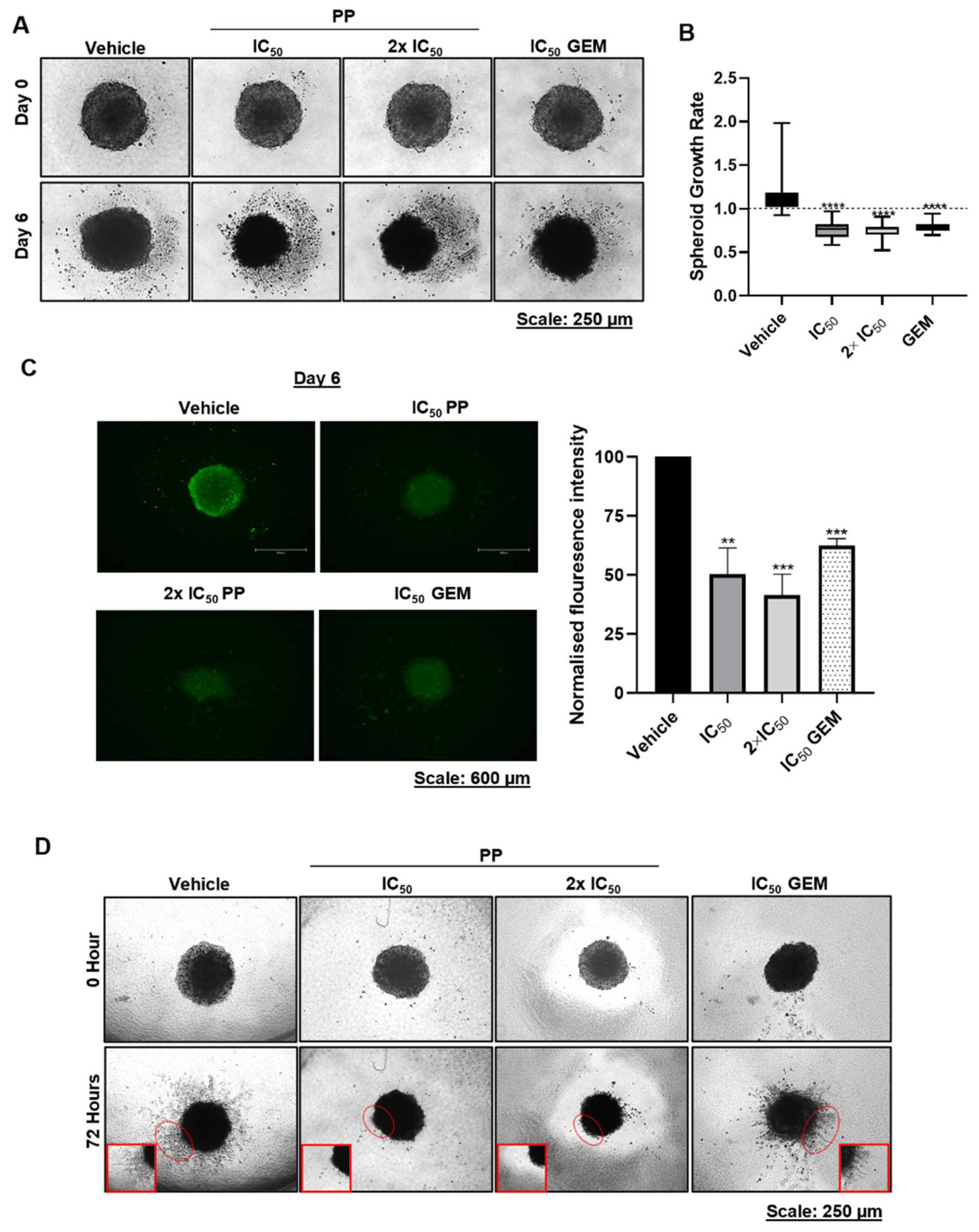
Figure 7.
Synergistic effects of pyrvinium pamoate and gemcitabine on PDAC cell viability, DNA damage and PDAC spheroid size/viability. (A) MTT assays of PANC-1 and CFPAC-1 cells post 72h treatment with ⅛ IC50, ⅟4 IC50, and ½ IC50 of PP or GEM and their combinations. Experiments were performed in quadruplicate and the graphs show mean cell viability ± SEM for each treatment condition. (B) Highest single agent (HSA) model synergy and antagonism surface maps generated using the Combenefit software by analysing the MTT cell viability assay results in (A). (C) Combination index (CI) plot and values generated using the CompuSyn software based on the results from (A). (D) Western blot analysis of γH2AX levels in PANC-1 treated for 72h with ⅛ IC50 PP or ⅛ IC50 GEM or their combination (PP+GEM). β-actin was used as a loading control and band intensities used for densitometry readings were obtained using ImageJ the software. Protein levels are represented as a ratio of the protein of interest/β-actin normalised to the first lane with a band. (E) Representative light microscopy images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) showing the impact of ⅛ IC50 PP or ⅛ IC50 GEM or PP+GEM on the morphology of PANC-1 cells. (F) Representative light microscope images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) of PANC-1 spheroids treated for 6 days with as indicated. The graph represents the mean spheroid growth rate ± SEM calculated by dividing each spheroid area on day 6 and by its respective area on day 0 pooled from three independent experiments performed in hexaplicate. (G) Representative fluorescence microscope images (4X; EVOS M5000 Imaging System; scale bars = 600 μm) of the spheroids in (F) stained with Calcein-AM on day 6. The accompanying graph show the mean calcein-AM fluorescence intensity measured using the ImageJ software and expressed as the percentage of the vehicle control. Data are represented as mean ± SEM; n=3. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Figure 7.
Synergistic effects of pyrvinium pamoate and gemcitabine on PDAC cell viability, DNA damage and PDAC spheroid size/viability. (A) MTT assays of PANC-1 and CFPAC-1 cells post 72h treatment with ⅛ IC50, ⅟4 IC50, and ½ IC50 of PP or GEM and their combinations. Experiments were performed in quadruplicate and the graphs show mean cell viability ± SEM for each treatment condition. (B) Highest single agent (HSA) model synergy and antagonism surface maps generated using the Combenefit software by analysing the MTT cell viability assay results in (A). (C) Combination index (CI) plot and values generated using the CompuSyn software based on the results from (A). (D) Western blot analysis of γH2AX levels in PANC-1 treated for 72h with ⅛ IC50 PP or ⅛ IC50 GEM or their combination (PP+GEM). β-actin was used as a loading control and band intensities used for densitometry readings were obtained using ImageJ the software. Protein levels are represented as a ratio of the protein of interest/β-actin normalised to the first lane with a band. (E) Representative light microscopy images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) showing the impact of ⅛ IC50 PP or ⅛ IC50 GEM or PP+GEM on the morphology of PANC-1 cells. (F) Representative light microscope images (10X; EVOS M5000 Imaging System; scale bars = 250 μm) of PANC-1 spheroids treated for 6 days with as indicated. The graph represents the mean spheroid growth rate ± SEM calculated by dividing each spheroid area on day 6 and by its respective area on day 0 pooled from three independent experiments performed in hexaplicate. (G) Representative fluorescence microscope images (4X; EVOS M5000 Imaging System; scale bars = 600 μm) of the spheroids in (F) stained with Calcein-AM on day 6. The accompanying graph show the mean calcein-AM fluorescence intensity measured using the ImageJ software and expressed as the percentage of the vehicle control. Data are represented as mean ± SEM; n=3. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



