
Submitted:
01 March 2024
Posted:
04 March 2024
You are already at the latest version
Abstract
Phagocytosis (and endocytosis) is an unusual cellular process which results in the formation of a novel subcellular organelle, the phagosome. This phagosome contains not only the internalised target of phagocytosis, but also the external medium, creating a new border between extracellular and intracellular environments. The boundary at the plasma membrane is, of course, tightly controlled and exploited in ionic cell signalling events. Although there has been much work on the control of phagocytosis by ions, notable Ca2+ ions influxing across the plasma membrane, increasing our understanding of the mechanism enormously, very little work has been done exploring the phagosome/cytosol boundary. In this paper, we have explored the changes in the intra-phagosomal Ca2+ ion content which occur during phagocytosis and phagosome formation in human neutrophils. Measuring Ca2+ ion concentration in the phagosome is potentially prone to artefacts as the intra-phagosomal environment experiences changes in pH and oxidation. However, by excluding such artefacts, we conclude that there are open Ca2+ channels on the phagosome, which allow Ca2+ ions to ‘drain’ into the surrounding cytosol. This conclusion was confirmed by monitoring the translocation of intracellularly expressed YFP-tagged C2 domain of PKC-γ. This approach marked regions of membrane at which Ca2+ influx occurred, the earliest being the phagocytic cup, and then the whole cell. This paper therefore presents data which has novel implications for understanding phagocytic Ca2+ signalling events, such as peri-phagosomal Ca2+ hotspots, and other phenomena.
Keywords:
intra-phagosomal Ca2+
; phagocytosis
; Ca2+channels
; phagosome
; neutrophil
1. Introduction
Phagocytosis, the process whereby an extracellular object is internalised by a cell (phagocyte), is a complex process, involving the extension of small pseudopodia which attached to the extracellular target, initially immobilising the particle within a ‘phagocytic cup’; and then, after a large cytosolic Ca2+ signal, a rapid extension of the pseudopodia fully encloses the target and draws it into the cell interior [1,2,3]. This results in the formation of an intracellular vesicle, the phagosome [4,5] whose membrane originates from the plasma membrane. The relationship between cytosolic Ca2+ signalling and phagocytosis is complex, the cytosolic Ca2+ signal which occurs during phagocytosis by neutrophils regulating both the rate of extension of pseudopodia around the target [1,2,3] and activation of the bactericidal oxidase system in the phagosomal membrane [6,7]. Ca2+ signalling within these cells involves both Ca2+ influx from the extracellular environment and the release from stored Ca2+ from within the cell [8]. Hots spots of Ca2+ in the cytosol near the closed phagosome have also been reported in some phagocytic cells such as mouse embryonic fibroblasts and dHL60 cells [9] and RAW 264.7 cells [10]. It is suggested that these Ca2+ microdomains (hot spots) arise from ‘leaky’ interactions of STIM-1 on the endoplasmic reticulum (ER), the Ca2+ storage organelle with channels on the phagosomal membrane [9]. These Stim-1 mediated Ca2+ hotspots are necessary for efficient phagocytosis and oxidase actvation [11,12]. However, neutrophils, which are highly efficient phagocytes, have very little or no endoplasmic reticulum [13]. This suggests that the source of the peri-phagosome Ca2+ rise in these cells may be the phagosome itself. However, very little is known about the intra-phagosomal Ca2+ concentration and how it changes during phagocytosis. Pioneering work by Dahlgren’s group more than 20years ago [14] using fura2 as a Ca2+ indicator, showed that intra-phagosomal Ca2+ rapidly decreased during phagocytosis. However, the kinetics and relationship to the process of phagocytosis were not investigated. The Ca2+ probe used, fura 2, was susceptible to ‘bleaching effects’ [15] making measurements within the phagosome difficult [14]. However advances in imaging and Ca2+ sensing fluor design has enabled these early observation to be extended and to further investigate intra-phagosomal Ca2+ changes. In this paper, we have used a Ca2+-sensor, fluo4, which has the more robust fluorescein-like component, covalently coupled to a phagocytic target. When phagocytosed by neutrophils, this indicator dynamically reported the intra-phagosomal Ca2+concentration and enabled a correlate of changes in phagosomal Ca2+ with binding, uptake and phagosomal closure. Using this approach, we report here that there was an initially high Ca2+ concentration within the phagosome which was reduced to the cytosolic concentration within 200s of phagosomal closure. This is consistent with the incorporated into the phagosomal membrane, of opened plasma membrane Ca2+ channel, so that the open phagocytic cup acts as a conduit for local Ca2+ elevation, but that once closed, phagosomal Ca2+ is ‘drained’ into the cytosol.
2. Results
2.1. Fluo4-Zymosan Targets Report Ca2+ Concentration
In order to monitor intra-phagosomal Ca2+, the Ca2+ sensitive fluorescent probe, fluo4, was coupled to C3bi-opsonised zymosan particles as a phagocytic target. The sensitivity of the fluo4-zymosan particles to Ca2+ concentration was established in vitro by measuring the fluo4 intensity at various Ca2+ concentrations (Figure 1). There were two micro-anatomical zones of the zymosan particle [16], the outer zone and the central core, the central core having a brighter fluorescence signal as a result of fluo4 plus a strong auto-fluorescent signal. . The fluorescence intensity of the outer zone acted as a true the Ca2+ indicator, with little autofluorescence. The central fluorescent signal was, however, useful for identifying the location of the target during phagocytosis. Each zone also had a different apparent kd for Ca2+ (Figure 1), fluo4 in the core zone having an apparent of approximately kd 300nM, similar to that reported for free fluo4 ie 345nM at pH7.2, [17]; whereas fluo4 in the outer zone had an apparently higher kd of approx. 1mM Ca2+(Figure 1). However, in both zones of the zymosan particle, fluo4 had maximum intensity in 1.3mM Ca2+ (ie the extracellular Ca2+ concentration) and near zero intensity at 100 nM , the cytosol free Ca2+ concentration, (Figure 1). The difference in apparent Kds, fortuitously, also gave the opportunity for a double estimation of intra-phagosomal Ca2+ to be made from the same zymosan particle within the same phagosome. It should be noted that fluo4 at both locations was almost totally saturated with Ca2+ above 10mM, so the effective measurement range was thus below 10mM Ca2+.
2.2. Phagosomal fluo4 Signal is Decreased
Having established that the fluo4-zymosan particles reported Ca2+ concentration, the effect of phagocytosis was examined. The fluorescence of zymosan particles within phagosomes was significantly reduced after internalisation compared with non-phagocytosed particles in the same microscopic field (Figure 2a,c). It is unlikely that this was an optical effect caused by the interference from the cytoplasm surrounding the phagocytic target because, (i) during the process of internalisation, no partial or zonal reduction in the fluorescent signal from zymosan was observed as would be expected from optical obstruction by the encroaching pseudopodia and (ii) although there is an effect of light scattering through cytoplasm which attenuates the detected fluorescent signals from intracellular fluors [18], this would be very small. The light scattering effect of cytoplasmic projections covering the phagosome depends on the number of cytosolic granules per unit volume (density), the granule refractive index and their size (19,20). The engulfing pseudopodia were thin and excluded light-scattering granules, such that cytosolic fluorescence at these locations often has increased excitation efficiency and appear brighter (18). In a worst-case scenario, taking the published values for bulk cytosolic granules (density, and their refractive index and their size), Mie scattering calculations give an estimate of the attenuation coefficient for 488nm laser light through neutrophil cytoplasm of about 700 mm-1 . As the approximate thickness of the cytoplasm covering the zymosan particle within the phagosome is less than 0.5 mm, less than 30% of the laser light entering the pseudopodia would be scattered and thus unavailable for excitation of fluo4, ie the intensity would be a minimum of 70% of its maximum. This is insufficient to account for the reduction in intensity of internalised fluo4-zymosan, which was less than about 5% of the non-internalised particles (Figure 2c). Thus, such a decrease in intensity of phagosomal signal (>95%) cannot be accounted for by an optical effect caused by the attenuation of light scattering effects of the pseudopodal cytoplasm. On the contrary, the efficiency of excitation and emission of fluors in this pseudopodia and near the forming phagosome is increase [18]. The elimination of the possibility that an optical effect decreased the fluo4 signal was confirmed by the use of a non-Ca2+ sensitive fluor (fluorescein) conjugated to zymosan, unlike fluo4-zymosan, the intensity of fluorescein was not significantly reduce when internalised (Figure 2b,c) .
Other possible causes of the reduced emission of phagosomal fluo4 were also excluded (Figure 3b). Changes in the phagosomal pH caused by activation of the oxidase/proton pump system [21] has been shown to cause a small reduction (approx. 10%) in fluorescence of non-Ca2+ sensing but pH-sensitive, probe, FITC-zymosan at the time when fluoe4-zymosan fluorescence was almost zero. At longer times, when the intra-phagosomal pH falls significantly, a similar reduction of fluorescence of intra-phagosomal fluo4-zymosan was also observed. This however was only evident at longer times after internalisation (5-10 mins) when the intra-phagosomal pH decreases significantly, stabilising at about pH5 after 15-20 mins [20]. Interestingly, the intensity of the fluo4-zymosan core was often seen to increase at low pH (Figure 3b). Thus these effects of pH cannot account for the decrease in fluo4 fluorescence observed within 0-30s after internalisation. It was thus concluded that pH changes could not explain the decrease of intra-phagosomal fluo4 signal.
2.3. Phagosomal fluo4 Signal Decrease not due to Oxidants
In addition to pH changes, the intra-phagosome environment is exposure to oxidants, notably short-lived superoxide which forms semi-stable hydrogen peroxide. These are generated by activation of the phagosomal oxidase at the phagosomal membrane [22]. The oxidative effect of H2O2 produced is enhanced by myeloperoxidase, a lysosomal enzyme in azurophilic granules of the neutrophil which are released into the phagosome by fusion with the phagosome. We have previously used zymosan-conjugated dichlorodihydrofluorescein (DCDHF) as an indicator of intraphagosomal oxidants, [23] and have shown that the oxidation within the phagosome begins after phagosomal closure and continues with similar kinetics to those of the fluo4 intensity decreased reported here. However, no immediate in vitro effect of H2O2 and myeloperoxidase was observed and fluo4 zymosan intensity was not significantly reduced before approx. 100s of contact (Figure 3a). Even by 100s, the intensity was reduced only be approx. 15%. It was thus concluded that intra-phagosomal oxidation could not account for the decrease in fluo4-zymosan intensity observed during phagocytosis It should also be noted that fluorescein (a chemically related molecule) was also resistant to the bleaching effect of the intra-phagosomal environment as it remained at the same brightness whether outside or inside the phagosome (Figure 2b).
2.4. Phagosomal fluo4 Zymosan REMAINED Responsive to Ca2+ Changes
In order to test whether fluo4 attached to zymosan remained responsive to Ca2+ after contact with the intra-phagosomal environment, three approaches were adopted. The first approach was simply to liberate the zymosan particle from within the phagosome by lysis of the cells with Triton X-100 in order to allow contact of the released particles with the extracellular high Ca2+ (1.3mM). This procedure significantly increased the fluorescent signal, consistent with a retained Ca2+ sensitivity (Figure 3c). As Triton X-100 destroyed the cell morphology, this approach could not be undertaken whilst single cell imaging. Instead, cell populations were mixed with zymosan particles, centrifuged to bring the particles and cells in close contact, then after 5 mins at 37oC, resuspended and 3-cycles of sedimentation at 1g used to separate free zymosan particles from cell associated zymosan. The resultant cell pellet was sampled for imaging to confirm that all zymosan particles were within phagosomes and their fluorescence intensities measured. Triton X-100 was then added to a portion of the cell pellet and sampled for imaging to confirm that no zymosan particles were within phagosomes and their fluorescence intensity measured. There was a highly significant increase in the fluorescence intensity of the zymosan after Triton X-100 treatment (Figure 3c). This data was consistent with retention of Ca2+-sensitivity by the phagosomal fluo4-zymosan particles, so that when released into the high Ca2+ extracellular environment their fluorescence increased. The second approach was to use saponin (digitonin), a plant detergent-like molecule which selectively permeabilizes the cell membranes [24] and collapses the transmembrane Ca2+ gradient, so that intracellular fluorescent Ca2+ indicators become saturated with Ca2+ [25] but leaves the cell morphology recognisable [24]. Saponin-permeabilisation significantly increased the fluorescent signal from fluo4-zymosan within phagosomes (Figure 3c), confirming that after phagocytosis, fluo4-zymosan retained its Ca2+ sensitivity. However, in both the previous approaches, the intra-phagosomal fluo4 zymosan particles was exposed to high concentrations of Ca2+. A third approach was therefore also adopted to exclude the possibility that fluo4 remained only weakly sensitive to Ca2+ and responded to only high Ca2+. The third approach was to artificially elevate cytosolic Ca2+, using a Ca2+ ionophore, ionomycin. Elevation of cytosolic Ca2+ by ionomycin caused a significant rise fluo-4 fluorescence from particles within phagosomes (Figure 3c,d). This was consistent with a rise in intra-phagosomal Ca2+ being detected by the functional Ca2+ probe whilst within the phagosome. The kinetics of the intra-phagosomal Ca2+ rise also suggested that the Ca2+ channels on the phagosomal membrane were open. However it cannot be excluded that ionomycin partitioned across all intracellular membranes and that Ca2+ entered the phagosme via this route. Whatever the mechanism for the rise in intra-phagosomal Ca2+, the effect showed that the intra-phagosomal fluo4 remained sensitive to physiological changes in Ca2+ concentration. It was thus concluded that the decrease in fluo4 intensity during phagocytosis was the result of a decrease in intra-phagosomal Ca2+conentraion from an initially saturating concentration of Ca2+ (probably the extracellular concentration of 1.3mM), to the cytosolic Ca2+ concentration (approx. 100nM),
2.5. Kinetics of INTRA-phagosomal Ca2+ Decrease
By monitoring the intensity of fluo4-zymosan particles during internalisation by the phagocyte, the relationship of the decrease in intra-phagosomal Ca2+ with the stages of phagocytosis were established. On engaging the zymosan particle, the cells form a “phagocytic cup” at the base of the zymosan, the “cup” then extends until the particle is enclosed within the phagosome [1,26]. Although the time of contact of the particle with the cell could be determined, the exact time of complete phagosome closure was more difficult to establish but could be estimated within a few frames (about 0.2-0.5 s) from phase contrast imaging. At the time of phagosome closure, the intensity of fluo4-zymosan was unchanged and equal to free zymosan particles in high Ca2+ (1.3mM). However, within 5-10 seconds of closure, the intensity of the internalised fluo4-zymosan particles abruptly decreased, reaching its low equilibrium level within 20 seconds (Figure 4 and movie 1). Presumably, the high (millimolar) phagosomal Ca2+ at the time of phagosome closure leaked out of the phagosome into the cytosol over this time. From knowledge of the phagosome water volume, it can be estimated that the phagosome initially contained about 2x107 Ca2+ions, and thus that it would be lost at an average rate of about 106 ions/sec over 20 seconds to reduce this to the cytosolic concentration to c.100nM. After the time when phagosomal Ca2+ had been reduced to below the fluo4 saturation level (10mM), there was an approximately linear decline in fluo4 intensity equivalent to a decrease in intra-phagosomal Ca2+ from 10mM to 0.1mM over 5s, suggesting a slower loss of Ca2+ (as expected if the outward leak were driven by the Ca2+ concentration gradient across the phagosomal membrane) over this time of about 0.2x104 Ca2+ ions/s. The plasma membrane Ca2+channels found on the neutrophil phagosomes is TRPM2 (27) carry currents of about 1pA (28) or about 107 ions/sec suggesting that only a few channels need to be open on the phagosome membrane (eg 5 channels each open for 0.2s would drain the phagosomal Ca2+ at that rate). It is thus possible that that plasma membrane Ca2+ channels opened by the phagocytic stimulus, which gives rise to the well-document phagocytic cytosolic Ca2+ signal [1,2,23,26], remain open in the phagosomal membrane and “drain” the enclosed phagosome of its Ca2+.
2.6. Location of Open Ca2+ Channels
In order to test the conclusion that Ca2+ channels remain open on the phagosomal membrane, the translocation of the YFP-tagged C2 domain of PKC-γ (YFP–C2-γ) from the cytosol to the plasma membrane was used as a real time spatial marker of Ca2+ influx [29,30] into the phagocytic cell line RAW 264.7 macrophages. When presented with mouse C3bi-opsonised-zymosan (without fluorescence tags), the translocation of YFP–C2-γ to the plasma membrane was observed after phagocytic cup formation, reflecting the large cytosolic Ca2+ signal expected. Initally, there was localised translation to the phagocytic cup, indicating that Ca2+ channels were open in this region (Figure 5a-1 sec). Within the next second (labelled ‘2 sec’ on Figure 5a) translocation of YFP–C2-γ) was observed at all plasma membrane location (Figure 5a-2 sec). This rapid opening of Ca2+ channels remote from the phagocytic site explains the difficulty in observing localised cytosolic Ca2+ rises during phagocytosis as the global open Ca2+ channels overwhelmed the effect of a localised influx of Ca2+ (23). Clearly, Ca2+ channels remain open on the forming phagosome after phagosome closure. In the example shown, the cell also contained a previously formed phagosome (Figure 5b marked ‘x’ ) which initially had no YFP–C2-γ on its surface indicating the absence of a Ca2+ flux from the phagosome. However, when the cytosolic Ca2+ signal was triggered by the second phagocytotic event, it was seen that YFP–C2-γ also translocated to the membrane of the previous phagosome (especially obvious at 20 sec, in Figure 5c). This can be explained if either (i) the Ca2+ channels on the previously formed phagosome had remained open, so that the elevated cytosolic Ca2+ began to refill the previously drained phagosome which begun leaking out (detected by YFP–C2-γ translocation) or (ii) the channels were previously closed by were re-opened as result of diffusible Ca2+ opening stimuli (eg IP3). Thus the use of YFP–C2-γ has demonstrated that Ca2+channels on the forming phagosome were open during phagosome formation and were open or operational after phagosome closure.
3. Discussion
The results presented in this paper show that the open phagosome entraps extracellular Ca2+ which ‘empties’ into the cytosol once the phagosome has closed. The concentration of phagosomal Ca2+ within the phagocyte equilibrates with the cytosolic Ca2+ within 20 seconds of phagosome closure. This sequence is shown as a cartoon (Figure 6). Thus the phagosome cannot act as a long term store of Ca2+ for later signalling events. Also, it cannot act as a reservoir for loading lysosomes with Ca2+ for other signalling events [31]. The rapid decline of intra-phagosomal Ca2+ was reported over 20 years ago by Dahlgren’s group, who monitored Ca2+ in a similar way to that described here but using fura2 as the Ca2+ reporter. Although fura2 was bleached during the experiments (both 340 and 380nm decreasing), the group found that the intra-phagosomal Ca2+ level dropped rapidly. Little notice has been given to this early observation which is confirmed (and extended ) here. However, Anke et al [27] have more recently shown that in phagosomes from phagocytes taken from Trpm2−/− mice ie lacking the TRPM2 Ca2+ channel, intra-phagosomal Ca2+ rose slowly (over a 10 min time period) compared to the intra-phagosomal Ca2+ expressing TRPM2. They suggested that the phagosomes devoid of TRPM2, lacked a leakage channel and so Ca2 accumulated within the phagosome. This conclusion is similar to that which was drawn from the data shown here.
From the work presented here it seems unlikely that Ca2+ events could be driven by phagosomal Ca2+ close to the time of completion of phagocytosis. However, if the Ca2+ channels closed slowly (over minutes rather than seconds), and the phagosomal membrane contained functional plasma membrane Ca2+ pumps, it is expected that the phagosome would re-load with Ca2+ over a longer time periods. The equilibrium generated across the plasma membrane would thus also be generated across the phagosomal membrane. Under these circumstances, phagosomal Ca2+could be important for driving peri-phagosomal Ca2+ events. The earliest report of such a Ca2+ cloud localised to near the phagosome [32] is usually dismissed as almost certainly an optical artefact caused by the thinner cytoplasm around the pseudopodia which increased the excitation and efficiency in that area [18]. However, the work presented here opens the possibility that in phagocytes with endoplasmic reticulum, the phagosome itself may be a source of Ca2+ which adds to or generates peri-phagosomal Ca2+ events such as Ca2+ hotspots (9,10).
4. Materials and Methods
4.1. Cell Preparation
Human neutrophils, isolated from the blood of healthy volunteers whole had given informed consent as described previously [10] were suspended in Krebs medium (NaCl 120 mM, KCl, 4.9 mM KH2PO4, 1.2 mM MgSO4, 1.2 mM CaCl2, 1.3 mM, HEPES 25 mM and bovine serum albumin, 0.1% adjusted to pH 7.4 with NaOH).
4.2. Properties of the Phagocytic Target
Zymosan particles (killed yeast cells) of approximately ellipsoid shape, having radii in the x, y and z planes (or semi-axes) of 1:1.5 and 1mm were opsonised with iC3b by incubation with human or mouse serum as previously described [11]. The particles have a cell wall enclosing an outer space and a dense central core [12]. The water space in the zymosan-containing phagosome is approximately 50% of the total volume [12], and so would each contain 6.3mm3of extracellular medium (1.3mM Ca2+). A cadaverine-derivative of the fluorescent Ca2+ indicator fluo4 (Fluo4 cadaverine (F36201 fluo-4 cadaverine, pentapotassium salt, Molecular Probes; Thermo Scientific) was linked to amines in the zymosan particles by carbodiimde linkage using (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride, Thermo Scientific).The reagents were used at 1mg/ml and incubated with a suspension of zymosan particles at room temperature for 4 hours before separation of the reactants from the solid particles by 3 rounds of centrifugation and resuspension in fresh medium to remove the reactants. A similar procedure was used to produce non-Ca2+ sensing fluorescein-linked zymosan particles by reacting the particles with fluorescein isothiocyanate (FITC, Sigma) as described previously (34).
4.3. Imaging and Intra-Phagosomal Ca2+ Monitoring
The fluo4-zymosan particles were sedimented onto a glass coverslip mounted onto a thermostatically control stage (37 ± 0.1 °C) in Krebs medium. The coverslip was then washed with medium to remove particles which had not made contact with the coverslip and were loosely adhered. Neutrophils were then allowed to adhere to the same glass coverslip whilst observing microscopically and selecting a microscopic field in which neutrophils and particles were close and expected to undergo phagocytosis. The field was then imaged using a resonant laser scanning head of the LeicaSP5 confocal inverted microscope (Leica Microsystems, Heidelberg) and 63x objective. Fluorescence (excited by 488nm laser scanning) and transmitted light (phase contrast images) were acquired simultaneously. Image analysis and presentation was achieved using ImageJ software (https://imagej.nih.gov/ij/)
4.4. Estimation of Intraphagosomal Ca2+ Concentration
The concentration of intra-phagosomal Ca2+ was estimated from the fluorescence intensity of the zymosan particles during the experiment (F). The intensity of zymosan particles outside the cell (Zo) in 1.3mM Ca2+ was taken as the Ca2+ saturation maximum (Fmax) and the minimum fluorescence (Fmin) was estimated as being the autofluorescence signal from unlabelled zymosan particles, as in the in vitro calibration curves (see Figure 1). Using the kd values from the calibration curves the intra-phagosomal Ca2+concentration was estimated using a variant of the standard equation for single wavelength fluors [25], was used to estimate:- : Ca2+= Kd (α−1)/(β−α), where α =F/Fmin and β =Fmax/Fmin.
4.5. Raw 264.7 Cell Transfection
RAW 264.7 cells were electroporated to introduce the C2-γ-YFP plasmid (3 µg plasmid DNA per 2×106 cells) using the Cell Line Nucleofector (Lonza) as described previously (33). Cells were incubated at 37°C in 5% CO2 for 3–4 h to enable expression of the newly introduced DNA, before imaging on a Leica SP5 confocal microscope. Fluorescent protein expression in transfected RAW 264.7 cells was detected by ∼1 h post transfection, but expression was optimal at ∼4 h post transfection. The plasmid encoding for C2-γ-YFP was a kind gift from Theodorus W. Gadella (Swammerdam Institute for Life Sciences, University of Amsterdam, The Netherlands)
Acknowledgments
The ‘cartoon’ was constructed using pre-made elements from Servier Medical Art (http://www.servier.fr/servier-medical-art).
Appendix A
MOVIE 1
MOVIE 1 shows a typical experiment, in which the process of phagocytosis of a fluo4-coupled zymosan particle (from contact to completion) was recorded using phase contrast microscopy (lower panel) and confocal fluorescence imaging (upper panel). The timing of the decrease in phagosomal fluo4-zymosan fluorescence after closure of the phagosome can be seen.
References
- Dewitt, S.; Hallett, M.B. (2002) Cytosolic free Ca2+ changes and calpain activation are required for beta integrin-accelerated phagocytosis by human neutrophils. J. Cell Biol. 159, 181-189.
- Nunes, P.; Demaurex, N. (2010) The role of calcium signaling in phagocytosis. J. Leuk. Biol. 88, 57–68. [CrossRef]
- Francis, E.A., Heinrich, V., ( 2017) Single-cell investigation of the role of calcium bursts in human immune cells. Biophysical J. 112 , 400A-400A. [CrossRef]
- Jaumouillé, V., Grinstein, S. (2016 ) Molecular Mechanisms of Phagosome Formation Microbiology Spectrum 4. [CrossRef]
- Uribe-Querol, E., Rosales, C. ( 2020) Phagocytosis: Our current understanding of a universal biological process. Frontiers Immunol. 11. [CrossRef]
- Hallett, M.B. Davies, E.V. . Campbell A.K. (1990) Oxidase activation in individual neutrophils is dependent on the onset and magnitude of the Ca2+ signal. Cell Calcium, 11 , 655-663. [CrossRef]
- Bei. L., Hu, T., Qian, Z.M., Shen, X. (1998) Extracellular Ca2+ regulates the respiratory burst of human neutrophils. Biochim. Biophys. Acta – Mol. Cell Res. 1404, 475-483. [CrossRef]
- Westman, J., Grinstein, S., Maxson, M.E. (2019) Revisiting the role of calcium in phagosome formation and maturation. J Leuk, Biol, 106, 837-851. [CrossRef]
- Nunes, P., Cornut, D., Demaurex, N. (2012) STIM1 juxtaposes ER to phagosomes, generating Ca2+ hotspots that boost phagocytosis. Curr. Biol. 22 , 1990-1997. [CrossRef]
- Roberts, R.E., Vervliet, T., Bultynck, G., Parys, J.B., Hallett, M.B. (2020) EPIC3, a novel Ca2+ indicator located at the cell cortex and in microridges, detects high Ca2+ subdomains during Ca2+ influx and phagocytosis. Cell Calcium 92 Article Number:102291. [CrossRef]
- Zhang, H., Clemens, R,A, Lowell, C.A.(2014) STIM1 calcium sensor is required for activation of the phagocyte oxidase during inflammation and host defense. Blood 123, 2238-2249. [CrossRef]
- Guido, D., Demaurex, N., Nunes, P. (2015) Junctate boosts phagocytosis by recruiting endoplasmic reticulum Ca2+ stores near phagosomes. J. Cell Sci. 128, 4074-4082. [CrossRef]
- Bessis, M. (1973) Living Blood Cells and Their Ultrastructure. Springer, Berlin.
- Lundqvist-Gustafsson, H., Gustafsson,M., Dahlgren C. (2020) Dynamic Ca(2+)changes in neutrophil phagosomes. A source for intracellular Ca(2+)during phagolysosome formation? Cell Calcium 27, 353-362. [CrossRef]
- Becker P.L., Fay F.S. (1987) Photobleaching of fura-2 and its effect on determination of calcium concentrations. Am. J. Physiol. 253: C613–C618. [CrossRef]
- Soto E.R, . Ostroff G.R. (2008) Characterization of multilayered nanoparticles encapsulated in yeast cell wall particles for DNA delivery. Bioconjugate Chem. 19, 840–848. [CrossRef]
- Gee K.R., Brown, K. A., Chen, W. N., Bishop-Stewart, J., Gray, D., Johnson I. (2000) Chemical and physiological characterization of fluo-4 Ca(2+)-indicator dyes Cell Calcium 27, 97-106. [CrossRef]
- Dewitt, S., Darley, R.L., Hallett, M.B. (2009) Translocation or just location? Pseudopodia affect fluorescent signals. J. Cell Biol. 184, 197-203. [CrossRef]
- Meyer, R.A. (1979) Light-scattering from biological cells-dependence of backscatter radiation on membrane thickness and refractive index. Applied Optics 18, 585-588. [CrossRef]
- Mie, G. (1908) Articles on the optical characteristics of turbid tubes, especially colloidal metal solutions. Ann. Der Physik 25, 377-445.
- Mantegazza, A.R., Savina, A., Vermeulen, M., Pérez, L. et al (2008) NADPH oxidase controls phagosomal pH , Blood 112 : 4712–4722. [CrossRef]
- Segal, A.W. (2005) How neutrophils kill microbes. Ann. Rev. Immunol. 23 , 197-223. [CrossRef]
- Dewitt, S., Laffafian I., Hallett M.B. (2003) Phagosomal oxidative activity during β2 integrin (CR3)-mediated phagocytosis by neutrophils is triggered by a non-restricted Ca2+signal: Ca2+ controls time not space. J. Cell Sci. 116, 2857–2865. [CrossRef]
- Jacob M.C., Favre, M., Bensa J.C. (1991) Membrane cell permeabilization with Saponin and multiparametric flow cytometry. Cytometry 12, 550-558. [CrossRef]
- Hallett MB, Al-Jumaa, Dewitt S. (2014) Optical methods for the measurement and manipulation of cytosolic calcium signals in neutrophils. Methods in Molecular Biology 1124, 107-120: Neutrophil Methods and Protocols (Ed Quinn MT, DeLeo FR), Humana Press.
- Francis, E.A. Heinrich. V. (2018) Mechanistic understanding of single-cell behavior is essential for transformative advances in biomedicine. Yale J Biol Med. 2018, 91, 279–289.
- Anke D., Kiya, T., Gong, H., Gao, X., Malik, A.B. (2017) Role of the phagosomal redox-sensitive TRP channel TRPM2 in regulating bactericidal activity of macrophages J. Cell Sci. 130, 735–744. [CrossRef]
- Starkus, J.G., Fleig A., Reinhold, P. (2010) The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular. J Physiol. 588. 1227–1240. [CrossRef]
- Oancea, E., Meyer, T. (1998). Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell 95, 307-318. [CrossRef]
- Teruel,M.N., Meyer T. (2002). Parallel single-cell monitoring of receptor-triggered membrane translocation of a calcium-sensing protein module. Science 295 1910-1912. [CrossRef]
- Kilpatrick, B.S., Eden, E.R., Schapira, A.H., Futter, C.F., Patel, S. (2013) Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci.126, 60-66. [CrossRef]
- Sawyer, D.W.; Sullivan, J.A.; Mandell, G.L. (1985) Intracellular free calcium localization in neutrophils during phagocytosis. Science. 230, 663–666. [CrossRef]
- Roberts, R.E., Martin,M., Marion, S., Elumalai, G.L., Lewis, K., Hallett, M.B. (2020) Ca2+-activated cleavage of ezrin visualised dynamically in living myeloid cells during cell surface area expansion. J. Cell Sci. 133, art no jcs236968. [CrossRef]
- Morris, M. R., Dewitt, S., Laffafian, I. and Hallett, M. B.(2003). Phagocytosis by inflammatory phagocytes: experimental strategies for stimulation and quantification. In Inflammation Protocols. Methods in Molecular Biology, Vol.225, pp. 35-46. New Jersey: Humana Press.
Figure 1.
Properties of fluo4-zymosan particles. (a) Shows the phase contrast (upper) and fluorescent (lower) appearance of zymosan particles with the dense ‘core’ and the transparent periphery indicated. (b) the relationship between free Ca2+ concentration and fluorescence intensity from the core and peripheral regions of individual zymosan particles. Individual experimental data is shown with the theoretical relationship (lines) shown for two dissociation constants (kd). (c) A sample experiment showing the same microscopic field; top=phase contrast; then florescence images at the same field at the Ca2+ concentrations indicated.
Figure 1.
Properties of fluo4-zymosan particles. (a) Shows the phase contrast (upper) and fluorescent (lower) appearance of zymosan particles with the dense ‘core’ and the transparent periphery indicated. (b) the relationship between free Ca2+ concentration and fluorescence intensity from the core and peripheral regions of individual zymosan particles. Individual experimental data is shown with the theoretical relationship (lines) shown for two dissociation constants (kd). (c) A sample experiment showing the same microscopic field; top=phase contrast; then florescence images at the same field at the Ca2+ concentrations indicated.
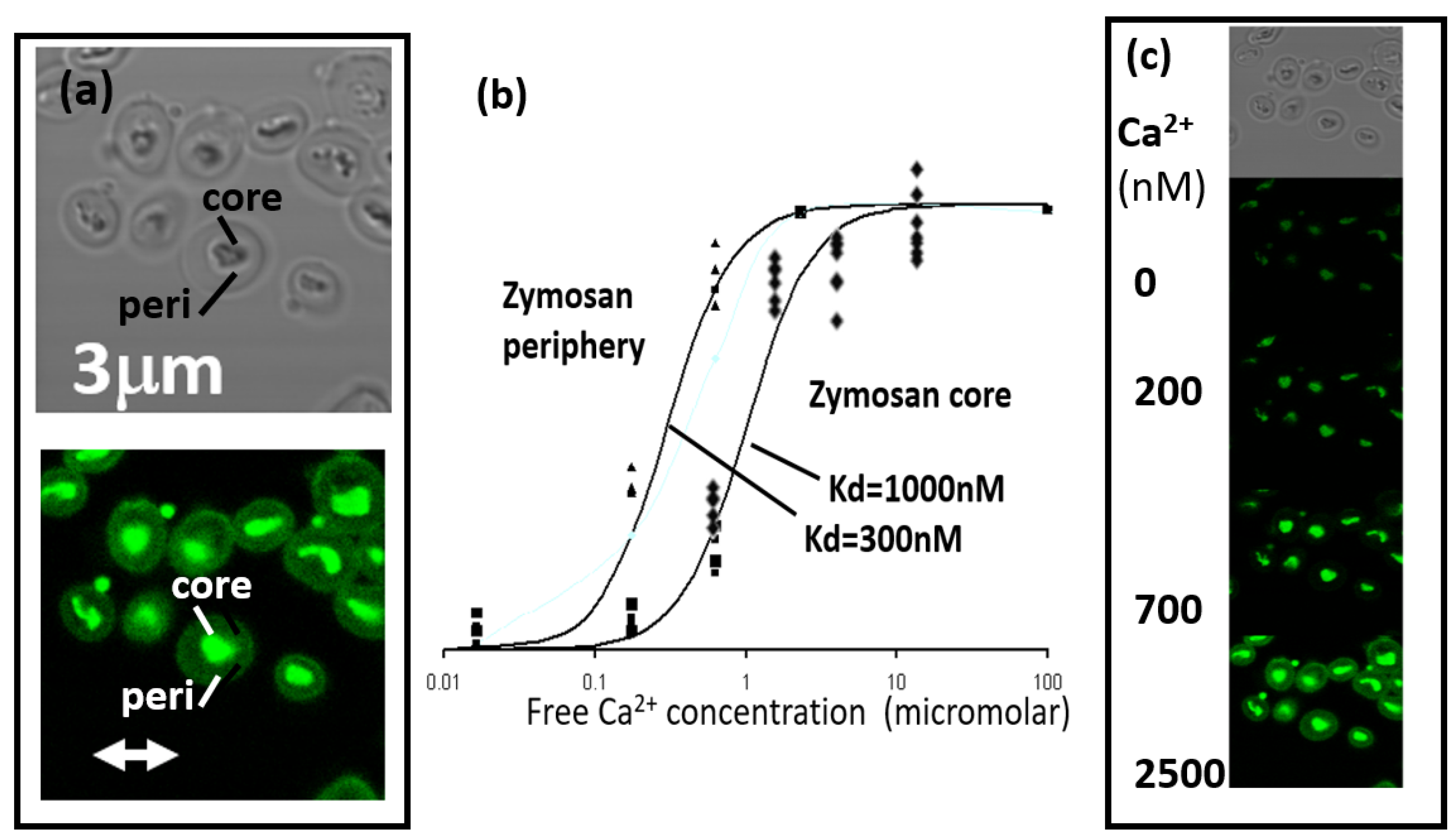
Figure 2.
Comparison of fluorescein and fluo4-coupled zymosan particles. In (a) and (b) the phase contrast and corresponding fluorescent images are shown for (a) fluo4-coupled zymosan and (b) fluorescein-coupled zymosan. In both sets of images show the internalised particles, marked by a ‘+’ and external particles by a ‘*’. (c) shows the quantitation of fluorescence from fluo4 and fluorescein particles either outside the cell (ZO) or inside a phagosome (Zph). The fluorescence units were arbitrary, but were comparable as measurements were taken from the same microscopic fields with the same excitation strength and detection sensitivity. The bars show the mean and the vertical line shows the range for at least 50 determinations.
Figure 2.
Comparison of fluorescein and fluo4-coupled zymosan particles. In (a) and (b) the phase contrast and corresponding fluorescent images are shown for (a) fluo4-coupled zymosan and (b) fluorescein-coupled zymosan. In both sets of images show the internalised particles, marked by a ‘+’ and external particles by a ‘*’. (c) shows the quantitation of fluorescence from fluo4 and fluorescein particles either outside the cell (ZO) or inside a phagosome (Zph). The fluorescence units were arbitrary, but were comparable as measurements were taken from the same microscopic fields with the same excitation strength and detection sensitivity. The bars show the mean and the vertical line shows the range for at least 50 determinations.
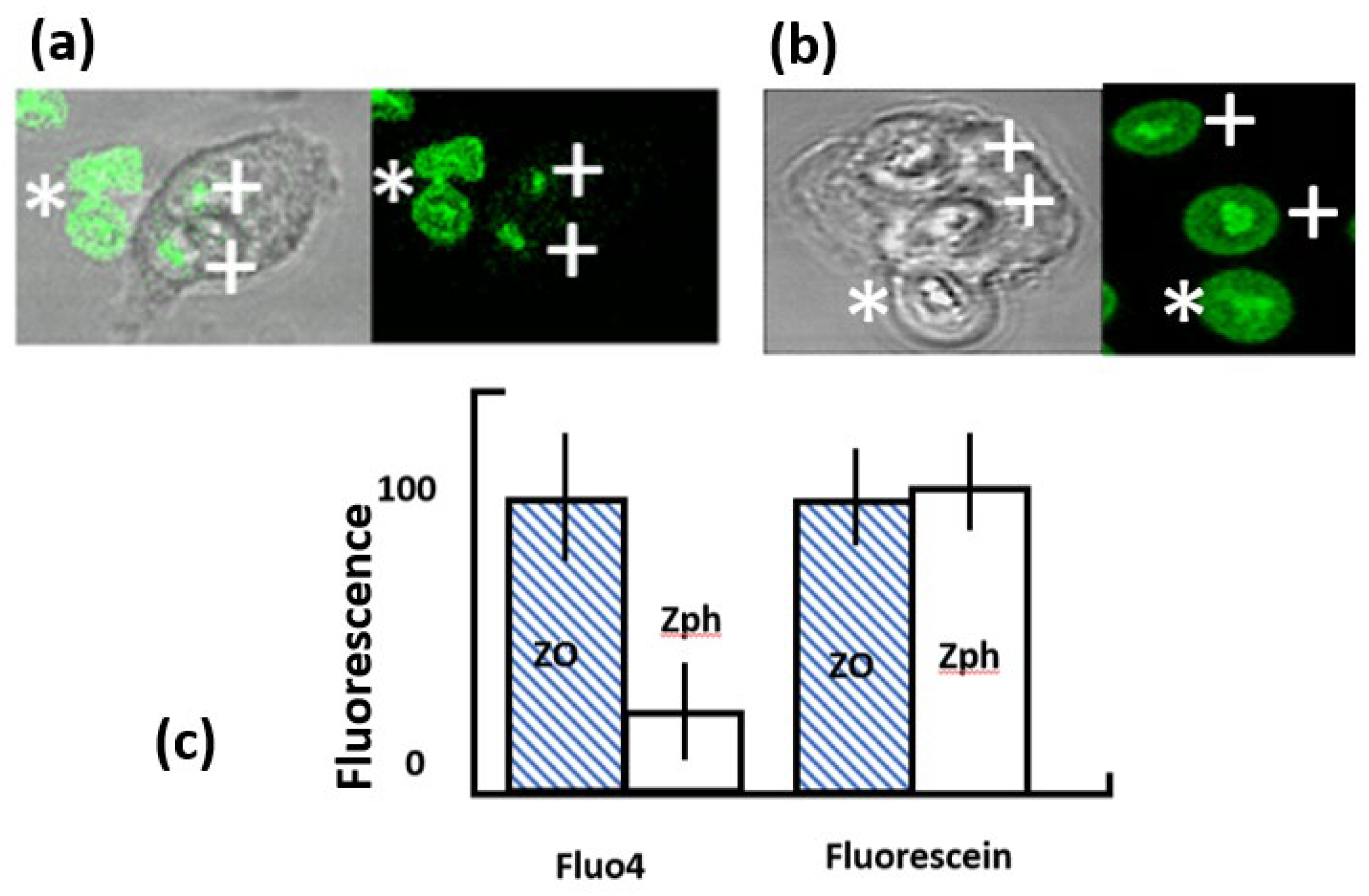
Figure 3.
The effect of expertimental manipulations on fluo4- zymosan fluorscence. (a) shows the effect of H2O2 on fluo4- zymosan-fluorescence incubated for the times shown. The top panel shows a typical experiment and the graph below the combined data from 3 separate experiment (n= 100 particles). The bars show the S.E.M. for the 3 experiments. (b) shows a typical experimentdemonstraing the effect of acidification on fluo4- zymosan-fluorescence at the pH shown. (c) The intensity of Fluo4-zymosan within the phagosome (open bars) and after treatment (cross-hatched bars), the pair marked ‘TX’ is before and after TritonX-100 (0.1%) treatment; the pair marked ‘Sap’ is before and after saponin (10%w/v) treatment, The pair marked Iono is for the same zymosan particles before and after addition of ionomycin (1 mM). In each case the bars show the mean and the vertical lines, the range, of replicate experiments for 5 experiments. (d) shows the time course of the change in fluo4-zymosan intensity after the addition of ionomycin, the star indicating the time point at which ionomycin was added and the images above the same cell, containing two fluo4-ztmosan particles before and after ionomycin treatment.
Figure 3.
The effect of expertimental manipulations on fluo4- zymosan fluorscence. (a) shows the effect of H2O2 on fluo4- zymosan-fluorescence incubated for the times shown. The top panel shows a typical experiment and the graph below the combined data from 3 separate experiment (n= 100 particles). The bars show the S.E.M. for the 3 experiments. (b) shows a typical experimentdemonstraing the effect of acidification on fluo4- zymosan-fluorescence at the pH shown. (c) The intensity of Fluo4-zymosan within the phagosome (open bars) and after treatment (cross-hatched bars), the pair marked ‘TX’ is before and after TritonX-100 (0.1%) treatment; the pair marked ‘Sap’ is before and after saponin (10%w/v) treatment, The pair marked Iono is for the same zymosan particles before and after addition of ionomycin (1 mM). In each case the bars show the mean and the vertical lines, the range, of replicate experiments for 5 experiments. (d) shows the time course of the change in fluo4-zymosan intensity after the addition of ionomycin, the star indicating the time point at which ionomycin was added and the images above the same cell, containing two fluo4-ztmosan particles before and after ionomycin treatment.
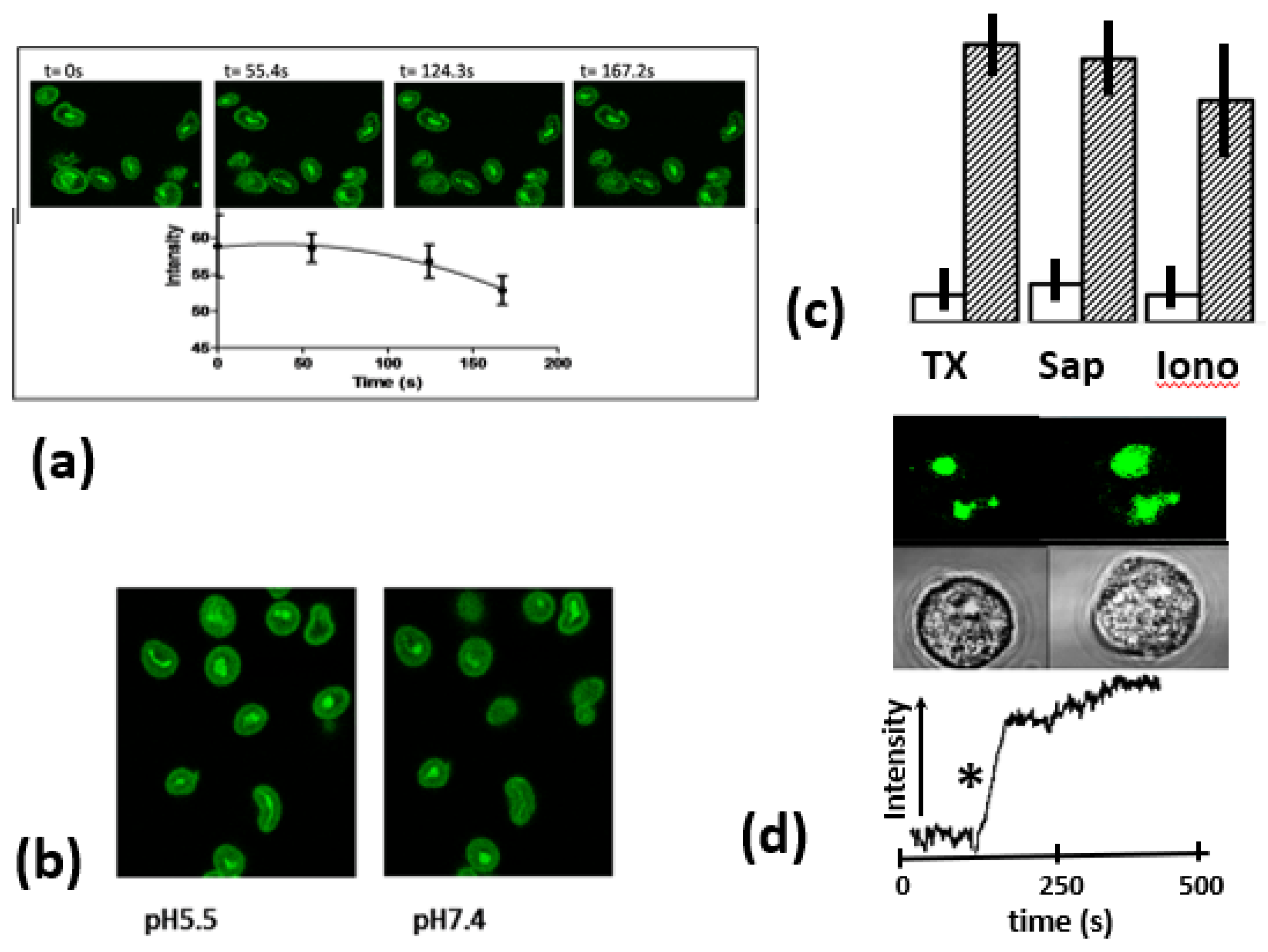
Figure 4.
Changes in fluo4-zymosan intensity during phagocytosis. (i) A time sequence the fluorescence and phase contrast images of a neutrophil as it internalised a zymosan particle. The particle which is internalised is labelled (a) and (b) the ‘control’ particle, which remain external. Each image shows the progress of phagocytosis at the times indicated. (ii) The lower graph shows the complete time sequence of the decrease in fluo4-zymosan intensity with key events (cup formation, closure of the phagosome and internalisation) marked. The sequence was typical of at least 14 other phagocytotic events. The movie in the Appendix A (MOVIE 1) shows a different experiment, in which a similar complete process of phagocytosis and the accompanying decrease in fluo4-zymosan fluorescence can be seen.
Figure 4.
Changes in fluo4-zymosan intensity during phagocytosis. (i) A time sequence the fluorescence and phase contrast images of a neutrophil as it internalised a zymosan particle. The particle which is internalised is labelled (a) and (b) the ‘control’ particle, which remain external. Each image shows the progress of phagocytosis at the times indicated. (ii) The lower graph shows the complete time sequence of the decrease in fluo4-zymosan intensity with key events (cup formation, closure of the phagosome and internalisation) marked. The sequence was typical of at least 14 other phagocytotic events. The movie in the Appendix A (MOVIE 1) shows a different experiment, in which a similar complete process of phagocytosis and the accompanying decrease in fluo4-zymosan fluorescence can be seen.
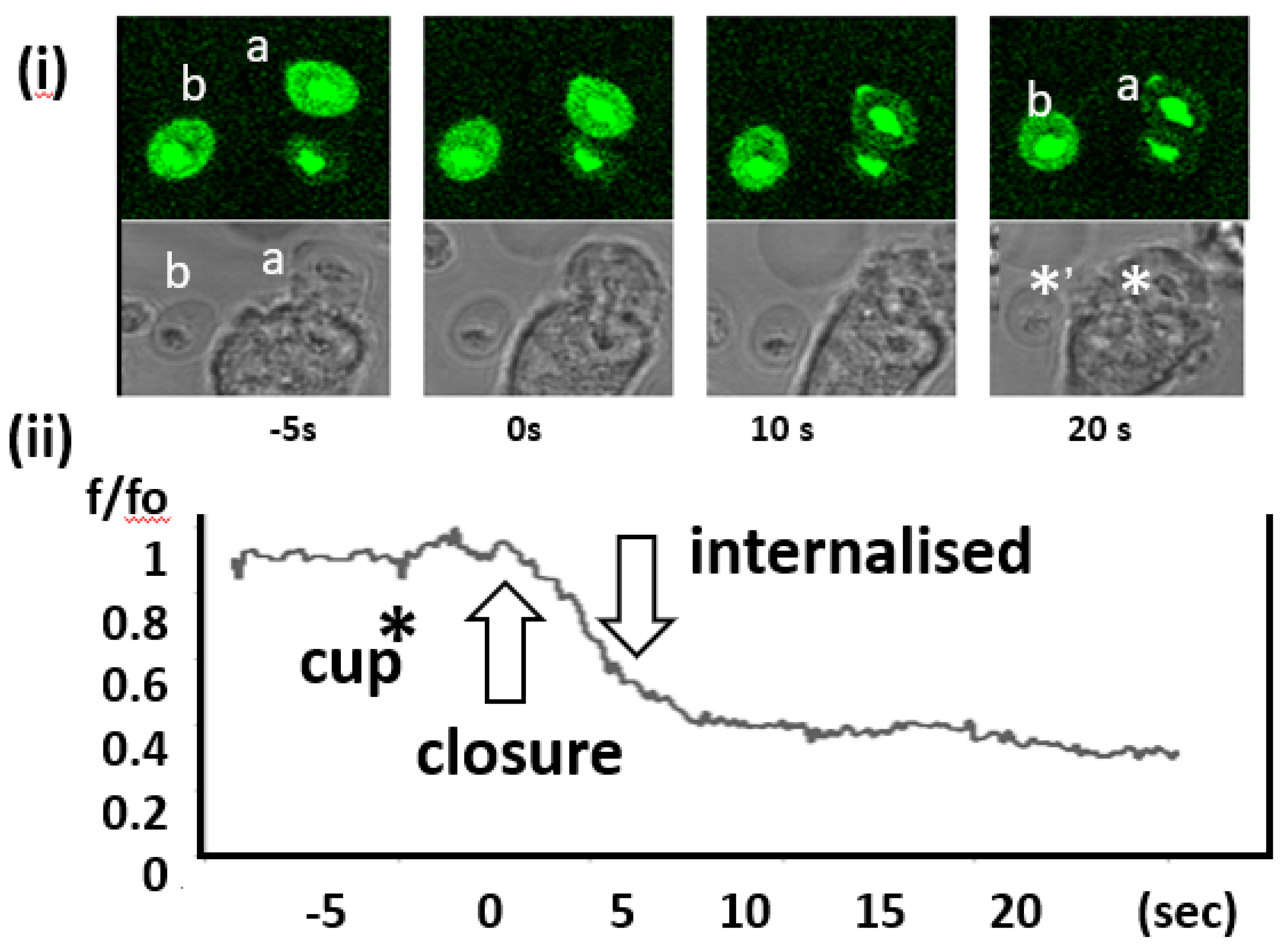
Figure 5.
Sites of Ca2+ influx marked by YFP-tagged C2 domain of PKC-γ. (a) A sequence of images of a RAW 264.7 cell expressing YFP–C2-γ. The phagocytic target is indicated by the crosshatched circle. At time zero (as indicated) a phagocytic cup formed at the base of the zymosan without translocation of YFP–C2-γ from the cytosol. After 1s, translocation of YFP–C2-γ from the cytosol to the plasma membrane of the phagocytic cup is seen. This is also shown in the enlarged image (b). At 2s, translocation of YFP–C2-γ to the phagocytic cup and the rest of the plasma membrane is obvious, as can also be seen at in the image marked ‘3s’. (b) Enlarged images, showing a previous old phagosome marked by an ‘X’ which has no translocated YFP–C2-γ on its membrane at 1s, but in the lower image at 3s, some translation had occurred. In image (c) at 20s, translocation of YFP–C2-γ had remained globally on the plasma membrane and can also be seen more clearly on the previously internalised phagosome. (d) shows the regions at which the measurements of fluorescence intensity at the plasma membrane (PM) and cytosol (within the ‘box’) were taken.
Figure 5.
Sites of Ca2+ influx marked by YFP-tagged C2 domain of PKC-γ. (a) A sequence of images of a RAW 264.7 cell expressing YFP–C2-γ. The phagocytic target is indicated by the crosshatched circle. At time zero (as indicated) a phagocytic cup formed at the base of the zymosan without translocation of YFP–C2-γ from the cytosol. After 1s, translocation of YFP–C2-γ from the cytosol to the plasma membrane of the phagocytic cup is seen. This is also shown in the enlarged image (b). At 2s, translocation of YFP–C2-γ to the phagocytic cup and the rest of the plasma membrane is obvious, as can also be seen at in the image marked ‘3s’. (b) Enlarged images, showing a previous old phagosome marked by an ‘X’ which has no translocated YFP–C2-γ on its membrane at 1s, but in the lower image at 3s, some translation had occurred. In image (c) at 20s, translocation of YFP–C2-γ had remained globally on the plasma membrane and can also be seen more clearly on the previously internalised phagosome. (d) shows the regions at which the measurements of fluorescence intensity at the plasma membrane (PM) and cytosol (within the ‘box’) were taken.
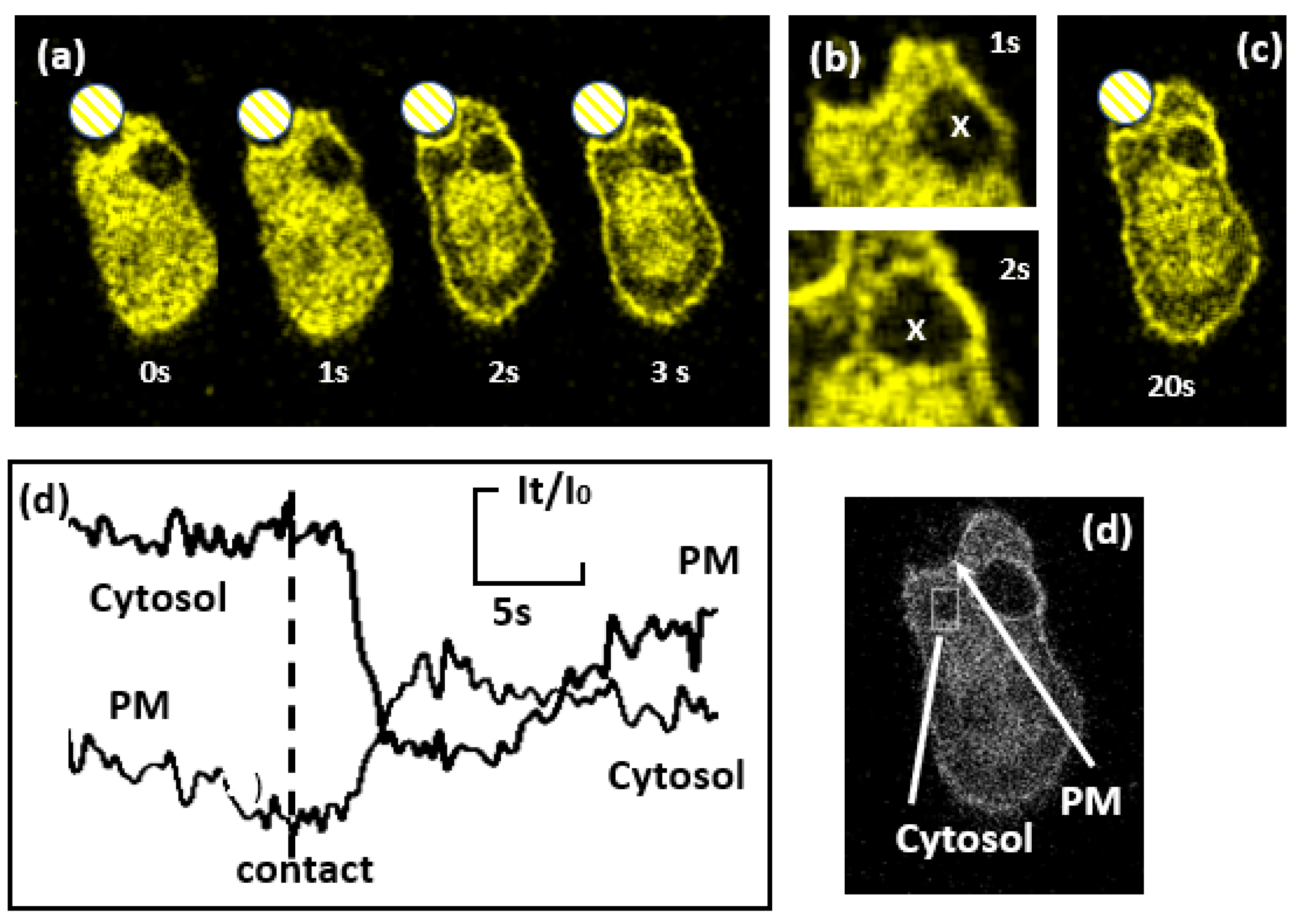
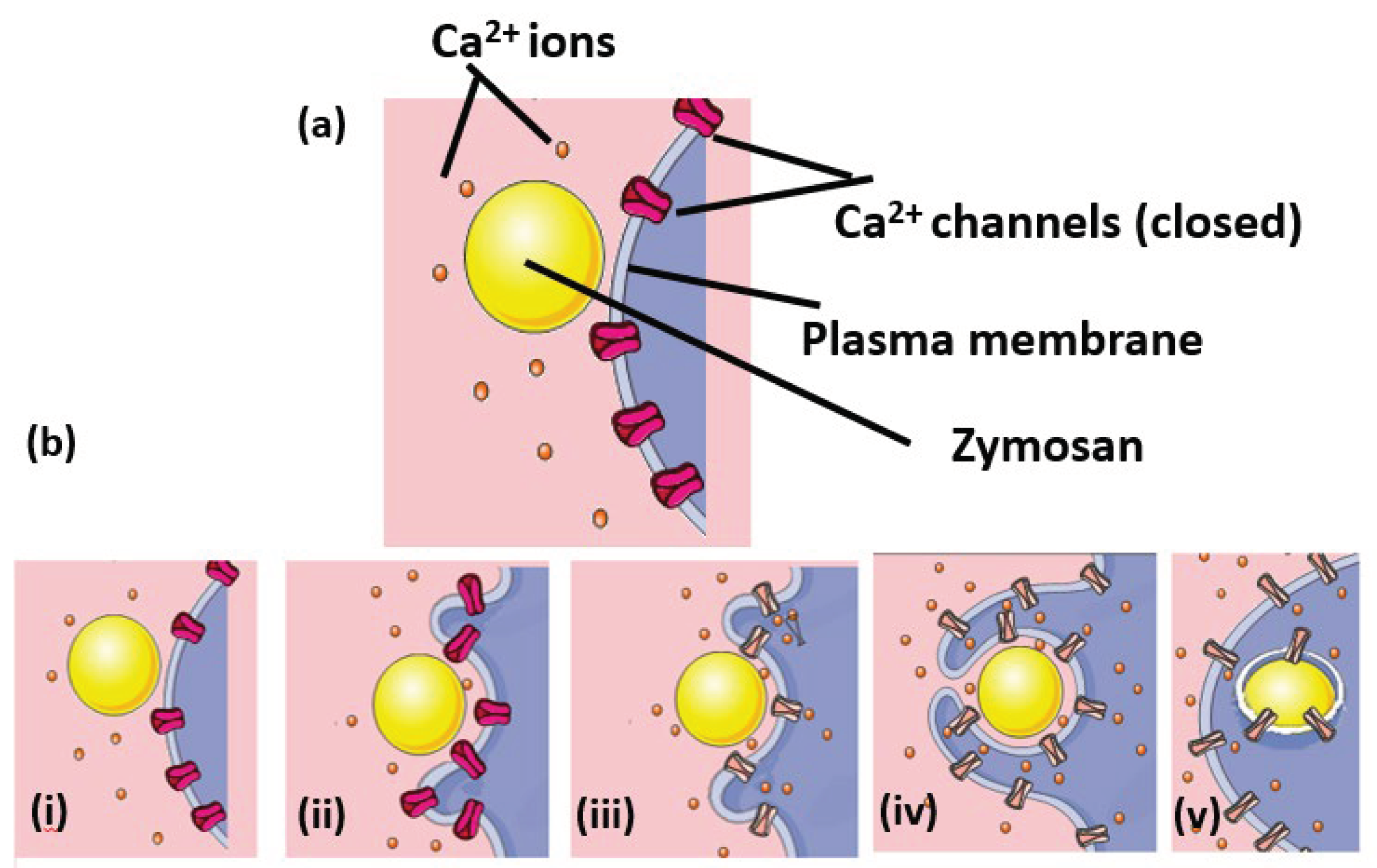
Figure 6.
Proposed sequence of Ca2+ channel opening on the phagosomal membrane. (a) Shows a ‘cartoon’ of some of the components of proposed model system. The phagocyte plasma membrane with Ca2+ ion channels (closed), before contact with the phagocytic target (zymosan particle) and Ca2+ ions are labelled. (b) The proposed sequence of events during phagocytosis are shown as follows. (i) before contact between the phagocyte and the target, where the cytosolic free Ca2+ion concentration is low: (ii) contact between the particle and the phagocyte resulting in the formation of a phagocytic cup. The Ca2+ ion channels remain closed and the cytosolic free Ca2+ion concentration is still low: (iii) A critical number of receptors are engaged and the Ca2+ channels open and the cytosolic free Ca2+ion concentration begins to rise. Note that the location of the open Ca2+ channels includes the portion of plasma membrane which forms the base of the forming phagosome and so provides a mechanism for preferentially elevating Ca2+ near the forming phagosome. (iv) In response to the elevated cytosolic free Ca2+ion concentration, the pseudopodia around the target nearly encloses it, trapping Ca2+ions in the forming phagosome. (v) Phagocytosis is complete but Ca2+ channels remain open on the phagosomal membrane, such that intra-phagosomal Ca2+ leaks out into the surrounding cytosol. Once ‘drained’ of Ca2+, the phagosome plays no further role in Ca2+ signalling.
Figure 6.
Proposed sequence of Ca2+ channel opening on the phagosomal membrane. (a) Shows a ‘cartoon’ of some of the components of proposed model system. The phagocyte plasma membrane with Ca2+ ion channels (closed), before contact with the phagocytic target (zymosan particle) and Ca2+ ions are labelled. (b) The proposed sequence of events during phagocytosis are shown as follows. (i) before contact between the phagocyte and the target, where the cytosolic free Ca2+ion concentration is low: (ii) contact between the particle and the phagocyte resulting in the formation of a phagocytic cup. The Ca2+ ion channels remain closed and the cytosolic free Ca2+ion concentration is still low: (iii) A critical number of receptors are engaged and the Ca2+ channels open and the cytosolic free Ca2+ion concentration begins to rise. Note that the location of the open Ca2+ channels includes the portion of plasma membrane which forms the base of the forming phagosome and so provides a mechanism for preferentially elevating Ca2+ near the forming phagosome. (iv) In response to the elevated cytosolic free Ca2+ion concentration, the pseudopodia around the target nearly encloses it, trapping Ca2+ions in the forming phagosome. (v) Phagocytosis is complete but Ca2+ channels remain open on the phagosomal membrane, such that intra-phagosomal Ca2+ leaks out into the surrounding cytosol. Once ‘drained’ of Ca2+, the phagosome plays no further role in Ca2+ signalling.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



