
Submitted:
02 March 2024
Posted:
04 March 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Since the beginning of the COVID-19 pandemic, extensive drug repurposing efforts sought to identify small molecule antivirals with various mechanisms of action. Here we aim to review research progress on small molecule viral entry and fusion inhibitors that directly bind to the SARS CoV-2 Spike protein. Early in the pandemic, numerous small molecules were identified in drug repurposing screens and reported to be effective in vitro SARS CoV-2 viral entry or fusion inhibitors. However, given minimal experimental information regarding the exact location of small molecule binding sites on Spike, it was unclear what the specific mechanism of action was, or where the exact binding sites were on Spike for some inhibitor candidates. The work of countless researchers has yielded great progress, with the identification of many viral entry inhibitors that target elements on the S1 receptor binding domain (RBD) or N-terminal domain (NTD) and disrupt S1 receptor binding function. In this review, we will also focus on highlighting fusion inhibitors that target inhibition of the S2 fusion function, either by disrupting the formation of the postfusion S2 conformation or alternatively by stabilizing structural elements of the prefusion S2 conformation to prevent conformational changes associated with S2 function. We highlight experimentally validated binding sites on the S1/S2 interface and on the S2 subunit. While most mutations to the Spike protein to date in variants of concern (VOCs) have been localized to the S1 subunit, the S2 subunit sequence is more conserved with only a few observed mutations in proximity to S2 binding sites. Several recent small molecules targeting S2 have been shown to have robust activity over numerous recent VOC mutant strains and/or greater broad spectrum antiviral activity for other more distantly related coronaviruses.
Keywords:
Arbidol
; Toremifene
; Entry Inhibitor
; Fusion Inhibitor
; Spike-dependent
; SARS CoV-2
; S2 segment
; S2 subunit
; Surface Plasmon Resonance
; Molecular Docking
1. Introduction
The world has made significant strides since the emergence of the novel coronavirus in 2019 [1,2]. The following years saw collective efforts throughout the world as scientists and health policy makers initially attempted to contain the spread of the disease. As time wore on, it has become apparent that COVID-19, and the virus that causes it, SARS CoV-2 will remain with us. Additionally, we have had to contend with new variants that can evade current treatments and vaccines, necessitating further resources and effort to keep up with evolution [3].
While we have come a long way in our clinical understanding of the disease and how to treat it, the cost of the pandemic in human lives and economic output, have highlighted the need to be prepared for future pandemics. We also have seen that there will not likely be a one-size-fits-all approach. While immunoglobulin-based therapeutic approaches have been beneficial for outpatient treatment [4], patients at higher risk for severe disease [5,6] as well as in patients hospitalized with severe disease [7,8,9,10], these therapies have limitations [11,12,13 and 14]. They require a great deal of invested capital and healthcare infrastructure that may not be available to rural communities or developing parts of the world, and may otherwise be prohibitively expensive. Small molecules can overcome many of these hurdles, and when inhibitors are repurposed from existing drugs, the costs (and therefore a large barrier to access) are substantially reduced. But to optimize the efficacy of small molecule inhibitors, we need to look outside of some of the current strategies for small molecule drugs. In some of our earlier research we were attracted to non-structural protein 13, the helicase [15], for its highly conserved nature among coronaviruses. The same concept, of targeting a highly conserved structural element, would also lead us to focus our attention on the S2 segment of the Spike protein (Figure 1) found on the surface of the viral envelope of SARS CoV2. This region, which consists of 585 amino acids, makes up its constituent fusion peptide and two heptad repeats (HR1 and HR2) [2] and offers an alternative target for small molecule inhibitors than the S1 receptor binding domain (RBD) that is currently the primary focus of treatment.
2. The S2 subunit sequence has remained very conserved in variants (VOCs)
During the summer of 2021, there was increasing concern regarding the emergence of the Omicron variant. There were concerns due to its being more transmissible compared to earlier variants. This enhanced transmissibility was the result of numerous mutations to the Spike protein, specifically the S1 segment, the region which is responsible for binding with the angiotensin converting enzyme 2 (ACE2) receptor on host cells [16,17]. The S1 segment is highly subject to mutation and the majority of observed mutations to Spike observed to date in Variants of Concern (VOCs) have been found on S1. Indeed, some immunoglobulin treatments themselves put selective pressure on variants, favoring those with altered S1 segments [18,19]. This has resulted in some of the immunoglobulin-based therapies and earlier vaccines losing their efficacy against some variants [16,17]. Thankfully the development of resistance is not insurmountable. If the S1 segment is the tip of the spear, the S2 segment is the shaft, which is highly conserved due to its structure being crucial for function. Indeed, the S2 segment is not only conserved among SARS CoV-2 variants, but among other coronaviruses such as Middle East Acute Respiratory Syndrome (MERS), SARS CoV-1, and even among zoonotic sarbecoviruses [20,21,22]. It is likely that this is due to the crucial role that structural rearrangement of S2 in coronaviruses has in stabilizing the postfusion conformation, allowing the delivery of viral RNA into the host cell.
With respect to specific targetable elements of the S2 subunit, the fusion peptide, stem helix, and the heptad repeat 1- heptad repeat 2 bundle (HR1- HR2 bundle) there exists little variation between different sarbecoviruses [20-22]. In a recent sequence-structure analysis of the mutations from the Omicron variant and several related sublineages (BA.1, BA.2 and BA.3) [23], mutations were observed at 31 sequence positions
(84%) on the S1 subunit, while mutations were only observed at 6 sequence positions (16%) on the S2 subunit. Three mutations Q954H, N967K, and L981F are found on the HR1 motif, leaving the CH, CD, SH, and HR2 motifs entirely conserved among these lineages. The fusion peptide is remarkably conserved, which initiates the fusion event by inserting itself into the target cell membrane. The HR1- HR2 bundle specifically has few observed mutations, while a recent functional mutation study demonstrated that the specific HR1 motif mutation L981F reduced fusion activity as a single point mutation [24], where allosteric effects of other mutations compensate for this effect in Omicron [24]. We outline below that small molecule inhibitors that disrupt the function of the postfusion 6HB structure are found to exhibit broad-spectrum activity against a range of coronavirus strains. While protein-based immunoglobulin treatments are not the focus of this review, a recent article by Guo et al., reviews and highlights S2-specific epitopes that are targeted by some new second generation immunoglobulins, resulting in effective neutralization and a wider-spectrum of activity [25]. Rather, we focus on small molecules and highlight where they putatively bind to the Spike protein to inhibit Spike function.
3. Conformational changes and S2 subunit function
Upon binding with the ACE2 receptor, the S1 subunit will dissociate from the S2 at the S1/S2 junction [26,27]. This is mediated by the endopeptidase furin. This will then expose the S2’ site which is then cleaved by the host’s transmembrane protease serine 2 via the plasma pathway, though it may also be mediated by lysosomal cathepsins during virion endocytosis [28,29]. This cleavage releases the structural constraints on the fusion peptide region and allow the flexibility for fusion to occur [30].
Once membrane fusion occurs, this leads to further conformational changes in the S2 subunit. The first of these is that the HR1 undergoes a jack-knife refolding change that allows the fusion protein insertion into the host cell membrane [29,30]. In the metastable prefusion conformation, the HR1 motif folds into four individual α-helix segments that pack against various structural elements including the long structural 3-helix bundle formed by the CH domain. In the postfusion conformation, the HR1 motif refolds into a long central α-helical coiled coil as shown in Figure 1C and forms a 6-helix bundle (6HB) with highly conserved residues from the HR2 motif [29,30]. The extended stem HR2 element packs against the long central HR1 coiled coil which induces the binding of the stem helix to the outer region of the central helix, and HR2 into the HR1 binding groove. This serves to strengthen the long central helical bundle formed by SH-HR2 with CH-HR1, and this structure becomes the fusion core in S2, which provides enough stability to the membrane and forming the fusion pore in the postfusion state [30,31].
4. Small molecule entry inhibitors of SARS CoV-2 Spike
Early in the pandemic, numerous drug-repurposing efforts performed screening for antiviral activity using various experimental strategies given what was possible for a novel biological safety level 3 (BSL-3) virus. Since assays using pseudotyped viral particles displaying the Spike protein (and not the viral genome) may be performed in BSL-2 facilities, these assays are much more amenable to high-throughput screening (HTS) efforts. In one of the first HTS drug-repurposing efforts reported (Oct, 2020) using a viral entry assay with pseudotyped particles [32], Chen et al. identified seven Spike-dependent entry inhibitors, where entry inhibitor activity was also measured in multiple cell types (Vero E6, Huh7, and Calu-3 cells). For these entry inhibitors, when antiviral activity was also assessed in cytopathic effect (CPE) assays in Vero E6 cells, it was interesting that very few of the entry inhibitors were found to be full inhibitors with (100% efficacy). In CPE assays cepharanthine was able to demonstrate 92.5% efficacy, compared to abemaciclib (68.7%) and four others that were partial inhibitors: osimertinib (60%), trimipramine (48%), NKH477 (45.6%), and ingenol (38.2%). Of these cepharanthine and abemaciclib exhibited the greatest efficacy and the most robust coronavirus entry activity in multiple cell types (Vero E6, Huh7, and Calu-3 cells) [32]. Given the diverse structural classes of these drugs, the data revealed that it was difficult to identify lower molecular weight (MW) small molecules with 100% entry inhibitor activity. From the structural and medicinal chemistry point of view, the assay data suggested that deciphering the structural requirements of the various compound’s activity would be quite interesting. Why are some compounds full inhibitors and others are not? Can this be explained or rationalized by: (1) differences in 2D structural features on a compound, (2) differences in binding sites or mechanism of action or (3) differences in the strength of binding ΔGbind at the same binding site? From our point of view, answers to these questions will increasingly reveal the design principles for next generation inhibitors with improved efficacy.
While the observed entry inhibitor activity is “Spike-dependent” in a pseudotyped virus assay, the small molecule drugs obviously have other known mechanisms of action. However, entry inhibitor activity may or may not necessarily be related to previously known “host-dependent” mechanisms of action. While numerous viral entry inhibitors have been identified that target a host protein and exhibit a “host-dependent” mechanisms of action, we do not aim to summarize those which have been recently reviewed elsewhere [33,34]. Rather, we aim to focus on small molecules that act by binding directly to the Spike protein with various “Spike-dependent” mechanisms of action. Other types of experimental approaches including biophysical binding studies, mutagenesis, and experimental CryoEM or X-ray structure determination are able to characterize more precisely how these small molecules bind to Spike and inhibit viral entry. We also highlight the role of molecular modeling approaches in predicting the location of entry inhibitor binding and the effects on Spike structure and dynamics.
As viral entry is dependent on the structure and function of the trimeric Spike protein expressed on the surface of a mature virus particle, it is not surprising that many viral entry inhibitors target the S1 subunit and disrupt receptor binding function. Alternatively, we highlight that numerous viral entry inhibitors that have been shown to target the S2 subunit and inhibit S2 conformational changes related to S2 function and viral membrane fusion.
4.1. S1-targeted small molecule entry inhibitors
As the S1 subunit function is to bind to the host (ACE2) receptor through the Spike receptor-binding-domain (RBD), numerous immunoglobulins, peptides and small molecules have been identified that putatively interact with the RBD of Spike [35]. Among numerous research achievements in this area, the designed miniprotein inhibitor from David Baker and colleagues (Oct, 2020), Cao et al., stands out as an impressive milestone in computational molecular design [36]. While we do not aim to review all of these efforts here, we highlight three major small molecule binding sites in (Figure 2) that are located on the S1 subunit. Numerous small molecules have been reported to bind in proximity of Site 1 which is found on the RBD in proximity of (res: F342, F374 and F377). As the majority of protein-ligand interactions formed in Site 3 involve RBD residues (res: 390, 517, 519, 546, 565), small molecules that bind to either Site 1 or Site 3 are both considered to bind to the RBD. As much effort has been focused on these sites, numerous structural classes of small molecules that bind to the RBD have been identified and are reviewed in detail by Sabbah et al. [37].
As the SARS CoV-2 Spike protein is large and dynamic, until recently there had been very little experimental structural information of the SAR CoV-2 Spike protein bound to small molecules. Here we review major proposed small molecule binding sites on the Spike prefusion conformation (Figure 2) that have not yet been confirmed by X-ray or CryoEM complex structures, but are supported by molecular docking studies, biophysical studies, or confirmatory mutations (discussed in detail below). Initial CryoEM complexes of Spike were first reported of the RBD in complex with linoleic acid (Figure 2.) (Site 1) [38]. Then the biliverdin binding site (Site 2) was found on the S1 N-terminal domain (NTD) [39]. Linoleic acid has now been shown in numerous structures to bind to the S1 RBD, in the free fatty acid (FFA) binding site (Site 1) [38]. The experimental determination of a complex between Spike and the small molecule SPC14 is a breakthrough in showing how a small molecule may bind to FFA binding site (Site 1) [40]. The CryoEM structure of Spike in complex with the molecule SPC14 (8h3e.pdb), shows how a small molecule containing a heptanoic acid moiety may bind to the FFA site and shift the conformational equilibrium to the closed state affecting receptor binding function. The Site 1 binding site on the RBD is defined by (res: Y365, L368, F374, F377, L513, F515, V524). In contrast the biliverdin site is defined by (res: W104, N121, V126, R190, F192).
Site 3 is another major RBD site that has not yet been confirmed by experimental structural biology, but is strongly supported by numerous molecular modeling, biophysical studies, and virtual screening efforts. The SSRI sertraline is a representative entry inhibitor of SARS CoV-2 that is proposed to bind to Site 3, where sertraline has been shown to bind to the S1 RBD according to biophysical binding data [41]. In the proposed binding model from docking, sertraline is proposed to bind to Site 3 with key protein-ligand interactions on the RBD (res: 390, 517, 519, 546, 565) where small molecule binding is on an allosteric interface between the S1 structural domains and parts of the S2 segment in close proximity (res:968-984) and (res:747-757). Numerous groups have modeled basic amine containing compounds at this site, including chloroquine and derivatives binding to the RBD [42]. Site 3 may also be a possible binding site for other basic amines identified as entry inhibitors including trimipramine [32]. Other examples of repurposed antivirals that bind to the RBD, include tilorone and pyronaridine which were shown to bind to the RBD by biophysical microscale thermophoresis (MST) experiments [43]. Interestingly, in the same study another basic amine with somewhat similar structure, quinacrine was not found to bind to the RBD by MST but retained entry inhibitor activity [43]. The most recent structural information for other RBD interacting entry inhibitors was a recently published a series of thiophenyl tryptophan-based trimers that were found to interact with some RBD contacts in an experimental CryoEM structure [44].
4.2. S2-tarteted fusion inhibitors that interfere with HR1-HR2 bundle assembly
From previous knowledge and research on trimeric viral class I fusion proteins, particularly HIV and influenza, the strategy of inhibiting Spike-mediated viral fusion with peptides that interfere with postfusion assembly had been established. For HIV-1, cleavage results in an N-terminal gp120 containing the RBD and the C-terminal fusion gp41 fragment. For HIV-1, the postfusion 6-helix bundle (6HB) of gp41 is formed from interactions between the heptad regions NHR and CHR. A body of research has shown that peptides based on these NHR and CHR region are able to prevent 6HB formation [45], but CHR derived peptides typically have better solubility and often are more potent fusion inhibitors [45].
Following the same rationale, For SARS-CoV and SARS CoV-2, in the S2 subunit the equivalent heptad regions HR1 and HR2 form the postfusion 6HB (Figure 1C). Prior to the SARS CoV-2 outbreak, it had been established that HR2 mimicking peptides of SARS-CoV were effective fusion inhibitors. Subsequently, numerous HR2 based peptides such as EK1 (Mar, 2020) [46,47] are proposed to bind to the HR1 grove in intermediate states of S2 conformational changes prior to formation of the final low energy post-fusion conformation. Peptides such as the 5-HB protein are proposed to bind to the HR2 structural element in the prefusion conformation prior to S2 conformational changes [48,49,50]. Recent advances in peptide-based fusion inhibitors based on HR1 or HR2 sequence peptides has been extensively reviewed recently [51]. We continue our focus on small molecule fusion inhibitors in the next section, starting with small molecule fusion inhibitors that mimic the action of these HR1 targeted peptides and interfere with 6HB formation.
4.3. S2-targeted small molecules that interfere with HR1-HR2 bundle assembly
One of the first reported (Aug, 2020) small molecule fusion inhibitors of SARS CoV-2 was salvianolic acid C [52], which is a natural product derived from the traditional Chinese medicine (TCM) product danshen. Salvianolic acid C is a small molecule natural product inhibitor proposed to bind to the HR1 grove and prevent assembly of the 6-helix bundle (6HB) postfusion core [52]. Using native-polyacrylamide gel electrophoresis (N-PAGE) and biophysical circular-dichroism (CD) experiments with HR1 and HR2 peptides, salvianolic acid C was shown to interfere with the formation of the 6HB fusion core. Docking studies show that salvianolic acid C likely binds to HR1 residues (940-949) interfering with HR2 peptide docking to the HR1 postfusion core grove. Thus, the mechanism of action of salvianolic acid C is similar to HR2 based peptides, such as EK1 that bind to the HR1 groove as anti-HR1 inhibitors.
Recently, posaconazole was identified as a potent inhibitor and has been proposed to bind to the E1182-L1186-L1193 motif on the HR2 domain and prevent assembly of the 6HB postfusion core [53]. Using mutations R1185A and N1192A from HR2 fusion core were shown to reduce the inhibitory effects of posaconazole in pseudotyped virus entry assays. Structurally related itraconazole [54] had also been previously identified as a fusion inhibitor of the 6HB post-fusion core [54]. Navitoclax has also been identified as a fusion inhibitor that is reported to target HR1 in S2 segment [55]. Using an N-PAGE gel-based assay, they authors concluded that navitoclax bound to HR1 peptides and interfered with HR1/HR2 assembly of the 6HB postfusion core. Navitoclax has also been demonstrated to exhibit broad spectrum activity against a range of VOCs, but also retains some activity against SARS-CoV, MERS-CoV and HCoV-299E [55]. Numerous studies of both peptides and small molecules that target disrupting the 6HB have demonstrated promising broad-spectrum activity against sequence variants or more distantly related coronaviruses [56,57]. These results highlight the advantages of targeting the HR1/HR2 assembly of the 6HB postfusion core.
4.4. S2-targeted small molecules that bind to the S2 prefusion conformation
The small molecule influenza HA inhibitor arbidol was one of the earliest (May, 2020) identified SARS-CoV-2 entry inhibitors [58]. The antiviral activity of arbidol was subsequently verified [59] and published in numerous other studies [60,61]. Our laboratory was very interested in identifying thermodynamically favorable small molecule binding sites on the S2 subunit and published (Feb, 2021) a map of the TOP50 most favorable binding sites on S2 [15] and compared the results to the crystal structure of Arbidol bound to influenza HA2 [15]. Prior to our study, the first report (Aug, 2020) to successfully predict the binding site of Arbidol on the SARS-CoV-2 S2 subunit was a computational docking study of Vankadari, where molecular docking was performed on the Spike monomer alone [62]. Nevertheless, the prediction of the binding site location was ultimately correct. Our laboratory also identified and predicted the same binding site [15,63] for arbidol on S2 trimers (Dec, 2021), using molecular docking and structure-activity-relationship (SAR) modeling to model the experimental activity of derivatives [63]. A nearly simultaneous (Dec, 2021) publication from Shuster et al [64], experimentally confirmed the binding site of arbidol on the S2 segment of Spike. Shuster et al [64] identified the site experimentally using a chemical biology approach and then corroborated the exact predicted binding site [15,62,63] by mutational studies [64]. Thus, while the location of the arbidol binding site on the S2 segment had been unclear to that point, simultaneous publications from two laboratories [63,64] identified and independently confirmed the site initially reported by Vankadari [62] using entirely different approaches. Thus, arbidol is the best representative of small molecule fusion inhibitors that bind to Site 5 on the S2 segment as shown in Figure 2A. In a chemical series of OA saponin derivative fusion inhibitors, the representative compound 12f was shown to bind to the S2 segment by SPR [65]. Subsequent molecular modeling studies of the structure-activity-relationship (SAR) suggested that the series of OA saponin derivatives including 12f was best modeled binding to the arbidol binding site [63].
Subsequently, numerous other molecules have been reported to bind at or in proximity to the arbidol site. NBCoV1 and NBCoV2 are small molecules were shown to bind to the Spike trimer by SPR with an affinity of 1.56 μM and 5.37 μM respectively. However, they were found to bind much weaker to constructs of the S1 segment, thus the authors concluded that the molecules may bind to S2 [66]. The NBCoV1 and NBCoV2 compounds were tested using a number of mutants from VOCs, B1.b.7 UK triple mutant (d67-70/N501Y/P681H) and B.1.617.2 Delta single and triple mutants in particular. Interestingly, NBoV2 (WT 22.8 μM) was found to lose the most activity with the WT D950N single mutant (88.7 μM) (from the B.1.617.2 Delta), while the WT D614G mutant (61 μM) and E484K (45 μM) also lost activity. While D950N is the only S2 mutation that was shown to demonstrate the largest shift, other mutations that are either in medium proximity ~27 Angstroms (D614G) or are quite distant from the site, 70-80 Angstroms (E484K), are both able to show significant effects in dose-responses, illustrating obvious long-range allosteric effects that have been studied by other structural and biophysical techniques to probe structure and dynamics. In another study NBCoV63 [67] was identified as a fusion inhibitor that is similar in structure to NBCoV1 in that it contains a 3-(furan-2-yl) benzoic acid substructure. While NBCoV63 exhibited lower potency it exhibits much better drug-like properties in that it does not contain a functional group implicated as a frequent-hitter [68,69]. In the docking study the NBCoV63 benzoic acid formed a salt bridge with K947 of HR1, which forms a salt bridge with E1182 of HR2 in the postfusion conformation. In the docking model, electrostatic interactions of the NBCoV63 acid (COO-) with K776 and K947 were key to the binding mode, very similar to the binding mode of arbidol. NBCoV63 was also shown to retain activity for Omicron (BA.4/BA.5) variants in pseudovirus assays in both (293T/ACE2) and (A549/ACE2/TMPRSS2) cell lines [67]. Next, we will summarize small molecules that are proposed to bind at other sites on S2.
4.5. Other Proposed Fusion Inhibitor binding sites on the S2 segment
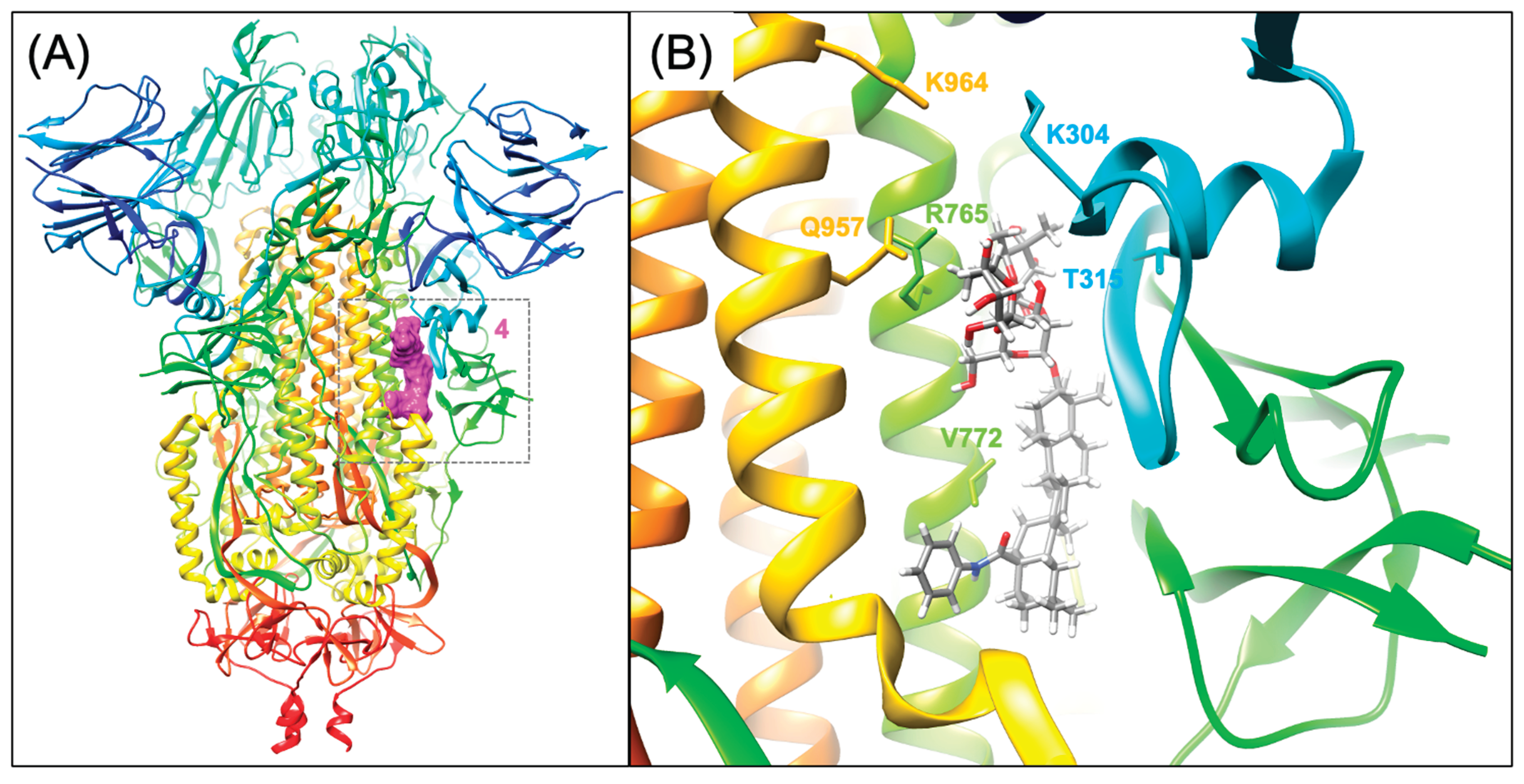
Site 4 is found on the interface of S1 and S2 as shown in (Figure 2) and numerous entry inhibitors have been modeled there by various groups. Nelfinavir was one of the first (May, 2020) small molecules that was reported to bind at this site according to molecular docking [70]. Subsequently (Sept, 2020) other entry inhibitors such as toremifene have also been proposed to bind at Site 4 according to molecular docking [71]. Interestingly, prior to the pandemic it was known that some natural product saponin derivatives (glycyrrhizin, aescin, α-hederin) exhibited entry inhibitor activity [72], and several new series of saponin derivative natural products have been shown to be SARS CoV-2 inhibitors [73,74]. As discussed previously, OA saponin derivative 12f was found to bind to the S2 subunit by SPR [65]. Similar in structure ursolic acid derivatives, such as UA-30 have also been reported to bind at the Site 4 S1/S2 interface site [73]. In a new series of 3-O-b-chacotriosyl ursolic acid derivatives, the derivative UA-18 was shown to bind to Spike by SPR and is shown to be best modeled as binding to this S1 and S2 interface site [73]. In this study, the double mutants (N764A/R765A), (Q957A/K964A), and quadruple mutant (N764A/R765A/Q957A/K964A) [73] were used to confirm the UA-18 binding site. Interestingly, in another study of betulonic acid saponin derivatives, BA-4 is a representative molecule that shows strong binding to S2 by SPR and does not bind to S1 [74]. Shown in Figure 3 is an example model of OA saponins binding this site, very similar to published models [73,74]. However, molecular modeling studies from our laboratory has also concluded that at some specific OA saponins such as 12a, 12f, are predicted to bind more favorably at the arbidol Site 5 rather than Site 4 [63]. In summary, toremifene, nelfinavir and a variety of natural product saponin derivatives are proposed to bind at Site 4 and presumably small molecule binding at this site acts to stabilize the prefusion conformation.
Moving our attention to another proposed binding site, as shown in Figure 1, Site 6 is another proposed binding site on the S2 segment of the SARS CoV-2 Spike is analogous to the proposed camphecene binding site on influenza HA2 [75] confirmed by mutagenesis and resistance mutations [76,77]. For a novel series of borneol ester derivatives, Yarovaya et al., identified that their derivatives 11 and 21 with the greatest activity in pseudovirus infections in both original Wuhan strain and Delta (B.1.617.2) strains [78]. The authors conclude that these derivatives 11 and 21 are well-modeled binding to an allosteric site that is analogous to the camphecene binding site on influenza HA [75]. On the SARS CoV-2 Spike protein, this binding site for borneol esters 11 and 12 is in close proximity to the fusion peptide helical conformation binding site in the prefusion conformation of Spike [78]. Key protein-ligand interactions that define this site using Wuhan strain numbering include S2 residues that are just C-terminal of the fusion peptide (F823, L826 and F833) and residues (P1057, H1058) from the S2 connector domain (CD) (res:986-1035). The borneol ester derivative binding site on the SARS CoV-2 Spike is supported from sequence analysis, extensive molecular docking, MD simulations and supported by the rationale of how camphecene derivatives are proposed to bind to influenza HA [75,76,77].
Last, we would also like to highlight Site 7, an additional putative binding site on the S2 segment for both peptides and small molecules. This Site 7 on the S2 subunit has been reported to be thermodynamically favorable for binding small molecules in several reports [15,63,79,80]. In this binding site, a key hydrophobic contact from residue W886 is an important structural element of a long helix-turn-helix (res:867-890) which is N-terminal to the HR1 and undergoes major conformational changes upon the transition from the prefusion to the postfusion conformation. In the prefusion conformation, this site is formed from W886, four residues from the N-terminus of the CD (Q1036, K1038, V1040, Y1047), and residues R1107 and N1108 which are N-terminal to the SH domain also undergo major refolding in the prefusion to postfusion transition. To our knowledge, the thermodynamic favorability for this site was first reported from a virtual screening study that was focused on the S2 segment that identified chitosan to bind in proximity to this site [79]. In our study using a pharmacophore mapping, docking and free energy approach, we identified this specific site as one of the most thermodynamically favorable binding sites on the S2 segment in the prefusion conformation [15,63] as well as the arbidol site (discussed above). In another study, a fusion inhibitor tripeptide VFI was also proposed to dock to this site, based on docking the VFI tripeptide to the entire Spike protein [80]. While this site has not yet been validated by mutagenesis, our laboratory and others will aim to see if point mutants effect fusion or small molecule binding. With recent advances in CryoEM characterization of Spike conformational states, it may be possible to characterize small molecule complexes of representative fusion inhibitors bound to Spike. As with other viruses in the past such as Influenza and HIV, prior to small molecule binding sites being experimentally confirmed by either mutagenesis or X-ray crystallography, it is important to remember that there is an important role for molecular modeling approaches to predict likely binding sites and to guide experimental approaches towards confirming them.
5. Conclusions
We reviewed a range of small molecules that act by binding directly to the Spike protein with various “Spike-dependent” mechanisms of action. While numerous viral entry inhibitors targeting the S1 subunit have been identified, we highlighted small molecules targeting the S2 subunit. S2-targeted peptides that interfere with HR1-HR2 bundle assembly are very promising and have been shown to be effective and exhibit broad-spectrum activity. Small molecules such as salvianolic acid C, posaconazole, itraconazole and navitoclax act using a similar mechanism of action to block the HR1/HR2 postfusion 6HB assembly. We summarized 7 proposed small molecule binding sites on the SARS CoV-2 Spike protein in the prefusion conformation. While Site 1 and Site 2 on the S1 subunit are confirmed by experimental CryoEM structure determination, Site 4 and Site 5 involving the S2 subunit are strongly supported by experimental point mutations and more than one independent study identifying the same binding site. Site 4 on the allosteric interface of the S1 and the S2 segment is where reference inhibitors nelfinavir, toremifene, and natural product saponin derivatives have been proposed to bind. Site 5 is found on the S2 subunit where arbidol is proposed to bind [62,63], as well as other small molecules such as NBCoV63 [67]. Small molecule binding at Site 4 and Site 5 on the S2 subunit are proposed to inhibit S2 conformational changes related to S2 function and viral membrane fusion. Future efforts to confirm the binding site location of representative small molecule inhibitors will elucidate how specific small molecule inhibitors inhibit S2 conformational changes. Future mutagenesis efforts interrogating these sites (pseudovirus fusion assay activity, biophysical small molecule binding, biophysical structure and function studies) should be able to delineate how small molecule binding prevents conformational changes in S2. Efforts in this direction may reveal new attractive strategies to target S2 and may also identify previously unknown functions of these binding sites. While significant attention has gone to therapeutic strategies focused on targeting S1, we highlighted the potential advantages of S2-targeted peptide and small molecule inhibitors.
Author Contributions
Conceptualization, M.F. and R.A.; M.F. and R.A.; writing—original draft preparation, M.F and R.A.; writing—review and editing, R.A.; supervision, R.A.; project administration, R.A.; funding acquisition, R.A. All authors have read and agreed to the published version of the manuscript.
Funding
Roger Armen was partially supported by grants 1P01HL114471 and R01 AR077666 and R01 HL153602 from the National Institutes of Health. There was no additional external funding received for this study.
Data Availability Statement
All relevant data are shown in figures or included in the text.
Acknowledgments
Roger Armen was partially supported by grants 1P01HL114471 and R01 AR077666 and R01 HL153602 from the National Institutes of Health. There was no additional external funding received for this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Li, G.; Rolf Hilgenfeld, R.; Whitley, R.; De Clercq, E. Therapeutic strategies for COVID-19: progress and lessons learned. Nat Rev Drug Discov. 2023, 22, 449–475. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, N.D.; Mohan, J.; Kushwaha, B.; Ghazi, T.; Nwabuife, J.C.; Koorbanally, N.; Chuturgoon, A.A. A comprehensive review on the global efforts on vaccines and repurposed drugs for combating COVID-19. Eur J Med Chem. 2023, 260, 115719. [Google Scholar] [CrossRef] [PubMed]
- Pitsillou, E.; Yu, Y.; Beh, R.C.; Liang, J.J.; Hung, A.; Karagiannis, T.C. Chronicling the 3-year evolution of the COVID-19 pandemic: analysis of disease management, characteristics of major variants, and impacts on pathogenicity. Clin Exp Med. 2023, 23, 3277–3298. [Google Scholar] [CrossRef] [PubMed]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Xiao, J.; Hooper, A.T.; Hamilton, J.D.; Musser, B.J.; Rofail, D.; Hussein, M.; J, I,; Atmodjo, D.Y.; Perry, C.; Pan, C.; Mahmood, A.; Hosain, R.; Davis, J.D.; Turner, K.C.; Baum, A.; Kyratsous, C.A.; Kim, Y.; Cook, A.; Kampman, W.; Roque-Guerrero, L.; Acloque, G.; Aazami, H.; Cannon, K.; Simón-Campos, J.A.; Bocchini, J.A.; Kowal, B.; DiCioccio, A.T.; Soo, Y.; Geba, G.P.; Stahl, N.; Lipsich, L.; Braunstein, N.; Herman, G.; Yancopoulos, G.D. REGEN-COV Antibody Combination and Outcomes in Outpatients with Covid-19. N Engl J Med. 2021, 385, e81.
- Takemoto, K. Retrospective Case-Control Study of REGEN-COV (casirivimab and imdevimab) Therapy for Patients with COVID-19 and Cancer Using the United States MarketScan® Database. Oncology. 2023, Sep 4.
- Li, D.; Xu, M.; Hooper, A.T.; Rofail, D.; Mohammadi, K.A.; Chen, Y.; Ali, S.; Norton, T.; Weinreich, D.M.; Musser, B.J.; Hamilton, J.D.; Geba, G.P. Casirivimab + imdevimab accelerates symptom resolution linked to improved COVID-19 outcomes across susceptible antibody and risk profiles. Sci Rep. 2023, 13, 12784. [Google Scholar] [CrossRef] [PubMed]
- Katsuo, C.; Kubota, K.; Tanaka, K.; Kurita, Y.; Nakajima, A. Combined REGN-COV2 Antibody Therapy Immediately Prevented a Patient with Refractory Type 1 Autoimmune Pancreatitis from Contracting SARS-CoV-2 during the Sixth Wave in Japan. Intern Med. 2023, 62, 1765–1770. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, S.K.; Tharwat, S.; Hassan, A.H. Comparing the efficacy of regen-cov, remdesivir, and favipiravir in reducing invasive mechanical ventilation need in hospitalized COVID-19 patients. World J Clin Cases 11(26):6105-6121. 2023. [Google Scholar] [CrossRef]
- Somersan-Karakaya, S.; Mylonakis, E.; Mou, J.; Oviedo-Orta, E.; O'Brien, M.P.; Mas Casullo, V.; Mahmood, A.; Hooper, A.T.; Hussein, M.; Ali, S.; Marty, F.M.; Forleo-Neto, E.; Bhore, R.; Hamilton, J.D.; Herman, G.A.; Hirshberg, B.; Weinreich, D.M. Effectiveness of Casirivimab and Imdevimab Antibody Combination in Immunocompromised Hospitalized Patients With Coronavirus Disease 2019: A Post Hoc Analysis in a Phase 1/2/3 Double-Blind Trial. Open Forum Infect Dis. 2023, 10, ofad211. [Google Scholar] [CrossRef]
- Bahakel, H.; Murphy, C.; Frenck Jr, R.W.; Grimley, M.S.; Marsh, R.A.; Paulsen, G.C.; Haslam, D.B.; Phillips, C.L.; Courter, J.; Spearman, P.; Schulert, G.; Danziger-Isakov, L. Single Site Experience of the use of Monoclonal Antibodies for the Treatment of COVID-19 in High-risk Pediatric and Young Adult Patients. Pediatr Infect Dis J. 2022, 41, 985–988. [Google Scholar] [CrossRef]
- Cruz-Teran, C.; Tiruthani, K.; McSweeney, M.; Ma, A.; Pickles, R.; Lai, S.K. Challenges and opportunities for antiviral monoclonal antibodies as COVID-19 therapy. Adv Drug Deliv Rev. 2021, 169, 100–117. [Google Scholar] [CrossRef]
- Johnson, A.M.; Barigye, R.; Saminathan, H. Perspectives on the use and risk of adverse events associated with cytokine-storm targeting antibodies and challenges associated with development of novel monoclonal antibodies for the treatment of COVID-19 clinical cases. Hum Vaccin Immunother. 2021, 17, 2824–2840. [Google Scholar] [CrossRef] [PubMed]
- Addetia, A.; Park, Y.J.; Starr, T.; Greaney, A.J.; Sprouse, K.R.; Bowen, J.E.; Tiles, S.W.; Van Voorhis, W.C.; Bloom, J.D.; Corti, D.; Walls, A.C.; Veesler, D. Structural changes in the SARS-CoV-2 spike E406W mutant escaping a clinical monoclonal antibody cocktail. Cell Rep. 2023, 42, 112621. [Google Scholar] [CrossRef] [PubMed]
- Ragonnet-Cronin, M.; Nutalai, R.; Huo, J.; Dijokaite-Guraliuc, A.; Das, R.; Tuekprakhon, A.; Supasa, P.; Liu, C.; Selvaraj, M.; Groves, N.; Hartman, H.; Ellaby, N.; Sutton, J.M.; Bahar, M.W.; Zhou, D.; Fry, E.; Ren, J.; Brown, C.; Klenerman, P.; Dunachie, S.J.; Mongkolsapaya, J.; Hopkins, S.; Chand, M.; Stuart, D.I.; Screaton, G.R.; Rokadiya, S. Generation of SARS-CoV-2 escape mutations by monoclonal antibody therapy. Nature Commun. 2023, 14, 3334. [Google Scholar] [CrossRef] [PubMed]
- Freidel, M.R.; Armen, R.S. Mapping major SARS-CoV-2 drug targets and assessment of druggability using computational fragment screening: Identification of an allosteric small-molecule binding site on the Nsp13 helicase. PLoS One. 2021, 16, e0246181. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.B.; Porrot, F.; Staropoli, I.; Lemoine, F.; Péré, H.; Veyer, D.; Puech, J.; Rodary, J.; Baele, G.; Dellicour, S.; Raymenants, J.; Gorissen, S.; Geenen, C.; Vanmechelen, B.; Wawina-Bokalanga, T.; Martí-Carreras, J.; Cuypers, L.; Sève, A.; Hocqueloux, L.; Prazuck, T.; Rey, F.A.; Simon-Loriere, E.; Bruel, T.; Mouquet, H.; André, E.; Schwartz, O. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2022, 602, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Krüger, N.; Schulz, S.; Cossmann, A.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Moldenhauer, A.S.; Winkler, M.S.; Lier, M.; Dopfer-Jablonka, A.; Jäck, H.M.; Behrens, G.M.N.; Pöhlmann, S. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell. 2022, 185, 447–456.e11. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Bruel, T.; Staropoli, I.; Guivel-Benhassine, F.; Porrot, F.; Maes, P.; Grzelak, L.; Prot, M.; Mougari, S.; Planchais, C.; Puech, J.; Saliba, M.; Sahraoui, R.; Fémy, F.; Morel, N.; Dufloo, J.; Sanjuán, R.; Mouquet, H.; André, E.; Hocqueloux, L.; Simon-Loriere, E.; Veyer, D.; Prazuck, T.; Péré, H.; Schwartz, O. Resistance of Omicron subvariants BA.2.75.2, BA.4.6, and BQ.1.1 to neutralizing antibodies. Nat Commun. 2023, 14, 824. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; McConnell, S.; Sullivan, D.J.; Casadevall, A. Analysis of SARS-CoV-2 mutations associated with resistance to therapeutic monoclonal antibodies that emerge after treatment. Drug Resist Updat. 2023, 71, 100991. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Temmam, S.; Vongphayloth, K.; Baquero, E.; Munier, S.; Bonomi, M.; Regnault, B.; Douangboubpha, B.; Karami, Y.; Chrétien, D.; Sanamxay, D.; Xayaphet, V.; Paphaphanh, P.; Lacoste, V.; Somlor, S.; Lakeomany, K.; Phommavanh, N.; Pérot, P.; Dehan, O.; Amara, F.; Donati, F.; Bigot, T.; Nilges, M.; Rey, F.A.; van der Werf, S.; Brey, P.T.; Eloit, M. ; Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature. 2022, 604, 330–336. [Google Scholar] [CrossRef]
- Zhou, H.; Ji, J.; Chen, X.; Bi, Y.; Li, J.; Wang, Q.; Hu, T.; Song, H.; Zhao, R.; Chen, Y.; Cui, M.; Zhang, Y.; Hughes, A.C.; Holmes, E.C.; Shi, W. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell. 2021, 184, 4380–4391.e14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA.1) and sub-variants (BA.1.1, BA.2, and BA.3) of SARS-CoV-2 spike infectivity and pathogenicity: A comparative sequence and structural-based computational assessment. J Med Virol. 2022, 94, 4780–4791. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Delipan, R.; Chakraborty, D.; Kanjo, K.; Singh, R.; Singh, N.; Siddiqui, S.; Tyagi, A.; Jha, V.; Thakur, K.G.; Pandey, R.; Varadarajan, R.; Ringe, R.P. Mutations in S2 subunit of SARS-CoV-2 Omicron spike strongly influence its conformation, fusogenicity, and neutralization sensitivity. J Virol. 2023, 97, e0092223. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lin, S.; Chen, Z.; Cao, Y.; He, B.; Lu, G. Targetable elements in SARS-CoV-2 S2 subunit for the design of pan-coronavirus fusion inhibitors and vaccines. Signal Transduct Target Ther. 2023, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yuan, H.; Li, X.; Wang, H. Spike protein mediated membrane fusion during SARS-CoV-2 infection. J Med Virol. 2023, 95, e28212. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zheng, X.; Zhou, B.; Li, J.; Chen, M.; Deng, R.; Wong, G.; Lavillette, D.; Meng, G. SARS-CoV-2 spike engagement of ACE2 primes S2' site cleavage and fusion initiation. Proc Natl Acad Sci U S A. 2022, 119, e2111199119. [Google Scholar] [CrossRef] [PubMed]
- Strobelt, R.; Adler, J.; Shaul, Y. The Transmembrane Protease Serine 2 (TMPRSS2) Non-Protease Domains Regulating Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Spike-Mediated Virus Entry. Viruses. 2023, 15, 2124. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.; Zhu, G.; Yang, M.; Cao, L.; Xing, X.; Yin, G.; Chan, C.; Qin, C.; Rao, Z.; Wang, X.; Sun, F.; Zhu, Y. Nanometer-resolution in situ structure of the SARS-CoV-2 postfusion spike protein. Proc Natl Acad Sci U S A. 2021, 118, e2112703118. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, Y.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh Jr, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Yang, K.; Wang, C.; White, K.I.; Pfuetzner, R.A.; Esquivies, L.; Brunger, A.T. Structural conservation among variants of the SARS-CoV-2 spike postfusion bundle. Proc Natl Acad Sci U S A. 2022, 119, e2119467119. [Google Scholar] [CrossRef]
- Chen, C.Z.; Xu, M.; Pradhan, M.; Gorshkov, K.; Petersen, J.D.; Straus, M.R.; Zhu, W.; Shinn, P.; Guo, H.; Shen, M.; Klumpp-Thomas, C.; Michael, S.G.; Zimmerberg, J.; Zheng, W.; Whittaker, G.R. Identifying SARS-CoV-2 Entry Inhibitors through Drug Repurposing Screens of SARS-S and MERS-S Pseudotyped Particles. ACS Pharmacol. Transl. Sci. 2020, 3, 1165. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Shi, K.; Wang, J.; Zhang, R.; Wang, Y. Targeting SARS-CoV-2 entry processes: The promising potential and future of host-targeted small-molecule inhibitors. Eur J Med Chem. 2024, 263, 115923. [Google Scholar] [CrossRef]
- Steiner, S.; Kratzel, A.; Barut, G.T.; Lang, R.M.; Moreira, E.A. Thomann, L.; Kelly, J.N.; Thiel, V. SARS-CoV-2 biology and host interactions. Nat Rev Microbiol. 2024, Jan 15.
- Liu, H.T.; Cheng, T.; Liu, B.Y.; Chi, J.; Shu, T.; Wang, T. Structures of the SARS-CoV-2 spike glycoprotein and applications for novel drug development. Front Pharmacol. 2022, 13, 955648. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; Strauch, E.M.; Stewart, L.; Diamond, M.S.; Veesler, D.; Baker, D. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science. 2020, 370, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Hajjo, R.; Bardaweel, S.K.; Zhong, H.A. An Updated Review on Betacoronavirus Viral Entry Inhibitors: Learning from Past Discoveries to Advance COVID-19 Drug Discovery. Curr Top Med Chem. 2021, 21, 571–596. [Google Scholar] [CrossRef] [PubMed]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Williamson, M.K.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; Capin, J.; Mulholland, A.J.; Spatz, J.; Fitzgerald, D.; Berger, I.; Schaffitzel, C. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science. 2020, 370, 725–730. [Google Scholar] [CrossRef]
- Rosa, A.; Pye, V.E.; Graham, C.; Muir, L.; Seow, J.; Ng, K.W.; Cook, N.J.; Rees-Spear, C.; Parker, E.; Silva Dos Santos, M.; Rosadas, C.; Susana, A.; Rhy, H.; Nans, A.; Masino, L.; Roustan, C.; Christodoulou, E.; Ulferts, R.; Wrobel, A.G.; Short, C.E.; Fertleman, M.; Sanders, R.W.; Heaney, J.; Spyer, M.; Kjær, S.; Riddell, A.; Malim, M.H.; Beale, R.; MacRae, J.I.; Taylor, G.P.; Nastouli, E.; van Gils, M.J.; Rosenthal, P.B.; Pizzato, M.; McClure, M.O.; Tedder, R.S.; Kassiotis, G.; McCoy, L.E.; Doores, K.J.; Cherepanov, P. SARS-CoV-2 can recruit a heme metabolite to evade antibody immunity. Sci Adv. 2021, 7, eabg7607. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Meng, F.; Xie, Y.; Wang, W.; Meng, Y.; Li, L.; Liu, T.; Qi, J.; Ni, X.; Zheng, S.; Huang, S.; Huang, N. In Silico Discovery of Small Molecule Modulators Targeting the Achilles' Heel of SARS-CoV-2 Spike Protein. ACS Cent Sci. 2023, 9, 252–265. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Y.; Chen, S.; Zhan, Q.; Wu, D.; Yang, C.; He, X.; Qiu, M.; Zhang, N.; Li, Z.; Guo, Y.; Wen, M.; Lu, L.; Ma, C.; Guo, J.; Xu, W.; Li, X.; Li, L.; Jiang, S.; Pan, X.; Liu, S.; Tan. S. Sertraline Is an Effective SARS-CoV-2 Entry Inhibitor Targeting the Spike Protein. J Virol. 2022, 96, e0124522. [Google Scholar] [CrossRef]
- Sinha, M.; Gupta, A.; Gupta, S.; Singh, P.; Pandit, S.; Chauhan, S.S.; Parthasarathi, R. Analogue discovery of safer alternatives to HCQ and CQ drugs for SAR-CoV-2 by computational design. Comput Biol Med. 2021, 130:104222.
- Puhl, A.C.; Fritch, E.J.; Lane, T.R.; Tse, L.V.; Yount, B.L.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Tavella, T.A.; Maranhão Costa, F.T.; Weston, S.; Logue, J.; Frieman, M.; Premkumar, L.; Pearce, K.H.; Hurst, B.L.; Andrade, C.H.; Levi, J.A.; Johnson, N.J.; Kisthardt, S.C.; Scholle, F.; Souza, T.M.L.; Moorman, M.J.; Baric, R.S.; Madrid, P.B.; Ekins, S. Repurposing the Ebola and Marburg Virus Inhibitors Tilorone, Quinacrine, and Pyronaridine: In Vitro Activity against SARS-CoV-2 and Potential Mechanisms. ACS Omega. 2021, 6, 7454–7468. [Google Scholar] [CrossRef]
- Gargantilla, M.; Francés, C.; Adhav, A.; Forcada-Nadal, A.; Martínez-Gualda, B.; Martí-Marí, O.; López-Redondo, M.L.; Melero, R.; Marco-Marín, C.; Gougeard, N.; Espinosa, C.; Rubio-Del-Campo, A.; Ruiz-Partida, R.; Del Pilar Hernández-Sierra, M.; Villamayor-Belinchón, L.; Bravo, J.; Llacer, J.L.; Marina, A.; Rubio, V.; San-Félix, A.; Geller, R.; Pérez-Pérez, M.J. C-2 Thiophenyl Tryptophan Trimers Inhibit Cellular Entry of SARS-CoV-2 through Interaction with the Viral Spike (S) Protein. J Med Chem. 2023, 66, 10432–10457. [Google Scholar] [CrossRef] [PubMed]
- Pattnaik, G.P.; Chakraborty, H. Entry Inhibitors: Efficient Means to Block Viral Infection. J Membr Biol. 2020, 253, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, S.; Bao, L.; Du, L.; Liu, S.; Qin, C.; Sun, F.; Shi, Z.; Zhu, Y.; Jiang, S.; Lu, L. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Xia, S.; Lan, Q.; Zhu, Y.; Wang, C.; Xu, W.; Li, Y.; Wang, L.; Jiao, F.; Zhou, J.; Hua, C.; Wang, Q.; Cai, X.; Wu, Y.; Gao, J.; Liu, H.; Sun, G.; Munch, J.; Kirchhoff, F.; Yuan, Z.; Xie, Y.; Sun, F.; Jiang, S.; Lu, L. Structural and functional basis for pan-CoV fusion inhibitors against SARS-CoV-2 and its variants with preclinical evaluation. Signal Transduct Target Ther. 2021, 6, 288. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Guo, L., Lin, S.; Chen, Z.; Yang, F., Yang, J., Wang, L.; Wen, A.; Duan, Y.; Zhang, X.; Dai, Y.; Yin, K.; Yuan, X.; Yu, C.; He, B.; Cao, Y.; Dong, H.; Li, J.; Zhao, Q.; Lu, G. An engineered 5-helix bundle derived from SARS-CoV-2 S2 pre-binds sarbecoviral spike at both serological- and endosomal-pH to inhibit virus entry. Emerg Microbes Infect. 2022, 11, 1920–1935.
- Xing, L.; Xu, X.; Xu, W.; Liu, Z.; Shen, X.; Zhou, J.; Xu, L.; Pu, J.; Yang, C.; Huang, Y.; Lu, L.; Jiang, S.; Liu, S. A Five-Helix-Based SARS-CoV-2 Fusion Inhibitor Targeting Heptad Repeat 2 Domain against SARS-CoV-2 and Its Variants of Concern. Viruses. 2022, 14, 597. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Chen, G.; Dang, B. Novel Engineered SARS-CoV-2 HR1 Trimer Exhibits Improved Potency and Broad-Spectrum Activity against SARS-CoV-2 and Its Variants. J Virol. 2022, 96, e0068122. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Li, Y.; Li, R.; Ma, Y.; Na, H. Structure and Function of the SARS-CoV-2 6-HB Fusion Core and Peptide-Based Fusion Inhibitors: A Review. Curr Med Chem. 2023. Nov 24. [Google Scholar] [CrossRef]
- Yang, C.; Pan, X.; Xu, X.; Cheng, C.; Huang, Y.; Li, L.; Jiang, S.; Xu, W.; Xiao, G.; Liu, S. Salvianolic acid C potently inhibits SARS-CoV-2 infection by blocking the formation of six-helix bundle core of spike protein. Signal Transduct Target Ther. 2020, 5, 220. [Google Scholar] [CrossRef] [PubMed]
- Jana, I.D.; Bhattacharya, P.; Mayilsamy, K.; Banerjee, S.; Bhattacharje, G.; Das, S.; Aditya, S.; Ghosh, A.; McGill, A.R.; Srikrishnan, S.; Das, A.K.; Basak, A.; Mohapatra, S.S.; Chandran, B.; Bhimsaria, D.; Mohapatra, S.; Roy, A.; Mondal, A. argeting an evolutionarily conserved "E-L-L" motif in spike protein to identify a small molecule fusion inhibitor against SARS-CoV-2. PNAS Nexus. 2022, 1, pgac198. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Pan, X.; Huang, Y.; Cheng, C.; Xu, X.; Wu, Y.; Xu, Y.; Shang, W.; Niu, X.; Wan, Y.; Li, Z.; Zhang, R.; Liu, S.; Xiao, G.; Xu, W. Drug Repurposing of Itraconazole and Estradiol Benzoate against COVID-19 by Blocking SARS-CoV-2 Spike Protein-Mediated Membrane Fusion. Adv Ther (Weinh). 2021. 4(5):2000224. [Google Scholar] [CrossRef]
- Jiao, F.; Andrianov, A.M.; Wang, L.; Furs, K.V.; Gonchar, A.V.; Wang, Q.; Xu, W.; Lu, L.; Xia, S.; Tuzikov, A.V.; Jiang, S. Repurposing Navitoclax to block SARS-CoV-2 fusion and entry by targeting heptapeptide repeat sequence 1 in S2 protein. J Med Virol. 2023, 95, e29145. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Zhang, X.; Huang, Y.; Cheng, W.; Qu, J.; Gui, S.; Li, L.; Li, S. Peptide-based inhibitors hold great promise as the broad-spectrum agents against coronavirus. Front Microbiol. 2023, 19, 1093646. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Lan, Q.; Zhu, Y.; Wang, C.; Xu, W.; Li, Y.; Wang, L.; Jiao, F.; Zhou, J.; Hua, C.; Wang, Q.; Cai, X.; Wu, Y.; Gao, J.; Liu, H.; Sun, G.; Münch, J.; Kirchhoff, F.; Yuan, Z.; Xie, Y.; Sun, F.; Jiang, S.; Lu, L. Structural and functional basis for pan-CoV fusion inhibitors against SARS-CoV-2 and its variants with preclinical evaluation. Signal Transduct Target Ther. 2021, 6, 288. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; Yang, X.; Shi, Z.; Deng, F.; Hu, Z.; Zhong, W.; Wang, M. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, C.; Chang, D.; Wang, Y.; Dong, X.; Jiao, T.; Zhao, Z.; Ren, L.; Dela Cruz, C.S.; Sharma, L.; Lei, X.; Wang, J. Identification of Potent and Safe Antiviral Therapeutic Candidates Against SARS-CoV-2. Front. Immunol. 2020, 11, 586572. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski, T.; Chen, L.; Eastman, RT.; Itkin, Z.; Shinn, P.; Chen, C.Z.; Guo, H.; Zheng, W.; Michael, S.; Simeonov, A.; Hall, M.D.; Zakharov, A.V.; Muratov, E.N. Synergistic and Antagonistic Drug Combinations against SARS-CoV-2. Mol Ther. 2021, 29, 873. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, A.; Padey, B.; Dubois, J.; Julien, T.; Traversier, A.; Dulière, V.; Brun, P.; Lina, B.; Rosa-Calatrava, M.; Terrier, O. In vitro evaluation of antiviral activity of single and combined repurposable drugs against SARS-CoV-2. Antiviral Res. 2020, 181, 104878. [Google Scholar] [CrossRef]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents. 2020, 56, 105998. [Google Scholar] [CrossRef]
- Freidel, M.R.; Armen, R.S. Modeling the Structure-Activity Relationship of Arbidol Derivatives and Other SARS-CoV-2 Fusion Inhibitors Targeting the S2 Segment of the Spike Protein. J Chem Inf Model. 2021, 61, 5906. [Google Scholar] [CrossRef]
- Shuster, A.; Pechalrieu, D.; Jackson, C.B.; Abegg, D.; Choe, H.; Adibekian, A. Clinical Antiviral Drug Arbidol Inhibits Infection by SARS-CoV-2 and Variants through Direct Binding to the Spike Protein. ACS Chem Biol. 2021, 16, 2845. [Google Scholar] [CrossRef]
- Li, H.; Cheng, C.; Li, S.; Wu, Y.; Liu, Z.; Liu, M.; Chen, J.; Zhong, Q.; Zhang, X.; Liu, S.; Song, G. Discovery and structural optimization of 3-O-β-chacotriosyl oleanane-type triterpenoids as potent entry inhibitors of SARS-CoV-2 virus infections. Eur J Med Chem. 2021, 215, 113242. [Google Scholar] [CrossRef] [PubMed]
- Curreli, F.; Ahmed, S.; Victor, S.M.B.; Drelich, A.; Panda, S.S.; Altieri, A.; Kurkin, A.K.; Tseng, C.T.K.; Hillyer, C.D.; Debnath., A.K. Discovery of Highly Potent Fusion Inhibitors with Potential Pan-Coronavirus Activity That Effectively Inhibit Major COVID-19 Variants of Concern (VOCs) in Pseudovirus-Based Assays. Viruses. 2021, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Curreli, F.; Chau, K.; Tran, T.T.; Nicolau, I.; Ahmed, S.; Das, P.; Hillyer, C.D.; Premenko-Lanier, M.; Debnath, A.K. Discovery of Highly Potent Small Molecule Pan-Coronavirus Fusion Inhibitors. Viruses. 2023, 15, 1001. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010, 53, 2719–40. [Google Scholar] [CrossRef] [PubMed]
- Tomašić, T.; Mašič, L.P. Rhodanine as a scaffold in drug discovery: a critical review of its biological activities and mechanisms of target modulation. Expert Opin Drug Discov. 2012, 7, 549–60. [Google Scholar] [CrossRef] [PubMed]
- Musarrat, F.; Chouljenko, V.; Dahal, A.; Nabi, R.; Chouljenko, T.; Jois, S.D.; Kousoulas, K.G. The anti-HIV drug nelfinavir mesylate (Viracept) is a potent inhibitor of cell fusion caused by the SARSCoV-2 spike (S) glycoprotein warranting further evaluation as an antiviral against COVID-19 infections. J Med Virol. 2020, 92, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.R.; Cheng, F. Repurposing of FDA-Approved Toremifene to Treat COVID-19 by Blocking the Spike Glycoprotein and NSP14 of SARS-CoV-2. J Proteome Res. 2020, 19, 4670–4677. [Google Scholar] [CrossRef]
- Wu, C.Y.; Jan, J.T.; Ma, S.H.; Kuo, C.J.; Juan, H.F.; Cheng, Y.S.E.; Hsu, H.H.; Huang, H.C.; Wu, D.; Brik, A.; Liang, F.S.; Liu, R.S.; Fang, J.M.; Chen, S.T.; Liang, P.H.; Wong, C.H. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc Natl Acad Sci U S A. 2004, 101, 10012–7. [Google Scholar] [CrossRef]
- Li, H.; Cheng, C.; Shi, S.; Wu, Y.; Gao, Y.; Liu, Z.; Liu, M.; Li, Z.; Huo, L.; Pan, X.; Liu, S.; Song, G. Identification, optimization, and biological evaluation of 3-O-β-chacotriosyl ursolic acid derivatives as novel SARS-CoV-2 entry inhibitors by targeting the prefusion state of spike protein. Eur J Med Chem. 2022, 238, 114426. [Google Scholar] [CrossRef]
- Liu, M.; Wang, J.; Wan, X.; Li, B.; Guan, M.; Ning, X.; Hu, X.; Li, S.; Liu, S.; Song, G. Discovery and structural optimization of 3-O-β-Chacotriosyl betulonic acid saponins as potent fusion inhibitors of Omicron virus infections. Bioorg Chem. 2023, 131, 106316. [Google Scholar] [CrossRef]
- Borisevich, S.S.; Zarubaev, V.V.; Shcherbakov, D.N.; Yarovaya, O.I.; Salakhutdinov, N.F. Molecular Modeling of Viral Type I Fusion Proteins: Inhibitors of Influenza Virus Hemagglutinin and the Spike Protein of Coronavirus. Viruses. 2023, 15, 902. [Google Scholar] [CrossRef]
- Zarubaev, V.V.; Pushkina, E.A.; Borisevich, S.S.; Galochkina, A.V.; Garshinina, A.V.; Shtro, A.A.; Egorova, A.A.; Sokolova, A.S.; Khursan, S.L.; Yarovaya, O.I.; Salakhutdinov, N.F. Selection of influenza virus resistant to the novel camphor-based antiviral camphecene results in loss of pathogenicity. Virology. 2018, 524, 69–77. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Baranova, D.V.; Galochkina, A.V.; Shtro, A.A.; Kireeva, M.V.; Borisevich, S.S.; Gatilov, Y.V.; Zarubaev, V.V.; Salakhutdinov, N.F. Quaternary ammonium salts based on (-)-borneol as effective inhibitors of influenza virus. Arch Virol. 2021, 166, 1965–1976. [Google Scholar] [CrossRef]
- Yarovaya, O.I.; Shcherbakov, D.N.; Borisevich, S.S.; Sokolova, A.S.; Gureev, M.A.; Khamitov, E.M.; Rudometova, N.B.; Zybkina, A.V.; Mordvinova, E.D.; Zaykovskaya, A.V.; Rogachev, A.D.; Pyankov, O.V.; Maksyutov, R.A.; Salakhutdinov, N.F. Borneol Ester Derivatives as Entry Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses. Viruses. 2022, 14, 1295. [Google Scholar] [CrossRef]
- Kalathiya, U.; Padariya, M.; Mayordomo, M.; Lisowska, M.; Nicholson, J.; Singh, A.; Baginski, M.; Fahraeus, R.; Carragher, N.; Ball, K.; Haas, J.; Daniels, A.; Hupp, T.R.; Alfaro, J.A. Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site. J Clin Med. 2020, 9, 1473. [Google Scholar] [CrossRef]
- Zannella, C.; Chianese, A.; Greco, G.; Santella, B.; Squillaci, G.; Monti, A.; Doti, N.; Sanna, G.; Manzin, A.; Morana, A.; De Filippis, A.; D'Angelo, G.; Palmieri, F.; Franci, G.; Galdiero., M. Design of Three Residues Peptides against SARS-CoV-2 Infection. Viruses. 2022, 14, 2103. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure of the SARS-CoV-2 Spike protein showing: (A) the prefusion conformation (6xr8.pdb) full-length trimeric protein (B) the prefusion conformation of the S2 subunit and (C) the postfusion conformation (6xra.pdb) of the S2 subunit showing the HR1/HR2 6-helix bundle. On Spike, residues are colored gray S1 NTD (res:14-304), black RBD (res:331-528), cyan S2 NTD (res:685-816), green HR1 (res:910-985), seafoam green CH (res:985-1035), dark blue CD (res:1035-1068), red (res:112-134), SH pink (res:1140-1161) and HR2 (res:1163-1211).
Figure 1.
Structure of the SARS-CoV-2 Spike protein showing: (A) the prefusion conformation (6xr8.pdb) full-length trimeric protein (B) the prefusion conformation of the S2 subunit and (C) the postfusion conformation (6xra.pdb) of the S2 subunit showing the HR1/HR2 6-helix bundle. On Spike, residues are colored gray S1 NTD (res:14-304), black RBD (res:331-528), cyan S2 NTD (res:685-816), green HR1 (res:910-985), seafoam green CH (res:985-1035), dark blue CD (res:1035-1068), red (res:112-134), SH pink (res:1140-1161) and HR2 (res:1163-1211).
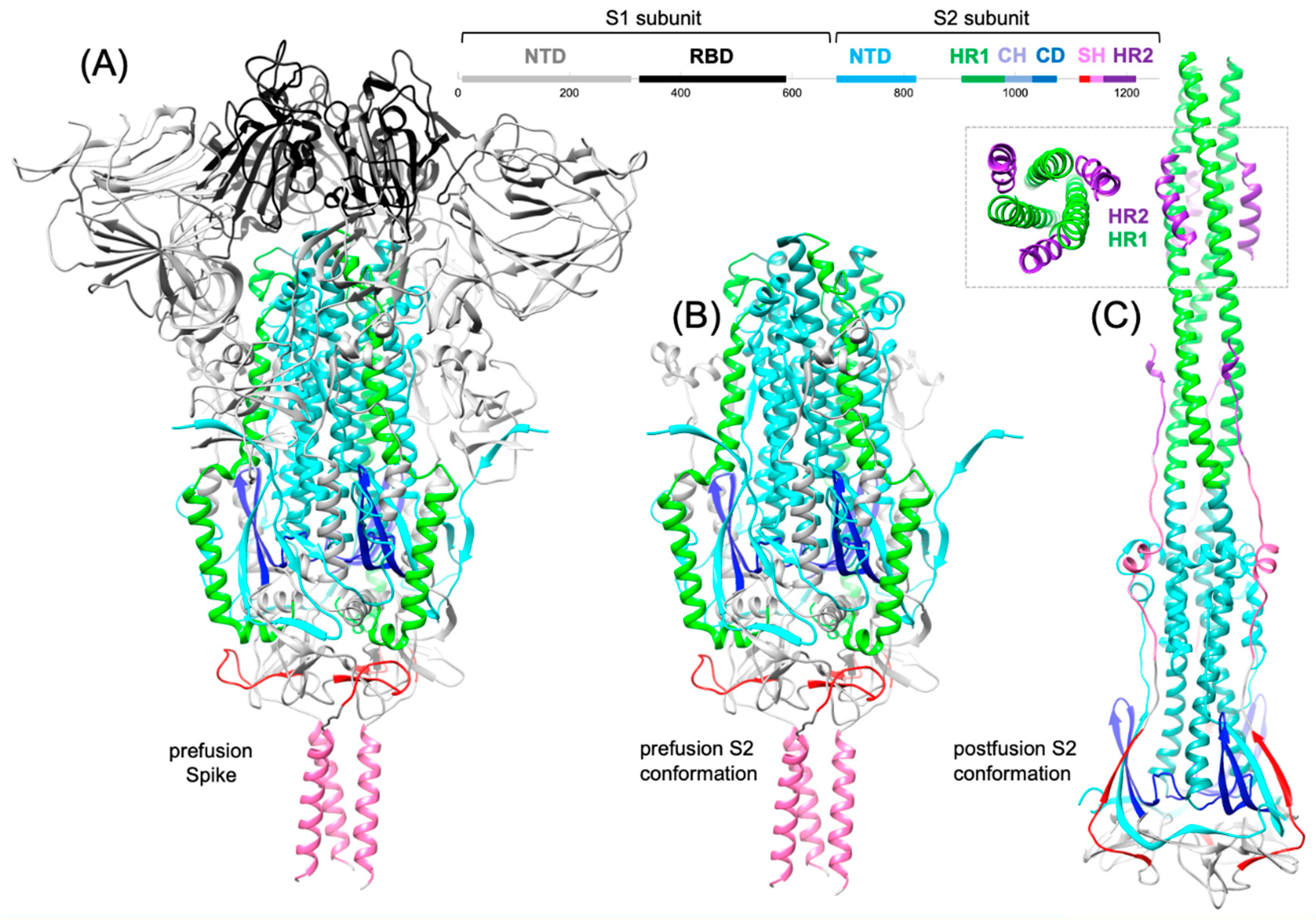
Figure 2.
Small molecule binding sites on the SARS-CoV-2 Spike protein. (A) Seven proposed or experimentally determined small molecule binding sites are shown as magenta (molecular surfaces) on the ribbon structure of the full-length trimeric protein (6xr8.pdb). (B) On Spike, small molecule binding sites are shown in magenta and residue positions implicated in a major SARS-CoV-2 VOCs are shown in red. Most mutations in the Spike protein in VOCs to date have been found on the S1 segment.
Figure 2.
Small molecule binding sites on the SARS-CoV-2 Spike protein. (A) Seven proposed or experimentally determined small molecule binding sites are shown as magenta (molecular surfaces) on the ribbon structure of the full-length trimeric protein (6xr8.pdb). (B) On Spike, small molecule binding sites are shown in magenta and residue positions implicated in a major SARS-CoV-2 VOCs are shown in red. Most mutations in the Spike protein in VOCs to date have been found on the S1 segment.
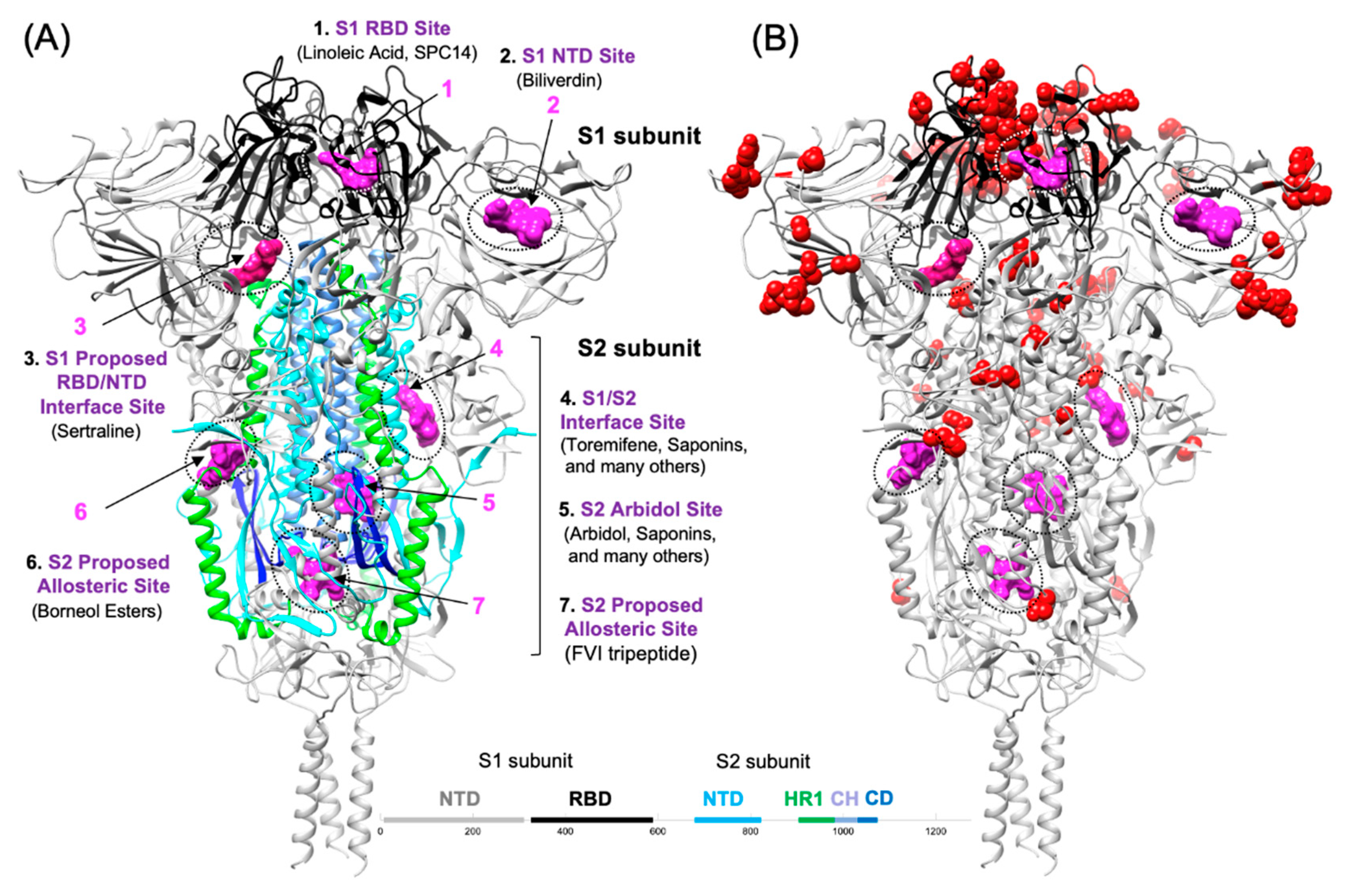
Figure 3.
Small molecule binding site 4 on the S1/S2 interface. (A) Molecular surface of modeled OA saponin 12a shown in magenta binding to Spike (6xr8.pdb) colored rainbow from blue (N-term) to red (C-term). (B) Model of OA saponin 12a binding to site 4.
Figure 3.
Small molecule binding site 4 on the S1/S2 interface. (A) Molecular surface of modeled OA saponin 12a shown in magenta binding to Spike (6xr8.pdb) colored rainbow from blue (N-term) to red (C-term). (B) Model of OA saponin 12a binding to site 4.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



