
Submitted:
02 March 2024
Posted:
04 March 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Clofazimine and Arbidol have both been reported to be effective in vitro SARS-CoV-2 fusion inhibitors. Both are promising drugs repurposed for treatment of COVID-19 and have been used in several previous and ongoing clinical trials. Small-molecule binding to expressed constructs of the trimeric S2 segment of Spike and the full-length SARS-CoV-2 spike protein were measured using a Surface Plasmon Resonance (SPR) binding assay. We demonstrate that Clofazimine, Toremifene, Arbidol and other Arbidol derivatives bind to the S2 segment of the Spike protein. Clofazimine provided the most reliable and highest quality SPR binding data to S2 over the conditions explored. A molecular docking approach was used to identify the most favorable binding sites on the S2 segment in the prefusion conformation, highlighting two possible small-molecule binding sites for fusion inhibitors. Results from molecular docking and modeling the structure-activity-relationship (SAR) of a newly reported series of Clofazimine derivatives supports the proposed Clofazimine binding site on the S2 segment. When the proposed Clofazimine binding site is superimposed with other experimentally determined coronavirus structures in structure-sequence alignments, the changes in sequence and structure may rationalize the broad-spectrum antiviral activity of Clofazimine in closely related coronavirus such as (SARS-CoV, MERS, hCoV-229E, hCoV-OC43).
Keywords:
Clofazimine
; Arbidol
; Toremifene
; Fusion Inhibitor
; Spike-dependent
; SARS-CoV-2
; S2 segment
; S2 subunit
; Nsp13 helicase
; Surface Plasmon Resonance
; Molecular Docking
; CHARMM
1. Introduction
Early in the COVID-19 pandemic, both Arbidol 1 [1,2], and Clofazimine 2 [2,3,4,5,6,7,8,9,10], were identified to be clinical drugs that were effective in vitro inhibitors of SARS-CoV-2 from drug repurposing screens. Clofazimine 2, was found in at least eight independent drug repurposing efforts [2,3,4,5,6,7,8,9,10] and was commonly identified among the most promising compounds in those studies based on screening and antiviral assay data [2,3,4,5,6,7,8,9,10]. Drug repurposing efforts are important to identify possible drugs available for clinical use when patients are not able to receive “standard of care” drug treatment [11,12]. In addition to that, ongoing studies of effective direct-acting antivirals [13,14] are important to understand the direct mechanism of action and the molecular basis for broad-spectrum activity. Both Arbidol 1, and Clofazimine 2 shown in Figure 1, have been reported to act as SARS-CoV-2 viral fusion inhibitors [1,5,6,9] and have demonstrated synergistic antiviral activity with the Nsp12 inhibitor Remdesivir [4,5,15]. Small-molecule fusion inhibitors [14] with this profile are attractive as they may be expected to exhibit antiviral activity with either Nsp12 replication inhibitors such as Remdesivir (Veklury) [16] or Nsp5 Main Protease (MPro) inhibitors such as nirmatrelvir (Paxlovid) [17,18].
Our previous studies focused on the mode of action of Arbidol 1 as a fusion inhibitor [19,20]. These studies showed that it was only a partial inhibitor of SARS-CoV-2 in cytopathic effect (CPE) assays [20], in comparison to full inhibitors such as Remdesivir. An early drug repurposing screen using psudotyped virus particles by Chen et al., reported similar results where a range of viral entry inhibitors including NKH477 and trimipramine were found to only be partial inhibitors in similar SARS-CoV-2 infection CPE assays in Vero E6 cells [21]. In comparison, several studies have shown that Clofazimine 2 is a full inhibitor of SARS-CoV-2 in a range of infection assay cell types (Vero E6, Huh7, and Caco-2 cells) [4,5,6] including physiologically relevant cell lines (cardiomyocytes, Calu-3 and human primary airway epithelial cells) [5,8]. In addition to this, 2 has also shown impressive preclinical antiviral activity in a golden Syrian hamster animal model [5]. As a clinical repurposing drug, Clofazimine 2 is a drug with clinical drawbacks and is associated with numerous adverse side effects for systemic use (abdominal pain, gastrointestinal track disturbances) [22]. Some specific adverse effects (skin discoloration, tissue accumulation) are directly due to the physiochemical properties of the drug due to its high lipophilicity (cLogP = 7.1) which results in accumulation in various lipophilic tissues. Thus, recent synthetic medicinal chemistry efforts have been aimed at generating improved derivatives of Clofazimine 2 with lower (cLogP) values and greater solubility to reduce these side effects [23]. However, despite numerous adverse effects, there is much world-wide clinical experience with Clofazimine to treat various forms of leprosy and drug-resistant tuberculosis and the systemic toxicity can be significantly reduced by delivering drug through inhalation [24,25]. Clofazimine has also been considered an experimental drug for other infections and is on the World Health Organization’s List of Essential Medicines [26]. Clofazimine 2 remains an important clinical drug that may be considered for treatment of COVID-19 [27]. While the fusion inhibitor activity of Clofazimine 2 has been shown to be Spike-dependent [5,23], to our knowledge, the precise mechanism of action by which it acts as a fusion inhibitor has yet to be determined. In this paper we demonstrate that Clofazimine 2 binds to the S2 segment of spike using a Surface Plasmon Resonance (SPR) binding assay [28,29], which helps to delineate how it acts as a potent fusion inhibitor.
An overview of the SPR direct binding assay setup (Figure 2A) shows how a full-length Spike protein construct may be immobilized on a SPR biosensor flow cell surface (FC2) and tested against a reference cell (FC1). A purified construct of the Spike S2 segment is immobilized on (FC4) and tested against a reference cell (FC3). This assay design allows simultaneous determination of small-molecule binding to both the full-length Spike protein and the S2 segment. Shown in Figure 2A are four sample surfaces of the biosensor chip corresponding to flow cells (FC1 to FC4) where protein samples may be immobilized for measuring small-molecule binding. During a binding experiment, the reference subtracted signal of FC2-1 and FC4-3 provides simultaneous measurement of binding to the full-length Spike and the S2 segment.
We demonstrate that Arbidol 1 and Clofazimine 2 bind to the S2 segment of Spike. As molecular docking and structure-activity-relationship (SAR) modeling work from our laboratory successfully predicted the binding site of Arbidol 1 on the S2 segment of Spike [19,20], as had been done previously by Vankadari with docking alone [30], another publication from Shuster et al [31], experimentally confirmed the binding site of 1 on the S2 segment of Spike as shown in Figure 3A and 3C. Elegant work from Shuster et al [31], independently identified the site using a chemical biology approach and then corroborated the exact predicted binding site [20] by mutational studies [31]. Thus, while the Arbidol 1 binding site on S2 has been determined experimentally [31], the binding site of Clofazimine 2 on the S2 segment remains unknown to the best of our knowledge. Along the lines of our previous efforts to predict small-molecule binding sites on S2 [19,20], we identify the most thermodynamically favorable binding site for Clofazimine 2 using reliable CHARMM-based molecular docking methods. Using a recently published series of Clofazimine 2 derivatives [23], we model the structure-activity-relationship (SAR) and demonstrate that the data for 18 derivative compounds are better modeled at the proposed Site 2 rather than Site 1, which is the Arbidol binding site (Figure 3). We demonstrate experimentally that Arbidol 1 and Clofazimine 2 bind to the S2 segment. Finally, we use molecular modeling to identify the most favorable binding site for Clofazimine 2 and illustrate how SAR data is best modeled at this proposed site.
2. Materials and Methods
Samples of Arbidol 1 and derivatives were purchased from ChemDiv, including 1 (1635-0087), 1b (8015-5742) and 1c (H027-0218C) and 1d (H027-0205C). Other compounds were purchased from Selleck Chemicals, including Clofazimine 2 (S4107), Toremifene 3 (S1776), and Ecliptasaponin A 4 (S9403). While Toremifene Citrate (S1776) came as a stock solution in 10 mM DMSO, all other purchased compounds were prepared to a standard 10 mM DMSO stock solution from an exact known weight (mg) of each compound. Compound DMSO stock solutions were prepared from the same source of DMSO as used in SPR experiments to minimize observed bulk responses in compound injections.
All SPR experiments were performed at Reaction Biology Corporation (Malvern, PA, USA) using a Biacore 8K+ (Cytiva) instrument with high sensitivity [32]. For immobilizations, a “series S” and “SA” sensor chips (Cytiva) were used to capture Avi-tag biotinylated protein samples of the SARS-CoV-2 Spike protein. Two biotinylated recombinant samples of the Spike protein were purchased from Acro Biosystems: a full-length trimeric SARS-CoV-2 Spike construct with the D614G mutation (Biotinylated SARS-CoV-2 S protein, catalog number SPN-C82E3) and a trimeric construct of the SARS-CoV-2 S2 segment (Biotinylated SARS-CoV-2 S2 protein, catalog number S2N-C52E8). As the D614G mutation is contained within S1, rather than in the S2 segment, the S2 construct would correspond exactly to the maturated S2 segment from either WT or the D614G full-length Spike protein. Numerous attempts were made to prepare the biosensor surface and optimize the assay conditions to improve the quality of the binding data (See Results). The protein samples were immobilized on the SA chips using a 5 (mL/min) flow rate with a running buffer composed of PBS with 0.05% Tween 20, resulting in ranges of 3000 – 5000 RU. Following immobilization, both the sample surface and the reference channel surface was blocked with biotin to attempt to minimize non-specific binding.
In the first round of assay development and initial data collection on the surfaces described above, small-molecule analytes were analyzed using running buffer composed of PBS with 0.05% Tween 20 and either a 1% DMSO or 2% DMSO solutions. Titrations of each analyte were performed using multi-cycle kinetics mode, with 200 mM as the highest concentration for a 2-fold serial dilution of 10 concentrations. In this first round of data collection, serial dilutions were performed on a plate, where the 200 mM concentration was prepared by mixing 10 mM DMSO stock solution with 0% DMSO running buffer to achieve either a 1% or 2% DMSO solution of analyte to match the running buffer.
In a final round of compound characterization using optimized conditions, a new SA chip was prepared aiming to facilitate collection of duplicate sensorgrams from two surfaces. Given the high lipophilicity of some of the compounds, rather than performing the dilution on the plate, the serial dilution was performed in DMSO first to avoid solubility issues. 10 concentrations were prepared in DMSO at 50x the concentration used in the assay and then transferred to the plate and mixed with the DMSO-free running buffer to achieve a 2% DMSO solution of analyte to match the 2% DMSO running buffer. Independent duplicates were compared for each compound performing two separate concentration series with a starting concentration of 100 mM and 50 mM respectively. Titrations of each analyte were performed using multi-cycle kinetics mode. All SPR data was appropriately solvent corrected [33], reference subtracted and analyzed while fitted to a steady state affinity model using Biacore Insight Evaluation Software.
All molecular docking and free energy calculations were performed using CHARMM [34] and previously described CHARMM-based computational methods established by our laboratory [35,36]. Molecular docking utilized the LPDB CHARMm force field for modeling small-molecule potential functions and resulting protein-ligand interactions [37,38]. As previously described, a two-step scoring approach is utilized to rank the final TOP5 poses from any docking attempt. For the final TOP5 docking poses, a final energy minimization of the protein-ligand complex is performed using the Generalized Born using Molecular Volume (GBMV) implicit solvent method [39,40]. Starting from the minimized complex, minimizations of the bound and free state are performed where potential energy components (VDW), (ELEC), and solvation (SOLV) are calculated in order to approximate the free energy of binding (ΔGbind) using a linear interaction energy scoring approach with previously determined empirical generalized parameters [35]. Results using the predicted (ΔGbind) values for the TOP5 poses of each individual docking “trials” are pooled and sorted by (ΔGbind), where the top-ranked members of a geometric cluster (RMSD < 2.0 Å) are identified. Statistics for (ΔGbind) are calculated from the average and standard deviation from the three top-ranked members of a geometric cluster (RMSD < 2.0 Å) or a triplicate representing the geometric cluster. For all work performed in this study, independent docking “trials” were initiated from either 20 generated conformations of a given small-molecule ligand, such that the initial geometry is entirely independent of any CHARMM-based procedure. MarvinSketch version 15.8.31 is a publicly available 3D conformation generator that was used to generate non-identical low energy conformations [41].
Our laboratory had previously used a pharmacophore procedure to identify the most favorable TOP50 binding sites on the SARS-CoV-2 Spike protein S2 segment [19]. This structural analysis had been performed using the 3.2 Å CryoEM structure (6vyb.pdb) of the full-length Spike protein where the ectodomain was in the “closed” state (6vxx.pdb) [42]. From this model (6vxx.pdb), molecular docking was performed using a hierarchical approach, such that 10 conformations of Clofazimine 2, were initially docked to all 50 sites on the Spike S2 segment. Then after identification of the TOP5 most favorable sites from this first step, more extensive sampling was used to refine the ranking of the TOP5 sites, 20 conformations of Clofazimine 2 were docked. From this model (6vxx.pdb), the consensus binding mode for Cofazimine, 2 binding to the SARS-CoV-2 Spike S2 segment (Figure 3B) was used for docking 18 derivatives of Clofazimine 2 [23]. These derivatives were modeled in five binding sites, Site 1 and Site 2 on the S2 segment as shown in (Figure 3) as well as for a lowest energy binding sites [19] in the Nsp5 Main Protease (6w63.pdb) [43], Nsp13 Helicase (6jyt.pdb) [44], and the Nsp16 2’-O methyltransferase (6wkq.pdb) [45]. The results for docking the series into the Nsp5 Main Protease and Nsp16 are expected to represent negative controls, where Clofazimine series SAR data would not be expected to be well-modeled binding to these two sites. As our previous work has highlighted, the Nsp5 Main Protease and Nsp16 binding sites in particular [19] are thermodynamically favorable for a variety of small-molecule fragment ligands to bind. This makes them more challenging negative control “decoy” binding sites, particularly compared to most of the possible binding sites on the S2 segment which are less thermodynamically favorable “decoy” binding sites. The physical basis for this is that the specific molecular shape and the hydrophobicity of the Nsp5 and Nsp16 binding sites are favorable for binding hydrophobic small-molecules and result in more thermodynamically favorable and more negative (ΔGbind) values when performing virtual screening of a library of compounds. All molecular graphics images of protein structures and molecular interactions are generated with UCSF Chimera [46].
3. Results
3.1. Biosensor Chip Preparation
Numerous attempts were made to prepare the biosensor surface and optimize the assay conditions to improve the quality of the binding data. The protein samples were immobilized on SA chips using a 5 (mL/min) flow rate with a running buffer composed of PBS with 0.05% Tween 20. For the full-length Spike protein on two separate channels, an injection of 20 (mg/mL) protein and a 200 sec contact time resulted in an immobilization level of 2831.6 RU, where an injection of 40 (mg/mL) protein and a 600 sec contact time resulted in an immobilization level of 5102.0 RU. For the S2 protein on two separate channels, an injection of 10 (mg/mL) protein and a 200 sec contact time resulted in an immobilization level of 2831.6 RU, where an injection of 20 (mg/mL) protein and a 600 sec contact time resulted in an immobilization level of 4721.0 RU. Even though both the sample surface and the reference surface were blocked with biotin to attempt to minimize non-specific binding, some non-specific binding was observed. While this non-specific analyte binding to the reference surface precluded characterization of some compounds, this effect was compound specific and could be rationalized to specific compounds that are more hydrophilic and may contain more hydrogen bonding opportunities with the reference biotinylated surface. In comparison, one of the most lipophilic compounds, Clofazimine (cLogP = 7.1) exhibited the least non-specific binding to the reference surface and reproducibly resulted in the highest quality SPR binding data over all conditions explored.
In a final round of compound characterization using optimized conditions, a new SA chip was prepared aiming to facilitate collection of duplicate sensorgrams from two duplicate surfaces. The protein samples were immobilized on the SA chips using a 5 (mL/min) flow rate with a running buffer composed of PBS with 0.05% Tween 20. For the full-length Spike protein, an injection of 40 (mg/mL) protein and a 600 sec contact time resulted in duplicate immobilization levels of 4692.0 and 4754.6 RU. For the S2 segment, an injection of 20 (mg/mL) protein and a 600 sec contact time resulted in duplicate immobilization levels of 3640.3 and 3758.0 RU.
3.2. Surface Plasmon Resonance (SPR) Binding Data
In preliminary rounds of assay development and initial data collection on the surfaces described above, small-molecule analytes were analyzed using running buffer composed of PBS with 0.05% Tween 20 and either a 1% or 2% DMSO solution. Titrations of each analyte were performed with 200 μM as the highest concentration for a 2-fold serial dilution of 10 concentrations. The best representative data for compounds 1, 1b, 1d, and 2 that fits to the steady state affinity model are shown in Table 1. Some of the best representative data showing simultaneous SPR sensorgrams binding to Spike and S2 with good quality fits are shown in Figure 4. For SPR data presented in Figure 4, all compounds demonstrate definitive sensorgram evidence for binding from concentration dependent change in response units (RU) and good quality fits to the steady state affinity models. Figure 4 demonstrates compounds of four different structural classes that bind to the S2 segment as well as the full-length Spike protein. While some of the lower affinity compounds 1c, 1d, and 4 exhibited greater observed differences in affinities between S2 and Spike, the higher affinity reference compounds 1, 2, and 3, all had reproducibly similar SPR sensorgrams and fits with lower differences in affinity between S2 and Spike, so for 1 (S2 = 5.9 μM) (Spike = 7.4 μM), for 2 (S2 = 6.3 μM) (Spike = 5.4 μM), and for 3 (S2 = 4.1 μM) (Spike = 4.1 μM). This binding data strongly suggests that the binding site of the small-molecules is found on the S2 segment of the full-length protein.
The observed affinities in this round (Kd = 5.9 μM to 7.4 μM) for Arbidol 1, were well within the range of reported antiviral activities for Arbidol 1 (EC50s = 4.1 μM to 10.0 μM) reported in the literature1,2 and from our previous work (EC50 = 5.6 μM) [20]. For 1 and derivatives, 1c and 1d, it was generally observed that the affinity for binding to the S2 segment was slightly more favorable than to the full-length Spike protein, as observed in each individual compound. This is shown in Figure 2C where 1 binds with a slightly higher affinity to the S2 segment (Kd = 5.9 μM) compared to full length Spike (Kd = 7.4 μM) shown in Figure 2B. The same trend (Kd S2 < Kd Spike) can be shown for derivatives 1c and 1d in Figure 4A and Figure 4B respectively. While the observed trend of (Kd S2 < Kd Spike) was not necessarily expected, it may be possible to rationalize the observation if the cleaved S2 segment small-molecule binding sites may be more dynamic or amenable to complementary induced-fit binding compared to the much larger full-length trimer.
An important observation was that comparing the SPR data for either the full-length Spike, or the S2 segment, the binding data exhibited the expected structure-activity-relationship (SAR) for the derivative series such that for Spike (1 < 1c) and (1 < 1d) [20]. Thus, 1 had a higher affinity than either derivative 1c or 1d as expected from both virtual screening data and experimental CPE data for 1 (EC50 = 5.6 μM) and 1b (EC50 = 29.5 μM) [20]. In addition, for only the data on the S2 segment the same was observed, 1 had a higher affinity than either derivative 1c or 1d as expected, providing additional confidence in the interpretation of the SPR data observed for different compounds. The observed affinity (Kd = 4.1 μM) for Toremifene 3, was very close to the reported antiviral activity for Toremifene 3, in live SARS-CoV-2 infections (EC50 = 3.58 μM) [47] and (EC50 = 1.92 μM) for SARS-CoV-2 pseudovirus entry assays [48]. Observing the expected SAR for (1, 1c and 1d) and being in reasonable agreement with reported antiviral activity for 1 and 3 helps to establish that we are able to interpret the SPR results from more than one perspective.
In a final round of compound characterization, a new SA chip was prepared aiming to facilitate collection of duplicate sensorgrams using optimized conditions and 2% DMSO running buffer. Independent duplicates were compared for each compound performing two separate concentration series with a starting concentration of 100 μM and 50 μM respectively. This dataset resulted in five compounds with quality duplicates binding to the S2 segment. The best SPR duplicates for binding to the S2 segment are shown in Figure 5. The best representative data for compounds 1, 1c, 2, 3, and 4 and fits to the steady state affinity model are summarized in Table 2, statistics are presented for duplicates. While the duplicate affinity of 1 binding to S2 as reported in Table 2 is slightly lower (Kd = 2.1 ± 0.2 μM) in this dataset than (Kd = 5.9 μM) as reported in Table 1, it remains much lower than the derivative 1c (Kd = 11.4 ± 1.3 μM). Thus, the binding data in the duplicates also follow the expected SAR for the derivative series such that for binding to S2, 1 had a higher affinity than derivative 1c as expected [20]. Again, in this dataset of duplicate measurements, the observed affinity for Spike (Kd = 4.1 mM) for Toremifene 3, were very close to several reported antiviral activities (EC50s = 1.9 μM to 3.6 μM) [47,48].
As mentioned previously, Clofazimine 2, was found to provide the most reliable and highest quality SPR binding data to S2 over the conditions explored. In a recently published series of Clofazimine derivatives, the affinity of 2 to full-length Spike by SPR was found to be (Spike = 3.82 μM) [23], very close to the best representative data for 2 (S2 = 3.9 μM) (Spike = 2.9 μM) from the first round as presented in Table 1. The best duplicate data for 2 (S2 = 6.5 ± 0.3 μM) (Spike = 4.6 ± 1.2 μM) is presented in Table 2. While the affinity is a bit lower in the duplicate dataset, both data sets show that Clofazimine 2 definitively binds to the S2 segment under these conditions.
While Clofazimine 2, has been reported to have a range of potent antiviral activity in CPE assays ranging from (EC50 ~ 0.08 μM to 0.56 μM) [4,5,6,7] with a consensus of (EC50 = 0.31 μM) [4,5], the antiviral activity of 2 has been reported to result as a combination of Spike-dependent fusion inhibitor activity as well as Nsp13 helicase unwinding activity [5]. The dose-response curves of 2 in the same study [5], suggest that the micromolar viral fusion activity (EC50 ~ 2.5 μM to 5.0 μM) is slightly more potent than the Nsp16 helicase unwinding activity (EC50 ~ 7.5 μM to 10 μM) [5].
Interestingly, in other independent assays that report SARS-CoV-2 Spike-mediated fusion activity [49], the activity values are also lower potency for Clofazimine 2 (EC50 = 2.56 μM) [49], which is much closer to the current measured affinity for full length Spike (Kd = 2.9 μM to 4.6 μM) by SPR and another reported value (Kd = 3.82 μM) for full length Spike by SPR [23]. In summary, while the observed affinity by SPR binding assay for Clofazimine 2 is in the micromolar range (Kd = 2.9 μM to 4.6 μM) rather than the more potent observed antiviral activity (EC50 = 0.31 μM) [4,5], this is in agreement with observations from viral fusion assays [5,49], SPR binding [23], and the concept that the resulting antiviral activity is a result of dual-targeted drug action on at least Spike and the Nsp13 helicase [5]. While Clofazimine has been reported to be a viral fusion inhibitor, to our knowledge it has yet to be reported that Clofazimine binds to the S2 segment of Spike. As Clofazimine is an important clinical candidate, narrowing down its mode of action as a direct-acting fusion inhibitor is important. The SPR data show that Clofazimine 2 binds to a well-formed binding site on the S2 segment trimer.
3.3. Predicting the Clofazimine Binding Site on S2 with Molecular Docking
While the Arbidol 1 binding site has been experimentally determined by Shuster et al [31], there still has yet to be any published experimental structure of a small-molecule fusion inhibitor bound to the Spike S2 segment solved by either X-ray crystallography or CryoEM techniques. From docking 2 into all the TOP50 binding sites predicted on the S2 segment [19], the top two favorable sites were identified and shown in Figure 6A. Site 2 shown in (Figure 3B), is the only feasible binding site for 2 according to our modeling data (Figure 6B), where Site 1, is predicted to be much less thermodynamically favorable for binding of 2. From analysis of molecular docking and calculated (ΔGbind) values at all 50 sites[19,20], Site 2 is easily identified as being the most favorable site, also from the identification of two other structurally related 3-fold symmetric sites. In terms of predicted (ΔGbind) values from the statistics of the top-ranked cluster (as a triplicate), Site 2 (ΔGbind = -9.4 ± 0.3 kcal/mol) is much more thermodynamically favorable than Site 1 (ΔGbind = -7.6 ± 0.2 kcal/mol), the Arbidol 1 binding site. The protein-ligand interactions of Clofazimine 2, modeled at Site 2, are complementary and favorable as described in more detail in the next section. In summary, as shown from docking and predicted (ΔGbind) values (Figure 6B), Clofazimine 2 and 3 are predicted to bind more favorably at Site 2 compared to Site 1. Ecliptasaponin A 4 is predicted to bind favorably at Site 1, the Arbidol binding site (Figure 6A). We have previously demonstrated how a series of oleanolic acid (OA) Saponin derivatives are best modeled at Site 1 rather than Site 2 on the S2 segment [20], and Ecliptasaponin A 4 is closely related in structure to these OA Saponin derivatives such as 12a. Both 4 and 12a are predicted to bind more favorably at Site 1, the Arbidol 1 binding site as shown in Figure 6.
3.4. Modeling a Series of Clofazimine Derivatives Binding to the S2 Segment
Beyond the fact that Clofazimine 2 is predicted to bind much more favorably to Site 2 than Site 1 according to calculated (ΔGbind) values, another independent line of evidence from modeling also strongly corroborates Site 2. Recently, a new series of chemical derivatives of Clofazimine 2, were published with antiviral activity data against SARS-CoV-2.23 Using the same methods, 18 derivatives were modeled binding to Site 1 and Site 2. For each derivative in the series, a TOP-ranked cluster was determined independently from docking numerous initial starting conformations, rather than simply modeling all derivatives exactly as the binding mode of the reference compound.
In modeling the series of 18 derivatives at Site 2, the “untrained” predicted ΔGbind values exhibited some correlation with the experimental EC50 values. The Pearson’s R2 correlation coefficient was R2 = 0.264 for all 18 compounds (Figure 7A) modeled at Site 2. In comparison, 18 compounds (Figure 7A) modeled at Site 1 exhibited a positive correlation but with a very low calculated correlation coefficient R2 = 0.029. Thus, from modeling all 18 compounds the “untrained” predicted ΔGbind values had much greater correlation at Site 2 (R2 = 0.264) compared to Site 1 (R2 = 0.029). Compared to previous benchmark studies characterizing this scoring function method and performance against datasets of diverse protein binding site architechtures and protein-ligand interactions, these levels of R2 correlation are adequate to establish confidence in the binding model as reflecting experimental SAR data [20,35,36] compared to models with zero correlation (R2 = 0.0). While the robustness of this correlation analysis may be determined rigorously using a cross-validation approach [20], this is not required in this situation, as the series may also be easily modeled as two separate series of derivatives. One series is structurally related to reference compound Clofazimine 2 (6d, 6e, 7a, 7b, 7c, 7d, 7e, 7f, 7g, 7i, 7k, 7m, 7o) and the other series is based on a different reference compound substructure (15a, 15b, 15f, 15b, 15h). Pearson’s R2 correlation values range from R2 = 0.247 for all 18 compounds (Figure 7A) modeled at Site 2 where even higher correlation coefficients of R2 = 0.306 to 0.311 were achieved modeling the data set as these two separate series of “untrained” predicted ΔGbind rankings as shown in (Figure 7B) with similar slopes. Compared to previous studies using this approach, the observed R2 correlation and slope for the two series are sufficiently similar [20,35,36].
As shown in Figure 8 in more detail, the derivatives from both series are well modeled at Site 2 and the binding mode can rationalize the SAR functional group substitutions at all three R groups (R, R1 and R2). The model can rationalize the SAR relationship at R where (O-CH3 > Cl > F). The reference Cl atom forms not hydrophobic interactions, but rather close and favorable hydrophilic interactions with the positively charged NZ atom from the side chain of K1038 where the phenyl ring forms favorable hydrophobic interactions with the hydrophobic side chain of K1038 atoms (CB, CG, CD, CE). Thus, the R group is partially solvent exposed in close proximity of electrostatic interactions with the NZ atom side chain of K1038. The substitution O-CH3 forms favorable interactions, but the F atom exhibits a weaker molecular interaction with a positively charged NZ atom than Cl. Thus, the model is able to rationalize the most important substitutions leading to favorable R groups.
Next, the model can explain the series of substitutions at R1, where the phenyl ring is buried in a hydrophobic pocket formed primarily by the side chain of A890 and Y1047 on one side and V1040 on the other side. For the position of the R1, para Cl or F substitutions are found in more favorable derivatives such as 15g. The favorability of F over Cl is easily rationalized by its proximity at the back of a hydrophobic pocket with close interactions with dipolar backbone atoms 2.54 Å from (G1046@HN) and 3.19 Å from (D141@OD1). Other R1 substitutions are also rationalized in this binding mode, such as O-CF3 (7i) being more favorable than O-CH3 (7g), where one of the CF3 electronegative fluorine atoms of (7i) forms a favorable electrostatic interaction with a backbone amide H1048@HN, such that the isosteric CH3 substitution is less favorable. Finally, the model is also able to rationalize the series of substitutions at R2, namely that the isopropyl group is more favorable than the cyclopropyl as demonstrated with derivatives 7a and 15h. For derivatives 7a and 15h, the cyclopropyl group carbon atoms are more unfavorable as they are closer in distance to the polar side chain atoms of R1107 and N1108. The smaller isopropyl group lacks these unfavorable interactions and the carbon atoms bind a bit closer in distance to the aromatic carbon atoms of W886. In summary, the derivative series when modeled at Site 2 forms complementary protein-ligand interactions that are able to explain substitutions at R, R1 and R2.
3.5. Modeling a Series of Clofazimine Derivatives Binding to Other SARS-CoV-2 Targets
To increase confidence in our comparison with the derivative series SAR data, the series of derivatives were also independently docked at other binding sites of other SARS-CoV-2 target proteins. As mentioned previously, Clofazimine 2 has been reported to be a dual-targeted SARS-CoV-2 antiviral [5], with Spike-dependent fusion inhibition activity as well as Nsp13 helicase unwinding antiviral activity [5]. Interestingly, the same research group also measured zero activity for Clofazimine 2 in an assay for the Nsp5 Main protease (Mpro) activity [5]. As we had previously published maps of thermodynamically favorable binding sites for Nsp5 Mpro, Nsp13 helicase and Nsp16 2’-O methyltransferase [19], we selected to model the derivative series at the most favorable site identified on these targets for Clofazimine 2. Thus, Nsp5 Mpro and Nsp16 are “negative control” proteins, where we would expect no correlation with experimental SAR data, particularly since 2 has been reported to have no inhibition activity for Nsp5 Mpro. As expected, modeling the series of 18 derivatives at both Nsp5 Mpro and Nsp16 as “negative control” binding sites resulted in poor agreement with the experimental SAR data as well as less favorable predicted (ΔGbind) values. Modeling the series at Nsp5 Mpro, the “untrained” predicted ΔGbind values exhibited a negative correlation (a negative slope) with a very low correlation coefficient R2 = 0.014 for all 18 compounds (Figure 7A). This agrees with the observation that Clofazimine 2 has been reported to have no inhibition activity for Nsp5 Mpro [5]. Modeling the series at Nsp16, the “untrained” predicted ΔGbind values exhibited a negative correlation with a low correlation coefficient R2 = 0.056 for all 18 compounds (Figure 7A). Interestingly, the results in Figure 7A show that in modeling the series of 18 derivatives at the most favorable site identified for Clofazimine 2 on the Nsp13 helicase (see Supplementary Figure S1), the “untrained” predicted ΔGbind values did exhibit some correlation (R2 = 0.141) with the experimental EC50 values, but not as much correlation as Site 2 on the S2 segment (R2 = 0.264).
To summarize, the comparison of the docking data at other target proteins “decoy” binding sites also strengthens the conclusion that the series of Clofazimine 2 derivatives are best modeled at Site 2 on the S2 segment, rather than Site 1 on the S2 segment. When the series is modeled at all 5 binding sites, the only sites that have reasonable R2 correlation values and positive slopes are for binding at Site 2 on the S2 segment (R2 = 0.264) and at the Nsp13 helicase site (R2 = 0.141).
4. Discussion
4.1. Possible Implications for Broad-Spectrum Antiviral Activity
As the COVID-19 pandemic progressed, it was not surprising that most of the observed mutations to the SARS-CoV-2 Spike protein were found in the S1 segment of Spike, which contains the receptor binding domain (RBD). In comparison, fewer mutations have been found on the S2 segment, which exhibits greater conservation in sequence across coronavirus strains [51]. The result that Clofazimine 2 binds to the S2 segment of Spike might have been anticipated from sequence alone, as 2 has been shown to exhibit some broad-spectrum activity against other coronavirus strains such as MERS [5]. When the proposed Clofazimine 2 binding site is superimposed with other experimentally determined coronavirus structures in structure-sequence alignments, the changes in sequence and structure are able to rationalize broad-spectrum antiviral activity of 2 in closely related coronavirus, including SARS CoV, MERS, hCoV-229E, and hCoV-OC43.
Figure 9B shows the backbone superposition in a structure-sequence alignment of SARS-CoV-2 and MERS structures, where Figure 9D shows the complementary fit of the molecular surface. Figure 9E shows how the MERS binding site is still formed, with minimal atom-clashes with the model of bound Clofazimine. Figure 9F shows the sequence conservation in the residue segments that form the binding site. In the highly conserved SARS-CoV-2 sequence 1036-1040 (QSKRV), Q1036 is the most conserved residue that forms the Clofazimine binding site with the (i, i+2) residue K1038, which is a key residue forming important hydrophobic and hydrophilic interactions with 2 in the model. In following the sequence conservation of the position K1038, the residue is the same for the most closely related viruses (SARS-WH20, SARS-BJ01, and MERS) and then begins to diverge, with the mutation K1038S for Human coronavirus OC43 (7sb3.pdb) [52], and K1038P for human coronavirus HKU1 (8ohn.pdb) [53]. Interestingly, Cofazimine 2 has been shown to have some activity [2,5] for the strain hCoV-229E (6u7h.pdb) [54], and this sequence retains the K1038 residue which is key to the binding site [54]. Clofazimine has been shown to have some activity for the strain human coronavirus OC43 [2,5], where the binding site is perturbed from the substitution K1038S and the binding site model would predict a much lower activity for the OC43 strain compared to SARS-CoV-2. This trend has been observed experimentally [2], where 2 was found to be less potent in infections with hCoV-OC43 (EC50 = 0.35 μM) compared to SARS-CoV-2 (EC50 = 0.01 μM) in the same study [2]. As the sequence diverges further for the residues forming the binding site, the model would predict diminishing activity in other more distantly related coronaviruses such as Rhinolophus bat coronavirus HKU2 (6m15.pdb) [55]. Put simply, the proposed binding model seems to be able to account for current information from numerous experimentally determined structures of Spike and available antiviral activity for 2 against several coronavirus strains (SARS-CoV-2 compared to MERS or OC43).
While it is quite possible that several other research groups have independently identified this putative binding site on the S2 segment; to the best of our knowledge, this binding site was first highlighted in the literature by our studies [19,20] and was also recently independently identified by Zannella et al [56], using an entirely different genetic approach to identify short peptide inhibitors of SARS-CoV-2 [56]. The short tripeptide inhibitor VFI was identified experimentally by Zannella et al. They proposed that VFI binds to the current site on S2 and demonstrated greater antiviral effects in pre-treatment assays; similar to a fusion inhibitor [56]. Interestingly, unlike the other peptides identified in that study, the VFI peptide exhibited broad-spectrum antiviral activity for both SARS-CoV-2 and hCoV-OC43 [56], similar to Clofazimine 2. When the VFI tripeptide is docked to Site 2, it binds in a very similar binding mode as Clofazimine 2 with impressive superposition of the three major hydrophobic peptide side chain pharmacophores (Figure 9C).
4.2. Possible Implications for Spike-Dependent Mechanism of Action as a Fusion Inhibitor
For the proposed Clofazimine 2 binding site (Site 2), a structural comparison of the experimentally determined structures of the prefusion and postfusion conformation [57], provides a model for the direct-action of Clofazimine on S2. Shown in Figure 10A, depicts a model of Clofazimine 2 bound to the experimental structure of the prefusion conformation and then superimposed on the experimentally determined post fusion structure of S2 [57]. Figure 10B illustrates how the Clofazimine 2 binding site undergoes a significant conformational change associated with hydrophobic collapse of the binding pocket in the post fusion conformation of S2. While Clofazimine 2 has favorable and complementary protein-ligand interactions in the prefusion conformation, the modeled conformation of 2 in the post fusion structure results in significant atom clashes as shown in Figure 10B, from the resulting refolding and hydrophobic collapse of the local elements of protein structure. Clofazimine 2 binding at this site is highly favorable in the prefusion conformation due to complementary hydrophobic interactions, which should result in a ligand-induced stabilization of the prefusion conformation of S2. The hydrophobic collapse of this site in the postfusion conformation should prevent binding of 2 according to our modeling. The model suggests that Clofazimine 2 binding in the prefusion conformation is the most likely mode of action, stabilizing the prefusion state and preventing conformational changes within S2 that are required for membrane fusion.
5. Conclusions
Clofazimine 2, has been shown to be a potent SARS-CoV-2 fusion inhibitor with robust activity in numerous antiviral assays [2,3,4,5,6,7,8,9,10]. 2 has also demonstrated promising preclinical antiviral activity in a Syrian hamster animal model of SARS-CoV-2 infection [5]. As 2 has demonstrated synergistic antiviral activity with other direct-acting antivirals such as Remdesivir [5], we hope that fusion inhibitors with this mechanism of action may be considered for future synergistic drug combination therapies [15]. More recent mechanistic studies have also shown that Clofazimine 2 is able to inhibit Spike-induced activation of TMEM16 and subsequent procoagulant activity [58]. This observation may increase clinical interest in using Clofazimine 2 as an experimental drug in the treatment of COVID-19 infections with significant pulmonary thrombosis, or in treatment of a range of other Spike-induced pathologies, potentially including the treatment of “long COVID” [59,60,61,62,63,64,65].
While Clofazimine 2, has been shown previously to bind to the SARS-CoV-2 Spike protein [23], it has yet to be reported that Clofazimine 2 binds to the S2 segment of Spike. This experimental result is important in understanding the effects of fusion inhibitors of different structural classes and their specific mechanisms of inhibiting viral fusion. Aiming towards a strategy of avoiding viral resistance mutations, the binding sites described on the S2 segment are composed of very conserved residues that seem to be required for S2 fusion activity function [51]. While it has yet to be experimentally determined exactly where Clofazimine 2 binds to S2, we provide several lines of evidence that Clofazimine 2 is best modeled as binding at Site 2. While it is only a model, it is a plausible structural hypothesis that is very useful to guide our next round of experimental design, including mutagenesis with complementary biophysical and pseudovirus entry assays. As Clofazimine has been demonstrated to be one of the most promising clinical fusion inhibitors of Spike, we hope that this work provides important structural insight for developing improved fusion inhibitors that target S2 and elucidating the mechanism of direct drug action.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Binding Site of Clofazimine 2 on the SARS-CoV-2 Nsp16 Helicase.; Table S1: Comparison of Clofazimine 2 derivatives predicted binding free energy at Site1 and Site 2 to those approximated from experimental EC50 values.; SUPP_INFO.zip: Summary PDB coordinate files for lowest energy binding modes of Clofazimine 2 modeled at Site 1, Site 2 on the Spike protein, as well as on Nsp13 helicase, Nsp5 Mpro and Nsp16 (ZIP).
Author Contributions
Conceptualization, M.F. and R.A.; methodology, M.F and R.A.; software, R.A.; validation, P.V., S.S. and R.A.; formal analysis, P.V., S.S. and R.A.; investigation, R.A.; resources, R.A.; data curation, P.V., S.S and R.A.; writing—original draft preparation, M.F and R.A.; writing—review and editing, M.F and R.A.; visualization, M.F., P.V., S.S., and R.A.; supervision, R.A.; project administration, R.A.; funding acquisition, R.A. All authors have read and agreed to the published version of the manuscript.
Funding
Roger Armen was partially supported by grants 1P01HL114471 and R01 AR077666 and R01 HL153602 from the National Institutes of Health. There was no additional external funding received for this study.
Data Availability Statement
The program CHARMM is publicly available under academic license for research (https://www.charmm.org). MarvinSketch is publicly available under academic license for research (http://www.chemaxon.com). UCSF Chimera is publicly available under academic license for research (https://www.cgl.ucsf.edu/chimera). All relevant data are shown in figures, listed in tables or included in supporting information. The docking ΔGbind data and 2D compound information for all compounds may be found in the manuscript and in Table S1. PDB files output files are provided in supporting information as a .ZIP file.
Acknowledgments
The authors thank Reaction Biology Corporation and the biophysical SPR team for assay support. Roger Armen was partially supported by grants 1P01HL114471 and R01 AR077666 and R01 HL153602 from the National Institutes of Health. There was no additional external funding received for this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; Yang, X.; Shi, Z.; Deng, F.; Hu, Z.; Zhong, W.; Wang, M. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, C.; Chang, D.; Wang, Y.; Dong, X.; Jiao, T.; Zhao, Z.; Ren, L.; Dela Cruz, C. S.; Sharma, L.; Lei, X.; Wang, J. Identification of Potent and Safe Antiviral Therapeutic Candidates Against SARS-CoV-2. Front. Immunol. 2020, 11, 586572. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y. Y.; Peng, T. T.; Yeh, T. K.; Huang, W. Z.; Chang, S. E.; Wu, S. H.; Hung, H. C.; Hsu, T. A.; Lee, S. J.; Song, J. S.; Lin, W. H.; Chiang, T. J.; Lin, J. H.; Sytwu, H. K.; Chen, C. T. Artificial intelligence approach fighting COVID-19 with repurposing drugs. Biomed J. 2020, 43, 355. [Google Scholar] [CrossRef] [PubMed]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P. D.; Teriete, P.; Hull, M. V.; Chang, M. W.; Chan, J. F.; Cao, J.; Poon, V. K.; Herbert, K. M.; Cheng, K.; Nguyen, T. H.; Rubanov, A.; Pu, Y.; Nguyen, C.; Choi, A.; Rathnasinghe, R.; Schotsaert, M.; Miorin, L.; Dejosez, M.; Zwaka, T. P.; Sit, K. Y.; Martinez-Sobrido, L.; Liu, W. C.; White, K. M.; Chapman, M. E.; Lendy, E. K.; Glynne, R.J.; Albrecht, R.; Ruppin, E.; Mesecar, A. D.; Johnson, J. R.; Benner, C.; Sun, R.; Schultz, P. G.; Su, A. I.; García-Sastre, A.; Chatterjee, A. K.; Yuen, K. Y.; Chanda, S. K. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature. 2020, 586, 113. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Yin, X.; Meng, X.; Chan, J. F.; Ye, Z. W.; Riva, L.; Pache, L.; Chan, C. C.; Lai, P. M.; Chan, C. C.; Poon, V. K.; Lee, A.C.; Matsunaga, N.; Pu, Y.; Yuen, C. K.; Cao, J.; Liang, R.; Tang, K.; Sheng, L.; Du, Y.; Xu, W.; Lau, C. Y.; Sit, K. Y.; Au, W.K.; Wang, R.; Zhang, Y. Y.; Tang, Y. D.; Clausen, T. M.; Pihl, J.; Oh, J.; Sze, K. H.; Zhang, A. J.; Chu, H.; Kok, K. H.; Wang, D.; Cai, X. H.; Esko, J. D.; Hung, I. F.; Li, R. A.; Chen, H.; Sun, H.; Jin, D. Y.; Sun, R.; Chanda, S. K.; Yuen, K. Y. Clofazimine broadly inhibits coronaviruses including SARS-CoV-2. Nature. 2021, 593, 418. [Google Scholar] [CrossRef]
- Mirabelli, C.; Wotring, J. W.; Zhang, C. J.; McCarty, S. M.; Fursmidt, R.; Pretto, C. D.; Qiao, Y.; Zhang, Y.; Frum, T.; Kadambi, N. S.; Amin, A. T.; O'Meara, T. R.; Spence, J. R.; Huang, J.; Alysandratos, K. D.; Kotton, D. N.; Handelman, S. K.; Wobus, C. E.; Weatherwax, K. J.; Mashour, G. A.; O'Meara, M. J.; Chinnaiyan, A. M.; Sexton, J. Z. Morphological cell profiling of SARS-CoV-2 infection identifies drug repurposing candidates for COVID-19. Proc Natl Acad Sci U S A. 2021, 118, e2105815118. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Zhu, S.; Li, S.; Shang, W.; Zhang, R.; Li, H.; Liu, W.; Xiao, G.; Peng, K.; Zhang, L. High-Throughput Screening of an FDA-Approved Drug Library Identifies Inhibitors against Arenaviruses and SARS-CoV-2. ACS Infect Dis. 2021, 7, 1409. [Google Scholar] [CrossRef] [PubMed]
- Le, B. L.; Andreoletti, G.; Oskotsky, T.; Vallejo-Gracia, A.; Rosales, R.; Yu, K.; Kosti, I.; Leon, K. E.; Bunis, D. G.; Li, C.; Kumar, G. R.; White, K. M.; García-Sastre, A.; Ott, M.; Sirota, M. Transcriptomics-based drug repositioning pipeline identifies therapeutic candidates for COVID-19. Sci Rep. 2021, 11, 12310. [Google Scholar] [CrossRef] [PubMed]
- Ginex, T.; Garaigorta, U.; Ramírez, D.; Castro, V.; Nozal, V.; Maestro, I.; García-Cárceles, J.; Campillo, N. E.; Martinez, A.; Gastaminza, P.; Gil, C. Host-Directed FDA-Approved Drugs with Antiviral Activity against SARS-CoV-2 Identified by Hierarchical In Silico/In Vitro Screening Methods. Pharmaceuticals (Basel). 2021, 14, 332. [Google Scholar] [CrossRef]
- Aherfi, S.; Pradines, B.; Devaux, C.; Honore, S.; Colson, P.; Scola, B.; Raoult, D. Drug repurposing against SARS-CoV-1, SARS-CoV-2 and MERS-CoV. Future Microbiol. 2021, 16, 1341. [Google Scholar] [CrossRef]
- Kushwaha, N. D.; Mohan, J.; Kushwaha, B.; Ghazi, T.; Nwabuife, J. C.; Koorbanally, N.; Chuturgoon, A. A. A comprehensive review on the global efforts on vaccines and repurposed drugs for combating COVID-19. Eur J Med Chem. 2023, 260, 115719. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hilgenfeld, R.; Whitley, R.; De Clercq, E. Therapeutic strategies for COVID-19: progress and lessons learned. Nat Rev Drug Discov. 2023, 22, 449. [Google Scholar] [CrossRef] [PubMed]
- Chan, S. W. Current and Future Direct-Acting Antivirals Against COVID-19. Front Microbiol. 2020, 11, 587944. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Stefanelli, I.; Cerchia, C.; Beccari, A. R.; Pelliccia, S.; Summa, V. SARS-CoV-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus or Host Cells. Int J Mol Sci. 2020, 21, 5707. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski, T.; Chen, L.; Eastman, RT.; Itkin, Z.; Shinn, P.; Chen, C. Z.; Guo, H.; Zheng, W.; Michael, S.; Simeonov, A.; Hall, M. D.; Zakharov, A. V.; Muratov, E. N. Synergistic and Antagonistic Drug Combinations against SARS-CoV-2. Mol Ther. 2021, 29, 873. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Fang, J.; Chen, S.; Rajaofera, M. J. N.; Li, X.; Wang B, Xia Q. The efficacy and safety of remdesivir alone and in combination with other drugs for the treatment of COVID-19: a systematic review and meta-analysis. BMC Infect Dis. 2023, 23, 672. [Google Scholar] [CrossRef] [PubMed]
- Tian H, Yang C, Song T, Zhou K, Wen L, Tian Y, Tang L, Xu W, Zhang X. Efficacy and safety of paxlovid (nirmatrelvir/ritonavir) in the treatment of COVID-19: An updated meta-analysis and trial sequential analysis. Rev Med Virol. 2023, 33, e2473. [CrossRef] [PubMed]
- Akinosoglou K, Schinas G, Gogos C. Oral Antiviral Treatment for COVID-19: A Comprehensive Review on Nirmatrelvir/Ritonavir. Viruses 2022, 14, 2540. [Google Scholar] [CrossRef]
- Freidel, M. R.; Armen, R. S. Mapping major SARS-CoV-2 drug targets and assessment of druggability using computational fragment screening: Identification of an allosteric small-molecule binding site on the Nsp13 helicase. PLoS One. 2021, 16, e0246181. [Google Scholar] [CrossRef]
- Freidel, M. R.; Armen, R. S. Modeling the Structure-Activity Relationship of Arbidol Derivatives and Other SARS-CoV-2 Fusion Inhibitors Targeting the S2 Segment of the Spike Protein. J Chem Inf Model. 2021, 61, 5906. [Google Scholar] [CrossRef]
- Chen, C. Z.; Xu, M.; Pradhan, M.; Gorshkov, K.; Petersen, J. D.; Straus, M. R.; Zhu, W.; Shinn, P.; Guo, H.; Shen, M.; Klumpp-Thomas, C.; Michael, S.G.; Zimmerberg, J.; Zheng, W.; Whittaker, G. R. Identifying SARS-CoV-2 Entry Inhibitors through Drug Repurposing Screens of SARS-S and MERS-S Pseudotyped Particles. ACS Pharmacol. Transl. Sci. 2020, 3, 1165. [Google Scholar] [CrossRef]
- Riccardi, N.; Giacomelli, A.; Canetti, D.; Comelli, A.; Intini, E.; Gaiera, G.; Diaw, M. M.; Udwadia, Z.; Besozzi, G.; Codecasa, L.; Biagio, A. D. Clofazimine: an old drug for never-ending diseases. Future Microbiol. 2020, 15, 557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, Y.; Guo, Z.; Zhao, X.; Wu, J.; Cao, S.; Liu, Y.; Li, Y.; Huang, W.; Wang, Y.; Liu, Q.; Li, Y.; Song, D. Clofazimine derivatives as potent broad-spectrum antiviral agents with dual-target mechanism. Eur J Med Chem. 2022, 234, 114209. [Google Scholar] [CrossRef]
- Stadler, J. A. M.; Maartens, G.; Meintjes, G.; Wasserman, S. Clofazimine for the treatment of tuberculosis. Front Pharmacol. 2023, 14, 1100488. [Google Scholar] [CrossRef] [PubMed]
- Mirnejad, R.; Asadi, A.; Khoshnood, S.; Mirzaei, H.; Heidary, M.; Fattorini, L.; Ghodousi, A.; Darban-Sarokhalil, D. Clofazimine: A useful antibiotic for drug-resistant tuberculosis. Biomed Pharmacother. 2018, 105, 1353. [Google Scholar] [CrossRef]
- Falzon, D.; Schünemann, H. J.; Harausz, E.; González-Angulo, L.; Lienhardt, C.; Jaramillo, E.; Weyer, K. World Health Organization treatment guidelines for drug-resistant tuberculosis, 2016 update. Eur Respir J. 2017, 49, 1602308. [Google Scholar] [CrossRef]
- Egiz, A.; Gala, D. Clofazimine: another potential magic bullet for the treatment of COVID-19? Postgrad Med J. 2022, 98, e124. [Google Scholar] [CrossRef]
- Wang, D.; Loo, J. F. C.; Chen, J.; Yam, Y.; Chen, S.C.; He, H.; Kong, S. K.; Ho, H. P. Recent Advances in Surface Plasmon Resonance Imaging Sensors. Sensors (Basel). 2019, 19, 1266. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H. H.; Park, J.; Kang, S.; Kim, M. Surface plasmon resonance: a versatile technique for biosensor applications. Sensors (Basel). 2015, 15, 10481. [Google Scholar] [CrossRef]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents. 2020, 56, 105998. [Google Scholar] [CrossRef]
- Shuster, A.; Pechalrieu, D.; Jackson, C. B.; Abegg, D.; Choe, H.; Adibekian, A. Clinical Antiviral Drug Arbidol Inhibits Infection by SARS-CoV-2 and Variants through Direct Binding to the Spike Protein. ACS Chem Biol. 2021, 16, 2845. [Google Scholar] [CrossRef] [PubMed]
- O'Connell, N. Protein Ligand Interactions Using Surface Plasmon Resonance. Methods Mol Biol. 2021, 2365, 3. [Google Scholar]
- Frostell-Karlsson, A.; Remaeus, A.; Roos, H.; Andersson, K.; Borg, P.; Hämäläinen, M.; Karlsson, R. Biosensor analysis of the interaction between immobilized human serum albumin and drug compounds for prediction of human serum albumin binding levels. J Med Chem. 2000, 43, 1986. [Google Scholar] [CrossRef]
- Brooks, B. R.; Brooks 3rd, C. L.; Mackerell Jr, A. D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; Caflisch, A.; Caves, L.; Cui, Q.; Dinner, A. R.; Feig, M.; Fischer, S.; Gao, J.; Hodoscek, M.; Im, W.; Kuczera, K.; Lazaridis, T.; Ma, J.; Ovchinnikov, V.; Paci, E.; Pastor, R. W.; Post, C. B.; Pu, J. Z.; Schaefer, M.; Tidor, B.; Venable, R. M.; Woodcock, H. L.; Wu, X.; Yang, W.; York, D. M.; Karplus, M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545. [Google Scholar] [CrossRef]
- Rahaman, O.; Estrada, T. P.; Doren, D. J.; Taufer, M.; Brooks 3rd, C. B.; Armen, R. S. Evaluation of several two-step scoring functions based on linear interaction energy, effective ligand size, and empirical pair potentials for prediction of protein-ligand binding geometry and free energy. J. Chem. Inf. Model. 2011, 51, 2047. [Google Scholar] [CrossRef]
- Armen, R. S.; Chen, J.; Brooks 3rd, C. B. An Evaluation of Explicit Receptor Flexibility in Molecular Docking Using Molecular Dynamics and Torsion Angle Molecular Dynamics. J. Chem. Theory Comput. 2009, 5, 2909. [Google Scholar] [CrossRef]
- Momany, F. A.; Rone, R. Validation of the general purpose QUANTA® 3.2/CHARMm® force field. J Comput Chem. 1992, 13, 888. [Google Scholar] [CrossRef]
- Roche, O.; Kiyama, R.; Brooks, C. L. 3rd. Ligand-protein database: linking protein-ligand complex structures to binding data. J Med Chem. 2001, 44, 3592. [Google Scholar] [CrossRef] [PubMed]
- Lee, M. S.; Feig, M.; Salsbury, Jr., F. R.; Brooks, C. L. 3rd. New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J. Comput. Chem. 2003, 24, 1348. [Google Scholar] [CrossRef]
- Feig, M.; Im, W.; Brooks, C. L. 3rd. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures J. Chem. Phys. 2004, 2, 903. [Google Scholar]
- Available at: http://www.chemaxon.com.
- Walls, A. C.; Park, Y.-J.; Tortorici, M. A.; Wall, A.; McGuire, A. T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020, 181, 281–292. [Google Scholar] [CrossRef]
- https://www.rcsb.org/structure/6W63.
- Jia, Z.; Yan, L.; Ren, Z.; Wu, L.; Wang, J.; Guo, J.; Zheng, L.; Ming, Z.; Zhang, L.; Lou, Z.; Rao, Z. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019, 47, 6538. [Google Scholar] [CrossRef]
- https://www.rcsb.org/structure/6WKQ.
- Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004, 25, 1605. [Google Scholar] [CrossRef]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S. Y.; Park, S.; Shum, D.; Kim, S. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob Agents Chemother. 2020, 64, e00819–20. [Google Scholar] [CrossRef]
- Yang, L.; Pei, R. J.; Li, H.; Ma, X. N.; Zhou, Y.; Zhu, F. H.; He, P. L.; Tang, W.; Zhang, Y, C; Xiong, J.; Xiao, S. Q.; Tong, X. K.; Zhang, B.; Zuo, J. P. Identification of SARS-CoV-2 entry inhibitors among already approved drugs. Acta Pharmacol Sin. 2021, 42, 1347. [Google Scholar] [CrossRef]
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeño, J. M.; Ortega-Prieto, A. M.; Bussani, R.; Cannatà, A.; Rizzari, G.; Collesi, C.; Schneider, E.; Arosio, A.; Shah, A. M.; Barclay, W. S.; Malim, M. H.; Burrone, J.; Giacca, M. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 2021, 594, 88. [Google Scholar] [CrossRef]
- Sakkal, L. A.; Rajkowski, K. Z.; Armen, R. S. Prediction of consensus binding mode geometries for related chemical series of positive allosteric modulators of adenosine and muscarinic acetylcholine receptors. J Comput Chem. 2017, 38, 1209. [Google Scholar] [CrossRef]
- Guo, L.; Lin, S.; Chen, Z.; Cao, Y.; He, B.; Lu, G. Targetable elements in SARS-CoV-2 S2 subunit for the design of pan-coronavirus fusion inhibitors and vaccines. Signal Transduct Target Ther. 2023, 8, 197. [Google Scholar] [CrossRef]
- Bangaru, S.; Antanasijevic, A.; Kose, N.; Sewall, L. M.; Jackson, A. M.; Suryadevara, N.; Zhan, X.; Torres, J. L.; Copps, J.; Torrents de la Peña, A.; Crowe Jr, J. E.; Ward, A. B. Structural mapping of antibody landscapes to human betacoronavirus spike proteins. Sci Adv. 2022, 8, eabn2911. [Google Scholar] [CrossRef]
- Pronker, M. F.; Creutznacher, R.; Drulyte, I.; Hulswit, R. J. G.; Li, Z.; van Kuppeveld, F. J. M.; Snijder, J.; Lang, Y.; Bosch, B. J.; Boons, G. J.; Frank, M.; de Groot, R. J.; Hurdiss, D. L. Sialoglycan binding triggers spike opening in a human coronavirus. Nature. 2023. [Google Scholar] [CrossRef]
- Li, Z.; Tomlinson, A. C.; Wong, A. H.; Zhou, D.; Desforges, M.; Talbot, P. J.; Benlekbir, S.; Rubinstein, J.L.; Rini, J. M. The human coronavirus HCoV-229E S-protein structure and receptor binding. Elife. 2019, 8, e51230. [Google Scholar] [CrossRef]
- Yu, J.; Qiao, S.; Guo, R.; Wang, X. Cryo-EM structures of HKU2 and SADS-CoV spike glycoproteins provide insights into coronavirus evolution. Nat Commun. 2020, 11, 3070. [Google Scholar] [CrossRef]
- Zannella, C.; Chianese, A.; Greco, G.; Santella, B.; Squillaci, G.; Monti, A.; Doti, N.; Sanna, G.; Manzin, A.; Morana, A.; De Filippis, A.; D'Angelo, G.; Palmieri, F.; Franci, G.; Galdiero, M. Design of Three Residues Peptides against SARS-CoV-2 Infection. Viruses. 2022, 14, 2103. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S. M.; Walsh Jr, R. M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020, 369, 1586. [Google Scholar] [CrossRef]
- Cappelletto, A.; Allan, H. E.; Crescente, M.; Schneider, E.; Bussani, R.; Ali, H.; Secco, I.; Vodret, S.; Simeone, R.; Mascaretti, L.; Zacchigna, S.; Warner, T. D.; Giacca, M. SARS-CoV-2 Spike protein activates TMEM16F-mediated platelet procoagulant activity. Front Cardiovasc Med. 2023, 9, 1013262. [Google Scholar] [CrossRef]
- Kakarla, V.; Kaneko, N.; Nour, M.; Khatibi, K.; Elahi, F.; Liebeskind, D. S.; Hinman, J. D. Pathophysiologic mechanisms of cerebral endotheliopathy and stroke due to Sars-CoV-2. J Cereb Blood Flow Metab. 2021, 41, 1179. [Google Scholar] [CrossRef]
- Albornoz, E. A.; Amarilla, A. A.; Modhiran, N.; Parker, S.; Li, X. X.; Wijesundara, D. K.; Aguado, J.; Zamora, A. P.; McMillan, C. L. D.; Liang, B.; Peng, N.Y.G.; Sng, J.D.J.; Saima, F.T.; Fung, J.N.; Lee, J. D.; Paramitha, D.; Parry, R.; Avumegah, M. S.; Isaacs, A.; Lo, M.W.; Miranda-Chacon, Z.; Bradshaw, D.; Salinas-Rebolledo, C.; Rajapakse, N.W.; Wolvetang, E.J.; Munro, T.P.; Rojas-Fernandez, A.; Young, P. R.; Stacey, K. J.; Khromykh, A. A.; Chappell, K. J.; Watterson, D.; Woodruff, T. M. SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike protein. Mol Psychiatry. 2023, 28, 2878. [Google Scholar] [CrossRef]
- Peng, Q.; Zhou, R.; Liu, N.; Wang, H.; Xu, H.; Zhao, M.; Yang, D.; Au, K. K.; Huang, H.; Liu, L.; Chen, Z. Naturally occurring spike mutations influence the infectivity and immunogenicity of SARS-CoV-2. Cell Mol Immunol. 2022, 19, 1302. [Google Scholar] [CrossRef]
- Fernández-de-Las-Peñas, C.; Cancela-Cilleruelo, I.; Rodríguez-Jiménez, J.; Arias-Navalón, J.A.; Martín-Guerrero, J. D.; Pellicer-Valero, O. J.; Arendt-Nielsen, L.; Cigarán-Méndez, M. Trajectory of post-COVID brain fog, memory loss, and concentration loss in previously hospitalized COVID-19 survivors: the LONG-COVID-EXP multicenter study. Front Hum Neurosci. 2023, 17, 1259660. [Google Scholar] [CrossRef]
- Volk, P.; Manesh, M. R.; Warren, M. E.; Besko, K.; Gonçalves de Andrade, E.; Wicki-Stordeur, L. E.; Swayne, L. A. Long-term neurological dysfunction associated with COVID-19: Lessons from influenza and inflammatory diseases? J Neurochem. 2023. [Google Scholar] [CrossRef]
- Zhang, Q.; Tang, W.; Stancanelli, E.; Jung, E.; Syed, Z.; Pagadala, V.; Saidi, L.; Chen, C. Z.; Gao, P.; Xu, M.; Pavlinov, I.; Li, B.; Huang, W.; Chen, L.; Liu, J.; Xie, H.; Zheng, W.; Ye, Y. Host heparan sulfate promotes ACE2 super-cluster assembly and enhances SARS-CoV-2-associated syncytium formation. Nat Commun. 2023, 14, 5777. [Google Scholar] [CrossRef] [PubMed]
- Baldari, C. T.; Onnis, A.; Andreano, E.; Del Giudice, G.; Rappuoli, R. Emerging roles of SARS-CoV-2 Spike-ACE2 in immune evasion and pathogenesis. Trends Immunol. 2023, 44, 424. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure of fusion inhibitor derivatives used in this study.
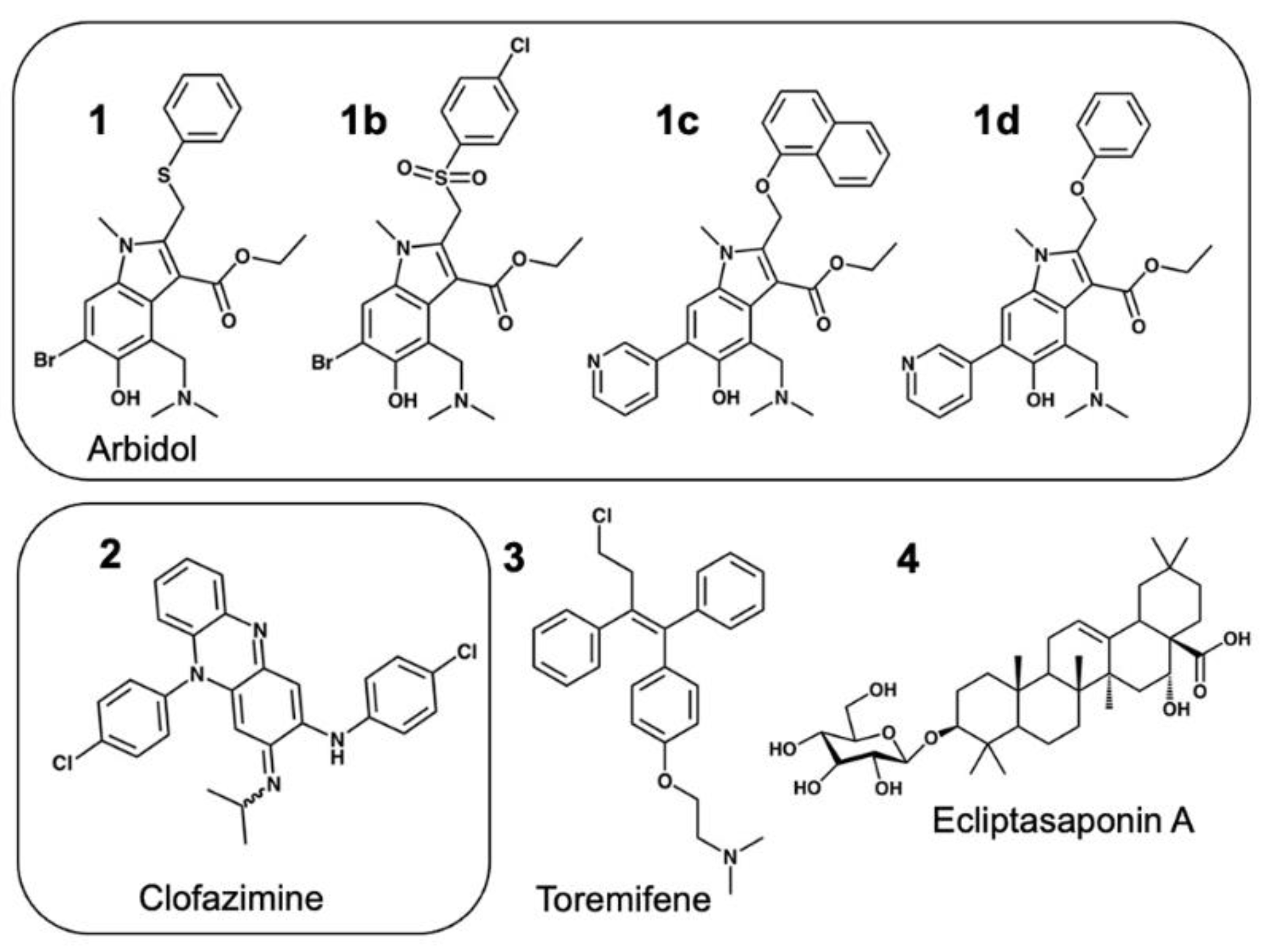
Figure 2.
A direct SPR binding assay measures simultaneous binding to full-length Spike and the S2 segment. (A) Flow cell (FC) surfaces shown with attached Spike (FC2), and reference FC1 and the S2 segment (FC4) and reference FC3. (B) Arbidol binding to Spike and the S2 segment showing both SPR sensorgrams and steady-state-affinity models. .
Figure 2.
A direct SPR binding assay measures simultaneous binding to full-length Spike and the S2 segment. (A) Flow cell (FC) surfaces shown with attached Spike (FC2), and reference FC1 and the S2 segment (FC4) and reference FC3. (B) Arbidol binding to Spike and the S2 segment showing both SPR sensorgrams and steady-state-affinity models. .
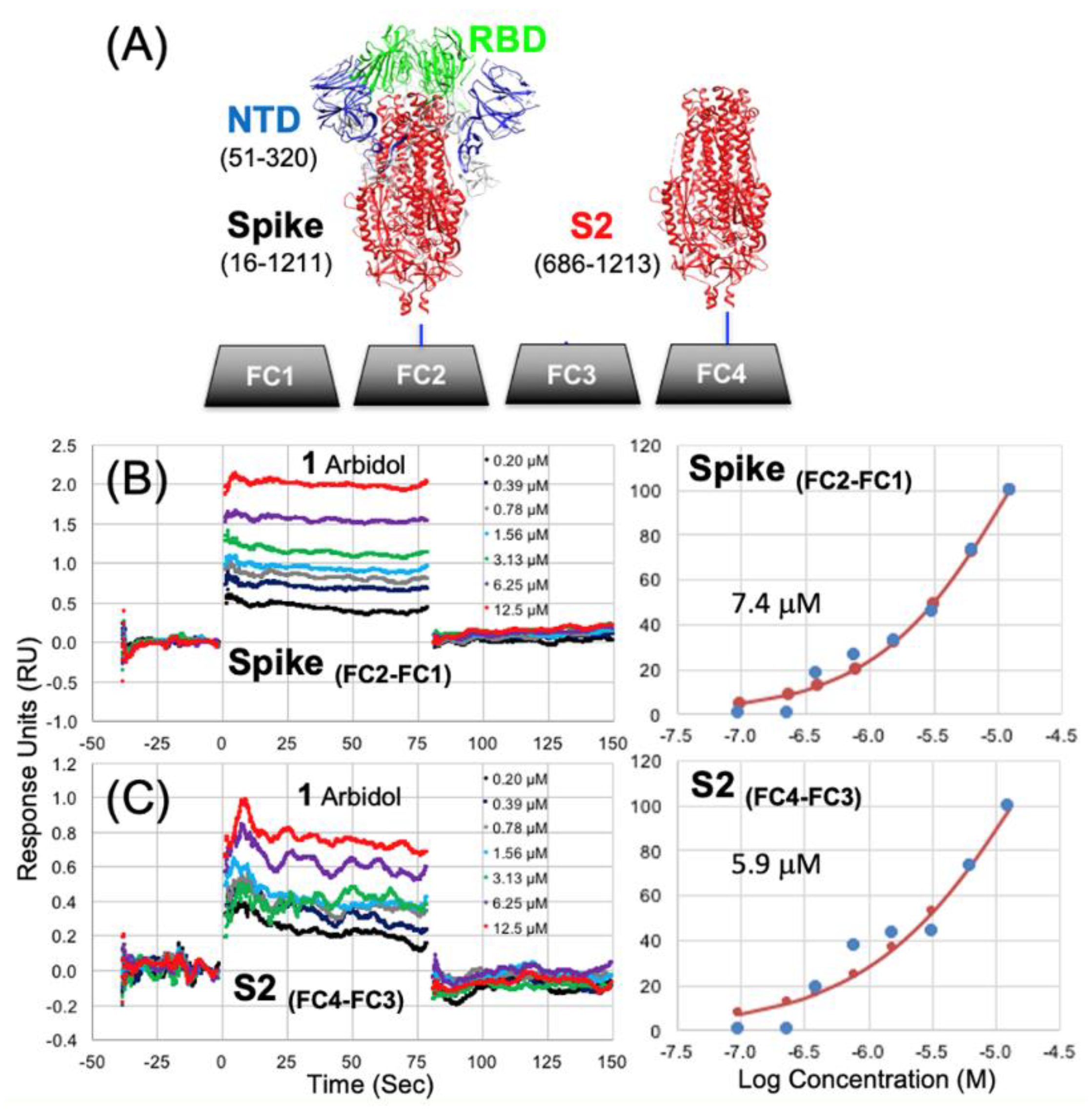
Figure 3.
Structure of the SARS-CoV-2 Spike S2 segment showing two possible fusion inhibitor binding sites. (A) The trimeric S2 segment in a pre-fusion conformation showing (B) Site 2 with Clofazimine 2 bound and (C) Site 1 with Arbidol 1 bound.
Figure 3.
Structure of the SARS-CoV-2 Spike S2 segment showing two possible fusion inhibitor binding sites. (A) The trimeric S2 segment in a pre-fusion conformation showing (B) Site 2 with Clofazimine 2 bound and (C) Site 1 with Arbidol 1 bound.
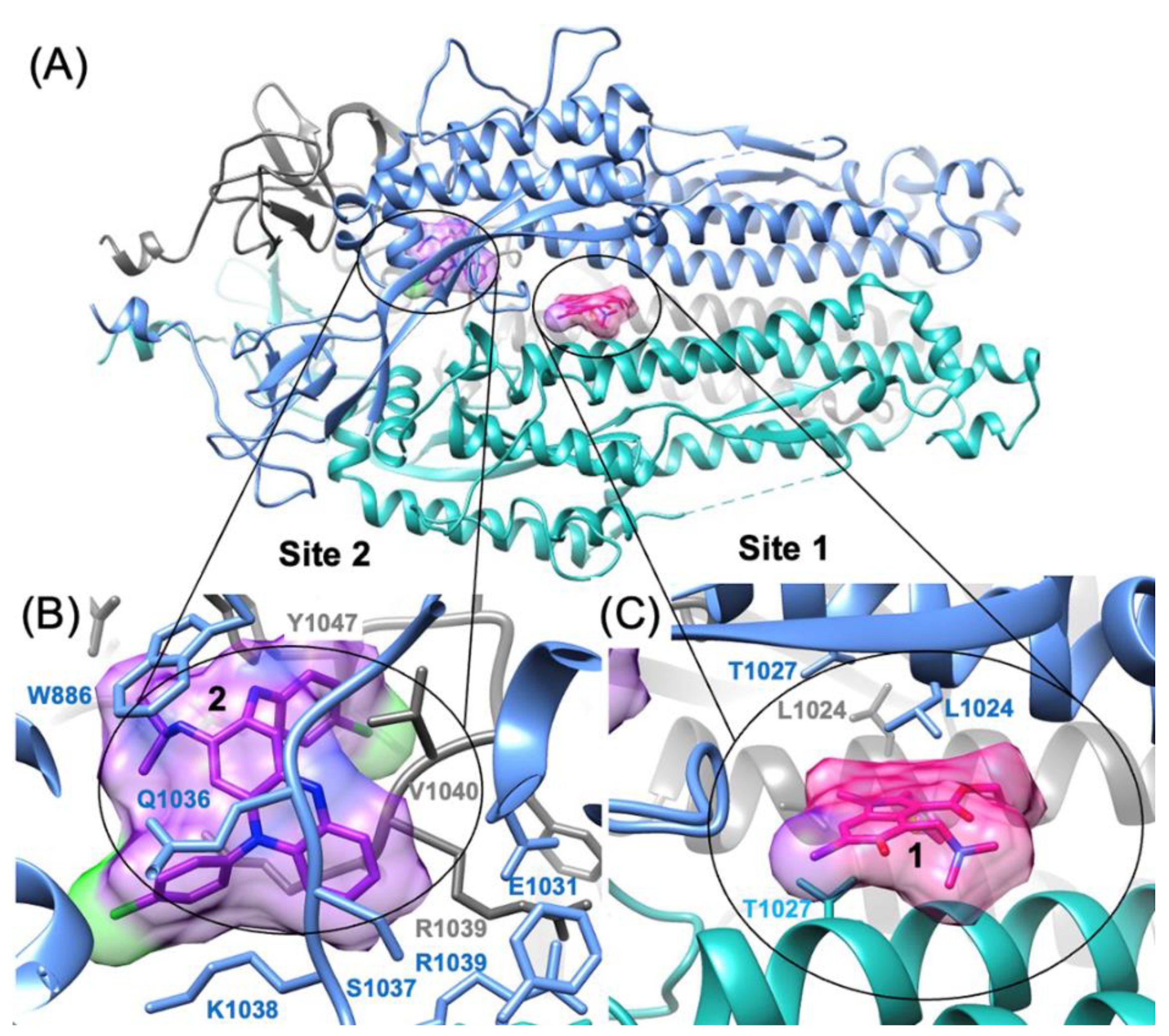
Figure 4.
SPR data for fusion inhibitors binding to Spike and the S2 segment. Compounds from five different structural classes were shown to bind to the S2 segment. (A) Arbidol derivative 1c, (B) Arbidol derivative 1d, (C) Clofazimine 2 (D) Toremifene 3.
Figure 4.
SPR data for fusion inhibitors binding to Spike and the S2 segment. Compounds from five different structural classes were shown to bind to the S2 segment. (A) Arbidol derivative 1c, (B) Arbidol derivative 1d, (C) Clofazimine 2 (D) Toremifene 3.
Figure 5.
SPR duplicates for fusion inhibitors binding to the S2 segment. Duplicate binding curves for each compound are shown comparing only binding the S2 segment of Spike for (A) Arbidol 1, (B) Clofazimine 2 (C) Toremifene 3 and (D) Ecliptasaponin A 4.
Figure 5.
SPR duplicates for fusion inhibitors binding to the S2 segment. Duplicate binding curves for each compound are shown comparing only binding the S2 segment of Spike for (A) Arbidol 1, (B) Clofazimine 2 (C) Toremifene 3 and (D) Ecliptasaponin A 4.
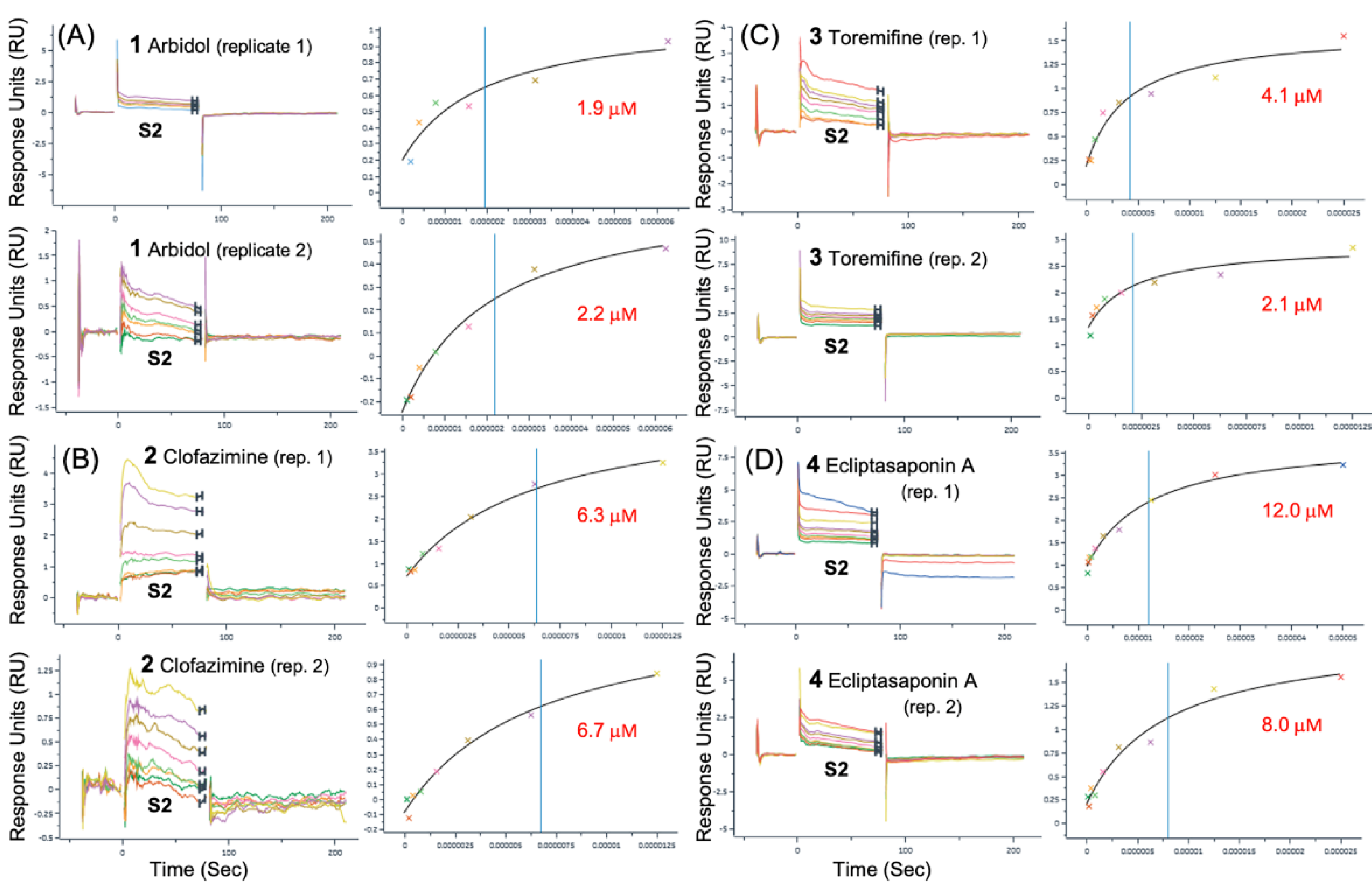
Figure 6.
Modeling fusion inhibitors at two possible binding sites on the S2 Segment. (A) Shown is the trimeric S2 segment in a prefusion conformation showing Site 2 with Clofazimine bound highlighted in purple and Site 1 with Arbidol bound highlighted in magenta. (B) For each fusion inhibitor, calculated ΔGbind values from the lowest energy cluster modeled at Site 1 and Site 2, shown in magenta and purple respectively. Clofazimine 2 and Toremifene 3 are predicted to bind more favorably to site 2, while Ecliptasaponin A 4, and OA Saponin 12a are predicted to bind more favorable to Site 1.
Figure 6.
Modeling fusion inhibitors at two possible binding sites on the S2 Segment. (A) Shown is the trimeric S2 segment in a prefusion conformation showing Site 2 with Clofazimine bound highlighted in purple and Site 1 with Arbidol bound highlighted in magenta. (B) For each fusion inhibitor, calculated ΔGbind values from the lowest energy cluster modeled at Site 1 and Site 2, shown in magenta and purple respectively. Clofazimine 2 and Toremifene 3 are predicted to bind more favorably to site 2, while Ecliptasaponin A 4, and OA Saponin 12a are predicted to bind more favorable to Site 1.
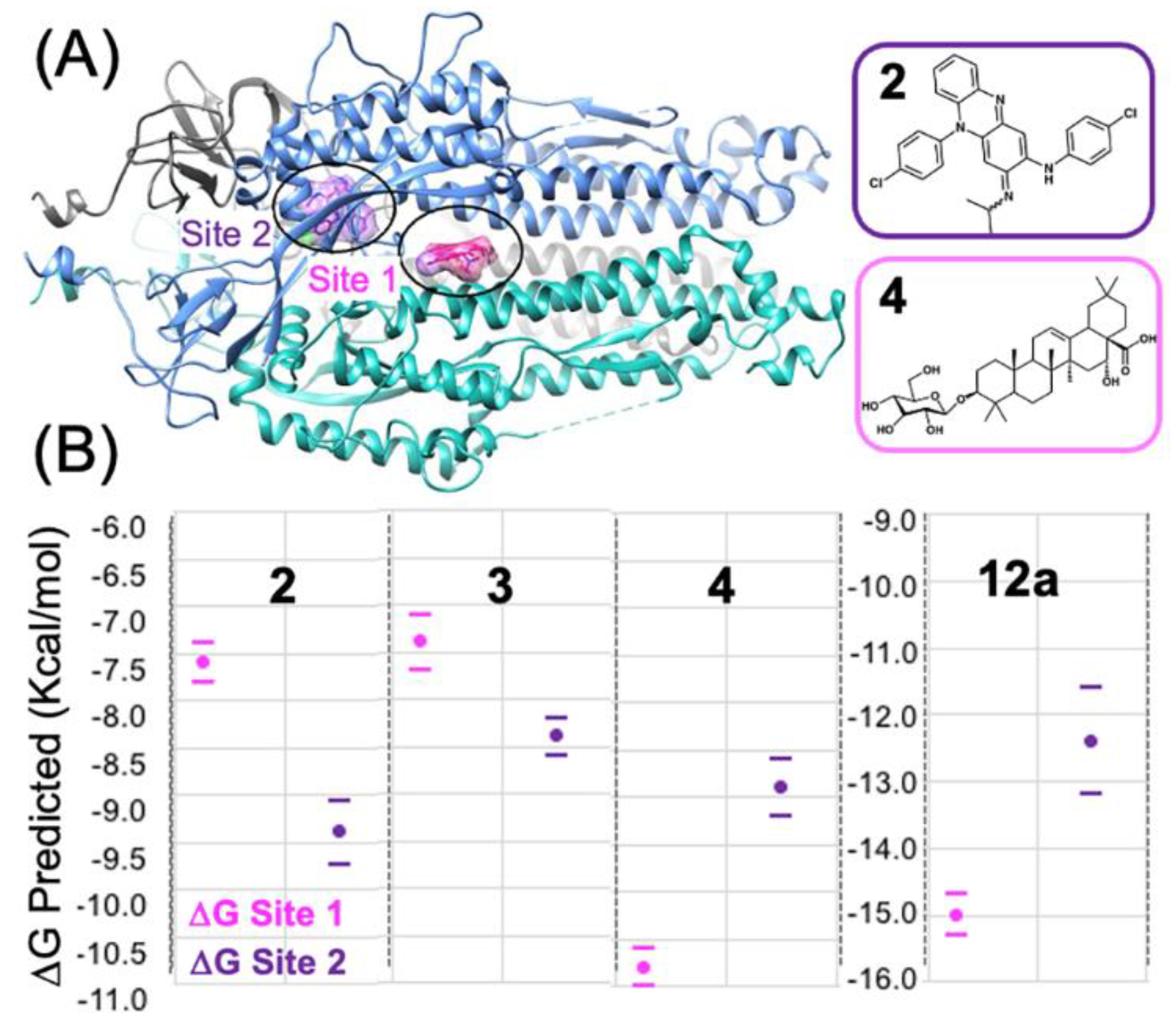
Figure 7.
Predicted ΔGbind values from Site 2 exhibit correlation with experimental SAR data for a series of Clofazimine 2 derivatives. A series of 18 Clofazimine derivatives were well-modeled binding to Site 2, where sufficient linear correlation is achieved either comparing (A) all 18 derivatives (R2 = 0.264) or (B) two separate groups of compound series (R2=0.311) and (R2=0.306) with experimental SAR data. Poor correlation is observed when the compounds are modeled at Site 1 of S2, Nsp5, or Nsp16 “decoy” binding sites. When the series is modeled at the most favorable site on the Nsp13 helicase, the predicted ΔGbind values exhibited some level of correlation for all 18 derivatives (R2 = 0.141) and quite reasonable correlation for the series of (15a, 15b, 15f, 15b, 15h) (R2 = 0.533).
Figure 7.
Predicted ΔGbind values from Site 2 exhibit correlation with experimental SAR data for a series of Clofazimine 2 derivatives. A series of 18 Clofazimine derivatives were well-modeled binding to Site 2, where sufficient linear correlation is achieved either comparing (A) all 18 derivatives (R2 = 0.264) or (B) two separate groups of compound series (R2=0.311) and (R2=0.306) with experimental SAR data. Poor correlation is observed when the compounds are modeled at Site 1 of S2, Nsp5, or Nsp16 “decoy” binding sites. When the series is modeled at the most favorable site on the Nsp13 helicase, the predicted ΔGbind values exhibited some level of correlation for all 18 derivatives (R2 = 0.141) and quite reasonable correlation for the series of (15a, 15b, 15f, 15b, 15h) (R2 = 0.533).
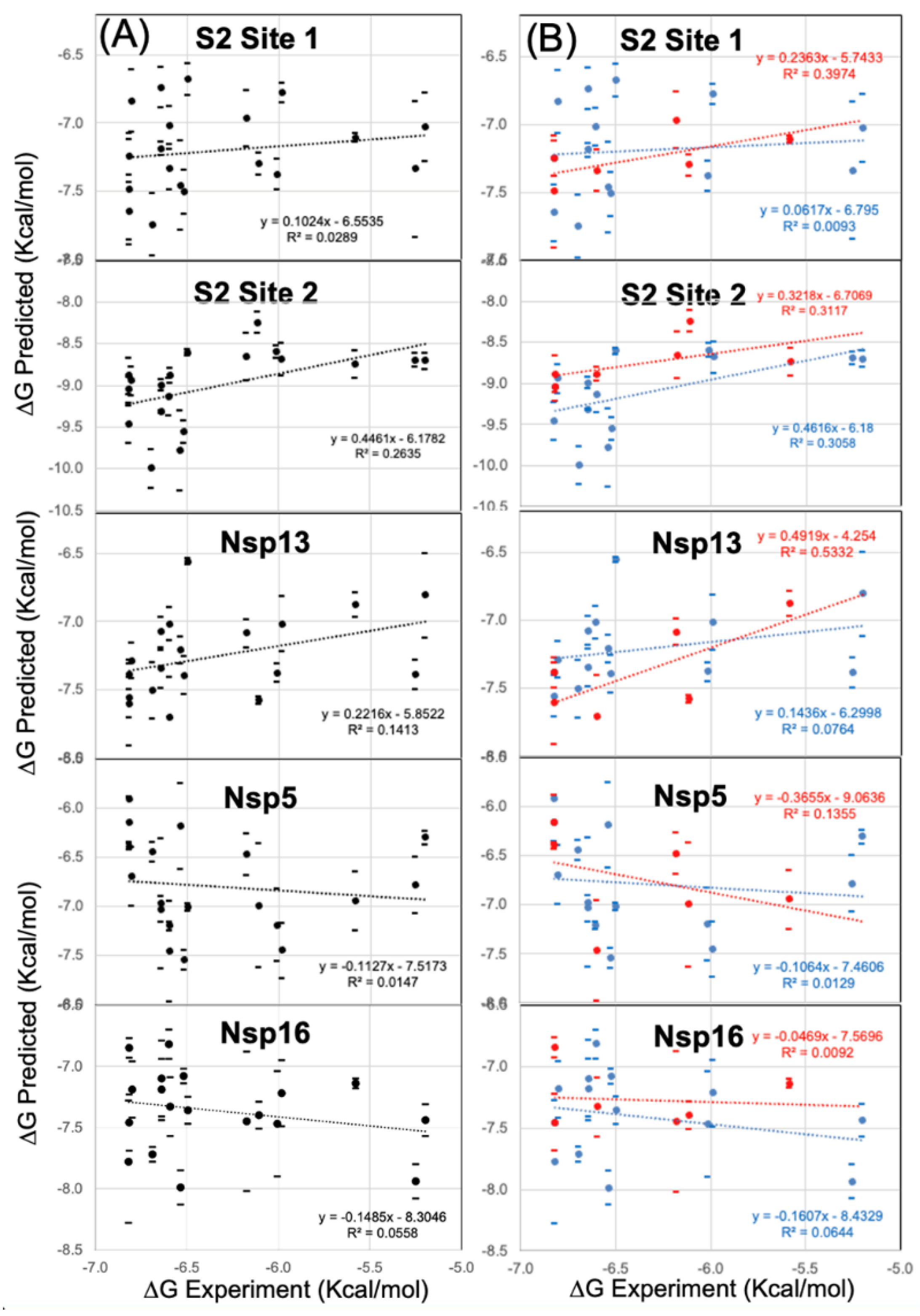
Figure 8.
Predicted ΔGbind values from Site 2 correlate with experimental SAR data and explain SAR substitutions for all three R groups. A series of 18 Clofazimine derivatives were well-modeled binding to Site 2 as two separate groups of compound series where the first series (A) shown in blue (6d, 6e, 7a, 7b, 7c, 7d, 7e, 7f, 7g, 7i, 7k, 7m, 7o) with correlation (R2=0.306) and the second series (B) shown in red (15a, 15b, 15f, 15b, 15h) had a slightly greater correlation (R2=0.311) with experimental SAR data. (C) A diagram to illustrate how the series modeled at Site 2 rationalizes R group substitutions at R (purple), R1 (green) and R2 (cyan).
Figure 8.
Predicted ΔGbind values from Site 2 correlate with experimental SAR data and explain SAR substitutions for all three R groups. A series of 18 Clofazimine derivatives were well-modeled binding to Site 2 as two separate groups of compound series where the first series (A) shown in blue (6d, 6e, 7a, 7b, 7c, 7d, 7e, 7f, 7g, 7i, 7k, 7m, 7o) with correlation (R2=0.306) and the second series (B) shown in red (15a, 15b, 15f, 15b, 15h) had a slightly greater correlation (R2=0.311) with experimental SAR data. (C) A diagram to illustrate how the series modeled at Site 2 rationalizes R group substitutions at R (purple), R1 (green) and R2 (cyan).
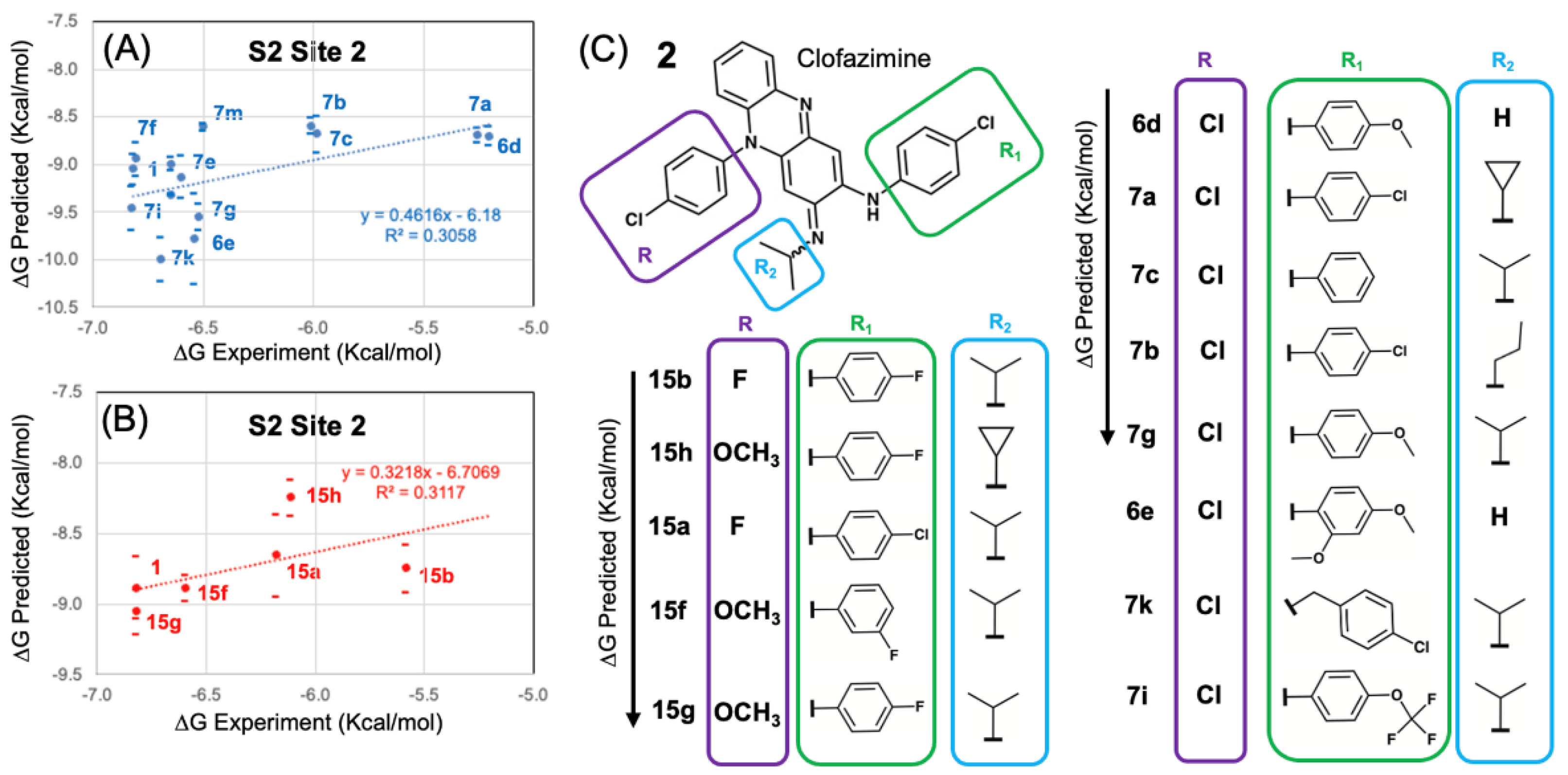
Figure 9.
The predicted Site 2 is consistent with available structural information and may rationalize broad-spectrum activity of Clofazimine 2 for MERS and other coronaviruses. (A) Predicted binding site for Clofazimine is shown illustrating the key binding site residues (W886, Q1036, K1038, V1040, and Y1047) shown below in the structure-sequence alignment. (B) A ribbon diagram is shown from a structure-sequence alignment between SARS-CoV-2 (6vxx.pdb) and MERS (8sak.pdb). (C) When docked at Site 2, the VFI tripeptide shows pharmacophore overlap with Clofazimine 2 and the three major hydrophobic peptide side chain pharmacophores. The surface model of the binding site is shown in (D) for SARS-CoV-2 showing a highly complementary binding surface for bound Clofazimine 2 in blue, where (E) shows that the binding surface shown in red is very similar in MERS with few atom clashes with the Clofazimine 2 binding mode. (F) Shows a sequence alignment derived from structure-sequence alignments with experimentally determined structures of the Spike protein from 15 different coronavirus strains. The sequence conservation of the SARS-CoV-2 residues that form the binding site (W886, A890, Q1036, K1038, V1040, Y1047, and R1107) are highlighted, where Q1036 is the most conserved of these residues.
Figure 9.
The predicted Site 2 is consistent with available structural information and may rationalize broad-spectrum activity of Clofazimine 2 for MERS and other coronaviruses. (A) Predicted binding site for Clofazimine is shown illustrating the key binding site residues (W886, Q1036, K1038, V1040, and Y1047) shown below in the structure-sequence alignment. (B) A ribbon diagram is shown from a structure-sequence alignment between SARS-CoV-2 (6vxx.pdb) and MERS (8sak.pdb). (C) When docked at Site 2, the VFI tripeptide shows pharmacophore overlap with Clofazimine 2 and the three major hydrophobic peptide side chain pharmacophores. The surface model of the binding site is shown in (D) for SARS-CoV-2 showing a highly complementary binding surface for bound Clofazimine 2 in blue, where (E) shows that the binding surface shown in red is very similar in MERS with few atom clashes with the Clofazimine 2 binding mode. (F) Shows a sequence alignment derived from structure-sequence alignments with experimentally determined structures of the Spike protein from 15 different coronavirus strains. The sequence conservation of the SARS-CoV-2 residues that form the binding site (W886, A890, Q1036, K1038, V1040, Y1047, and R1107) are highlighted, where Q1036 is the most conserved of these residues.
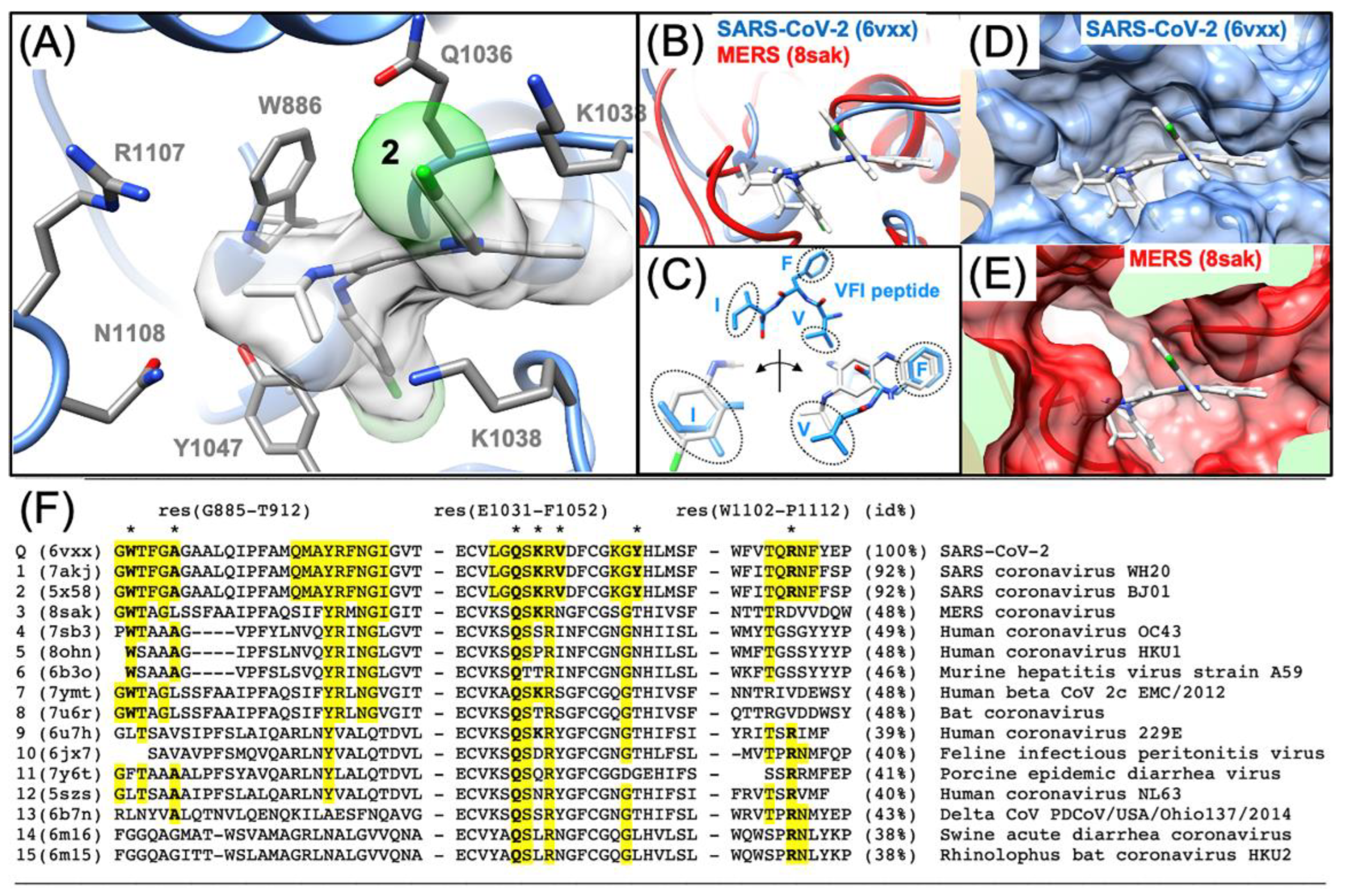
Figure 10.
A possible mechanism of action for binding at Site 2 is to stabilize the prefusion conformation and prevent conformational changes required for fusion. (A) Ribbon diagrams of the experimentally determined structures of the prefusion conformation and the superimposed post fusion structure (6xra.pdb) of S2. The proposed binding site for Clofazimine 2 is shown highlighted with a magenta molecular surface. To visualize local conformational changes, four residue segments (943-1034), (1035-1070), (1078-1120) and (1121-1141) are shown as green, blue, cyan and red respectively. (B) A zoom-in molecular surface diagram showing the superimposed structure of the prefusion conformation in medium blue showing complementary molecular surface and interactions where in the post fusion structure, the magenta atoms and surface of 2 clash with the teal and yellow molecular surface that has undergone local hydrophobic collapse during the conformational transition.
Figure 10.
A possible mechanism of action for binding at Site 2 is to stabilize the prefusion conformation and prevent conformational changes required for fusion. (A) Ribbon diagrams of the experimentally determined structures of the prefusion conformation and the superimposed post fusion structure (6xra.pdb) of S2. The proposed binding site for Clofazimine 2 is shown highlighted with a magenta molecular surface. To visualize local conformational changes, four residue segments (943-1034), (1035-1070), (1078-1120) and (1121-1141) are shown as green, blue, cyan and red respectively. (B) A zoom-in molecular surface diagram showing the superimposed structure of the prefusion conformation in medium blue showing complementary molecular surface and interactions where in the post fusion structure, the magenta atoms and surface of 2 clash with the teal and yellow molecular surface that has undergone local hydrophobic collapse during the conformational transition.
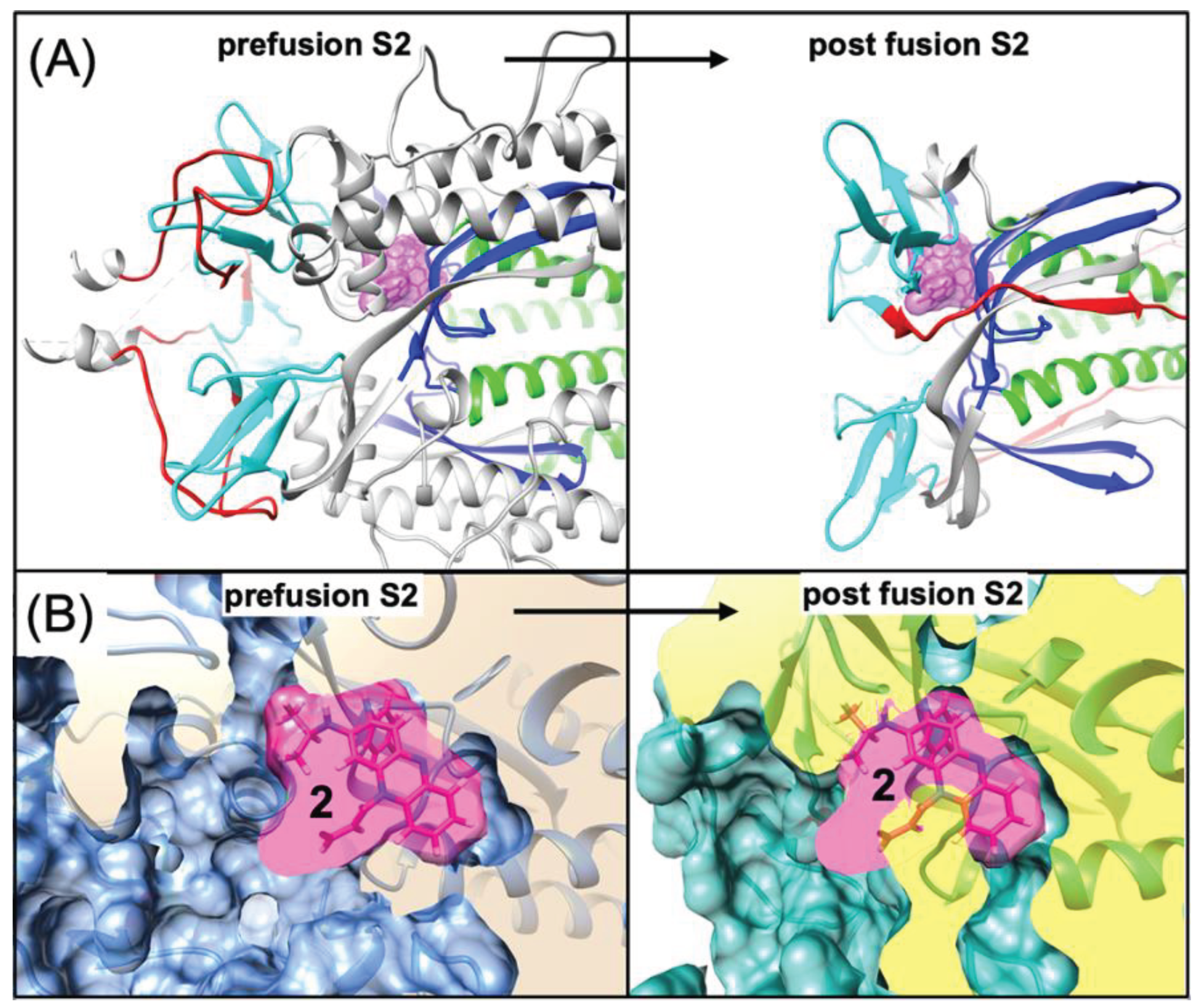

Table 1.
Preliminary SPR binding data for binding to Spike and to the S2 segment. The best representative binding data from round 1 are shown.
Table 1.
Preliminary SPR binding data for binding to Spike and to the S2 segment. The best representative binding data from round 1 are shown.
| Spike | Spike | S2 | S2 | |
| cmp | Kd (mM) |
Affinity Chi² (RU²) | Kd (mM) |
Affinity Chi² (RU²) |
| 1 | 7.44 | 1.04E-02 | 5.9 | 4.10E-03 |
| 1b | N/A | N/A | 31.2 | 4.06E-03 |
| 1d | 10 | 2.47E-02 | 27 | 2.35E-02 |
| 2 | 2.9 | 9.46E-03 | 3.9 | 4.93E-02 |
Table 2.
SPR binding data duplicates for binding to the S2 segment. The best representative binding data from round 2 are shown with the calculated standard deviation for duplicates.
Table 2.
SPR binding data duplicates for binding to the S2 segment. The best representative binding data from round 2 are shown with the calculated standard deviation for duplicates.
| Spike | Spike | S2 | S2 | |
| cmp | Kd (mM) |
Affinity Chi² (RU²) | Kd (mM) |
Affinity Chi² (RU²) |
| 1 | N/A | N/A | 2.1 ± 0.2 | 1.43E-03 |
| 1c | 40.4 ± 1.5 | 6.10E-03 | 11.4 ± 1.3 | 9.86E-03 |
| 2 | 4.6 ± 1.2 | 5.34E-03 | 6.5 ± 0.3 | 1.30E-02 |
| 3 | 4.1 | 2.70E-03 | 3.1 ± 1.4 | 1.69E-02 |
| 4 | 73.8 ± 8.3 | 2.69E-03 | 10.0 ± 2.8 | 1.04E-02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



