
Submitted:
26 February 2024
Posted:
04 March 2024
You are already at the latest version
Abstract
Wide bandgap semiconductors-based photocatalysts are usually limited by their low solar energy conversion efficiency due to their limited absorption solar wavelength, surface’s fast recombination of photoelectron–hole pairs and low charge-carrier mobility. Here we report a new stepwise solution synthesis for making a new photocatalytic core/shell of titanate nanowire/reduced graphene oxide shell (or titanate/rGO) 1D-nanocomposite. The new core/shell nanocomposite maximized the specific surface area, largely reduced the charge transfer resistance and reaction energy barrier, and significantly improved the absorption of the visible light. The core/shell nanocomposites’ large on/off current ratio and rapid photo-responses boosted the photocurrent by 30.0%, the photocatalysis rate by 50.0%, and the specific surface area by 16.4%, when comparing with the pure titanate nanowire core. Our numerical simulations support the effective charge separation on the new core-shell nanostructure, which can help further advance the new photocatalysis.
Keywords:
Core-shell nanostructure
; graphene
; Titanate nanowire
; photocatalysis
; wide bandgap
1. Introduction
Developing highly efficient artificial photoconversion systems over a photocatalyst with a wide wavelength range of light is very challenging.[1] Titanate and titanate-based wide bandgap semiconductor photocatalysts have been investigated for decades due to their good abundance, biocompatibility, cost effectiveness, photoelectron separation, and long-term thermal stability.[2,3,4,5,6,7] The photocatalytic performance of titanate is strongly dependent on its morphology and structure, as the good nanoscale titanate photocatalysts usually show the superior functional properties in the form of nanorods,[8,9] nanoparticles (NPs),[10,11] and hollow spheres[12]. Specifically, 1-dimensional (1D) titanate nanostructures, such as the photocatalytic nanowires (NWs), attract lots of attention because of their large specific surface area and the ease for the photogenerated charge carriers to transfer along their longitudinal surface.[13,14,15,16,17] However, the large bandgap of titanate (3.0 ~ 3.2 eV) limits the usability of the wide wavelengths of solar energy.[18,19,20,21] This limitation is further exacerbated by the fast recombination of photoelectron-hole pairs and the slow transfer of charge carriers in the bulk and on the surface.[10] A promising solution has been found in the use of 2D graphene on titanate,[22,23] which can significantly increase the photocatalysis activity owing to its large surface area, excellent mobility of charge carriers, and controllable bandgap. The resultant narrow bandgaps, short transport distances and accelerated charge mobility have improved the photocatalytic performance of the graphene-on- titanate.[24,25] This improvement can help optimize the nanosynthesis for improving the nanocomplex architecture with better optoelectric and photocatalytic properties.
Despite these advancements, the optimal integration of graphene with titanate nanostructures remains a sophisticated endeavor, demanding meticulous control over the composite’s nanoarchitectural design and synthesis. The interfacial interaction between graphene and titanate plays a inportant role in determining the efficiency of charge transfer mechanisms, which are essential for enhancing the photocatalytic activity.[26] By finely tuning the surface properties and the morphology of the titanate nanostructures, it is possible to achieve a more effective spatial separation of photo-generated electron-hole pairs, thus reducing recombination rates. The selection of graphene oxide (GO) or reduced graphene oxide (rGO) as a component in the composite influences the electronic properties of the nanocomposite, including band alignment and charge carrier dynamics.[27,28] Through strategic engineering of these nanocomposites, researchers aim to harness the full potential of visible light absorption, thereby overcoming the inherent limitations associated with the wide bandgap of titanate. This approach not only aims to improve the photocatalytic efficiency under a broader spectrum of light but also contributes to the sustainability of the photocatalytic process by utilizing solar energy more effectively.
In this study, a new core/shell nanostructure of titanate NW/GO and a titanate NW/ rGO were made by design as photoelectrodes and photocatalysts, respectively. The new NWs were successfully synthesized by hydrothermal methods and showed a large surface area, high light absorption (UV-Vis-NIR), reduced charge transfer impedance, and decreased reaction energy barrier. The superior optoelectronic and photocatalytic performance of the nanocomposites was fully demonstrated by the photocurrent and degradation of methylene blue (MB, a blue-colored drug molecule) under ultraviolet (UV) and visible (vis) light, with the rGO-titanate NWs exhibiting the highest photocurrent, photo-response, and degradation efficiency, comparing with the titanate and GO-titanate NWs. Numerical analysis revealed that the unique structure and chemical assembly were responsible for the active photoresponse and photocatalysis. The findings of this study provide new insights into the development of highly efficient and stable photocatalysts for environmental cleaning.
2. Results and Discussion
The process of synthesizing Titanate/GO NWs and Titanate/rGO NWs is clearly illustrated in Schematic 1. Initially, Titanate/GO NWs with a core-shell structure were formed hydrothermally by suspending GO and titania powders in NaOH aqueous solution, then reduced to rGO-Titanate NWs at elevated temperatures in the Ar gas environment, with approximately 35-40 μm in length and 60 nm in diameter with a smooth surface and uniform distribution, as shown in Figure 1 (a) and Figure S2. The energy dispersive X-ray (EDX) spectrum in Figure S2 (a2) confirmed the elemental components of Ti and O, with a small amount of background C present on the specimen, and that of Si from the substrate that supports the specimen. The annealing method in an inert gas atmosphere has been proven in the literature to be a simple and effective way to reduce GO to rGO. Moreover, the SAED pattern in Figure S3 revealed the mixed diffraction patterns of the crystalline titanate. The X-ray diffraction (XRD) peaks of titanate NWs (Figure 1 (b)) at 9.8°, 11.2°, 24.4°, and 29.7° can be assigned to (001), (200), (110), and (003), respectively (JCPDS card No.: 47-0561).[29] Evidently, a broad peak at 18 - 30° can be attributed to the amorphous phase of rGO, indicating that the GO-titanate NWs have been successfully and effectively reduced to rGO-Titanate NWs by the calcination in the inert gas atmosphere.[30] As shown in Figure 1 (c) and (d), the rGO shell thickness of rGO-titanate NWs was reduced from 10 nm to approximately 6 nm, and the morphology was unchanged during the calcination.
Schematic 1.
The process to make rGO-Titanate nanowires.
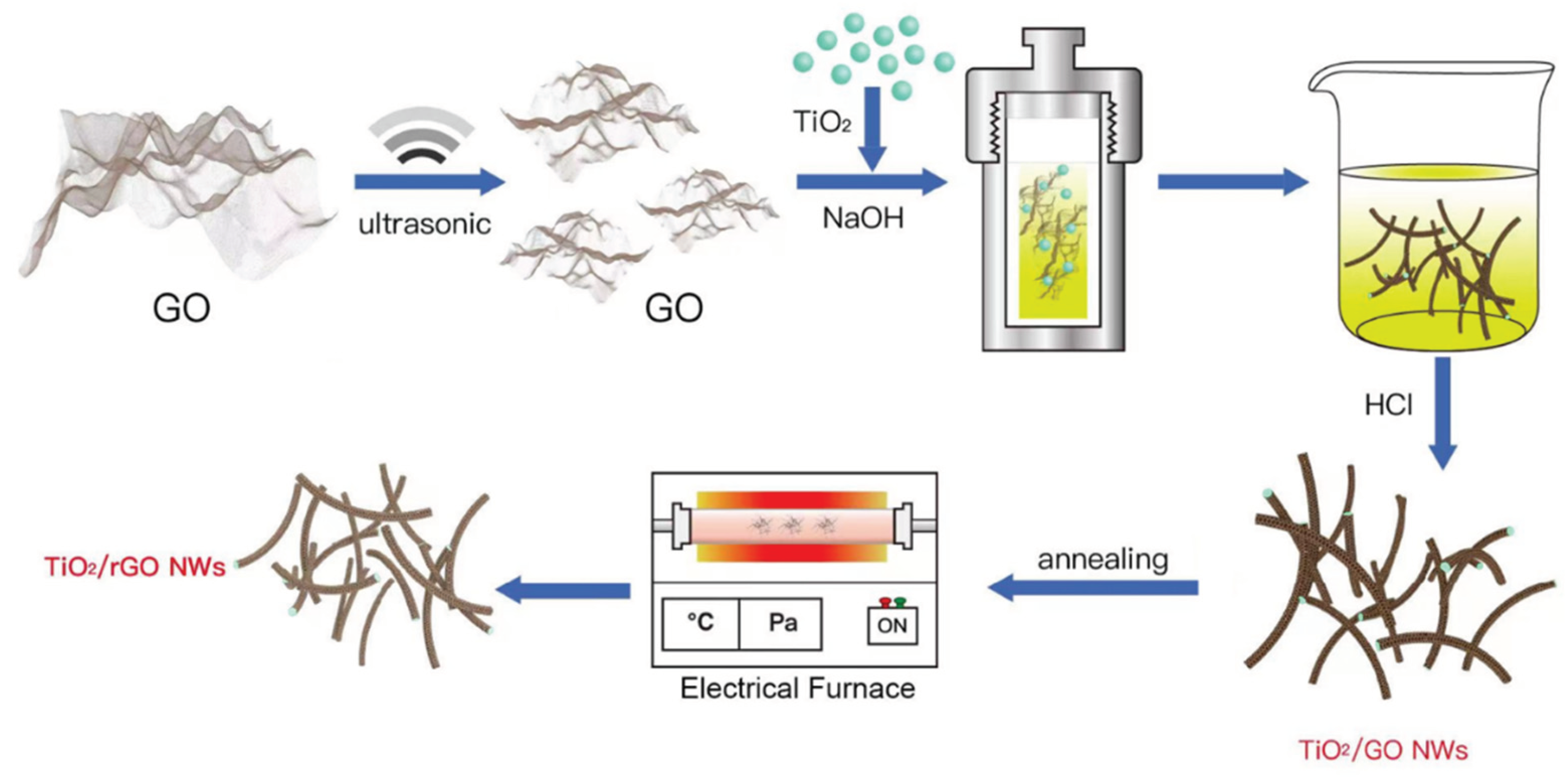
Figure 1.
(a) SEM images of titanate, GO-titanate, and rGO-titanate nanowires. (b) XRD of titanate, GO-titanate, and rGO-titanate nanowires. (c) and (d) the TEM images for GO-titanate and rGO-Titanate nanowires.
Figure 1.
(a) SEM images of titanate, GO-titanate, and rGO-titanate nanowires. (b) XRD of titanate, GO-titanate, and rGO-titanate nanowires. (c) and (d) the TEM images for GO-titanate and rGO-Titanate nanowires.

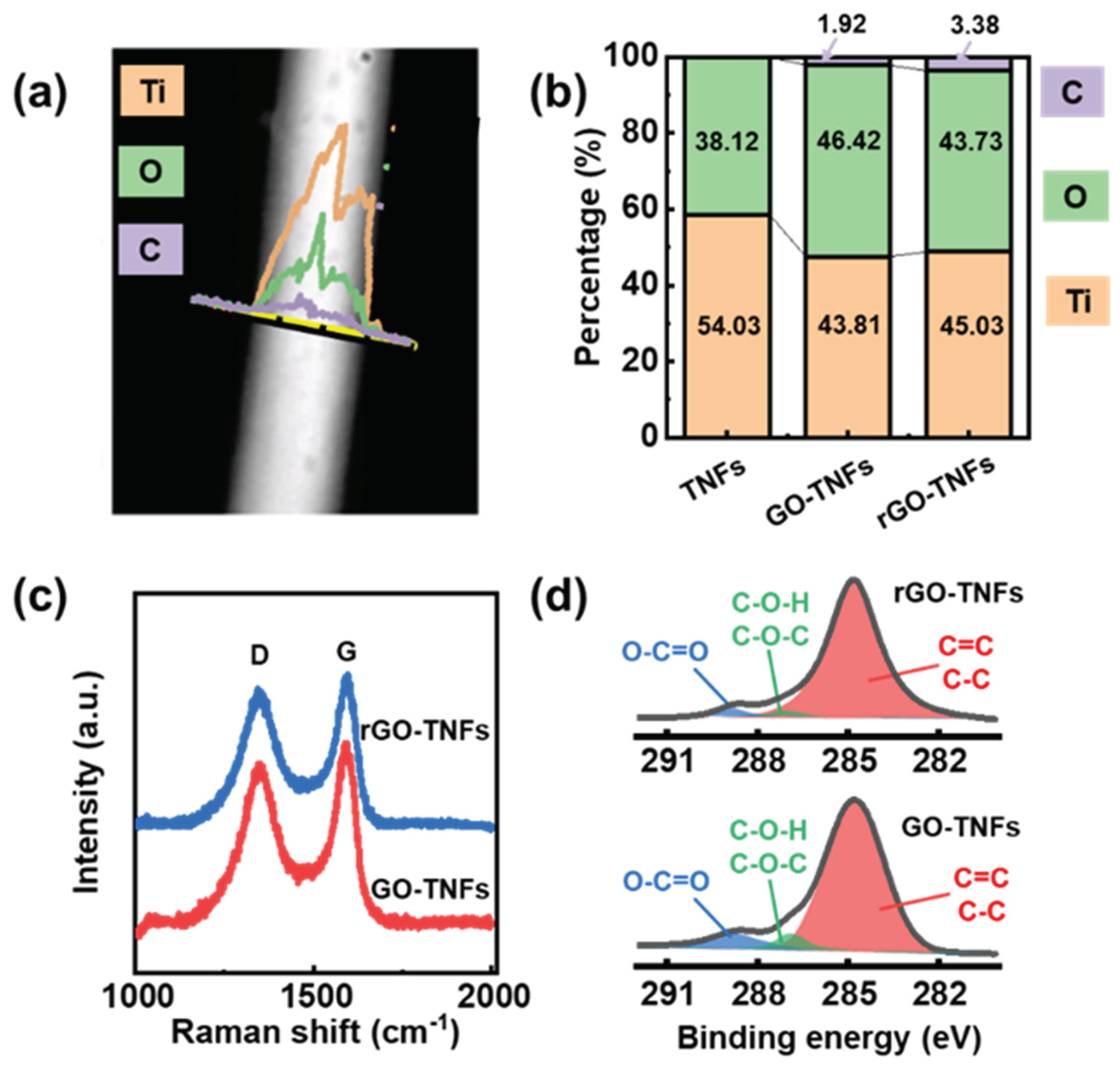
On this basis, the core-shell NWs were analyzed using the EDX, FTIR spectroscopy, XPS and microRaman techniques to further confirm that the GO on titanate NWs was successfully reduced to rGO. From the EDX elemental analysis data in Figure 2 (a) and (b), the O element ratio decreased from 46.42% (for the GO-Titanate NWs) to 43.73% (for the rGO-Titanate NWs), while the C ratio increased from 1.92% to 3.38%. Consistently, the FTIR peaks in Figure S4 displayed the IR spectra of the samples. The broad and strong absorption band at 400 - 700 (cm−1) belonged to the stretching vibration of Ti-O-Ti and Ti-O. The bands at 930 and 1250 (cm−1) were attributed to the C-O and C-OH groups, respectively, while the band at 1720 - 1750 (cm−1) was from the carbonyl groups on the GO and rGO edge. The peak at 1630 (cm−1) was ascribed to the C=O bonds and deformation vibrations of the absorbed water. The peak at 2900 cm−1 corresponded to the stretching and bending vibrations of hydroxyl group in the basal plane and the edge, and the broad band from 3300 to 3500 (cm−1) was from the surface hydroxyl group or the absorbed water molecules. On the TiO2/rGO NWs, these modes from the oxygen-containing functional groups were mostly diminished, which supports the reduction of GO to rGO, which is in line with others’ reports[31]. The microRaman spectra of the samples are presented in Figure S5. The peaks at 142, 194, 400, 517, and 645 cm-1 correspond to the Raman modes of Eg(1), Eg(2), B1g(1), A1g+B1g(2), and Eg(3) of the titanate, respectively. Additionally, two broad bands were observed at approximately 1348 cm-1 and 1592 cm-1 (Figure 2 (c)), which were attributed to the disordered amorphous carbon attributed to the disordered amorphous (the D band) and graphitic sp2 carbon (the G band). The ID/IG ratio was approximately 0.9 (with ID and IG correspond to the intensity of the D band and G band, respectively), which confirmed the presence of the GO or rGO layer on TiO2 NWs. The low content of GO or rGO in the hybrid composites resulted in no significant shift of the D band and G band. In parallel, the XPS was employed to analyze the element chemical state. The spectra demonstrated the presence of O, Ti, and C in the Titanate NWs, Titanate/GO NWs, and Titanate/rGO NWs (Figure S6). The O 1s spectra exhibited two distinct peaks. Specifically, for Titanate NWs, the two peaks at 531.2 eV and 529.9 eV can be attributed to the C-O and Ti-O groups, respectively. The incorporation of GO and rGO shells results in a blue shift of the Ti-O band at 530.1 eV, indicating the interaction and binding between the GO (rGO)-shell and the Titanate-core. In the C 1s spectra (Figure 2 (d)), there are three distinct peaks: the 284.7 (eV) from the C-C and C=C, the 286.8 (eV) from C-O-C and C-O-H, and the 288.6 (eV) from O-C=O and C-O-Ti, respectively. Apparently, the C-O-C, C-O-H, O-C=O, and C-O-Ti peaks of the rGO-titanate NWs were reduced significantly compared with those of the GO-titanate NWs. Based on the EDX, FTIR and XPS data, the GO was largely reduced to the rGO during the calcination, and titanate nanowires were well wrapped with the GO or rGO, which agreed with the XRD and Raman analyses. Figure S6 (c) showed the Ti 2p spectra of all the specimens, indicating that the titanium elements in the samples were in the form of Ti4+ value, as evidenced by the energy difference between Ti 2p3/2 and Ti 2p1/2 peaks of 5.7 eV. Comparing Titanate NWs with Titanate/GO and Titanate/rGO NWs, the GO and rGO shells caused a blue shift of 0.3 eV and 0.1 eV, respectively for the two bands, implying chemical bonding between the core and the shell. Overall, the XPS data provided a new insight into the elemental composition and chemical state of the specimens, revealing the interaction and binding between the core and shell.
Figure 2.
(a) EDX spectra of rGO-titanate nanowires. (b) EDX element ratios of C, O, and Ti for titanate, GO-titanate, and rGO-titanate nanowires. (c) Raman spectroscopy of GO-titanate and rGO-titanate nanowires. (d) XPS spectroscopy of GO-titanate and rGO-titanate nanowires.
Figure 2.
(a) EDX spectra of rGO-titanate nanowires. (b) EDX element ratios of C, O, and Ti for titanate, GO-titanate, and rGO-titanate nanowires. (c) Raman spectroscopy of GO-titanate and rGO-titanate nanowires. (d) XPS spectroscopy of GO-titanate and rGO-titanate nanowires.
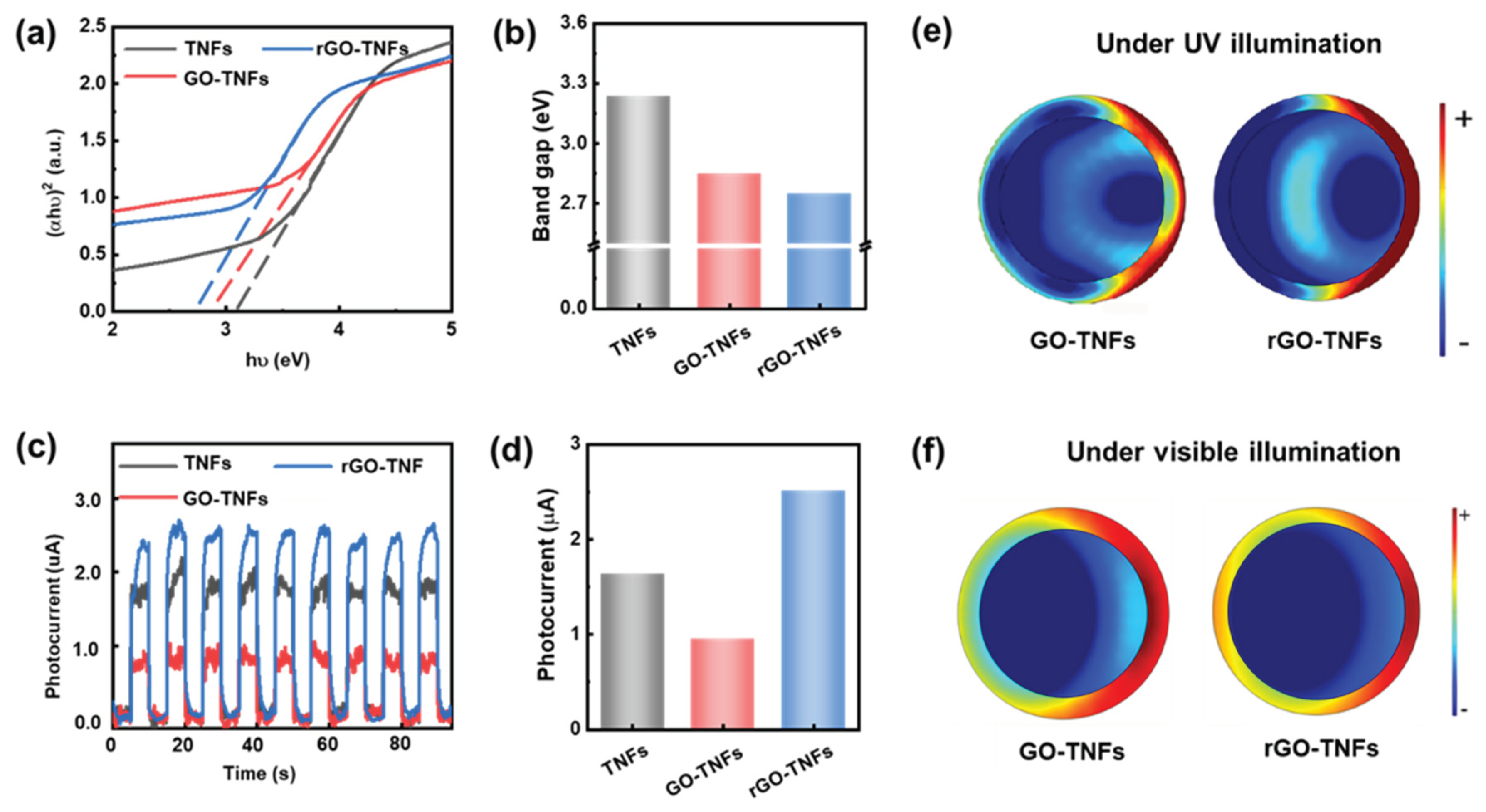
To further support the photocatalysis performance of NWs, the optoelectronic properties were characterized by means of the UV-Vis absorption and photocurrent response experiments, which are supported using the finite element analysis-based theoretical simulation. The theoretical analysis indicates that the oxidation level of GO is related to the GO bandgap nature.[17] GO is a non-crystalline material with different oxidation-containing functional groups, so a sharp adsorption edge with a precise bandgap energy cannot be observed in the conversion plots. Therein, the Tauc plots derived from the UV-vis absorption spectra were employed to determine the NW bandgap (as depicted in Figure 3 (a) and (b)), showing the rGO-titanate NW’s bandgap was significantly reduced to 2.75 eV (vs. titanate’s 3.23 eV and GO-titanate 2.98 eV), which further expanded the light absorption range of wavelengths to improve the photocatalytic performance. As shown by the UV-vis absorption spectra of the specimens in Figure S7, where the absorption in the ultraviolet region for all the samples confirmed the band absorption of anatase TiO2. Moreover, the rGO flakes, Titanate, and Titanate/rGO core/shell were further characterized by hyper spectrometer imaging (HIS) in the visible and near-IR (NIR) wavelengths from 400 nm to 1000 nm (Figure S8), revealing the absorption of Titanate/rGO to be significantly greater more than that of Titanate NWs. This new HIS data suggest that the core/shell nanocomposite of Titanate/rGO can help better utilize the diffusive solar energy in the photocatalysis. The increased sunlight absorption might generate more electron-hole pairs, thereby leading to improved photoresponse and photocatalytic performance.
The photocurrent response of the nanocomposites under intermittent UV light was shown in Figure 3 (c) and (d), where all the photoanodes exhibit a rapid photoresponse and as the photocurrent (which is almost negligible in the dark) increases instantly to its maximum within 0.1 – 0.2 s upon exposure to light. The photocurrent of rGO-titanate is about 4 and 1.3 times greater than that for GO-titanate and titanate NWs, respectively. The reduction in the photocurrent for Titanate/GO NWs is attributed to the insulating characteristics of the GO layer, whereas the increase in the photocurrent for Titanate/rGO NWs is apparently due to the conductive properties and suppression of charge recombination by the rGO layer. These findings are consistent with the bandgap variation in Figure 3(b). To further understand the active photoresponse and photocatalytic behavior, the charge distribution of the NWs under the light irradiation was studied by the finite element analysis, providing the charge density at the cross section of the NWs under the incident light (254 and 420 nm, respectively), as illustrated in Figure 3 (e) and (f). Intuitively, the negative charges should be confined to the naked cations on the titanate core surface, allowing the positive charges to be dissipated along the rGO shell more quickly than that along the GO shell. This could explain why the photocurrent charges’ separation on the rGO-titanate being better than that on the GO coated NWs, which boosted the photocatalysis performance of the rGO-titanate.
Figure 3.
(a) Tauc plots of the UV-Vis absorbance spectra for titanate, GO-titanate, and rGO-titanate nanowires. (b) The bandgap of titanate, GO-titanate, and rGO-titanate nanowires from Tauc plot. (c) and (d) photocurrent responses curves and average values. (e) and (f) Charge distribution of the nanowires as induced by the UV and Visible light.
Figure 3.
(a) Tauc plots of the UV-Vis absorbance spectra for titanate, GO-titanate, and rGO-titanate nanowires. (b) The bandgap of titanate, GO-titanate, and rGO-titanate nanowires from Tauc plot. (c) and (d) photocurrent responses curves and average values. (e) and (f) Charge distribution of the nanowires as induced by the UV and Visible light.
To enhance our understanding of the charge transfer kinetics and physiochemical properties of the NWs, electrochemical methods were employed. The Nyquist plots of the impedance spectra in Figure 4 (a) were fitted well with an equivalent circuit to obtain the bulk resistances and charge transfer resistances of the NWs. The bulk resistances of GO-titanate and rGO-titanate NWs (Figure 4 (b)) were significantly decreased compared with the pure titanate NWs, because of the conductivity improvement from the reduction of GO. As shown in Figure 4 (c), there was a progressive decrease in the charge transfer resistances from the titanate to the GO-titanate and to the rGO-titanate NWs, indicating the rGO coating significantly enhance the photocatalysis performance by accelerating the charge transfer rate across the interface between the solid nanowire surface and liquid solutions. Table 1 presents the pore size, volume, and surface area obtained from N2 adsorption–desorption isotherms of the specimens depicted in Figure S9. Furthermore, Figure S9 showed that Titanate/GO NWs had the largest specific surface area of 15.32 m2/g, which improves 23% over Titanate NWs and was attributed to the high specific surface area of the GO layer. However, the annealing decreased the specific surface area, presumably due to the collapse of the three-dimensional porous structure. From a chemical perspective, the weakened inter-bonding of the rGO flakes (much weaker than that of the GO flakes) could minimize the 3D-stacking of the NW/rGO type of 1D-nanocomposites, which matched their pore size and pore volume. Nevertheless, the surface area of Titanate/rGO NWs (14.55 m2/g) was still larger than that of pure Titanate NWs (12.50 m2/g), which could provide more active sites for dye molecule absorption and photocatalytic degradation. Meanwhile, the rGO shell on the NWs provides a large surface area with lots of defects and dangling bonds (Figure 4e) to better the dye molecules physical adsorption and kinetic mass-transfer[32] and efficiently boosting the photocatalysis kinetics. Evidently, the energy barrier of charge transfer on the rGO-titanate NWs was significantly decreased by reducing the reversible potential and increasing the exchanged current density, with respect to that on the pure titanate and the GO-titanate NWs, as shown in the Figure 4 (d) and (f), which further helped the charge transfer process and improved the photocatalysis efficiency.
Figure 4.
(a) Nyquist plots of the impedance spectra for titanate, GO-titanate, and rGO-titanate nanowires and (b) the values of bulk resistance and (c) the charge transfer resistance. (d) Tafel plot of titanate, GO-titanate, and rGO-titanate nanowires and the values of (e) Diffusion coefficient and (f) exchanged current density.
Figure 4.
(a) Nyquist plots of the impedance spectra for titanate, GO-titanate, and rGO-titanate nanowires and (b) the values of bulk resistance and (c) the charge transfer resistance. (d) Tafel plot of titanate, GO-titanate, and rGO-titanate nanowires and the values of (e) Diffusion coefficient and (f) exchanged current density.
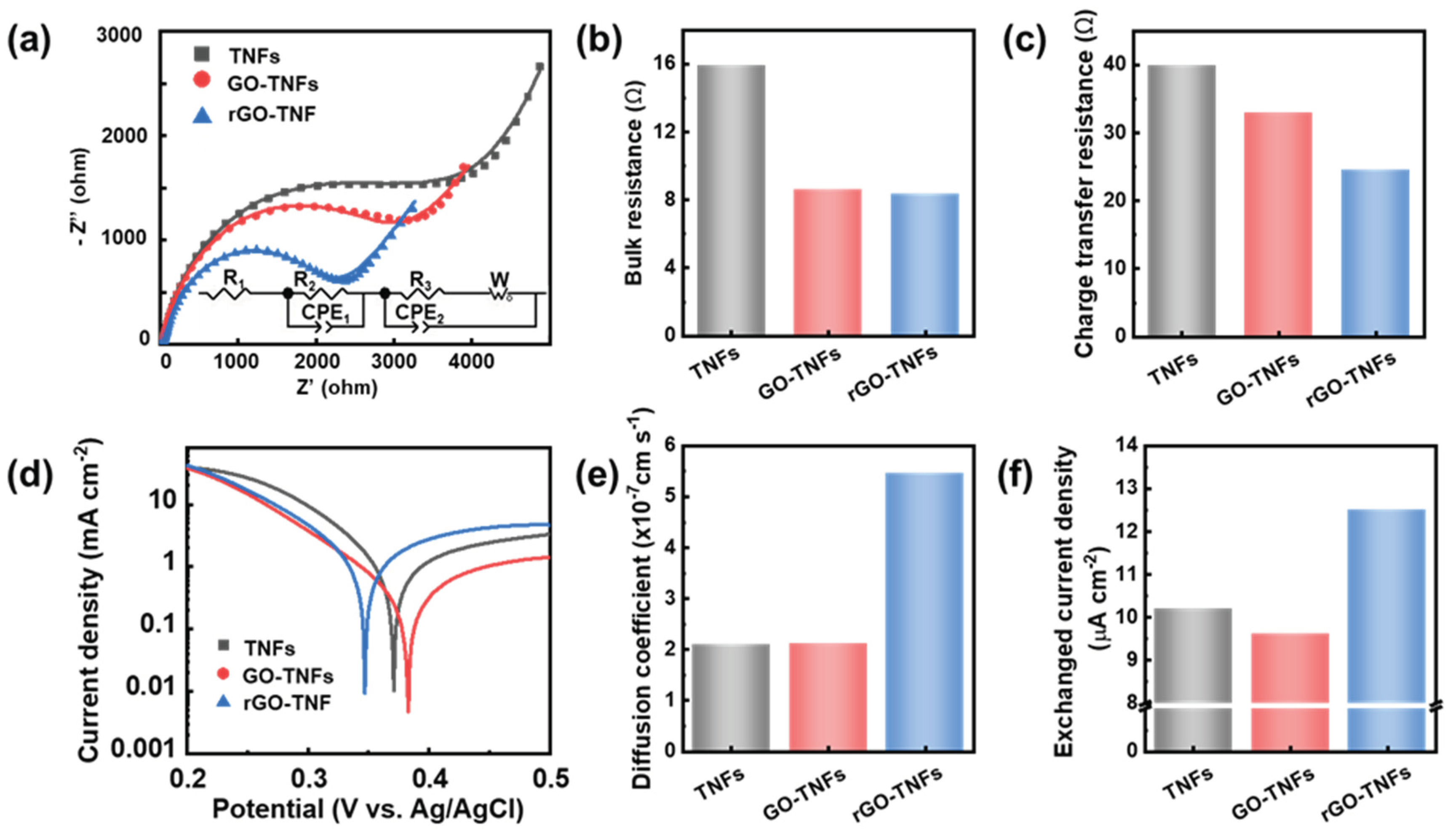

Table 1.
Structural parameters of the specimens as achieved from the N2 adsorption–desorption isotherm in Figure S9.
Table 1.
Structural parameters of the specimens as achieved from the N2 adsorption–desorption isotherm in Figure S9.
| TiO2 | TiO2/GO | TiO2/rGO | |
| Pore size (nm) | 11.84 | 10.95 | 11.27 |
| Pore volume (cm³/g) | 0.036982 | 0.041934 | 0.040996 |
| Surface area (m²/g) | 12.50 | 15.32 | 14.55 |
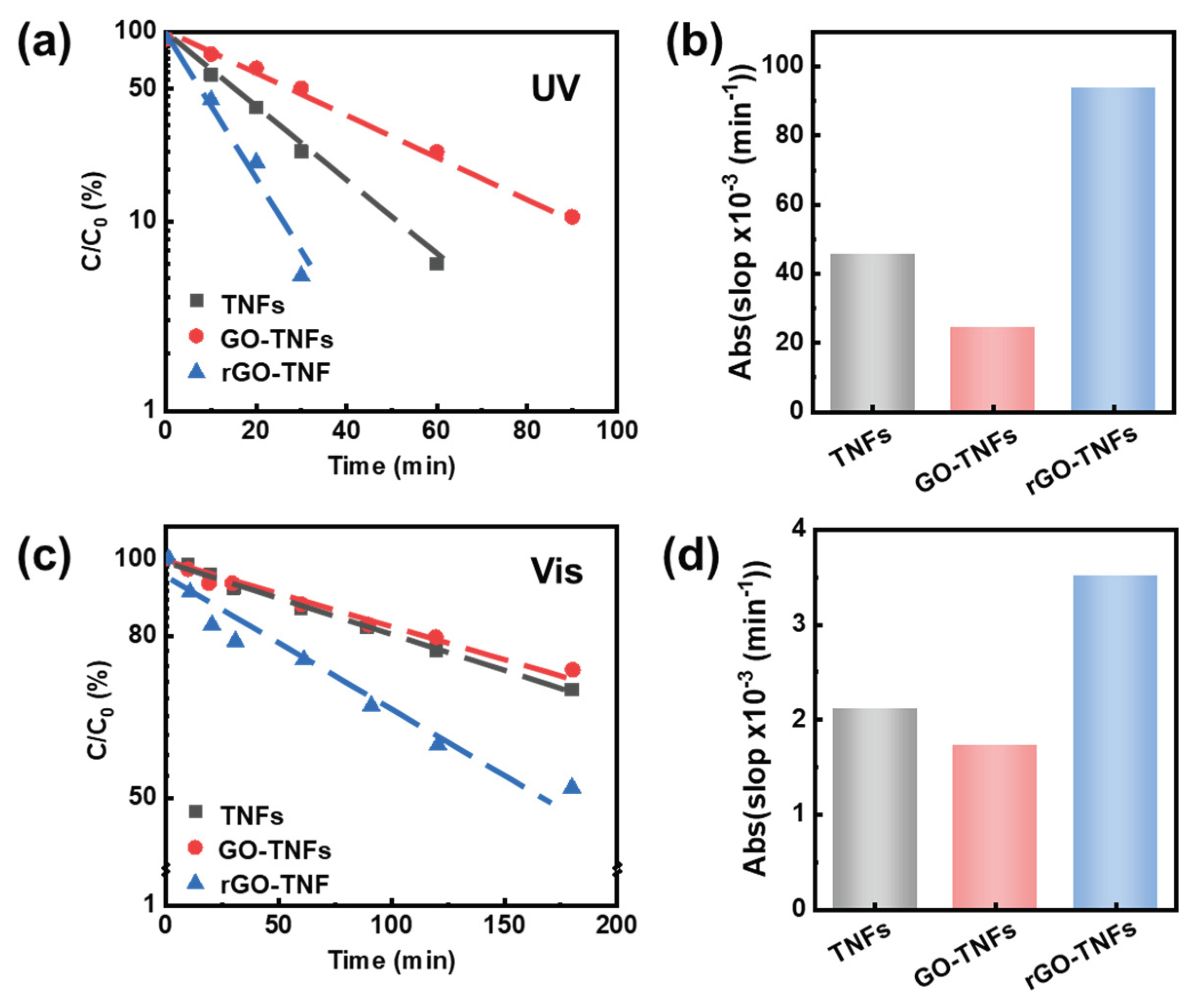
To assess photocatalytic activity of the specimens, the degradation of methylene blue (MB) dyes as catalyzed by the specimens under both visible and UV light, was carried out. The intensity of the absorption band of the MB aqueous solutions continuously decreases with increasing irradiation time (see Figure S10), indicating the photocatalytic ability of all the TiO2-based specimens. Figure 5 (a) presented the logarithmic percentage of the residual dye molecules were linearly fitted with the UV illumination time, indicating that the MB degradation process was the first-order kinetic process. The rGO-titanate core-shell exhibits the most effective photocatalytic performance (Figure 5(b)). Notably, compared with titanate NWs and GO-titanate NWs under visible illumination, the photocatalytic performance of rGO-titanate core-shell NW for MB degradation under UV light was significantly improved (Figure 5 (c) and (d)). This time the dye solution is completely bleached in ~ 30 min by the Titanate/rGO, 60 min by the Titanate NWs, and >90 min by the Titanate/GO. This confirms the best photocatalytic activity of the NW/rGO, by design.
Figure 5.
(a) First-order linear plot of C/C0 versus time of MB degradation with logarithmic y coordinate under UV illumination. (b) Absolute values of slops from first-order linear plot under UV light. (c) First-order linear plot of C/C0 versus time of MB degradation with logarithmic y coordinate under visible illumination. (d) Absolute values of slopes from first-order linear plot under visible light.
Figure 5.
(a) First-order linear plot of C/C0 versus time of MB degradation with logarithmic y coordinate under UV illumination. (b) Absolute values of slops from first-order linear plot under UV light. (c) First-order linear plot of C/C0 versus time of MB degradation with logarithmic y coordinate under visible illumination. (d) Absolute values of slopes from first-order linear plot under visible light.
Mechanistically, the novel structure and well-integrated surface chemistry and physics resulted in the high kinetic charge transportation, low surface energy barrier, and large surface area for the rGO-titanate NW to enhance the dye molecules sorption and mass transfer to boost the dye photocatalytic degradation (Figure. S11). Evidently, the tight bonding between the conductive rGO shell and wide bandgap titanate NW core has facilitated the photo-excited charges separation, docking and transport. The excited photoelectrons migrated to the graphene surface via the bonding between the rGO and titanate, letting the rGO act as an electron acceptor to effectively suppress the charge recombination. The photoelectrons could also be scavenged by O2 to produce reactive superoxide radicals (•O2−), while the holes can be trapped by H2O to produce hydroxyl radicals (•OH), docking on the core (to quench the quick combination), and react with (or decompose) the dye molecules, which should be verified using future experimental measurements in the follow-up work.
3. Materials and Methods
3.1. Materials
The chemical reagents used in this study include the graphene oxide (GO, 98%, Tanfeng Tech. Co. Ltd., Suzhou, China), titanium dioxide (TiO2 in rutile, 99.5%, Shanghai Keyan Industrial Co. Ltd., Shanghai, China), Nafion solution (5%, DuPont, USA), sodium hydroxide (NaOH, 96%, Xilong Chemical Co. Ltd., Guangdong, China) and methylene blue (MB, 98.5%, Xilong Chemical Co. Ltd., Guangdong, China). The distilled water was purified to show the resistivity higher than 18.0 MΩ.cm, by means of a Hi-tech laboratory water purification system. All reagents were directly used as received without further purification.
3.2. Nanosynthesis Process
The core-shell Titanate/GO NWs were made by a hydrothermal method at elevated temperature and pressure. Typically, 0.10 g GO power was exfoliated in 40 mL H2O by ultrasonic treatment for 1 h, then 1.0 g TiO2 powder and 16.0 g NaOH were added and stirred for 2 h, then sealed in a Teflon-lined autoclave and heated at 200-250 ℃ for 2-4 days,[29,33,34,35,36] forming the new core-shell Titanate/GO NWs. For comparison, the titanate NWs were synthesized using the same process in the absence of GO. The achieved product was cleaned by vacuum filtration in HCl solution (0.1 M) and DI water 7-8 times. The as-synthesized titanate/GO NWs and the titanate NWs were thermally treated at 680-750 ℃ for 1-3 hours with Ar gas as protection atmosphere to, respectively, crystallize the titanate NWs and to reduce the GO into rGO through the pyrolysis.
3.3. Characterizations
Morphology, structure and chemical composition of the as-synthesized samples were respectively characterized by means of the ZEISS Sigma scanning electron microscope (SEM) and Tecnai F30 high resolution transmission electron microscope (HRTEM), both equipped with the energy dispersive spectroscopes (EDS), the X’Pert PRO X-ray diffractometer (XRD) with Cu Ka radiation (λ = 1.54056 Å), and a PHI Quantum-2000 X-ray photoelectron spectroscope (XPS) with a nonmonochromatized Al Kα X-ray (1486.6 eV) as the excitation source and C 1s as the reference line. Then, the FT-IR spectra in the range of 400 ~ 4000 cm-1 were collected on the Nicolet iS50 FT-IR spectrophotometer using pellets of the samples being mixed in KBr. The resonant microRaman scattering spectra were collected in back-scattering geometry on a Renishaw LabRAM-HR Raman microscope using a 325 nm (He-Cd) laser excitation source. The diffuse reflection and absorption spectra were tested on the UV-Vis-NIR spectrophotometer (Agilent Carry-5000). The surface area and pore size distribution of the samples were measured using the surface area analyzer (Micromeritics ASAP2420), and the pore-size distribution (PSD) was obtained from the density functional theory (DFT) model based on the N2 sorption isotherm.
3.4. Photoelectrochemical Measurement
Photoelectrochemical properties, including photoresponse and AC impedance, were conducted on an electrochemical workstation (Chenhua CHI660E, China) using a three-electrode cell. Specifically, the prepared nanomaterials coated on FTO electrode as a working electrode, a Pt wire worked as a counter electrode, and Ag/AgCl was used as a reference electrode. For the transient photocurrent experiments, a low-power UV-LED (3 W, 365-370 nm, 4 mW/cm2) was used as a light source, and a 0.1 M Na2SO4 aqueous solution was employed as the electrolyte. Frequency signals of the UV light were modulated by a function waveform generator (AFG-2225, Taiwan) with 0.1 Hz square wave at a fixed potential of 1 V. For the working electrodes, the prepared samples (5 and 20 g/L in 1% Nafion solution) were drop-cast smoothly on a cleaned FTO (1×2 cm2) glass plate surface for about 0.05 mL/cm2 and then dried at 60 ℃ for 30 min. The AC impedance spectra were measured in a 0.5 M Na2SO4 aqueous solution in a frequency range of 0.01 ~ 105 Hz with a bias of ~ -0.4 V. For cyclic voltammetry (CV), the working electrode was glassy carbon electrode coated with 4μg nanofibers, then tested in 10 mM K3Fe(CN)6 and 1M KNO3 solution after drying with the scanning rate of 10mV/s, 20mV/s, 50mV/s, and 100mV/s, to obtain Tafel plot, diffusion coefficient, and exchanged current density.
The Tafel equation is an equation in electrochemical kinetics relating the rate of an electrochemical reaction to the overpotential, which is used to plot the Tafel-diagram. The Tafel equation can be stated as:
where the plus sign under the exponent refers to an anodic reaction, and a minus sign to a cathodic reaction, η is overpotential (V), A is Tafel slope (V), i is current density (A/cm2), i0 is exchange current density (A/m2). The exchange current density was obtained by from Tafel plot.
The Randles–Sevcik equation was used to calculate the diffusion coefficient.
where ip is current maximum in amps; n is number of electrons transferred in the redox event; A is electrode area in cm2; D is diffusion coefficient in cm2/s, C is concentration of K3Fe(CN)6 in mol/cm3, ν is scan rate in V/s.
3.5. Photocatalytic Performance
The photocatalytic ability of the specimens was tested in a home-built photocatalytic workstation (see Figure S1). The photoreactor was designed in a dark chamber with a striped light source surrounded by a quartz sleeve, where the suspension including nano-catalyst and MB aqueous solution (130 mL, 10 mg/L) was well irradiated by the light. The suspension was stirred for 30 min in dark, then a UV lamp (500 W high-pressure mercury lamp with a primary emission wavelength of 365 nm) and a sunlight simulator (high-pressure xenon lamp with the main emission wavelengths of 466.7, 506.9, 630.9, 687.9 and 711.1 nm) were used respectively for the irradiation. The light intensity in the solution was adjusted to be ~138.2 mW cm-2, which was measured using a light dynamometer (FZ-A) from Photoelectric Instrument Factory of Beijing Normal University (Beijing, China). Under the light irradiation, the solution was stirred by a magnetic stirring, and the temperature of the solution was maintained at 24 °C by the water circulating through an external cooling system. At a given time interval, a sample of the reaction solution was taken out, centrifuged, and filtered out. Finally, the filtrate concentration was determined by measuring the absorbance on an Agilent Carry-5000 UV-Vis-NIR spectrophotometer.
3.6. Simulation
The polarized charge density of the core/shell specimens as caused by the electromagnetic field of the incident light was calculated using the finite element analysis. The length of NWs was set to be infinite, while the size of the core and the shell was set to be the mean value of our experiment. The titanate NW’s core diameter, GO shell thickness, and rGO shell thickness were 70 nm, 9 nm, and 6 nm, respectively. The specimens were irradiated by the longitudinal magnetic plane wave in the air. The complex refractive indices of air, titanate, GO and rGO were from the database of the simulation solution and the literature.[10] The periodic incident light source port was used as the boundary condition in the simulation.
5. Conclusions
In summary, core-shell structures of the photocatalytic GO-titanate NWs and rGO-titanate NWs were successfully synthesized via the simple hydrothermal methods and then evaluated using the photocatalytic degradation of the MB dye. The rGO-shell increased the visible light absorption by ~75% and the specific surface area, reduced the impedance by ~50% and the charge transfer resistance by ~63%, and helped charge separation. The rGO-titanate based photoelectrode, with the highest on/off current ratio and fastest photo-response, supported the highest photocatalytic degradation efficiency. The simulations of the active photoresponse and photocatalysis, focusing on the charge distribution, can help us to study the new photocatalysis mechanism further.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: image of the photocatalytic workstation; Figure S2: SEM images and EDS spectra; Figure S3: SAED pattern; Figure S4: FTIR spectra; Figure S5: Raman spectra; Figure S6: XPS; Figure S7: UV-Vis spectra; Figure S8: Hyperspectroscopy; Figure S9: Nitrogen adsorption–desorption isotherm; Figure S10 nitrogen adsorption–desorption isotherm, and Figure S11: photocatalytic mechanism.
Author Contributions
The manuscript was written through contributions of all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (Grant #61974125), and the National Science Foundation (Grant #2230853) and National Institute of Standards and Technology (Grant #70NANB22H010) of USA.
Data Availability Statement
All data are available upon reasonable request.
Acknowledgments
The team would like to thank Paula Prescott and Connie Dixon for ordering lab supplies and managing financial reimbursement. The team also would like to thank Kz Shein, Zay Lynn, and David N. Parette for the technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kumar, D.; Lee, A.; Lee, T.; Lim, M.; Lim, D.-K. Ultrafast and Efficient Transport of Hot Plasmonic Electrons by Graphene for Pt Free, Highly Efficient Visible-Light Responsive Photocatalyst. Nano Lett. 2016, 16, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cao, H.; Xu, L.; Fu, H.; Sun, S.; Xiao, Z.; Sun, C.; Long, X.; Xia, Y.; Wang, S. Design and Preparation of Highly Active TiO2 Photocatalysts by Modulating Their Band Structure. Journal of Colloid and Interface Science 2023, 629, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, G.; Liu, F.; Wu, J.; Yang, S. Synergetic Effect of CQD and Oxygen Vacancy to TiO2 Photocatalyst for Boosting Visible Photocatalytic NO Removal. Journal of Hazardous Materials 2023, 452, 131237. [Google Scholar] [CrossRef]
- Higashimoto, S. Titanium-Dioxide-Based Visible-Light-Sensitive Photocatalysis: Mechanistic Insight and Applications. Catalysts 2019, 9, 201. [Google Scholar] [CrossRef]
- Guo, Q.; Zhou, C.; Ma, Z.; Yang, X. Fundamentals of TiO2 Photocatalysis: Concepts, Mechanisms, and Challenges. Advanced Materials 2019, 31, 1901997. [Google Scholar] [CrossRef]
- Li, W.; Wu, Z.; Wang, J.; Elzatahry, A.A.; Zhao, D. A Perspective on Mesoporous TiO2 Materials. Chem. Mater. 2014, 26, 287–298. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, M.; Yu, H.; Zuo, Y.; Gao, J.; He, G.; Sun, Z. Facile Fabrication of Ag/Graphene Oxide/TiO2 Nanorod Array as a Powerful Substrate for Photocatalytic Degradation and Surface-Enhanced Raman Scattering Detection. Applied Catalysis B: Environmental 2019, 252, 174–186. [Google Scholar] [CrossRef]
- Barim, G.; Dhall, R.; Arca, E.; Kuykendall, T.R.; Yin, W.; Takeuchi, K.J.; Takeuchi, E.S.; Marschilok, A.C.; Doeff, M.M. Heterostructured Lepidocrocite Titanate-Carbon Nanosheets for Electrochemical Applications. ACS Appl. Nano Mater. 2022, 5, 678–690. [Google Scholar] [CrossRef]
- Guo, H.; Jiang, N.; Wang, H.; Shang, K.; Lu, N.; Li, J.; Wu, Y. Enhanced Catalytic Performance of Graphene-TiO2 Nanocomposites for Synergetic Degradation of Fluoroquinolone Antibiotic in Pulsed Discharge Plasma System. Applied Catalysis B: Environmental 2019, 248, 552–566. [Google Scholar] [CrossRef]
- Verma, V.; Al-Dossari, M.; Singh, J.; Rawat, M.; Kordy, M.G.M.; Shaban, M. A Review on Green Synthesis of TiO2 NPs: Photocatalysis and Antimicrobial Applications. Polymers 2022, 14, 1444. [Google Scholar] [CrossRef]
- Wang, M.; Han, J.; Xiong, H.; Guo, R.; Yin, Y. Nanostructured Hybrid Shells of R-GO/AuNP/m-TiO2 as Highly Active Photocatalysts. ACS Appl. Mater. Interfaces 2015, 7, 6909–6918. [Google Scholar] [CrossRef]
- Tian, J.; Zhao, Z.; Kumar, A.; I. Boughton, R.; Liu, H. Recent Progress in Design, Synthesis, and Applications of One-Dimensional TiO 2 Nanostructured Surface Heterostructures: A Review. Chemical Society Reviews 2014, 43, 6920–6937. [Google Scholar] [CrossRef]
- Ramgir, N.; Bhusari, R.; Rawat, N.S.; Patil, S.J.; Debnath, A.K.; Gadkari, S.C.; Muthe, K.P. TiO2/ZnO Heterostructure Nanowire Based NO2 Sensor. Materials Science in Semiconductor Processing 2020, 106, 104770. [Google Scholar] [CrossRef]
- Tian, W.-C.; Ho, Y.-H.; Chen, C.-H.; Kuo, C.-Y. Sensing Performance of Precisely Ordered TiO2 Nanowire Gas Sensors Fabricated by Electron-Beam Lithography. Sensors 2013, 13, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Deb, P.; Dhar, J.C. Graphene Oxide Charge Blocking Layer with High K TiO2 Nanowire for Improved Capacitive Memory. Journal of Alloys and Compounds 2021, 868, 159095. [Google Scholar] [CrossRef]
- Fei, Y.; Ye, X.; Al-Baldawy, A.S.; Wan, J.; Lan, J.; Zhao, J.; Wang, Z.; Qu, S.; Hong, R.; Guo, S.; et al. Enhanced Photocatalytic Performance of TiO2 Nanowires by Substituting Noble Metal Particles with Reduced Graphene Oxide. Current Applied Physics 2022, 44, 33–39. [Google Scholar] [CrossRef]
- Ben-Jaber, S.; Peveler, W.J.; Quesada-Cabrera, R.; Cortés, E.; Sotelo-Vazquez, C.; Abdul-Karim, N.; Maier, S.A.; Parkin, I.P. Photo-Induced Enhanced Raman Spectroscopy for Universal Ultra-Trace Detection of Explosives, Pollutants and Biomolecules. Nat Commun 2016, 7, 12189. [Google Scholar] [CrossRef]
- Man, T.; Lai, W.; Xiao, M.; Wang, X.; Chandrasekaran, A.R.; Pei, H.; Li, L. A Versatile Biomolecular Detection Platform Based on Photo-Induced Enhanced Raman Spectroscopy. Biosensors and Bioelectronics 2020, 147, 111742. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Z.; Lan, J.; Khan, I.; Ye, X.; Wan, J.; Fei, Y.; Huang, S.; Li, S.; Kang, J. Recent Advances and Perspectives in Photo-Induced Enhanced Raman Spectroscopy. Nanoscale 2021, 13, 8707–8721. [Google Scholar] [CrossRef]
- Park, H.; Goto, T.; Han, D.H.; Cho, S.; Nishida, H.; Sekino, T. Low Alkali Bottom-Up Synthesis of Titanate Nanotubes Using a Peroxo Titanium Complex Ion Precursor for Photocatalysis. ACS Appl. Nano Mater. 2020, 3, 7795–7803. [Google Scholar] [CrossRef]
- Xu, J.; Chen, D.; Wu, J.; Wu, J.; Zhou, J.; Zhou, T.; Wang, X.; Cheng, J. Nanowires-Assembled TiO2 Nanorods Anchored on Multilayer Graphene for High-Performance Anodes of Lithium-Ion Batteries. Nanomaterials 2022, 12, 3697. [Google Scholar] [CrossRef] [PubMed]
- Kusiak-Nejman, E.; Morawski, A.W. TiO2/Graphene-Based Nanocomposites for Water Treatment: A Brief Overview of Charge Carrier Transfer, Antimicrobial and Photocatalytic Performance. Applied Catalysis B: Environmental 2019, 253, 179–186. [Google Scholar] [CrossRef]
- Yu, X.; Lin, D.; Li, P.; Su, Z. Recent Advances in the Synthesis and Energy Applications of TiO2-Graphene Nanohybrids. Solar Energy Materials and Solar Cells 2017, 172, 252–269. [Google Scholar] [CrossRef]
- Kusiak-Nejman, E.; Wanag, A.; Kowalczyk, Ł.; Kapica-Kozar, J.; Colbeau-Justin, C.; Mendez Medrano, M.G.; Morawski, A.W. Graphene Oxide-TiO2 and Reduced Graphene Oxide-TiO2 Nanocomposites: Insight in Charge-Carrier Lifetime Measurements. Catalysis Today 2017, 287, 189–195. [Google Scholar] [CrossRef]
- Minh, V.C.; Dat, P.T.; Thuy, P.T.; Sang, N.X.; Tuan, N.T.; Tung, T.T.; Losic, D. Effect of Large Graphene Particle Size on Structure, Optical Property and Photocatalytic Activity of Graphene-Titanate Nanotube Composites. Optical Materials 2021, 122, 111662. [Google Scholar] [CrossRef]
- Luo, J.; Wang, Z.; Jiang, H.; Liu, S.; Xiong, F.-Q.; Ma, J. Localized Building Titania–Graphene Charge Transfer Interfaces for Enhanced Photocatalytic Performance. Langmuir 2020, 36, 4637–4644. [Google Scholar] [CrossRef] [PubMed]
- Giannakoudakis, D.A.; Vikrant, K.; LaGrow, A.P.; Lisovytskiy, D.; Kim, K.-H.; Bandosz, T.J.; Carlos Colmenares, J. Scrolled Titanate Nanosheet Composites with Reduced Graphite Oxide for Photocatalytic and Adsorptive Removal of Toxic Vapors. Chemical Engineering Journal 2021, 415, 128907. [Google Scholar] [CrossRef]
- Cole, P.; Tian, Y.; Thornburgh, S.; Malloy, M.; Roeder, L.; Maulding, M.; Huang, Y.; Tian, Z.R. Hydrothermal Synthesis of Valve Metal Zr-Doped Titanate Nanofibers for Bone Tissue Engineering. Nano and Medical Materials 2023, 3, 249. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Z. The Enhanced Photoactivity of Hydrogenated TiO 2 @reduced Graphene Oxide with p–n Junctions. RSC Advances 2015, 5, 26328–26334. [Google Scholar] [CrossRef]
- Yeh, T.-F.; Syu, J.-M.; Cheng, C.; Chang, T.-H.; Teng, H. Graphite Oxide as a Photocatalyst for Hydrogen Production from Water. Advanced Functional Materials 2010, 20, 2255–2262. [Google Scholar] [CrossRef]
- Martin, W.; Tian, Y.; Xiao, J. Understanding Diffusion and Electrochemical Reduction of Li+ Ions in Liquid Lithium Metal Batteries. J. Electrochem. Soc. 2021, 168, 060513. [Google Scholar] [CrossRef]
- Xiao, Y.; Tian, Y.; Zhan, Y.; Zhu, J. Degradation of Organic Pollutants in Flocculated Liquid Digestate Using Photocatalytic Titanate Nanofibers: Mechanism and Response Surface Optimization. Front. Agr. Sci. Eng. 2023, 10, 492–502. [Google Scholar] [CrossRef]
- Dong, W.; Zhang, T.; Epstein, J.; Cooney, L.; Wang, H.; Li, Y.; Jiang, Y.-B.; Cogbill, A.; Varadan, V.; Tian, Z.R. Multifunctional Nanowire Bioscaffolds on Titanium. Chem. Mater. 2007, 19, 4454–4459. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, L.; Xiao, Y.; Collins, T.; Akhter, A.; Huang, Y.; Tian, Z.R. Mo-Doped Titanate Nanofibers from Hydrothermal Syntheses for Improving Bone Scaffold. Characterization and Application of Nanomaterials 2024, 7, 3587. [Google Scholar] [CrossRef]
- Cole, P.; Tian, Y.; Thornburgh, S.; Malloy, M.; Roeder, L.; Zhang, L.; Patel, M.; Xiao, Y.; Huang, Y.; Tian, Z.R. Hydrothermal Synthesis of Valve Metal Ta-Doped Titanate Nanofibers for Potentially Engineering Bone Tissue. Characterization and Application of Nanomaterials 2024, 6, 3606. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



