
Submitted:
06 March 2024
Posted:
06 March 2024
You are already at the latest version
Abstract
The treatment of the bulky Rind-based dibromosilanes, (Rind)2SiBr2 (2) [Rind = 1,1,7,7-tetra-R1-3,3,5,5-tetra-R2-s-hydrindacen-4-yl: EMind (a: R1 = Et, R2 = Me) and Eind (b: R1 = R2 = Et)], with two equivalents of tBuLi in Et2O at low temperatures resulted in the formation of blue solutions derived from the diarylsilylenes, (Rind)2Si: (3). Upon warming the solutions above −20 °C, the blue color gradually faded, accompanying the decomposition of 3 and yielding the cyclic hydrosilanes (4) via intramolecular C−H bond insertion at the Si(II) center. The molecular structures of the bulky Eind-based 3b and 4b were confirmed by X-ray crystallography. Thus, at −20 °C, blue crystals were formed (Crystal-A), which were identified as mixed crystals of 3b and 4b. Additionally, colorless crystals of 4b as a singular component were isolated (Crystal-B), whose structure was also determined by an X-ray diffraction analysis. Although the isolation of 3 was difficult due to their thermally-labile nature, their structural characteristics and electronic properties were discussed based on the experimental findings complemented by computational results. We also examined the hydrolysis of 3b to afford the silanol, (Eind)2SiH(OH) (5b).
Keywords:
silicon
; silylenes
; hydrosilanes
; C−H bond activation
; silanols
1. Introduction
In 1989, Jutzi and co-workers reported the synthesis of decamethylsilicocene, (Me5C5)2Si:, as the first thermally-stable divalent silicon species [1]. Since then, various neutral Si(II) compounds, known as silylenes, have been synthesized employing bulky substituents for steric protection and/or coordination with Lewis bases [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16]. Notably, the isolable cyclic two-coordinate silylenes, N-heterocyclic silylene (A) and dialkylsilylene (B), were reported by West’s and Kira’s groups in 1994 and 1999, respectively (Figure 1) [17,18]. Subsequent advancements led to the synthesis and characterization of heteroatom-substituted acyclic silylenes, represented by a boron- and nitrogen-substituted silylene (C) [19], a sulfur-substituted silylene (D) [20], and a nitrogen- and silicon-substituted silylene (E) [21]. Moreover, new members of the two-coordinate silylenes family, such as a cyclic (alkyl)(amino)silylene (F) [22], a cyclic (amino)(ylide)silylene (G) [23], an acyclic (imino)(siloxy)silylene (H), and an acyclic (vinyl)(silyl)silylene (I) [25], have been successfully isolated by introducing substituents with distinctive electronic effects. Also, a newly-modified cyclic dialkylsilylene (J) was developed by Iwamoto’s group, which is allowed to transform into a genuine silanone [26]. Despite the increasing interest in the synthesis and reaction chemistry of silylenes due to their inherent unsaturated nature, the isolation of aryl-substituted silylenes, i.e., diarylsilylenes, as stable compounds at room temperature remains an elusive goal.
Historically, in 1981, West, Fink, and Michl reported the generation of a transient blue dimesitylsilylene, (Mes)2Si: (K) (Mes = 2,4,6-trimethylphenyl), in a 3-methylpentane (3-MP) matrix at −196 °C by photolysis of a trisilane, (Mes)2Si(SiMe3)2 [Scheme 1(a)] [27]. When the matrix is melted at higher temperatures, the dimerization of the silylene K affords a tetramesityldisilene, (Mes)2Si=Si(Mes)2, as the first stable disilene molecule. As shown in Scheme 1(b), Tokitoh, Suzuki, and Okazaki found in 1993 that the cis- and trans-disilenes, (Tbt)(Mes)Si=Si(Mes)(Tbt) (Tbt = 2,4,6-tris[bis(trimethylsilyl)methyl]phenyl), undergo thermal cleavage of the Si=Si double bond in solution to form a diarylsilylene, (Tbt)(Mes)Si: (L), establishing an equilibrium between dissociation and association with the disilene at temperatures exceeding 70 °C [28]. However, to the best of our knowledge, no crystalline diarylsilylene has been known until now.
For more than about 15 years, our research has been centered on investigating the chemistry of low-coordinated element-organic compounds using the bulky aryl groups derived from the rigid fused-ring 1,1,3,3,5,5,7,7-octa-R-substituted s-hydrindacene skeleton, called the “Rind” groups (Figure 2) [29,30]. The readily accessible Rind groups offer several advantages over other bulky groups, including adjustability of the physical properties through the outer R1 groups and control of the steric bulkiness via the inner R2 groups [31].
During the course of our studies, we have synthesized a series of isolable two-coordinate Ge(II) compounds, diarylgermylenes, (Rind)2Ge: (M), featuring the bulky Rind groups [Rind = EMind (a: R1 = Et, R2 = Me), Eind (b: R1 = R2 = Et), and MPind (c: R1 = Me, R2 = nPr)] [32,33,34]. These compounds were obtained as purple to blue crystals at room temperature by the reaction of GeCl2·dioxane with two equivalents of (Rind)Li, and their structures were determined by an X-ray crystallographic analysis. Notably, the bulky Eind-based germylene, (Eind)2Ge: (M-b), reacted with trimethylamine N-oxide (Me3N+–O–) or N2O gas to afford an isolable monomeric germanone, (Eind)2Ge=O, with the planar three-coordinate Ge(IV) atom and a terminal oxygen atom [33,35]. Additionally, we have successfully isolated a Sn(II) analogue, diarylstannylene, (EMind)2Sn: (N-a), as purple crystals by a similar reaction of SnBr2·dioxane with two equivalents of (EMind)Li [36]. These experimental findings prompted us to further investigate the “missing” diarylsilylenes through the incorporation of the bulky Rind groups.
In this article, we report the formation and characterization of diarylsilylenes, (Rind)2Si: (3), bearing the less bulky EMind (a) and the bulky Eind (b) groups. Although the isolation of 3 posed challenges due to their thermally-labile nature, we extensively examined their structural attributes and electronic properties, drawing insights from both the experimental observations and computational data. We also report the thermal reaction and hydrolysis of 3b, resulting in the formation of the cyclic hydrosilane 4b and silanol 5b through activation of the C−H and O−H bonds at the Si(II) center.
2. Results and Discussion
In a previous study, we reported the synthesis and structures of the sterically- congested dihydrosilanes, (Rind)2SiH2 (1), and dibromosilanes, (Rind)2SiBr2 (2), with the two large Rind groups [Rind = EMind (a) and Eind (b)] attached to the Si(IV) center [37]. We then conducted the modified coupling reaction between the Eind-substituted chlorodihydrosilane, (Eind)SiH2Cl, and (Eind)Li, on a larger scale than before, resulting in the formation of colorless crystals of (Eind)2SiH2 (1b) with an isolated yield of up to 73%. Following the bromination of the Si–H bonds in 1b using N-bromosuccinimide (NBS), (Eind)2SiBr2 (2b) was obtained in 64% yield.
After obtaining sufficient quantities of the two types of sterically-congested molecules of 2, i.e., the less bulky EMind-based 2a and the bulky Eind-based 2b, we proceeded to investigate the reduction of 2 for the transformation to the diarylsilylenes, (Rind)2Si: (3). As illustrated in Scheme 2, the treatment of 2 with two equivalents of tBuLi in Et2O at lower temperatures yielded blue solutions originating from 3 [Figure 3(a)]. After attempting various reducing agents, we finally found that employing two equivalents of tBuLi towards 2 in Et2O at lower temperatures proved the most efficient for the conversion to 3 [38]. However, unfortunately, as the solutions warmed above approximately −20 °C, the vibrant blue hue began to fade, signaling the thermal degradation of 3. Visually, the bulky Eind-based 3b appeared to be more stable at –20 °C compared to the less bulky EMind-based 3a. It is conceivable that differences in the steric bulkiness of the two Rind groups influence the thermal stability of the Si(II) compounds in solution.
Confirmation of the formation of the diarylsilylenes 3 was achieved by UV-vis spectroscopy. Following stirring of the blue solutions for 2 hours at temperatures below −20 °C, the reaction mixture was evaporated to dryness while maintaining temperatures below −20 °C. Precooled toluene was then added to the blue residues, and the resulting suspensions were centrifuged to eliminate any insoluble materials. The obtained blue supernatants were used for the UV-vis measurements. The UV-vis spectra of these supernatants in toluene exhibited an absorption maximum (λmax) at 605 nm for 3a and 618 nm for 3b. These observed values are close to the previously reported absorption peaks of dimesitylsilylene, (Mes)2Si: (K), in a low-temperature matrix (λmax = 577 nm) [39], and in cyclohexane at 25 °C by laser flash photolysis (LFP) (λmax = 580 nm) [40]. In addition, the absorption peaks of these diarylsilylenes are significantly red-shifted compared to those of diphenylsilylene, Ph2Si:, in a low-temperature matrix (λmax = 505 nm) [39], and in hexane at 25°C by LFP (λmax = 520 nm) [41]. The origin of these absorption bands is ascribed to the forbidden n → 3p transition, which may be affected by the introduction of electron-donating alkyl substituents on the phenyl groups bound to the Si(II) center. The above experimental observations are consistent with the theoretical calculations of 3 in a singlet state (vide infra).
Upon standing the toluene solution containing 3b at −20°C, blue crystals precipitated (Crystal-A) [Figure 3(b)]. The single-crystal X-ray diffraction (SC-XRD) analysis elucidated the composition of a mixed crystal containing 3b and a cyclic hydrosilane (4b) formed via intramolecular C−H bond activation at the Si(II) center (Figure 4). Consequently, the central silicon atom (Si1) and the carbon atoms (C55 and C56) from one of the proximate ethyl groups are disordered over the two positions in Crystal-A with the occupancy factor of 0.289(3) for 3b and 0.711(3) for 4b. The position of the hydrogen atom (H1) attached to the silicon atom of 4b was determined using difference Fourier maps and isotropically refined. Unfortunately, attempts to crystallize 3a in a pure form were unsuccessful. Also, despite efforts to induce crystallization by cooling the solution containing 3a to temperatures below −20 °C, no crystals were formed.
The ORTEP drawing of 3b and 4b are shown in Figure 4. Despite the challenges in refining accurate metric parameters due to the inherent disorder of the Si and C atoms, the structures and connectivity of both 3b and 4b were confirmed. In the solid state, the diarylsilylene 3b exhibits a discrete monomeric structure characterized by a bent two-coordinate geometry, attributed to the presence of a nonbonding pair of electrons at the Si(II) center. The C−Si−C bond angle in 3b [112.3(2)°] is smaller than those observed in the related Si(IV) compounds, 1b [123.60(17)°] and 2b [116.60(13)°] (Table 1) [37]. A similar relationship is observed in the C−Ge−C bond angles between the bulky Eind-based Ge(II) compound, germylene, (Eind)2Ge: (M-b), [111.98(5)°] and Ge(IV) compounds, including the germanone, (Eind)2Ge=O [124.27(2)°], germanol, (Eind)2GeH(OH) [123.04(6)°], and dibromogermane, (Eind)2GeBr2 [120.4(6)°] [33,42]. It is emphasized that while 3b constitutes a minor fraction in the mixed crystal, we have achieved the first determination of the crystal structure of the diarylsilylene.
As already mentioned, upon heating the solutions containing 3 from −20 °C to room temperature, they underwent a gradual color change from blue to colorless due to the thermal decomposition of 3 (Scheme 2). In the case of the bulky Eind-based 3b, we obtained a complex mixture mainly containing 4b and (Eind)H, together with some unidentified silicon compounds. Colorless crystals of 4b were obtained in 11% yield, and the structure was characterized by NMR spectroscopy and SC-XRD analysis. In the 1H NMR spectrum of 4b in C6D6, a broad doublet signal due to the newly formed Si−H bond was found at δ = 6.43 ppm with a vicinal coupling with the methine proton [3J(1H−1H) = 4.2 Hz]. The 29Si NMR resonance was observed at δ = −11.9 ppm, appearing as a doublet with the 1J(29Si−1H) coupling constant of 201 Hz. In the 13C NMR spectrum of 4b, each carbon displayed non-equivalence, which is consistent with the asymmetrical structure induced by the intramolecular cyclization. In the thermal decomposition reaction of the less bulky EMind-based 3a in solution, a more complex mixture was generated compared to that of 3b. In the 29Si NMR spectrum of the reaction mixture, a doublet signal appeared at δ = −14.8 ppm [1J(29Si−1H) = 196 Hz], comparable to that of 3b, suggesting the formation of a cyclic hydrosilane 4a. However, due to the overly complex mixture, it was not feasible to isolate 4a.
A similar intramolecular C−H bond insertion reaction was previously reported for the diarylsilylenes such as (Mes)2Si: (K) [43] and (Tbt)(Mes)Si: (L) [28]. In the cases of K and L, the relatively reactive benzylic C−H bond of the Mes and Tbt groups are incorporated into the silylene center. On the other hand, in the case of 3, the less reactive alkyl group’s C−H bond of the Rind groups is inserted into the silylene center, thus indicating that the Si(II) center of the diarylsilylenes possesses an extremely high reactivity. It is worthy of note that such an intramolecular C−H bond insertion has not been observed in the Eind-based germylene M-b at room temperature.
The structure of 4b was ultimately characterized by the SC-XRD analysis, as shown in Figure 5 (Crystal-B). The H atom bound to the Si atom was located from the difference Fourier maps and isotropically refined. The crystal systems and space groups are different between Crystal-A [monoclinic, C2/c (#15)] and Crystal-B [triclinic, P−1 (#2)]. In the two crystal structures of 4b [Figure 4(b) and Figure 5], while the orientation of the ethyl groups differs, the molecular frameworks are identical to each other. Thus, the molecule of 4b possesses three chiral centers located at the Si1A, C55A, and C39 atoms for Crystal-A and Si1, C13, and C3 atoms for Crystal-B. In both Crystal-A and Crystal-B, a pair of enantiomers is present in the unit cell, thus being a racemic compound. Based on these results, the Si(II) center was intramolecularly inserted into one of the methylene C−H bonds of the inner ethyl group with stereoselectivity, arising from the severe steric congestion between the bulky Eind groups. The selected structural parameters are summarized in Table 1. The Si−C bond lengths and C−Si−C bond angles of 4b exhibit slight differences between Crystal-A and Crystal-B, probably due to the disorder analysis in the co-crystal and/or the crystal packing configuration.
We also tried to measure the 29Si NMR signal of 3b at low temperature. Following a procedure similar to the UV-vis spectrum measurements of 3, precooled deuterated toluene (C7D8, toluene-d8) was introduced into the blue reaction mixture containing 3b. The resultant blue suspension in C7D8 was then subjected to the low-temperature 29Si NMR measurements. In the 29Si NMR spectrum measured at −20 °C, the distinctive downfield resonance was observed at δ = 513.1 ppm assignable to 3b, accompanied by some additional signals in the upfield region ranging from δ = 0 to −35 ppm, attributable to unidentified Si(IV) compounds (Figure 6). The 29Si NMR chemical shift of 3b (δ = 513.1 ppm) is between those of the two-coordinate cyclic dialkylsilylenes B (δ = 567.4 ppm) [18] and acyclic heteroatom-substituted silylenes C (δ = 439.7 ppm in C6D6 at 25 °C) [19] and E (δ = 438.2 and 467.5 ppm in C6D6 at 20 °C) [21]. Notably, the experimental chemical shift value aligns with the GIAO calculated value of 3b in a singlet state (vide infra).
To clarify the electronic properties of the diarylsilylenes, DFT computations at the (U)B3LYP-D3/6-31G(d,p) level were performed for 3 using the Gaussian 09 suite of programs [44]. The singlet states exhibit a lower energy level compared to the triplet states; the singlet-triplet energy differences (ΔEST) were calculated to be 20.3 kcal mol−1 for 3a and 21.5 kcal mol−1 for 3b, respectively. The optimized structure of 3b in the singlet state is analogous to the X-ray crystal structure (Table 1). Thus, the C−Si−C bond angle of the calculated singlet state of 3b (112.00°) is comparable to that of the experimental XRD analysis in 3b [112.3(2)°]. Furthermore, DFT calculations were conducted for 4b, an isomer of 3b. The optimized geometry of 4b almost matches the X-ray crystal structures (Table 1). The Si(IV) compound 4b was found to be 56.1 kcal mol−1 more stable than the Si(II) compound 3b, accompanying the formation of the new Si−C and Si−H bonds. The calculated C(sp2)−Si−C(sp2) bond angle increased with the C−H bond insertion, 119.50° for 4b versus 112.00° for 3b. The calculated Si−C(sp2) bond distances in 4b [1.9099 and 1.9012 Å] are slightly shorter than those in 3b [1.9470 and 1.9489 Å]. These data are consistent with the increased s-character of the Si atomic orbitals directed to the aryl groups by the intramolecular C−H bond insertion reaction involving the singlet silylene [45].
The frontier molecular orbitals (MOs) of the singlet state of 3b are displayed in Figure 7. The HOMO mainly represents the non-bonding pair of electrons on the Si atom, while the LUMO involves the empty 3p orbital of the Si atom. The HOMO−LUMO energy gaps (ΔE) are estimated to be 2.886 eV for 3a and 2.962 eV for 3b. According to the TD-DFT calculations, the weak absorption peaks were evaluated to be 620 nm (f = 0.0207; n−p) for 3a and 590 nm (f = 0.0210; n−p) for 3b, corresponding to the forbidden HOMO → LUMO transition, which align well with the UV-vis data (λmax = 605 nm for 3a and λmax = 618 nm for 3b) (vide supra). The 29Si chemical shift of 3b was calculated to be δ = 511.1 ppm by the gauge-independent atomic orbital (GIAO) method, which is in good agreement with the experimental value for the 29Si chemical shift (δ = 513.1 ppm) (vide supra).
As a preliminary step, investigations into the reactivity of the diarylsilylenes 3 have also been initiated. For example, upon treatment of the Et2O solution containing 3b with oxygen-free water at −20 °C, a silanol, (Eind)2SiH(OH) (5b), was obtained as the major product in 44% isolated yield (Scheme 2) [46,47]. The identification of 5b was supported by the spectroscopic data, while the molecular structure was validated by the SC-XRD analysis (Figure 8). In the 1H NMR spectrum of 5b in CDCl3, the Si−H signal was found at δ = 6.75 ppm along with satellite signals [1J(29Si−1H) = 216 Hz]. The O−H signal due to the silanol moiety (SiO−H) also appeared at δ = 2.02 ppm. The 29Si NMR resonance of 5b was observed at δ = −23.2 ppm, appearing as a doublet with the 1J(29Si−1H) coupling constant of 216 Hz. In the IR spectrum of 5b (KBr, pellet), a relatively sharp SiO−H vibration peak appears at 3672 cm−1, together with a weak Si−H stretching vibration at 2227 cm−1. These observed stretching frequencies for 5b are higher than those reported for (Mes)2SiH(OH) at 3201 cm−1 (SiO−H) and 2157 cm−1 (Si−H) [48].
The ORTEP drawing of 5b is shown in Figure 8. The hydroxy group is disordered over the two positions in the crystal with the occupancy factors of 0.525(3) and 0.475(3). Nevertheless, the H atoms bound to the Si and O atoms were located from the difference Fourier maps and isotropically refined. The silanol 5b exhibits a monomeric structure with no intermolecular O−H···O hydrogen bonding in the crystal, which is in sharp contrast to the fact that the tetrameric molecular arrangement was observed for (Mes)2SiH(OH) via intermolecular hydrogen bonding in the crystal [48]. The difference in the steric bulkiness between the Eind group and Mes group significantly influences the aggregation behavior of the silanols in the solid state. Refining the accurate metric parameters of 5b was difficult due to the disorder of the O atom. The C−Si−C bond angle of 5b [122.05(5)°] is similar to that of 1b [123.60(17)°] and larger than that of 2b [116.60(13)°] [37], contingent upon the steric environments surrounding the Si(IV) center (Table 1). These bond angles of the Si(IV) compounds are larger than that of the Si(II) compound 3b [112.3(2)°], because of the disappearance of the non-bonding pair of electrons on the silicon atom. DFT studies were also performed to evaluate the structure of 5b. As summarized in Table 1, the optimized structure reproduces the X-ray structure. The SiO−H and Si−H stretching frequencies are calculated as 3882 and 2321 cm−1 for 5b, in good qualitative agreement with the experimental values.
3. Materials and Methods
3.1. General Procedures
The air- and/or moisture-sensitive compounds were handled using either standard Schlenk-line techniques or within a glove box under an inert argon atmosphere. Anhydrous diethylether (Et2O), THF, hexane, and toluene were dried by passage through columns of activated alumina and supported copper catalyst supplied by Nikko Hansen & Co., Ltd. (Osaka, Japan). Deuterated benzene (C6D6, benzene-d6) and toluene (C7D8, toluene-d8) were dried and degassed over a potassium mirror in vacuo prior to use. 4-Bromo-1,1,3,3,5,5,7,7-octaethyl-s-hydrindacene, (Eind)Br [31], (Eind)SiH2Cl [37], and (EMind)2SiBr2 [37] were prepared by the literature procedures. All other chemicals and gases were used as received.
The nuclear magnetic resonance (NMR) measurements were carried out using JEOL RESONANCE JNM-ECS400 and JNM-AL400 spectrometers (399.8 MHz for 1H, 100.5 MHz for 13C and 79.4 MHz for 29Si) and BRUKER AVANCE NEO 600 OneBay spectrometer (600.1 MHz for 1H, 150.9 MHz for 13C and 119.2 MHz for 29Si). Chemical shifts (δ) are given as dimensionless numbers and relative to 1H or 13C NMR chemical shifts of the solvents (residual CHCl3 in CDCl3, 1H(δ) = 7.26 and 13C(δ) = 77.16; residual C6D5H in C6D6, 1H(δ) = 7.16 and 13C(δ) = 128.06). The 29Si NMR spectra were referenced using the external standard of tetramethylsilane (Me4Si) [29Si(δ) = 0.0]. The absolute values of the coupling constants are given in Hertz (Hz), regardless of their signs. Multiplicities are abbreviated as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). The UV-vis spectra were obtained using a Shimadzu UV-3101(PC)S spectrometer. The infrared spectra were measured by a JASCO FT/IR-4100 spectrometer. The elemental analyses (C and H) were performed at the Materials Characterization Support Team, RIKEN Center for Emergent Matter Science and the Microanalytical Laboratory at the Institute for Chemical Research (Kyoto University). Melting points (mp) were determined by a Stanford Research Systems OptiMelt instrument and a Yazawa BY-2 instrument.
3.1.1. Synthesis of (Eind)2SiH2 (1b)
To a solution of (Eind)Br (3.49 g, 7.56 mmol) in Et2O (40 mL) was added tBuLi (1.62 M in pentane, 10.3 mL, 16.7 mmol) at –78 ºC. The reaction mixture was allowed to warm to room temperature and concentrated in vacuo. To a solution of the resulting (Eind)Li in THF (15 mL) was added a solution of (Eind)SiH2Cl (3.38 g, 7.56 mmol) in THF (15 mL) at 0 °C. The mixture was allowed to warm to room temperature and stirred overnight. After the volatiles were removed in vacuo, hexane (200 mL) was added to the residual solid. The resulting suspension was filtered through a plug of Celite®. After the filtrate was concentrated on a rotary evaporator, the residual solid was recrystallized from hexane to afford 1b as colorless crystals (4.37 g, 5.51 mmol, 73%). The NMR data are consistent with those previously reported [37].
3.1.2. Synthesis of (Eind)2SiBr2 (2b)
To a solution of 1b (460 mg, 0.580 mmol) in hexane (20 mL) was added N-bromosuccinimide (NBS) (220 mg, 1.24 mmol) at 0 ºC under an argon atmosphere. The mixture was allowed to warm to room temperature. After stirring overnight at room temperature, the resulting suspension was filtered to remove any insoluble materials. The filtrate was concentrated, and the residual solid was recrystallized from hexane to afford 2b as colorless crystals (355 mg, 0.373 mmol, 64%). The NMR data are consistent with those previously reported [37].
3.1.3. Reduction of (EMind)2SiBr2 (2a) for UV-Vis Measurement of 3a
To a solution of 2a (328 mg, 0.391 mmol) in Et2O (8 mL) was added tBuLi (1.62 M in pentane, 0.51 mL, 0.83 mmol) at –78 ºC. After stirring for 2 hours at –40 ºC, the volatiles were removed in vacuo while maintaining temperatures below –20 ºC. Precooled toluene (5 mL) was added to the residue, and the resulting suspension was centrifuged to remove any insoluble materials. The obtained supernatant was used for the UV-vis measurement. 3a: UV-vis (toluene, –20 ºC) λmax = 605 nm.
3.1.4. Reduction of (Eind)2SiBr2 (2b) for UV-Vis Measurement of 3b
To a solution of 2b (391 mg, 0.411 mmol) in Et2O (10 mL) was added tBuLi (1.62 M in pentane, 0.49 mL, 0.79 mmol) at –78 ºC. After stirring for 2 hours at –40 ºC, the volatiles were removed in vacuo while maintaining temperatures below –20 ºC. Precooled toluene (10 mL) was added to the residue, and the resulting suspension was centrifuged to remove any insoluble materials. The obtained supernatant was used for the UV-vis measurement. 3b: UV-vis (toluene, –20 ºC) λmax = 618 nm.
3.1.5. Reduction of (Eind)2SiBr2 (2b) for XRD Measurement of Crystal-A
To a solution of 2b (373 mg, 0.392 mmol) in Et2O (11 mL) was added tBuLi (1.62 M in pentane, 0.48 mL, 0.78 mmol) at –78 ºC. After stirring for 2 hours at –40 ºC, the volatiles were removed in vacuo while maintaining temperatures below –20 ºC. Precooled toluene (5 mL) was added to the residue, and the resulting suspension was centrifuged to remove any insoluble materials. The obtained supernatant was concentrated to about 2 mL and upon standing at –20 ºC, blue crystals precipitated (Crystal-A), which was used for XRD measurement.
3.1.6. Reduction of (Eind)2SiBr2 (2b) for 29Si NMR Measurement of 3b
To a solution of 2b (307 mg, 0.323 mmol) in Et2O (5 mL) was added tBuLi (1.62 M in pentane, 0.38 mL, 0.62 mmol) at –78 ºC. After stirring for 1 hour at –40 ºC, the volatiles were removed in vacuo while maintaining temperatures below –30 ºC. After precooled deuterated toluene (C7D8, 1.0 mL) at –50 ºC was added to the residue, the resulting suspension was used for the 29Si NMR measurement. 3b: 29Si NMR (119.2 MHz, C7D8, –20 ºC) δ = 513.1.
3.1.7. Reduction of (EMind)2SiBr2 (2a) for Thermal Reaction of 3a
To a solution of 2a (107 mg, 0.128 mmol) in Et2O (3 mL) was added tBuLi (1.62 M in pentane, 0.20 mL, 0.32 mmol) at –78 ºC. The mixture was allowed to warm to room temperature and stirred overnight. After the volatiles were removed in vacuo, hexane (5 mL) was added to the residual solid. The resulting suspension was filtered to remove any insoluble materials. The filtrate was concentrated, and the residual solid was used for the NMR measurements, indicating the formation of a complex mixture containing 4a and (EMind)H, together with some unidentified silicon compounds. 4a: 29Si NMR (119.2 MHz, C6D6, 25 ºC) δ = −14.8 ppm (d, 1JSi−H = 196 Hz).
3.1.8. Reduction of (Eind)2SiBr2 (2b) for Thermal Reaction of 3b
To a solution of 2b (310 mg, 0.326 mmol) in Et2O (7 mL) was added tBuLi (1.62 M in pentane, 0.39 mL, 0.63 mmol) at –78 ºC. The mixture was allowed to warm to room temperature and stirred overnight. After the volatiles were removed in vacuo, hexane (7 mL) was added to the residual solid. The resulting suspension was filtered to remove any insoluble materials. The filtrate was concentrated, and the residual solid was used for the NMR measurements, indicating the formation of a complex mixture mainly containing 4b and (Eind)H, together with some unidentified silicon compounds. After the Kugelrohr distillation, the obtained solid was recrystallized from a mixed solvent of hexane and ethanol to afford 4b as colorless crystals (27.5 mg, 34.7 mmol, 11%). 4b: 1H NMR (600.1 MHz, C6D6, 25 ℃) δ = 0.40–3.30 (m, 87 H, CH2CH3, CH2CH3, CH2, CH), 6.43 (d, J = 4.2 Hz, 1 H, SiH), 6.81 (s, 1 H, ArH), 6.85 (s, 1 H, ArH). 13C NMR (150.9 MHz, C6D6, 25 ℃) δ = 9.21, 9.24, 9.39, 9.45, 9.50, 9.57, 9.66, 9.79, 9.90, 10.08, 10.16, 10.46, 10.73, 10.77, 12.10, 12.19, 30.23, 30.64, 30.72, 30.81, 31.24, 31.26, 32.53, 33.22, 33.23, 33.51, 33.69, 34.40, 34.62, 35.56, 35.60, 36.36, 41.98, 43.40, 43.81, 45.09, 47.27, 47.97, 49.24, 51.38, 52.89, 53.74, 53.89, 54.42, 121.18, 122.80, 129.48, 129.93, 142.74, 149.18, 149.34, 150.05, 153.62, 156.66, 157.07, 166.07. 29Si NMR (119.2 MHz, C6D6, 25 ºC) δ = −11.9 ppm (d, 1JSi−H = 201 Hz). Anal. Calcd. for C56H90Si: C, 84.99; H, 11.16. Found: C, 84.43; H, 11.28. Melting point 141−143 ºC (dec.).
3.1.9. Hydrolysis of 3b for Synthesis of 5b
To a solution of 2b (206 mg, 0.217 mmol) in Et2O (9 mL) was added tBuLi (1.62 M in pentane, 0.34 mL, 0.55 mmol) at –78 ºC. After stirring for 2 hours at –30 ºC, degassed H2O (5.00 mL, 277 mmol) was added to the mixture at –30 ºC. The mixture was allowed to warm to room temperature, then the volatiles were removed in vacuo. The residual solid was dissolved in hexane and filtered through a short silica gel column. After the filtrate was concentrated, the residue was recrystallized from hexane to afford 5b as colorless crystals (77.0 mg, 95.1 mmol, 44%). 5b: 1H NMR (600.1 MHz, CDCl3, 25 ℃) δ = 0.60 (br. s, 24 H, CH2CH3), 0.80 (br. s, 24 H, CH2CH3), 1.25 (br. s, 4 H, CH2CH3), 1.51–1.55 (m, 8 H, CH2CH3), 1.60–1.74 (m, 20 H, CH2CH3, CH2), 1.80 (br. s, 8 H, CH2CH3), 2.02 (br. s, 1 H, OH), 6.75 (s, 1 H, SiH, satellite, 1J(Si–H) = 216 Hz), 6.69 (s, 2 H, ArH). 13C NMR (150.9 MHz, CDCl3, 25 ℃) δ = 9.07, 9.09, 10.06, 10.14, 32.27, 32.44, 32.78, 32.85, 41.82, 47.46, 54.01, 122.55, 135.42, 149.32, 154.08. 29Si NMR (119.2 MHz, CDCl3, 25 ºC) δ = −23.2 ppm (d, 1JSi−H = 216 Hz). IR (KBr, cm−1): ν = 3672 (SiO−H), 2227 (Si−H). Melting point 166–169 ºC (dec.).
3.2. X-ray Crystallographic Studies of Crystal-A, Crystal-B, and 5b.
Single crystals of Crystal-A (co-crystal of 3b and 4b) were grown from toluene at −20 ºC for the X-ray diffraction measurements. Single crystals suitable for the X-ray diffraction analysis were obtained from a mixed solvent of hexane and ethanol for Crystal-B (4b) and from hexane for 5b. Intensity data were collected using a Rigaku AFC-10 diffractometer with a Saturn724+ CCD detector for Crystal-A and a Rigaku XtaLAB P200 with a PILATUS200 K detector for Crystal-B and 5b. The diffraction data were collected using MoKα radiation (λ = 0.71073 Å). The specimens were cooled at 100 K in a cold nitrogen stream during the measurements. Bragg spots were integrated and scaled with the programs of CrystalClear [49] for Crystal-A and CrysAlisPro [50] for Crystal-B and 5b. The structure was solved by a direct method with the programs of SIR2008 [51] for Crystal-A and SHELXT-2018/2 [52] for Crystal-B and 5b. All the structures were refined with a least-squares method on F2 using SHELXL-2019/3 software [53]. The anisotropic temperature factors were applied to all the non-hydrogen atoms. The Si–H and O–H hydrogen atoms were located on the difference Fourier maps and refined isotropically. Positions of all the C–H hydrogen atoms in ordered moieties were also located on the difference Fourier maps, while those in disordered moieties were calculated geometrically. All the C–H hydrogen atoms were refined as riding models. The detailed crystallographic data have been deposited at the Cambridge Crystallographic Data Centre: Deposition code CCDC 2336975 (co-crystal of 3b and 4b), 2336976 (4b), and 2336978 (5b). A copy of the data can be obtained free of charge via www.ccdc.cam.ac.uk/data–request.
3.2.1. Crystal-A (Co-Crystal of 3b and 4b)
C56H90Si, M = 791.36, crystal size 0.28 × 0.18 × 0.11 mm, monoclinic, space group C2/c (#15), a = 32.720(7) Å, b = 18.594(4) Å, c = 16.662(4) Å, β = 104.756(3)º, V = 9803(4) Å3, Z = 8, Dx = 1.072 g cm−3, μ(Mo Kα) = 0.082 mm−1, 40049 reflections collected, 11249 unique reflections, and 540 refined parameters. The final R(F) value was 0.0757 [I > 2σ(I)]. The final Rw(F2) value was 0.1823 (all data). The goodness-of-fit on F2 was 1.098.
3.2.2. Crystal-B (4b)
C56H90Si, M = 791.36, crystal size 0.26 × 0.09 × 0.04 mm, triclinic, space group P−1 (#2), a = 10.1298(6) Å, b = 10.1400(6) Å, c = 24.4558(13) Å, α = 84.557(5)º, β = 85.566(5)º, γ = 78.612(5)º, V = 2447.0(2) Å3, Z = 2, Dx = 1.074 g cm−3, μ(Mo Kα) = 0.083 mm−1, 20736 reflections collected, 11460 unique reflections, and 534 refined parameters. The final R(F) value was 0.0768 [I > 2σ(I)]. The final Rw(F2) value was 0.1914 (all data). The goodness-of-fit on F2 was 1.023.
3.2.3. (Eind)2SiH(OH) (5b)
C56H92OSi, M = 809.38, crystal size 0.27 × 0.18 × 0.12 mm, monoclinic, space group P21/c (#14), a = 12.9236(3) Å, b = 18.9633(4) Å, c = 20.6577(5) Å, β = 102.518(2)º, V = 4942.3(2) Å3, Z = 4, Dx = 1.088 g cm−3, μ(Mo Kα) = 0.084 mm−1, 122903 reflections collected, 12940 unique reflections, and 563 refined parameters. The final R(F) value was 0.0455 [I > 2σ(I)]. The final Rw(F2) value was 0.1147 (all data). The goodness-of-fit on F2 was 1.048.
4. Conclusions
We investigated the synthesis of the diarylsilylenes, (Rind)2Si: (3), supported by two bulky Rind groups at the Si(II) center. The Rind-based dibromosilanes, (Rind)2SiBr2 (2), reacted with two equivalents of tBuLi in Et2O at lower temperatures to produce the blue colored solutions containing 3 below –20 ºC. The formation of 3 was characterized by the UV-vis spectra with the absorption peaks appearing at λmax = 605 nm (3a) and 618 nm (3b) in toluene at –20 ºC, attributed to the forbidden n → 3p transition. Moreover, the 29Si NMR signal assignable to 3b was observed at δ = 513.1 ppm in C7D8 at –20 ºC. These experimental data are in good agreement with the theoretical calculations of 3 in the singlet state. While isolating 3 posed challenges due to its thermally-unstable nature, we obtained co-crystals of 3b with the cyclic hydrosilane 4b, formed through the intramolecular C–H bond insertion into the silylene center. The molecular structures of 3b and 4b were confirmed by X-ray crystallography, with the former being the first determination of the crystal structure of the diarylsilylene. Additionally, we examined the hydrolysis reaction of 3b, resulting in the formation of the bulky silanol compound, (Eind)2SiH(OH) (5b), which exhibits a monomeric structure in the solid state.
The accessibility of 3 offers a new perspective for understanding the electronic nature of the divalent Si(II) species and the diverse transformations originating from unsaturated, two-coordinate silylenes, thereby unveiling a new facet of low-valent silicon chemistry. Further studies about the reactivities of the diarylsilylenes are currently underway.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. UV-vis spectra of 3a and 3b (Figures S1 and S2), NMR spectra of 3b, 4b and 5b (Figures S3–S13), and IR spectrum of 5b (Figure S14) (PDF), crystallographic details for Crystal-A (co-crystal of 3b and 4b), Crystal-B (4b), and 5b (CIF), and CIF-checked files (PDF).
Author Contributions
T.M. (Tsukasa Matsuo) conceived the ideas of this study. K.M. and T.M. (Tatsuto Morimoto) synthesized the silicon compounds. K.M., T.M. (Tatsuto Morimoto), and S.M. performed the measurements. K.M., T.M. (Tatsuto Morimoto), and K.O. performed the calculations. K.M., T.M. (Tatsuto Morimoto), K.O., and D.H. analyzed the crystal structures. All the authors contributed to the discussion of the paper. K.M., T.M. (Tatsuto Morimoto), K.O., D.H., and T.M. (Tsukasa Matsuo) contributed to the writing of the paper. K.O., D.H., and T.M. (Tsukasa Matsuo) supervised the work.
Funding
This research was supported by Grants-in-Aid for Scientific Research on Innovative Areas “Stimuli-responsive Chemical Species for the Creation of Functional Molecules (No. 2408)” (JSPS KAKENHI Grant Nos. JP20109003 for Tsukasa Matsuo and JP15H00964 for Daisuke Hashizume), Scientific Research (B) (No. JP15H03788) and (C) (Nos. 18K05160 and 21K05091) for Tsukasa Matsuo, and Young Scientists (Start-up) (No. 22K20561) for Kei Ota. This study was partially supported by a MEXT-Supported Program for the Strategic Research Foundation at Private Universities 2014–2018 subsidy from MEXT and Kindai University. We thank the Collaborative Research Program of The Institute for Chemical Research, Kyoto University (grants #2016-94, #2017-99, #2018-110, #2019-120, #2020-126, #2021-130, and #2022-134).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
We are grateful to the Materials Characterization Support Team, RIKEN Center for Emergent Matter Science (CEMS), for the elemental analyses. We thank Dr. N. Hayakawa and Dr. Tomoharu Tanikawa for their initial contributions to this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jutzi, P.; Holtmann, U.; Kanne, D.; Krüger, C.; Blom, R.; Gleiter, R.; Hyla-Kryspin, I. Decamethylsilicocene—The first stable silicon(II) compound: Synthesis, structure, and bonding. Chem. Ber. 1989, 122, 1629–1639. [CrossRef]
- Wang, L.; Li, Y.; Li, Z.; Kira, M. Isolable silylenes and their diverse reactivity. Coord. Chem. Rev. 2022, 457, 214413. [CrossRef]
- Shan, C.; Yao, S.; Driess, M. Where silylene–silicon centres matter in the activation of small molecules. Chem. Soc. Rev. 2020, 49, 6733–6754. [CrossRef]
- Fujimori, S.; Inoue, S. Small Molecule Activation by Two-Coordinate Silylenes. Chem. Eur. J. 2020, 3131–3142. [CrossRef]
- Zhou, Y.-P.; Driess, M. Isolable Silylene Ligands Can Boost Efficiencies and Selectivities in Metal-Mediated Catalysis. Angew. Chem. Int. Ed. 2019, 58, 3715–3728. [CrossRef]
- Weetman, C.; Inoue, S. The Road Travelled: After Main-Group Elements as Transition Metals. ChemCatChem 2018, 10, 4213–4228. [CrossRef]
- Chu, T.; Nikonov, G.I. Oxidative Addition and Reductive Elimination at Main-Group Element Centers. Chem. Rev. 2018, 118, 3608–3680. [CrossRef]
- Hadlington, T.J.; Driess, M.; Jones, C. Low-valent group 14 element hydride chemistry: towards catalysis. Chem. Soc. Rev. 2018, 47, 4176–4197. [CrossRef]
- Tacke, R.; Ribbeck, T. Bis(amidinato)- and bis(guanidinato)silylenes and silylenes with one sterically demanding amidinato or guanidinato ligand: synthesis and reactivity. Dalton Trans. 2017, 46, 13628–13659. [CrossRef]
- Lee, V.Y. Organosilicon Compounds: Theory and Experiment (Synthesis); Elsevier: London, UK, 2017.
- Yadav, S.; Saha, S.; Sen, S.S. Compounds with Low-Valent p-Block Elements for Small Molecule Activation and Catalysis. ChemCatChem 2016, 8, 486–501. [CrossRef]
- Scheschkewitz, D. Functional Molecular Silicon Compounds II: Low Oxidation States; Springer: Basel, Switzerland, 2014.
- Asay, M.; Jones, C.; Driess, M. N-Heterocyclic Carbene Analogues with Low-Valent Group 13 and Group 14 Elements: Syntheses, Structures, and Reactivities of a New Generation of Multitalented Ligands. Chem. Rev. 2011, 111, 354–396. [CrossRef]
- Lee, V.Y.; Sekiguchi, A. Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb; Wiley: West Sussex, UK, 2010.
- Kira, M. An isolable dialkylsilylene and its derivatives. A step toward comprehension of heavy unsaturated bonds. Chem. Commun. 2010, 46, 2893–2903. [CrossRef]
- Mizuhata, Y.; Sasamori, T.; Tokitoh, N. Stable heavier carbene analogues. Chem. Rev. 2009, 109, 3479–3511. [CrossRef]
- Denk, M.; Lennon, R.; Hayashi, R.; West, R.; Belyakov, A.V.; Verne, H.P.; Haaland, A.; Wagner, M.; Metzler, N. Synthesis and Structure of a Stable Silylene. J. Am. Chem. Soc. 1994, 116, 2691–2692. [CrossRef]
- Kira, M.; Ishida, S.; Iwamoto, T.; Kabuto, C. The First Isolable Dialkylsilylene. J. Am. Chem. Soc. 1999, 121, 9722–9723. [CrossRef]
- Protchenko, A.V.; Birjkumar, K.H.; Dange, D.; Schwarz, A.D.; Vidovic, D.; Jones, C.; Kaltsoyannis, N.; Mountford, P.; Aldridge, S. A Stable Two-Coordinate Acyclic Silylene. J. Am. Chem. Soc. 2012, 134, 6500–6503. [CrossRef]
- Rekken, B.D.; Brown, T.M.; Fettinger, J.C.; Tuononen, H.M.; Power, P.P. Isolation of a Stable, Acyclic, Two-Coordinate Silylene. J. Am. Chem. Soc. 2012, 134, 6504–6507. [CrossRef]
- Protchenko, A.V.; Schwarz, A.D.; Blake, M.P.; Jones, C.; Kaltsoyannis, N.; Mountford, P.; Aldridge, S. A Generic One-Pot Route to Acyclic Two-Coordinate Silylenes from Silicon(IV) Precursors: Synthesis and Structural Characterization of a Silylsilylene. Angew. Chem. Int. Ed. 2013, 52, 568 –571. [CrossRef]
- Kosai, T.; Ishida, S.; Iwamoto, T. A Two-Coordinate Cyclic (Alkyl)(amino)silylene: Balancing Thermal Stability and Reactivity. Angew. Chem. Int. Ed. 2016, 55, 15554–15558. [CrossRef]
- Alvarado-Beltran, I.; Baceiredo, A.; Saffon-Merceron, N.; Branchadell, V.; Kato, T. Cyclic Amino(Ylide) Silylene: A Stable Heterocyclic Silylene with Strongly Electron-Donating Character, Angew. Chem. Int. Ed. 2016, 55, 16141–16144. [CrossRef]
- Wendel, D.; Reiter, D.; Porzelt, A.; Altmann, P.J.; Inoue, S.; Rieger, B. Silicon and Oxygen’s Bond of Affection: An Acyclic Three-Coordinate Silanone and Its Transformation to an Iminosiloxysilylene. J. Am. Chem. Soc. 2017, 139, 17193–17198. [CrossRef]
- Roy, M.M.D.; Ferguson, M.J.; McDonald, R.; Zhou, Y.; Rivard, E. A vinyl silylsilylene and its activation of strong homo- and heteroatomic bonds. Chem. Sci. 2019, 10, 6476–6481. [CrossRef]
- Kobayashi, R.; Ishida, S.; Iwamoto, T. An Isolable Silicon Analogue of a Ketone that Contains an Unperturbed Si=O Double Bond. Angew. Chem. Int. Ed. 2019, 58, 9425–9428. [CrossRef]
- West, R.; Fink, M. J.; Michl, J. Tetramesityldisilene, a Stable Compound Containing a Silicon-Silicon Double Bond. Science 1981, 214, 1343–1344. [CrossRef]
- Tokitoh, N.; Suzuki, H.; Okazaki, R. Synthesis, Structure, and Reactivity of Extremely Hindered Disilenes: The First Example of Thermal Dissociation of a Disilene into a Silylene. J. Am. Chem. Soc. 1993, 115, 10428–10429. [CrossRef]
- Matsuo, T.; Hayakawa, N. π-Electron systems containing Si=Si double bonds. Sci. Technol. Adv. Mater. 2018, 19, 108–129. [CrossRef]
- Matsuo, T.; Tamao, K. Fused-Ring Bulky “Rind” Groups Producing New Possibilities in Elemento-Organic Chemistry. Bull. Chem. Soc. Jpn. 2015, 88, 1201–1229. [CrossRef]
- Matsuo, T.; Suzuki, K.; Fukawa, T.; Li, B.; Ito, M.; Shoji, Y.; Otani, T.; Li, L.; Kobayashi, M.; Hachiya, M.; Tahara, Y.; Hashizume, D.; Fukunaga, T.; Fukazawa, A.; Li, Y.; Tsuji, H.; Tamao, K. Synthesis and Structures of a Series of Bulky “Rind-Br” Based on a Rigid Fused-Ring s-Hydrindacene Skeleton. Bull. Chem. Soc. Jpn. 2011, 84, 1178–1191. [CrossRef]
- Ohno, R.; Numata, Y.; Konaka, S.; Yagura, S.; Kuroda, A.; Harada, M.; Fujita, N.; Hayakawa, N.; Nakai, H.; Rosas-Sánchez, A.; Hashizume, D.; Matsuo, T. Synthesis and Characterization of a Series of Diarylgermylene and Dihalodigermenes Having Fused-Ring Bulky “Rind” Groups. Bull. Chem. Soc. Jpn. 2021, 94, 1931–1939. [CrossRef]
- Suzuki, K.; Numata, Y.; Fujita, N.; Hayakawa, N.; Tanikawa, T.; Hashizume, D.; Tamao, K.; Fueno, H.; Tanaka, K.; Matsuo, T. A stable free tetragermacyclobutadiene incorporating fused-ring bulky EMind groups. Chem. Commun. 2018, 54, 2200–2203. [CrossRef]
- Li, L.; Fukawa, T.; Matsuo, T.; Hashizume, D.; Fueno, H.; Tanaka, K.; Tamao, K. A stable germanone as the first isolated heavy ketone with a terminal oxygen atom, Nat. Chem. 2012, 4, 361–365. [CrossRef]
- Kuroda, A.; Fujita, N.; Horita, T.; Ota, K.; Rosas-Sánchez, A.; Hoshino, M.; Hashizume, D.; Matsuo, T. Formation and Reactions of Ge=O Double-bonded Species Bearing EMind Groups. Chem. Lett. 2022, 51, 828–831. [CrossRef]
- Numata, Y.; Nishikawa, Y.; Inoue, K.; Ohnishi, H.; Konaka, S.; Tanikawa, T.; Hashizume, D.; Matsuo, T. A Series of Room-Temperature Thermally Stable Bromostannylenes Bearing the Bulky Rind Group: Synthesis, Characterization, and Crystal Structures. Organometallics 2021, 40, 1956–1965. [CrossRef]
- Hayakawa, N.; Morimoto, T.; Takagi, A.; Tanikawa, T.; Hashizume, D.; Matsuo, T. Synthesis and Structures of Sterically Congested Diarylsilanes Bearing Two Bulky Rind Groups. Chem. Lett. 2016, 45, 409–411. [CrossRef]
- Sasamori, T.; Hironaka, K.; Sugiyama, Y.; Takagi, N.; Nagase, S.; Hosoi, Y.; Furukawa, Y.; Tokitoh, N. Synthesis and Reactions of a Stable 1,2-Diaryl-1,2-dibromodisilene: A Precursor for Substituted Disilenes and a 1,2-Diaryldisilyne. J. Am. Chem. Soc. 2008, 130, 13856–13857. [CrossRef]
- Michalczyk, M.J.; Fink, M.J.; DeYoung, D.J.; Carlson, C.W.; Welsh, K.M.; West, R.; Michl, J. Organosilylenes and Their Dimerization to Disilenes. Silicon, Germanium, Tin Lead Compd. 1986, 9, 75−83.
- Conlin, R.T.; Netto-Ferreira, J.C.; Zhang, Scaiano, J.C. Kinetic study of dimesitylsilylene by laser flash photolysis. Organometallics 1990, 9, 1332−1334. [CrossRef]
- Moiseev, A.G.; Leigh, W.J. Diphenylsilylene. J. Am. Chem. Soc. 2006, 128, 14442–14443. [CrossRef]
- Hayakawa, N.; Sugahara, T.; Numata, Y.; Kawaai, H.; Yamatani, K.; Nishimura, S.; Goda, S.; Suzuki, Y.; Tanikawa, T.; Nakai, H.; Hashizume, D.; Sasamori, T.; Tokitoh, N.; Matsuo, T. 1,2-Dihalodigermenes bearing bulky Eind groups: synthesis, characterization, and conversion to halogermylenoids, Dalton Trans. 2018, 47, 814−822. [CrossRef]
- Ando, W.; Hamada, Y,; Sekiguchi, A. Reactions of Oxasilacyclopropane. Generation of Silanediyl by Photo and Thermal Induced Cycloelimination. J. Chem. Soc., Chem. Commun., 1983, 952−954. [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.A., Jr.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N.J.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, Ö; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian 09, Rev. D.01; Gaussian, Inc.: Wallingford, CT, 2010.
- Kosa, M.; Karni, M.; Apeloig, Y. Were Reactions of Triplet Silylenes Observed? J. Am. Chem. Soc. 2013, 135, 9032–9040. [CrossRef]
- Guthardt, R.; Jacob, H.L.; Bruhn, C.; Siemeling, U. A complete series of N-heterocyclic tetrylenes (Si–Pb) with a 1,1’-ferrocenediyl backbone enabled by 1,3,2-diazaborolyl N-substituents. Dalton Trans. 2023, 52, 14380–14389. [CrossRef]
- Ishida, S.; Abe, T.; Hirakawa, F.; Kosai, T.; Sato, K.; Kira, M.; Iwamoto, T. Persistent Dialkylsilanone Generated by Dehydrobromination of Dialkylbromosilanol. Chem. Eur. J. 2015, 21, 15100−15103. [CrossRef]
- Krempner, C.; Martens, K.; Reinke, H. Synthesis and structure of silyl acetonitriles. J. Organomet. Chem. 2007, 692, 5799−5803. [CrossRef]
- CrystalClear; Rigaku/MSC. Inc.: The Woodlands, TX, USA, 2005.
- CrysAlisPro; Agilent Technologies Ltd.: Yarnton, Oxfordshire, UK, 2014.
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.: Caro, L.D.; Giacovazzo, C.; Polidori, G.; Siliqi, D.; Spagna, R. IL MILIONE: a suite of computer programs for crystal structure solution of proteins. J. Appl. Cryst. 2007, 40, 609−613. [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, A71, 3–8. [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [CrossRef]
Figure 1.
Examples of isolable two-coordinate silylenes A−J.
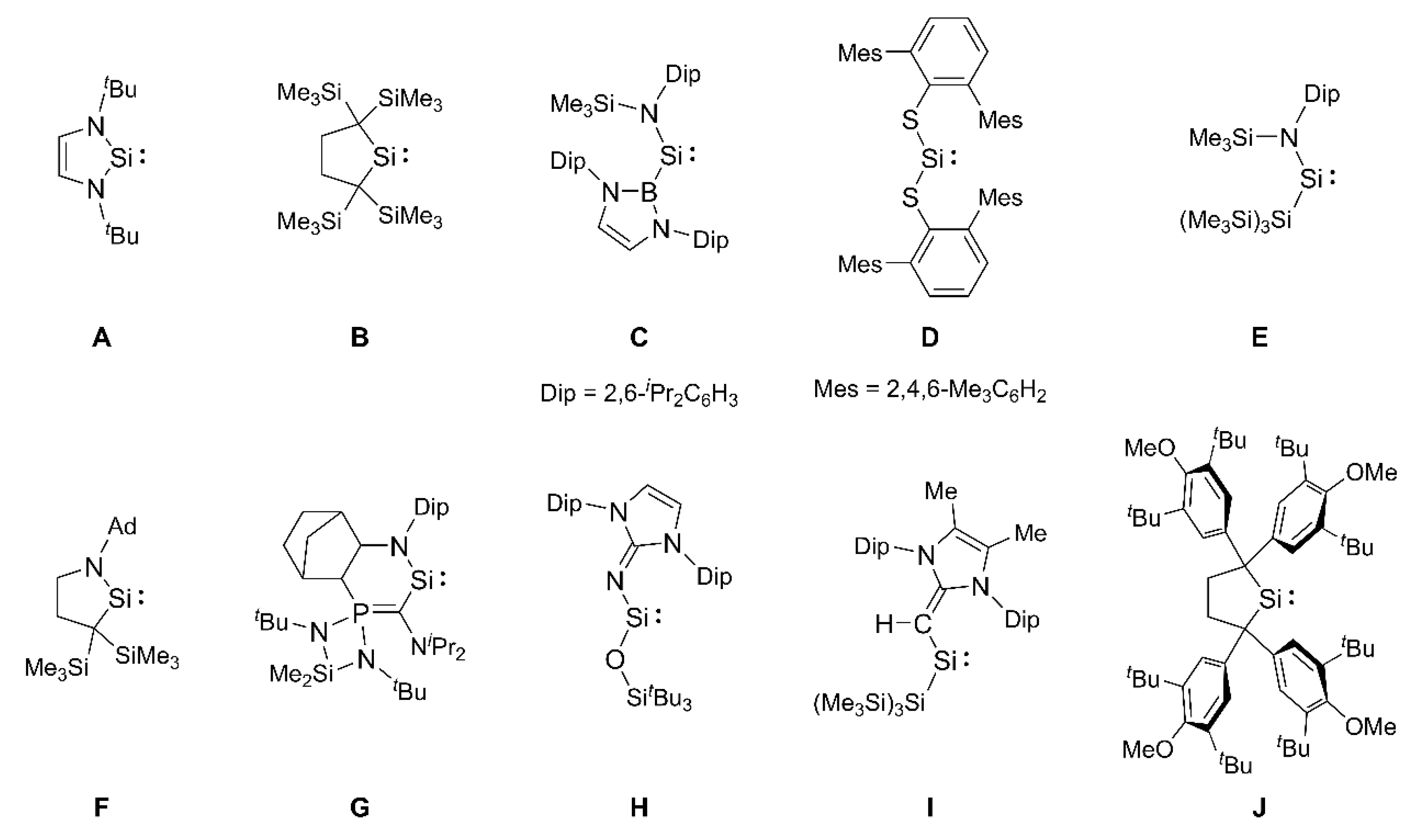
Scheme 1.
Photochemically-generated dimesitylsilylene K (a) and thermally-generated diarylsilylene L (b).
Scheme 1.
Photochemically-generated dimesitylsilylene K (a) and thermally-generated diarylsilylene L (b).
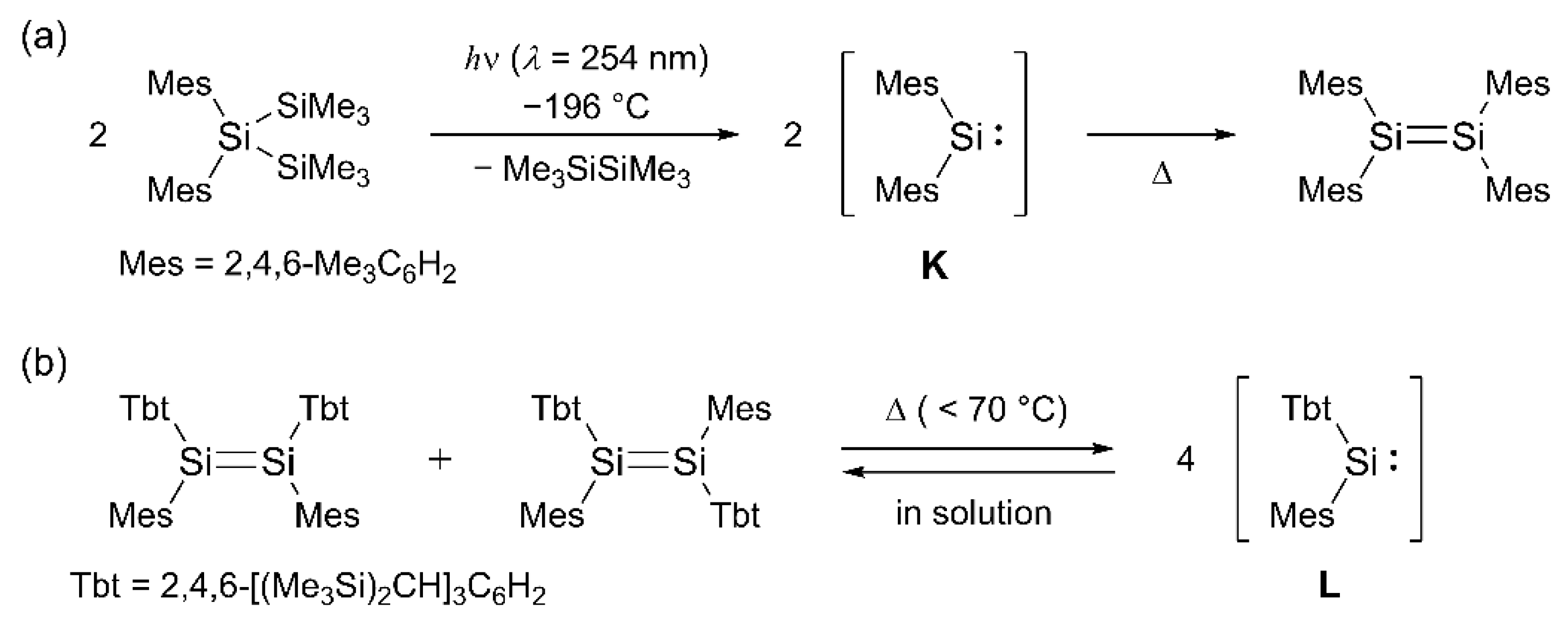
Figure 2.
Diarylsilylenes 3 (this study), diarylgermylenes M, and diarylstannylenes N with Rind groups.
Figure 2.
Diarylsilylenes 3 (this study), diarylgermylenes M, and diarylstannylenes N with Rind groups.
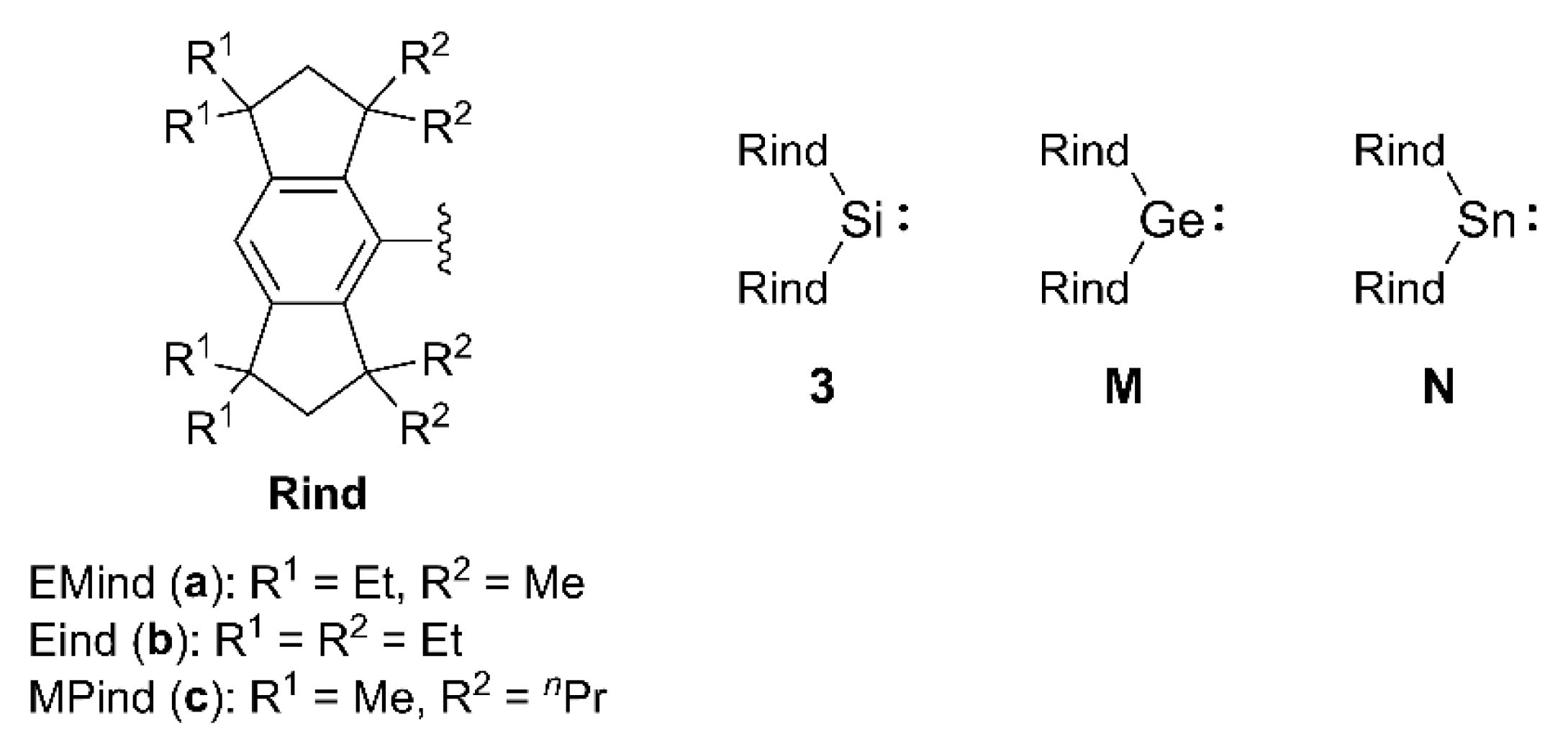
Scheme 2.
Formation, thermal reaction, and hydrolysis of 3.
Figure 3.
Pictures of the toluene solution containing 3b (a) and the crystals containing 3b (Crystal-B) (b).
Figure 3.
Pictures of the toluene solution containing 3b (a) and the crystals containing 3b (Crystal-B) (b).

Figure 4.
Molecular structures of 3b (a) and 4b (b) (Crystal-A). Thermal ellipsoids are drawn at 50% probability level. The hydrogen atoms, except for those on the Si1A, C55A, and C56A atoms in 4b, are omitted for clarity.
Figure 4.
Molecular structures of 3b (a) and 4b (b) (Crystal-A). Thermal ellipsoids are drawn at 50% probability level. The hydrogen atoms, except for those on the Si1A, C55A, and C56A atoms in 4b, are omitted for clarity.
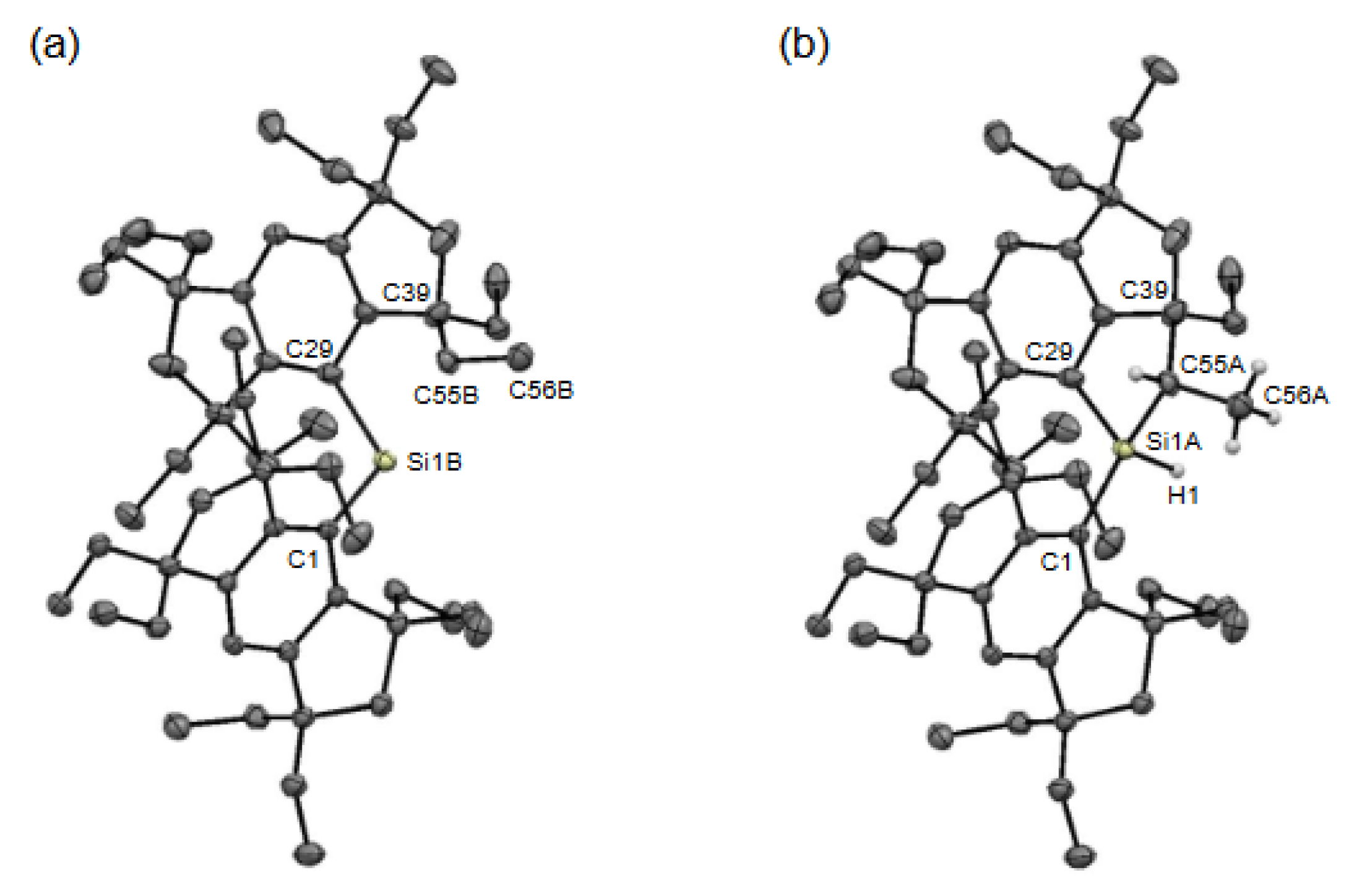
Figure 5.
Molecular structure of 4b (Crystal-B). Thermal ellipsoids are drawn at 50% probability level. The hydrogen atoms, except for those on the Si1, C13, and C3 atoms, are omitted for clarity.
Figure 5.
Molecular structure of 4b (Crystal-B). Thermal ellipsoids are drawn at 50% probability level. The hydrogen atoms, except for those on the Si1, C13, and C3 atoms, are omitted for clarity.
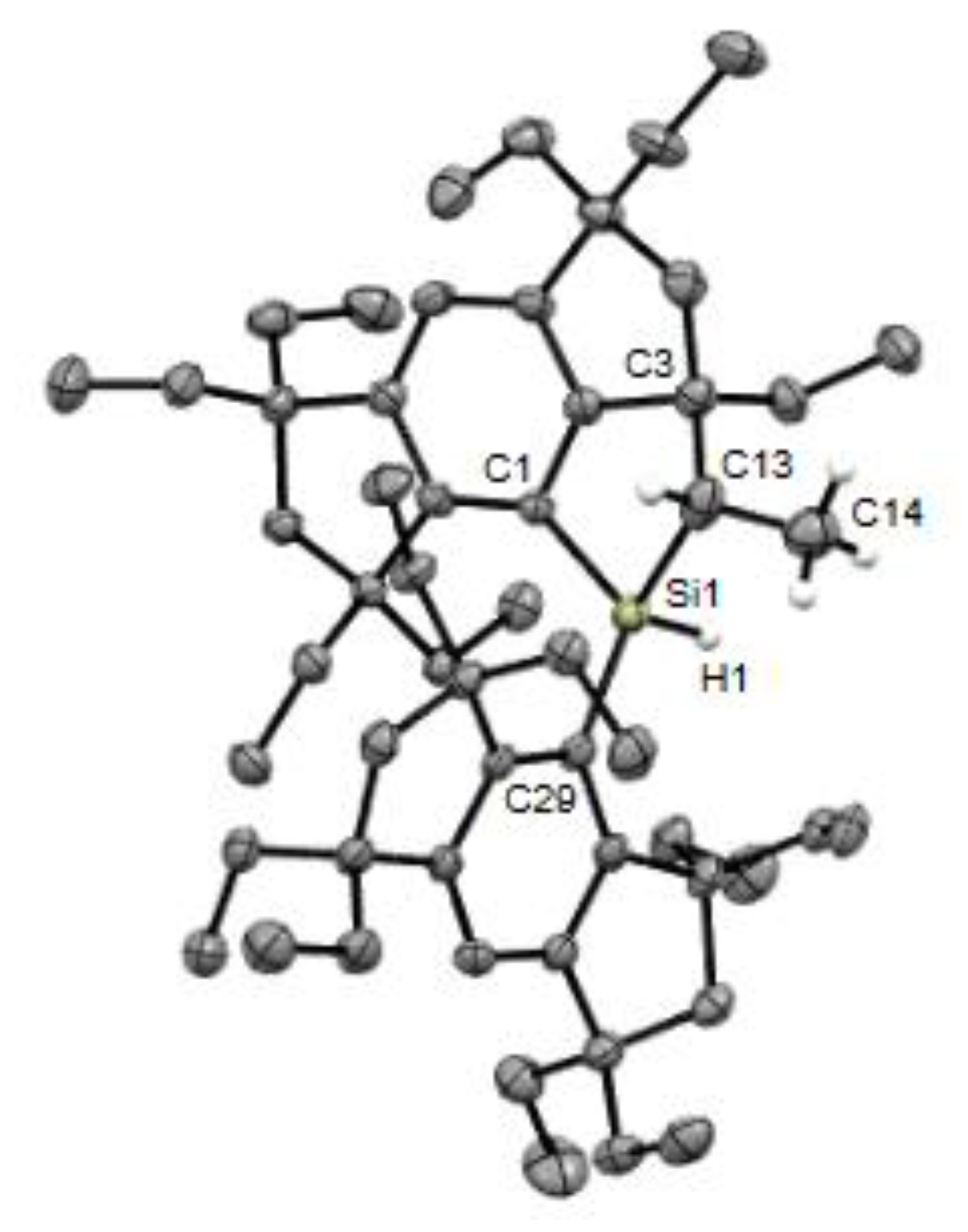
Figure 6.
29Si NMR spectrum of the solution containing 3b in C7D8 at −20 °C.
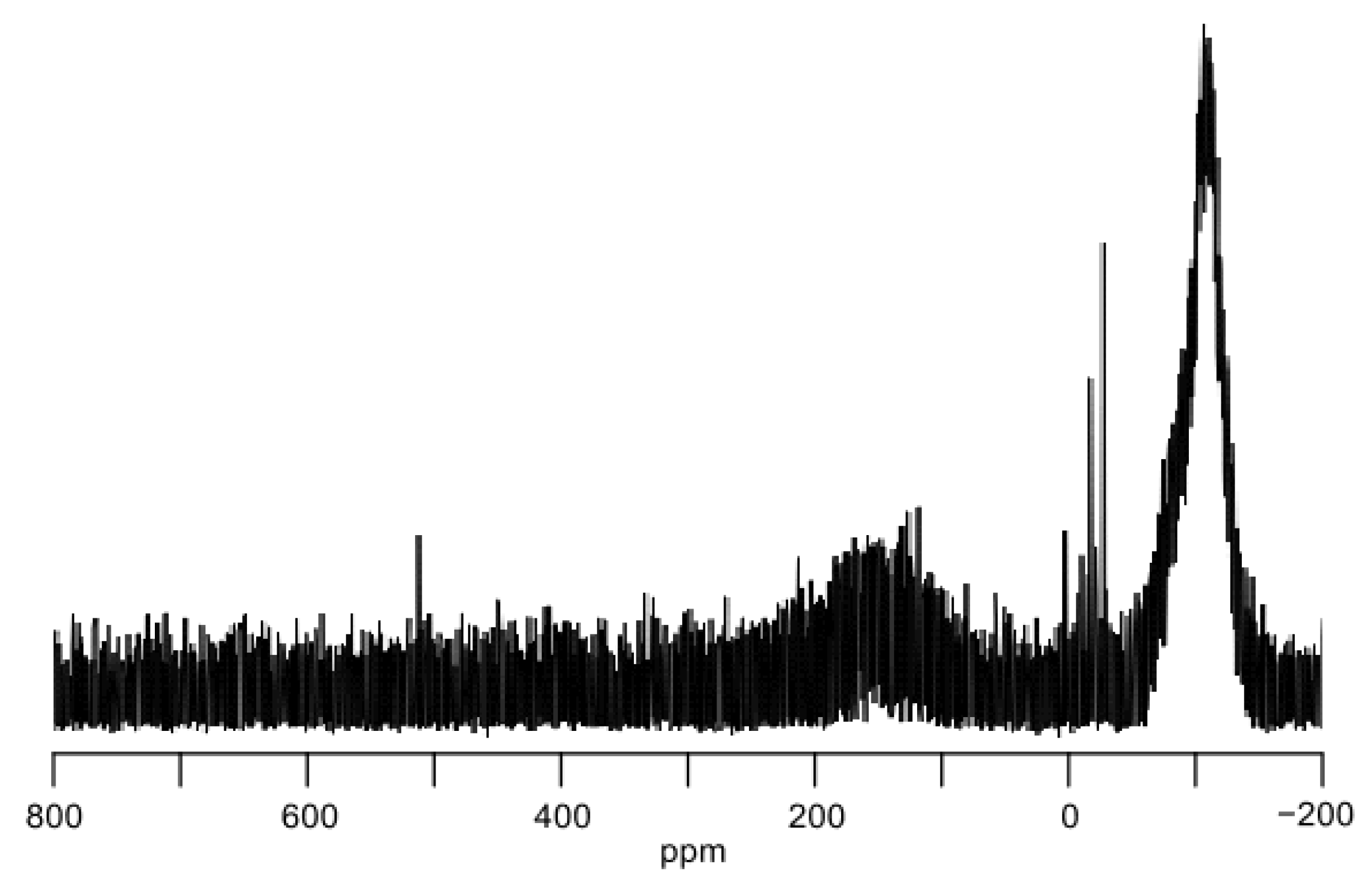
Figure 7.
Selected MOs of 3b in the singlet state (front views), together with the energy levels.
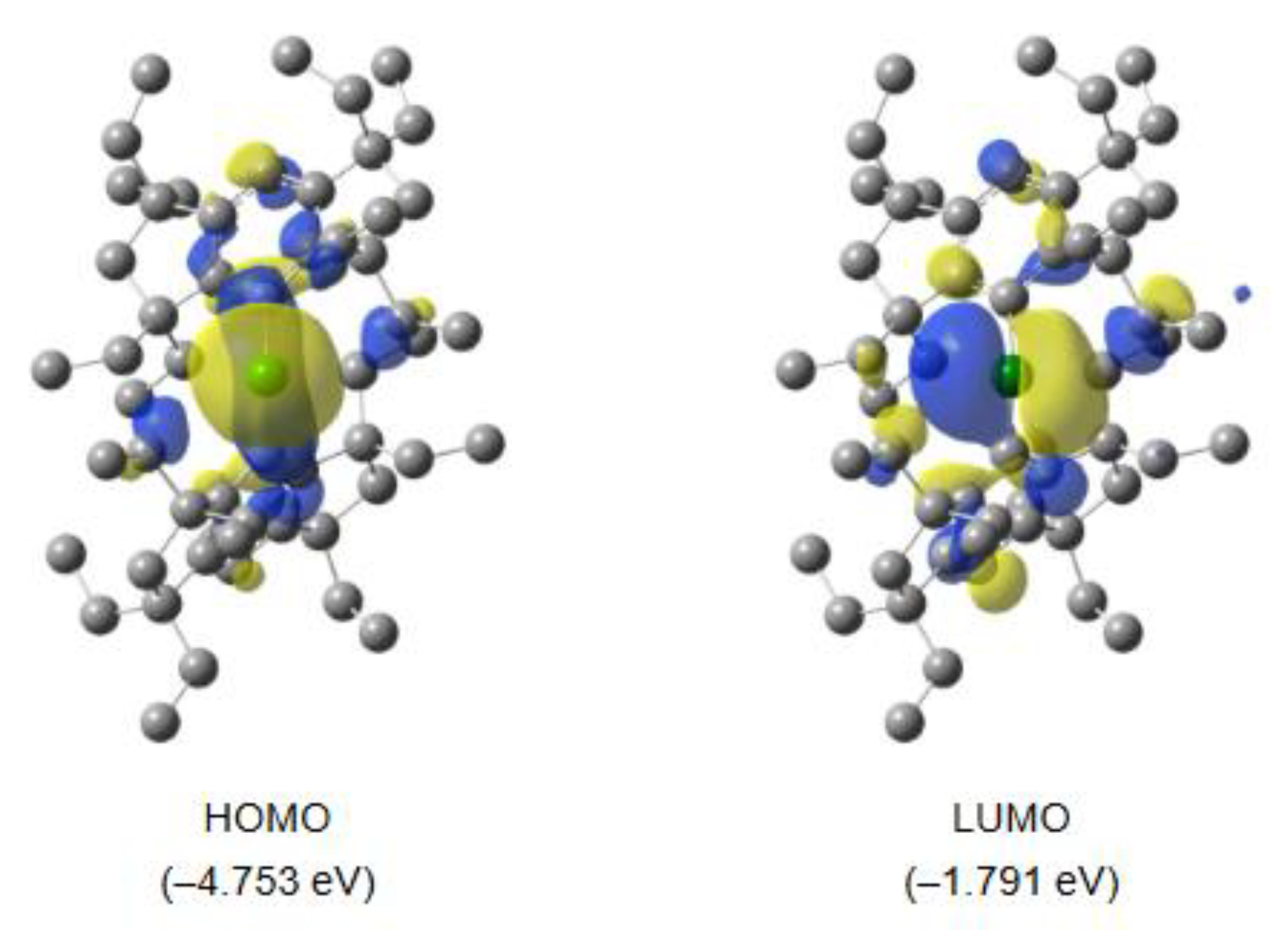
Figure 8.
Molecular structure of 5b. Thermal ellipsoids are drawn at 50% probability level. The hydrogen atoms, except for those on the Si1 and O1A atoms, and disordered O atom are omitted for clarity.
Figure 8.
Molecular structure of 5b. Thermal ellipsoids are drawn at 50% probability level. The hydrogen atoms, except for those on the Si1 and O1A atoms, and disordered O atom are omitted for clarity.
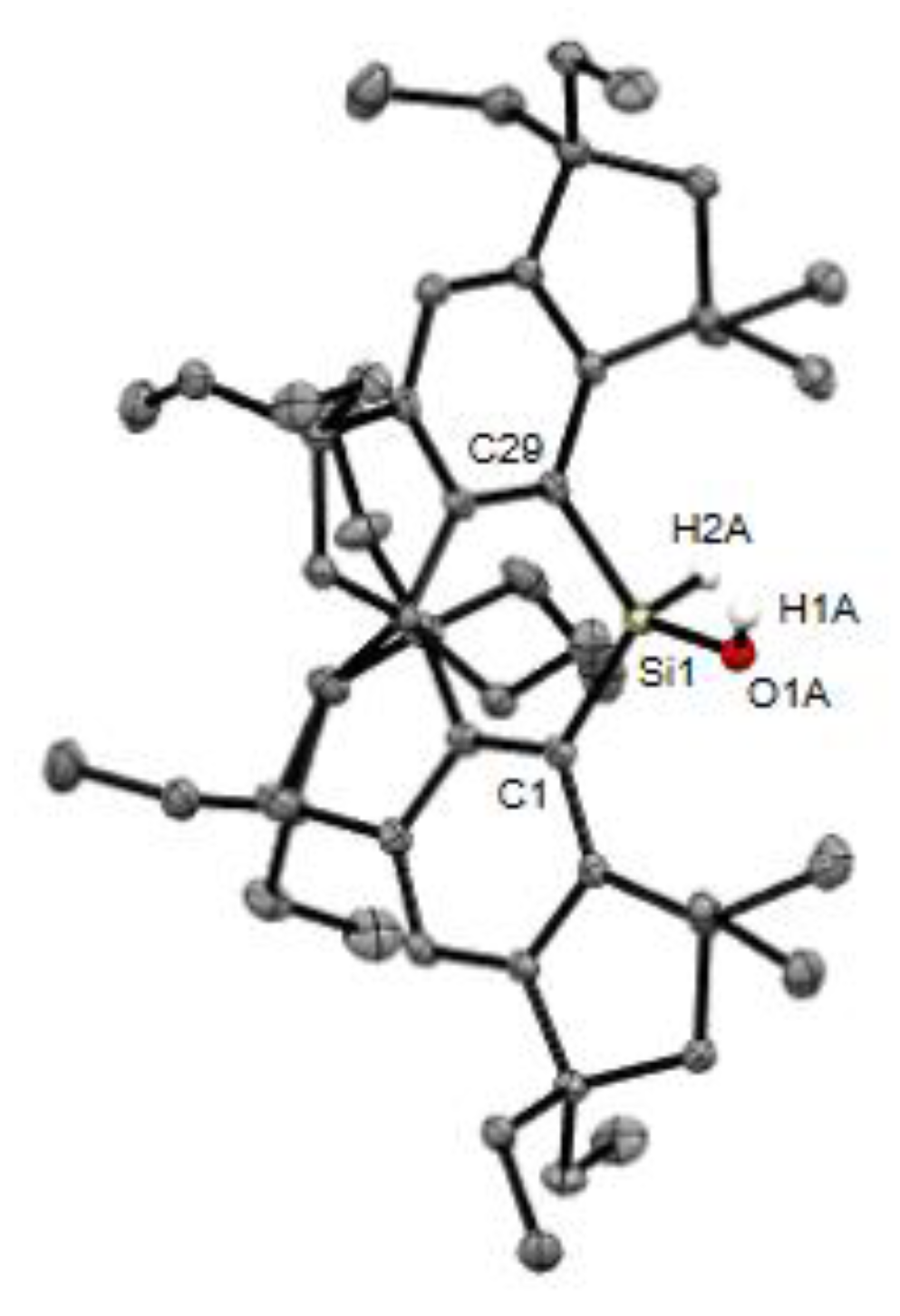

Table 1.
Selected structural parameters.
| Compounds | Si−C (Å) | C(sp2)−Si−C(sp2) (°) |
|---|---|---|
| experimental | ||
| 1b [37] | 1.950(4), 1.938(5) | 123.60(17) |
| 2b [37] | 1.934(3) | 116.60(13) |
| 3b (Crystal-A) | 1.941(5), 2.004(5) | 112.3(2) |
| 4b (Crystal-A) | 1.919(2), 1.873(3), | 119.57(12) |
| 1.904(4) | ||
| 4b (Crystal-B) | 1.899(3), 1.909(2), | 122.11(11) |
| 1.964(3) | ||
| 5b | 1.9090(11), 1.9228(12) | 122.05(5) |
| calculations | ||
| 3b (singlet) | 1.9470, 1.9489 | 112.00 |
| 3b (triplet) | 1.8980, 1.8926 | 127.95 |
| 4b | 1.9099, 1.9012, | 119.50 |
| 1.9358 | ||
| 5b | 1.9294, 1.9174 | 121.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



