
Submitted:
06 March 2024
Posted:
07 March 2024
You are already at the latest version
Abstract
Spinocerebellar ataxia type 3 is a neurodegenerative disease caused by an abnormal expansion of CAG repeats in the disease gene ATXN3, leading to prolonged polyglutamine (polyQ) tracts in the respective protein ataxin-3. The polyQ-expanded protein forms polyQ-containing aggregates in several brain regions which leads to neuronal cell loss. Autophagy is thought to play a significant role in clearing these polyQ-containing protein aggregates. Therefore, the autophagy mechanism was investigated in SCA3 with focus on the autophagic protein p62/sequestosome-1. Human and mouse RNA sequencing data were analyzed to determine RNA expression level of autophagic genes. Additionally, autophagy was studied in the SCA3 304Q knock-in mouse model and through several cell culture experiments using HEK-293T cells transfected with ATXN3 of differ-ent polyQ lengths. For cell culture experiments, autophagy was induced with rapamycin treat-ment, proteasomal degradation was blocked with MG132 and degradation of autophagic pro-teins was investigated using cycloheximide treatment. Here, we report an increased autophagy impairment with SCA3 disease progression seen in HEK-293T cells transfected with ATXN3 plasmid with expanded polyQ tracts, higher aged 304Q-knock-in mice, or human post-mortem brain material. Furthermore, co-localized aggregation of the autophagic protein p62 and the dis-ease protein ATXN3 was observed in the course of SCA3 disease. Therefore, we suggest a signifi-cant role for p62 in aggregate formation seen in the neurodegenerative disease SCA3.
Keywords:
Spinocerebellar ataxia type 3
; ataxin-3
; p62/sequestosome-1
; autophagy
; autophagy impairment
; co-localized aggregation
1. Introduction
Polyglutamine (polyQ) diseases include nine neurodegenerative diseases with spinocerebellar ataxia type 3 (SCA3) being one of the most common [1,2]. SCA3 is caused by an abnormal expansion of CAG repeats in the coding region of the disease gene ATXN3, generating elongated polyQ tracts in the ataxin-3 (ATXN3) protein [2]. An important characteristic of SCA3 is the formation of protein aggregates in neurons of several brain regions [3]. The expansion of the polyQ repeat causes misfolding of the affected protein ATXN3, resulting in polyQ-containing aggregates that are associated with disease progression [3]. Affected brain regions of SCA3 are cerebellum, substantia nigra, striatum, thalamus, pontine nuclei and other areas of the central nervous system [4]. PolyQ-containing aggregates do not exist in isolation but sequester other proteins such as chaperones and proteins of the ubiquitin-proteasome-system [5]. Additionally, Sittler and colleagues identified aggregates as nuclear accumulations positive for ATXN3, ubiquitin and the autophagic protein p62/sequestosome-1 in post-mortem brains of SCA3 patients [6]. Moreover, further autophagic proteins like LC3, ATG5, ATG12 or ATG16L has been shown to be recruited into aggregates of polyQ proteins like ATXN3 [7]. Autophagy, a process of cellular degradation and recycling, could play a significant role in clearance of protein aggregates in polyQ diseases [8,9]. However, it was shown that polyQ-expanded ATXN3 leads to an impairment of autophagy already in early disease stages of SCA3 [10]. Previous studies in zebrafish, patient-derived induced pluripotent stem (iPSCs) cells and a transgenic mouse model indicated that an induction of autophagic activity being beneficial for disease progression of SCA3 [10,11,12]. The ubiquitin-binding protein p62, also called sequestosome-1 (SQSTM1), normally functions as a receptor for selective autophagic degradation of ubiquitinated proteins [9]. Inhibiting autophagy activity leads to accumulation of p62 inclusions [9]. Since p62 interacts with the autophagic protein LC3, p62 itself is a substrate of autophagy whose levels seem to increase due to autophagy impairment [9]. To date it is unclear whether aggregation of polyQ-containing proteins is a consequence of autophagy dysregulation or whether autophagy is enhanced as a defense mechanism in polyQ disorders [9].
In the current study, the autophagy mechanism was studied in cell culture experiments using HEK-293T cells transfected with ATXN3 plasmids of different polyQ lengths, in a SCA3 304Q knock-in (KI) mouse model [13], and in post-mortem human brain material of SCA3 patients. In cell culture experiments, autophagy was induced by inhibition of the main regulator mTOR using rapamycin treatment [8]. Furthermore, proteasomal degradation was blocked with the specific inhibitor MG132 and to investigate degradation of the autophagic proteins cycloheximide treatment was performed [14,15]. RNA sequencing data from human post-mortem brain material of SCA3 patients and of SCA3 KI mice revealed differential RNA expression of autophagic genes including p62.
Here we show that the receptor for selective autophagy, p62, plays a significant role in aggregate formation developing in the course of SCA3. With SCA3 disease progression, aggregates containing only ATXN3 or p62, as well as co-localized aggregates comprising both proteins, were found. We concluded that, the increased aggregation of p62 and ATXN3 is thought to be caused by an increase of autophagy impairment.
2. Materials and Methods
2.1. Cell Culture and Transfection
HEK-293T cells (ATCC: CRL-3216) were cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco, Thermo Fisher Scientific, Waltham, USA) supplemented with 10 % fetal bovine serum (Roche Diagnostics GmbH, Mannheim, DE) and 1 % antibiotic-antimycotic (Gibco, Thermo Fisher Scientific) at 37 °C and 5 % CO2. Cells were passaged at 80 – 90 % confluence.
HEK-293T cells were transfected with plasmids encoding V5-tagged ATXN3 and different polyQ repeat length (V5ATXN3-15Q, V5ATXN3-77Q or V5ATXN3-148Q). Transfection was performed using the Attractene transfection reagent (Qiagen, Hilden, DE) following the standard transfection protocol. Briefly, 24 h before transfection, 400 000 cells per well were seeded on a six-well tissue culture plate in DMEM medium. Cells were transfected with 1.2 µg plasmid DNA and incubated 72 h at 37°C and 5 % CO2.
2.2. Pharmacological Treatments and Harvest of HEK-293T Cells
To induce autophagy in HEK-293T cells transfected with ATXN3 plasmids, cells were treated with 100 nM rapamycin for 6 h prior to harvesting. For inhibition of proteasomal degradation, cells were treated with 10 µM MG132 for 6 h prior to harvesting. HEK-293T cells transfected with ATXN3 and were treated with 10 µM cycloheximide for 0 h or 1 h, 5 h and 12 h prior to harvesting to inhibit protein translation. For all treatment conditions dimethyl sulfoxide (DMSO) was used as treatment control.
After incubation, treated HEK-293T cells were harvested using cold DPBS (Thermo Fisher Scientific). A cell pellet was generated by centrifuging the harvested cells at 350 g for 5 min. Cells were lysed in RIPA buffer (50 mM Tris pH 7.45; 150 mM NaCl; 0.1 % (w/v) SDS; 0.5 % (w/v) sodium deoxycholate; 1 % (v/v) Triton X-100;) supplemented with cOmplete protease inhibitor cocktail (Merck KGaA, Darmstadt, D) on ice for 30 min with vortexing every 10 min.
2.3. Sample Preparation for Western Blot, Filter Trap and DD-AGE Analysis
For Western Blot, Filter Trap and Denaturing Detergent-Agarose Gel Electrophoresis (DD-AGE) analysis, protein extracts from cell culture experiments and animal tissue were used. Brain tissue samples (cerebellum) of 3- and 18-months old male and female wildtype (WT), heterozygous SCA3 (WT/304Q) and homozygous SCA3 (304Q/304Q) KI mice were obtained from a previous study [13].
Prior to Western Blot analysis samples were mixed with 25 % (v/v) 4X LDS-buffer (2.5 M Tris pH 8.5; 50 % glycerol; 2.5 % phenol red; 2.1 mM EDTA; 294 mM LDS) and 100 mM DTT to a final protein concentration of 30 µg. Samples were heat denatured for 10 min at 70°C and 600 rpm in a thermoshaker.
In order to perform a Filter Trap analysis, samples were mixed with 1X DPBS, 2 % (w/v) SDS and 50 mM DTT to a final protein concentration of 1 µg and afterwards heated for 5 min at 95°C.
DD-AGE samples were mixed with 25 % (v/v) 4X DD-AGE buffer containing 2X TAE buffer (40 mM Tris; 20 mM acetic acid; 1mM EDTA 0.1 % SDS) and 50 % (v/v) glycerol, 8 % (w/v) SDS, and 0.01 g Orange G leading to a final protein concentration of 25 µg.
2.4. Western Blot Analysis
Western Blot analysis was performed according to standard procedures [16]. Reduced proteins were separated according to their size by SDS-PAGE using a Mini-PROTEAN® Tetra Cell (Bio-Rad Laboratories, Hercules, CA, USA). For stacking gel 6 % (w/v) acrylamide/bis-acrylamide (29:1) and for separating gel 12 % (w/v) acrylamide/bis-acrylamide (29:1) was used. SDS-PAGE was performed using a 1X MOPS running buffer (50 mM MOPS, 50 mM Tris, 0.1% (w/v) SDS, 1 mM EDTA at 100 V and 250 mA.
For protein transfer, a nitrocellulose membrane with 0.2 µm pore size was used (Amersham Protran Premium, Cytiva, Marlborough, MA, USA). A wet transfer was performed using 1X Bicine/Bis-Tris transfer buffer (25 mM Bicine, 25 mM Bis-Tris, 1 mM EDTA) containing 15 % (v/v) methanol. Transfer was carried out at 80 V and 250 mA for 2 h.
After transferring the proteins, the membrane was blocked for 1 h using 5 % (w/v) milk powder in 1X TBST buffer (10 mM Tris pH 7.5; 150 mM NaCl; 0.1 % (v/v) Tween 20). For protein detection, the membrane was incubated with a primary antibody diluted in TBST buffer overnight at 4°C. Primary antibodies used for Western Blot detection are listed in Table 1. Subsequently, the membrane was incubated with a fluorescence-coupled secondary antibody for 1 h at RT. Depending on the primary antibody species, IRDye 800CW-labelled goat anti-rabbit (926-32211) or IRDye 800CW-labelled goat anti-mouse (926-32210) secondary antibodies (both from LI-COR Biosciences, Lincoln, NE, USA) were used, diluted 1:5000 in 1X TBST buffer. Immunodetection was performed with the Odyssey FC instrument and Image Studio 4.0 software (both LI-COR Biosciences).
2.5. Filter Trap Analysis
Filter Trap analysis was performed according to standard protocols using the Minifold II Slot-Blot System (Schleicher & Schuell, Düren, DE) [16]. Nitrocellulose membrane with 0.45 µm pore size (Amersham Protran, Cytiva) were used. The membrane was first equilibrated two times with equilibration buffer (1X DPBS; 0.1 % (w/v) SDS). Afterwards, the samples were loaded onto the membrane applying a vacuum. The membrane was washed twice with 1X DPBS. Blocking and immunodetection were performed according to Western Blot analysis. As a primary antibody against ATXN3, Ataxin-3 Monoclonal Antibody (clone 2SCA-1H9) was used (MA3-082, Thermo Fisher Scientific) with a dilution of 1:2500 in 1X TBST. For fluorescence detection the IRDye 800CW-labbeled goat anti-mouse (926-32210) secondary antibody (LI-COR Biosciences) with a 1:5000 dilution in 1X TBST was used. Immunodetection was performed with the Odyssey FC instrument and Image Studio 4.0 software (both LI-COR Biosciences).
2.6. DD-AGE Analysis
The DD-AGE analysis was performed to detect p62- and ATXN3-aggregates according to the SDD-AGE protocol of Halfmann [17]. Modifications of the protocol are mentioned below.
First, an agarose gel with 1 % (w/v) agarose in 50 mL 1X TAE buffer (40 mM Tris; 20 mM acetic acid; 1 mM EDTA) and 0.1 % (w/v) SDS was poured in a PerfectBlueTM Gel System Mini S from Peqlab. A 1X TAE running buffer was used containing 0.1 % (w/v) SDS. Gel electrophoresis was performed at 40 V for approximately 1.5 h until the running front migrated 4 cm. Transfer of the proteins onto the nitrocellulose membrane was performed according to Western Blot analysis except for the Bicine/Bis-Tris transfer buffer containing 10 % methanol. Blocking of the membrane and immune detection was identical to Western Blot analysis. For p62 detection, the primary antibody 5114S (Cell Signaling Technology) was used in a 1:1000 dilution in 1X TBST. ATXN3 was detected with the Ataxin-3 Monoclonal Antibody (clone 2SCA-1H9, MA3-082, Thermo Fisher Scientific) diluted 1:2500 in TBST.
2.7. Immunohistochemistry Staining
Immunohistochemistry (IHC) staining was performed to stain the autophagic proteins p62 and BECN1 as well as the SCA3 disease protein ATXN3 in brain slices of wildtype (WT/WT) and homozygous 304Q-KI (304Q/304Q) mice. First, brain slices were deparaffinized and washed 3-times for 5 min with 1X PBS. Then, a 3 min microwave treatment for epitope retrieval was performed using 1X citric acid and 1X sodium citrate. Brain slices were washed again 3-times with 1X PBS for 5 min each. Afterwards, brain slices were blocked with 5 % (v/v) normal goat serum and 0.3 % (v/v) Triton X-100 in 1X PBS (770 mM NaCl; 319 mM NaH₂PO₄ * H₂O; 99.7 mM Na₂HPO₄ * 2 H₂O) and washed with 1X PBS prior to immunostaining. Primary antibodies were diluted in 1X PBS with 15 % (v/v) normal goat serum and incubated overnight at 4°C in a humidity chamber on the slices. Primary antibodies used for staining are listed in Table 2. Brain slices were washed 3-times with 1X PBS for 5 min each. Secondary antibodies were also diluted in 1X PBS with 15 % (v/v) normal goat serum and incubated for 1 h at RT. Depending on the species, the goat-anti-rabbit IgG (H+L) diluted 1:250 or the goat-anti-mouse IgG (H+L) diluted 1:200 (both from Vector Laboratories) was used. Staining was carried out incubating an Avidin–Biotin Complex (ABC) (Vectastain ABC KIT, Elite PK-6100 Standard; Vector Laboratories) for 2 h on the brain slices and using 3,3'-Diaminobenzidine (Sigma-Aldrich) staining. The reaction was stopped by adding water. Brain slices were dehydrated after staining and sealed with the Leica CV ultra-mounting medium (Leica Biosystems, Nussloch, DE), coverslips and nail polish.
2.8. Immunofluorescence Staining
Immunofluorescence (IF) staining was performed to stain the autophagic protein p62 and ATXN3 on brain slices of WT/WT and 304Q/304Q mice. Deparaffinization, microwave treatment and blocking were identical with IHC staining. After blocking, the brain slices were washed 3-times for 10 min with 1X PBS. Primary antibody against ATXN3 (clone 1H9, MAB5360, Merck) was diluted 1:500 and mixed with an antibody against p62 (5114S, Cell Signaling Technology) that was diluted 1:250 in 1X PBS and 3 % normal goat serum. Primary antibodies were incubated overnight at 4°C in a humid chamber. Afterwards brain slices were washed 3-times for 10 min each with 1X PBS. Brain slices were incubated with fluorescent secondary antibodies in the following order: goat-anti-mouse-Alexa488 antibody (ab150077, Abcam) was added followed by the goat-anti-rabbit-Alexa555 (A-10667, Thermo Fisher Scientific), both diluted 1:500 in 1X PBS with 3 % (v/v) normal goat serum and each incubated for 1 h at RT. The brain slices were sealed using VECTASHIELD® Antifade Mounting Medium (Vector Laboratories) containing DAPI to stain cell nuclei, coverslips, and nail polish. Stained brain slices were stored at 4°C until microscopy.
2.9. Microscopy
Images of the stained brain slices were taken with the Axioplan 2 Imaging fluorescence microscope containing an Apotome, a Plan-Neofluar 20 x /0.50 objective and the AxioXam MRm camera (Zeiss, Oberkochen, DE). Software used was the AxioVision SE64 Rel. 4.9.1 (Zeiss).
2.10. RNA Sequencing of Human and Mouse Cerebellar RNA
RNA isolation, RNA sequencing protocol and bioinformatic analyses of RNA sequencing were already described in Haas et al., 2021. SCA3 mouse raw sequencing files are available through GEO under accession number: GSE145613. Human RNA-seq data set has been deposited at the European Genome-phenome Archive (EGA) under the accession number: EGAS00001004241.
2.11. Statistical Analysis
Quantification of Western Blot, Filter Trap and DD-AGE results was performed with the LI-COR Image Studio program (Image Studio Lite Ver 5.2; Image Studio Ver 2.1; LI-COR® Odyssey® Fc Imaging System).
Statistical analysis was performed with the GraphPad Prism software 6 using one-way ANOVA, Student’s t-test or Tukey´s multiple comparison test. The data are presented as bar charts showing means ± standard error of the mean (SEM). Statistical significance was indicated by asterisks representing p-values ≤ 0.05 (*), ≤ 0.01 (**), and ≤ 0.001 (***).
3. Results
3.1. Downregulation of p62 RNA Expression in SCA3 Patients
Dysregulation of autophagic proteins was already shown in post-mortem brain samples of SCA3 patients compared to age- and sex-matched controls [6]. Increasing evidence suggest that the dysregulation is not only present on protein but also already exist on transcriptional level [18]. Therefore, we statistically evaluated RNA sequencing data of human post-mortem cerebellar brain samples from SCA3 mutation carriers compared to respective controls based on a previously published dataset [13]. SCA3 mutation carriers demonstrated a significant reduced BECN1 expression (p = 0.048), but no RNA expression changes of mTOR, LC3 (MAP1LC3B), and p62 compared to respective controls (p > 0.1; Figure 1A). Further autophagy genes which demonstrated dysregulation at protein levels in SCA3 [6] including ATG7, ATG12, ATG16L12, and RAB1A showed no changes in RNA expression (p > 0.1), whereas the lysosome specific protein LAMP2 (p = 0.009) and a key regulator of autophagy, RAB7A (p = 0.0087), revealed a significant downregulation in SCA3 (Supplementary Figure S1). Evaluation of RNA expression of autophagic genes in homozygous SCA3 KI mice compared to wildtype mice revealed no RNA expression changes of BECN1, mTOR, LC3, and p62 in young animals (2 months old), but an increased expression of p62 in 12 months old SCA3 KI mice (Table 3). Interestingly, comparing young (2 months old) and old (12 months) wildtype as well as young and old SCA3 KI animals revealed a strong dysregulation of autophagic genes including BECN1, mTOR, LC3, and p62 during aging in both genotypes (Table 3). Similar effects were observed for the beforehand mentioned important autophagy regulators (Supplementary Table S1). These results suggest an autophagy impairment in aged mice independent of the genotype.
Several studies reported on the important role of p62 in protein aggregation processes [19,20,21] and highlighted that several domains of p62 are involved in protein-protein interaction mediating protein aggregation [22]. The two main protein coding isoforms of p62 differ at the start codon position: (I) reference isoform p62-202 (ENST00000389805) is a 440 amino acid (aa) large protein, whereas p62 isoform p62-201 (ENST00000360718) use another translation initiation at methionine 85 and therefore resulting in a truncated p62 variant lacking parts of the PB1 domain, which is essential for p62 self-oligomerization and therefore critical in the formation of autophagosomes [23,24]. Therefore, we analyzed the expression of different human p62 isoforms in our human post-mortem RNA sequencing data and found a significant downregulation of the reference isoform p62-202 in SCA3 patients compared to controls. The truncated p62-201 isoform showed similar levels in both genotypes (Figure 1B,C). Of the 16 known p62 isoforms annotated at Ensembl (Ensembl, last visit 11.01.2014), only 5 isoforms (including 3 protein coding, 2 CDS not defined and two retained intron transcripts) were detected in the majority of analyzed probands. Another 4 isoforms were not found in any of the post-mortem brain sample (Figures 1B,C and Supplementary Figure S2).
Figure 1.
Total RNA expression profile of selected autophagic marker genes in post-mortem cerebellar brain samples of SCA3 patients. (A) Total BECN1, mTOR, LC3 and p62 gene expression determined in cerebella post-mortem brain of SCA3 patients and unaffected controls using RNA sequencing. (B) Schematic representation of p62 isoforms based on Ensembl annotation. (C) p62 mRNA isoform expression in cerebellar post-mortem brain samples comparing 5 male healthy controls and 5 male SCA3 patients. TPM = transcript per million reads. * p < 0.05.
Figure 1.
Total RNA expression profile of selected autophagic marker genes in post-mortem cerebellar brain samples of SCA3 patients. (A) Total BECN1, mTOR, LC3 and p62 gene expression determined in cerebella post-mortem brain of SCA3 patients and unaffected controls using RNA sequencing. (B) Schematic representation of p62 isoforms based on Ensembl annotation. (C) p62 mRNA isoform expression in cerebellar post-mortem brain samples comparing 5 male healthy controls and 5 male SCA3 patients. TPM = transcript per million reads. * p < 0.05.
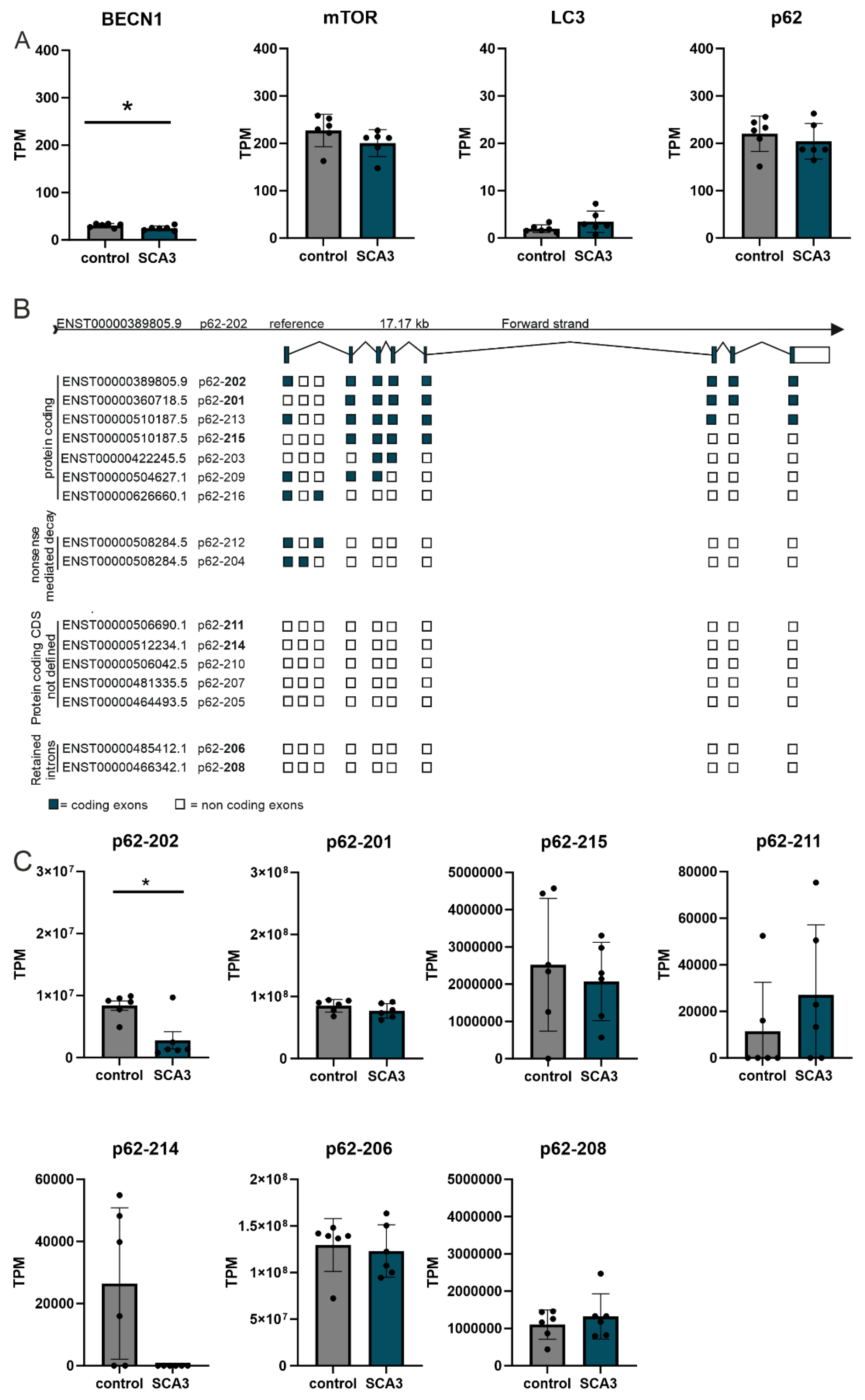

Table 3.
RNA expression of autophagic genes in SCA3 304Q KI mice at 2 and 12 months of age and in human SCA3 post-mortem cerebellum samples comparing healthy controls to SCA3 disease mice or patients. For analyzing aging effects, regulation of autophagic genes were evaluated by comparing young (2 months) vs old WT or SCA3 mice (12 months), respectively. All data from RNA sequencing data from cerebellum, all n=5-6 per genotype and age, all from male mice/subjects, M = months. Negative values implicate a downregulation in SCA3 contrasts and positive values an up-regulation always compared to respective controls.
Table 3.
RNA expression of autophagic genes in SCA3 304Q KI mice at 2 and 12 months of age and in human SCA3 post-mortem cerebellum samples comparing healthy controls to SCA3 disease mice or patients. For analyzing aging effects, regulation of autophagic genes were evaluated by comparing young (2 months) vs old WT or SCA3 mice (12 months), respectively. All data from RNA sequencing data from cerebellum, all n=5-6 per genotype and age, all from male mice/subjects, M = months. Negative values implicate a downregulation in SCA3 contrasts and positive values an up-regulation always compared to respective controls.
| SCA3 304Q 2 months compared to WT |
SCA3 304Q 12 months compared to WT |
aged WT 2M vs. 12M aging WT |
aged SCA3 304Q 2M vs. 12M aging knock-in (KI) |
human SCA3 post-mortem compared to CNTR |
|
|---|---|---|---|---|---|
| BECN1 | logFC = -0.033 | logFC = 0.057 | logFC = -0.684 | logFC = -0.6645 | logFC = -0.297 |
| p = -0.58 | p = 0.35 | p = 3.82E-22 | p = 1.24E-21 | p = 0.049 | |
| mTOR | logFC = -0.069 | logFC = 0.003 | logFC = 0.366 | logFC = 0.286 | logFC = -0.162 |
| p = -0.151 | p = 0.93 | p = 2.86E-06 | p = 0.00014 | p = 0.21 | |
| LC3 = MAP1LC3B | logFC = -0.013 | logFC = -0.060 | logFC = -1.038 | logFC = 0.085 | logFC = 0.800 |
| p = -0.778 | p = 0.198 | p = 3.75E-11 | p = 0.086 | p = 0.087 | |
| p62 = SQSTM1 | logFC = 0.019 | logFC = 0.110 | logFC = -0.441 | logFC = -0.396 | logFC = -0.088 |
| p = -0.76 | p = 0.002 | p = 9.83E-09 | p = 1.29E-07 | p = 0.52 |
3.2. Impairment of Autophagy with Increasing Age of SCA3 KI Mice
To investigate autophagy in a SCA3 mouse model several autophagy proteins were examined in wildtype (WT/WT), heterozygous (WT/304Q), and homozygous (304Q/304Q) mice of the 304Q-KI mouse line using Western Blot and DD-AGE analysis (Figure 2). Investigated autophagic proteins included the receptor for selective autophagy of ubiquitinated proteins, p62, and the autophagy regulator BECN1 [9,25]. Furthermore, a ribosomal protein phosphorylated und activated by mTOR, phospho-S6, as well as LC3, the regulator of expansion and closure of the phagophore during autophagy [26,27]. Western Blot analyses and quantification in 3 months old SCA3 mice revealed a significant reduction of soluble p62 between WT/WT and WT/304Q (p = 0.0464), whereas a tendency was observed between WT/WT and 304Q/304Q mice (p = 0.2959, Figure 2B,C). This soluble p62 protein reduction was also present in 18 months old SCA3 mice with a significant reduction from WT/304Q to 304Q/304Q animals (p = 0.0311, Figure 2E,F). Since p62 itself is a substrate of autophagy, it is present at higher levels in the cell when autophagy is impaired [9]. Several studies imply an increase in p62-positive aggregates with progression of SCA3 as well as a co-localization of ATXN3- and p62-positive aggregates [6,28]. Therefore, a DD-AGE analysis was performed to investigate the aggregation of both proteins in the 304Q-KI mice at 18 months of age (Figure 2G,H). For ATXN3, there were no detection of ATXN3-positive aggregates in WT/WT mice. The number of ATXN3-positive aggregates increased significantly between WT/WT and WT/304Q mice (p ≤ 0.0001) and WT/304Q and 304Q/304Q animals (p ≤ 0.0001). No significant increase in p62-positive aggregates were observed between the different genotypes, but a tendency between WT/WT and WT/304Q (p = 0.12) and WT/WT and 304Q/304Q mice (p = 0.1478, Figure 2H) could be observed. Further Western Blot analyses revealed a slight, non-significant reduction of BECN1 in 3 months old 304Q-KI mice compared to WT/WT (Figure 2B,C). However, a significant decrease in BECN1 was observed between WT/WT and WT/304Q (p = 0.0108) and WT/304Q and 304Q/304Q (p = 0.0067) at 18 months of age (Figure 2E,F). For the phospho-S6 protein expression, no changes were detectable in 3 months old SCA3 mice (Figure 2B,C). In the 18 months old animals, only WT/304Q showed a significant increase of phospho-S6 levels to 304Q/304Q mice (p = 0.0038) and WT/WT (p = 0.0154) (Figure 2E,F). In both, 3- and 18-months old 304Q-KI mice, LC3-I (orange arrow) significantly decreased in the homozygous (304Q/304Q) mice compared to WT/WT animals (3 months: p = 0.0283); 18 months: p = 0.0125, Figure 2B,C,E,F). Additionally, homozygous (304Q/304Q) 18 months old mice also showed significant decrease of LC3-I compared to WT/304Q 304Q-KI mice (p = 0.0054, Figure 2E,F). Signals for LC3-II (blue arrow), the form of LC3 which is associated with autophagosome formation, were too low for further quantification in all genotypes and ages (Figure 2B,E). Expression of normal and elongated ATXN3 was confirmed for all mice at 3- and 18-months (Figure 2A,D).
3.3. Aggregation and Co-Localization of ATXN3- and p62-Positive Aggregates in SCA3 Mice
The detected ATXN3- and p62-positive aggregates in DD-AGE analyses were further investigated using IHC and IF staining in 3-, 12- and 18-months old homozygous 304Q-KI mice (WT/WT vs 304Q/304Q) compared to wildtype controls. Pons (Figure 3) and cerebellum (Supplementary Figure S3) were evaluated as the main pathogenesis areas of SCA3 in the brain. For the 3 months old mice, no p62-positive aggregates were detectable in WT/WT or 304Q/304Q animals. 18 months old WT mice showed nor ATXN3- positive neither p62- positive aggregates. However, in 304Q/304Q mice numerous ATXN3- and p62-positiv aggregates were seen. Here, higher number of ATXN3-positive aggregates but larger p62 accumulations were observed. To investigate the localization of these aggregates and eventually co-aggregation of ATXN3 and p62, IF staining of 18 months old WT and 304Q-KI mice were performed (Figure 3, Supplementary Figure S3). IF analyses confirmed that WT/WT animals showed no detectable ATXN3- or p62- positive aggregates, whereas 304Q/304Q animals demonstrated a strong aggregation of both proteins. IF analyses confirmed that aggregates the presence of double-positive aggregates, next to single positive ATXN3 (green) or p62 aggregates (red) (Figure 3). Similar results were observed in the cerebellum, but with lower numbers of ATXN3- and p62-containing aggregates (Supplementary Figure S3).
3.4. Rapamycin Treatment Decreases Expression of Autophagic Proteins
As our in vivo data indicate an impairment of autophagy with SCA3 disease progression, cell culture experiments with rapamycin were performed to induce autophagy. HEK-293T cells were transfected with ATXN3 15Q plasmids as a non-pathological form or ATXN3 77Q and 148Q plasmids with a pathological elongated repeat length. Protein levels and aggregation of the SCA3 disease protein ATXN3 were investigated as well as the protein levels of the autophagic proteins BECN1, p62, phospho-S6 and LC3. Western Blot analyses revealed no significant changes in the respective protein levels between the different genotypes after rapamycin (Rapa) treatment (Figure 4A,B). For ATXN3 and BECN1 there was no change in the protein levels between rapamycin treated and untreated cells (DMSO), also independent from the repeat length (Figure 4A). p62 expression showed a decrease according to the rapamycin treatment, but independent from the repeat length and therefore indicating a successful induction of autophagy (Figure 4B). Additionally, the phospho-S6 expression decreased strongly because of the treatment (short and long exposure is shown for better visualization of rapamycin treated cell expressions, Figure 4B). Since phospho-S6 is phosphorylated and activated by mTOR, the strong downregulation represented a successful autophagy induction and inhibition of mTOR. A standard indicator for autophagy is LC3-II, therefore the LC3-II/LC3-I ratio was analyzed showing an increase in autophagosome formation because rapamycin decreased LC3-I expression and increased LC3-II expression (Figure 4B) [29]. Overall, the demonstrated change in protein level of p62, phospho-S6 and LC3 confirmed an induction of autophagy after rapamycin treatment in all ATXN3 transfected HEK-293T cells independent from the ATXN3 repeat length. Unfortunately, rapamycin treatment did not change the amount of ATXN3-positive aggregates compared to DMSO-treated cells after 6h of treatment (Figure 4C).
3.5. Cycloheximid Treatment Increases Aggregation of ATXN3
Due to the detected autophagic impairment, degradation of the autophagic proteins was examined using cycloheximide (CHX), an inhibitor of the eukaryotic cytosolic translation. Protein expression of ATXN3 showed no changes between cycloheximide treated and untreated cells but the ATXN3 15Q HEK-293T cells (green arrow) had a significant higher expression than the cells transfected with longer ATXN3 polyQ repeats after normalization to respective DMSO controls (p = 0.0082 between ATXN3 15Q and 148Q) (Figure 5A). Considering the longer exposure (LE), in the stacking gel high molecular weight aggregates in ATXN3 77Q and 148Q HEK-293T cell samples were present. Regarding these results a DD-AGE analysis was performed investigating ATXN3 aggregation in these samples (Figure 5D). No ATXN3-positive aggregates were found in CHX treated and untreated ATXN3 15Q HEK-293T cells. ATXN3 77Q HEK-293T cells of CHX and DMSO (control) treatment showed a strong aggregation with small (~460 kDa) ATXN3-positive aggregates. In CHX treated and untreated ATXN3 148Q HEK-293T cells small (~460 kDa) and large ATXN3-positve aggregates were detected. Comparison of CHX treated and untreated cells showed that the CHX treated cells had a stronger ATXN3 aggregation in 77Q and 148Q transfected HEK-293T cells indicating an accumulation of ATXN3 protein aggregates. Western Blot analyses of phospho-S6 showed overall an unequal expression but also an increased expression in the CHX treated ATXN3 HEK-293T cells of all three genotypes (Figure 5A). p62 expression displayed no significant change between CHX treated and untreated cells whereas CHX treated ATXN3 148Q cells had a significant lower expression of p62 (p = 0.0304) compared to the other genotypes (Figure 5B). As shown before for the ATXN3 detection, p62 detection showed a signal in the stacking gel of ATXN3 77Q and 148Q HEK-293T cells, also indicating the presence of high molecular species in these samples. Quantification of these high molecular species showed significantly more p62-specific accumulations in ATXN3 148Q HEK-293T cells than in ATXN3 15Q cells (p = 0.0226, Figure 5B). Unfortunately, DD-AGE analysis investigating p62 aggregation was not successful (data not shown). BECN1 as well as the ratio of LC3-II/LC3-I expression showed no significant changes between CHX treated and untreated cells or the different genotypes (Figure 5B). A CHX treatment timeline (0, 1, 5 and 12 h) was performed to assess whether longer incubation time triggers a more pronounced effect (Figure 5C). After 1 h of treatment p62 and LC3 expression showed a reduction in the expression compared to 0 h CHX treatment. However, 5 h and 12 h CHX treatment had no significant stronger effect on the p62 or LC3 expression of the ATXN3 HEK-293T cells indicating that 1 h of CHX treatment is sufficient to recognize effects at protein level (Figure 5C).
3.6. Proteasomal Inhibition Induces Autophagic Degradation
To further investigate the implication of autophagy in ATXN3 (15Q, 77Q, 148Q) transfected HEK-293T cells, the proteasomal degradation was blocked using the specific inhibitor MG132. ATXN3 15Q cells in MG132 treated and untreated cells showed a lower ATXN3 expression compared to ATXN3 77Q and 148Q cells, indicating a faster autophagic degradation of ATXN3 in ATXN3 15Q HEK-293T cells (Figure 6A), however, without reaching significance. Compared to untreated cells, p62 expression was decreased in MG132 treated cells for all three genotypes, suggesting an increased autophagic degradation of p62 according to MG132 treatment (Figure 6A). BECN1 expression was increased after MG132 treatment compared to untreated cells in ATXN3 15Q and 77Q cells, indicating a BECN1-dependent autophagic activation in these cells (Figure 6A). Interestingly, ATXN3 15Q and 77Q cells demonstrated a higher BECN1 expression due to blocking the proteasomal degradation compared to their respective controls. This was not the case for ATXN3 148Q cells where the BECN1 expression remained the same in MG132 treated and untreated cells. This indicates no autophagic activation via the BECN1 pathway in ATXN3 148Q cells in comparison to ATXN3 15Q and 77Q cells. Regarding phospho-S6 expression no significant changes were detected between MG132 treated and untreated cells as well as the different genotypes (Figure 6A). LC3-I and LC3-II showed a strong increase in protein expression following the MG132 treatment, confirming the autophagic activation in response to the proteasomal inhibition (Figure 6A). The previous findings about autophagy activation were validated with a polyubiquitin detection (Figure 6B). Levels of total ubiquitinated proteins were upregulated in response to MG132 in all genotypes compared to untreated cells, with the strongest expression in ATXN3 148Q HEK-293T cells (p = 0.0921 between ATXN3 15Q and 77Q; p = 0.0414 between ATXN3 77Q and 148Q). In order to further investigate polyubiquitination, K63-specific immunodetection (autophagic degradation signal) and K48-specific immunodetection (proteasomal degradation signal) was performed. K63-specific polyubiquitination showed a similar result compared to polyubiquitin detection with a significant strong increase in ATXN3 148Q HEK-293T cells (p = 0.0039 between 15Q and 148Q; p = 0.0007 between 77Q and 148Q) (Figure 6B). An increase in K48-specific polyubiquitination independent of the genotype validated the successful inhibition of proteasomal degradation (Figure 6C). Therefore, autophagic degradation as well as proteasomal degradation was elevated after treating ATXN3 overexpressing HEK-293T cells with MG132.
4. Discussion
SCA3 is a disease where protein aggregation plays an important role in disease progression, caused by the expansion of polyQ repeats in the mutated protein ATXN3 [3]. The polyQ-expanded protein forms inclusion that sequester a variety of other proteins [5,6,7]. An important protein found in these aggregates is the receptor for selective autophagic degradation of ubiquitinated proteins p62/ sequestosome-1 [6,9]. In this study we demonstrate decreasing p62 expression in the 304Q-KI mouse model of SCA3 with disease progression, which is consisting with previous studies [10,28]. This reduced soluble p62 protein expression is probably caused by aggregation of the soluble p62 protein in inclusions. Analysis of port-mortem brains by Sittler et al. revealed an accumulation of p62-positive aggregates together with misfolded ATXN3 and ubiquitin [6]. In our study, the strongest co-aggregation of p62 proteins with ATXN3 was observed in homozygous SCA3 mice, which was not present in WT animals. Interestingly, homozygous SCA3 mice showed strong aggregation with aggregates positive for both ATXN3 and p62 or only for one of the proteins, indicating a possible co-localization for some extend of ATXN3 and p62. The interaction and co-aggregation of p62 with disease protein ATXN3 was previously shown in other studies, indicating a role of p62 in aggregate formation in SCA3 disease [30,31]. Aggregation of ATXN3 and p62 was not only shown for the 304Q-KI mouse model but also in HEK-293T cells transfected with ATXN3 plasmids and treated with cycloheximide, a drug which inhibits the eukaryotic cytosolic translation. HEK-293T cells transfected with ATXN3 plasmids with expanded polyQ tracts (77Q, 148Q) demonstrated aggregates positive for both ATXN3 and p62, validating the ATXN3 and p62 co-localization.
Autophagy is a well-regulated physiological process which is involved in several important cellular processes including development, metabolism, immunity, neuronal health, and tumorigenesis [18]. Transcriptional regulation of autophagy is linked to the central modulator of autophagy, TOR by controlling the nuclear localization of nutrient-regulated transcriptions factors [32,33]. An increase in mRNA levels of certain ATG genes and p62/SQSTM1 regulated by different transcription factors is demonstrated to induce or suppress autophagy [34]. In this study, RNA sequencing results of SCA3 human and mouse cerebella of aged animals revealed selective dysregulation of autophagic genes. Importantly, the most striking effects were observed in aged controls and aged SCA3 mouse cerebella, underlining the already described mRNA dysregulation of autophagic genes in neurodegenerative diseases and aging [35,36]. In 12-month-old SCA3 KI mice only ATG12, an ubiquitin-like protein which get conjugated to ATG5 (ATG5-ATG12 complex important for extension and closure of autophagosome [37]) and the selective autophagy receptor p62/SQSTM1 were significantly dysregulated compared to controls. For p62/SQSTM1 it is known that several of its protein domains including Phox and Bem1 (PB1) and ubiquitin-associated domain (UBA) are involved in protein-protein interaction and may play a critical role in formation of autophagosomes and cytoplasmic accumulations [23,24]. Currently, up to 16 p62 isoforms are annotated (Ensembl, last visit 11.01.2024). The two best characterized isoforms, reference p62-202 (ENST00000389805) and p62-201 (ENST00000360718), are differing at the 5´-UTR. Isoform p62-201 (ENST00000360718) employs an alternative start codon corresponding to amino acid position 85, resulting in a truncated mRNA and protein lacking large parts of the PB1 domain [23]. Isoform expression analyses in SCA3 human post-mortem RNA sequencing datasets revealed a significant down regulation of the p62 reference isoform p62-202 in SCA3 but not of the truncated isoform p62-201. Earlier studies demonstrated that the PB1 domain of p62 is important for the binding of several interacting partners and essential for the polymerization scaffold of p62 and aggresome formation [23,24]. Interestingly, studies revealed differences in the behavior of both p62 isoforms and demonstrated that the truncated isoform p62-201 forms larger aggregates compared to reference isoform p62-202 [23]. The finding that in human SCA3 post-mortem datasets the reference isoform p62-202 is lower expressed may explain the very large p62-positive aggregates in the SCA3 KI mouse.
In addition to protein aggregation, autophagy impairment plays a major role in the development of SCA3. For instance, Watchon et al. demonstrated decreased levels of LC3-II, a marker for autophagosomes and autolysosomes, in cells expressing ATXN3 with expanded polyQ tract [10,27]. Our current study confirms autophagy impairment with increasing age of 304Q-KI mice. The previously discussed RNA sequencing data of mouse cerebellar RNA demonstrated strong dysregulation of autophagic genes (BECN1, p62, mTOR, LC3) comparing young and old SCA3 KI mice. Additionally, the protein expression of LC3-I which is responsible for the autophagosome formation in autophagy showed decreased levels in homozygous 304Q-KI mice. In 18-months old animals, BECN1 protein expression was significantly reduced with increasing length of ATXN3 polyQ tract. This BECN1 reduction is possibly caused by an aberrant interaction with the polyQ-expanded ATXN3 protein which has been reported in a previous study [38].
Since autophagy is impaired during SCA3, activation of autophagy and reduction of aggregate formation was shown to ameliorate SCA3 disease progression [11,39,40]. In the current study, autophagy was activated with the inhibitor of mTOR, rapamycin. Rapamycin treatment of ATXN3 transfected HEK-293T cells showed a significant reduction of soluble p62. These results suggest a successful autophagic activation, since p62 itself is an autophagy substrate [9]. Additionally, this was confirmed by the increased LC3-II and reduced phospho-S6 expression due to rapamycin treatment. LC3-II activation indicates formation of autophagosomes whereas inhibiting mTOR also leads to blocking protein S6 phosphorylation [26,27]. Since no significant effects were seen regarding BECN1 expression, autophagy in SCA3 is regulated in a mTOR-dependent manner. Unfortunately, no reduction of protein aggregation was achieved in ATXN3 HEK-293T cells using rapamycin treatment. Previous studies of Ravikumar et al. showed that autophagy induction due to rapamycin treatment reduced aggregate formation and cell death in a Huntington’s Disease (HD) cell model [41]. However, rapamycin treatment did only reduce protein aggregation 9 h after transfection of the cells [41]. Rapamycin treatment at later time point showed no significant reduction in aggregation or cell death in the HD cell model [41]. Since in the current study treatment with rapamycin was performed 72 h after transfection of HEK-293T cells, an earlier time point should be tested for reducing protein aggregates.
Due to the detected autophagic impairment in 304Q-KI mice as well as ATXN3 HEK-293T cells transfected with Atxn3-15Q, Atxn3-77Q or Atxn3-148Q, degradation of the autophagic proteins was examined using CHX. ATXN3 as well as p62 expression was significantly lower in ATXN3 148Q HEK-239T cells treated with CHX compared to ATXN3 15Q or 77Q HEK-293T cells. This could be caused by a faster degradation of ATXN3 and p62 in cells transfected with longer ATXN3 polyQ tracts. However, DD-AGE analysis demonstrated an increased aggregation of ATXN3 in HEK-293T cells transfected with longer ATXN3 polyQ tracts (77Q, 148Q). Therefore, CHX treatment leads to an increased aggregation of ATXN3 and possibly of p62. CHX treatment of ATXN3 HEK-293T cells transfected with 15Q, 77Q or 148Q increased phospho-S6 levels compared to untreated ATXN3 HEK-293T cells of all genotypes. Since increased phospho-S6 indicates an activation of mTOR, CHX treatment led to an inhibition of autophagy in ATXN3 HEK-293T cells transfected with 15Q, 77Q or 148Q, which is consistent with former studies [42,43]. A study of Dang et al. where the impact of CHX on autophagic pathways was investigated, demonstrated that CHX-mediated inhibition of protein synthesis leads to a problem in autophagosome-lysosome fusion [43]. Therefore, treating cells with CHX triggers an autophagic impairment, which explains the increased aggregation of the disease protein ATXN3 and the possible aggregation of p62.
For further investigation of the autophagy pathway in ATXN3 HEK-293T cells with different repeat length (15Q, 77Q, 148Q), MG132 was used to block the proteasomal degradation. Expression of p62 was reduced in all genotypes according to MG132 treatment compared to control cells (DMSO treatment), which indicates an activation of autophagy. Since phospho-S6 showed no significant decrease in MG132-treated ATXN3 HEK-293T cells (15Q, 77Q, 148Q) and BECN1 expression increased due to treatment with MG132 in ATXN3 HEK-293T cells transfected with 15Q or 77Q, blocking the proteasome activates BECN1-dependent autophagy. These observations are confirmed by two studies where inhibiting the proteasomal degradation led to an autophagy activation via BECN1 [44,45]. However, BECN1 activation after MG132 was not found in ATXN3 148Q cells, suggesting no BECN1-dependent autophagic activation in HEK-293T cells transfected with very long ATXN3 polyQ tracts. This supports the assumption of strong autophagic impairment according to longer polyQ tracts in the ATXN3 protein.
An inhibition of the proteasomal degradation with MG132 led to increased levels of K48-specific and K63-specific polyubiquitination in ATXN3 HEK-293T cells of all genotypes compared to DMSO controls. Therefore, we detected an elevated autophagic degradation (K63) and proteasomal degradation (K48) in ATXN3 15Q, 77Q or 148Q HEK-293T cells. The different ATXN3 polyQ lengths showed no impact on K48-polyubiquitination and thereby on proteasomal degradation. However, ATXN3 148Q HEK-293T cells treated with MG132 showed significantly higher amounts of K63-polyubiquitin chains compared to ATXN3 15Q and 77Q HEK-293T cells. Since the deubiquitinase protein ATXN3 preferentially cleaves K63-linked polyubiquitin chains, increased levels of K63 chains in ATXN3 148Q HEK-392T cells indicate impaired binding of mutant ATXN3 protein to polyubiquitin chains [46].
The present study demonstrates a role of the receptor for selective autophagy, p62, in aggregate formation seen in the neurodegenerative disease SCA3. Investigation of the SCA3 304Q-KI mouse model as well as cell culture experiments with overexpression of ATXN3 plasmids with different polyQ lengths (15Q, 77Q, 148Q) confirmed a co-localized aggregation of p62 and ATXN3 with increasing polyQ length of ATXN3. The overserved elevated aggregation of the autophagic protein p62 or the disease protein ATXN3 is probably caused by the increase of autophagy impairment triggered by disease progression of SCA3 304Q-KI mice or by elevated polyQ tract length in ATXN3 plasmids.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, J.H-S, O.R and A.J.Z.; methodology, A.J.Z., C.B., J.M.S, J.J.W; software, A.J.Z, C.B., J.M.S; validation, A.J.Z., J.H-S. and O.R.; formal analysis, J.H-S.; investigation, A.J.Z.; resources, J.H-S.; data curation, A.J.Z.; writing—original draft preparation, A.J.Z.; writing—review and editing, O.R., J.J.W, J.H-S; visualization, A.J.Z.; supervision, O.R., J-H-S; project administration, J.H-S.; funding acquisition, J.H-S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of the University of Tübingen (human: 911/2019BO2 date: 12.12.2019; animals: HG3/13 date 20.06.2013.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Human and Mouse RNA sequencing data are available in a publicly accessible repository. The other data are available upon reasonable request from the corresponding author.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Narendra, D.P., and Youle, R.J. (2012). Neurodegeneration: Trouble in the cell’s powerhouse. Nature 483, 418–419. [CrossRef]
- Fan, H.-C., Ho, L.-I., Chi, C.-S., Chen, S.-J., Peng, G.-S., Chan, T.-M., Lin, S.-Z., and Harn, H.-J. (2014). Polyglutamine (PolyQ) diseases: genetics to treatments. Cell Transplant 23, 441–458. [CrossRef]
- Huang, S., Zhu, S., Li, X.-J., and Li, S. (2019). The Expanding Clinical Universe of Polyglutamine Disease. Neuroscientist 25, 512–520. [CrossRef]
- Matos, C.A., de Macedo-Ribeiro, S., and Carvalho, A.L. (2011). Polyglutamine diseases: the special case of ataxin-3 and Machado-Joseph disease. Prog Neurobiol 95, 26–48. [CrossRef]
- Gatchel, J.R., and Zoghbi, H.Y. (2005). Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet 6, 743–755. [CrossRef]
- Sittler, A., Muriel, M.-P., Marinello, M., Brice, A., den Dunnen, W., and Alves, S. (2018). Deregulation of autophagy in postmortem brains of Machado-Joseph disease patients: Autophagy in Machado-Joseph disease. Neuropathology 38, 113–124. [CrossRef]
- Iwata, A., Christianson, J.C., Bucci, M., Ellerby, L.M., Nukina, N., Forno, L.S., and Kopito, R.R. (2005). Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci U S A 102, 13135–13140. [CrossRef]
- Levine, B., and Kroemer, G. (2008). Autophagy in the Pathogenesis of Disease. Cell 132, 27–42. [CrossRef]
- Jimenez-Sanchez, M., Thomson, F., Zavodszky, E., and Rubinsztein, D.C. (2012). Autophagy and polyglutamine diseases. Progress in Neurobiology 97, 67–82. [CrossRef]
- Watchon, M., Luu, L., Plenderleith, S.K., Yuan, K.C., and Laird, A.S. (2023). Autophagy Function and Benefits of Autophagy Induction in Models of Spinocerebellar Ataxia Type 3. Cells 12, 893. [CrossRef]
- Ou, Z., Luo, M., Niu, X., Chen, Y., Xie, Y., He, W., Song, B., Xian, Y., Fan, D., OuYang, S., et al. (2016). Autophagy Promoted the Degradation of Mutant ATXN3 in Neurally Differentiated Spinocerebellar Ataxia-3 Human Induced Pluripotent Stem Cells. Biomed Res Int 2016, 6701793. [CrossRef]
- Menzies, F.M., Huebener, J., Renna, M., Bonin, M., Riess, O., and Rubinsztein, D.C. (2010). Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 133, 93–104. [CrossRef]
- Haas, E., Incebacak, R.D., Hentrich, T., Huridou, C., Schmidt, T., Casadei, N., Maringer, Y., Bahl, C., Zimmermann, F., Mills, J.D., et al. (2022). A Novel SCA3 Knock-in Mouse Model Mimics the Human SCA3 Disease Phenotype Including Neuropathological, Behavioral, and Transcriptional Abnormalities Especially in Oligodendrocytes. Mol Neurobiol 59, 495–522. [CrossRef]
- Guo, N., and Peng, Z. (2013). MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia Pac J Clin Oncol 9, 6–11. [CrossRef]
- Buchanan, B.W., Lloyd, M.E., Engle, S.M., and Rubenstein, E.M. (2016). Cycloheximide Chase Analysis of Protein Degradation in Saccharomyces cerevisiae. J Vis Exp, 53975. [CrossRef]
- Weber, J.J., Golla, M., Guaitoli, G., Wanichawan, P., Hayer, S.N., Hauser, S., Krahl, A.-C., Nagel, M., Samer, S., Aronica, E., et al. (2017). A combinatorial approach to identify calpain cleavage sites in the Machado-Joseph disease protein ataxin-3. Brain 140, 1280–1299. [CrossRef]
- Halfmann, R., and Lindquist, S. (2008). Screening for Amyloid Aggregation by Semi-Denaturing Detergent-Agarose Gel Electrophoresis. JoVE, 838. [CrossRef]
- Ma, Q., Long, S., Gan, Z., Tettamanti, G., Li, K., and Tian, L. (2022). Transcriptional and Post-Transcriptional Regulation of Autophagy. Cells 11, 441. [CrossRef]
- Stumptner, C., Heid, H., Zatloukal, K., Fuchsbichler, A., Hauser, H., and Denk, H. (1999). Identification of p62, a phosphotyrosine independent ligand of p56lck kinase, as a major component of intracytoplasmic hyaline bodies in hepatocellular carcinoma. Verh Dtsch Ges Pathol 83, 254–259.
- Strnad, P., Nuraldeen, R., Guldiken, N., Hartmann, D., Mahajan, V., Denk, H., and Haybaeck, J. (2013). Broad Spectrum of Hepatocyte Inclusions in Humans, Animals, and Experimental Models. In Comprehensive Physiology, Y. S. Prakash, ed. (Wiley), pp. 1393–1436. [CrossRef]
- Zatloukal, K., Stumptner, C., Fuchsbichler, A., Heid, H., Schnoelzer, M., Kenner, L., Kleinert, R., Prinz, M., Aguzzi, A., and Denk, H. (2002). p62 Is a Common Component of Cytoplasmic Inclusions in Protein Aggregation Diseases. The American Journal of Pathology 160, 255–263. [CrossRef]
- Birgisdottir, Å.B., Lamark, T., and Johansen, T. (2013). The LIR motif – crucial for selective autophagy. Journal of Cell Science 126, 3237–3247. [CrossRef]
- Somlapura, M., Gottschalk, B., Lahiri, P., Kufferath, I., Pabst, D., Rülicke, T., Graier, W.F., Denk, H., and Zatloukal, K. (2021). Different Roles of p62 (SQSTM1) Isoforms in Keratin-Related Protein Aggregation. IJMS 22, 6227. [CrossRef]
- Cabe, M., Rademacher, D.J., Karlsson, A.B., Cherukuri, S., and Bakowska, J.C. (2018). PB1 and UBA domains of p62 are essential for aggresome-like induced structure formation. Biochemical and Biophysical Research Communications 503, 2306–2311. [CrossRef]
- Hayat, M.A. ed. (2016). Autophagy: cancer, other pathologies, inflammation, immunity, infection, and aging (Amsterdam: Boston : Elsevier, Academic Press).
- Kudchodkar, S.B., Yu, Y., Maguire, T.G., and Alwine, J.C. (2004). Human Cytomegalovirus Infection Induces Rapamycin-Insensitive Phosphorylation of Downstream Effectors of mTOR Kinase. J Virol 78, 11030–11039. [CrossRef]
- Runwal, G., Stamatakou, E., Siddiqi, F.H., Puri, C., Zhu, Y., and Rubinsztein, D.C. (2019). LC3-positive structures are prominent in autophagy-deficient cells. Sci Rep 9, 10147. [CrossRef]
- Onofre, I., Mendonça, N., Lopes, S., Nobre, R., de Melo, J.B., Carreira, I.M., Januário, C., Gonçalves, A.F., and de Almeida, L.P. (2016). Fibroblasts of Machado Joseph Disease patients reveal autophagy impairment. Sci Rep 6, 28220. [CrossRef]
- Mizushima, N., and Yoshimori, T. (2007). How to Interpret LC3 Immunoblotting. Autophagy 3, 542–545. [CrossRef]
- Seidel, K., den Dunnen, W.F.A., Schultz, C., Paulson, H., Frank, S., de Vos, R.A., Brunt, E.R., Deller, T., Kampinga, H.H., and Rüb, U. (2010). Axonal inclusions in spinocerebellar ataxia type 3. Acta Neuropathol 120, 449–460. [CrossRef]
- Zhou, L., Wang, H., Chen, D., Gao, F., Ying, Z., and Wang, G. (2014). p62/Sequestosome 1 Regulates Aggresome Formation of Pathogenic Ataxin-3 with Expanded Polyglutamine. IJMS 15, 14997–15010. [CrossRef]
- Beck, T., and Hall, M.N. (1999). The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402, 689–692. [CrossRef]
- Cardenas, M.E., Cutler, N.S., Lorenz, M.C., Di Como, C.J., and Heitman, J. (1999). The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev 13, 3271–3279.
- Füllgrabe, J., Klionsky, D.J., and Joseph, B. (2014). The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol 15, 65–74. [CrossRef]
- Park, H., Kang, J.-H., and Lee, S. (2020). Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. IJMS 21, 3369. [CrossRef]
- Markaki, M., Metaxakis, A., and Tavernarakis, N. (2017). The Role of Autophagy in Aging. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging (Elsevier), pp. 123–138. [CrossRef]
- Guo, F., Liu, X., Cai, H., and Le, W. (2018). Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathology 28, 3–13. [CrossRef]
- Ashkenazi, A., Bento, C.F., Ricketts, T., Vicinanza, M., Siddiqi, F., Pavel, M., Squitieri, F., Hardenberg, M.C., Imarisio, S., Menzies, F.M., et al. (2017). Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 545, 108–111. [CrossRef]
- Moore, L.R., Keller, L., Bushart, D.D., Delatorre, R.G., Li, D., McLoughlin, H.S., do Carmo Costa, M., Shakkottai, V.G., Smith, G.D., and Paulson, H.L. (2019). Antisense oligonucleotide therapy rescues aggresome formation in a novel spinocerebellar ataxia type 3 human embryonic stem cell line. Stem Cell Research 39, 101504. [CrossRef]
- Liu, S.-W., Chang, J.-C., Chuang, S.-F., Liu, K.-H., Cheng, W.-L., Chang, H.-J., Chang, H.-S., Lin, T.-T., Hsieh, C.-L., Lin, W.-Y., et al. (2019). Far-infrared Radiation Improves Motor Dysfunction and Neuropathology in Spinocerebellar Ataxia Type 3 Mice. Cerebellum 18, 22–32. [CrossRef]
- Ravikumar, B., Vacher, C., Berger, Z., Davies, J.E., Luo, S., Oroz, L.G., Scaravilli, F., Easton, D.F., Duden, R., O’Kane, C.J., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36, 585–595. [CrossRef]
- Watanabe-Asano, T., Kuma, A., and Mizushima, N. (2014). Cycloheximide inhibits starvation-induced autophagy through mTORC1 activation. Biochem Biophys Res Commun 445, 334–339. [CrossRef]
- Dang, T.T., and Back, S.H. (2021). Translation Inhibitors Activate Autophagy Master Regulators TFEB and TFE3. Int J Mol Sci 22, 12083. [CrossRef]
- Liu, C., Yan, X., Wang, H.-Q., Gao, Y.-Y., Liu, J., Hu, Z., Liu, D., Gao, J., and Lin, B. (2012). Autophagy-independent enhancing effects of Beclin 1 on cytotoxicity of ovarian cancer cells mediated by proteasome inhibitors. BMC Cancer 12, 622. [CrossRef]
- Ge, P.-F., Zhang, J.-Z., Wang, X.-F., Meng, F.-K., Li, W.-C., Luan, Y.-X., Ling, F., and Luo, Y.-N. (2009). Inhibition of autophagy induced by proteasome inhibition increases cell death in human SHG-44 glioma cells. Acta Pharmacol Sin 30, 1046–1052. [CrossRef]
- Winborn, B.J., Travis, S.M., Todi, S.V., Scaglione, K.M., Xu, P., Williams, A.J., Cohen, R.E., Peng, J., and Paulson, H.L. (2008). The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J Biol Chem 283, 26436–26443. [CrossRef]
Figure 2.
Activation of autophagy with disease progression in SCA3 mice. (A-E) Protein levels of ATXN3, p62, BECN1, phospho-S6 and LC3 were measured in 3- and 18-months old 304Q-KI mice. Beta-Actin (ACTB) is shown as loading control. All expression levels were normalized to WT/WT levels. Statistical analysis was performed with one-way ANOVA and Tukey´s multiple comparison test. Green arrow represents endogenous ATXN3, while red arrow represents expanded ATXN3. Orange arrow symbolizes LC3-I while blue arrow label LC3-II. (G) Aggregation of ATXN3 and p62 were analyzed in 18 months old 304Q-KI mice using DD-AGE. All aggregation values were normalized to WT/WT. Statistical analysis was performed with one-way ANOVA and Tukey´s multiple comparison test. * p < 0.05, ** p < 0.01, *** p < 0.001 by one-way ANOVA. Values are shown as mean +/− SEM. N = 3 for each experiment, mixed sex of 304Q-KI mice.
Figure 2.
Activation of autophagy with disease progression in SCA3 mice. (A-E) Protein levels of ATXN3, p62, BECN1, phospho-S6 and LC3 were measured in 3- and 18-months old 304Q-KI mice. Beta-Actin (ACTB) is shown as loading control. All expression levels were normalized to WT/WT levels. Statistical analysis was performed with one-way ANOVA and Tukey´s multiple comparison test. Green arrow represents endogenous ATXN3, while red arrow represents expanded ATXN3. Orange arrow symbolizes LC3-I while blue arrow label LC3-II. (G) Aggregation of ATXN3 and p62 were analyzed in 18 months old 304Q-KI mice using DD-AGE. All aggregation values were normalized to WT/WT. Statistical analysis was performed with one-way ANOVA and Tukey´s multiple comparison test. * p < 0.05, ** p < 0.01, *** p < 0.001 by one-way ANOVA. Values are shown as mean +/− SEM. N = 3 for each experiment, mixed sex of 304Q-KI mice.
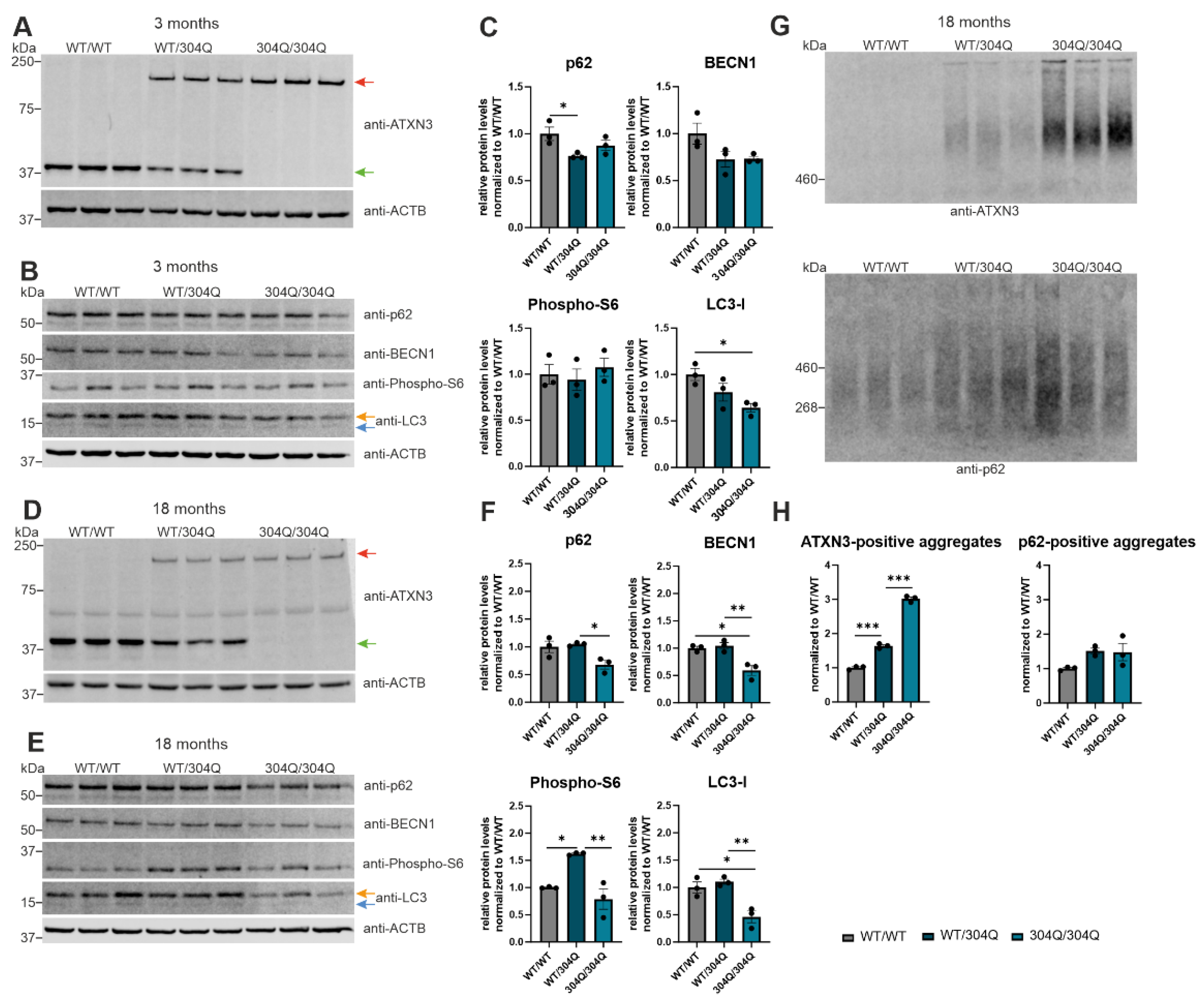
Figure 3.
Co-localization of ATXN3- and p62-positive accumulations in the pons of SCA3 mice. First three rows show IHC staining of 3- and 18-months old WT and 304Q/304Q SCA3 mice pons sections. Staining was performed with ATXN3 or p62- specific antibodies. Last row represents IF staining of 18 months old SCA3 and WT/WT mice pons sections labeled with antibodies against ATXN3 and p62. Green = ATXN3, red = p62, orange = ATXN3 and p62-double-positive aggregates. Scale bar indicates 20 µm.
Figure 3.
Co-localization of ATXN3- and p62-positive accumulations in the pons of SCA3 mice. First three rows show IHC staining of 3- and 18-months old WT and 304Q/304Q SCA3 mice pons sections. Staining was performed with ATXN3 or p62- specific antibodies. Last row represents IF staining of 18 months old SCA3 and WT/WT mice pons sections labeled with antibodies against ATXN3 and p62. Green = ATXN3, red = p62, orange = ATXN3 and p62-double-positive aggregates. Scale bar indicates 20 µm.
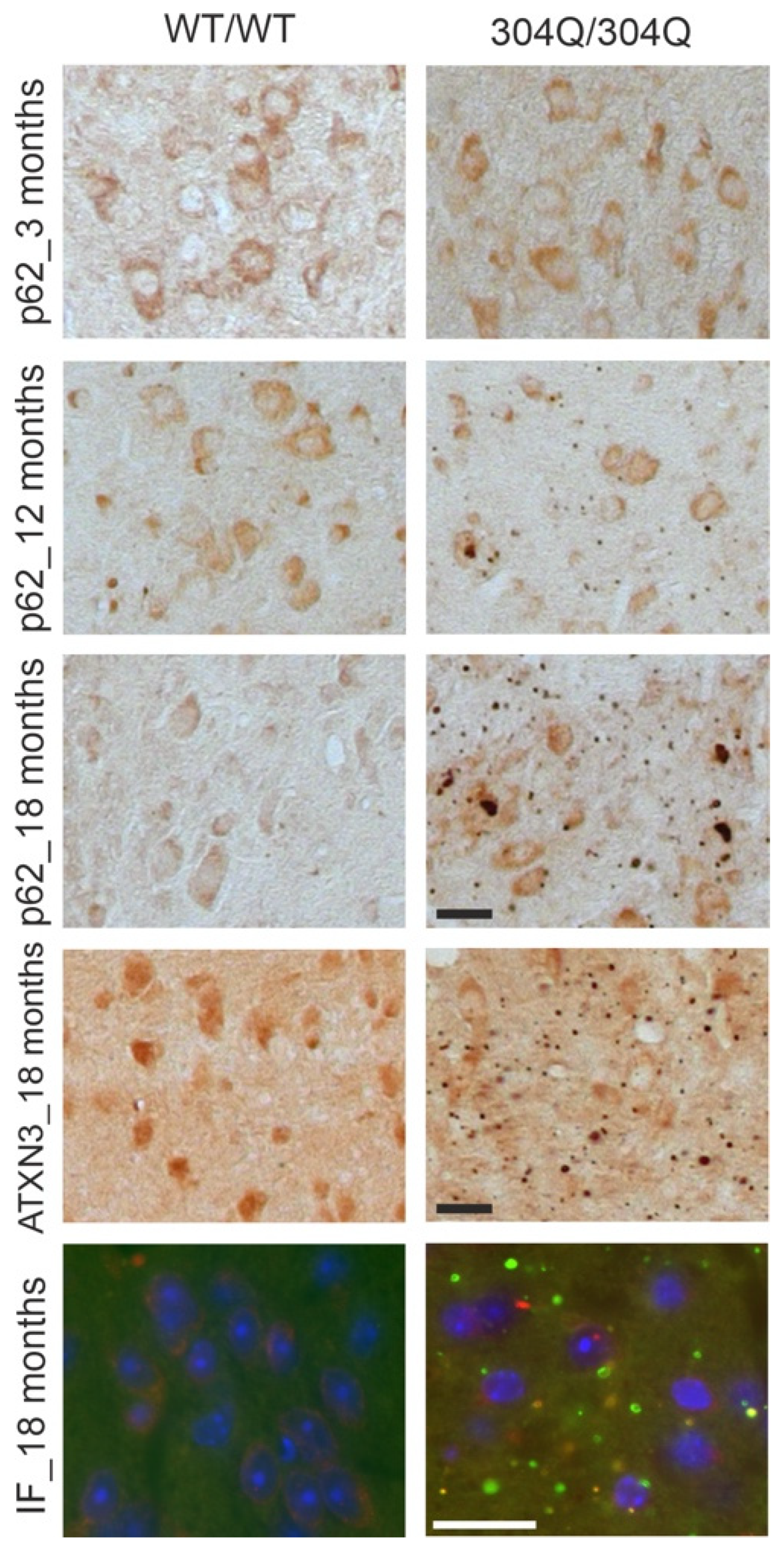
Figure 4.
Rapamycin activated autophagy in ATXN3 expressing HEK-293T cells did not reduce ATXN3 aggregation. (A, B) Protein levels of ATXN3, BECN1, p62, phospho-S6 and LC3 were measured in HEK-293T cells transfected with ATXN3 (15Q, 77Q, 148Q) and treated with rapamycin and compared to untreated controls (DMSO). Beta-Actin (ACTB) is shown as loading control. Protein levels were normalized to the respective ATXN3 15Q protein expression. Statistical analyses were performed from three independent experiments with one-way ANOVA and Tukey´s multiple comparison test. Green arrow represents 15Q ATXN3, yellow arrow 77Q ATXN3 and red arrow 148Q ATXN3. Orange arrow symbolizes LC3-I while blue arrow label LC3-II. (C) Aggregation of ATXN3 was investigated in rapamycin treated ATXN3 transfected HEK-293T cells compared to untreated cells (DMSO) using filter trap analysis. Amount of ATXN3-positive aggregates was normalized to the respective ATXN3 15Q expression values. Statistical analysis was performed from three independent experiments with one-way ANOVA and Tukey´s multiple comparison test. * p < 0.05 by one-way ANOVA. Values are shown as mean +/− SEM.
Figure 4.
Rapamycin activated autophagy in ATXN3 expressing HEK-293T cells did not reduce ATXN3 aggregation. (A, B) Protein levels of ATXN3, BECN1, p62, phospho-S6 and LC3 were measured in HEK-293T cells transfected with ATXN3 (15Q, 77Q, 148Q) and treated with rapamycin and compared to untreated controls (DMSO). Beta-Actin (ACTB) is shown as loading control. Protein levels were normalized to the respective ATXN3 15Q protein expression. Statistical analyses were performed from three independent experiments with one-way ANOVA and Tukey´s multiple comparison test. Green arrow represents 15Q ATXN3, yellow arrow 77Q ATXN3 and red arrow 148Q ATXN3. Orange arrow symbolizes LC3-I while blue arrow label LC3-II. (C) Aggregation of ATXN3 was investigated in rapamycin treated ATXN3 transfected HEK-293T cells compared to untreated cells (DMSO) using filter trap analysis. Amount of ATXN3-positive aggregates was normalized to the respective ATXN3 15Q expression values. Statistical analysis was performed from three independent experiments with one-way ANOVA and Tukey´s multiple comparison test. * p < 0.05 by one-way ANOVA. Values are shown as mean +/− SEM.
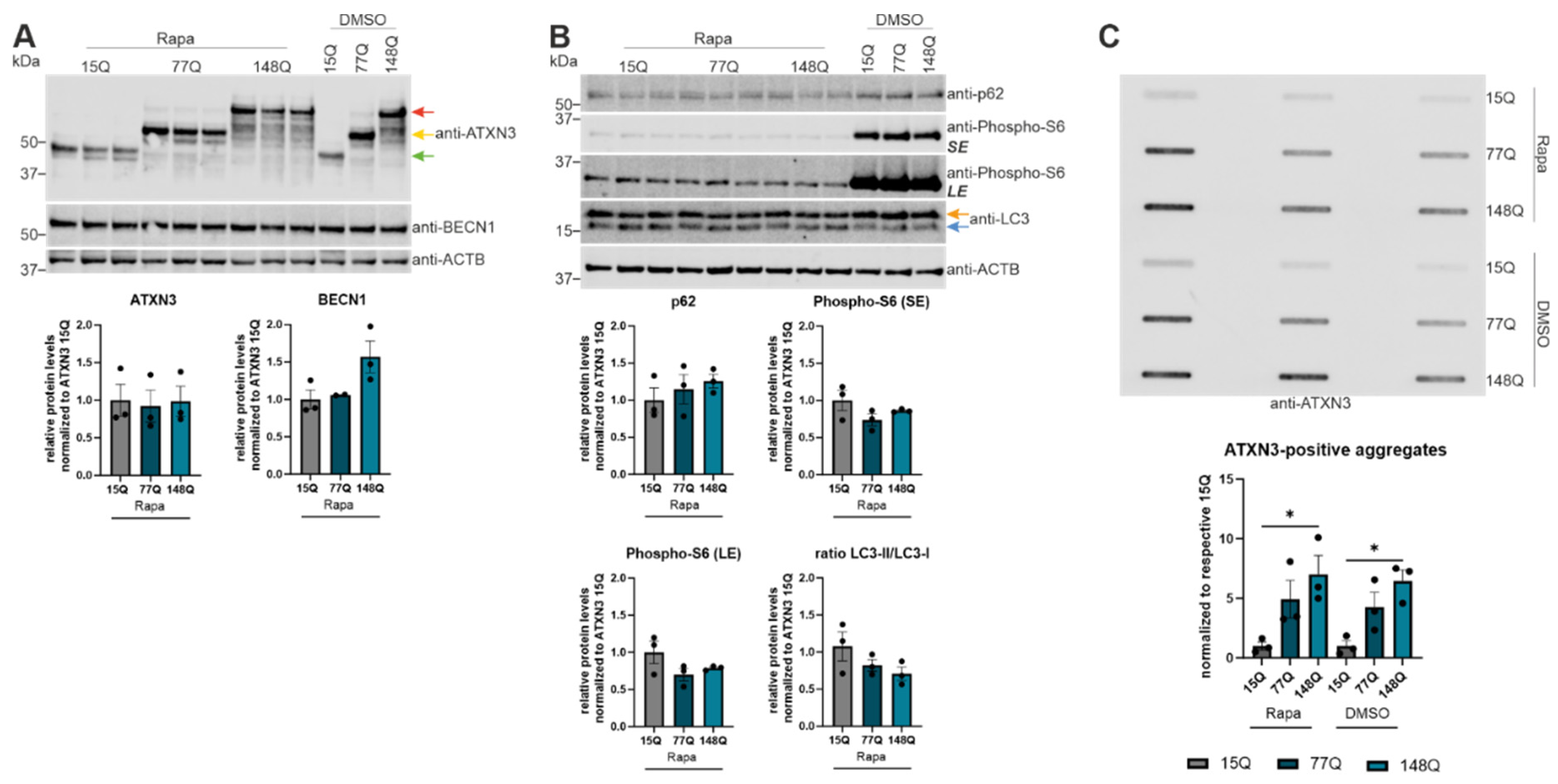
Figure 5.
Increased aggregation of ATXN3 according to cycloheximide (CHX) treatment. (A, B) Protein levels of ATXN3, phospho-S6, p62, BECN1 and LC3 were measured in HEK-293T cells transfected with ATXN3 (15Q, 77Q, 148Q) and treated with cycloheximide or untreated controls (DMSO) by Western Blotting. GAPDH or Beta-Actin (ACTB) are shown as loading control. Protein levels were normalized to the respective ATXN3 15Q protein expression. Statistical analyses were performed from three independent experiments with one-way ANOVA and Tukey´s multiple comparison test. Green arrow represents 15Q ATXN3, yellow arrow 77Q ATXN3 and red arrow 148Q ATXN3. Orange arrow symbolizes LC3-I while blue arrow labels LC3-II. (C) Protein levels of ATXN3, p62, and LC3 were measured in ATXN3 (15Q, 77Q, 148Q) transfected HEK-293T cells treated with CHX for 1 h, 5 h, and 12 h or untreated controls (DMSO) by Western Blotting. GAPDH or Beta-Actin (ACTB) are shown as loading control. Green arrow represents 15Q ATXN3, yellow arrow 77Q ATXN3 and red arrow 148Q ATXN3. Orange arrow symbolizes LC3-I while blue arrow labels LC3-II. (D) Aggregation of ATXN3 was analyzed in ATXN3 (15Q, 77Q, 148Q) transfected HEK-293T cells treated with CHX or untreated controls (DMSO) using DD-AGE analysis. Aggregation values were normalized to the respective ATXN3 15Q aggregation. Statistical analysis was performed with one-way ANOVA and Tukey´s multiple comparison test. * p < 0.05, ** p < 0.01 by one-way ANOVA. Values are shown as mean +/− SEM.
Figure 5.
Increased aggregation of ATXN3 according to cycloheximide (CHX) treatment. (A, B) Protein levels of ATXN3, phospho-S6, p62, BECN1 and LC3 were measured in HEK-293T cells transfected with ATXN3 (15Q, 77Q, 148Q) and treated with cycloheximide or untreated controls (DMSO) by Western Blotting. GAPDH or Beta-Actin (ACTB) are shown as loading control. Protein levels were normalized to the respective ATXN3 15Q protein expression. Statistical analyses were performed from three independent experiments with one-way ANOVA and Tukey´s multiple comparison test. Green arrow represents 15Q ATXN3, yellow arrow 77Q ATXN3 and red arrow 148Q ATXN3. Orange arrow symbolizes LC3-I while blue arrow labels LC3-II. (C) Protein levels of ATXN3, p62, and LC3 were measured in ATXN3 (15Q, 77Q, 148Q) transfected HEK-293T cells treated with CHX for 1 h, 5 h, and 12 h or untreated controls (DMSO) by Western Blotting. GAPDH or Beta-Actin (ACTB) are shown as loading control. Green arrow represents 15Q ATXN3, yellow arrow 77Q ATXN3 and red arrow 148Q ATXN3. Orange arrow symbolizes LC3-I while blue arrow labels LC3-II. (D) Aggregation of ATXN3 was analyzed in ATXN3 (15Q, 77Q, 148Q) transfected HEK-293T cells treated with CHX or untreated controls (DMSO) using DD-AGE analysis. Aggregation values were normalized to the respective ATXN3 15Q aggregation. Statistical analysis was performed with one-way ANOVA and Tukey´s multiple comparison test. * p < 0.05, ** p < 0.01 by one-way ANOVA. Values are shown as mean +/− SEM.
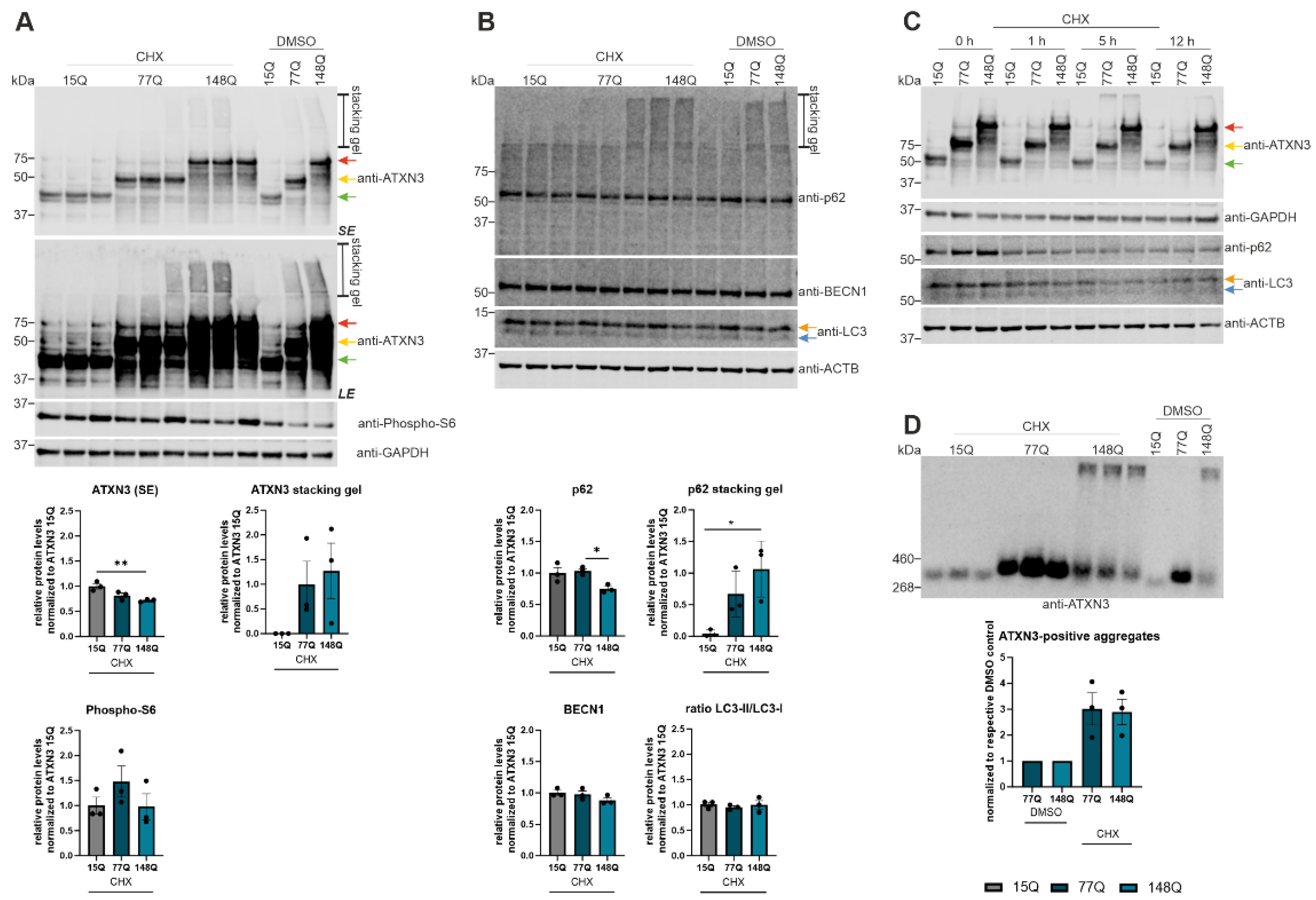
Figure 6.
BECN1-dependent autophagic activation caused by proteasomal inhibition. (A, B, C) Protein levels of ATXN3, p62, BECN1, phospho-S6, LC3, total polyubiquitin, K63- and K48-specific polyubiquitin were measured in ATXN3 (15Q, 77Q, 148Q) HEK-293T cells treated with MG132 or untreated controls (DMSO). Beta-Actin (ACTB) is shown as loading control. Protein levels were normalized to the respective ATXN3 15Q protein expression. Statistical analyses were performed from three independent experiments with one-way ANOVA and Tukey´s multiple comparison test. Green arrow represents 15Q ATXN3, yellow arrow 77Q ATXN3 and red arrow 148Q ATXN3. Orange arrow symbolizes LC3-I while blue arrow labels LC3-II. * p < 0.05, ** p < 0.01, *** p < 0.001 by one-way ANOVA. Values are shown as mean +/− SEM.
Figure 6.
BECN1-dependent autophagic activation caused by proteasomal inhibition. (A, B, C) Protein levels of ATXN3, p62, BECN1, phospho-S6, LC3, total polyubiquitin, K63- and K48-specific polyubiquitin were measured in ATXN3 (15Q, 77Q, 148Q) HEK-293T cells treated with MG132 or untreated controls (DMSO). Beta-Actin (ACTB) is shown as loading control. Protein levels were normalized to the respective ATXN3 15Q protein expression. Statistical analyses were performed from three independent experiments with one-way ANOVA and Tukey´s multiple comparison test. Green arrow represents 15Q ATXN3, yellow arrow 77Q ATXN3 and red arrow 148Q ATXN3. Orange arrow symbolizes LC3-I while blue arrow labels LC3-II. * p < 0.05, ** p < 0.01, *** p < 0.001 by one-way ANOVA. Values are shown as mean +/− SEM.
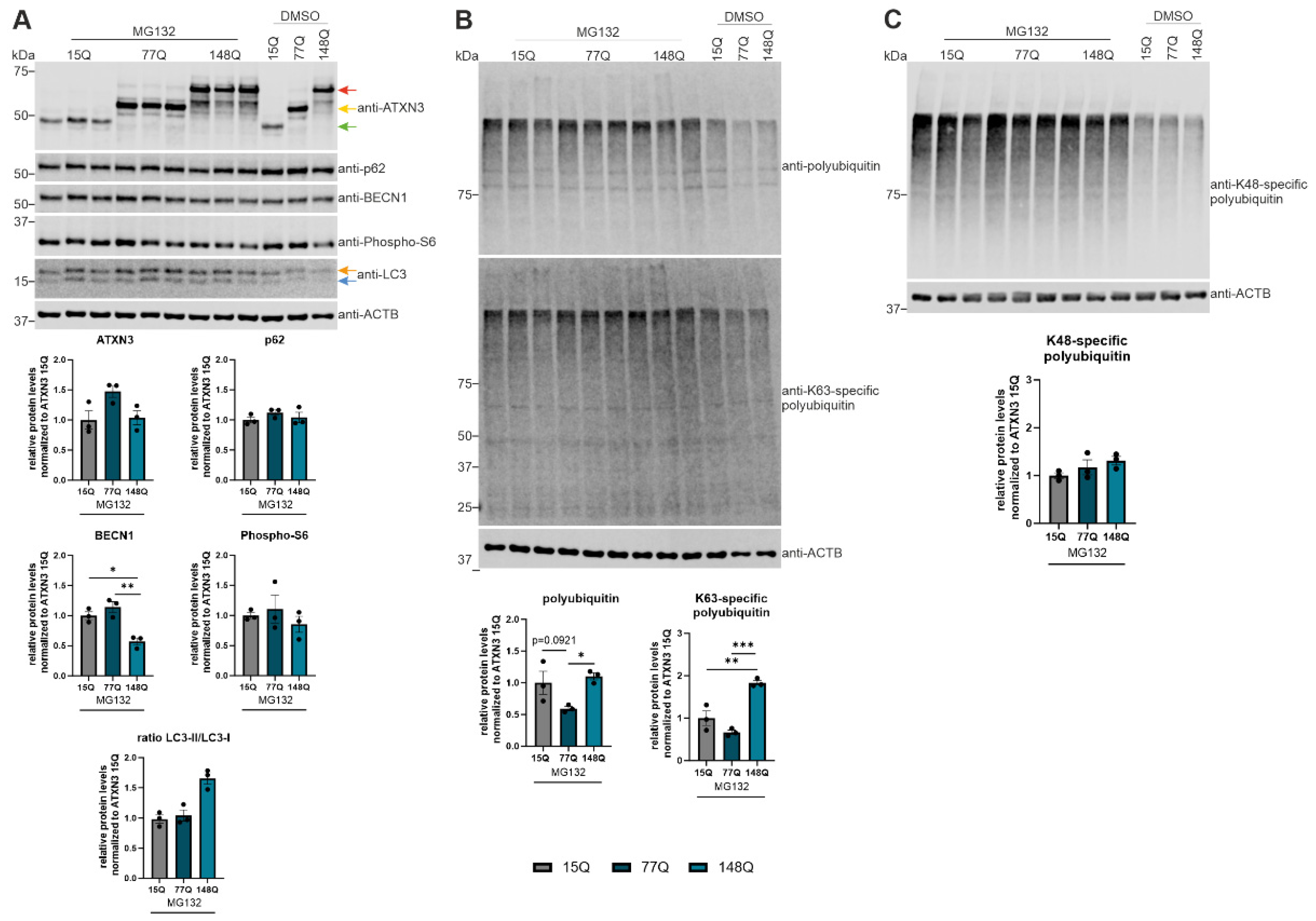
Table 1.
Antibodies used for Western Blot.
| Target Protein | Product number | Species | Manufacturer | Dilution |
|---|---|---|---|---|
| ACTB | A2228 | mouse | Merck | 1:5000 |
| ATXN3 | clone 13H9L9 702788 |
rabbit | Thermo Fisher Scientific | 1:1000 |
| BECN1 | 3738S | rabbit | Cell Signaling Technology, Danvers, MA, USA | 1:1000 |
| GAPDH | clone GA1R ab125247 |
mouse | Abcam, Cambridge, UK | 1:2000 |
| K63-polyubiquitin | 5621S | rabbit | Cell Signaling Technology | 1:500 |
| K48-polyubiquitin | 8081S | rabbit | Cell Signaling Technology | 1:1000 |
| LC3B | 2775S | rabbit | Cell Signaling Technology | 1:500 |
| SQSTM1/p62 | 5114S | rabbit | Cell Signaling Technology | 1:1000 |
| phospho-S6 ribosomal protein (Ser235/236) | 2211S | rabbit | Cell Signaling Technology | 1:1000 |
| Ubiquitin | clone P4D1 sc-8017 |
mouse | Santa Cruz Biotechnology, Inc., Texas, USA |
1:1000 |
Table 2.
Primary antibodies used for Immunohistochemistry staining.
| Target Protein | Product number | Species | Manufacturer | Dilution |
|---|---|---|---|---|
| ATXN3 | clone 1H9 MAB5360 |
mouse | Merck | 1:1000 |
| p62 | 5114S | rabbit | Cell Signaling Technology | 1:500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



