
Submitted:
07 March 2024
Posted:
08 March 2024
You are already at the latest version
Abstract
.Vitamin A (retinol) and its derivatives (retinoids) assume critical roles in neural development, cellular differentiation, axon elongation, programmed cell apoptosis and various fundamental cellular processes. Retinoids function by binding to specific nuclear receptors, such as retinoic acid receptors (RARs) and retinoid X receptors (RXRs), activating specific signaling pathways in the cells. Disruption of the retinoic acid signaling pathway can result in neuroinflammation, ox-idative stress, mitochondrial dysfunction and neurodegenerative processes, and has been asso-ciated with a range of neurodegenerative diseases. The present study explores the potential therapeutic application of our innovative synthetic retinoid, Ellorarxine, also known as DC645 and NVG0645, for the treatment of neurodegenerative disorders in vitro. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) tetrazolium assay, lactate dehy-drogenase (LDH) assay, enzyme-linked immunosorbent assay (ELISA), senescence-associated (SA) β-galactosidase (β-gal) staining and immunofluorescence staining were performed. The re-sults showed that no cytotoxicity was detected at the experimental concentrations of Ellorarxine. Ellorarxine significantly reduced cell death, increased mitochondrial viability, reduced the num-ber of senescent cells, modulated cytokine release and regulated cellular autophagy. Furthermore, Ellorarxine also increased Cyp26 and selectively RARβ expression. These results make Ellorarxine a promising drug candidate that should be further investigated in the treatment of neurodegenerative diseases.
.
Keywords:
Ellorarxine
; DC645
; NVG0645
; retinoid
; mitochondrial dysfunction
; neuroinflammation
; neurodegeneration
; neuroprotective effects
1. Introduction
Vitamin A and its derivatives, known as retinoids, are specific modulators for neural differentiation, motor neuron outgrowth and immunology in vertebrates [1,2,3,4]. Retinoids have gained considerable attention in the context of their ability to regulate the gene expression of varieties of encoded enzymes, neurotransmitter transporter proteins and receptors, transcription factors, cell surface receptors and neuropeptide hormones [5]. All-trans-retinoic acid (RA), a metabolite of Vitamin A, performs physiological function by binding to and activating RA receptors (RARs) and retinoid X receptors (RXRs), which each have three subtypes (α, β and γ) with several isoforms [6]. RA translocates across the nuclear membrane through RARs, interacts with retinoic acid response elements (RAREs), and participates in the mechanism of gene regulation [7]. RAR and RXR exhibit broad expression across nearly all tissues especially the brain, although the distribution of each isotype varies [8]. Apart from the genomic effect, retinoids have also been emphasized that have important non-genomic effects, mediating homeostatic synaptic plasticity and neurotransmitter release [9,10].
RARs have been proven to be closely related to neurodegenerative diseases. Studies have shown that in vitamin A-deficient rats, the expression of RARα is inhibited, leading and the deposition of amyloid beta (Aβ) peptide in cerebral blood vessels [11]; RARβ and RXRβ/RXRγ mRNA in the hippocampus are also been downregulated, made young animals with VAD showed cognitive decline as those of aged animals [12]. RARβ is involved in the neuroprotection of striatal medium spiny neurons (spMSNs), a cell type affected in different neuropsychiatric diseases and particularly susceptible to degeneration in Huntington's disease (HD) [13]. Retinoid deficiency or mutations in the RARβ and RXRγ genes are associated with inhibition of spatial learning and memory and the development of depression in animals [14]. RA is often associated with and modulates regions of high neuroplasticity, and in the hippocampus, RA signaling is regulated by the availability of RALDH1 and RALDH2 synthetases and the CYP26B1 catabolic enzyme [15]. Retinoids are critical for long-term potentiation (LTP) and long-term depression (LTD) of neuroplasticity associated with learning and memory, as well as homeostatic synaptic plasticity (HSP) [16]. Retinoids play an important role in preventing neuroinflammatory responses to provide neuroprotection, and retinoids can downregulate the expression of cytokines and inflammatory molecules in microglia [17]. Retinoids also regulate the expression of tyrosine hydroxylase, dopamine β-hydroxylase, and dopamine D2 receptors [18]. Reduced acetylcholine (ACh) in neurodegenerative diseases has also been implicated in RA-mediated reductions in ChAT production and neuronal cell death [19]. The functional neuroprotective effects of synthetic retinoids in neurodegenerative diseases are being widely studied, and some have been developed as potential drugs for the treatment of neurodegenerative diseases and have achieved good results [20,21,22]. Therefore, selective RXR and RAR modulators become one of the promising therapies against neurodegenerative diseases [23].
In this study, we aimed to evaluate the efficacy of a synthetic retinoid (Nevrargenic’s lead drug, RAR modulator, Ellorarxine, also known as DC645 and NVG0645) on neurodegenerative symptoms. C6 is an established cell line derived from rat glioma that can differentiate into astrocyte-like cells, express GFAP under specific conditions, and has been used to culture astrocyte models [24]. SH-SY5Y is a human neuroblastoma cell line that can differentiate into neuron-like cells and can serve as a neuronal model for neurodegenerative diseases [25]. Microglia are related to a series of neurodegenerative diseases (such as attention deficit disorder and Parkinson's disease) and can have neuroprotective or neurotoxic effects [26]. Hence, a human microglial clone 3 cell line (HMC3) is used as a microglial model [27].
2. Results
2.1. Ellorarxine Upregulates the Expression of Cyp26b1 and RARβ
RARs are expressed in a variety of cells, including C6, SH-SY5Y and HMC3. Regulations of RAR-expression regulate cellular functions [28,29,30]. To investigate the regulation of RAR expression by Ellorarxine, immunofluorescence was used to detect the expression of Cyp 26B1 and RARs. The expression level of Cyp26b1 and RARs was determined using the average fluorescence intensity [31].
The results showed that Ellorarxine significantly upregulated the expression of Cyp26b1 (Figure 1) (C6: 118%, SH-SY5Y: 47%, HMC3: 36%) and RARβ (C6: 35%, differentiated SH-SY5Y: 71%, HMC3: 46%) , but not RARα or RARγ (Figure 2). The regulation of the distribution of RARα and RARγ by Ellorarxine was more pronounced: Ellorarxine caused RARα in C6 cells to migrate towards the ends of the nucleus (Figure 2A, white arrow), and RARγ in differentiated SH-SY5Y to migrate towards the cell membrane (Figure 2D, yellow arrow).
2.2. Ellorarxine Pretreatment Alleviates Mitochondrial Dysfunction
In cultured neuronal cells, MTT assay, measuring the amount of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium in the cell cytoplasm that converted to MTT formazan, is one of the most commonly used methods to determine mitochondrial viability and activity of NAD-dependent oxidoreductases [32]. To investigate the effect of Ellorarxine on mitochondrial viability, we examined the percentage of mitochondrial viability with or without 4 h Ellorarxine pretreatment of glia, neurons and microglia under oxidative stress or PBS control using an MTT assay. To induce mitochondrial dysfunction, oxidative stress was applied to the cells using 100 mM hydrogen peroxide (H2O2).
Results showed that Ellorarxine has a significant enhancement effect (10%) on mitochondrial function in SH-SY5Y (Figure 3A). The mitochondrial function after the stress of 100 mM H2O2 with mitochondrial activity being significantly reduced in both cases compared to Figure 3A where mitochondrial dysfunction was induced (Figure 3B). In this case, Ellorarxine had a significant enhancing effect on mitochondrial function in SH-SY5Y by improving mitochondrial viability by 17% (Figure 3B).
2.3. Ellorarxine Pretreatment Reduced Cell Death
LDH assay, measuring lactate dehydrogenase (LDH) release, is more valid for the determination of cell death in cultured neuronal cells than the MTT assay [33]. To investigate the effect of Ellorarxine on cell death, we performed an LDH assay on C6, SH-SY5Y and HMC3 with or without 4 h Ellorarxine pretreatment. To induce cell death, a final concentration of either 200 mM H2O2 or 15 μg/mL of LPS was applied.
Results showed that Ellorarxine had a significant protective effect of 19% on SH-SY5Y (Figure 4A). The percentage of cell death after the stress of 200 mM H2O2. Ellorarxine significantly protected C6 from death by 10% (Figure 4B). The percentage of cell death after the use of 10 μg/mL LPS. Ellorarxine showed a significant protective effect (14%) on SH-SY5Y (Figure 4C).
2.4. Ellorarxine Pretreatment Modulated Inflammatory Cytokine Release
The enzyme-linked immunosorbent assay (ELISA) can detect the antigens and cytokines accurately and sensitively, which is one of the most widely used cytokine measurement methods [34]. To investigate the effect of Ellorarxine on neuroinflammation, ELISAs were performed on HMC3 with or without 4 h Ellorarxine pretreatment. To induce inflammation, LPS was added to make a final concentration of 10 μg/mL.
2.5. Ellorarxine Treatment Reduced the Number of Senescent Cells
Senescence-associated β-Galactosidase (SA-β-Gal) is one of the most commonly used markers for senescence and has been used in cultured neuronal cells [35,36]. To investigate the anti-senescence effects of Ellorarxine, we treated the cells with 20 mM H2O2 for 24 h before treatment with Ellorarxine. Using SA-β-Gal staining to determine the number of senescent cells.
We found that Ellorarxine post-treatment significantly reduced the proportion of senescent cells by 60% under inflammatory stress (Figure 6B). At the same time, the proportion of senescent cells in the Ellorarxine post-treatment group in the control situation was also reduced, though not statistically significant (Figure 6).
2.6. Ellorarxine Treatment Regulated Cellular Autophagy
The distinctive feature of cellular events during macroautophagic/autophagic induction is characterized by the conjugation of LC3B, a mammalian homolog of Atg8, with phosphatidylethanolamine [37]. To investigate the regulation of autophagy by Ellorarxine, we cultured the cells with DMEM/F12 without serum for 24 h before the Ellorarxine treatment. Immunocytochemical staining was used to determine the expression of LC3BII in the cells. The expression level of LC3BII was determined using average optical density (AOD) [38].
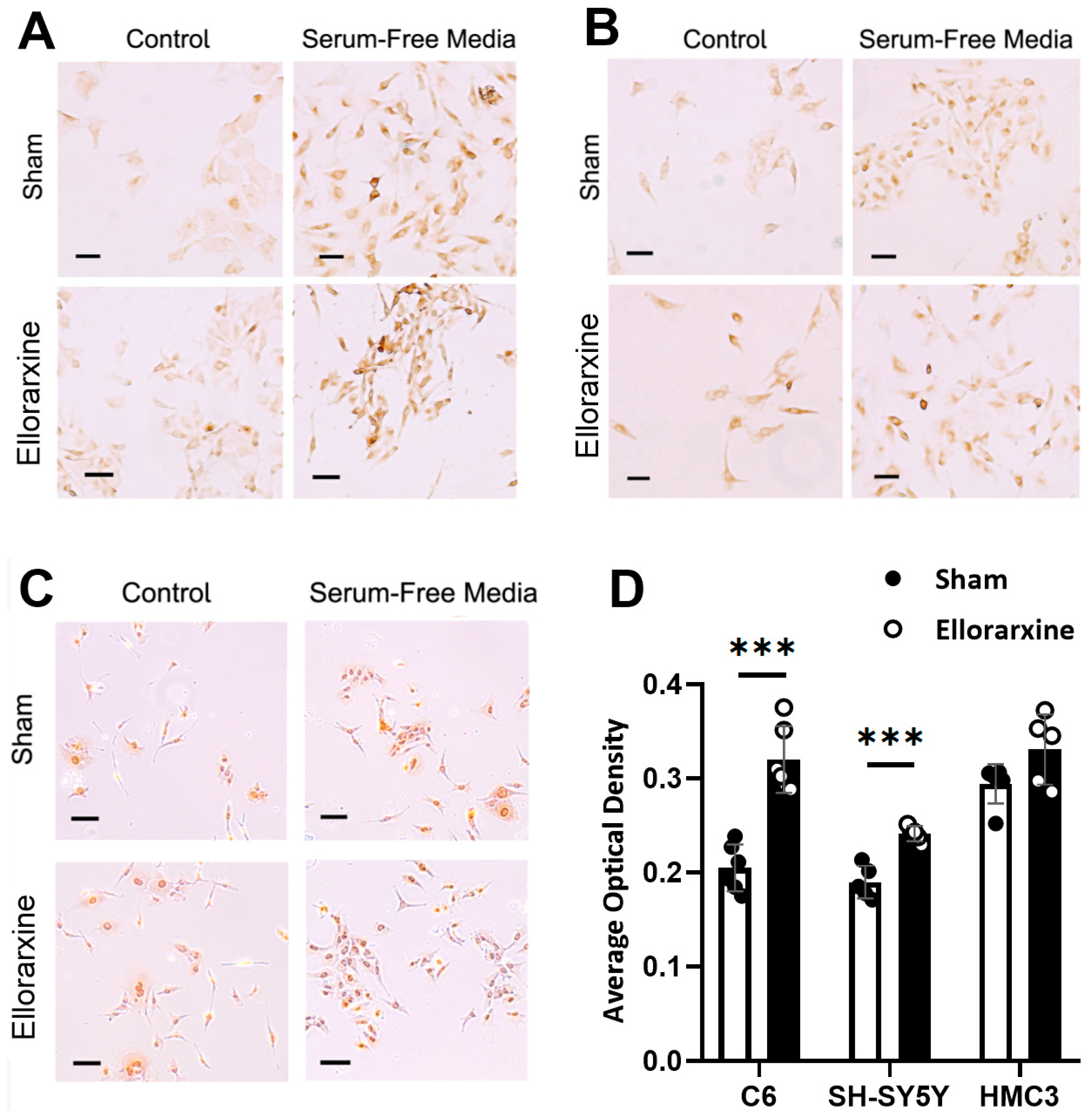
We found that Ellorarxine treatment significantly elevated the level of autophagy in C6, SH-SY5Y and HMC3 in serum-free media (Figure 7D) (C6: 56%, SH-SY5Y: 27%), hence showing that Ellorarxine was able to regulate cellular autophagy.
3. Discussion
Mitochondrial dysfunction plays a causative role in neurodegenerative diseases. Defects in mitochondrial respiratory chain function, oxidative stress, morphology/kinetics, and calcium handling capacity can induce neurodegenerative diseases represented by Parkinson's disease [39,40]. Mitochondrial dysfunction can also induce a variety of motor neurodegenerative diseases [41]. Studies have shown that RARβ plays an important role in controlling neurotransmission, energy metabolism and transcription, and especially in G protein-coupled receptors, cAMP and calcium signaling [42]. Many of the identified RARβ target genes associated with these pathways have been implicated in various neurodegenerative diseases such as Alzheimer's and Parkinson's diseases [43,44]. Studies have shown that loss of RARβ can lead to mitochondrial dysfunction in mice, and that RARβ agonists can prevent mitochondrial failure induced by mitochondrial toxins, and reduce mitochondrial fragmentation and cell death [13,45]. Our results show that Ellorarxine can significantly alleviate mitochondrial dysfunction in neurons induced by oxidative stress. Under control conditions, Ellorarxine can also significantly improve neuronal mitochondrial function. This may be because Ellorarxine can upregulate the expression of RARβ and activate the neuroprotective effect of RARβ. Our immunofluorescence semi-quantitative analysis results showed that Ellorarxine can significantly upregulate the expression of RARβ in three cell types without affecting the expression of RARα and RARγ. This suggests that Ellorarxine exhibits a level of selectivity for RARβ, and therefore, can act as an RARβ agonist to enhance the function of RARβ, thereby exerting a neuroprotective effect [46,47,48]. This may be due to the up-regulation by Ellorarxine of the expression of Cyp26b1. The Cyp26 family of enzymes (CYP26A1, B1 and C1) are key proteins that regulate the internal levels of RA in cells, and retinoids are the only substrates of this enzyme family. Cyp26b1 plays an important role in establishing the RA gradient. The RA metabolite 4-oxo-RA produced by Cyp26b1 catabolism was previously shown to be a potent agonist specifically targeting RARβ [15,49,50,51,52,53]. Therefore, the selectivity of Ellorarxine for RARβ may be derived from this metabolite produced by its hydroxylation of RA.
It is widely recognized that oxidative stress is important in the etiology of several late-onset neurodegenerative diseases [54,55,56]. Oxidative stress is a condition in which the balance between ROS production and antioxidant levels is severely disrupted and results in excessive ROS damage to cells, leading to apoptosis [57,58,59]. In this study, we found that Ellorarxine could significantly reduce the apoptosis of glial cells caused by H2O2 and was statistically insignificant, but showed a tendency to reduce the apoptosis of neuronal cells and microglia. RA has been shown to reduce the sensitivity of various cells to apoptosis caused by oxidative stress [60,61]. Studies have shown that RA protects mesangial cells from H2O2-induced apoptosis by inhibiting the activator protein 1 (AP-1) pathway, inhibiting c-fos and c-jun expression and inhibiting c-Jun N-terminal kinase (JNK) [62,63]. It has also been reported that RA reduces apoptosis through the inhibition of oxidative stress and the preservation of superoxide dismutase (SOD) protein levels [60]. RA has been shown to improve the antioxidant defense system [61].
In a variety of neurodegenerative diseases, the inflammatory response triggered by xenobiotics, chemicals and beta-amyloid, etc. is driven by inflammatory and pro-inflammatory cytokines and chemokines (TNF-alpha, IL-6, etc.) [64,65]. Microglia exert neuroprotective or neurotoxic effects depending on the intensity of the stimulus and the extent of the inflammatory response. Excessive cytokine release can overactivate microglia, resulting in neurotoxicity [66]. NF-κB is widely present in mammals and is a key factor in cellular inflammatory responses and neuroprotection. Inhibition of NF-κB activation plays a neuroprotective role in microglia in lipopolysaccharide (LPS)-induced neurodegenerative diseases [67]. RAR in microglia inhibits the NF-κB signalling pathway and suppresses their production. Ellorarxine was able to activate RAR in microglia, modulate microglial function and reduce the production of pro-inflammatory cytokines and chemokines [68,69]. Our results showed that neither TNFα nor IL-6 release was stimulated under baseline conditions from Ellorarxine treatment. This showed a hint that Ellorarxine does not upregulate an acute inflammatory response because these pro-inflammatory cytokines of the innate immune system are unaffected following Ellorarxine treatment. Under LPS exposure conditions, Ellorarxine reduced the production of IL-6 by 40% compared with the control group. At the same time, we found that Ellorarxine can significantly reduce neuronal apoptosis induced by LPS exposure. These results indicate that Ellorarxine can reduce neuroinflammation and provide neuroprotection.
In the normal state, damaged organelles and protein aggregates reach the lysosome through endosomal and autophagosomal delivery, where they are digested and recycled through cellular autophagy [70]. In a variety of neurodegenerative diseases, defects occur at different stages of the autophagic pathway, causing neurons to degenerate due to the accumulation of ubiquitinylated protein aggregates [71,72]. Lysosomal storage disorders are also often characterised by a severe neurodegenerative phenotype. ATRA has been successfully used to treat acute promyelocytic leukemia (APL) and its induced differentiation of the APL cell line NB4 involves the induction of autophagy [73]. Recent studies showed that RA can improve autophagy through depression of the PI3K-Akt-mTOR signaling pathway via RARα [74]. Ellorarxine was able to induce cellular autophagy by activating RARα, enhancing and restoring cellular autophagy function. In the present study, our results showed that Ellorarxine was able to significantly upregulate the autophagy levels of glial cells, neurons and microglia under induced stress conditions.
The pathogenesis of neurodegeneration involves many processes, including protein misfolding and aggregation, abnormalities in kinase signaling pathways, neuronal calcium dysregulation and impaired synaptic transmission [75,76]. Senescence has been identified as one of the important risk factors for common neurodegenerative diseases, Alzheimer's and Parkinson's diseases [77,78]. Unlike the normal programmed terminal differentiation process, senescence is a unique pro-inflammatory fate in which cells acquire a unique secretome of cytokines, chemokines, proteases and growth factors, collectively known as the senescence-associated secretory phenotype (SASP). arious stressors, such as DNA damage, reactive oxygen species (ROS), neuroinflammation, robust mitogenic/oncogenic signaling, loss of specific tumor suppressors, mitotic stress, DNA replication arrest, and chromatin disruption, can lead to cell senescence [79]. Eliminating senescent cells, inhibiting SASP and reversing cellular senescence are effective in modulating neuroinflammatory diseases. These senolytic approaches (mouse models and treatments targeting senescent cells) can eliminate senescent cells, and the accompanying SASP, may have beneficial effects on the development of neurodegeneration in the brain [80,81]. This study found that Ellorarxine can significantly reduce the amount of cell senescence induced by oxidative stress. Although it is not yet certain whether retinoid exposure reduces the number of senescent cells due to reversing cell senescence or eliminating senescent cells, studies have pointed out that combined treatment with vitamin A and quercetin can inhibit the senescence response after acute liver injury. This may be related to the antioxidant and anti-inflammatory effects of vitamin A.
In this study, we mainly found that Ellorarxine protected C6, SH-SY5Y and HMC3 from oxidative stress and neuroinflammation, and reduced the number of senescent cells. Specifically, mitochondrial dysfunction and cell death induced by hydrogen peroxide and LPS in SH-SY5Y and neuroinflammation in HMC3, could be alleviated by Ellorarxine pretreatment. Cellular senescence induced by hydrogen peroxide in C6, and autophagy disorders induced in SH-SY5Y and C6 could be reversed by Ellorarxine treatment.
4. Materials and Methods
4.1. Reagent and Resources
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Recombinant Anti-Retinoic Acid Receptor alpha antibody [EPR23871-271] (ab275745) | Abcam | https://www.abcam.com/products/primary-antibodies/retinoic-acid-receptor-alpha-antibody-epr23871-271-ab275745.html |
| Anti-Retinoic Acid Receptor beta antibody (ab5792) | Abcam | https://www.abcam.com/products/primary-antibodies/retinoic-acid-receptor-beta-antibody-ab5792.html |
| Anti-Retinoic Acid Receptor gamma antibody (ab97569) | Abcam | https://www.abcam.com/products/primary-antibodies/retinoic-acid-receptor-gamma-antibody-ab97569.html |
| Anti-Cyp26B1 antibody (ab113236) | Abcam | https://www.abcam.com/products/primary-antibodies/cyp26b1-antibody-ab113236.html |
| Goat Anti-Mouse IgG H&L (Alexa Fluor® 488) (ab150113) | Abcam | https://www.abcam.com/products/secondary-antibodies/goat-mouse-igg-hl-alexa-fluor-488-ab150113.html |
| Chemicals, peptides, and recombinant proteins | ||
| Phosphate buffered saline (P5368-10pak) | Sigma-Aldrich | https://www.sigmaaldrich.com/GB/en/search/p5368-10pak?focus=products&page=1&perpage=30&sort=relevance&term=p5368-10pak&type=product |
| Triton X-100 | ||
| BSA | ||
| Tween 20 | ||
| Thiazolyl Blue Tetrazolium Bromide | Sigma-Aldrich | https://www.sigmaaldrich.com/GB/en/search/m2128?focus=products&page=1&perpage=30&sort=relevance&term=m2128&type=product |
| H2O2 | ||
| LPS | ||
| Critical commercial assays | ||
| CytoTox 96® Non-Radioactive Cytotoxicity Assay | Promega | https://www.promega.co.uk/products/cell-health-assays/cell-viability-and-cytotoxicity-assays/cytotox-96-non_radioactive-cytotoxicity-assay/?catNum=G1780 |
| Human IL-6 ELISA Kit (ab178013) | Abcam | https://www.abcam.com/products/elisa/human-il-6-elisa-kit-ab178013.html |
| Human TNF alpha ELISA Kit (ab46087) | Abcam | https://www.abcam.com/products/elisa/human-tnf-alpha-elisa-kit-ab46087.html |
| Senescence Cells Histochemical Staining Kit | Sigma-Aldrich | https://www.sigmaaldrich.com/GB/en/search/cs0030-1kt?focus=products&page=1&perpage=30&sort=relevance&term=cs0030-1kt&type=product |
| VECTASTAIN® Elite® ABC-HRP Kit (Peroxidase, Universal) (PK-6200) | VECTASTAIN | https://vectorlabs.com/products/vectastain-elite-abc-hrp-kit-universal |
| ImmPACT® DAB Substrate Kit, Peroxidase (HRP) (SK-4105) | VECTASTAIN | https://vectorlabs.com/products/immpact-dab-hrp-substrate |
| Experimental models: Cell lines | ||
| Rat glioma C6 cells | ||
| Human neuroblastoma SH-SY5Y cells | ||
| Human microglia clone 3 HMC3 cells | ||
| Software and algorithms | ||
| ZEN software | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
| Prism8 | GraphPad | https://www.graphpad.com/ |
| ImageJ | LOCI | https://imagej.net/ |
4.2. Cell Lines and Culture
C6 (rat glioma), HMC3 (human microglial clone 3), and SH-SY5Y (human neuroblastoma) were obtained from Durham University and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supple-mented with 10% fetal bovine serum (FBS, Gibco) and 1% Penicillin Streptomycin Solution (Pen-Strep, Lonza) at 37 °C in a humidified 5% CO2 incubator. The growth medium was changed every 2 days. When the culture reached 80% confluence, trypsin-EDTA was added and incubated for 3-5 min to make adherent cells detached. Triturated cells were seeded 1:2 into 24-well plates or T75 flasks for further growth.
4.3. Cell Differentiation
The cells were differentiated by adding retinoic acid (RA) to Dulbecco’s modified Eagle’s medium (DMEM, Gibco) with 1% Penicillin Streptomycin Solution (Pen-Strep, Lonza) to a final concentration of 10 μM, 24 h after subculturing. Cultures were differentiated for 6 days. The medium was changed every 2 days. After being differentiated, cells were cultured under normal conditions for two days to eliminate the effects of RA. Differentiated cultures were used for all the treatments mentioned hereafter [25].
4.4. Preparation of Ellorarxine
Ellorarxine (1mM in DMSO) was synthesized following Nevrargenic’s patent of DC645. and was stored at -20°C. The drug was prepared to 1μM stock solution using dH2O and was stored at 4°C.
4.5. Pre-Treatments
After trypsinization, cells were plated (40000 cells/mL) in 24-well plate chambers and left to grow for 24 hours at 37°C and 5% CO2 before being treated with 10% DMSO (Sham) or 10nM Ellorarxine for 4 hours before being stressed.
4.6. Methyl Thiazolyl-Diphenyl-Tetrazolium Bromide (MTT) Assay
50 μl of 5mg/ml MTT (M2128, Sigma) was added to each well and left to incubate for 4 hours at 37°C and 5% CO2. Subsequently, the media was removed and 200 μl DMSO was added to each well to dissolve the formazan crystals. Finally, 100 μl from each well was transferred to a 96-well tissue culture plate and the absorbance was measured at 595nm, using a microplate reader.
4.7. Lactate Dehydrogenase (LDH) Release Assay
LDH release was measured using CytoTox 96 kit (ADG1781, Promega). 100 μl of the supernatant was taken out of each well and transferred to a 96-well tissue culture plate. 100 μl of the cytotoxicity detection kit LDH solution was added to each well and incubated for 30 minutes in the dark at room temperature. The reaction was stopped by adding 50 μl of stop solution. Subsequently, the optical density was measured at 490nm. This assay was normalised by freezing the remaining plate and later thawing it, then pipetting the contents of each well into Eppendorf tubes, centrifuging those for 10 minutes for the cells to settle down, and then taking out 100μl of the supernatant from each Eppendorf tube and following the same proce-dure as described above. This gave an indication of total LDH and allowed normalisation.
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
24 hours after stressing the cells, 100μl of the supernatant was collected from each well and ELISA was car-ried out using the Human IL-6 ELISA kit (ab178013, Abcam) and Human TNF-α ELISA kit (ab46087) accord-ing to the manufacturer’s protocol. The standard curve generated was used to calculate concentrations from the absorbance measurements.
4.9. Senescence-Associated β-Galactosidase (SA-β-Gal) Staining
Cells were plated 8000/mL in 6-well (35mm) chambers onto 15mm×15mm coverslips. 24 hours after stressing the cells, Senescence-Associated β-Galactosidase (SA-β-Gal) Staining was carried out using Se-nescence Cells Histostaining Kit (Sigma-Aldrich, CS0030-1KT) according to the manufacturer’s protocol. Cell nuclei were then stained by DAPI. The stained cells were counted and compared to the total number of cells, evaluated by counting the DAPI-stained nuclei.
4.10. Immunocytochemistry Staining
Cells were plated 8000/mL in 6-well (35mm) chambers onto 15mm×15mm coverslips. 24 hours after stressing the cells, immunocytochemistry staining was carried out using the VECTASTAIN Elite ABC Univer-sal Kit (PK-6200) and ImmPACT DAB Substrate Kit, Peroxidase (SK-4105) according to the manufacturer’s protocol.
4.11. Immunofluorescence Staining
Cells were plated 8000/mL in 6-well (35mm) chambers onto 15mm×15mm coverslips. 24 hours after treatment, the cells were fixed in 4% paraformaldehyde (PFA) for 10 minutes at room temperature. Cells were washed three times 5 min with PBS and then blocked in PBS containing 1% bovine serum albumin, 1% fish skin gelatin and 0.3% Triton X-100 at room temperature for 1 h. Then the cells were incubated with primary antibody for 1 h at room temperature. Primary antibodies were diluted as: RARα (1:100, Abcam, ab275745), RARβ (1:100, Abcam, ab5792), RARγ (1:100, Abcam, ab97569), Cyp26B1 (1:200, Abcam, ab113236). Cells were then washed three times 5 min in PBS and incubated with secondary antibodies (Goat Anti-Mouse IgG H&L Alexa Fluor® 488, 1:1000, Abcam, ab150113) for 1.5 h at room temperature. Cells were then washed three times 5min with PBS, and incubated with DAPI (1μg/mL) for 5 minutes at room temperature to stain the DNA for nuclear localization. Fluorescent images were captured by using a Zeiss fluorescent microscope (Zeiss ApoTome).
4.12. Quantification and Statistical Analysis
The semi-quantitative detection of immunofluorescence images and immunocytochemistry images were applied using ImageJ [31,38].
The data were obtained from at least three independent experiments for each experimental condition. Data are expressed as means ± SD. Two-tailed t-tests were used to analyze differences between two groups. p values <0.05 are considered significant. All these analyses were performed using Graphpad Prism 8. Source data used to plot graphs in each panel of main figures and Supplemental Figures are pro-vided in a Microsoft Excel file as Supplemental Material. Key statistical results for each panel of figures are shown in figure legends.
5. Patents
Whiting A, Valentine R, Chisholm DR, McCaffery P, Greig IR, Khatib T (2019) Synthetic retinoids for use in RAR activation. Patent No. GB1903242.4
Supplementary Materials
The following supporting information can be downloaded at: www.nevrargenics.com.
Author Contributions
Conceptualization, P.C. and A.W.; methodology, Y.Z.; validation, Y.Z.; formal analysis, Y.Z.; data curation, Y.Z. and L.G.; writing—original draft preparation, Y.Z.; writing—review and editing, P.C. and A.W.; visualization, Y.Z.; supervision, P.C.; project administration, P.C.; funding acquisition, P.C. and A.W. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lane, M.A.; Bailey, S.J. Role of Retinoid Signalling in the Adult Brain. Prog. Neurobiol. 2005, 75. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.; Wu, S.C.; Blumberg, B. Retinoic Acid Signaling and Neuronal Differentiation. Cell. Mol. Life Sci. 2015, 72. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Berciano, M.T.; Ruiz-Soto, M.; Berciano, J.; Landreth, G.; Lafarga, M. Retinoids and Motor Neuron Disease: Potential Role in Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2016, 360. [Google Scholar] [CrossRef] [PubMed]
- Gürbüz, M.; Aktaç, Ş. Understanding the Role of Vitamin A and Its Precursors in the Immune System. Nutr. Clin. Et. Metab. 2022, 36. [Google Scholar] [CrossRef]
- McCaffery, P.J.; Adams, J.; Maden, M.; Rosa-Molinar, E. Too Much of a Good Thing: Retinoic Acid as an Endogenous Regulator of Neural Differentiation and Exogenous Teratogen. Eur. J. Neurosci. 2003, 18. [Google Scholar] [CrossRef] [PubMed]
- Giguère, V. Retinoic Acid Receptors and Cellular Retinoid Binding Proteins: Complex Interplay in Retinoid Signaling. Endocr. Rev. 1994, 15. [Google Scholar] [CrossRef]
- Lavudi, K.; Nuguri, S.M.; Olverson, Z.; Dhanabalan, A.K.; Patnaik, S.; Kokkanti, R.R. Targeting the Retinoic Acid Signaling Pathway as a Modern Precision Therapy against Cancers. Front. Cell Dev. Biol. 2023, 11. [Google Scholar] [CrossRef]
- Li, B.; Cai, S.Y.; Boyer, J.L. The Role of the Retinoid Receptor, RAR/RXR Heterodimer, in Liver Physiology. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867. [Google Scholar] [CrossRef]
- Liao, Y.P.; Ho, S.Y.; Liou, J.C. Non-Genomic Regulation of Transmitter Release by Retinoic Acid at Developing Motoneurons in Xenopus Cell Culture. J. Cell Sci. 2004, 117. [Google Scholar] [CrossRef]
- Chen, L.; Lau, A.G.; Sarti, F. Synaptic Retinoic Acid Signaling and Homeostatic Synaptic Plasticity. Neuropharmacology 2014, 78. [Google Scholar] [CrossRef]
- Corcoran, J.P.T.; Po, L.S.; Maden, M. Disruption of the Retinoid Signalling Pathway Causes a Deposition of Amyloid β in the Adult Rat Brain. Eur. J. Neurosci. 2004, 20. [Google Scholar] [CrossRef]
- Etchamendy, N.; Enderlin, V.; Marighetto, A.; Pallet, V.; Higueret, P.; Jaffard, R. Vitamin A Deficiency and Relational Memory Deficit in Adult Mice: Relationships with Changes in Brain Retinoid Signalling. Behav. Brain Res. 2003, 145. [Google Scholar] [CrossRef]
- Ciancia, M.; Rataj-Baniowska, M.; Zinter, N.; Baldassarro, V.A.; Fraulob, V.; Charles, A.L.; Alvarez, R.; Muramatsu, S. ichi; de Lera, A.R.; Geny, B.; et al. Retinoic Acid Receptor Beta Protects Striatopallidal Medium Spiny Neurons from Mitochondrial Dysfunction and Neurodegeneration. Prog. Neurobiol. 2022, 212. [Google Scholar] [CrossRef]
- Chiang, M.Y.; Misner, D.; Kempermann, G.; Schikorski, T.; Giguère, V.; Sucov, H.M.; Gage, F.H.; Stevens, C.F.; Evans, R.M. An Essential Role for Retinoid Receptors RARβ and RXRγ in Long-Term Potentiation and Depression. Neuron 1998, 21. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C. Regulating Retinoic Acid Availability during Development and Regeneration: The Role of the CYP26 Enzymes. J. Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Walters, B.J.; Josselyn, S.A. Retinoic Acid Receptor Plays Both Sides of Homeostatic Plasticity. Proc. Natl. Acad. Sci. USA 2019, 116. [Google Scholar] [CrossRef]
- Sanjay, *!!! REPLACE !!!*; Kim, J.Y. Anti-Inflammatory Effects of 9-Cis-Retinoic Acid on β-Amyloid Treated Human Microglial Cells. Eur. J. Inflamm. 2022, 20. [Google Scholar] [CrossRef]
- Magalingam, K.B.; Somanath, S.D.; Md, S.; Haleagrahara, N.; Fu, J.Y.; Selvaduray, K.R.; Radhakrishnan, A.K. Tocotrienols Protect Differentiated SH-SY5Y Human Neuroblastoma Cells against 6-Hydroxydopamine-Induced Cytotoxicity by Ameliorating Dopamine Biosynthesis and Dopamine Receptor D2 Gene Expression. Nutr. Res. 2022, 98. [Google Scholar] [CrossRef]
- Pedersen, W.A.; Berse, B.; Schüler, U.; Wainer, B.H.; Blusztajn, J.K. All-trans- and 9-cis-Retinoic Acid Enhance the Cholinergic Properties of a Murine Septal Cell Line: Evidence That the Effects Are Mediated by Activation of Retinoic Acid Receptor-α. J. Neurochem. 1995, 65. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.P.; Casadesus, G.; Zhu, X.; Lee, H.G.; Perry, G.; Smith, M.A.; Gustaw-Rothenberg, K.; Lerner, A. All-Trans Retinoic Acid as a Novel Therapeutic Strategy for Alzheimer’s Disease. Expert. Rev. Neurother. 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, H.; Nakagomi, M.; Yamagata, N.; Katsuki, H.; Kawahara, K.; Kitaoka, K.; Miki, T.; Shudo, K. Tamibarotene: A Candidate Retinoid Drug for Alzheimer’s Disease. Biol. Pharm. Bull. 2012, 35. [Google Scholar] [CrossRef]
- Zhang, X.; Subbanna, S.; Williams, C.R.O.; Canals-Baker, S.; Smiley, J.F.; Wilson, D.A.; Das, B.C.; Saito, M. Anti-Inflammatory Action of BT75, a Novel RARα Agonist, in Cultured Microglia and in an Experimental Mouse Model of Alzheimer’s Disease. Neurochem. Res. 2023, 48. [Google Scholar] [CrossRef]
- le Maire, A.; Alvarez, S.; Shankaranarayanan, P.; R de Lera, A.; Bourguet, W.; Gronemeyer, H. Retinoid Receptors and Therapeutic Applications of RAR/RXR Modulators. Curr. Top. Med. Chem. 2012, 12. [Google Scholar] [CrossRef]
- Galland, F.; Seady, M.; Taday, J.; Smaili, S.S.; Gonçalves, C.A.; Leite, M.C. Astrocyte Culture Models: Molecular and Function Characterization of Primary Culture, Immortalized Astrocytes and C6 Glioma Cells. Neurochem. Int. 2019, 131. [Google Scholar] [CrossRef]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Exp. 2016, 2016. [Google Scholar] [CrossRef]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53. [Google Scholar] [CrossRef] [PubMed]
- Lepiarz, I.; Olajide, O. The Human Microglia (HMC-3) as a Cellular Model of Neuroinflammation. IBRO Rep. 2019, 6. [Google Scholar] [CrossRef]
- Hewson, Q.C.; Lovat, P.E.; Pearson, A.D.J.; Redfern, C.P.F. Retinoid Signalling and Gene Expression in Neuroblastoma Cells: RXR Agonist and Antagonist Effects on CRABP-II and RARβ Expression. J. Cell Biochem. 2002, 87. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chandra, V.; Rastinejad, F. Retinoic Acid Actions through Mammalian Nuclear Receptors. Chem. Rev. 2014, 114. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Storer, P.D.; Chavis, J.A.; Racke, M.K.; Drew, P.D. Agonists for the Peroxisome Proliferator-Activated Receptor-α and the Retinoid X Receptor Inhibit Inflammatory Responses of Microglia. J. Neurosci. Res. 2005, 81. [Google Scholar] [CrossRef]
- Fontenete, S.; Carvalho, D.; Lourenço, A.; Guimarães, N.; Madureira, P.; Figueiredo, C.; Azevedo, N.F. FISHji: New ImageJ Macros for the Quantification of Fluorescence in Epifluorescence Images. Biochem. Eng. J. 2016, 112. [Google Scholar] [CrossRef]
- Surin, A.M.; Sharipov, R.R.; Krasil’nikova, I.A.; Boyarkin, D.P.; Lisina, O.Y.; Gorbacheva, L.R.; Avetisyan, A. V.; Pinelis, V.G. Disruption of Functional Activity of Mitochondria during MTT Assay of Viability of Cultured Neurons. Biochemistry 2017, 82. [Google Scholar] [CrossRef] [PubMed]
- Lobner, D. Comparison of the LDH and MTT Assays for Quantifying Cell Death: Validity for Neuronal Apoptosis? J. Neurosci. Methods 2000, 96. [Google Scholar] [CrossRef] [PubMed]
- Leng, S.X.; McElhaney, J.E.; Walston, J.D.; Xie, D.; Fedarko, N.S.; Kuchel, G.A. ELISA and Multiplex Technologies for Cytokine Measurement in Inflammation and Aging Research. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2008, 63. [Google Scholar] [CrossRef]
- Geng, Y.Q.; Guan, J.T.; Xu, X.H.; Fu, Y.C. Senescence-Associated Beta-Galactosidase Activity Expression in Aging Hippocampal Neurons. Biochem. Biophys. Res. Commun. 2010, 396. [Google Scholar] [CrossRef] [PubMed]
- Struewing, I.T.; Durham, S.N.; Barnett, C.D.; Mao, C.D. Enhanced Endothelial Cell Senescence by Lithium-Induced Matrix Metalloproteinase-1 Expression. J. Biol. Chem. 2009, 284. [Google Scholar] [CrossRef]
- Hwang, H.J.; Kim, Y.K. The Role of LC3B in Autophagy as an RNA-Binding Protein. Autophagy 2023, 19. [Google Scholar] [CrossRef]
- Nabors, L.B.; Songu-Mize, E.; Mize, R.R. Quantitative Immunocytochemistry Using an Image Analyzer. II. Concentration Standards for Transmitter Immunocytochemistry. J. Neurosci. Methods 1988, 26. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of Mitochondrial ROS in the Brain: From Physiology to Neurodegeneration. FEBS Lett. 2018, 592. [Google Scholar] [CrossRef]
- Dey, K.; Bazala, M.A.; Kuznicki, J. Targeting Mitochondrial Calcium Pathways as a Potential Treatment against Parkinson’s Disease. Cell Calcium 2020, 89. [Google Scholar] [CrossRef]
- Catanesi, M.; D’angelo, M.; Tupone, M.G.; Benedetti, E.; Giordano, A.; Castelli, V.; Cimini, A. Micrornas Dysregulation and Mitochondrial Dysfunction in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Niewiadomska-Cimicka, A.; Krzyżosiak, A.; Ye, T.; Podleśny-Drabiniok, A.; Dembélé, D.; Dollé, P.; Krężel, W. Genome-Wide Analysis of RARβ Transcriptional Targets in Mouse Striatum Links Retinoic Acid Signaling with Huntington’s Disease and Other Neurodegenerative Disorders. Mol. Neurobiol. 2017, 54. [Google Scholar] [CrossRef]
- Marie, A.; Darricau, M.; Touyarot, K.; Parr-Brownlie, L.C.; Bosch-Bouju, C. Role and Mechanism of Vitamin A Metabolism in the Pathophysiology of Parkinson’s Disease. J. Park. Dis. 2021, 11. [Google Scholar] [CrossRef]
- Dheen, S.T.; Jun, Y.; Yan, Z.; Tay, S.S.W.; Ling, E.A. Retinoic Acid Inhibits Expression of TNF-α and INOS in Activated Rat Microglia. Glia 2005, 50. [Google Scholar] [CrossRef]
- Almaguer, J.; Hindle, A.; Lawrence, J.J. The Contribution of Hippocampal All-Trans Retinoic Acid (ATRA) Deficiency to Alzheimer’s Disease: A Narrative Overview of ATRA-Dependent Gene Expression in Post-Mortem Hippocampal Tissue. Antioxidants 2023, 12. [Google Scholar] [CrossRef]
- Kolarcik, C.L.; Bowser, R. Retinoid Signaling Alterations in Amyotrophic Lateral Sclerosis. Am. J. Neurodegener. Dis. 2012, 1. [Google Scholar] [CrossRef]
- Medina, D.X.; Chung, E.P.; Teague, C.D.; Bowser, R.; Sirianni, R.W. Intravenously Administered, Retinoid Activating Nanoparticles Increase Lifespan and Reduce Neurodegeneration in the SOD1G93A Mouse Model of ALS. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Ruiz-Soto, M.; Berciano, M.T.; Berciano, J.; Lafarga, M. Neuroprotective Effect of Bexarotene in the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Front. Cell Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Isoherranen, N.; Zhong, G. Biochemical and Physiological Importance of the CYP26 Retinoic Acid Hydroxylases. Pharmacol. Ther. 2019, 204. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Lichti, U.; Mamiya, S.; Aronova, M.; Zhang, G.; Yuspa, S.H.; Hamada, H.; Sakai, Y.; Morasso, M.I. Increased Retinoic Acid Levels through Ablation of Cyp26b1 Determine the Processes of Embryonic Skin Barrier Formation and Peridermal Development. J. Cell Sci. 2012, 125. [Google Scholar] [CrossRef]
- Topletz, A.R.; Thatcher, J.E.; Zelter, A.; Lutz, J.D.; Tay, S.; Nelson, W.L.; Isoherranen, N. Comparison of the Function and Expression of CYP26A1 and CYP26B1, the Two Retinoic Acid Hydroxylases. Biochem. Pharmacol. 2012, 83. [Google Scholar] [CrossRef] [PubMed]
- Spoorendonk, K.M.; Peterson-Maduro, J.; Renn, J.; Trowe, T.; Kranenbarg, S.; Winkler, C.; Schulte-Merker, S. Retinoic Acid and Cyp26b1 Are Critical Regulators of Osteogenesis in the Axial Skeleton. Development 2008, 135. [Google Scholar] [CrossRef]
- Stevison, F.; Jing, J.; Tripathy, S.; Isoherranen, N. Role of Retinoic Acid-Metabolizing Cytochrome P450s, CYP26, in Inflammation and Cancer. In Advances in Pharmacology; 2015; Volume 74.
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of Als. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Dorszewska, J.; Kowalska, M.; Prendecki, M.; Piekut, T.; Kozłowska, J.; Kozubski, W. Oxidative Stress Factors in Parkinson’s Disease. Neural Regen. Res. 2021, 16. [Google Scholar] [CrossRef]
- Rummel, N.G.; Butterfield, D.A. Altered Metabolism in Alzheimer Disease Brain: Role of Oxidative Stress. Antioxid. Redox Signal 2022, 36. [Google Scholar] [CrossRef]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive Oxygen Species, Toxicity, Oxidative Stress, and Antioxidants: Chronic Diseases and Aging. Arch. Toxicol. 2023, 97. [Google Scholar] [CrossRef] [PubMed]
- Poh Loh, K.; Hong Huang, S.; De Silva, R.H.; Tan, B.; Zhun Zhu, Y. Oxidative Stress: Apoptosis in Neuronal Injury. Curr. Alzheimer Res. 2006, 3. [Google Scholar] [CrossRef] [PubMed]
- Sastre, J.; Pallardö, F. V.; Viña, J. Mitochondrial Oxidative Stress Plays a Key Role in Aging and Apoptosis. IUBMB Life 2000, 49. [Google Scholar] [CrossRef]
- Kang, J. Bin; Park, D.J.; Shah, M.A.; Koh, P.O. Retinoic Acid Exerts Neuroprotective Effects against Focal Cerebral Ischemia by Preventing Apoptotic Cell Death. Neurosci. Lett. 2021, 757. [Google Scholar] [CrossRef]
- Ahlemeyer, B.; Bauerbach, E.; Plath, M.; Steuber, M.; Heers, C.; Tegtmeier, F.; Krieglstein, J. Retinoic Acid Reduces Apoptosis and Oxidative Stress by Preservation of SOD Protein Level. Free Radic. Biol. Med. 2001, 30. [Google Scholar] [CrossRef]
- Xu, Q.; Konta, T.; Kitamura, M. Retinoic Acid Regulation of Mesangial Cell Apoptosis. Exp. Nephrol. 2002, 10. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, M.; Ishikawa, Y.; Moreno-Manzano, V.; Xu, Q.; Konta, T.; Lucio-Cazana, J.; Furusu, A.; Nakayama, K. Intervention by Retinoic Acid in Oxidative Stress-Induced Apoptosis. Nephrol. Dial. Transplant. 2002, 17. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Denis Alexander, H.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kevin Howcroft, T.; Campisi, J.; Louis, G.B.; Smith, M.T.; Wise, B.; Wyss-Coray, T.; Augustine, A.D.; McElhaney, J.E.; Kohanski, R.; Sierra, F. The Role of Inflammation in Age-Related Disease. Aging 2013, 5. [Google Scholar] [CrossRef]
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.L. Role of Microglia in Neurological Disorders and Their Potentials as a Therapeutic Target. Mol. Neurobiol. 2017, 54. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Chen, B.; Kang, X.; Zhang, R.; Guo, Y.; Zhao, J.; Yang, H. Neuroprotective Effects of Natural Compounds on LPS-Induced Inflammatory Responses in Microglia. Am. J. Transl. Res. 2020, 12. [Google Scholar]
- Tian, Y.; Liu, B.; Li, Y.; Zhang, Y.; Shao, J.; Wu, P.; Xu, C.; Chen, G.; Shi, H. Activation of RARα Receptor Attenuates Neuroinflammation After SAH via Promoting M1-to-M2 Phenotypic Polarization of Microglia and Regulating Mafb/Msr1/PI3K-Akt/NF-ΚB Pathway. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Takaoka, Y.; Takahashi, M.; Kurauchi, Y.; Hisatsune, A.; Seki, T.; Shudo, K.; Katsuki, H. Retinoic Acid Receptor Agonist Am80 Inhibits CXCL2 Production from Microglial BV-2 Cells via Attenuation of NF-ΚB Signaling. Int. Immunopharmacol. 2016, 38. [Google Scholar] [CrossRef]
- Nixon, R.A. The Role of Autophagy in Neurodegenerative Disease. Nat. Med. 2013, 19. [Google Scholar] [CrossRef]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein Aggregation and Degradation Mechanisms in Neurodegenerative Diseases. Am. J. Neurodegener. Dis. 2013, 2. [Google Scholar]
- Lin, M.; Yu, H.; Xie, Q.; Xu, Z.; Shang, P. Role of Microglia Autophagy and Mitophagy in Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Visnjic, D.; Dembitz, V.; Lalic, H. The Role of AMPK/MTOR Modulators in the Therapy of Acute Myeloid Leukemia. Curr. Med. Chem. 2018, 26. [Google Scholar] [CrossRef]
- Long, C.; Zhou, Y.; Shen, L.; Yu, Y.; Hu, D.; Liu, X.; Lin, T.; He, D.; Xu, T.; Zhang, D.; et al. Retinoic Acid Can Improve Autophagy through Depression of the PI3K-Akt-MTOR Signaling Pathway via RARα to Restore Spermatogenesis in Cryptorchid Infertile Rats. Genes. Dis. 2022, 9. [Google Scholar] [CrossRef]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Ochneva, A.; Zorkina, Y.; Abramova, O.; Pavlova, O.; Ushakova, V.; Morozova, A.; Zubkov, E.; Pavlov, K.; Gurina, O.; Chekhonin, V. Protein Misfolding and Aggregation in the Brain: Common Pathogenetic Pathways in Neurodegenerative and Mental Disorders. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Kritsilis, M.; Rizou, S. V.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Masaldan, S.; Belaidi, A.A.; Ayton, S.; Bush, A.I. Cellular Senescence and Iron Dyshomeostasis in Alzheimer’s Disease. Pharmaceuticals 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Petersen, R.C. Cellular Senescence in Brain Aging and Neurodegenerative Diseases: Evidence and Perspectives. J. Clin. Investig. 2018, 128. [Google Scholar] [CrossRef]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular Senescence and Senolytics: The Path to the Clinic. Nat. Med. 2022, 28. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting Cellular Senescence with Senotherapeutics: Senolytics and Senomorphics. FEBS J. 2023, 290. [Google Scholar] [CrossRef]
Figure 1.
Ellorarxine upregulates the expression of Cyp26b1. (A) Immunofluorescence staining of Cyp26b1 (green) and DAPI (blue) in C6s, SH-SY5Ys and HMC3s. Scale bar, 50μm, (B) Average fluorescence intensity of Cyp26b1 in C6s, SH-SY5Ys and HMC3s, n=6 per group. Data are presented as mean ± SD. *p < 0.05. **p < 0.01. ***p < 0.001.
Figure 1.
Ellorarxine upregulates the expression of Cyp26b1. (A) Immunofluorescence staining of Cyp26b1 (green) and DAPI (blue) in C6s, SH-SY5Ys and HMC3s. Scale bar, 50μm, (B) Average fluorescence intensity of Cyp26b1 in C6s, SH-SY5Ys and HMC3s, n=6 per group. Data are presented as mean ± SD. *p < 0.05. **p < 0.01. ***p < 0.001.
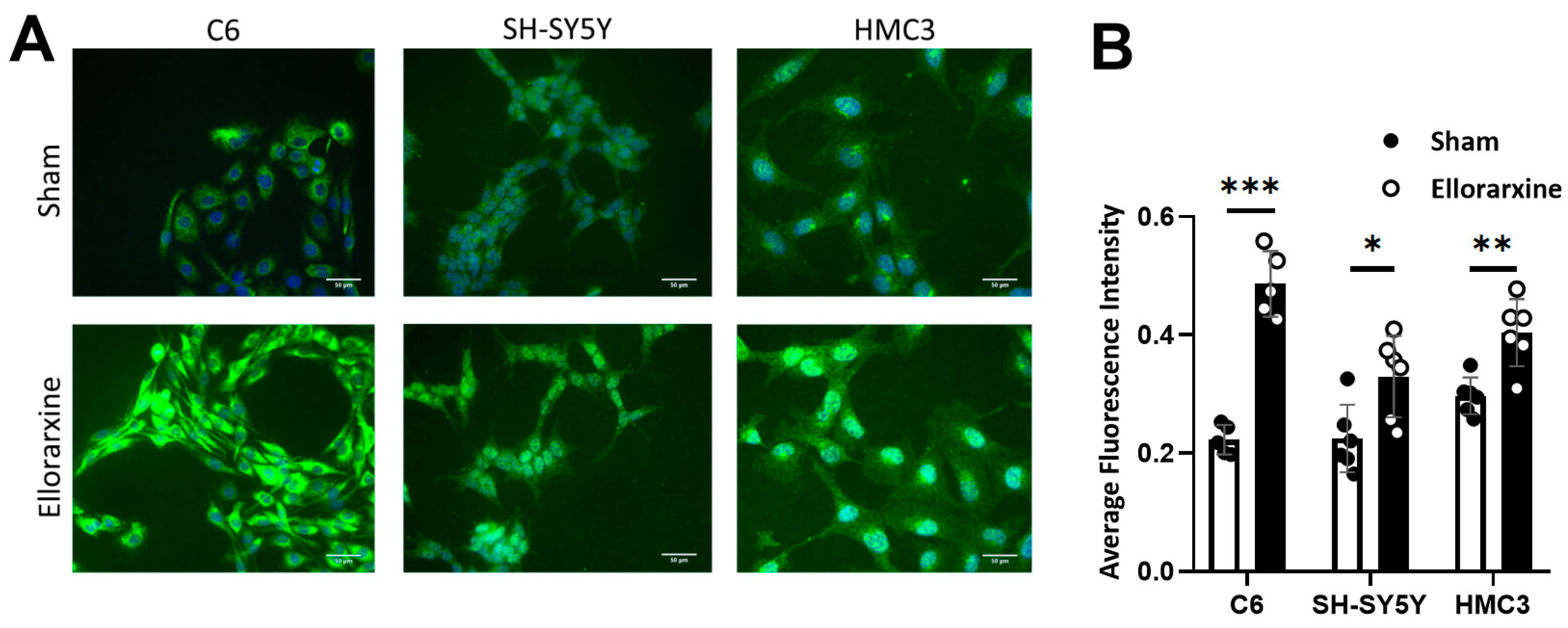
Figure 2.
Ellorarxine upregulates the expression of RARβ(A) Immunofluorescence staining of RARα, RARβ, RARγ (green) and DAPI (blue) in C6 cells. Scale bar, 10 μm. (B) Immunofluorescence staining of RARα, RARβ, RARγ (green) and DAPI (blue) in SH-SY5Y cells. Scale bar, 50 μm. (C) Immunofluorescence staining of RARα, RARβ, RARγ (green) and DAPI (blue) in HMC3 cells. Scale bar, 50 μm. (D) Immunofluorescence staining of RARα, RARβ, RARγ (green) and DAPI (blue) in differentiated SH-SY5Y cells. Scale bar, 50 μm.(E) Average fluorescence intensity of RARα, RARβ, RARγ in C6 cells, n=4 per group. Data are presented as mean ± SD. (F) Average fluorescence intensity of RARα, RARβ, RARγ in SH-SY5Y cells, n=4 per group. Data are presented as mean ± SD. (G) Average fluorescence intensity of RARα, RARβ, RARγ in HMC3 cells, n=4 per group. Data are presented as mean ± SD. (H) Average fluorescence intensity of RARα, RARβ, RARγ in differentiated SH-SY5Y cells, n=4 per group. Data are presented as mean ± SD. *p < 0.05.
Figure 2.
Ellorarxine upregulates the expression of RARβ(A) Immunofluorescence staining of RARα, RARβ, RARγ (green) and DAPI (blue) in C6 cells. Scale bar, 10 μm. (B) Immunofluorescence staining of RARα, RARβ, RARγ (green) and DAPI (blue) in SH-SY5Y cells. Scale bar, 50 μm. (C) Immunofluorescence staining of RARα, RARβ, RARγ (green) and DAPI (blue) in HMC3 cells. Scale bar, 50 μm. (D) Immunofluorescence staining of RARα, RARβ, RARγ (green) and DAPI (blue) in differentiated SH-SY5Y cells. Scale bar, 50 μm.(E) Average fluorescence intensity of RARα, RARβ, RARγ in C6 cells, n=4 per group. Data are presented as mean ± SD. (F) Average fluorescence intensity of RARα, RARβ, RARγ in SH-SY5Y cells, n=4 per group. Data are presented as mean ± SD. (G) Average fluorescence intensity of RARα, RARβ, RARγ in HMC3 cells, n=4 per group. Data are presented as mean ± SD. (H) Average fluorescence intensity of RARα, RARβ, RARγ in differentiated SH-SY5Y cells, n=4 per group. Data are presented as mean ± SD. *p < 0.05.
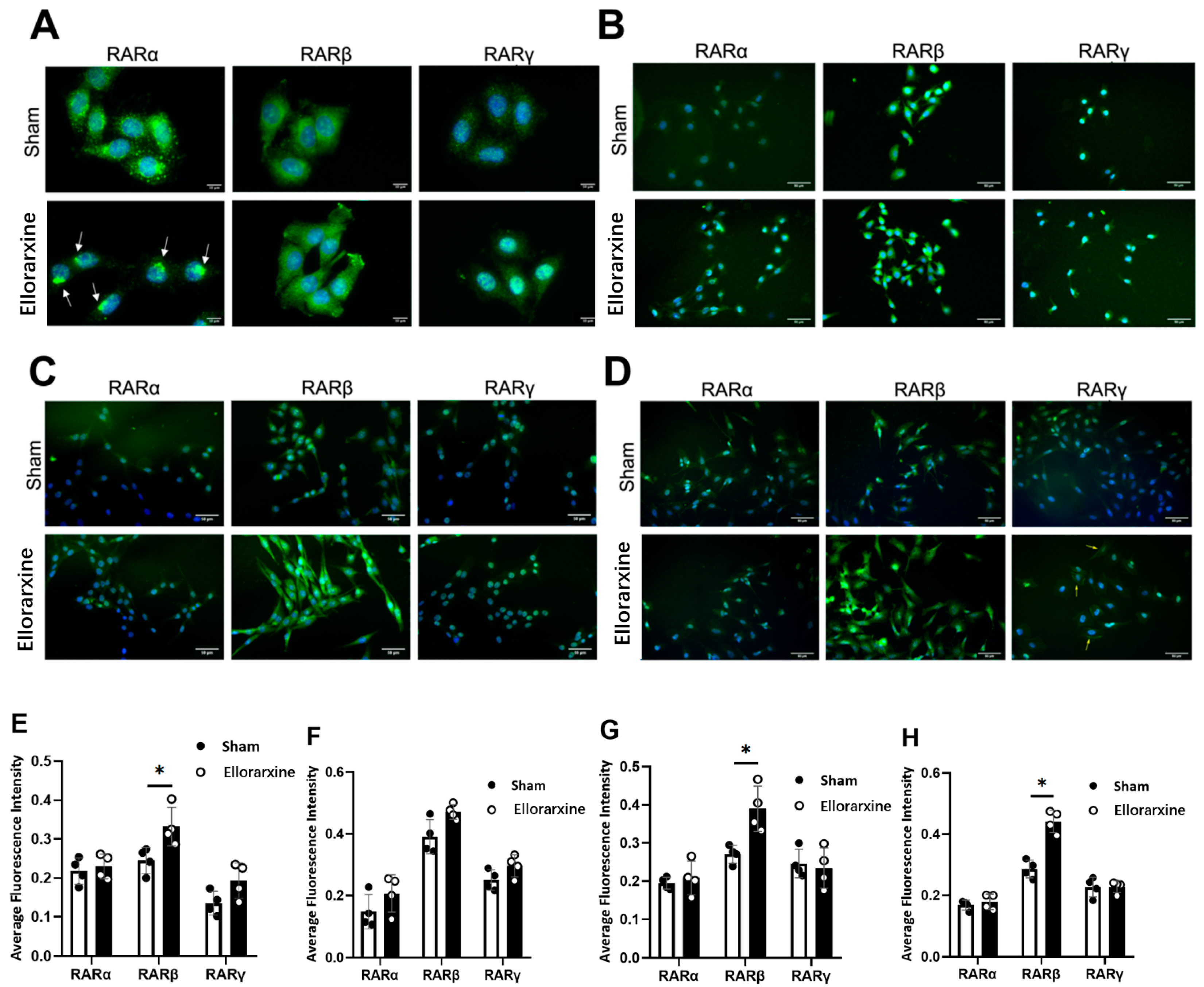
Figure 3.
Ellorarxine pretreatment alleviates mitochondrial dysfunction. (A) Mitochondrial viability in C6, SH-SY5Y, and HMC3 treated with 10% DMSO (Sham) and 10 nM Ellorarxine. n=4 per group. (B) Mitochondrial viability in C6, SH-SY5Y, and HMC3 pre-treated with 10% DMSO (Sham) and 10 nM Ellorarxine under 100 mM H2O2 stress. n=4 per group. Data are presented as mean ± SD. ** p < 0.01.
Figure 3.
Ellorarxine pretreatment alleviates mitochondrial dysfunction. (A) Mitochondrial viability in C6, SH-SY5Y, and HMC3 treated with 10% DMSO (Sham) and 10 nM Ellorarxine. n=4 per group. (B) Mitochondrial viability in C6, SH-SY5Y, and HMC3 pre-treated with 10% DMSO (Sham) and 10 nM Ellorarxine under 100 mM H2O2 stress. n=4 per group. Data are presented as mean ± SD. ** p < 0.01.
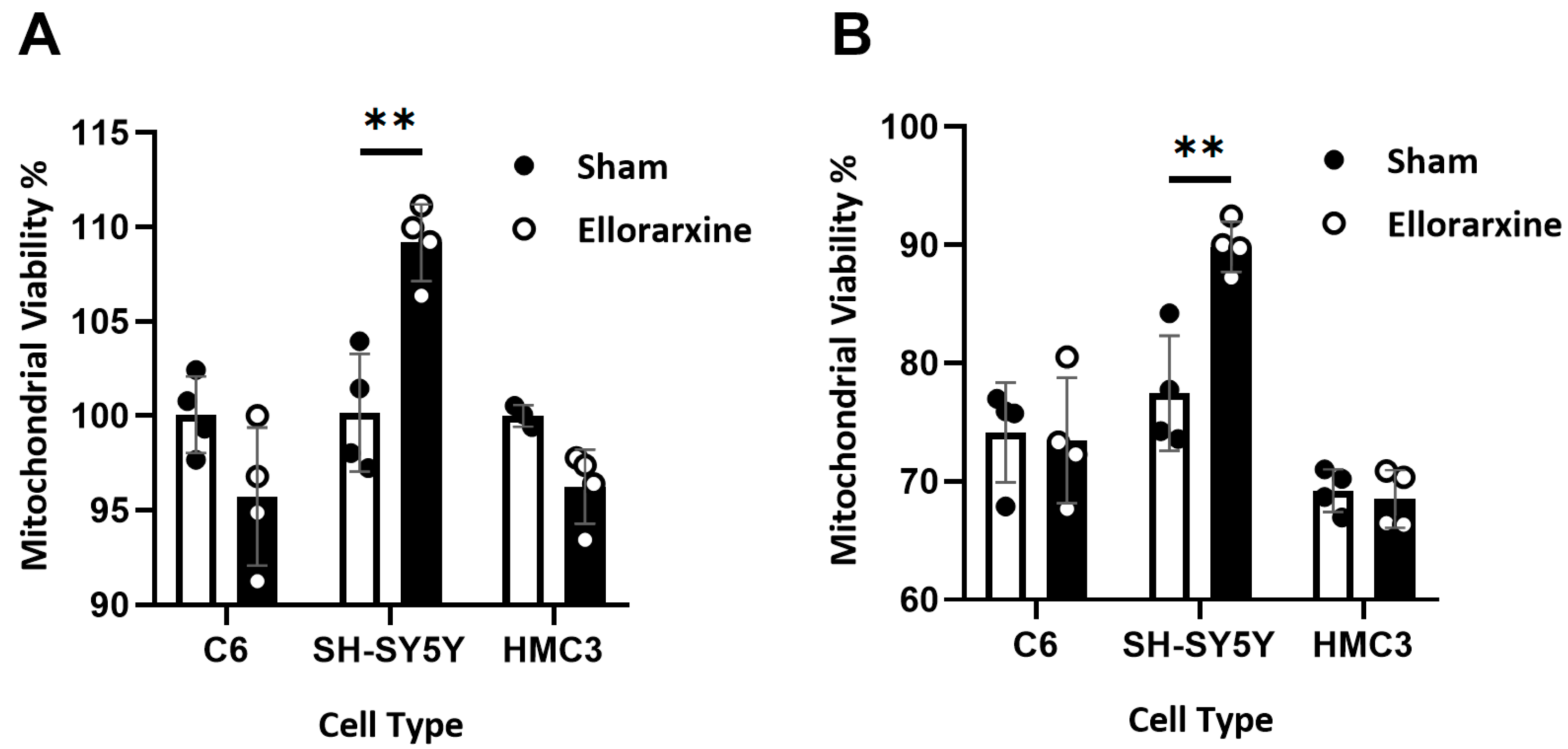
Figure 4.
Ellorarxine pretreatment reduced cell death. (A) Cell death in C6, SH-SY5Y, and HMC3 treated with 10% DMSO (Sham) and 10 nM Ellorarxine. n=4 per group.(B) Cell death in C6, SH-SY5Y, and HMC3 pre-treated with 10% DMSO (Sham) and 10nM Ellorarxine under 200 mM H2O2 stress. n=4 per group. (C) Cell death in C6, SH-SY5Y, and HMC3 pre-treated with 10% DMSO (Sham) and 10nM Ellorarxine under 10 μg/mL LPS stress. n=4 per group. Data are presented as mean ± SD. * p < 0.05.
Figure 4.
Ellorarxine pretreatment reduced cell death. (A) Cell death in C6, SH-SY5Y, and HMC3 treated with 10% DMSO (Sham) and 10 nM Ellorarxine. n=4 per group.(B) Cell death in C6, SH-SY5Y, and HMC3 pre-treated with 10% DMSO (Sham) and 10nM Ellorarxine under 200 mM H2O2 stress. n=4 per group. (C) Cell death in C6, SH-SY5Y, and HMC3 pre-treated with 10% DMSO (Sham) and 10nM Ellorarxine under 10 μg/mL LPS stress. n=4 per group. Data are presented as mean ± SD. * p < 0.05.
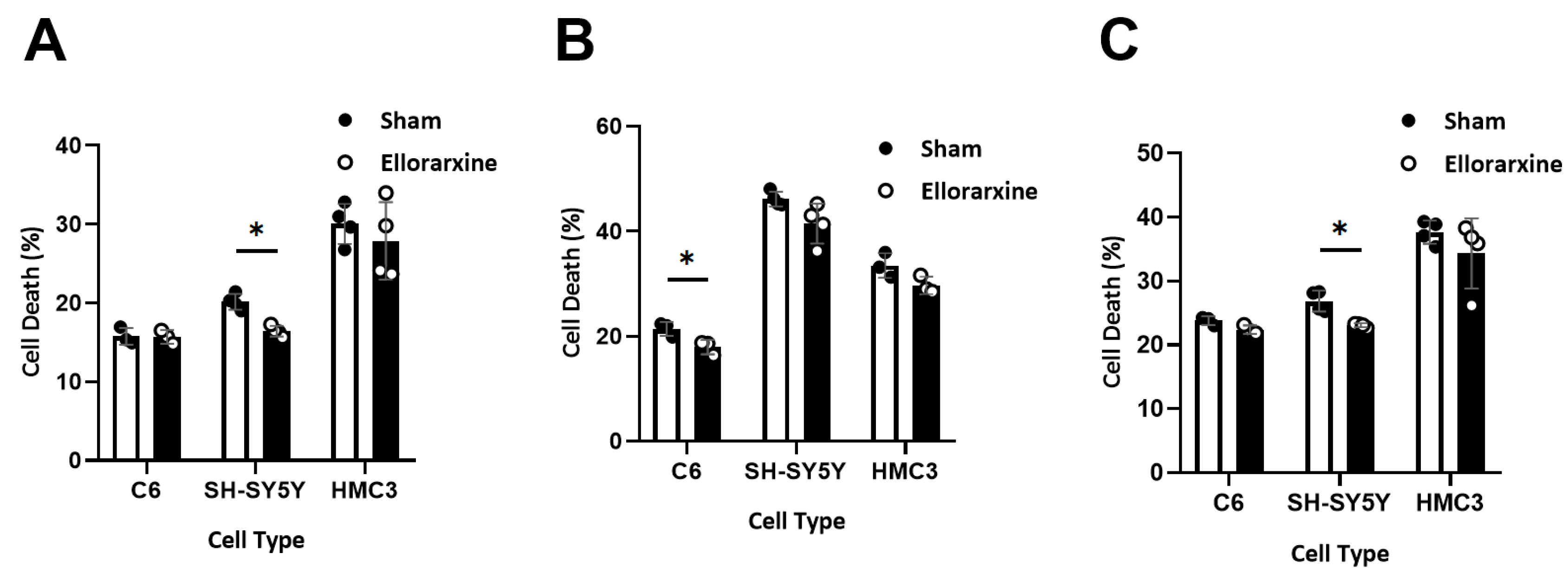
Figure 5.
Ellorarxine pretreatment modulated inflammatory cytokine release. (A) TNF-α release under LPS stress with 10% DMSO (Sham) and 10 nM Ellorarxine. n=4 per group. (B) IL-6 release under LPS stress with 10% DMSO (Sham) and 10 nM Ellorarxine. n=3 per group. Data are presented as mean ± SD. *** p < 0.001.
Figure 5.
Ellorarxine pretreatment modulated inflammatory cytokine release. (A) TNF-α release under LPS stress with 10% DMSO (Sham) and 10 nM Ellorarxine. n=4 per group. (B) IL-6 release under LPS stress with 10% DMSO (Sham) and 10 nM Ellorarxine. n=3 per group. Data are presented as mean ± SD. *** p < 0.001.
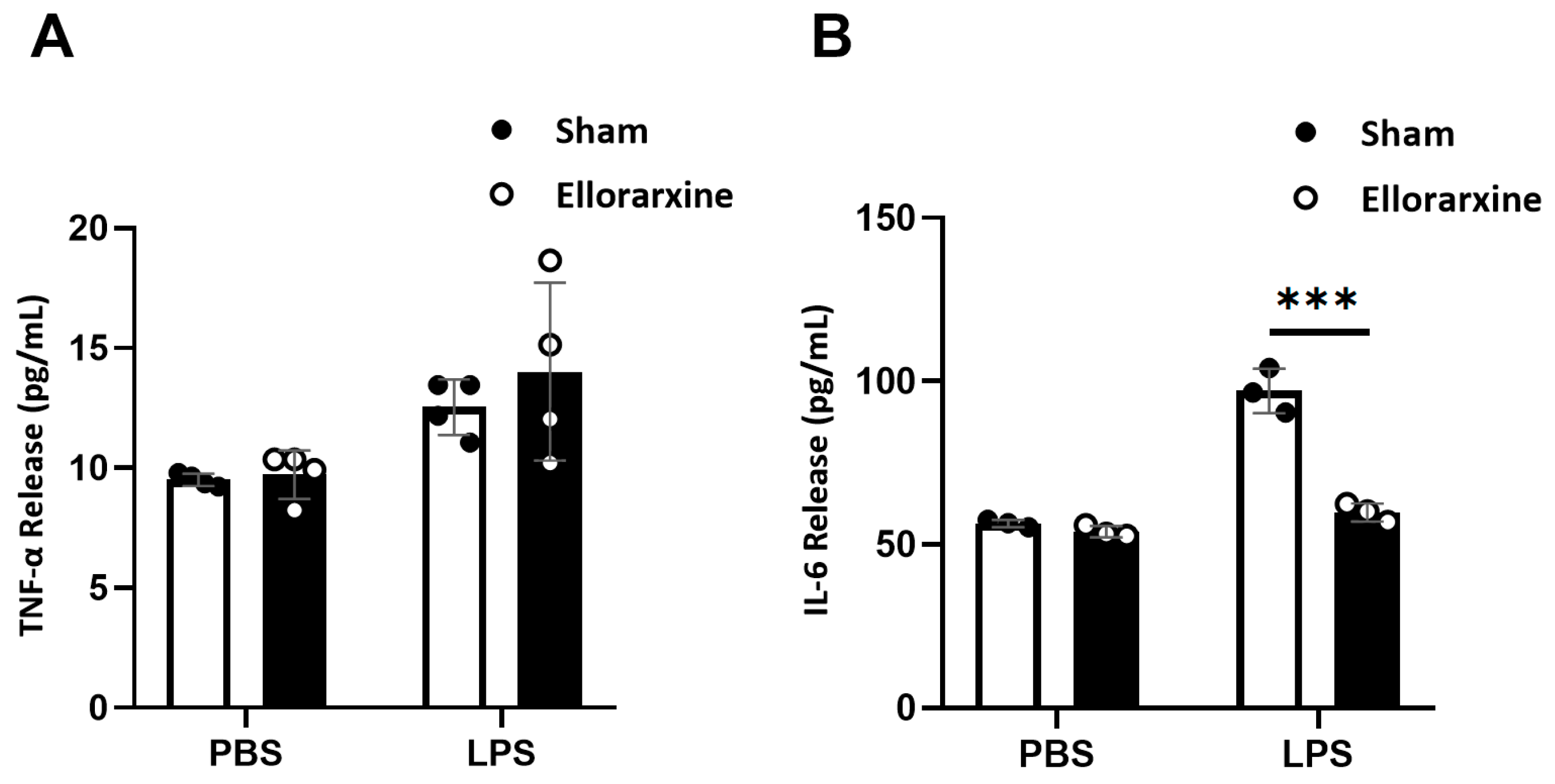
Figure 6.
Ellorarxine treatment reduced the number of senescent cells. (A) SA-β-Gal staining of senescent C6 cells. Scale bar, 50μm.(B) Percentage of senescent cells under 20 mM H2O2 stress with 10% DMSO (Sham) and 10 nM Ellorarxine. n=3 per group. Data are presented as mean ± SD. ** p < 0.01.
Figure 6.
Ellorarxine treatment reduced the number of senescent cells. (A) SA-β-Gal staining of senescent C6 cells. Scale bar, 50μm.(B) Percentage of senescent cells under 20 mM H2O2 stress with 10% DMSO (Sham) and 10 nM Ellorarxine. n=3 per group. Data are presented as mean ± SD. ** p < 0.01.
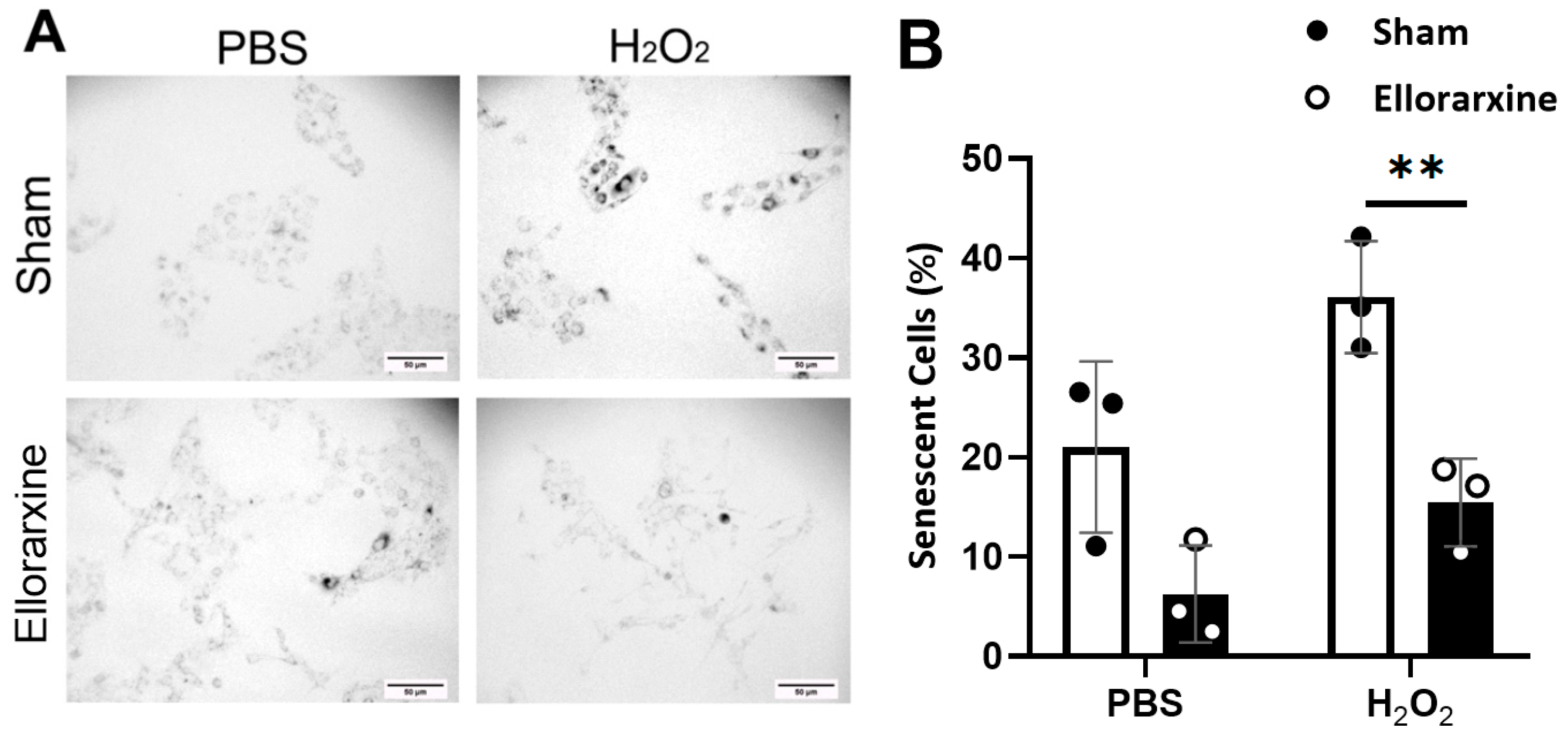
Figure 7.
Ellorarxine treatment regulated cellular autophagy. (A) LC3BII immunocytochemical staining of C6 cells. Scale bar, 20 μm. (B) LC3BII immunocytochemical staining of SH-SY5Y cells. Scale bar, 20 μm. (C) LC3BII immunocytochemical staining of HMC3 cells. Scale bar, 20μm. (D) Average Optical Density under serum-free media with 10% DMSO (Sham) and 10 nM Ellorarxine. n=6 per group. Data are presented as mean ± SD. *** p < 0.001.
Figure 7.
Ellorarxine treatment regulated cellular autophagy. (A) LC3BII immunocytochemical staining of C6 cells. Scale bar, 20 μm. (B) LC3BII immunocytochemical staining of SH-SY5Y cells. Scale bar, 20 μm. (C) LC3BII immunocytochemical staining of HMC3 cells. Scale bar, 20μm. (D) Average Optical Density under serum-free media with 10% DMSO (Sham) and 10 nM Ellorarxine. n=6 per group. Data are presented as mean ± SD. *** p < 0.001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



