
Submitted:
07 March 2024
Posted:
08 March 2024
You are already at the latest version
Abstract
Reactions of quinones with compounds containing an amino group can produce a wide variety of addition or substitution products that depend on reactivity of both quinone and amino derivative. 6,7-Dichloropyrido[1,2-a]benzimidazole-8,9-diones undergo selective nucleophilic substitution reaction with different benzohydrazides and α-hydroxy-p-quinone imine derivatives stabilized by strong intramolecular hydrogen bond were isolated. Synthesized compounds represent a combination of several structural motifs: benzimidazole core fused with α-hydroxy-p-quinone imine which contains a benzamido fragment. The protonation/deprotonation processes were investigated in a solution using UV-Vis spectroscopy and 1H NMR titration experiment. X-ray crystallography analysis revealed a set of weak non-covalent interactions such as intra- and intermolecular hydrogen bonds and π-π stacking. Additionally, the redox behavior of 6,7-dichloropyrido[1,2-a]benzimidazole-8,9-dione and its p-imino derivative was investigated in acidic and neutral environment using cyclic voltammetry measurements. Cathode material based on 6,7-dichloropyrido[1,2-a]benzimidazole-8,9-dione could act as potential effective active electrode in aqueous electrolyte batteries, however further optimization is required.
Keywords:
quinone
; quinone imine
; hydrogen bonding
; X-ray crystallography
; NMR titration
; redox.
1. Introduction
Quinones and quinone derivatives are well known due to the redox activity that is important in a wide range of biological processes such as photosynthesis [1] and cellular respiration [2]. Quinones represent a class of biologically active compounds with both cytotoxic and cytoprotective effects [3]. Considerable attention to redox active compounds in general and quinones in particular [4,5] can be explained by growing demands on energy storage devices for portable electronics and renewable energy-powered vehicles [6]. Quinones potentially can be used in different applications connected with energy storage due to the remarkable redox activity: as organic cathode materials for different kinds of rechargeable batteries [7] including redox flow batteries [8], and Zn-ion batteries [9], or as a redox mediators in lithium-sulfur batteries [10]. Physical properties of quinones can be modulated by the introduction of heteroaromatics fused with quinone core [8], and different substituents that affect solubility [11] or affect the form of quinone fragment [12] or can facilitate binding with metal cations [13]. Additionally, redox properties may be tuned to some extent by intra- and intermolecular hydrogen bonding [14].
Redox potentials, solubility, and stability in the case of small quinones can be affected by modification with electron donating or withdrawing functional groups or combination with a side chain that can form hydrogen bonds. Additionally, substituted o-quinones besides their "classical" form, can also exist in various forms [15] like quinone methides, quinone imines, and zwitterions. This ability to adopt different structures allows to modulate their properties for different applications. Despite the progress in the design of quinone derivativities and their wide application as redox active materials the limited information on molecular-level insights into the bulk properties (e.g., solubility, stability, redox activity etc.) is available. Investigation of quinone structure at the molecular level, self-assembly in solid state and behavior in solution will help to tune the performance of quinone-functionalized materials.
An approach to modulate redox properties of quinones is an introduction of nitrogen-containing redox-active groups (e.g., C≡N, C=N, and N=N) or incorporation of unsaturated carbon–nitrogen bond and π-conjugated aromatic fragment [16].
This work aimed to gain an understanding of the molecular structure and behavior of unsymmetrical heterocyclic o-quinones and their derivatives bearing imine moiety. Also, electrochemical studies of selected compounds were conducted to assess their potential applications.
2. Results and Discussion
2.1. Synthesis and Structural Studies of Quinone Derivatives 3a-g
6,7-Dichloropyrido [1,2-a]benzimidazole-8,9-dione (1a) is a representative of unsymmetrical heterocyclic o-quinones that contains a combination of two structural motifs: o-quinone fragment and imidazo [1,2-a]pyridine core that possess C=N bond (Scheme 1). It can be obtained in one-step synthesis from commercially available tetrachloro-1,4-benzoquinone and 2-aminopyridine [17]. During earlier studies [18,19,20] it was proved that quinone 1a and some of its derivatives are electrochemically active compounds. Investigation of the reactivity of heterocyclic quinones 1 with C- and N-nucleophiles (primary amines) indicated that the attack of the nucleophile proceeds selectively at C(6)-position of quinone. Interestingly, obtained compounds containing different acceptor groups at C(6)-position can exist in o-quinone form or as p-quinone methides depending on the introduced substituent.
To expand the scope of redox active heterocyclic o-quinone derivatives the modification of quinone 1a with different benzohydrazides were carried out as well as structural studies in solid state and in solution were conducted.
2.1.1. Synthesis of Quinone Derivatives 3a-g
Quinone derivatives 3a-g (Scheme 1a, atoms are numbered according to ORTEP diagram, vide infra) were obtained by nucleophilic substitution of a chlorine atom of quinone 1a,b by benzohydrazides 2a-d. Isolated compounds 3a-g have orange color in the solid state. Interestingly, in the case of aminoderivatives of quinones 1 (a merocyanine on the base of o-quinone form) deep-blue colored crystals were obtained [18,20]. In general, derivatives containing aroyl hydrazine fragment were expected as a result of such substitution [15,21], and a few tautomeric structures can be supposed for the products [22,23]. For the compounds obtained (3a-g) the structure determination of the quinone/substituent fragments (Scheme 1b) can explain the observed difference in color of crystals 3a-g in comparison to aminosubstituted derivatives of quinone 1a.
2.1.2. Single Crystal X-ray Analysis of Quinone Derivative 3a
The use of routine identification procedures (such as 1H-NMR and FTIR) to establish the molecular structure of compounds 3a-g left some room for doubt. To clarify the situation, crystals of compound 3a were grown from dichloromethane (DCM) solution and the molecular structure of them was established using the single-crystal X-ray crystallography. Crystal data and refinement details for the studied crystal are presented in Table 1.
Figure 1a shows a perspective view of molecule 3a with thermal ellipsoids and the atom-numbering scheme. p-Quinonimine form was confirmed by the inspection of the bond length: bonds between C(9)=O(23) and C(6)=N(11) have double bond character as well as O(22)-C(8) is a single bond. Also, an analysis of bond lengths shows that the structure of compound 3a can be represented as a superposition of mesomeric forms. The main forms are shown in Figure 1b; at that, the non-ionized form has the highest specific weight.
The molecules of compound 3a are characterized by a flattened conformation; only the phenyl group is slightly out of the plane of the heterocyclic system (angle between planes is 5.19°). In the structure of compound 3a intramolecular hydrogen bonds NH···N and OH···O were found (Figure 2). The hydroxy group of compound 3a forms bifurcated hydrogen bonds where O(22)-H···O(23) is an intramolecular bond (Figure 1a) and O(22)-H···O’(23) is an intermolecular one (Figure 2a). By means of these intermolecular H-bonds the centrosymmetric molecular dimers are formed in the crystal structure. Additional intermolecular interactions were found: stacking interaction between the planes of the molecules and a short intermolecular contact between heterocycle (C(3)-H) and the amide group of the substituent (d C(3)···O(14) = 3.112 Å, d C(3)-H···O(14) = 2.344 Å) (Figure 2b,c).
Hirshfeld surfaces and energy framework calculations (Figures S13 and S14) were obtained in a whole-of molecule approach using B3LYP/6-31G(d,p) energy model implemented in CrystalExplorer21.5 program [26]. Energy frameworks provide an opportunity to explore cooperative effects of intermolecular interactions in the crystal admitting the electrostatic, dispersion and total energy between pairs of the molecules [25]. In the case of the crystal of compound 3a strong stabilizing interlayer electrostatic interaction was found between molecules involving in formation of hydrogen bonded (O–H⋯O) dimers. On the other hand, dispersion energy was more dominant for the intercolumn stacking motif. Overall, energy framework analysis of the crystal revealed two distinct patterns of electrostatic and dispersion energies with each contributing similarly.
2.1.3.1. H NMR Spectroscopy Analysis of Quinone Derivatives 3a-g
To determine the structure of obtained products in solution compounds 3a-g 1H NMR spectroscopy data were analyzed, and a set of two broad signals corresponding to NH and OH protons were observed (Figures S1–S7). In DMSO-d6 solution signals appeared at 14.36–14.90 ppm can be assigned to the NH proton while signals of the OH group were observed at 10.89–11.41 ppm. Additionally, 1H NMR spectrum of compound 3a was also recorded in CDCl3 solution (a solvent in which hydrogen-bonding interactions are expected to be weaker [27]) (Figure S8), where NH proton was found at 14.68 ppm (versus 14.71 ppm in DMSO-d6 solution). It can be concluded that a strong intramolecular bond between NH group proton of substituent (benzamide group at imine bond) and nitrogen of heterocycle (N(12)-H···N(5)) can be found in solution as well as in solid state (vide supra).
It is known [28] that in the case of α-hydroxyquinone derivatives an intramolecular hydrogen bond was observed and OH proton signal appears at 7.30 ppm in CDCl3 solution. For compound 3a a distinguishable shift was observed for OH proton signal in CDCl3 solution (7.20 ppm) in comparison to DMSO-d6 solution (10.99 ppm), that can indicate the formation of the additional intermolecular interactions between the OH group and a solvent (DMSO-d6) with hydrogen bond acceptor abilities [29].
It was found that the most downfielded signal of NH proton (14.90 ppm in DMSO-d6 solution) was observed for compound 3c with NO2 group at the benzene ring (benzamide fragment); however, NO2 group at C(2) of the heterocycle led to the upfield shift of NH proton. The most upfielded signal of NH proton (14.36 ppm) was found for compound 3f with NO2 group at heterocyclic fragment and electron donating group at the benzene ring. In general, the more downfield shifted the NH proton signal the stronger is the intramolecular H-bond [30]. Thus, electron withdrawing group (EWG) at the benzene ring increases the acidity of the NH proton and increases the intramolecular H-bond strength as well as EWG at heterocyclic fragment (at C(2) atom) influences the electron density at N(5) affecting intramolecular H-bond in turn.
In the case of compounds 3a-g the moiety at C(6) position can be described as a structural analog of aroyl hydrazone (a different approach [23] to the naming of quinone imine derivatives was observed). Well known characteristic of compounds containing the carbon-nitrogen double bond is the ability to undergo E/Z isomerization in the solution activated by light and/or chemical inputs [31,32].
Compounds 3a and 3b were chosen for the investigation of the isomerization process. In general, in the case of E/Z isomerization additional set of signals [33,34] is expected to appear. No signals of the second form (isomerization products) were observed in 1H NMR spectra of compound 3a either in DMSO-d6 or in CDCl3 solution. Also, in the case of compound 3b configurational switching was not induced by the addition of the excess of trifluoracetic acid (TFA) and following irradiation by UV light (365 nm; the irradiation by a high-pressure mercury lamp at room temperature) judging from the 1H NMR spectra of compound 3b in DMSO-d6 solution (Figure S9). The formation of a strong intramolecular hydrogen bond N(12)-H···N(5) can explain the existence of a single configuration of substituted imine that agrees with stabilization of the only one form in the presence of intramolecular hydrogen bond. Additional stabilization of the molecule may be explained by excitation energy dissipation caused by zwitterionic structure.
It is known [35] that for redox properties tests (fabrication of electrodes) a mixture of organic compound, conductive additive and a binder is often prepared using N-methyl-2-pyrrolidone (NMP) [36] as a solvent (strongly basic solvent) [37]. This fact prompted us to investigate the influence of the base on the structure of the products 3a-g.
During preliminary solubility tests of compounds 3a-g the color change (from yellow to green or blue) was observed in NMP solution or in the presence of base. For better understanding of the effect, a few 1H NMR experiments were carried out. Upon addition an excess of the base (1,8-diazabicyclo(5.4.0)undec-7-ene, DBU) 1H NMR spectrum of compound 3b in DMSO-d6 solution showed some changes (Figure 3): the signal of OH proton completely disappeared; the sharpened signal of NH proton (the sharp line can indicate a dynamically stable state) shifted upfield. Additionally, a new minor proton signal at 13.44 ppm was observed. Simultaneously, the yellow-colored solution of compound 3b became dark blue. It should be noted that the same deprotonation behavior was observed for compound 3a in CDCl3 solution (Figure S10). After the excess of TFA was added the solution became yellow again, minor signal at 13.44 ppm disappeared as well as the signal of the OH proton restored. It can be concluded that deprotonation provides the formation of anionic polymethine dye structure [38] (blue) between atoms O(14) and O(22), protonation restores quinone imine form (yellow), consequently, the equilibrium between two forms is reversible.
To gain more information about the deprotonation process of compound 3b, 1H NMR titration experiment was carried out. As shown in Figure 4, in the case of compound 3b deprotonation upon sequential addition of the base (DBU) the 1H NMR spectra reveals several features. Broad signal of OH proton vanished upon addition of only 0.15 equivalents of the base that can be explained by dynamical process as well; color changes were immediate (Figure 4, highlighted in yellow). The signal of NH proton (benzamide fragment) sharpens and undergoes upfield shift from 14.70 to 14.51 ppm (Figure 4, highlighted in red).
A new signal appeared at 9.58 ppm (+ 0.15 eqv. of DBU) (Figure 4, highlighted in green), that can be explained by the formation of hydrogen-bonded complex of protonated DBU1 with the deprotonated compound 3b. Upon further addition of the base this signal was broadened and shifted downfield (9.86 ppm) due to the interaction of protonated DBU (DBUH+) with the anionic compound 3b through the N–H bond [39].
Upon addition of more than 1.05 eqv. of the base a second minor form of the compound appears (Figure 4, highlighted in blue); ratio between major and minor forms is 0.95:0.05 taking into consideration all proton signals. Moreover, the addition of excess amount of the base (4 and 8 eqv.) did not result in the change of 1H NMR spectra of compound 3b (additional processes as a function of the time and/or temperature in the solution should not be excluded as 1H NMR titration experiment was carried out within an hour after addition of DBU to the compound 3b at room temperature (T = 294 K)).
Also, the presence of two different species (one major and one minor) was detected from the changes in 1H NMR spectra of compound 3a and 3c upon deprotonation with DBU in DMSO-d6 solution (after mixed with more than 1 equivalent of DBU). For compound 3a 1H NMR spectrum was also recorded in the presence of NaOH, as a result, acquired spectrum was identical to the one with excess of DBU (Figure S12).
Unfortunately, low solubility of compounds 3a-g limited the possibility to obtain qualitative 13C NMR spectra.
2.1.4. Electronic Absorption
The UV-Vis absorption spectra of compound 3a were investigated in solution using solvents of various polarities (DCM and DMSO). Two absorption maxima were found at 381 nm and at 446-449 nm in the absorption spectra of compound 3a in both solutions (Figure 5a) that can be attributed to the neutral form of the compound. Upon addition of the base (DBU) to DCM solution of 3a, the solution instantaneously turned violet, and the absorption revealed a broad band centered at 556 nm. When a base was added to DMSO solution of compound 3a, the bathochromic shift was observed with absorption maxima at 607 nm accompanied by a blue coloration. The charge transfer character of the deprotonated molecule is probable as structure contains very polar groups.
It was noticed that an absorption band of deprotonated species was red-shifted and molar absorption coefficient increased together with the introduction of the electron-withdrawing substituent to the benzene ring (UV-Vis spectra of compound 3c, Figure 5b).
2.2. Electrochemistry/Redox Chemistry Studies of Quinone Derivatives 1a and 3a
Quinone imines contain structural fragments with potentially high redox activity. It was shown by Almeida, R., et. al. [40] that p-quinone imines are known to undergo a redox cycle through aminophenols [41]. To analyze redox properties of quinone imine 3a in comparison to initial o-quinone 1a (electrochemically active compound [42]), open circuit potential (OCP) and cyclic voltammetry (CV) measurements in solid state were carried out. Preliminary solubility tests showed limited solubility of compounds 1a and 3a in aqueous media; compound 1a was insoluble in water (whole pH range), at the same time compound 3a was insoluble in neutral and acidic environments. To the best of our knowledge p-quinone imines were not tested as electrode materials before.
2.2.1. Open Circuit Potential Measurements
To analyze the electrochemical properties of the compounds CV measurements and OCP measurements before and after CV were performed (Figure S15). Cathode materials CM-1a and CM-3a were prepared by combining compounds 1a and 3a with Vulcan XC72 CB, respectively (the detailed sample preparation is described in the Experimental section). OCP of freshly assembled half-cells for cathode materials CM-1a and CM-3a in the acidic (0.5 M H2SO4) electrolyte was 0.44 V and 0.37 V vs Ag/AgCl, however, in a neutral (0.5 M K2SO4) electrolyte potentials of both were 0.28 V vs Ag/AgCl. After the CV measurements the OCP of sample half-cells stabilize and for both samples in the acidic electrolyte were 0.41 V vs Ag/AgCl and in neutral electrolyte 0.37 V vs Ag/AgCl. The OCP for both samples are approximately the same, and with decreasing pH, there is a visible shift to higher potential values going from neutral to the acidic electrolyte.
2.2.2. Cyclic Voltammetry Measurements
CV results for samples CM-1a, CM-3a and sample without active material (substrate) in neutral and acidic electrolytes at varying scanning speeds are shown in Figure 6. For the substrate in neutral electrolyte (Figure 6a), no visible redox processes were observed. In the acidic electrolyte (Figure 6d), hydrogen evolution reaction can be observed around the potential of –0.3 V vs Ag/AgCl and one insignificant redox process at faster scan rates around 0.3 V vs Ag/AgCl. However, when scanning samples with active materials this substrate process cannot be observed and therefore has no electrochemical significance other than providing electrical conductivity.
For sample CM-3a in neutral electrolyte (Figure 6) no significant redox processes can be observed. Also, in acidic electrolyte for sample CM-3a (Figure 6) there is no significant processes, however, upon closer inspection (Figure S16) two reversible oxidation (at 0.40 V and 0.07 V vs Ag/AgCl) and reduction processes (at 0.23 V and –0.06 V vs Ag/AgCl) can be seen.
Sample CM-1a has two redox maxima in the scanned potential window from –0.4 V to 1.0 V vs Ag/AgCl electrode in both neutral and acidic electrolyte (Figure 6). In neutral electrolyte (Figure S17) oxidation peaks are at 0.27 V and 0.10 V vs Ag/AgCl, however, reduction peaks are at 0.14 V and –0.32 V vs Ag/AgCl. In addition, the oxidation peaks are found at 0.48 V and 0.26 V vs Ag/AgCl and reduction peaks - at 0.40 V and 0.23 V vs Ag/AgCl in acidic electrolyte (Figure S18). This indicates a shift in reaction potential to more positive values by increasing H+ ion concentration and thus lowering the pH level of the electrolyte. Both redox processes for sample CM-1a correspond to o-quinone fragment in the molecule. However, by comparing both sample CM-1a and CM-3a electrochemical performance, it can be concluded that converting o-quinone 1a to α-hydroxy-p-quinone imine 3a accompanied by the additional stabilization by intra- and intermolecular hydrogen bonds the electrochemical reactivity of cathode material has been greatly suppressed.
All CV measurement result development in time can be seen in Figure S19. At the start, samples were cycled at the potential window of –0.4 V to 1.0 V vs Ag/AgCl reference electrode. Observations indicate that all samples go through the surface activation phase where sample-specific capacity increases with each subsequent cycle. During the measurements at different scanning speeds, the stabilization of the system is observed. However, sample CM-1a in neutral electrolyte goes through an irreversible oxidation process at 0.11 V vs Ag/AgCl and reduction process at –0.32 V vs Ag/AgCl. This irreversible redox process can be observed during all scan speeds. At an increased potential window (from – 1.0 V to 1.5 V) another irreversible process at scan speed of 0.1 V/s for sample CM-1a in neutral electrolyte can be observed during the oxidation at –0.31 V and reduction at –0.56 V vs Ag/AgCl. A slight capacity decrease due to possible dissolution of active materials can be observed for sample CM-3a in neutral electrolyte and sample CM-1a in acidic electrolyte.
2.2.2. Raman Measurements
Raman measurements were performed on pure compounds 1a and 3a, prepared cathodes (CM-1a and CM-3a) and after cycling them in acidic and neutral electrolytes (Figure 7). For sample CM-3a, the spectra for prepared and cycled cathodes remain as for pure compound 3a, where amide band can be seen at 1600-1630 cm-1 as well as bands for aromatic/heteroaromatic ring at 1550 and 1470 cm-1 [43]. This indicates that compound 3a was preserved in the cathode forming process and did not go through any chemical changes. Also, after CV measurements the active material is unchanged and present in the samples. For sample CM-1a, the spectra for the prepared and cycled cathode in acidic electrolyte remain as for pure compound 1a (bands for aromatic/heteroaromatic ring at 1570 and 1450 cm-1, and band for carbonyl groups at 1650-1690 cm-1) [43]. However, for cathode CM-1a cycled in neutral electrolyte, only C and D bands of carbon [44,45]. can be seen. Since redox processes are visible for this sample in CV measurements (Figure 6), the active material could have dissolved from the electrode in the electrolyte and gone through the electrochemical reactions from the electrolyte.
2.2.3. Scanning Electron Microscopy Measurements
Scanning electron microscopy examinations of compounds 1a and 3a (Figure S21) were performed to assess the morphology of the products. Compound 1a consists of needle-like particles with sizes ranging from 1 µm to 10 µm in diameter and 3 µm to 30 µm in length. Compound 3a has smaller particles with an overall size of 1 µm in diameter and 5-20 µm in length.
Also, the prepared cathode surfaces with and without active materials before and after cyclic voltammetry are shown in Figure 8. Resemblance of the structures of compound 1a and 3a (as shown in Figure S20) can be seen in the images of cathode disks (samples CM-1a and CM-3a in Figure 8) before CV measurements. For sample CM-3a these structures also can be seen in images after CV in neutral and acidic electrolyte with some partial dissolution in acidic electrolyte as less structures can be seen. The formation of non-covalent interactions between compound 3a and substrate, probably, can explain greater stability of CM-3a in comparison to CM-1a. However, for sample CM-1a cycled in neutral electrolyte only a few original structures can be seen and for the sample cycled in acidic electrolyte no original particle structures can be seen. Thus, further suggesting findings from Raman spectroscopy (Figure 7b) that the active material 1a dissolves in electrolyte and goes though electrochemical reactions from it.
3. Materials and Methods
3.1. Materials and Instrumentation
Polyvinylidene fluoride (PVDF) (MW ~530,000) and Dimethylformamide (DMF) were purchased from Merck; Vulcan XC72 Carbon Black (CB) was used and 0.05 mm thick conductive graphite paper (RERAS, purchased from China and used as electrode substrate) were used to prepare cathode materials.
Melting points were measured on Kruess KSP 11 Melting Point Analyzer. 1H NMR spectra were recorded on a Brucker Avance 300 or 500 spectrometer at 300 or 500 respectively in DMSO-d6 or CDCl3 solutions. Chemical shifts were expressed in parts per million (δ, ppm) relative to solvent signal (DMSO-d6: 2.50 ppm CDCl3: 7.26 ppm for 1H NMR) [46]. Compounds 3a-g are too insoluble to record a qualitative 13C NMR spectrum. Elemental CHN analysis was carried on Euro Vector EA 3000 analyzer. FTIR spectra were recorded on a Perkin-Elmer Spectrum 100 FTIR spectrometer. The UV-Vis absorption spectra were acquired with Perkin-Elmer 35 UV/Vis spectrometer using 1 cm length quartz cuvettes with a concentration of compound c = 2.5∙10-5 M. Low resolution mass spectra were acquired on a Waters EMD 1000MS mass detector (ESI + mode, voltage 30 V) with Xterra MS C18 5 μm 2.1 100 mm column and gradient eluent mode using 0.1% HCOOH in deionized water and MeCN or MeOH.
3.2. X-ray Crystallography Analysis
For compound 3a diffraction data were collected at low temperature (T = 150.0(1) K) on Rigaku, XtaLAB Synergy, Dualflex, HyPix diffractometer using cupper monochromated Cu-Kα radiation (λ = 1.54184 Å). The crystal structure was solved with the help of ShelXT structure solution program [47] using the Intrinsic Phasing solution method. The model was refined with version of the program olex2.refine using Levenberg-Marquardt minimization [48]. All nonhydrogen atoms were refined in anisotropical approximation. For further details, see crystallographic data for compound 3a deposited at the Cambridge Crystallographic Data Centre as Supplementary Publications Numbers CCDC 2238663 (for compound 3a). This data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk). For crystal packing visualization program Mercury [49] was used.
3.3. Cathode Material Preparation
Quinone derivatives 1a or 3a were combined with Vulcan XC72 CB at a mass ratio of 5:4. The resulting powder was dried overnight at 80 °C. Then a binder solution of PVDF:DMF (mass ratio 1:9) was added, so the quinone to PVDF mass ratio would be 5:1. Stirring and ultra-sonication were used to create the ink slurry. Extra DMF was added to the slurry to form homogenous ink (the total weight ratio of DMF to quinone was approximately 12:1). Manual doctor blade coater (with a 25 µm gap size) was employed to apply the coating onto carbon paper that was pre-dried at 120 °C for an hour. Coated cathode substrates were then dried in air to evaporate DMF. For further material characterization cathode disks were cut out using a hollow punch.
3.4. Cyclic Voltammetry
To analyze the electrochemical properties of the different sample compounds cyclic voltammetry (CV) measurements were performed using a 3-electrode measuring cell “TSC Surface” (from rhd instruments). For different measurements, the prepared thin layer electrodes on carbon paper (with or without compound 1a or 3a) were used as working electrodes placed in 1 mL of electrolyte with the platinum counter electrode and Ag/AgCl (3 M KCl) reference electrode. Two different pH electrolyte solutions were used for measurements – neutral 0.5 M K2SO4 and acidic 0.5 M H2SO4 solutions. The CV measurements were performed from –0.4 to +1.0 V with the following program: (1) open circuit measurement (OCP) of freshly assembled half-cell before the CV measurements; (2) ten cycles with scan speed of 0.075 V/s to stabilize the half-cell; (3) 5 cycles of 5 scans with scan rates ranging from 0.005 V/s to 0.1 V/s; (4) OCP measurement after CV.
3.5. Raman Spectroscopy
Raman measurements were performed using a Renishaw In-ViaV727 spectrometer in a backscattering geometry at room temperature at 100x magnification. For phonon excitation red laser (He-Ne, λ = 633 nm, grating – 1200 mm–1, 125 µW) was used and the sample exposure time was 10 s.
3.6. Scanning Electron Microscopy
Hitachi TM3000 Tabletop scanning electron microscope (SEM) with an acceleration voltage of 5 kV was used to obtain surface information of the obtained electrode and sample materials. To characterize the obtained samples different magnifications were used.
3.7. Synthesis of Quinone Derivatives 1a,b and 3a-g
6,7-Dichloropyrido [1,2-a]benzimidazole-8,9-dione (1a) and 6,7-dichloro-2-nitropyrido [1,2-a]benzimidazole-8,9-dione (1b) were prepared according to the previously reported procedure [18,42].
General method for synthesis of compounds 3a-g. To a stirring solution of 6,7-dichloropyrido [1,2-a]benzimidazole-8,9-dione (1a, 1 eq) or 6,7-dichloro-2-nitropyrido [1,2-a]benzimidazole-8,9-dione (1b, 1 eq) in dichloromethane (DCM) at room temperature, a solution of benzhydrazide derivative (2a-d, 2 eq) in DCM or DMF was added. Triethylamine (1 eq) was added to the reaction mixture, which was then stirred at room temperature for 8 hours. After completion of the reaction, the reaction mixture was filtered through a filter paper and the solvent was distilled in vacuo to a residual volume of 20 ml. The resulting orange colored precipitate was collected, recrystallized from DCM/n-hexane, washed with hot ethanol (20 ml), and dried at room temperature.
Compound 3a. Prepared using 6,7-dichloropyrido [1,2-a]benzimidazole-8,9-dione (150 mg, 0.56 mmol, 1 eq), benzohydrazide (153 mg, 1.12 mmol, 2 eq) and triethylamine (d = 0.73 g/mL, v = 78 μL, 0.56 mmol, 1 eq). Yield: 59%, orange powder. M.P.: 258-260 °C. MS: C18H11ClN4O3 requires [M+H]+ 367.1; found [M+H]+ 367.2. 1H NMR (500 MHz, DMSO-d6): 14.71 (br.s., 1H, exchange with D2O, NH), 10.98 (br.s., 1H, exchange with D2O, OH), 9.29 (d, J = 6.6 Hz, 1H, H-1), 8.16 (d, J = 9.0 Hz, 1H, H-4), 8.11 (d, J = 7.4, 2H, CHPh), 7.89 (t, J = 8.0, 1H, H-3), 7.73 (m, 3H, CHPh), 7.51 (d, J = 6.8 Hz, 1H, H-2). FTIR (KBr, cm-1): 3331, 3094, 3033, 1708, 1628, 1604, 1548, 1437, 1351, 1247. Anal. Calcd. for C18H11ClN4O3: C, 58.95; H, 3.02; N, 15.28; found C, 58.82; H, 3.02; N, 15.31.
Compound 3b. Prepared using 6,7-dichloropyrido [1,2-a]benzimidazole-8,9-dione (150 mg, 0.56 mmol, 1 eq), 4-methoxybenzohydrazide (187 mg, 1.12 mmol, 2 eq) and triethylamine (d = 0.73 g/mL, v = 78 μL, 0.56 mmol, 1 eq). Yield: 36%, orange solid. M.P.: > 300 °C. MS: C19H13ClN4O4 requires [M+H]+ 397.1; found [M+H]+ 397.2. 1H NMR (500 MHz, DMSO-d6): 14.70 (br.s., 1H, exchange with D2O, NH), 10.95 (br.s., 1H, exchange with D2O, OH), 9.33 (d, J = 6.7 Hz, 1H, H-1), 8.24 (d, J = 9.0 Hz, 1H, H-4), 8.11 (d, J = 8.6 Hz, 2H, CHPh), 7.91 (m, 1H, H-3), 7.53 (t, J = 6.8 Hz, 1H, H-2), 7.25 (d, J = 8.5 Hz, 2H, CHPh), 3.91 (s, 3H, -OCH3). FTIR (KBr, cm-1): 3468, 3301, 3083, 3024, 2975, 2832, 1690, 1630, 1609, 1582, 1552, 1504, 1351, 1325, 1259. Anal. Calcd. for C19H13ClN4O4: C, 57.51; H, 3.30; N, 14.12; found C, 57.58; H, 3.35; N, 13.82.
Compound 3c. Yield: Prepared using 6,7-dichloropyrido [1,2-a]benzimidazole-8,9-dione (150 mg, 0.56 mmol, 1 eq), 4-nitrobenzohydrazide (204 mg, 1.12 mmol, 2 eq) and triethylamine (d = 0.73 g/mL, v = 78 μL, 0.56 mmol, 1 eq). 52%, orange solid. M.P.: 275-278 °C. MS: C18H10ClN5O5 requires [M+H]+ 412.1; found [M+H]+ 412.2. 1H NMR (300 MHz, DMSO-d6): 14.95 (br.s., 1H, exchange with D2O, NH), 11.07 (br.s., 1H, exchange with D2O, OH), 9.29 (d, J = 6.5, 1H, H-1), 8.53 (m, 2H, CHPh), 8.32 (d, J = 8.3, 3H, H-4 un CHPh), 7.91 (m, 1H, H-3), 7.53 (m, 1H, H-2). FTIR (KBr, cm-1): 3618, 3306, 3113, 3089, 3016, 1703, 1626, 1605, 1573, 1556, 1519, 1346, 1275. Anal. Calcd. for C18H10ClN5O5: C, 52.51; H, 2.45; N, 17.01; found C, 52.20; H, 2.58; N, 16.73.
Compound 3d. Prepared using 6,7-dichloropyrido [1,2-a]benzimidazole-8,9-dione (150 mg, 0.56 mmol, 1 eq), isonicotinohydrazide (154 mg, 1.12 mmol, 2 eq) and triethylamine (d = 0.73 g/mL, v = 78 μL, 0.56 mmol, 1 eq). Yield: 50%, orange crystals. M.P.: > 250 °C (decomp.). MS: C17H10ClN5O3 requires [M+H]+ 368.1; found [M+H]+ 368.2. 1H NMR (300 MHz, DMSO-d6): 14.52 (br.s., 1H, exchange with D2O, NH), 10.90 (br.s., 1H, exchange with D2O, NH), 9.31 (d, J = 6.7, 1H, H-1), 8.91 (d, J = 3.8, 2H, CHPy), 8.18 (d, J = 8.9, 1H, H-4), 7.97 (d, J = 4.6, 2H, CHPy), 7.89 (t, J = 8.0, 1H, H-3), 7.52 (t, J = 6.7, 1H, H-2). FTIR (KBr, cm-1): 3420, 3055, 1709, 1647, 1568, 1555, 1331, 1280. Anal. Calcd. for C17H10ClN5O3+0.5H2O: C, 54.20; H, 2.94; N, 18.59; found C, 54.29; H, 2.88; N, 18.30.
Compound 3e. Prepared using 6,7-dichloro-2-nitropyrido [1,2-a]benzimidazole-8,9-dione (150 mg, 0.48 mmol, 1 eq), benzohydrazide (131 mg, 0.96 mmol, 2 eq) and triethylamine (d = 0.73 g/mL, v = 67 μL, 0.48 mmol, 1 eq). Yield: 61%, yellow solid. M.P.: > 250 °C (decomp.). MS: C18H10ClN5O5 requires [M+H]+ 412.1; found [M+H]+ 412.4. 1H NMR (300 MHz, DMSO-d6): 14.42 (br.s., 1H, exchange with D2O, NH), 11.30 (br.s., 1H, exchange with D2O, OH), 10.06 (d, J = 2.3 Hz, 1H, H-1), 8.56 (dd, J = 9.8, 2.1 Hz 1H, H-3), 8.40 (d, J = 9.7 Hz, 1H, H-4), 8.12 (m, 2H, CHPh), 7.72 (d, J = 7.7, 3H, CHPh). FTIR (KBr, cm-1): 3397, 3091, 3033, 1690, 1642, 1552, 1525, 1351, 1311, 1268. Anal. Calcd. for C18H10ClN5O5: C, 52.51; H, 2.45; N, 17.01; found C, 52.21; H, 2.67; N, 16.76.
Compound 3f. Prepared using 6,7-dichloro-2-nitropyrido [1,2-a]benzimidazole-8,9-dione (150 mg, 0.48 mmol, 1 eq), 4-methoxybenzohydrazide (160 mg, 0.96 mmol, 2 eq) and triethylamine (d = 0.73 g/mL, v = 67 μL, 0.48 mmol, 1 eq). Yield: 42%, yellow crystals. M.P.: > 250 °C (decomp.). MS: C19H12ClN5O6 requires [M+H]+ 442.1; found [M+H]+ 442.2. 1H NMR (300 MHz, DMSO-d6): 14.36 (br.s., 1H, exchange with D2O, NH), 11.23 (br.s., 1H, exchange with D2O, OH), 10.05 (s, 1H, H-1), 8.56 (d, J = 9.7 Hz, 1H, H-3), 8.42 (d, J = 10.8 Hz, 1H, H-4), 8.10 (d, J = 7.1 Hz, 2H, CHPh), 7.24 (d, J = 8.3 Hz, 2H, CHPh), 3.90 (s, 3H, OCH3). FTIR (KBr, cm-1): 3399, 3085, 3028, 1687, 1640, 1605, 1555, 1523, 1350, 1310, 1266. Anal. Calcd. for C19H12ClN5O6: C, 51.66; H, 2.74; N, 15.85; found C, 51.35; H, 2.68; N, 15.61.
Compound 3g. Prepared using 6,7-dichloro-2-nitropyrido [1,2-a]benzimidazole-8,9-dione (150 mg, 0.48 mmol, 1 eq), 4-nitrobenzohydrazide (174 mg, 0.96 mmol, 2 eq) and triethylamine (d = 0.73 g/mL, v = 67 μL, 0.48 mmol, 1 eq). Yield: 36%, orange solid. M.P.: > 250 °C (decomp.). MS: C18H9ClN6O7 requires [M+H]+ 457.0; found [M+H]+ 457.1. 1H NMR (300 MHz, DMSO-d6): 14.61 (br.s., 1H, exchange with D2O, NH), 11.41 (br.s., 1H, exchange with D2O, OH), 10.03 (s, 1H, H-1), 8.58 (d, J = 10.8 Hz, 2H, H-4 and H-3) 8.51 (m, 2H, CHPh), 8.32 (d, J = 8.3 Hz, 2H, CHPh). FTIR (KBr, cm-1): 3340, 3114, 3081, 1707, 1626, 1602, 1547, 1518, 1344, 1306, 1266. Anal. Calcd. for C18H9ClN6O7: C, 47.33; H, 1.99; N, 18.40; found C, 47.24; H, 2.02; N, 18.14.
4. Conclusions
To summarize, a set of heterocyclic α-hydroxy-p-quinone imine derivatives was obtained via one-step nucleophilic substitution of 6,7-dichloropyrido [1,2-a]benzimidazole-8,9-diones 1a,b with different benzohydrazides 2a-d. α-Hydroxy-p-quinone imine form of the synthesized products was proved by X-ray crystallography analysis of compound 3a. The formation of a strong intramolecular hydrogen bond N(12)-H···N(5) in solid state and in the solution can explain the stabilization of the only one configuration of substituted imine. 1H NMR acid-base titration experiment showed that deprotonation/protonation processes are reversible. Deprotonation led to the electronic delocalization in the molecule that is accompanied by distinct changes in the UV-Vis spectra.
Attempts to modulate redox properties by incorporation of additional unsaturated carbon–nitrogen bond to the heterocyclic quinone 1a structure led to the changes in redox active fragment and formation of p-quinone imine 3a. As a result, the electrochemical behavior is changed, as it is no longer possible to observe pronounced redox peaks in CV measurements. Structural changes of quinone fragment (probably induced by the formation of intramolecular H-bond) decreased redox activity of the derivative despite the introduction of an additional C=N bond.
Sample CM-1a has a distinct maximum of two redox reactions. In an acidic environment both peaks are stable while in a neutral environment only one of them is stable and remains unchanged after several CV cycles. Moreover, for the stable redox reactions the potential difference is only up to 0.2 V. This indicates that the sample CM-1a could act as an effective active electrode in aqueous electrolyte batteries. However, the dissolution of the sample in the electrolyte was observed, so the potential application as cathode material for aqueous batteries would require compound 1a to be attached to the polymer backbone.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figures S1-S8: 1H NMR spectra (for compounds 3a-g in DMSO-d6 solution and for compound 3a in CDCl3 solution). Figure S9: Additional 1H NMR spectra for compound 3b upon acid addition and irradiation. Figure S10, S12: 1H NMR spectra for compounds 3a-c upon base addition. Figures S13, S14: Hirshfeld surfaces and energy frameworks calculated with CrystalExplorer software. Figures S15-S20: CV curves of samples CM-1a and CM-3a in neutral and acidic electrolyte at various scan speeds (PDF).
Author Contributions
Conceptualization, N.B.; formal analysis, A.G.; investigation, A.G., S.B., R.D., N.G., A.Z.; writing—original draft preparation, A.G., S.B., R.D., A.Z., N.B.; writing—review and editing, A.G., N.B..; visualization, A.G., R.D., N.G., A.Z.; supervision, N.B.; funding acquisition, A.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported by the European Social Fund within the Project No 8.2.2.0/20/I/008 «Strengthening of PhD students and academic personnel of Riga Technical University and BA School of Business and Finance in the strategic fields of specialization» of the Specific Objective 8.2.2 «To Strengthen Academic Staff of Higher Education Institutions in Strategic Specialization Areas» of the Operational Programme «Growth and Employment». This research/publication was supported by Riga Technical University's Doctoral Grant programme (DOK.LĶI/23).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- P. Cao et al., “Structural basis for the assembly and quinone transport mechanisms of the dimeric photosynthetic RC–LH1 supercomplex,” Nat. Commun., vol. 13, no. 1, pp. 1–12, 2022. [CrossRef]
- J. Gutiérrez-Fernández et al., “Key role of quinone in the mechanism of respiratory complex I,” Nat. Commun., vol. 11, no. 1, pp. 1–17, 2020. [CrossRef]
- J. L. Bolton and T. Dunlap, “Formation and Biological Targets of Quinones: Cytotoxic versus Cytoprotective Effects,” Chem. Res. Toxicol., vol. 30, no. 1, pp. 13–37, Jan. 2017. [CrossRef]
- M. Mansha et al., “Recent Developments on Electroactive Organic Electrolytes for Non-Aqueous Redox Flow Batteries: Current Status, Challenges, and Prospects,” Chem. Rec., vol. 24, no. 1, Jan. 2024. [CrossRef]
- C. Y. Go, J. Shin, M. K. Choi, I. H. Jung, and K. C. Kim, “Switchable Design of Redox-Enhanced Nonaromatic Quinones Enabled by Conjugation Recovery,” Adv. Mater., Dec. 2023. [CrossRef]
- P. Simon and Y. Gogotsi, “Perspectives for electrochemical capacitors and related devices,” Nat. Mater., vol. 19, no. 11, pp. 1151–1163, Nov. 2020. [CrossRef]
- J. J. Shea and C. Luo, “Organic Electrode Materials for Metal Ion Batteries,” ACS Appl. Mater. Interfaces, vol. 12, no. 5, pp. 5361–5380, Feb. 2020. [CrossRef]
- R. B. Jethwa, D. Hey, R. N. Kerber, A. D. Bond, D. S. Wright, and C. P. Grey, “Exploring the Landscape of Heterocyclic Quinones for Redox Flow Batteries,” ACS Appl. Energy Mater., Dec. 2023. [CrossRef]
- L. E. Blanc, D. Kundu, and L. F. Nazar, “Scientific Challenges for the Implementation of Zn-Ion Batteries,” Joule, vol. 4, no. 4, pp. 771–799, Apr. 2020. [CrossRef]
- Y. Tsao et al., “Designing a Quinone-Based Redox Mediator to Facilitate Li2S Oxidation in Li-S Batteries,” Joule, vol. 3, no. 3, pp. 872–884, Mar. 2019. [CrossRef]
- L. Sieuw et al., “A H-bond stabilized quinone electrode material for Li–organic batteries: the strength of weak bonds,” Chem. Sci., vol. 10, no. 2, pp. 418–426, 2019. [CrossRef]
- M. Sugumaran, “Reactivities of quinone methides versus o-Quinones in catecholamine metabolism and eumelanin biosynthesis,” Int. J. Mol. Sci., vol. 17, no. 9, pp. 1–23, 2016. [CrossRef]
- I. Poddel’sky, N. O. Druzhkov, G. K. Fukin, V. K. Cherkasov, and G. A. Abakumov, “Bifunctional iminopyridino-catechol and its o-quinone: Synthesis and investigation of coordination abilities,” Polyhedron, vol. 124, pp. 41–50, Mar. 2017. [CrossRef]
- T. V. Astaf’eva, M. V. Arsenyev, R. V. Rumyantcev, G. K. Fukin, V. K. Cherkasov, and A. I. Poddel’sky, “Imine-Based Catechols and o -Benzoquinones: Synthesis, Structure, and Features of Redox Behavior,” ACS Omega, vol. 5, no. 35, pp. 22179–22191, Sep. 2020. [CrossRef]
- R. C. B. Ribeiro, P. G. Ferreira, A. De A. Borges, L. Da S. M. Forezi, F. De Carvalho da Silva, and V. F. Ferreira, “1,2-Naphthoquinone-4-sulfonic acid salts in organic synthesis,” Beilstein J. Org. Chem., vol. 18, pp. 53–69, 2022. [CrossRef]
- Z. Wu et al., “Molecular and Morphological Engineering of Organic Electrode Materials for Electrochemical Energy Storage,” Electrochem. Energy Rev., vol. 5, no. s1, pp. 1–67, 2022. [CrossRef]
- N. Batenko, S. Belyakov, G. Kiselovs, and R. Valters, “Synthesis of 6,7-dichloropyrido [1,2-a]benzimidazole-8,9-dione and its analogues and their reactions with nucleophiles,” Tetrahedron Lett., vol. 54, no. 35, pp. 4697–4699, 2013. [CrossRef]
- N. Batenko, S. Belyakov, G. Kiselovs, and R. Valters, “Synthesis of 6,7-dichloropyrido [1,2-a]benzimidazole-8,9-dione and its analogues and their reactions with nucleophiles,” Tetrahedron Lett., vol. 54, no. 35, pp. 4697–4699, Aug. 2013. [CrossRef]
- Gaile, S. Belyakov, B. Turovska, and N. Batenko, “Synthesis of Asymmetric Coupled Polymethines Based on a 7-Chloropyrido [1,2- a ]benzimidazole-8,9-dione Core,” J. Org. Chem., vol. 87, no. 5, pp. 2345–2355, Mar. 2022. [CrossRef]
- Gaile, S. Belyakov, V. Rjabovs, I. Mihailovs, B. Turovska, and N. Batenko, “Investigation of Weak Noncovalent Interactions Directed by the Amino Substituent of Pyrido- and Pyrimido-[1,2- a ]benzimidazole-8,9-diones,” ACS Omega, vol. 8, no. 43, pp. 40960–40971, Oct. 2023. [CrossRef]
- T. YAMADA, T. YAMASHITA, M. NAKAMURA, H. SHIMAMURA, A. YAMAGUCHI, and M. TAKAYA, “Synthesis and Hemostatic Activity of 1, 2-Naphthoquinones,” YAKUGAKU ZASSHI, vol. 100, no. 8, pp. 799–806, 1980. [CrossRef]
- F. I. Carroll, H. W. Miller, and R. Meck, “Thiosemicarbazone and amidinohydrazone derivatives of some 1,4-naphthoquinones,” J. Chem. Soc. C Org., vol. 3, no. 15, p. 1993, 1970. [CrossRef]
- K. H. Dudley, H. W. Miller, P. W. Schneider, and R. L. McKee, “Potential Naphthoquinone Antimalarials. 2-Acylhydrazino-1,4-naphthoquinones and Related Compounds,” J. Org. Chem., vol. 34, no. 9, pp. 2750–2755, 1969. [CrossRef]
- M. B. Smith and J. March, March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure: Sixth Edition, vol. 9780471720. 2006.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka, and M. A. Spackman, “Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals,” Chem. Commun., vol. 51, no. 18, pp. 3735–3738, 2015. [CrossRef]
- P. R. Spackman et al., “CrystalExplorer : a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals,” J. Appl. Crystallogr., vol. 54, no. 3, pp. 1006–1011, Jun. 2021. [CrossRef]
- J. Catalán, “Toward a Generalized Treatment of the Solvent Effect Based on Four Empirical Scales: Dipolarity (SdP, a New Scale), Polarizability (SP), Acidity (SA), and Basicity (SB) of the Medium,” J. Phys. Chem. B, vol. 113, no. 17, pp. 5951–5960, Apr. 2009. [CrossRef]
- N. N. Shapet’ko and D. N. Shigorin, “NMR study of intramolecular hydrogen bond protons in quinoid structures,” Zhurnal Strukt. Khimii, vol. 8, no. 3, pp. 538–540, 1967.
- M. J. Kamlet and R. W. Taft, “Solvent Hydrogen-Bond Acceptor (HBA) Basicities,” J. Am. Chem. Soc., vol. 98, no. 2, pp. 377–383, 1975. [CrossRef]
- X. Su, M. Lõkov, A. Kütt, I. Leito, and I. Aprahamian, “Unusual para-substituent effects on the intramolecular hydrogen-bond in hydrazone-based switches,” Chem. Commun., vol. 48, no. 85, p. 10490, 2012. [CrossRef]
- X. Su and I. Aprahamian, “Hydrazone-based switches, metallo-assemblies and sensors,” Chem. Soc. Rev., vol. 43, no. 6, pp. 1963–1981, 2014. [CrossRef]
- J. E. Johnson, N. M. Morales, A. M. Gorczyca, D. D. Dolliver, and M. A. McAllister, “Mechanisms of Acid-Catalyzed Z / E Isomerization of Imines,” J. Org. Chem., vol. 66, no. 24, pp. 7979–7985, Nov. 2001. [CrossRef]
- X. Su and I. Aprahamian, “Switching Around Two Axles: Controlling the Configuration and Conformation of a Hydrazone-Based Switch,” Org. Lett., vol. 13, no. 1, pp. 30–33, Jan. 2011. [CrossRef]
- Ryabchun, Q. Li, F. Lancia, I. Aprahamian, and N. Katsonis, “Shape-Persistent Actuators from Hydrazone Photoswitches,” J. Am. Chem. Soc., vol. 141, no. 3, pp. 1196–1200, Jan. 2019. [CrossRef]
- J. Kim, J. Ling, Y. Lai, and P. J. Milner, “Redox-Active Organic Materials: From Energy Storage to Redox Catalysis,” ACS Mater. Au, 2023. [CrossRef]
- M. Wang, X. Dong, I. C. Escobar, and Y.-T. Cheng, “Lithium Ion Battery Electrodes Made Using Dimethyl Sulfoxide (DMSO)—A Green Solvent,” ACS Sustain. Chem. Eng., vol. 8, no. 30, pp. 11046–11051, Aug. 2020. [CrossRef]
- Laurence, J. Legros, A. Chantzis, A. Planchat, and D. Jacquemin, “A Database of Dispersion-Induction DI, Electrostatic ES, and Hydrogen Bonding α 1 and β 1 Solvent Parameters and Some Applications to the Multiparameter Correlation Analysis of Solvent Effects,” J. Phys. Chem. B, vol. 119, no. 7, pp. 3174–3184, Feb. 2015. [CrossRef]
- Y. Ooyama and S. Yagi, Progress in the Science of Functional Dyes. 2021. [CrossRef]
- M. S. Miran, H. Kinoshita, T. Yasuda, M. A. B. H. Susan, and M. Watanabe, “Hydrogen bonds in protic ionic liquids and their correlation with physicochemical properties,” Chem. Commun., vol. 47, no. 47, pp. 12676–12678, 2011. [CrossRef]
- R. G. Almeida et al., “Synthesis of quinone imine and sulphur-containing compounds with antitumor and trypanocidal activities: Redox and biological implications,” RSC Med. Chem., vol. 11, no. 10, pp. 1145–1160, 2020. [CrossRef]
- Klopčič and M. S. Dolenc, “Chemicals and Drugs Forming Reactive Quinone and Quinone Imine Metabolites,” Chem. Res. Toxicol., vol. 32, no. 1, pp. 1–34, 2019. [CrossRef]
- N. Batenko, A. Kricka, S. Belyakov, B. Turovska, and R. Valters, “A novel method for the synthesis of benzimidazole-based 1,4-quinone derivatives,” Tetrahedron Lett., vol. 57, no. 3, pp. 292–295, 2016. [CrossRef]
- E. Smith and G. Dent, Modern Raman Spectroscopy – A Practical Approach. Wiley, 2004. [CrossRef]
- N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud’homme, I. A. Aksay, and R. Car, “Raman spectra of graphite oxide and functionalized graphene sheets,” Nano Lett., vol. 8, no. 1, pp. 36–41, 2008. [CrossRef]
- M. Saravanan, M. Ganesan, and S. Ambalavanan, “An in situ generated carbon as integrated conductive additive for hierarchical negative plate of lead-acid battery,” J. Power Sources, vol. 251, pp. 20–29, 2014. [CrossRef]
- H. E. Gottlieb, V. Kotlyar, and A. Nudelman, “NMR chemical shifts of common laboratory solvents as trace impurities,” J. Org. Chem., vol. 62, no. 21, pp. 7512–7515, 1997. [CrossRef]
- G. M. Sheldrick, “Crystal structure refinement with SHELXL,” Acta Crystallogr. Sect. C Struct. Chem., vol. 71, no. Md, pp. 3–8, 2015. [CrossRef]
- J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard, and H. Puschmann, “The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment - Olex2 dissected,” Acta Crystallogr. Sect. A Found. Crystallogr., vol. 71, no. 1, pp. 59–75, 2015. [CrossRef]
- F. MacRae et al., “Mercury 4.0: From visualization to analysis, design and prediction,” J. Appl. Crystallogr., vol. 53, pp. 226–235, 2020. [CrossRef]
| 1 |
1H NMR spectrum of DBU and TFA mixture in DMSO-d6 solution was recorded; the NH+ signal appears at 9.69 ppm (Figure S11). |
Scheme 1.
(a) Synthesis of compounds 3a-g; (b) p-Quinone imine and benzamide fragments of obtained compounds 3a-g.
Scheme 1.
(a) Synthesis of compounds 3a-g; (b) p-Quinone imine and benzamide fragments of obtained compounds 3a-g.
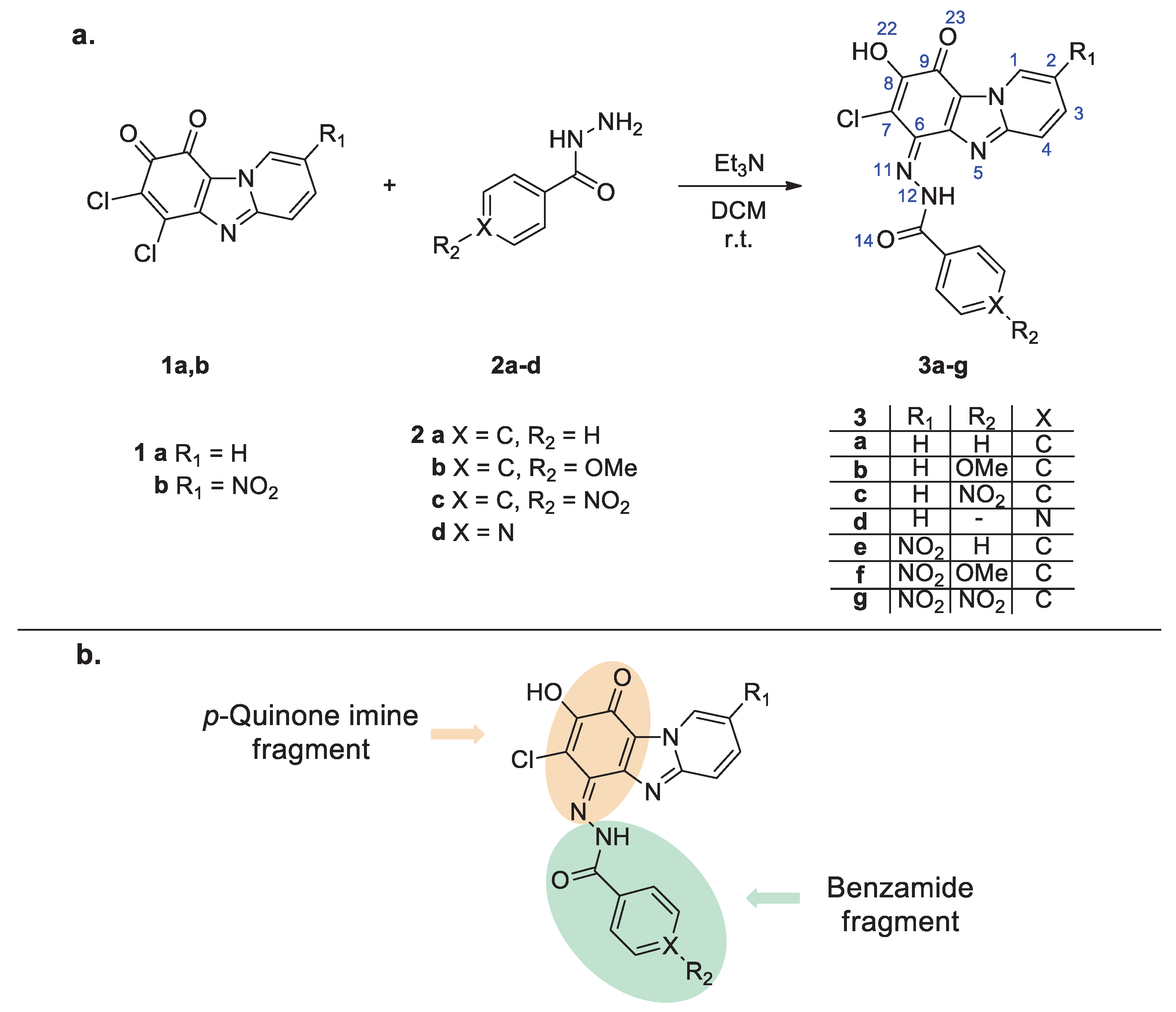
Figure 1.
(a) ORTEP diagram of compound 3a showing thermal ellipsoids at the 50% probability level; (b) Two mesomeric structures of compound 3a based on the bond distances in the crystal structure. Typical bond distances are listed according to the literature [24].
Figure 1.
(a) ORTEP diagram of compound 3a showing thermal ellipsoids at the 50% probability level; (b) Two mesomeric structures of compound 3a based on the bond distances in the crystal structure. Typical bond distances are listed according to the literature [24].
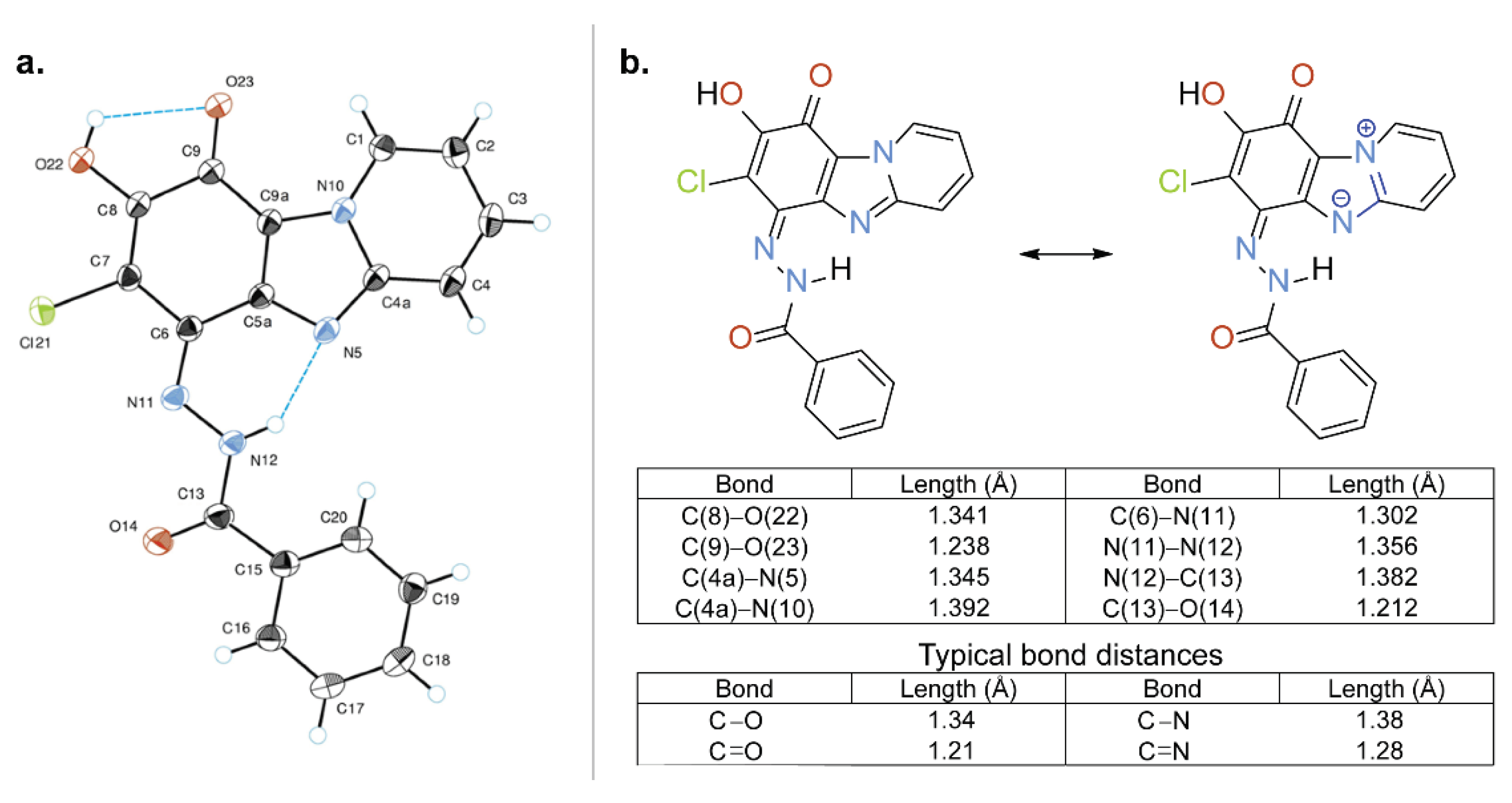
Figure 2.
(a) A dimer (highlighted) formed in the crystal structure of compound 3a. (b) Crystal packing of compound 3a along a axis. (c) Crystal packing of compound 3a along c axis.
Figure 2.
(a) A dimer (highlighted) formed in the crystal structure of compound 3a. (b) Crystal packing of compound 3a along a axis. (c) Crystal packing of compound 3a along c axis.
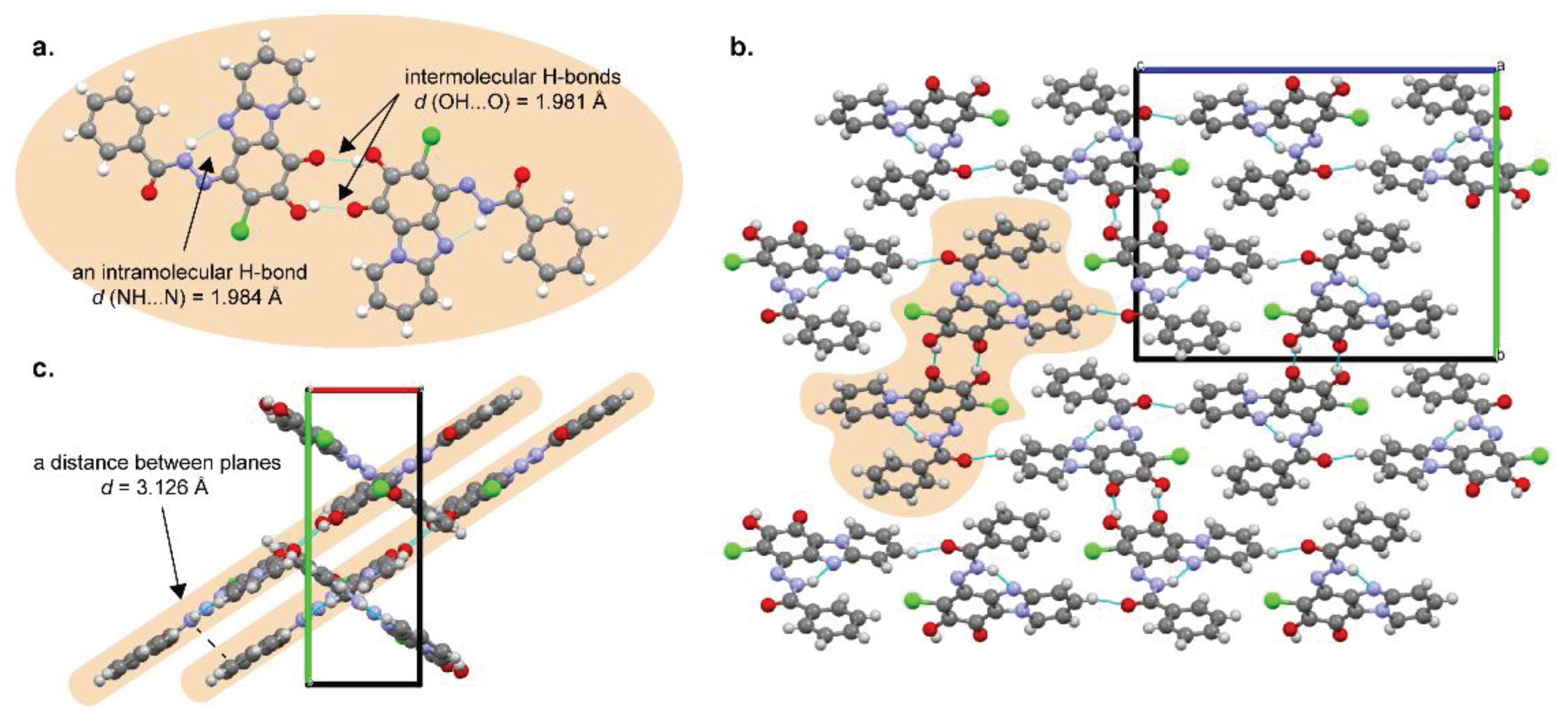
Figure 3.
An expansion of 1H NMR spectra of compound 3b in DMSO-d6 solution upon sequential addition of base (DBU) and acid (TFA).
Figure 3.
An expansion of 1H NMR spectra of compound 3b in DMSO-d6 solution upon sequential addition of base (DBU) and acid (TFA).
Figure 4.
1H NMR titration of compound 3b in DMSO-d6 solution with DBU (0 – 8 equivalents).
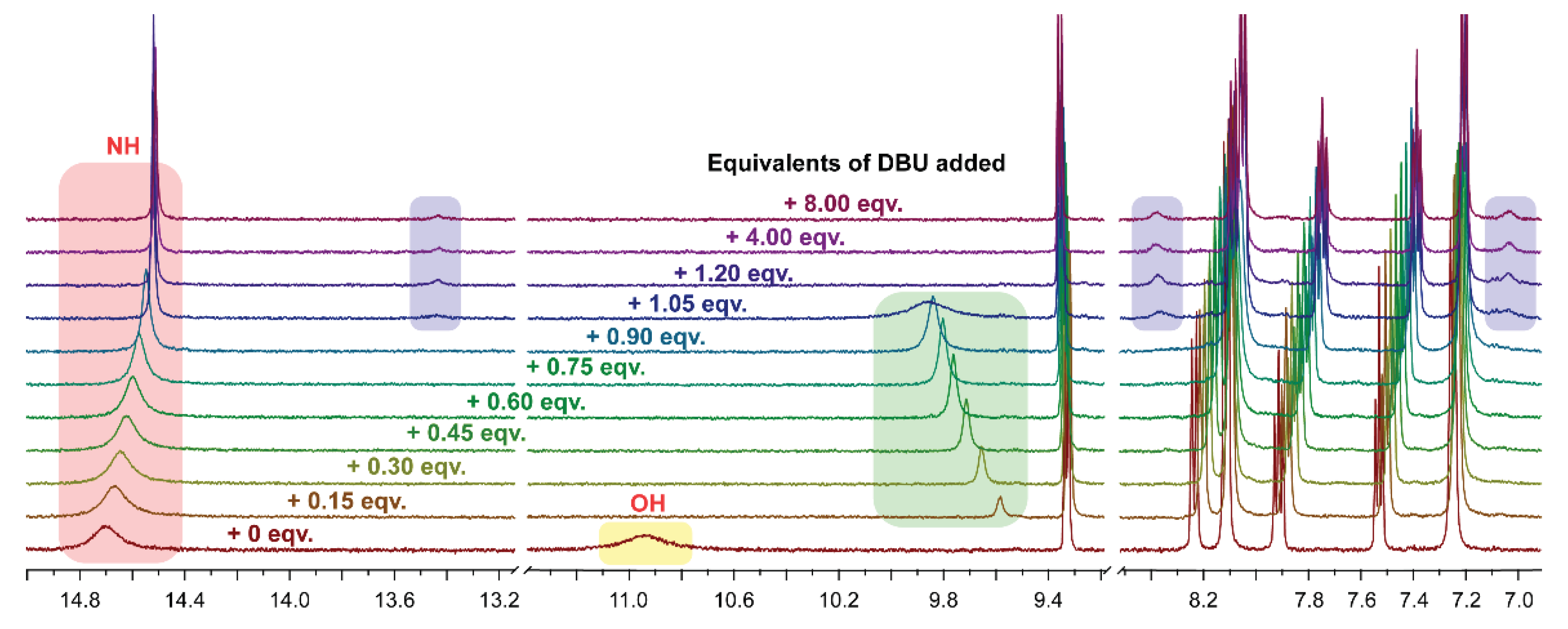
Figure 5.
(a) UV-Vis absorption spectra of neutral (in DMSO or DCM solution) and deprotonated (upon addition base to the DMSO or DCM solution) forms of compound 3a. (b) UV-Vis absorption spectra neutral (in DMSO solution) and deprotonated (upon addition a base to the DMSO solution) forms of compound 3a and 3c.
Figure 5.
(a) UV-Vis absorption spectra of neutral (in DMSO or DCM solution) and deprotonated (upon addition base to the DMSO or DCM solution) forms of compound 3a. (b) UV-Vis absorption spectra neutral (in DMSO solution) and deprotonated (upon addition a base to the DMSO solution) forms of compound 3a and 3c.
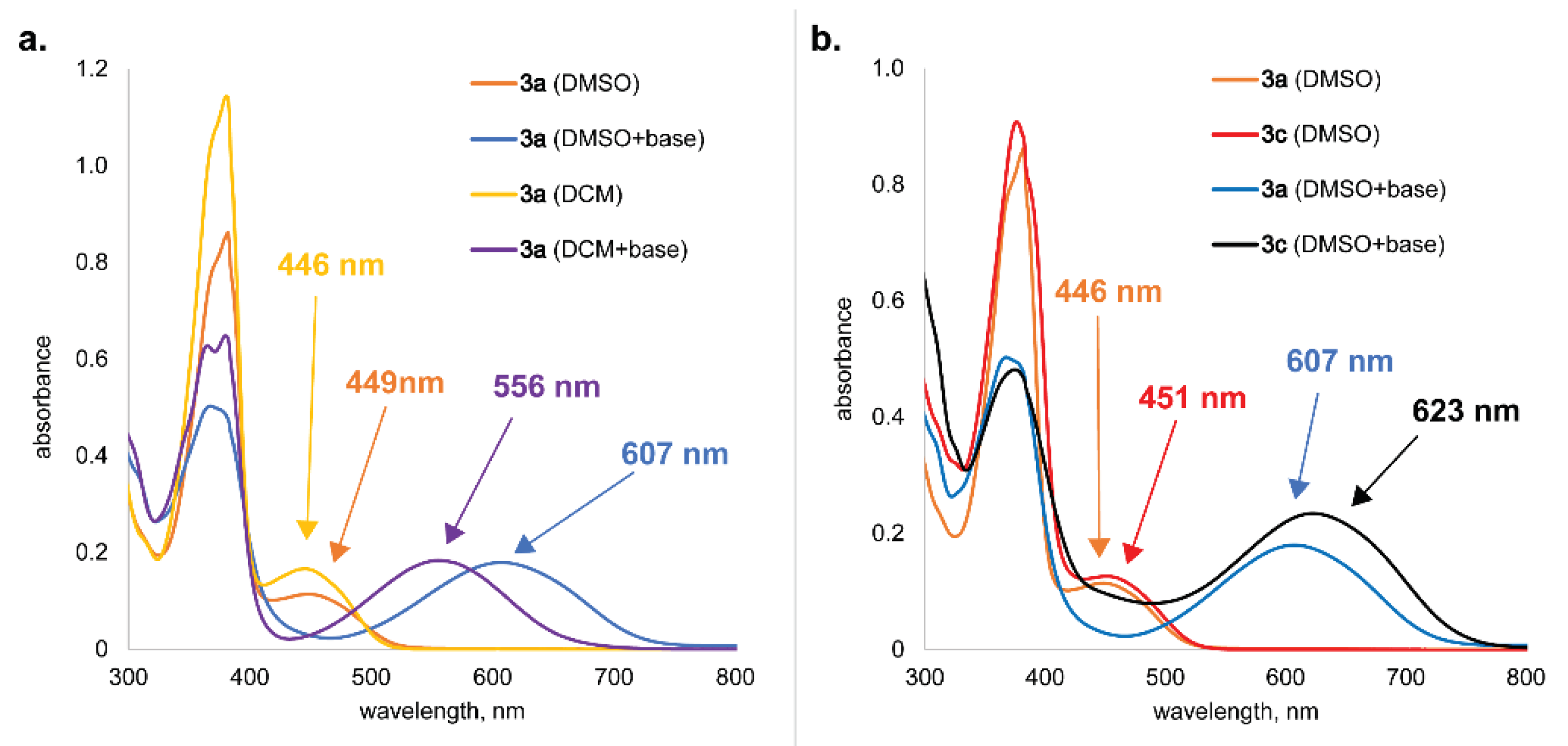
Figure 6.
CV results at varying scanning speeds for samples CM-1a, CM-3a and substrate in neutral (0.5 M K2SO4) electrolyte and in acidic (0.5 M H2SO4) electrolyte.
Figure 6.
CV results at varying scanning speeds for samples CM-1a, CM-3a and substrate in neutral (0.5 M K2SO4) electrolyte and in acidic (0.5 M H2SO4) electrolyte.
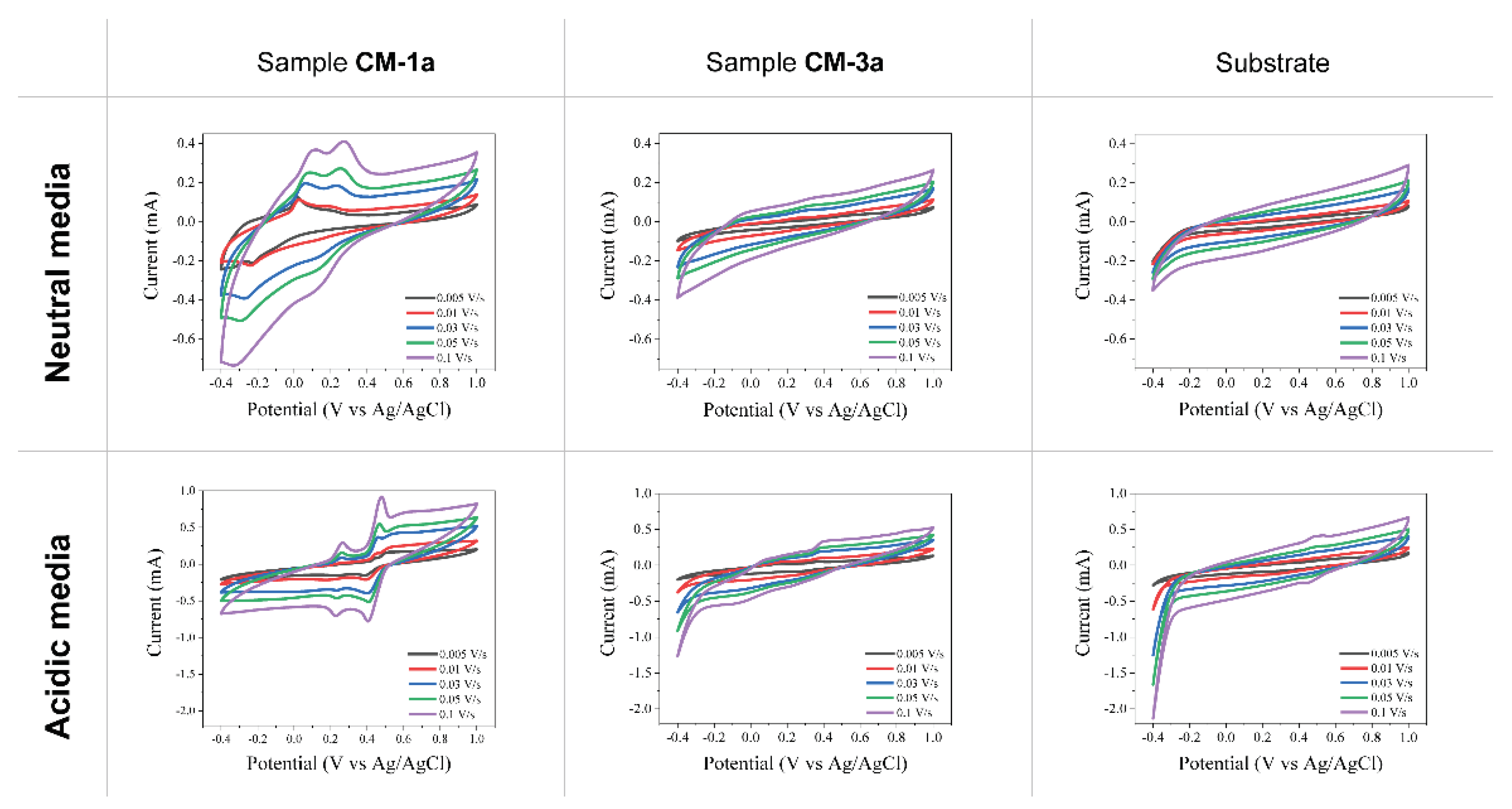
Figure 7.
Raman spectra of pure compounds 1a and 3a (green line), prepared cathodes (CM-1a and CM-3a) before (blue line) and after cycling them in acidic (red line) and neutral (grey line) electrolytes.
Figure 7.
Raman spectra of pure compounds 1a and 3a (green line), prepared cathodes (CM-1a and CM-3a) before (blue line) and after cycling them in acidic (red line) and neutral (grey line) electrolytes.
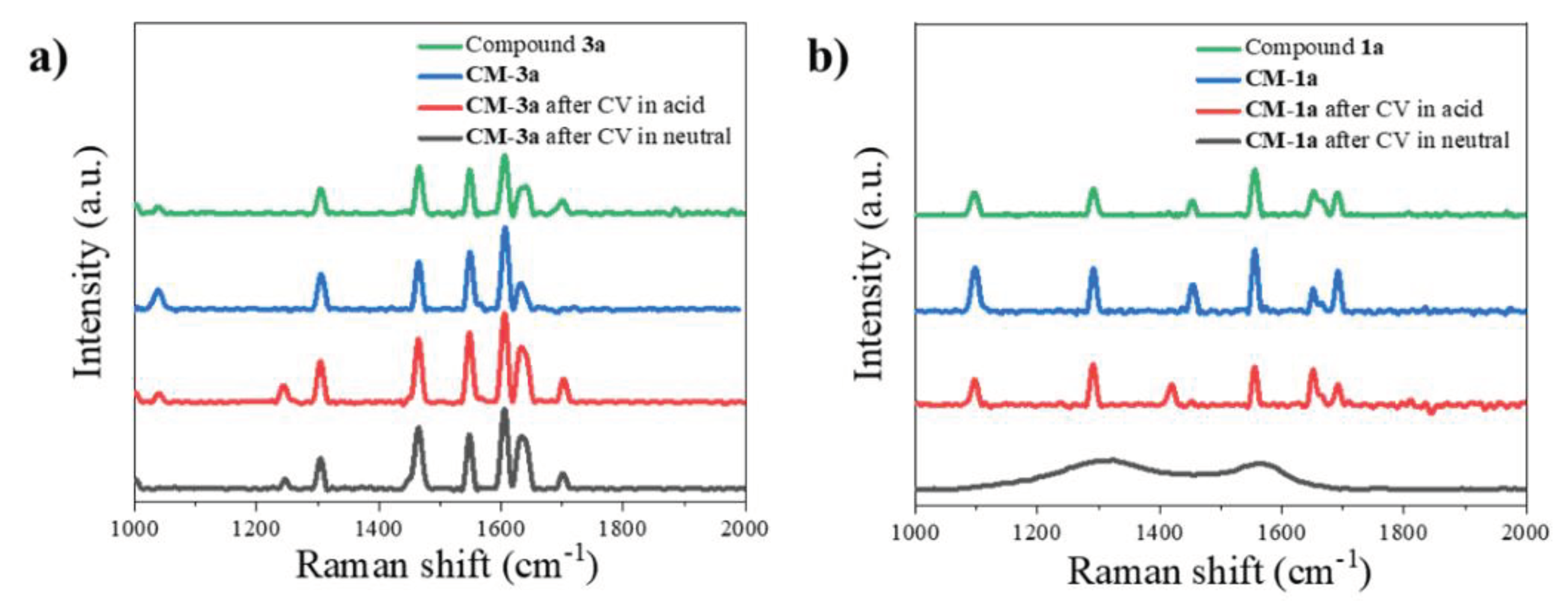
Figure 8.
Scanning electron microscopy images of samples CM-1a, CM-3a and substrate (coating without active material) before and after CV measurements in neutral and acidic electrolyte (magnification of x2500).
Figure 8.
Scanning electron microscopy images of samples CM-1a, CM-3a and substrate (coating without active material) before and after CV measurements in neutral and acidic electrolyte (magnification of x2500).


Table 1.
Crystal data and structures refinement details for compound 3a.
| Crystal parameter | Compound 3a |
| Empirical formula | C18H11ClN4O3 |
| Calculated density (g/cm3) | 1.556 |
| Formula Weight | 366.766 |
| Color | Red |
| Size/mm3 | 0.18×0.03×0.01 |
| Temperature/K | 150.0(1) |
| Crystal System | monoclinic |
| Space Group | P21/n |
| a/Å | 5.65994(5) |
| b/Å | 14.90948(19) |
| c/Å | 18.5815(2) |
| α/° | 90 |
| β/° | 93.1950(9) |
| γ/° | 90 |
| V/Å3 | 1565.60(3) |
| Wavelength/Å | 1.54184 |
| Radiation type | Cu Kα |
| Absorption coefficient (mm−1) | 2.419 |
| θmin/° | 3.8 |
| 2θmax/° | 155.0 |
| Measured reflections | 17875 |
| Number of independent reflections | 3322 |
| Reflections with I≥2σ(I) | 3089 |
| Rint | 0.0327 |
| Number of refined parameters | 243 |
| Restraints | 0 |
| Largest Peak | 0.3476 |
| Deepest Hole | -0.3143 |
| Goodness of fit | 1.0362 |
| wR2 (all data) | 0.0962 |
| wR2 | 0.0945 |
| R1 (all data) | 0.0367 |
| R1 | 0.0346 |
| CCDC deposition number | 2238663 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



