
Submitted:
07 March 2024
Posted:
08 March 2024
You are already at the latest version
Abstract
Previous studies showed that some lamellarin-resembling annelated azaheterocyclic carbaldehydes and related imino adducts, sharing the 1-phenyl-5,6-dihydropyrrolo[2,1-a]isoquinoline (1-Ph-DHPIQ) scaffold, are cytotoxic in tumor cells and may reverse multidrug resistance (MDR) mediated by P-glycoprotein (P-gp). Herein, we synthesized and evaluated for their antiproliferative activity in four tumor cell lines (RD, HCT116, HeLa, A549) and ability of inhibiting P-gp-mediated MDR some ten diversely substituted 1-Ph-DHPIQ derivatives bearing at position 2 carboxylate groups (COOH, COOEt), nitriles (CN) and Mannich bases (e.g., morpholinomethyl derivatives). Lipophilicity parametrization and molecular docking calculations improved the understanding of the structure-activity relationships in cytotoxicity and P-gp inhibition of the investigated 1-Ph-DHPIQs, which led us to disclose a novel Mannich base HCl salt (8b’), which proved to be cytotoxic to all the tested tumor cell lines in the low micromolar range (IC50 < 20 M) and to inhibit in-vitro the efflux pumps P-gp and MRP1 responsible for MDR, with IC50s of 0.45 and 12.1 M, respectively.
Keywords:
pyrrolo[2
; 1-a]isoquinolines
; Mannich bases
; cytotoxicity
; P-glycoprotein
; inhibition
; multidrug resistance reversal.
1. Introduction
Pyrrolo[2,1-a]isoquinoline is an azaheterocyclic structure recurring in natural alkaloids isolated from marine invertebrate or plants, which are endowed with diverse biological activities [1]. Among them, type I lamellarins, incorporating into their structure R-substituted (R = OH, OMe) 1-phenyl-5,6-dihydropyrrolo[2,1-a]isoquinoline (hereafter indicated as 1-Ph-DHPIQ, Figure 1), showed diverse biological activities, including anticancer and antiviral activities, as well as inhibition of the efflux pumps responsible for multidrug-resistance (MDR) [2]. In Figure 1, together with the general structure of lamellarins 1, the structures of synthetic bioactive 1-Ph-DHPIQ derivatives are shown, one of which (2) proved to be a potent topoisomerase I inhibitor [3] and the other (3), that merges the pharmacophore of tamoxifen (a well-known selective estrogen receptor modulator) with the 1-Ph-DHPIQ structure 2, acting as estrogen receptor (ER) modulator [4].
In previous studies, our research groups synthesized and tested for cytotoxicity against four tumor cells diverse R-substituted 1-arylpyrrolo[2,1-a]quinoline derivatives (4, Figure 1), along with 1-arylindolizines [5]. All the compounds held the aldehyde group at C2 and were tested as inhibitors of proliferation of rhabdomyosarcoma (RD), colon cancer (HCT116), adenocarcinoma of the cervix uterus (HeLa) and lung adenocarcinoma (A549). The most hydrophobic aldehyde derivatives, like 4a and 4c (data in Table 1 below), and some related imino adducts proved to be cytotoxic with IC50s mostly in the low μM range [5,6].
Interestingly, the 5,6-dehydro analog of 4c loses cytotoxicity (IC50s >> 100 μM), whereas several synthetic 1-Ph-DHPIQ-2-carbaldehydes were evaluated for their ability to inhibit P-glycoprotein (P-gp) and multidrug-resistance-associated protein-1 (MRP1) efflux pumps in MDCK-MDR1 (overexpressing P-gp protein) and MDCK-MRP1 (overexpressing MRP1 protein) cell lines and screened for their effects in drug combination assays with doxorubicin [7]. Compound 4c and closer analogs proved to be potent inhibitors of P-gp with IC50s < 0.5 μM and reversed MDR in tumor cells to doxorubicin even at low noncytotoxic concentrations. Data showed that lipophilicity of the substituents plays a role in increasing the P-gp inhibitory potency of these compounds. In addition, some Schiff bases of 1-Ph-DHPIQs were disclosed as hits addressing Alzheimer’s disease-related target proteins, such as cholinesterases (ChEs) and monoamine oxidases (MAOs) [6].
Herein, we focused on the evaluation of possible replacements of the electrophilic aldehyde group (and their imino adducts), synthesizing and screening in-vitro for their cytotoxicity in tumor cells (RD, HCT116, HeLa, A549), modulation of MDR efflux pumps (P-gp, MRP1) and ADME-related physicochemical properties (e.g., water solubility, lipophilicity) a number of R-substituted 1-Ph-DHPIQ derivatives bearing at C2 carboxylic groups (COOH, COOEt), nitrile (CN) and a morpholinomethyl moiety (as Mannich base). The carboxyl group (carboxylate, ester) and nitrile [8,9] were examined as electrophilic groups less reactive than the aldehyde carbonyl [10], whereas the morpholinomethyl derivative was synthesized for improving the aqueous solubility of the 1-Ph-DHPIQ derivative while maintaining the anticancer activity as demonstrated by others in several case studies [11]. These chemical transformations allowed us to gain new insights into the structure-activity relationships of this class of nature-inspired molecules.
2. Results and Discussion
2.1. Chemistry
The synthesis on new R1-3-substituted 1-Ph-DHPIQ derivatives (4b, 5a-b, 6a-b, 7), bearing diverse functional groups at C2 (X = CHO, CN, COOH, COOEt), was carried out via a domino reaction between 1-benzoyl-3,4-dihydroisoquinolines and electron-deficient alkenes (Scheme 1). As reported earlier [12], the synthesis of the aldehyde 4b, like the already reported 4a and 4c, was accomplished by reacting (3,4-dihydro-6,7-dimethoxyisoquinolin-1-yl)(4-methoxyphenyl)methanone with acrolein. Similarly, the carboxylic acids 5a and 5b were obtained by the reaction of the suitable 1-benzoyl-3,4-dihydroisoquinolines with p-nitrophenyl acrylate in 3,3,3-trifluoroethanol under microwave activation.
The carbonitriles 6a and 6b without the substituent R4 had been already synthesized from the suitable 1-benzoyl-3,4-dihydroisoquinolines in boiling trifluoroethanol [13]. The carboxymethyl ester 7 was prepared by the reaction of the isoquinoline in the presence of ZnO (10 mol%) in trifluoroethanol under microwave activation. Suitable 1-Ph-DHPIQ-2-carbaldehydes 4 were used as starting materials for synthesizing either further carbonitriles 6c and 6d, or the Mannich bases 8a and 8b (Scheme 1; R1-4 groups in Table 1). The one-pot reaction of aldehydes 4d (R1 = R2 = R3 = OEt; R4 = Me) and 4e (R1 = R2 = R3 = OEt; R4 = Ph) with hydroxylamine hydrochloride in the presence of sodium acetate in ethanol, followed by boiling in acetic anhydride, led to the formation of carbonitriles 6c and 6d, respectively. The 2-morpholinomethyl Mannich bases 8a and 8b were synthesized by one pot reaction of aldehydes 4b and 4c, respectively, with morpholine in acetonitrile followed by reduction of the iminium intermediate with NaBH4 in methanol. To increase the aqueous solubility of 8b, it was converted to the corresponding hydrochloride salt 8b’.
2.2. Biological Evaluation
2.2.1. In Vitro Cytotoxicity Screening
The newly synthesized DHPIQs, along with the previously reported derivatives 4a, 4c, 6a and 6b, with camptothecin and doxorubicin taken as positive controls, were tested at the maximum concentration of 100 μM for their antiproliferative activity, using MTT assay on four cancer cell lines, namely RD, HCT116, HeLa, A549. Due to solubility issues, the more lipophilic Mannich base 8b was tested only as hydrochloride salt 8b’. Compounds showing more than 50% inhibition were tested at lower scalar concentrations and IC50s calculated by interpolation of the dose-response curves (Table 1).
As shown by the inhibition data, among the newly synthesized compounds only 8a and 8b’ bearing the 2-morpholinomethyl group achieved noteworthy antiproliferative activity with IC50s << 100 μM. Of the two Mannich bases, the more lipophilic 8b’ achieved 50% antiproliferative activity in all four cancer lines in the low micromolar range (IC50s < 21 μM). The newly synthesized DHPIQ-2-aldehyde derivative 4b proved to be less active than the two more lipophilic congeners 4a and 4c.
It appears clear that the replacement of the 2-CHO group (or one of its adducts or derivatives, like the Mannich base in this study) with less electrophilic group, such as CN (6a-d), COOH (5a-b) and COOEt (7), induces a loss of activity.
2.2.2. P-gp and MRP1 Inhibitory Potency
The P-gp inhibitory potency of eight DHPIQs was evaluated by measuring the transport inhibition of calcein-AM, a profluorescent P-gp substrate, in the MDCK-MDR1 cell line overexpressing P-gp. Moreover, to evaluate the MRP1 inhibitor activity, the same compounds were screened in MDCK-MRP1 cells overexpressing MRP1. MC18 [14,15] and verapamil [16] were used as positive control for P-gp and MRP1 efflux pumps, respectively. The IC50 values (Table 2) show that five out of eight compounds tested (i.e., 5b, 6a, 6b, 7, 8a, 8b’), regardless the functional group at the C2 position, inhibited P-gp efflux pump with a potency in the submicromolar range of concentrations (IC50s < 0.5 μM).
Within the limits of the examined substituents R2 and R3 onto the 1-phenyl group, it can be inferred that OMe compared to Cl in para position does improve the inhibition activity toward both the efflux pumps, whereas the EtO substituents in meta and para do enhance potency toward P-gp (but not MRP1). Indeed, as for as P-gp/MRP1 selectivity is concerned, except for the tetraethoxy-substituted 1-Ph-DHPIQ carboxylic acid (5b) and nitrile (6b) which achieved a selectivity ratio toward P-gp of more than two orders of magnitude, the other compounds proved to be unselective (4a) or moderately P-gp-selective, with the tetraethoxy 1-Ph-DHPIQ Mannich base (8b’) approximately thirty-fold more active towards P-gp.
2.2.3. Binding Affinity to Human Serum Albumin (HSA)
The interactions between compounds and HSA were evaluated by surface plasmon resonance (SPR), using warfarin, a well-known strong HSA binder, as a standard reference [17,18,19]. The assessment of the binding affinity to HSA can help to estimate the bioavailability in the early stage of drug design and development. By plotting the response at equilibrium, which were quite rapidly reached for each tested compounds at any increasing concentration, the KD values were determined. Considering the average physiological HSA concentration in plasma (680 μM), at 10 μM concentration compounds 4-7 can be predicted highly bound to albumin, whilst the morpholinomethyl Mannich bases can be estimated HAS-bound for less than 40%.
2.3. Structure Activity Relationships
2.3.1. Solubility-Related Parameters
The solubility of the investigated compounds was evaluated in PBS (50 mM, pH 7.4, 0.15 M KCl) at 25°C using RP-HPLC as the analytical method (Table 2). The ester 7 and the Mannich base 8a were preliminarily monitored by RP-HPLC for the hydrolytic stability at pH 7.4 (50 mM PBS, 0.15 M KCl), and at pH 2 (0.01 M HClaq, 0.15 M KCl) as well, and found stable over 4 hours at 25 °C in both conditions. All the compounds proved to be poorly soluble in buffered water solution at neutral pH, even ‘practically insoluble’ (< 0.1 mg/mL) according to the solubility categorization adopted by US and European Pharmacopoeia [20]. Nevertheless, the transformation of the aldehydes 4 into ethyl carboxylate ester 7 or morpholinomethyl basic derivatives 8 significantly improves the water solubility of the 1-Ph-DHPIQ analogs. In particular, the transformation of the DHPIQ-2-aldehyde via Mannich reaction (8a,b) opens the road to future pharmaceutical development of new water-soluble antiproliferative DHPIQ derivatives endowed with the ability to inhibit P-gp and reverse MDR.
To quantitate the effect of hydrophobicity on the biological activity of the examined compounds, a relative lipophilicity scale was determined by RP-HPLC [21]. The polycratic capacity factor (log k’w) for each compound in Table 2 was experimentally determined (details in the experimental section) and compared with 1-octanol–water partition coefficient calculated (Clog P) with ACDLabs software (release 10.0; Advanced Chemistry Development, Inc., Toronto, Canada). Log k’w values are quite well correlated with Clog Ps for the whole series (r2 = 0.791). However, omitting from the regression the two Mannich bases 8a and 8b which, due to their basicity (pKas about 6.9, estimated by ACDLabs software), should be predominantly in the protonated forms at pH 4.7 (i.e., the pH of the aqueous component of the mobile phase in RP-HPLC), the correlation slightly improves (r2 = 0.859), providing a linear equation with a slope equals to +0.8 and an intercept of about -0.1.
No statistically significant linear or nonlinear correlation was derived between the measured biological activities and the lipophilicity descriptors determined or calculated in this study. As observed above, lipophilicity plays a secondary role in modulating the anticancer activity of the DHPIQ derivatives (Table 1). The replacement of the electrophilic and electron-withdrawing CHO group in 4a,c with a less reactive electrophilic but more electron-withdrawing CN in 6a-d resulted in a drop of cytotoxic activity, regardless of the hydrophobicity of the substituents onto the 1-Ph-DHPIQ scaffold. A drop of activity was also observed by replacing 2-CHO with COOH (5) and COOEt (7). Instead, with derivatives of the aldehyde group, such as the Mannich bases 8, an increase in antiproliferative activity was achieved on all the tumor cells tested. Between the two Mannich bases, the 2-4-fold greater activity of 8b’ compared to 8a apparently reflects the difference in lipophilicity of approximately 1.8 log P units. Similarly, the 3-4-fold greater cytotoxicity of 4a compared to 4b may be related to the difference of about one log P unit between the two aldehydes.
Even the inhibition data of the MDR-related efflux pumps P-gp and MRP1 (Table 2) did not significantly correlate with the lipophilicity parameter alone (either log k’w or Clog P). First, the inhibitory potency on P-gp/MRP1 appeared to be not affected at all by the properties (electrophilicity, lipophilicity, bulkiness/polarizability) of the X functional group (CHO, CN, COOH, COOEt, MM). Sometimes the effect of lipophilicity is apparent from pairwise comparisons of substituents onto the 1-Ph-DHPIQ scaffold, and such an effect is manifested differently on P-gp (direct correlation) and MRP1 (inverse correlation). For instance, all the compounds bearing as R1 (pos. 8 and 9), R2 (pos. 4′) and R3 (pos. 3′) the more lipophilic OEt groups, instead of OMe or Cl (as R1 and R2), achieved the higher inhibition potency against P-gp (IC50s < 0.5 μM). Besides these ‘local’ hydrophobic effects of the substituents, it is worth highlighting that the most water-soluble molecules 7 and 8a,b proved to be also the most potent P-gp inhibitors.
Modeling studies helped to understand the probable binding mode to P-gp binding site(s) of the 1-Ph-DHPIQ-containing ligands and to rationalize their IC50 data. The particular molecular shape of ligands in combination with some of their structural features emerged as critical pinching elements that tightly hamper the target molecular surface exposed to ligands.
2.3.2. Molecular Docking Calculation
Molecular modelling studies on P-gp binders are nowadays feasible thanks to many of performed Cryo-EM solved structures. We felled confident to achieve deep insights into the DHPIQ ligands by docking to the so called ‘inward-facing’ helices binding site, hampering the ‘outward-facing’ shift of the protein, that is the basis of the transmembrane domains flipping leading to the pulling out of the xenobiotics. The binding modes of compounds 4a-b, 5b, 6a-b, 7, 8a and 8b produced a sketch for a plausible interaction pattern of this type of ligands with the P-gp. As perceivable from Figure 2, the crevice located between the two six helices domain typical of the class of ABCB1 binding cassette subfamily is largely occupied by the whole molecular scaffold of the inhibitors able then to lock the in-out facing of the target protein.
Having proved this instance, additional insights were gained from ligand-residues interactions analysis for the most active compounds 7 and 8b which suggest the importance a cluster of aromatic residues. Indeed, in both the cases it is relevant π–π stacking of the diethoxy-phenyl moieties with Phe336 and Phe983, engaging at the same time van der Waals contacts enrolling the Phe732, Phe978 and Leu975. Similar evidence is also gained by the two ligands pendants close to Tyr950. It is worth noting the hydrogen bonds involving sidechains of Tyr310 and Tyr95, strongly assisting the binding of both 7 and 8b (Figure 3).
As an additional figure of merit, a significant (r2 = 0.716; r2 = 0.844, omitting from regression 6b) linear relationship between the estimated free energy of binding and pIC50 was obtained considering the dockings of all the inhibitors (Table 3).
3. Materials and Methods
3.1. Chemistry
All reagents and solvents were purchased from Merck, J.T. Baker, or Sigma-Aldrich Chemical Co. and, unless specified, used without further purification. The melting points (mp) of all the compounds were determined on a SMELTING POINT 10 apparatus in open capillaries. IR spectra were recorded on an Infralum FT-801 FTIR spectrometer. The sample was analyzed as solid KBr disk, and the most important frequencies are expressed in cm-1. 1H and 13C NMR spectra were recorded in chloroform-d (CDCl3) or dimethylsulfoxide-d6 (DMSO-d6) solutions at 25°C, with a 600 MHz NMR spectrometer. Peak positions are given in parts per million (ppm, δ) referenced to the appropriate solvent residual peak and signal multiplicities are collected as: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), br.s. (broad singlet), m (multiplet). MALDI mass spectra were recorded using Bruker autoflex speed instrument operating in positive reflectron mode. Elemental analyses were carried out on Euro Vector EA-3000 Elemental Analyzer for C, H and N; experimental data agreed to within 0.4 % of the theoretical values.
The synthetic procedures of compounds 4a, 4c-e [12] and 6a-b [13] were recently described. Compounds 4b, 5a-b, 6c-d, 7, 8a-b and 8b’ were synthesized according to the following procedures.
3.1.1. Synthesis of (1-(4-Methoxyphenyl)-8,9-dimethoxy-5,6-dihydropyrrolo[2,1-a]isoquinoline-2-carbaldehyde (4b)
Acrolein (118 mg, 2.10 mmol) was added to a solution of (6,7-dimethoxy-3,4-dihydroisoquinolin-1-yl)(4-methoxyphenyl)methanone (400 mg, 1.22 mmol) in trifluoroethanol (10 ml). The reaction was stirred at 40oC for three hours. The reaction progress was monitored by TLC (sorbfil, EtOAc/hexane, 1:2). Than the solvent was removed under vacuum; the residue was crystallized from diethyl ether to afford compound 4b as a beige powder (363 mg, 82%): mp 132-133°C; 1H NMR (600 MHz, CDCl3,): δ = 3.04 (t, 2H, J = 6.3 Hz, 6-CH2), 3.38 (s, 3H, ОCH3), 3.83 (s, 3H, ОCH3), 3.85 (s, 3H, ОCH3), 4.11 (t, 2H, J = 6.3 Hz, 5-CH2), 6.59 (s, 1H, 7-H), 6.68 (s, 1H, 10-H), 6.97 (d, 2H, J = 8.6Hz, H-Ar), 7.35 (d, 2H, J = 8.6 Hz, H-Ar), 7.38 (s, 1H, 3-Н), 9.62 (s, 1Н, CHO); 13C NMR (150 MHz, CDCl3) δ: 29.1, 45.3, 55.2, 55.3, 55.9, 107.6, 111.1, 113.9 (2C), 121.2, 121.5, 124.2, 124.2, 125.0, 126.2, 127.2, 131.9 (2C), 147.6, 147.6, 159.0, 186.5; MS (LCMS) m/z = 364 [M+Н]+; Anal. calcd for C22H21NO4 (%): C 72.71, H 5.82, N 3.85, found: C 72.83, H 5.63, N 3.93.
3.1.2. Synthesis of 1-Aryl-5,6-dihydropyrrolo[2,1-a]isoquinoline-2-carboxylic acids 5a-b
4-Nitrophenylacrylate (1.20 mmol) was added to a solution of corresponding 1-aroylisoquinolines (1.0 mmol) in trifluoroethanol (5 ml). The reaction proceeded under microwave activation for 1 hour at 140oC. The progress of the reaction was monitored by TLC (sorbfil, EtOAc/hexane, 1:1). The solvent was evaporated, the residue was crystallized from ether.
1-(4-Chlorophenyl)-8,9-dimethoxy-5,6-dihydropyrrolo[2,1-а]isoquinoline-2-carboxylic acid (5a): Yellow powder (345 mg, 90%): mp 210–215°C; 1H NMR (600 MHz, CDCl3): δ = 3.03 (t, 2H, J = 6.6Hz, 6-CH2), 3.34 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 4.10 (t, 2H, J = 6.6Hz, 5-CH2), 6.36 (s, 1H, 7-H), 6.67 (s, 1H, 10-H), 7.33 (d, 2H, J = 8.6Hz, H-Ar), 7.38 (d, 2H, J = 8.6Hz, H-Ar), 7.44 (s, 1Н, 3-H); 13C NMR (150 MHz, CDCl3): δ = 29.2, 45.2, 55.2, 56.0, 107.7, 111.2, 112.3, 120.3, 120.9, 124.3, 126.7, 127.8, 128.5 (2C), 132.2 (2C), 133.2, 134.1, 147.7, 147.8, 162.4. MS (LCMS) m/z = 384 [M+Н]+; Anal. calcd for C21H18ClNO4, (%): C, 65.71; H, 4.73; N, 3.65, found: C, 65.98; H, 4.94; N, 3.83.
1-(3,4-Diethoxyphenyl)-8,9-diethoxy-5,6-dihydropyrrolo[2,1-a]isoquinoline-2-carboxylic acid (5b): Beige powder (372 mg, 80%): mp 170–172°C; 1H NMR (600 MHz, CDCl3): δ = 1.16 (t, 3Н, J = 7.1Hz, OCH2CH3), 1.37–1.42 (m, 6H, OCH2CH3), 1.45 (t, 3Н, J = 7.1Hz, OCH2CH3), 3.00 (t, 2H, J = 6.6Hz, 6-CH2), 3.56 (q, 2H, J = 7.1Hz, OCH2CH3), 4.01–4.06 (m, 4H, OCH2CH3), 4.09 (t, 2H, J = 6.6Hz, 5-CH2), 4.11 (q, 2H, J = 7.1Hz, OCH2CH3), 6.53 (s, 1H, 7-H), 6.66 (s, 1H, 10-H), 6.88–6.92 (m, 3H, H-Ar), 7.41 (s, 1H, 3-H); 13C NMR (150 MHz, CDCl3): δ = 14.6, 14.9 (2C), 15.0, 29.2, 45.3, 63.8, 64.5, 64.7, 64.8, 109.3, 112.5, 113.3, 113.8, 116.1, 121.4, 121.5, 123.0, 123.9, 126.4, 127.7, 128.3, 147.2, 147.3, 148.0, 148.9, 162.5; MS (LCMS) m/z = 466 [M+Н]+; Anal. calcd for C27H31NO6, (%): C, 69.66; H, 6.71; N, 3.01, found: C, 70.23; H, 6.88; N, 3.12.
3.1.3. Synthesis of Carbonitriles 6c,d.
Nitriles 6c,d were obtained in two stages. In the first step, compounds 4d or 4e (0.3 mmol) were dissolved in 7 ml of ethanol. Hydroxylamine hydrochloride (2.0 mmol) and sodium acetate (3.0 mmol) were added to the resulting solution, the mixtures were refluxed for 14 hours. The reaction progress was monitored by TLC (sorbfil, EtOAc/hexane, 1:2). The solvent was removed under vacuum; the residue was crystallized from ether to afford oximes as a beige powder. After filtration and drying in the second stage, the obtained oximes were boiled in 3 ml of acetic anhydride until the initial spot disappeared on TLC. Ice was added to the mixture, sodium bicarbonate was added to pH 8, and the mixture was extracted with EtOAc (3×10 ml). After removing the solvent, the residue was recrystallized from EtOAc/hexane.
1-(3,4-Diethoxyphenyl)-8,9-diethoxy-3-methyl-5,6-dihydropyrrolo[2,1-a]isoquinoline-2-carbonitrile (6c): Beige powder (46 mg, 50%): mp 270–272°C; 1H NMR (600 MHz, CDCl3): δ = 1.20 (t, 3Н, J = 7.1Hz, OCH2CH3), 1.40 – 1.46 (m, 9H, OCH2CH3), 2.42 (s, 3H, CH3), 2.99 (t, 2H, J = 6.6Hz, 6-CH2), 3.61 (q, 2H, J = 7.1Hz, OCH2CH3), 3.92 (t, 2H, J = 6.6Hz, 5-CH2), 4.04 – 4.08 (m, 4H, OCH2CH3), 4.12 (q, 2H, J = 7.1Hz, OCH2CH3), 6.68 (s, 1H, 7-H), 6.80 (s, 1H, 10-H), 6.90 (d, 1H, J = 8.6Hz, H-Ar), 6.98 (br.s, 2Н, H-Ar); 13C NMR (150 MHz, CDCl3): δ = 11.3, 14.7 (2C), 14.9 (2C), 29.0, 41.8, 64.1, 64.6, 64.7, 64.8, 93.5, 109.4, 113.1, 114.0, 114.9, 117.2, 121.2, 121.4, 122.2 (2C), 123.9, 125.3, 126.7, 136.2, 147.5, 148.1, 149.0; MS (LCMS) m/z = 461 [M+Н]+; Anal. calcd for C28H32N2O4, (%): C, 73.02; H, 7.00; N, 6.08, found: C, 72.98; H, 6.93; N, 5.81.
1-(3,4- Diethoxyphenyl)-8,9- diethoxy -3-phenyl-5,6- dihydropyrrolo [2,1-a]isoquinoline-2-carbonitrile (6d): Brown powder (68 mg, 65%): mp 271–273°C; 1H NMR (600 MHz, CDCl3): δ = 1.22 (t, 3Н, J = 7.1Hz, OCH2CH3), 1.41 – 1.47 (m, 9H, OCH2CH3), 2.95 (t, 2H, J = 6.6Hz, 6-CH2), 3.64 (q, 2H, J = 7.1Hz, OCH2CH3), 4.06 (t, 2H, J = 6.6Hz, 5-CH2), 4.07 (q, 4H, J = 7.1Hz, OCH2CH3), 4.14 (q, 2H, J = 7.1Hz, OCH2CH3), 6.70 (s, 1H, 7-H), 6.84 (s, 1H, 10-H), 6.94 (d, 1H, J = 8.6Hz, H-Ar), 7.04 (d, 2H, J = 6.1Hz, H-Ar), 7.43–7.45 (m, 1H, H-Ar), 7.49-7.51 (m, 4Н, H-Ar); 13C NMR (150 MHz, CDCl3): δ = 14.7, 14.9 (2C), 29.3, 43.2, 64.1, 64.6, 64.7, 64.8, 94.2, 109.9, 112.9, 114.0, 115.1, 117.2, 121.0, 122.4 (2C), 122.9, 124.9, 126.3, 126.5, 128.9 (2C), 129.0, 129.1, 129.6 (2C), 139.3, 147.4, 147.7, 148.3, 149.1; MS (LCMS) m/z = 523 [M+Н]+; Anal. calcd for C33H34N2O4, (%): C, 75.84; H, 6.56; N, 5.36, found: C, 75.91; H, 6.86; N, 5.42.
3.1.4. Synthesis of Ethyl 1-(3,4-Diethoxyphenyl)-8,9-diethoxy-5,6-dihydro pyrrolo[2,1-a]isoquinoline-2-carboxylate (7)
Ethyl acrylate (150 mg, 1.5 mmol) and ZnO (8 mg, 10 mol%) were added to a solution of (6,7-diethoxy-3,4-dihydroisoquinolin-1-yl)(3,4-diethoxyphenyl)methanone (411 mg, 1.0 mmol) in trifluoroethanol (10 ml). The reaction was carried out at 140°C for 30 min. The progress of the reaction was monitored by TLC (sorbfil, EtOAc/hexane, 1:1). The solvent was removed; the residue was treated with sodium acetate solution and extracted with EtOAc (3 x 10 ml). After removal of the solvent, 5 ml of toluene was added and distilled to dryness to get rid of unreacted ethyl acrylate. The residue was crystallized from ether. White powder (296 mg, 60%): mp 170–172oC; 1H NMR (600 MHz, CDCl3): δ = 1.12 (t, 3Н, J = 7.1Hz, OCH2CH3), 1.15 (t, 3Н, J = 6.1Hz, OCH2CH3), 1.37–1.42 (m, 6H, OCH2CH3), 1.45 (t, 3Н, J = 7.1Hz, OCH2CH3), 2.98 (t, 2H, J = 6.6Hz, 6-CH2), 3.56 (q, 2H, J = 7.1Hz, OCH2CH3), 4.03 (q, 2H, J = 7.1Hz, OCH2CH3), 4.05–4.08 (m, 4H, 5-CH2, OCH2CH3), 4.11 (q, 4H, J = 7.1Hz, OCH2CH3), 6.53 (s, 1H, 7-H), 6.56 (s, 1H, 10-H), 6.88–6.90 (m, 3H, H-Ar), 7.34 (s, 1H, 3-H); 13C NMR (150 MHz, CDCl3): δ = 14.2, 14.8 (2C), 14.9 (2C), 28.1, 43.7, 60.2, 64.9, 65.7, 65.8, 65.9, 105.6, 112.0, 113.2, 113.9, 116.5, 120.4, 120.7, 126.1, 126.9, 129.2, 132.4, 132.5, 146.7, 148.9, 149.0, 152.1, 161.9; MS (LCMS) m/z = 494 [M+Н]+; Anal. calcd for C29H35NO6, (%): C, 70.57; H, 7.15; N, 2.84, found: C, 70.64; H, 6.95; N, 3.12.
3.1.5. Synthesis of 2-(Morpholin-4-yl-methyl)-5,6-dihydropyrrolo[2,1-a]isoquinolines 8a,b.
Morpholine (1.34 mmol) was added to a solution of pyrrolo[2,1-a]isoquinoline-2-carbaldehydes 4b,c (0.67 mmol) in acetonitrile (15 ml). The mixture was boiled for 20 hours. Then acetonitrile was replaced by methanol, sodium borohydride (2.68 mmol) was added in portions, and the mixture was boiled for 1 h. Then the reaction mass was cooled, and the solvent was evaporated. A saturated sodium bicarbonate solution (30 ml) was added to the dry residue and extracted with EtOAc (3×15 ml). The solvent was evaporated, compound 8a was isolated as oils, compound 8b was crystallized from a mixture of diethyl ether and EtOAc.
8,9-Dimethoxy-1-(4-methoxyphenyl)-2-(morpholin-4-ylmethyl)-5,6-dihydropyrrolo[2,1-a]isoquinoline (8a): Yellow oil (154 mg, 53%); 1H NMR (600 MHz, CDCl3): δ = 2.36 – 2.45 (m, 4H, CH2 (morpholinyl)), 2.99 (t, 2H, J = 6.6Hz, 6-CН2), 3.27 (s, 2H, CH2N), 3.36 (s, 3H, OCH3), 3.67 (t, 4H, J = 4.5Hz, CH2 (morpholinyl)), 3.82 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 4.01 (t, 2H, J = 6.6Hz, 5-CН2), 6.59 (s, 1H, 7-H), 6.64 (s, 1H, 10-H), 6.65 (s, 1H, 3-H), 6.92 (d, 2H, J = 8.6Hz, H-Ar), 7.36 (d, 2H, J = 8.6Hz, H-Ar); 13C NMR (150 MHz, CDCl3): δ = 29.3, 42.1, 47.8, 53.1 (2C), 54.5, 55.1, 55.6, 66.5, 66.6, 105.5, 111.4, 111.7, 112.8, 117.4, 122.8, 123.2, 123.9, 124.4, 125.4 (2C), 127.9, 132.3, 148.0, 148.9, 160.2; MS (LCMS) m/z = 435 [M+Н]+; Anal. calcd for C26H30N2O4, (%): C, 71.87; H, 6.96; N 6.45, found: C, 71.95; H, 7.03; N, 6.54.
1-(3,4-Diethoxyphenyl)-8,9-diethoxy-2-(morpholin-4-ylmethyl)-5,6-dihydropyrrolo[2,1-a]isoquinoline (8b): White powder (223 mg, 64%): mp 72-73oC; 1H NMR (600 MHz, CDCl3): δ = 1.17 (t, 3H, J = 7.1Hz, OCH2CH3), 1.40 – 1.43 (m, 6H, OCH2CH3), 1.46 (t, 3H, J = 7.1Hz, OCH2CH3), 2.35 – 2.45 (m, 4H, CH2 (morpholinyl), 2.97 (t, 2H, J = 6.2 Hz, 6-CН2), 3.26 (br.s., 2H, CH2N), 3.58 (q, 2H, J = 7.1Hz, OCH2CH3), 3.64 – 3.67 (m, 4H, CH2 (morpholinyl), 3.99 (t, 2H, J = 6.2Hz, 5-CН2), 4.02 – 4.08 (m, 4H, OCH2CH3), 4.11 (q, 2H, J = 7.1Hz, OCH2CH3), 6.62 (s, 1H, 7-H), 6.65 (s, 1H, 10-H), 6.67 (s, 1H, H-Ar), 6.88 (d, 1H, J = 8.1Hz, H-Ar), 6.92 (dd, 1H, J = 1.5, 8.1Hz, H-Ar), 7.07 (s, 1H, 3-H); 13C NMR (150 MHz, CDCl3): δ = 14.5, 15.2 (2C), 15.4, 29.2, 42.1, 46.9, 53.1, 53.2, 64.9, 65.0, 65.1, 65.8, 66.5 (2C), 110.1, 110.7, 113.3, 113.7, 117.0, 118.1, 123.1, 123.7, 124.4, 125.3, 125.9, 133.1, 148.5, 148.9, 149.0, 150.7; MS (LCMS) m/z = 521 [M+Н]+. Anal. calcd for C31H40N2O5, (%): C, 71.51; H, 7.74; N, 5.38, found: C, 71.33; H, 7.92; N, 5.21.
3.1.6. Synthesis of 4-{[1-(3,4-Diethoxyphenyl)-8,9-diethoxy-5,6-dihydropyrrolo[2,1-a]isoquinolin-2-yl]methyl}morpholin-4-ium chloride (8b’)
Concentrated hydrochloric acid was added dropwise to a solution of compound 8b (52 mg, 0.10 mmol) in DCM (2 ml) until pH 2. The solvent was removed and the residue was crystallized with ether to afford compound 8b’ as a white powder (49 mg, 87%): mp 90-92oC; 1H NMR (600 MHz, DMSO-d6): δ = 1.14 (t, 3H, J = 7.1Hz, OCH2CH3), 1.40 – 1.43 (m, 6H, OCH2CH3), 1.48 (t, 3H, J = 7.1Hz, OCH2CH3), 1.77 – 1.80 (m, 2H, CH2 (morpholinyl)), 2.51 – 2.52 (m, 2H, CH2 (morpholinyl)), 2.98 (br.s., 2H, CH2N), 3.17 – 3.20 (m, 2H, CH2 (morpholinyl)), 3.56 (q, 2H, J = 7.1Hz, OCH2CH3), 3.78 – 3.82 (m, 2H, 6-CН2), 3.98 – 4.07 (m, 8H, OCH2CH3, 5-CН2, CH2 (morpholinyl)), 4.11 (q, 2H, J = 7.1Hz, OCH2CH3), 6.51 (s, 1H, 7-H), 6.66 (s, 1H, 10-H), 6.76 – 6.78 (m, 2H, H-Ar), 6.93 (d, 1H, J = 8.1Hz, H-Ar), 7.38 (s, 1H, 3-H), 12.41 (br.s., 1H, NH+ (morpholinyl)); 13C NMR (150 MHz, DMSO-d6): δ = 14.6 (2C), 14.9 (2C), 15.0, 29.2 (2C), 44.9 (2C), 51.9, 63.7, 63.9, 64.7, 64.8 (2C), 108.8, 109.0, 113.4, 114.1, 118.0, 121.1, 121.5, 123.2, 123.4, 124.1, 126.3, 127.8, 147.1, 147.2, 148.3, 149.4; MS (LCMS) m/z = 521 [M-Cl]+; Anal. calcd for C31H40N2O5×HCl, (%): C, 66.83; H, 7.42; N, 5.03, found: C, 67.03; H, 7.55; N, 5.21.
3.2. Biological Evaluation
3.2.1. Cell Cultures
Human cell cultures RD (rhabdomyosarcoma), HCT116 (intestinal carcinoma), HeLa (cervical adenocarcinoma) and A549 (lung carcinoma) were grown in DMEM medium (for A549, HCT116 and RD) and EMEM (for HeLa) supplemented with 10% fetal calf serum, 2mM L-glutamine and 1% gentamicin as an antibiotic at 37oC and 5% CO2 in a humid atmosphere. MDCK-MDR1 and MDCK-MRP1 are a gift from Prof. P. Borst, NKI-AVL Institute, Amsterdam, The Netherlands. MDCK- MDR1 and MDCK-MRP1 were grown in high-glucose DMEM supplemented with fetal bovine serum (FBS; 10 %), glutamine (2 mM), penicillin (100 U mL-1), and streptomycin (100 mg mL-1) in a humidified incubator at 37 °C in a 5 % CO2 atmosphere.
3.2.2. Cytotoxicity Assay
The cytotoxicity was determined by the MTT test. The cells were seeded at a concentration of 1×10 4 cells/200 μl in a 96-well plate and cultured at 37 °C in a humidified atmosphere with 5% CO2. After 24 hours of incubation, different concentrations of test compounds (100 to 1.56 μM⋅L-1) were added to the cancer cell cultures and the cells were then cultured under the same conditions for 72 hours. Each concentration of the test compound was assayed in triplicate. All substances were dissolved in DMSO, whose final concentration in the well did not exceed 0.1% and was not toxic to the cells. Wells in which the co-solvent DMSO was added at a final concentration of 0.1% were used as controls. After incubation, 20 μl of MTT (3α-4,5-dimethylthiazol-2-yl-2,5-diphenyl tetrazolium bromide, 5 mg⋅mL-1) was added to each well and the plates were incubated for another 2 hours. Next, media was removed from the plates and 100 μL of DMSO was added to each well to dissolve the formed formazan crystals. Using a flatbed analyzer (Victor3, PerkinElmer), the optical density was determined at 530 nm, minus the measured background absorbance at 620 nm. The concentration value, which causes 50% inhibition of cell population growth (IC50), was determined from the dose-dependent curves using the OriginPro 9.0 software.
3.2.3. Inhibition Assays of P-Glycoprotein (P-gp) and Multidrug-Resistance-Associated Protein-1 (MRP1)
The experiments were carried out as already described [7]. MDCK-MDR1 and MDCK-MRP1 cell line (50,000 cells per well) was seeded into black CulturePlate 96/wells plate with 100 mL medium and allowed to become confluent overnight. 100 mL of different concentrations of test compounds (0.1-100 mM) were solubilized in culture medium and added to each well. The 96/wells plate was incubated at 37 °C for 30 min 100 μL of Calcein-AM, solved in phosphate buffered saline (PBS), was added to each well to yield a final concentration of 2.5 mM, and the plate was incubated for 30 min. The plate was washed three times with 100 mL ice cold PBS. Saline buffer (100 mL) was added to each well and the plate was read by a PerkinElmer Victor3 spectrofluorimeter at excitation and emission wavelengths of 485 nm and 535 nm, respectively. In these experimental conditions, Calcein cell accumulation in the absence and in the presence of tested compounds was evaluated and fluorescence basal level was estimated by untreated cells. In treated wells the increase of fluorescence with respect to basal level was measured. IC50 values were determined by fitting the fluorescence increase percentage versus log [dose].
3.2.4. Affinity to Human Serum Albumin (HSA) by Surface Plasmon Resonance (SPR)
The fatty acid-free HSA (A3782 from Sigma-Aldrich) was applied to functionalize surface on COOH V sensor chip (Pall FortèBio), by using amine coupling (EDC/NHS). HSA aqueous stock solution was diluted at the final concentration of 50 μg/mL in 10 mM sodium acetate buffer (pH 5.0), and immobilization in peripheral flow cells 1 and 3 (unmodified dextran surface in middle flow cell 2 used as a reference) at a final apparent level of about 5000 RU was achieved by applying the following protocol: injection of EDC/NHS (freshly mixed 0.4M EDC and 0.1M NHS) 1:1v/v at flow of 25 μL/min for 4 min; injection of HSA solution at flow 25 μL/min for 8 min; capping on unreacted activated carboxyl groups by injection of 1 M ethanolamine solution (pH 9) at flow 25 μL/min for 8 min. Functionalized surface was then treated with repeated injection of 4 M NaCl, as regenerating reagent, and conditioned overnight prior to use with pH 7.4 PBS (10 mM NaH2PO4 and 150 mM NaCl) as running buffer. Each cycle, without regeneration necessary, for binding assay to HSA was carried out in PBS-4% DMSO and consisted of run buffer injection (60 s), injection of analyte single concentration for 120 s (ranging from 1 to 200 μM) at a flow 20 μL/min, and dissociation phase for 150 s (flow 20 μL/min). Each measurement was performed at least in triplicate and analyzed by using QDAT software (non-linear regression analysis of 1:1 stoichiometric reversible binding model), and double referencing.
3.3. Solubility and Lipophilicity
3.3.1. Determination of Kinetic Solubility in PBS
Sample solution at 200 μM in PBS (50 mM, pH 7.4, KCl 0.15 M) from stock solution 10 mM in DMSO was incubated at room temperature (25±1 °C) for 2 h, following shaking of the suspension on an orbital shaker at 250 rpm, and then it was separated by centrifugation (2500 rpm for 3 min). Immediately after filtration step, 100 µL of filtrate was transferred into 100 µL of 1:1 (v/v) mixture of DMSO and PBS (50 mM, pH 7.4, KCl 0.15 M), to avoid precipitation from the saturated solution and analyzed by HPLC. The peak area was plotted against a calibration curve of tested compound in methanol [22]. Analytical conditions: Mobile Phase: MeOH/ammonium formate 20 mM pH 4.7 (70:30, 75:25); MeOH 0.1% TFA/ H2O 0.1% TFA (70:30, 60:40); stationary phase: Phenomenex, Kinetex 5 μ, C18, 100 Å (150×4.6 mm); flux: 1 mL/min; injection: 10 μL, 265, 290, 320 nm wavelength. HPLC analyses were performed on Agilent HPLC 1260 Infinity Series Integrated System (Agilent Technologies, Milan, Italy).
3.3.2. Determination of Lipophilicity by RP-HPLC
The lipophilicity parameters were determined by an RP-HPLC technique [21]. Methanol solution of DHPIQ derivatives (1 mg/mL) were analyzed by Agilent 1260 infinite HPLC system (Agilent Technologies, Milan, Italy) equipped with a diode array detector (DAD), and a Phenomenex, Kinetex 5 μ, C18, 100 Å (150×4.6 mm), and eluted with different percentage of mobile phase composition (0.05 increments of MeOH volume fractions in 20 mM ammonium formate buffer at pH 4.7 or water 0.1% TFA, ranging ϕ between 0.85 and 0.30). The chromatographic measurements were carried out at 25±1 °C at a flow rate of 1 mL/min, and at 265, 290, 320 nm wavelength. The logarithm of capacity factors (log k′ = log (tR − t0)/t0) of each compound at different mobile phase compositions have been calculated; tR represents the retention time of the solute and t0 is the column dead time, measured as the elution time of a KNO3 solution in MeOH. For each compound, the log k′ values increased linearly with decreasing MeOH volume fraction. The logarithm of the capacity factor extrapolated to 100% aqueous mobile phase (log k′w) were calculated from the linear regressions on at least five data points (R2 > 0.9984).
Lipophilicity was also computationally assessed as log P and log D values at pH 4.7 (pH of the aqueous buffer in the mobile phase for RP-HPLC) using ACDLabs software (release 10.0; Advanced Chemistry Development, Inc., Toronto, Canada). The values of calculated log P (Clog P), referred to lipophilicity of the neutral species, are listed in Table 2 together with the RP-HPLC parameters log k’w in Table 2. Log k’w and Clog P were reasonably correlated (r2 = 0.7911), but an inspection of the correlation plot suggested that, omitting from the regression analysis the two 2-morpholinomethyl DHPIQ derivatives 8a and 8b, which should be predominantly protonated (positively charged) at the tertiary amino group, all the other data points fit well the following linear equation:
Log k’w = 0.78( ± 0.16) Clog P – 0.12( ± 0.99)
n = 6, r2 = 0.8592, s = 0.3699, F = 24.41
where n is the number of data points, r2 the coefficient of determination, s the standard deviation of the regression equation, and F the F-value from the Fisher test for regression model significance (95% confidence intervals of the regression coefficients are given in parentheses).
3.4. Molecular Doocking Calculations
The ligands’ structures were built starting from the relative SMILES strings converted to three dimensional structures with OMEGA [23] followed by 10000 steps of steepest descent minimization using the UFF was performed with Open Babel [24]. The target molecule (P-gp) was prepared starting from the MDR1 Cryo-EM structure (Protein Data Bank entry 7A6C) [25] with the Protein Preparation Wizard interface of Maestro [26,27], removing the co-crystallized elacridar molecule, loading and optimizing hydrogen atoms position, and assigning the ionization states of acid and basic residues according to PROPKA prediction at pH 7.0. Electrostatic charges for protein atoms were loaded according to the AMBER UNITED force field [28], while the molcharge set of QUACPAC [29] was used to achieve Marsili-Gasteiger charges for the inhibitors.
Solvent was explicitly considered by means of the proper parametrization of water contribution according to the relative AUTODOCK hydration force field [30], and the population size and the number of energy evaluation figures were set to 300 and 10000000, respectively. Dockings were then performed throughout 1000 runs of the Lamarckian genetic algorithm (LGA) implemented in AUTODOCK 4.2.6 [31] using the GPU-OpenCL algorithm version [32], and the best energy/best cluster poses as scored by AUTODOCK were selected.
4. Conclusions
In our recent studies on the annelated azaheterocyclic core of the marine alkaloids lamellarins [6,7], we disclosed novel DHPIQ-containing carbaldehydes (and related imino adducts) showing anticancer activities, in some cases coupled with inhibition of P-gp efflux pump [2,6,7]. In-silico ligand-based approaches and molecular docking calculations supported (likely as a part of a possible multitarget activity) the propensity of some suitably substituted 1-Ph-DHPIQ-2-carbaldehydes to bind the DNA-topoisomerase I complex [5], likely blocking the DNA replication.
Herein, we investigated the role of the electrophilic 2-CHO group in modulating the anticancer activity, using a classical bioisosteric replacement approach. Firstly, CHO was replaced by CN, a less reactive electrophilic group that has gained importance in covalent drug discovery [8,9]. In addition, the less reactive carboxyl group (in 2-COOH and 2-COOEt derivatives) was investigated as a possible replacement of the more reactive carbonyl group.
The replacement of CHO with CN, COOH and COOEt resulted in a sharp decrease of cytotoxic activity against all four tumor cell lines tested (RD, HCT116, HeLa, A549). The comparison between CHO and CN, albeit possessing very similar stereoelectronic and lipophilic features, indirectly suggested that CHO, and not CN, may be involved in covalent reactions with nucleophiles of biological target (DNA, enzyme proteins) [10]. Lipophilicity of the substituents onto the 1-Ph-DHPIQ moiety appeared to play a secondary role in affecting cytotoxicity. In contrast, regardless the main functional group at C2 (CHO, CN, COOH, COOEt), as a trend the more lipophilic the more potent inhibitor of P-gp (and not MRP1). The P-gp inhibition SARs were favorably supported and complemented by in-silico docking calculation models.
Unfortunately, except the ester 7, all the above derivatives (4, 5 and 6) were found very poorly soluble in aqueous solutions at the physiological pHs. A successful attempt pursued in this study to improve the water solubility was the synthesis of a couple of DHPIQs bearing at C2 the basic 2- morpholinomethyl chain, which is estimated to be more than half in protonated form at neutral pH. As a matter of fact, a novel DHPIQ-based Mannich base prepared as HCl salt (8b’), stable at pHs 2 and 7.4 at room temperature, proved to be cytotoxic to all the tested tumor cell lines in the low micromolar range (IC50 < 20 μM) and to inhibit in-vitro the efflux pumps P-gp and MRP1 responsible for MDR, with IC50s of 0.45 and 12.1 μM, respectively. Compounds 8a and 8b’ resulted significantly (3-to-5-fold) more soluble than the related aldehydes, but still below the minimum solubility threshold of 0.1 mg/mL according to the categorization of the US and Eur. Pharmacopoeia [20]. Nevertheless, the synthesis of DHPIQs 8a-b’ provided a useful approach to prepare new water-soluble lamellarin-like antineoplastic substances, including more hydrophilic prodrugs and Mannich bases, endowed with the ability of overcoming P-gp-mediated MDR.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figures S1-S9: 1H and 13C-NMR of compounds 4b, 5a-b, 6c-d, 7, 8a-b and 8b’.
Author Contributions
Conceptualization, A.A.N., R.P., L.G.V. and C.D.A; methodology, A.A.N., R.P., L.V.A., M.N. and M.d.C.; software, A.C.; validation, A.O., T.B., M.C. and N.A.C.; formal analysis, A.A.N., R.P. and M.N.; investigation, A.A.N., R.P., A.O., L.V.A., M.N. and M.d.C.; resources, L.G.V. and C.D.A.; data curation, T.B., A.V.V., A.C. and M.C.; writing—original draft preparation, A.A.N., R.P., M.N. and A.C.; writing—review and editing, A.A.N., R.P. and C.D.A.; visualization, A.C., T.B. and N.A.C.; supervision, L.G.V. and C.D.A.; funding acquisition, L.G.V. and C.D.A. All authors have read and agreed to the published version of the manuscript.
Funding
C.D.A., M.C., A.C., M.d.C. and R.P. acknowledge the financial support of the Italian Ministry of Education, Universities and Research (PRIN, Grant 201744BNST_004).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained only within the article and supplementary materials.
Acknowledgments
R.P. is a researcher supported by Apulian Region “Research for Innovation (REFIN)”—POR PUGLIA FESR-FSE 2014/2020 (Project F88A1A13).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fukuda, T.; Ishibashi, F.; Iwao, M. Synthesis and biological activity of lamellarin alkaloids: an overview. Heterocycles 2011, 83, 491–529. [Google Scholar] [CrossRef]
- Matveeva, M.D.; Purgatorio, R.; Voskressensky, L.G.; Altomare, C.D. Pyrrolo[2,1- a ]isoquinoline scaffold in drug discovery: Advances in synthesis and medicinal chemistry. Future Med. Chem. 2019, 11, 2735–2755. [Google Scholar] [CrossRef] [PubMed]
- Kakhki, S.; Shahosseini, S.; Zarghi, A. Design, synthesis and cytotoxicity evaluation of new 2-aryl-5, 6-dihydropyrrolo[2,1-a]isoquinoline derivatives as topoisomerase inhibitors. Iran. J. Pharm. Res. 2014, 13, 71–77. [Google Scholar]
- Kakhki, S.; Shahosseini, S.; Zarghi, A. Design and synthesis of pyrrolo [2,1-a] isoquinoline-based derivatives as new cytotoxic agents. Iran J. Pharm. Res. 2016, 15, 743–751. [Google Scholar] [PubMed]
- Nevskaya, A.A.; Miftyakhova, A.R.; Anikina, L.V.; Borisova, T.N.; Varlamov, A.V.; Voskressensky, L.G. Synthesis and Cytotoxicity of Novel 1-Arylindolizines and 1-Arylpyrrolo[2,1-a]isoquinolines. Tetrahedron Lett. 2021, 87, 153552. [Google Scholar] [CrossRef]
- Nevskaya, A.A.; Anikina, L.V.; Purgatorio, R.; Catto, M.; Nicolotti, O.; de Candia, M.; Pisani, L.; Borisova, T.N.; Miftyakhova, A.R.; Varlamov, A.V.; et al. Homobivalent Lamellarin-Like Schiff Bases: In Vitro Evaluation of Their Cancer Cell Cytotoxicity and Multitargeting Anti-Alzheimer’s Disease Potential. Molecules 2021, 26, 359. [Google Scholar] [CrossRef]
- Nevskaya, A.A.; Matveeva, M.D.; Borisova, T.N.; Niso, M.; Colabufo, N.A.; Boccarelli, A.; Purgatorio, R.; de Candia, M.; Cellamare, S.; Voskressensky, L.G.; et al. A New Class of 1-Aryl-5,6-dihydropyrrolo[2,1-a]isoquinoline derivatives as reversers of P-glycoprotein-mediated multidrug resistance in tumor cells. ChemMedChem 2018, 13, 1588–1596. [Google Scholar] [CrossRef]
- Dufour, E.; Storer, A.C.; Menard, R. Peptide aldehydes and nitriles as transition state analog inhibitors of cysteine proteases. Biochemistry 1995, 34, 9136–9143. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307. [Google Scholar] [CrossRef]
- LoPachin, R.M.; Gavin, T. Molecular Mechanisms of Aldehyde Toxicity: A Chemical Perspective. Chem. Res. Toxicol. 2014, 27, 1081–1091. [Google Scholar] [CrossRef]
- Roman, G. Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 2015, 80, 743–816. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, M.; Borisova, T.; Titov, A.; Anikina, L.; Dyachenko, S.; Astakhov, G.; Varlamov, A.; Voskressensky, L. Domino Reactions of 1-Aroyl-3,4-Dihydroisoquinolines with α,β-Unsaturated Aldehydes. Synthesis 2017, 49, 5251–5257. [Google Scholar] [CrossRef]
- Astakhov, G.S.; Shigaev, R.R.; Borisova, T.N.; Ershova, A.A.; Titov, A.A.; Varlamov, A.V.; Voskressensky, L.G.; Matveeva, M.D. Facile Synthesis of Pyrrolo[2,1-a]Isoquinolines by Domino Reaction of 1-Aroyl-3,4-Dihydroisoquinolines with Conjugated Ketones, Nitroalkenes and Nitriles. Mol. Divers. 2021, 25, 2441–2446. [Google Scholar] [CrossRef] [PubMed]
- Colabufo, N.A.; Pagliarulo, V.; Berardi, F.; Contino, M.; Inglese, C.; Niso, M.; Ancona, P.; Albo, G.; Pagliarulo, A.; Perrone, R. Bicalutamide Failure in Prostate Cancer Treatment: Involvement of Multi Drug Resistance Proteins. Eur. J. Pharmacol. 2008, 601, 38–42. [Google Scholar] [CrossRef]
- Colabufo, N.A.; Berardi, F.; Cantore, M.; Perrone, M.G.; Contino, M.; Inglese, C.; Niso, M.; Perrone, R.; Azzariti, A.; Simone, G.M.; et al. Small P-Gp Modulating Molecules: SAR Studies on Tetrahydroisoquinoline Derivatives. Bioorg. Med. Chem. 2008, 16, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Trompier, D.; Chang, X.-B.; Barattin, R.; d’Hardemare, A.D.M.; Di Pietro, A.; Baubichon-Cortay, H. Verapamil and Its Derivative Trigger Apoptosis through Glutathione Extrusion by Multidrug Resistance Protein MRP1. Cancer Res. 2004, 64, 4950–4956. [Google Scholar] [CrossRef] [PubMed]
- Purgatorio, R.; De Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Denora, N.; Carrieri, A.; Nevskaya, A.A.; Voskressensky, L.G.; Altomare, C.D. Evaluation of Water-Soluble Mannich Base Prodrugs of 2,3,4,5-Tetrahydroazepino[4,3-b]indol-1(6H)-one as Multitarget-Directed Agents for Alzheimer’s Disease. ChemMedChem 2021, 16, 589–598. [Google Scholar] [CrossRef]
- Fabini, E.; Danielson, U.H. Monitoring Drug–Serum Protein Interactions for Early ADME Prediction through Surface Plasmon Resonance Technology. J. Pharm. Biomed. Anal. 2017, 144, 188–194. [Google Scholar] [CrossRef]
- Frostell-Karlsson, Å.; Remaeus, A.; Roos, H.; Andersson, K.; Borg, P.; Hämäläinen, M.; Karlsson, R. Biosensor Analysis of the Interaction between Immobilized Human Serum Albumin and Drug Compounds for Prediction of Human Serum Albumin Binding Levels. J. Med. Chem. 2000, 43, 1986–1992. [Google Scholar] [CrossRef]
- European Pharmacopoeia 10th Edition, 2019.
- Purgatorio, R.; De Candia, M.; De Palma, A.; De Santis, F.; Pisani, L.; Campagna, F.; Cellamare, S.; Altomare, C.; Catto, M. Insights into Structure-Activity Relationships of 3-Arylhydrazonoindolin-2-One Derivatives for Their Multitarget Activity on β-Amyloid Aggregation and Neurotoxicity. Molecules 2018, 23, 1544. [Google Scholar] [CrossRef]
- Purgatorio, R.; Kulikova, L.N.; Pisani, L.; Catto, M.; De Candia, M.; Carrieri, A.; Cellamare, S.; De Palma, A.; Beloglazkin, A.A.; Reza Raesi, G.; et al. Scouting around 1,2,3,4-Tetrahydrochromeno[3,2- c ]Pyridin-10-ones for Single- and Multitarget Ligands Directed towards Relevant Alzheimer’s Targets. ChemMedChem 2020, 15, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM Structures Reveal Distinct Mechanisms of Inhibition of the Human Multidrug Transporter ABCB1. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Schrödinger Release 2023-1, Maestro.
- Schrödinger, LLC: New York, NY, USA, 2023.
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Molecular Modeling Software | OpenEye Scientific. Available online: https://www.eyesopen.com (accessed on 20 September 2023).
- Forli, S.; Olson, A.J. A Force Field with Discrete Displaceable Waters and Desolvation Entropy for Hydrated Ligand Docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- El Khoury, L.; Santos-Martins, D.; Sasmal, S.; Eberhardt, J.; Bianco, G.; Ambrosio, F.A.; Solis-Vasquez, L.; Koch, A.; Forli, S.; Mobley, D.L. Comparison of Affinity Ranking Using AutoDock-GPU and MM-GBSA Scores for BACE-1 Inhibitors in the D3R Grand Challenge 4. J. Comput.-Aided Mol. Des. 2019, 33, 1011–1020. [Google Scholar] [CrossRef]
Figure 1.
Structure of the 1-phenyl-5,6-dihydropyrrolo[2,1-a]isoquinoline (1-Ph-DHPIQ) scaffold and natural (lamellarins) and synthetic derivatives (2-4).
Figure 1.
Structure of the 1-phenyl-5,6-dihydropyrrolo[2,1-a]isoquinoline (1-Ph-DHPIQ) scaffold and natural (lamellarins) and synthetic derivatives (2-4).
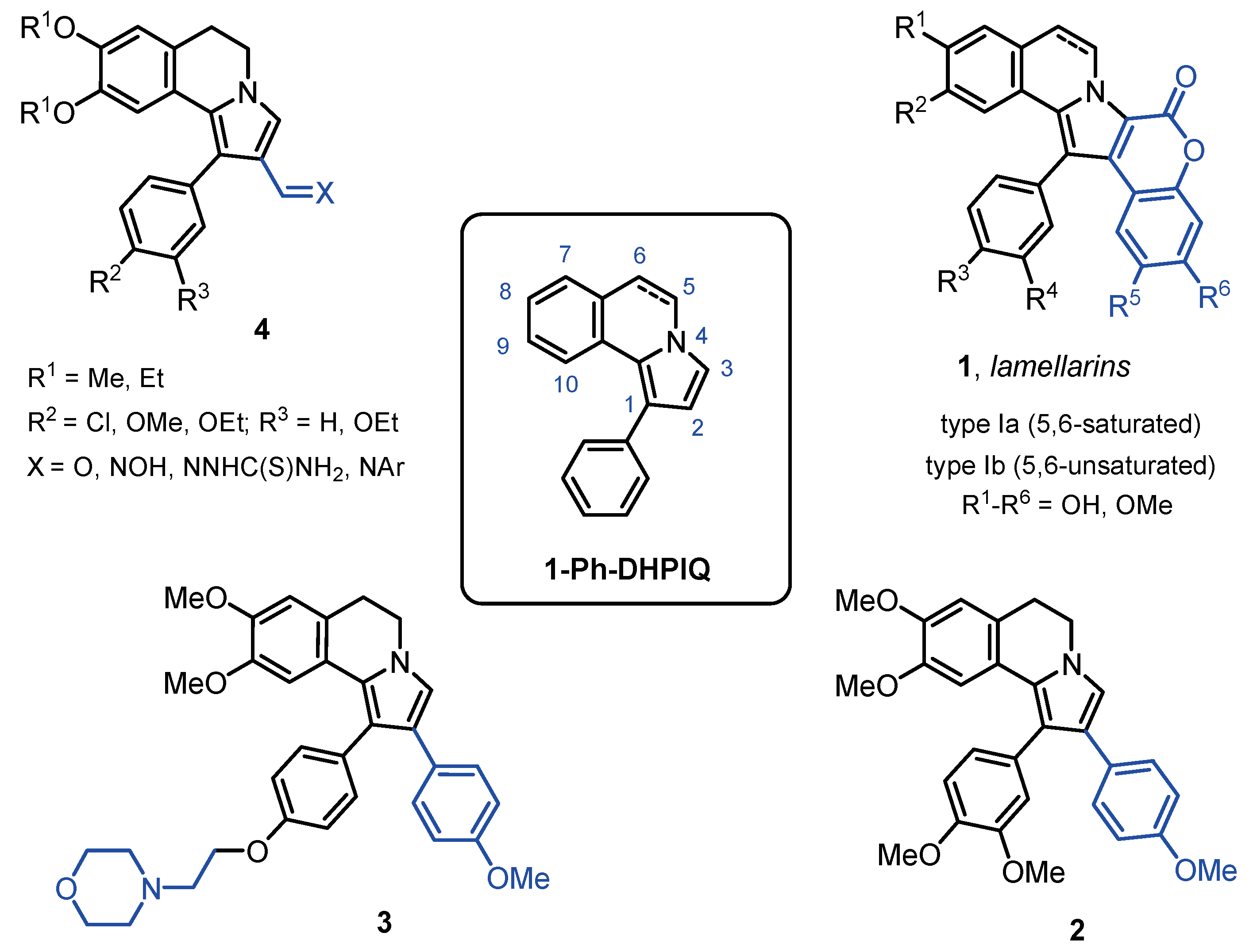
Scheme 1.
Synthesis of diversely R1-4-substituted 1-Ph-DHPIQ derivatives.
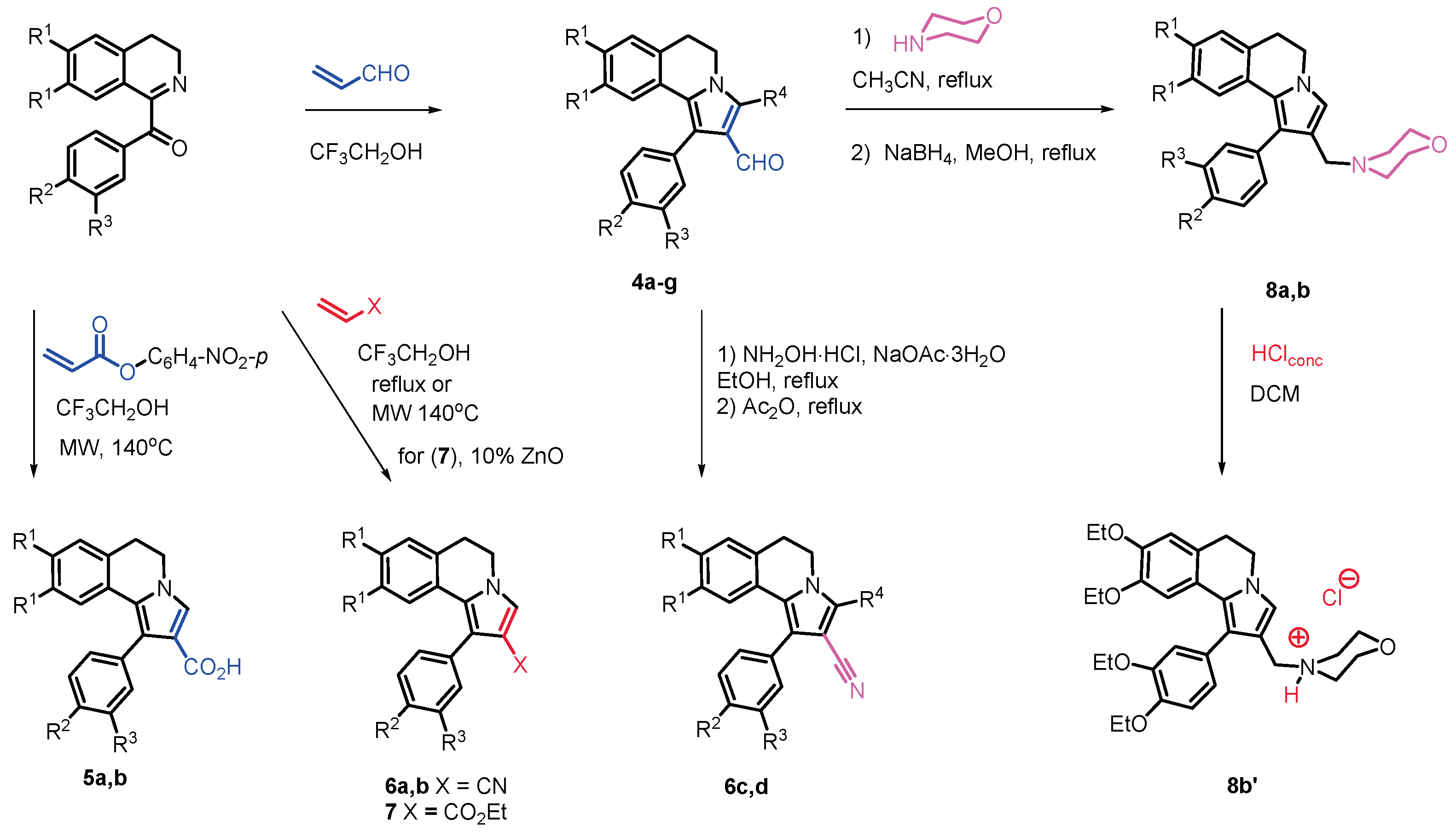
Figure 2.
Front (left) and extracellular view (right) of the binding mode of 7 to CryoEM P-gp structure. The transmembrane-spanning helices are depicted as orange, ligand in meshes.
Figure 2.
Front (left) and extracellular view (right) of the binding mode of 7 to CryoEM P-gp structure. The transmembrane-spanning helices are depicted as orange, ligand in meshes.
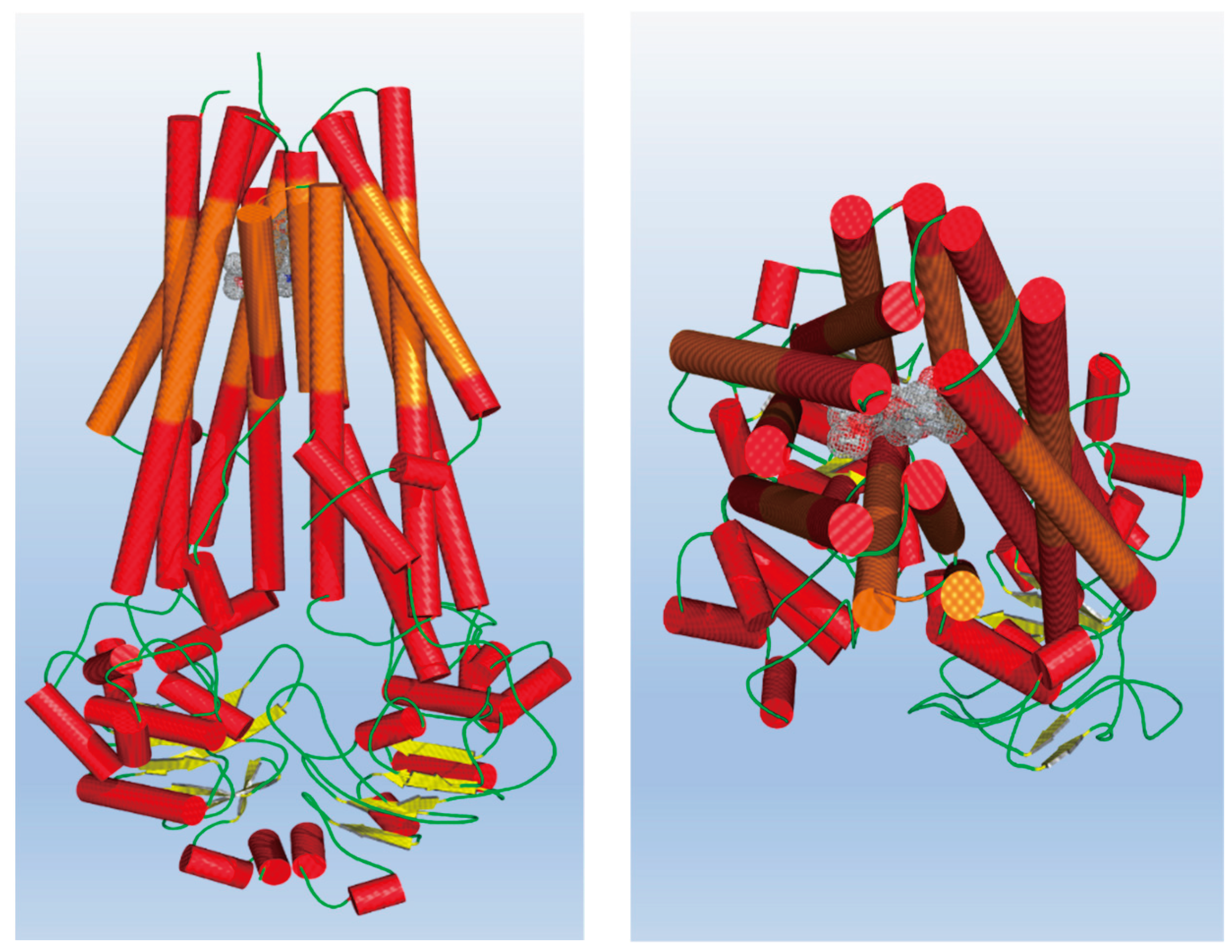
Figure 3.
Detailed view of 7 (left) and 8b’ (right) dockings. In the interaction pattern scheme, hydrogen bonds are depicted in cyan, Van der Waals contacts and π–π stackings in magenta, respectively.
Figure 3.
Detailed view of 7 (left) and 8b’ (right) dockings. In the interaction pattern scheme, hydrogen bonds are depicted in cyan, Van der Waals contacts and π–π stackings in magenta, respectively.

Table 1.
In vitro cytotoxicity data in human cancer cell cultures. a.
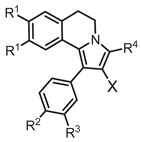
| N | X | R1 | R2 | R3 | R4 | RD | HCT116 | HeLa | A549 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4ab | CHO | OMe | Cl | H | H | 17.6 (3.2) | 22.0 (4.0) | 33.0 (4.5) | 38.6 (3.2) | ||
| 4b | CHO | OMe | OMe | H | H | 95.2 (7.1) | > 100 | > 100 | > 100 | ||
| 4cc | CHO | OEt | OEt | OEt | H | 21.3 (1.2) | 11.8 (0.2) | 44.5 (2.0) | 19.7 (0.3) | ||
| 5a | CO2H | OMe | Cl | H | H | > 100 | > 100 | > 100 | > 100 | ||
| 5b | CO2H | OEt | OEt | OEt | H | n.a. | n.a. | n.a. | n.a. | ||
| 6a | CN | OMe | Cl | H | H | > 100 | > 100 | > 100 | > 100 | ||
| 6b | CN | OEt | OEt | OEt | H | > 100 | > 100 | > 100 | > 100 | ||
| 6c | CN | OEt | OEt | OEt | Me | > 100 | > 100 | > 100 | > 100 | ||
| 6d | CN | OEt | OEt | OEt | Ph | > 100 | n.a. | n.a. | n.a. | ||
| 7 | CO2Et | OEt | OEt | OEt | H | n.a. | n.a. | n.a. | n.a. | ||
| 8a | MMd | OMe | OMe | H | H | 37.7 (1.6) | 56.4 (1.7) | 65.4 (4.5) | 66.0 (1.6) | ||
| 8b’ | MM·HCl | OEt | OEt | OEt | H | 18.6 (2.7) | 15.7 (0.7) | 17.5 (1.4) | 20.8 (3.6) | ||
| Camptothecin | 16.0 (0.2) | 12.3 (0.5) | 0.33 (0.07) | 3.32 (0.02) | |||||||
| Doxorubicin | 0.29 (0.02) | 0.14 (0.01) | 0.89 (0.01) | 0.38 (0.02) | |||||||
a RD: rhabdomyosarcoma; HCT116: colon cancer cells; HeLa: adenocarcinoma of the cervix uterus; A549: lung adenocarcinoma. Each experiment was performed in triplicate; data are expressed as the mean µM IC50 (SD), that is, the concentration value causing 50% inhibition of cell population growth. b Ref. 5. c Ref. 6. d MM = morpholinomethyl. n.a.: not active at 100 μM; > 100: less than 60% inhibition at 100 μM.
Table 2.
Inhibitory potencies of R1-3-substituted 1-Ph-DHPIQ toward P-gp and MRP1 drug efflux pumps, and ADME-related physicochemical parameters.
Table 2.
Inhibitory potencies of R1-3-substituted 1-Ph-DHPIQ toward P-gp and MRP1 drug efflux pumps, and ADME-related physicochemical parameters.
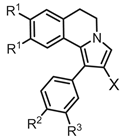
| N | X | R1 | R2 | R3 | P-gp, IC50 (μM)a |
MRP1, IC50 (μM)a |
HSA, KD (μM)b |
S (μM)c | CLog Pd | Log k’we |
|---|---|---|---|---|---|---|---|---|---|---|
| 4a | CHO | OMe | Cl | H | 25.3 (1.9) | 21.9 (1.5) | 1.60 (0.04) | 6.66 (0.09) | 5.62 | 4.32 |
| 4b | CHO | OMe | OMe | H | 4.48 (0.17) | 6.42 (0.27) | 1.95 (0.06) | 19.8 (0.1) | 4.86 | 3.39 |
| 5b | CO2H | OEt | OEt | OEt | 0.35 (0.04) | > 100 | 5.20 (0.20) | 14.6 (0.6) | 4.70 | 6.91 |
| 6a | CN | OMe | Cl | H | 5.89 (0.42) | 16.6 (0.5) | 4.60 (0.10) | 1.42 (0.01) | 5.75 | 4.42 |
| 6b | CN | OEt | OEt | OEt | 0.39 (0.06) | > 100 | 1.80 (0.20) | 1.06 (0.05) | 6.79 | 5.09 |
| 7 | CO2Et | OEt | OEt | OEt | 0.32 (0.08) | 3.23 (0.29) | 12.1 (0.1) | 41.7 (1.1)g | 7.75 | 5.49 |
| 8a | MMf | OMe | OMe | H | 0.36 (0.02) | 1.80 (0.31) | 19.3 (0.5) | 112 (4)g | 4.73 | 3.86 |
| 8b’ | MM·HCl | OEt | OEt | OEt | 0.45 (0.03) | 12.1 (2.1) | 26.9 (0.5) | 41.7 (1.2)g | 6.53 | 4.31 |
| MC18 | 1.20 (0.3) | |||||||||
| Verapamil | 4.53 (0.50) | |||||||||
| Warfarin | 5.30 (0.35) | |||||||||
a Inhibition potency data toward P-glycoprotein (P-gp) and multidrug-resistance-associated protein-1 (MRP1), expressed by mean IC50 (SD) values of at least two independent experiments each performed in triplicate; > 100: less than 60% inhibition at 100 μM. MC18 and verapamil were assayed as the P-gp-selective and MRP1-selective positive controls, respectively. b Binding to human serum albumin (HSA) determined by surface plasmon resonance technique, expressed by at equilibrium dissociation constants (KD values); the affinity of warfarin was reported as reference value. c Solubility of 1-Ph-DHIQ derivatives in PBS (50 mM, pH 7.4, 0.15 M KCl) at 25± 1 °C. d Log of 1-octanol/water partition coefficient calculated by ACDLabs software (release 10.0, Advanced Chemistry Development, Inc., Toronto, Canada). e Log of the polycratic capacity factor determined as lipophilicity parameter by a reversed phase (RP) HPLC technique, as described in Materials and Methods. f MM = morpholino-methyl. g Stable for 4 h in PBS (50 mM, pH 7.4, KCl 0.15M) and in HCl (0.01 M, pH 2.0, 0.15 M KCl) at 25 ± 1 °C.
Table 3.
Docking metrics of 1-Ph-DHPIQ derivatives 7 and 8b’.
| N | FEB(a) | ΔE(b) | EFF(c) | TAN(d) | POP(e) |
|---|---|---|---|---|---|
| 7 | – 11.04 | 0.08 | – 0.307 | 0.307 | 265/1000 |
| 8b | – 10.62 | 0.21 | – 0.279 | 0.276 | 121/1000 |
(a) FEB, Free Energy of Binding; (b) DE, Energy difference between the selected pose and the relative global minimum; (c) EFF, Ligand efficacy; (d) TAN, Tanimoto-Combo similarity coefficient with elacridar X-ray pose; (e) POP, Cluster members population.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



