
Submitted:
06 March 2024
Posted:
11 March 2024
You are already at the latest version
Abstract
C-terminal Src kinase (CSK) is the major inhibitory kinase for Src family kinases (SFKs) through phosphorylation of their C-tail tyrosine site, and regulates various cellular activities in association with SFKs functions. As a cytoplasmic protein, CSK need be recruited to the plasma membrane to regulate SFKs activities. The regulatory mechanism for CSK activity and its subcellular localization remains largely unclear. In this work, we developed a genetically encoded biosensor based on fluorescence resonance energy transfer (FRET) to visualize CSK activity in live cells. The biosensor with optimized substrate peptide confirmed crucial Arg107 site in CSK SH2 domain, and displayed sensitivity and specificity to CSK activity while little responses to co-transfected Src and Fyn. FRET measurements showed CSK having relatively mild level of kinase activity in comparison to Src and Fyn ones in rat airway smooth muscle cells. The biosensor tagged with different submembrane-targeting signals detected CSK activity at both non-lipid raft and lipid raft microregions, while showed higher FRET level at non-lipid ones. Co-transfected receptor-type protein tyrosine phosphatase alpha (PTPα) had inhibitory effect on the CSK FRET response. The biosensor didn’t detect obvious changes of CSK activity between metastatic cancer cells and normal ones. In conclusion, a novel FRET biosensor was generated to monitor CSK activity, and demonstrated CSK activity existing in both non-lipid and lipid raft membrane microregions while more present at non-lipid ones.
Keywords:
C-terminal Src kinase (CSK)
; fluorescence resonance energy transfer (FRET)
; Src family kinase
; PTPalpha
; submembrane kinase activity
Introduction
The discovery of Src, the first member of Src family kinases, dates back to the 1970s, when it was identified as a transformed gene in the Rous sarcoma virus (RSV) [1,2,3]. Subsequent studies identified other relevant kinases, collectively known as SFKs, including Yes, Fyn, Lyn, and Lck [4,5]. SFKs activity has been regulated by phosphorylation events. In the late 1980s, researchers were studying the mechanism of negative regulation of SFKs, and showed that phosphorylation of the C-tail tyrosine site inhibited SFKs activity. They focused on identifying kinases responsible for phosphorylation of inhibitory tyrosine residues. Through biochemical and genetic methods, they identified and characterized C-terminal Src kinase (CSK) as the kinase responsible for this inhibitory phosphorylation [6,7].
The CSK molecule weighs 50 kDa and consists of three functional domains: SH3, SH2, and kinase domains [8]. In contrast to SFKs structures [9], CSK does not contain N-terminal acylation groups and the regulatory tyrosine residues in kinase and C-terminal domains [10]. Since CSK lacks transmembrane domains and fatty acyl modifications, it is mainly present in the cytoplasm, while its substrate SFKs are immobilized on the membrane by the myristic and palmitate moieties of the N-terminus. Therefore, translocation of CSK to cell membranes that inactivate SFKs are considered a critical step in CSK regulation [11]. While the transfer of CSK to the cell membrane requires binding to its membrane anchor, Masato Okada. et al. successfully isolated a transmembrane adapter-like molecule CBP/PAG1 (CSK-binding protein/phosphoprotein associated with a glycophospholipid-rich membrane) from a cholesterol-rich membrane microdomain "lipid raft" [12,13],. At present, it has been shown that when CBP is phosphorylated by active SFKs, CSK is recruited to the lipid raft by binding to CBP, and CSK phosphorylates the C-terminal Y527 of SFKs, resulting in an intramolecular interaction between phosphorylated tyrosine and the SH2 domain of SFKs [14,15],. This interaction puts SFKs in an inactive conformation and prevents its activation [16]. Inactivated SFKs can be activated by PTPα dephosphorylation [17]. CSK primarily plays a pivotal role in numerous cellular processes, including apoptosis, cell proliferation, canner invasion, and angiogenesis, primarily through the regulation of SFKs; furthermore, it contributes to the inhibition of cancer progression, as SFKs have been linked to various cancer developments [16].
Although membrane localization is important in the regulation of CSK function, there is a lack of imaging tools that can directly visualize CSK activities at the plasma membrane and its submembrane microdomains. Fluorescence resonance energy transfer (FRET) technology and genetically encoded FRET biosensors provide powerful tools for visualizing signaling molecules in living cells with high spatiotemporal resolution [18]. The FRET donor and acceptor pair, consisting of enhanced cyan fluorescent protein (ECFP) and a variant of yellow fluorescent protein (YPet), has been optimized for FRET biosensor engineering, which can form weak dimers that enhance the efficiency and sensitivity of FRET [19]. This FRET pair has also been used to design a range of highly sensitive biosensors such as the Rac1, Syk and ZAP70 FRET [20].
While the choice of donor and acceptor fluorescent proteins has a great influence on the sensitivity of the biosensor, the SH2 domain and substrate tyrosine peptide are key factors in determining the specificity and sensitivity of the biosensor. Therefore, using the above design strategy and the ECFP/YPet FRET pair, we designed the CSK FRET biosensor and characterized the sensitivity and specificity in ASM cells to select the optimal biosensor. The activity of CSK is detected by FRET reactions of the biosensors in cancer cells. By localizing the biosensor to the membrane microregions of ASM cells, we observed that the non-lipid raft region has a large number of active CSK. Thus, our work provides a highly sensitive CSK FRET biosensor for live-cell imaging and further reveals the mechanism by which different membrane spacings in ASM cells regulate CSK activity.
Materials and Methods
DNA Constructs
The CSK biosensor was first constructed into the vector pRSETb for bacterial expression. The DNA fragment including SH2 domain, a 15 amino acids linker and the substrate peptide FTSTEPQYQPGENL was amplified by PCR based on the template of a Fyn biosensor [20]. The individual PCR fragment was ligated between ECFP and YPet fluorescent proteins from the Fyn biosensor by SphI/SacI restriction sites. The biosensor in pRSETb was further amplified by PCR and ligated into the mammalian expression vector pCAGGS [21] by using EcoRI/SalI sites. Two other substrate versions (FTATEPQYQPGENL & EEEIYFFF) of the biosensor and the Y/F mutants in substrates were constructed in the same way. The membrane-targeted biosensors were constructed by PCR amplification of the biosensor while adding the Lyn tag with 21aa (MGCIKSKRKDNLNDDGVDMKT) or the Fyn tag (MGCVQCKDKEATKLTEERDGSLNQ) at the N-terminus of the biosensor and then by ligation into the pCAGGS vector with EcoRI/SalI sites. The KRas tag (KKKKKKKSKTKCVI) [22] is added to the C-terminal of the biosensor in the same way.
Reagents and Cell Culture
ASM cells were maintained in low-glucose Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS, Thermo), 100 μg/ml penicillin, and 100 unit/ml streptomycin at 37°C with 5% CO2 in a humidified incubator. The MDA-MB-231 cells were cultured in RPMI-1640 (Thermo). The MCF-10A cells were cultured in advanced DMEM/F12 medium supplemented with 20 ng/mL epidermal growth factor (EGF), 10 μg/mL insulin, 10 ng/ml cholera toxin, and 250 ng/μL hydrocortisone. BEAS-2B cells were maintained in high-glucose DMEM (Thermo), and A549 cells in Ham’s F12 nutrient mixture (Thermo). The culture medium were supplemented with 10% fetal bovine serum (FBS, Thermo), penicillin (100 unit/ml) and streptomycin (100 ng/mL). The cells were cultured at 37 °C in a humidified incubator with 5% CO2.
Fibronectin was purchased from Thermo Fisher Scientific. Rat platelet-derived growth factor (PDGF-BB) and human epidermal growth factor (EGF) were purchased from Sigma.
Cell Transfection with DNA
The DNA constructs were transfected into ASM cells by Lipofectamine 3000 reagent (Invitrogen) 2-3 days before imaging experiments. Generally, 1 μg biosensor DNA was transfected into cells per well in 24-well plate. For comparison of the FRET responses to different kinases in cells, 1 μg biosensor construct was co-transfected with plasmids encoding the different kinases (in the indicated amount later). During co-transfection, the different DNA plasmids were mixed thoroughly by pipetting up and down 10-20 times in an Eppendorf tube, and then mixed with Lipofectamine 3000 reagent at the ratio of 1:2 (μg DNA : μL Lipofectamine). After transfection for 8-16 hours, cells were maintained in low-serum DMEM containing 0.5% FBS, then seeded on 10 μg/ml fibronectin-coated glass-bottom dishes in low-serum DMEM for about 24 h before FRET imaging.
Microscope, and Image Acquisitions
The processes of FRET imaging and quantification were similar to our recent descriptions [20,23],. Briefly, the Zeiss microscopy system (Zeiss Cell Observer) was equipped with the functions of multi-positions, fine auto-focusing, and automatic-switchable dichroic rotator. The scope stage was supplemented with an incubator box (Zeiss) to maintain temperature at 37°C and 5% CO2 for live cell samples. For FRET image acquisitions through ECFP and FRET (YPet) channels, the parameters of excitation filter and dichroic mirror were 436 ± 10 and 455 nm, respectively, and the emission filters of ECFP and FRET (YPet) channels were 475 ± 20 nm and 535 ± 15 nm, respectively. Twenty to thirty positions from each sample dish were selected and imaged under the same condition. For PDGF or EGF stimulation during the FRET imaging, 1 mL of DMEM containing the growth factor was injected into the sample dish by a syringe guided through a microtube, which didn’t disrupt the imaging process.
FRET Quantification
FRET quantifications were processed using the Wang Lab (UCSD) developed software package FluoCell in MATLAB (available on http://github.com/lu6007/fluocell) [24]. Fluorescence signals from ECFP and FRET (YPet) images were measured after background subtractions, and the ratio of the two channels was calibrated in the pixel-to-pixel manner. Data procession and statistical analysis were done by the software of GraphPad Prism 6, and Excel. The quantified FRET data from a group of cells was expressed in time-course curves (Mean ± S.E.M.), and scattering dots (Mean ± S.D.). *, **, *** and **** indicate p < 0.05, 0.01, 0.001, and 0.0001 from Student’s t-test, which was applied for significant difference analysis. Multiple times of t-test analysis done between the control and one experimental group were carried for variable experimental conditions using GraphPad Prism 6. All described FRET experiments have been repeated independently on different days with similar conclusions, and statistical quantifications were performed based on the data acquired from different time.
Results
1. Biosensor Design, and Characterization of the Sensitivity and Specificity of CSK Biosensor in Mammalian Cells
In this study, we constructed a genetically encoded FRET biosensor to measure the activity of CSK kinase. The design of this biosensor is similar to the previously reported Fyn FRET biosensor [19,20]. The biosensor consists of an N-terminal ECFP, followed by the SH2 domain from Src, a flexible linker of 15 amino acids, a substrate peptide, and a C-terminal YPet (Figure 1A). We designed three different substrate peptides for the CSK biosensor: 1) from the Src C-terminal tail (FTSTEPQYQPGENL) [25]; 2) from the Fyn C-terminal tail (FTATEPQYQPGENL); 3) the optimal peptide substrate for CSK determined through screening a random peptide library (EEEIYFFF) [26].
As shown in Figure 1A, when the substrate is not phosphorylated, due to the spatial proximity between N- and C-termini of SH2 domain in the three-dimensional structure [27], the donor ECFP is positioned closely to the acceptor YPet, resulting in high FRET efficiency. Upon phosphorylation of the tyrosine by the kinase, the phosphorylated substrate binds to the SH2 pocket located at the opposite site of its two termini, then YPet and ECFP separate, leading to a decrease in FRET efficiency. The phosphorylation and FRET changes in the biosensor are reversible when the substrate is dephosphorylated. Therefore, the ECFP/FRET emission ratio of this biosensor can be used to indicate the activity of CSK kinase in vitro and in live cells.
The purified biosensor proteins expressed in E. coli showed a predicted molecular size around 70 kDa (Figure S1), indicating appropriate folding of the recombinant biosensor consisting of multiple fragments. Furthermore, we characterized the biosensor containing the three different substrate peptides in mammalian cells. Since tyrosine kinase activations and the subsequent substrate phosphorylation are relatively fast reactions, typically reaching plateau in growth factors-stimulated cells within 10−15 min [28], we focus on this physiologically relevant period and monitor the biosensor within 30 min of activation in live cells (Figure 1B). Biosensors for three different substrate peptides were separately transfected into rat airway smooth muscle (ASM) cells, and the changes of ECFP/FRET emission ratio represented the activity of CSK kinase. These experimental results suggest that the activity of CSK kinase is relatively low in the cytoplasm. Interestingly, the biosensor with the substrate peptide from screening a random peptide library (EEEIYFFF) showed an increased ECFP/FRET emission ratio change of approximately 26%, while the other two biosensors showed minimal changes (Figure 1D). Similar results were achieved for the three substrates-based biosensors in HeLa cells with EGF stimulation (Figure S2A&B). Therefore, The EIY version was chosen to continue with the research of this work.
2. Detection of CSK Activity with FRET Biosensor at Membrane Microregions
According to reports, CSK is a cytoplasmic protein because it does not contain acylation sites, so it needs to be recruited to the plasma membrane to exert its inhibitory function [29,30]. Therefore, adapter proteins are required. CSK binding protein (CBP) is a transmembrane protein localized in lipid rafts [31], also known as a lipid-associated phosphoprotein. In T cells, CBP binds to the SH2 domain of CSK, which inhibits SFKs [30]. When CBP is dephosphorylated, for example, upon activation of the T cell receptor, it dissociates from CSK, enabling activating SFK activity as SFK is no longer inhibited by CSK [32].
To observe the activity of CSK in the DRM (detergent-resistant membrane) region, Fyn-tagged CSK and Lyn-tagged CSK biosensors were constructed by fusing a DRM-targeting motif containing myristoylation and palmitoylation sites (glycine and cysteine) derived from Fyn and Lyn, respectively, at the N-terminus of “EEEIYFFF” substrate-based cytosolic CSK (Cyto-CSK) biosensor [33]. Additionally, a polybasic motif (Arginine) adapted from K-Ras was fused at the carboxyl terminus of the Cyto-CSK biosensor to generate the KRas-tagged CSK biosensor, enabling its localization to the general membrane region outside of the DRM [33]. Similar results have confirmed the proper localization of proteins modified by Fyn, Lyn, and KRas [33,34]. Confocal images showed membrane localizations for the three versions of the biosensor (Figure S3). Along with previous studies, Fyn, Lyn, and KRas-tagged CSK biosensors can be targeted to different microdomains on the plasma membrane successfully through distinct lipid modifications [22].
We transfected the three types of membrane-targeted biosensors and the selected EIY-cytosolic biosensor into ASM cells (Figure 1C). Upon stimulation with PDGF, all four types of biosensors showed an immediate increase in ECFP/FRET ratio (Figure 1E, Movies S1-S4). The ECFP/FRET ratio of the KRas-CSK biosensor was significantly higher than that reported by the other two membrane-targeted biosensors and the cytosolic one, when the FRET ratio of each group of cells was measured before and after PDGF treatment (0, 20 min) (Figure 1F). As control, the cells expressing KRas-CSK biosensor didn’t show FRET change when without PDGF stimulation during the same imaging process (Figure S4). The cytosolic biosensor can diffuse three-dimensionally throughout the entire cell, while biosensors localized to specific cell membrane regions are restricted to 2D membrane microdomains [20]. The differences in the local topology of the biosensors may contribute to their response dynamics. This indicates that the CSK kinase activity is primarily localized to non-DRM regions. Therefore, the KRas-CSK version was selected as the optimal CSK biosensor and referred to as the CSK biosensor for the remaining parts of this study.
Compared with wild-type CSK-FRET biosensors, mutant biosensors with monotyrosine (Y) mutated to non-phosphorylated phenylalanine (F) in their polypeptide substrate showed a significantly lower increase in the ECFP/FRET ratio under PDGF stimulation (Figure 1G, H). The KRas-tagged Y/F mutant wasn’t generated successfully yet. This result suggests that the FRET response of the biosensor is dependent on tyrosine phosphorylation of the designed CSK substrate sequence (Figure 1A) and that the FRET response of the CSK biosensor is primarily attributable to intracellular CSK kinase activity. These results further demonstrate the specificity of the designed biosensor to CSK kinase.
3. Demonstration of SH2 Domain Crucial for CSK Activation
In order to further validate the specificity of the CSK biosensor, wild-type CSK (WT), SH2-inactive CSK (R107E), and CSK lacking the SH3 domain (ΔSH3) plasmids [35] were co-transfected into ASM cells. The activity of CSK was detected using the CSK-FRET biosensor, which was co-transfected with the CSK plasmids into ASM cells. PDGF was used to rapidly activate CSK in the cells. The results showed that after PDGF stimulation, the ECFP/FRET ratio in the cells immediately increased (Figure 2A, B). By analysis of the effect of different CSK plasmids on CSK activity before and after PDGF treatment, CSK(WT) and CSK(ΔSH3) led to increase of the activity in the cells both at the basal level and upon activation, whereas the SH2-inactive CSK(R107E) did not have this effect (Figure 2C). These experiments demonstrate that the CSK-FRET biosensor has good specificity for CSK kinase.
Yaqub et al. [36] suggested that the SH3 domain is crucial for maintaining CSK activity, whereas this conclusion has not been validated in this study. Furthermore, this experiment further proves the essential role of the SH2 domain for CSK activation, which cannot be replaced or lacking.
4. To Characterize the Specificity of CSK Biosensor to CSK Kinase.
To further examine the specificity of the CSK biosensor in mammalian cells, we introduced CSK biosensors into ASM cells along with wild-type Src (Src-WT) and wild-type Fyn (Fyn-WT) and measured the ECFP/YPet emission ratio (Figure 3A). The time curves of quantitative analysis indicates that CSK activity was effectively activated under both Src WT and Fyn WT expression conditions (Figure 3B). Further statistical comparison showed that CSK activity was significantly higher in cells expressing Src-WT and Fyn-WT than in the control group before PDGF stimulation. It is speculated that CSK is a negative regulator of SKFs, and overexpression of Src kinase and Fyn kinase in cells promotes the activation of CSK kinase. However, after PDGF stimulation, there was no significant difference between the experimental group expressing Fyn-WT and the control group. Interestingly, CSK activity was reduced in the experimental group expressing Src-WT, which needs further study (Figure 3C).
In considering cells expressing endogenous CSK, the same co-transfection method was used to simultaneously transfect inhibitory CSK(R107E) on the basis of the above experiments (Figure 3D). After PDGF stimulation, the time curve of quantitative analysis indicates that CSK activity could be effectively activated under the different conditions (Figure 3E). Further statistical comparison of the difference showed that before PDGF stimulation, there was no significant difference between the experimental group and the control group (pcDNA3.1), while after PDGF stimulation, intracellular CSK activity was significantly inhibited in the experimental group (Figure 3F). These results suggest that in mammalian cells, the substrate tyrosine is mostly phosphorylated by CSK, leading to changes in FRET of the biosensor.
5. Comparison of the Activity Levels of Different Kinases in ASM Cells.
Biosensors based on FRET technology have been developed to detect the activities of Src, Fyn, and focal adhesion kinase (FAK), which are very mature biosensors with verified specificity and sensitivity. We compared the FRET responses of constructed CSK biosensor with the three biosensors to study the activity differences of these kinases in membrane microregions of the cells. The four biosensor plasmids were transfected in ASM cells including KRas-CSK FRET, FAK-FRET [22], Src-FRET [37], and Fyn-FRET [20], respectively, and FRET responses were stimulated with PDGF in the cells (Figure 4A, B). The quantification data showed that the ECFP/FRET emission ratio change of KRas-CSK-FRET was 19%, FAK-FRET 3%, Src-FRET 44%, and Fyn-FRET was 60% (Figure 4C, D). This indicates that in the membrane microregions, the activity of CSK kinase is lower than that of Fyn kinase and Src kinase, but higher than that of FAK kinase. Hence, the activity of CSK kinase in the membrane microregion is relatively mild in comparison to the high activity level of Src and Fyn kinases.
6. The Protein Tyrosine Phosphatase PTPα in Regulating CSK Kinase Activity.
The receptor-type protein tyrosine phosphatase alpha (PTPα) is a widely expressed 130 kDa transmembrane protein that contains a short extracellular glycosylated domain and two intracellular catalytic domains [38]. PTPα belongs to the PTP superfamily, which includes over 110 other receptor-type and intracellular PTPs, and it shares high homology with the closely related PTPε [39]. Previous studies have demonstrated that PTPα can regulate the invasiveness of colon cancer cells, as confirmed by in vitro experiments using the chicken chorioallantois membrane assay [40]. Additionally, PTPα acts as an activator of the oncogene c-Src and other Src family kinases (SFKs), through dephosphorylation of the C-terminal tyrosine residue (Tyr527) of c-Src both in vitro and in vivo [38,41]. Fibroblasts from PTPα gene knockout mice exhibited enhanced Src phosphorylation at Tyr527, resulting in decreased Src activity [41,42].
When investigating the mechanism by which PTPα regulates Src activity, researchers found that the Tyr789 residue of PTPα is essential for the binding of PTPα to Src and for Src dephosphorylation and activation. Tyr789 of PTPα binds to the Src SH2 domain [43,44], leading to the exposure of the phosphorylated Src carboxyl terminus and subsequent dephosphorylation, which activates Src [45]. RPTPα is compositionally phosphorylated on Ser180, Ser204 and Tyr789 [46]. In the interphase, phosphorylation of Ser204 inhibits the binding of Src, so Src is not activated. In mitosis, pSer204 is dephosphorylated due to activation of PP2A. Non-phosphorylated Ser204 binds to Src via regions that have not yet been identified in Src, and RPTPα while maintaining phosphorylation on Tyr789 binds to the adaptor protein GRB2. The binding of Src to RPTPα in mitosis leads to dephosphorylation of pTyr527 and pTyr416 in Src, resulting in moderate activation of Src kinase activity. After mitosis release, Ser204 is rapidly phosphorylated and Src phosphorylation returns [47].
The primary function of PTPα is to serve as a positive regulator of SFKs, while CSK acts as a negative regulator of SFKs. Therefore, this study examined the effect of PTPα on CSK kinase activity. Various PTPα plasmids with functional site mutations were used: wild-type PTPα (WT), PTPα with mutations in the catalytic active site (CCSS) and PTPα(S204A), and PTPα lacking key tyrosine phosphorylation sites in the C-terminus (Y789F). The time course of quantified analysis reflected effective activation of CSK activity under different conditions of PTPα expression (Figure 5A, B). Furthermore, statistical comparison of the FRET levels between the experimental and control groups (pcDNA3.1) revealed that all four PTPα plasmids had a certain degree of upregulating effect on CSK activity before PDGF stimulation, with PTPα(S204A) showing the most significant effect (Figure 5C). However, after PDGF stimulation, PTPα(CCSS) and PTPα(Y789F) exerted a certain inhibitory effect on CSK activity, while the inhibitory effect of wild-type PTPα(WT) and PTPα(S204A) lacking the catalytic domain for phosphatase activity was not significant (Figure 5C). Therefore, these results suggest that the expression of PTPα in cells has a certain inhibitory effect on CSK kinase activity upon PDGF stimulation.
Furthermore, this study investigated whether the inhibitory effect of PTPα acts directly on the FRET biosensor or inhibits intracellular CSK protein. Using the same co-transfection method as described above, CSK (R107E) was co-transfected, which acts as a CSK protein inhibitor. The time course of quantified analysis again reflected effective activation of CSK activity under different conditions of PTPα expression (Figure 5D, E). Moreover, a statistical comparison of the ECFP/FRET between the experimental groups and control (pcDNA3.1) revealed that all four PTPα plasmids exerted a certain inhibitory effect on CSK activity (Figure 5F), regardless of whether it was before or after PDGF stimulation. Therefore, this further confirms that PTPα has a certain inhibitory effect on CSK FRET responses.
In conclusion, the experimental results mentioned above demonstrate that PTPα has a certain inhibitory effect on CSK kinase activity, whether it acts on the FRET biosensor or inhibits intracellular CSK protein. These findings provide new insights into the regulatory relationship between PTPα and CSK and offer important clues for further investigation into this mechanism.
7. Detection of CSK Activation in Cancer Cells.
Since SFKs are involved in the signaling cascade of cell survival, proliferation, and migration, it is not surprising that abnormal SFK activity promotes tumorigenesis. In fact, SFK has been reported to have high levels and activity in different types of tumors, including lung, skin, colon, and breast cancer [48]. For example, Redin et al. have shown that in the case of non-small cell lung cancer (NSCLC), Yes1, as a member of SFKs, is involved in increasing the number of regulatory T cells (Treg) capable of infiltrating tumors, so Yes1 is one of the most important predictors of poor prognosis for this type of cancer. Subsequently, they tried using SFK inhibitors in this particular type of tumor, along with immune checkpoint inhibitors (ICIs), which are known to have beneficial effects, resulting in accelerated tumor regression compared to monotherapy because the SFK inhibitor dasatinib works synergistically with ICIs while also reducing the number of Tregs [49]. This also hints at the possible function of CSK as a tumor suppressor. Masaki et al. observed reduced CSK levels in human hepatocellular carcinoma, suggesting that CSK has this antitumor effect [50]. CSK is effective in inhibiting cancer progression, while its downregulation supports tumorigenesis. The mechanism dysfunction of CSK function in normal cells promotes the development of cancer [16].
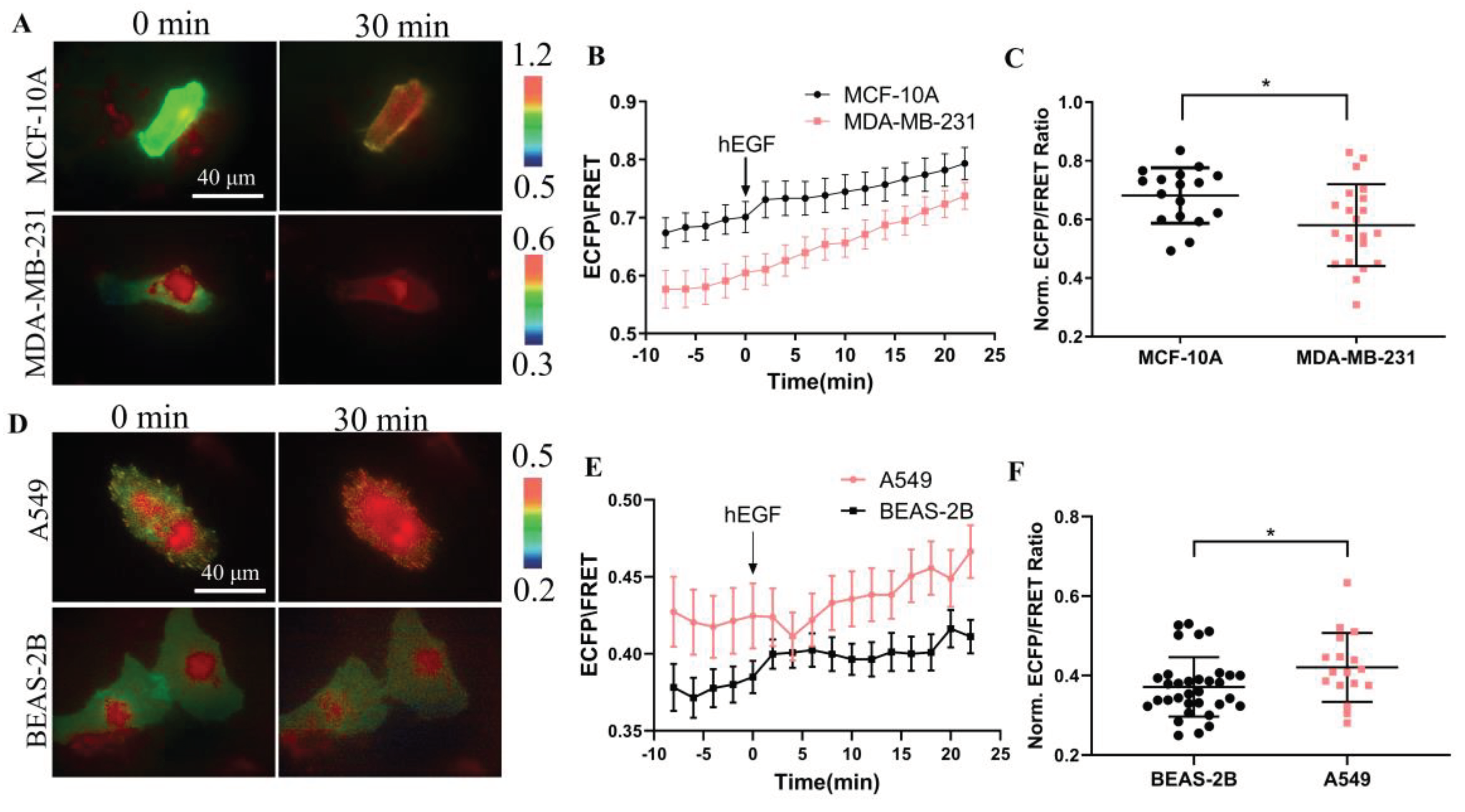
Therefore, we used the designed biosensor to detect the activity of CSK protein in normal cells and cancer cells and performed a comparative analysis. As shown in Figure 6A, KRas-CSK biosensors were transfected in breast cells MCF-10A and malignant breast cancer cells MDA-MB-231, respectively, and we found that the ECFP/FRET ratio in these two cells was between 0.55~0.7 (Figure 6B), and after stimulation with EGF, there was a certain significant difference in the activity of CSK protein in the two cells, and in normal breast cells, the activity of CSK protein was higher (Figure 6C). When KRas-CSK biosensors were transfected in human normal lung epithelial cells BEAS-2B and human non-small cell lung cancer cell A549 (Figure 6D), respectively, and we found that the ECFP/FRET ratio in these two cells was between 0.35~0.5 (Figure 6E), and after stimulation with EGF, the CSK protein activity was also different, but the activity of the CSK protein in cancer cells A549 was significantly higher than that of normal cells BEAS-2B (Figure 6F). In summary, the difference of CSK activity in cancer cells and non-cancer cells is not very obvious, and the relevance between CSK protein and cancer development needs to be further explored.
Discussion
C-terminal Src kinase (CSK) is a cytosolic tyrosine-protein kinase with an important role in regulating critical cellular decisions, such as cellular apoptosis, survival, proliferation, cytoskeletal organization and many others. Current knowledge of the mechanisms, regulation, and function of CSK is still in its early stages, and most of the known roles and functions of CSK are known by inhibitory regulation of Src family kinases (SFKs). Since SFKs play a vital role in apoptosis, cell proliferation, and survival regulation, the inhibition of SFK by CSK has a pro-apoptotic effect, which is mediated by inhibiting the cell signaling cascade controlled by SFKs, such as the MAPK/ERK, STAT3, and PI3K/AKT signaling pathways. Abnormal activation of CSK and SFK can lead to cancer, cardiovascular, and neurological disorders [16].
Our work developed and characterized the first CSK FRET biosensor for sensitive monitoring of CSK kinase activity in living cells. Using the SH2 domain with a tunable binding affinity to tyrosine phosphorylated substrate peptides, an optimized biosensor containing SH2 from Fyn-FRET and a piece of CSK substrate randomly screened from the peptide library was designed. The optimal polypeptide as a substrate is more sensitive in FRET response to that of a peptide that mimics the tail of the Src C-terminal, and bears little resemblance to a physiological phosphorylation site. By comparison in ASM cells, the biosensor showed a clear preference for CSK activity than Src and Fyn kinases.
The effect of CSK on SFK is largely dependent on the CSK-binding protein CBP/PAG-1, which binds to the SH2 domain of CSK using its phosphotyrosine-containing motif. This binding not only activates CSK, but also recruits CSK into SFK-enriched membrane microregions. This allows CBP to play an important role in CSK-mediated phosphorylation/inactivation of SFKs. Here, we place the CSK biosensor close to the CBP on the cell membrane via the Fyn, Lyn, KRas tags to monitor CSK activity in different membrane microregions. We observed the film localization images of Lyn-tag and KRas-tag CSK biosensors by confocal microscopy, while the film localization of Fyn-tag one was less clear (Figure S3). This design strategy highlights the spatial resolution achievable by genetically targeted and subcell-anchored CSK FRET biosensors, which should be difficult for cytosolic biosensor because the reported signals are averaged across spatially.
CSK kinases are known to be located in these membrane microregions by binding proteins on the cell membrane. Biosensors labeled KRas tag had more and stronger FRET responses to PDGF stimuli than sensors labeled Fyn and Lyn tags (Figure 1D-F), although the three membrane-targeted biosensors showed similar dynamic ranges in FRET changes (Figure 1H). This is surprising, considering that these three versions of the biosensor are identical, except for small tags at the N- or C-terminal. Previous studies have shown CSK is recruited by CBP which is located at lipid raft regions, and then activated CSK phosphorylates SFKs to result in SFKs inhibitions [14,15]. Study has also shown that Src kinase is mainly located at the non-lipid raft region through N-terminal myristoylation while without N-terminal palmitoylation [51]. In order to keep Src kinase inactive, reasonably the Src-CSK complex may remain at the non-lipid raft region. At the same time, CSK may form complex with other SFK members such as Fyn and Lyn at the lipid raft regions. Actually, it has been reported that CSK is located at both raft and non-raft membrane fractions in cells, although CBP is mostly located at rafts [52]. Together with our observations based on FRET measurements, the activity of CSK kinase exists in both lipid raft and non-lipid raft regions, while is more dominant in the non-lipid raft regions. It will also be interesting to learn more about how these microdomains are classified into different segments based on their specific functional molecules. Therefore, biosensors with specific targeted markers may be useful in characterizing subtle differences between different membrane microdomains and investigating the underlying mechanisms of local molecular regulation.
In 1992, Zheng et al. established a rat embryo fibroblast (REF) model of rat embryo fibroblast (REF) overexpressing RPTPα, and found that overexpression of RPTPα could specifically catalyze the dephosphorylation of tyrosine residue at position 527 (corresponding to human c-Src 530) of the c-Src C-terminus, which continuously activated C-Src and induced malignant transformation of cells [53]. With the deepening of the research, Zheng et al. further proposed a "displacement model": RPTPα pTyr789 binds to Grb2 SH2-c-SH3 at rest, while c-Src pTyr527 preferentially binds to self-SH2 [54]. Under certain activation signals, RPTPα pTyr789 dissociates from Grb2, and in turn binds to c-Src SH2 and dephosphorylates pTyr527, thereby activating c-Src. The protein tyrosine kinase CSK can phosphorylate the 527-position tyrosine at the C-terminal of Src, so we further explore the relationship between these two different proteins acting on the same site of Src. Our observations show that RPTPα has a certain inhibitory effect on CSK kinase (Figure 5). Because these two proteins are positive and negative regulators of Src kinase, respectively, there may be antagonism between the two, so when the activity of one protein is increased, the other protein is correspondingly reduced. However, how RPTPα inhibits CSK, the interaction sites and mechanisms between them need to be further explored.
Because SFKs are overexpressed or continuously activated in a variety of cancers [55], a series of studies have pointed to the role of SFKs in tumors. SFKs, especially Src, can participate in the tumorigenesis process by activating STAT transcription factors [56]. A variety of human tumors have high levels of Src kinase activity expression, for example, breast, colorectal, and pancreatic cancers [57]. The increase in Src kinase activity in this tumor may be caused by tyrosine phosphatase-mediated dephosphorylation of carboxyl terminal negative regulatory elements, or by increased protein levels and protein stability of Src, or by upregulation of upstream receptor tyrosine kinase activity and loss of key regulatory proteins. CSK is a negative regulator of Src, so we further investigated the expression of CSK kinase in cancer cells. Based on our observations, we found that there was no significant difference in the expression of CSK kinase in cancer cells and non-cancer cells (Figure 6). So in cancer cells, whether there is a correlation between the expression of the two kinases needs to be explored.
In summary, we have developed a FRET-based CSK biosensor that can detect CSK activity in living cells with high spatiotemporal resolution. Positioning the CSK biosensor in membrane microregions by acylation and prenylation can further provide a powerful tool for monitoring dynamic molecular activity in subcellular compartments. Our results show that CSK activity exist in both non-lipid and lipid rafts membrane while is more localized in the non-lipid raft region of the cell membrane. Hypothetically along with the previous studies, after the binding protein CBP/PAG-1 is phosphorylated by SFKs, the CSK kinase is recruited using the phosphotyrosine motif it contains, and binds to its SH2 domain, resulting in the CSK kinase being close to the activated Src kinase, phosphorylating the C-terminal tyrosine residue of the Src kinase, inhibiting Src activity, and the inactivated Src maintained to the non-lipid raft region together with the complex formed by the CSK kinase.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgements
We appreciate the advice for this work from Professor Yingxiao Wang (University of Southern California); the work was assisted with help from Dr. Jia Guo, Jingjing Li, and Lei Liu (Changzhou University). This project was supported financially by National Natural Science Foundation of China (NSFC 12372312, 11872129), Natural Science Foundation of Jiangsu Province (BK20181416), Projects of “Jiangsu Specially-appointed Professor” (M.O.); National Natural Science Foundation of China (12272063) (L.D.).
Author contributions
M.O. and L.D. designed the research; Y.X. performed the majority of experiments; Y.X. and M.O. carried the major data organization and analysis; S.Z. helped with project discussion and manuscript editing; Y.P. assisted with experiments; L.D. provided the setups of equipment; Y.X., M.O., and L.D. prepared the paper.
Statement for No Conflict of Interest
The authors declare no conflicts of interest with the contents of this article.
References
- Coffin, J.M. Genes responsible for transformation by avian RNA tumor viruses. Cancer Res 1976, 36(11 Pt. 2), 4282–8. [Google Scholar]
- Kawai, S., P.H. Duesberg, and H. Hanafusa, Transformation-defective mutants of Rous sarcoma virus with src gene deletions of varying length. J Virol 1977, 24, 910-4. [CrossRef]
- Purchio, A.F.; et al. Identification of a polypeptide encoded by the avian sarcoma virus src gene. Proc Natl Acad Sci U S A 1978, 75, 1567–71. [Google Scholar] [CrossRef]
- Roskoski, R., Jr., Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res 2015, 94: p. 9-25. [CrossRef]
- Roskoski, R., Jr. , Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Commun 2004, 324, 1155–64. [Google Scholar] [CrossRef]
- Okada, M. and H. Nakagawa, A protein tyrosine kinase involved in regulation of pp60c-src function. J Biol Chem 1989, 264, 20886–93. [Google Scholar] [CrossRef]
- Okada, M. and H. Nakagawa, Identification of a novel protein tyrosine kinase that phosphorylates pp60c-src and regulates its activity in neonatal rat brain. Biochem Biophys Res Commun 1988, 154, 796–802. [Google Scholar] [CrossRef]
- Okada, M. , Regulation of the Src Family Kinases by Csk. International Journal of Biological Sciences 2012, 8, 1385–1397. [Google Scholar] [CrossRef]
- Xu, W.; et al. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell 1999, 3, 629–38. [Google Scholar] [CrossRef]
- Sun, G. and M.K. Ayrapetov, Dissection of the catalytic and regulatory structure-function relationships of Csk protein tyrosine kinase. Front Cell Dev Biol 2023, 11: p. 1148352. [CrossRef]
- Nada, S.; et al. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell 1993, 73, 1125–35. [Google Scholar] [CrossRef]
- Kawabuchi, M.; et al. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature 2000, 404, 999–1003. [Google Scholar] [CrossRef]
- Svec, A. , Phosphoprotein associated with glycosphingolipid-enriched microdomains/Csk-binding protein: a protein that matters. Pathol Res Pract 2008, 204, 785–92. [Google Scholar] [CrossRef]
- Cartwright, C.A.; et al. Cell transformation by pp60c-src mutated in the carboxy-terminal regulatory domain. Cell 1987, 49, 83–91. [Google Scholar] [CrossRef]
- Cowan-Jacob, S.W.; et al. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure 2005, 13, 861–71. [Google Scholar] [CrossRef]
- Fortner, A., et al., Apoptosis regulation by the tyrosine-protein kinase CSK. Frontiers in Cell and Developmental Biology, 2022. 10. [CrossRef]
- Roskoski, R., Jr. , Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun 2005, 331, 1–14. [Google Scholar] [CrossRef]
- Bajar, B., et al., A Guide to Fluorescent Protein FRET Pairs. Sensors 2016, 16(9). [CrossRef]
- Ouyang, M.; et al. Determination of hierarchical relationship of Src and Rac at subcellular locations with FRET biosensors. Proc Natl Acad Sci U S A 2008, 105, 14353–8. [Google Scholar] [CrossRef]
- Ouyang, M.; et al. Sensitive FRET Biosensor Reveals Fyn Kinase Regulation by Submembrane Localization. ACS Sensors 2018, 4, 76–86. [Google Scholar] [CrossRef]
- Komatsu, N.; et al. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell 2011, 22, 4647–56. [Google Scholar] [CrossRef]
- Seong, J., et al., Detection of focal adhesion kinase activation at membrane microdomains by fluorescence resonance energy transfer. Nature Communications 2011, 2(1). [CrossRef]
- Yao, H., et al., Genetically Encoded FRET Biosensor Detects the Enzymatic Activity of Prostate-Specific Antigen. Molecular & Cellular Biomechanics, 2020. [CrossRef]
- Qin, Q., et al., Fluocell for Ratiometric and High-Throughput Live-Cell Image Visualization and Quantitation. Front Phys, 2019. 7. [CrossRef]
- Weijland, A.; et al. Src regulated by C-terminal phosphorylation is monomeric. Proc Natl Acad Sci U S A 1997, 94, 3590–5. [Google Scholar] [CrossRef]
- Lee, S.; et al. Determination of the substrate-docking site of protein tyrosine kinase C-terminal Src kinase. Proc Natl Acad Sci U S A 2003, 100, 14707–12. [Google Scholar] [CrossRef] [PubMed]
- Waksman, G. Crystal structure of the phosphotyrosine recognition domain SH2 of the Src oncogene product complexed with tyrosine-phosphorylated peptides. Cell Mol Biol (Noisy-le-grand) 1994, 40, 611–8. [Google Scholar]
- Hamamura, K.; et al. Functional activation of Src family kinase yes protein is essential for the enhanced malignant properties of human melanoma cells expressing ganglioside GD3. J Biol Chem 2011, 286, 18526–37. [Google Scholar] [CrossRef] [PubMed]
- Manz, B.N.; et al. Small molecule inhibition of Csk alters affinity recognition by T cells. Elife 2015, 4. [Google Scholar] [CrossRef]
- Potuckova, L., et al., Positive and Negative Regulatory Roles of C-Terminal Src Kinase (CSK) in FcεRI-Mediated Mast Cell Activation, Independent of the Transmembrane Adaptor PAG/CSK-Binding Protein. Front Immunol 2018, 9: p. 1771. [CrossRef]
- Davidson, D.; et al. Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol Cell Biol 2003, 23, 2017–28. [Google Scholar] [CrossRef]
- Torgersen, K.M.; et al. Release from tonic inhibition of T cell activation through transient displacement of Cterminal Src kinase (Csk) from lipid rafts. J Biol Chem 2001, 276, 29313–8. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, D.A.; et al. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 2002, 296, 913–6. [Google Scholar] [CrossRef]
- Gao, X. and J. Zhang, Spatiotemporal analysis of differential Akt regulation in plasma membrane microdomains. Mol Biol Cell 2008, 19, 4366–73. [Google Scholar] [CrossRef]
- Kazi, J.U.; et al. The tyrosine kinase CSK associates with FLT3 and c-Kit receptors and regulates downstream signaling. Cell Signal 2013, 25, 1852–60. [Google Scholar] [CrossRef]
- Yaqub, S., et al., Activation of C-terminal Src kinase (Csk) by phosphorylation at serine-364 depends on the Csk-Src homology 3 domain. Biochem J 2003, 372(Pt 1): p. 271-8. [CrossRef]
- Seong, J., et al., Visualization of Src Activity at Different Compartments of the Plasma Membrane by FRET Imaging. Chemistry & Biology 2009, 16, 48-57. [CrossRef]
- Pallen, C.J. , Protein tyrosine phosphatase alpha (PTPalpha): a Src family kinase activator and mediator of multiple biological effects. Curr Top Med Chem 2003, 3, 821–35. [Google Scholar] [CrossRef]
- Krueger, N.X., M. Streuli, and H. Saito, Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. Embo j 1990, 9, 3241-52. [CrossRef]
- Krndija, D.; et al. Substrate stiffness and the receptor-type tyrosine-protein phosphatase alpha regulate spreading of colon cancer cells through cytoskeletal contractility. Oncogene 2010, 29, 2724–38. [Google Scholar] [CrossRef]
- Ponniah, S.; et al. Targeted disruption of the tyrosine phosphatase PTPalpha leads to constitutive downregulation of the kinases Src and Fyn. Curr Biol 1999, 9, 535–8. [Google Scholar] [CrossRef] [PubMed]
- Su, J., M. Muranjan, and J. Sap, Receptor protein tyrosine phosphatase alpha activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr Biol 1999, 9, 505-11. [CrossRef]
- Su, J., L.T. Yang, and J. Sap, Association between receptor protein-tyrosine phosphatase RPTPalpha and the Grb2 adaptor. Dual Src homology (SH) 2/SH3 domain requirement and functional consequences. J Biol Chem 1996, 271, 28086-96. [CrossRef]
- den Hertog, J., S. Tracy, and T. Hunter, Phosphorylation of receptor protein-tyrosine phosphatase alpha on Tyr789, a binding site for the SH3-SH2-SH3 adaptor protein GRB-2 in vivo. Embo j 1994, 13, 3020-32. [CrossRef]
- Maksumova, L.; et al. Differential function of PTPalpha and PTPalpha Y789F in T cells and regulation of PTPalpha phosphorylation at Tyr-789 by CD45. J Biol Chem 2007, 282, 20925–32. [Google Scholar] [CrossRef] [PubMed]
- Tracy, S., P. van der Geer, and T. Hunter, The receptor-like protein-tyrosine phosphatase, RPTP alpha, is phosphorylated by protein kinase C on two serines close to the inner face of the plasma membrane. J Biol Chem 1995, 270, 10587-94. [CrossRef]
- Vacaru, A.M. and J. den Hertog, Serine dephosphorylation of receptor protein tyrosine phosphatase alpha in mitosis induces Src binding and activation. Mol Cell Biol 2010, 30, 2850–61. [Google Scholar] [CrossRef]
- Ishizawar, R. and S.J. Parsons, c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–14. [Google Scholar] [CrossRef] [PubMed]
- Redin, E., et al., SRC family kinase (SFK) inhibitor dasatinib improves the antitumor activity of anti-PD-1 in NSCLC models by inhibiting Treg cell conversion and proliferation. J Immunother Cancer 2021, 9(3). [CrossRef]
- Masaki, T.; et al. Reduced C-terminal Src kinase (Csk) activities in hepatocellular carcinoma. Hepatology 1999, 29, 379–84. [Google Scholar] [CrossRef] [PubMed]
- Seong, J.; et al. Visualization of Src activity at different compartments of the plasma membrane by FRET imaging. Chem Biol 2009, 16, 48–57. [Google Scholar] [CrossRef]
- Oneyama, C.; et al. The lipid raft-anchored adaptor protein Cbp controls the oncogenic potential of c-Src. Mol Cell 2008, 30, 426–36. [Google Scholar] [CrossRef]
- Zheng, X.M., Y. Wang, and C.J. Pallen, Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature 1992, 359, 336-9. [CrossRef]
- Zheng, X.M., R.J. Resnick, and D. Shalloway, A phosphotyrosine displacement mechanism for activation of Src by PTPalpha. Embo j 2000, 19, 964-78.
- Summy, J.M. and G.E. Gallick, Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev 2003, 22, 337–58. [Google Scholar] [CrossRef]
- Silva, C.M. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004, 23, 8017–23. [Google Scholar] [CrossRef]
- Harris, K.F.; et al. Ubiquitin-mediated degradation of active Src tyrosine kinase. Proc Natl Acad Sci U S A 1999, 96, 13738–43. [Google Scholar] [CrossRef]
Figure 1.
Characterizations of the designed CSK biosensors with different substrate peptides and submembrane localizations. (A) The schematic drawing depicting the conformational change of the biosensor upon tyrosine phosphorylation at the substrate. (B, C) The ECFP/FRET ratiometric images of ASM cells treated with 10 μg/mL PDGF. The multiple versions of the biosensor consist of different substrate peptides (B) or submembrane-targeting signal peptides (C). The color bar from blue to red represents the emission ratio (ECFP/FRET) of biosensors from low to high. (D, E) The time courses of the ECFP/FRET emission ratio quantified for the cells in (B, C). (F) The scatter plots (mean ± S.D.) compare the basal and maximal levels of ECFP/FRET ratio in cells with time courses shown in (D) and (E). (**** indicates p < 1.0e-4, n = 21, 25, 20, 19, 22, 23). In cells transfected with Src-tail, Fyn-tail, and EIY CSK-FRET (1.0 μg of DNA on a 24-well plate), the ECFP/FRET ratio values (mean ± S.D.) were 0.297±0.028, 0.373±0.057, or 0.36 ± 0.035 at peak after PDGF stimulation, respectively. For Fyn-tag, Lyn-tag, or KRas-tag CSK-FRET, the ratio values were 0.33 ± 0.031, 0.33 ± 0.033, or 0.44 ± 0.035 at peak, respectively. (G) ECFP/FRET ratiometric images of the Y/F mutant biosensors before and after treatment with 10 μg/mL PDGF. Multiple versions of the biosensor consist of Y/F mutant in different substrate peptides or in the EIY biosensor with submembrane targeted peptides. (H) The scatter plots show quantified FRET changes of the different CSK biosensors along with the negative Y/F mutants in response to 10 μg/mL PDGF stimulation. (**** indicates p < 1.0e-4).
Figure 1.
Characterizations of the designed CSK biosensors with different substrate peptides and submembrane localizations. (A) The schematic drawing depicting the conformational change of the biosensor upon tyrosine phosphorylation at the substrate. (B, C) The ECFP/FRET ratiometric images of ASM cells treated with 10 μg/mL PDGF. The multiple versions of the biosensor consist of different substrate peptides (B) or submembrane-targeting signal peptides (C). The color bar from blue to red represents the emission ratio (ECFP/FRET) of biosensors from low to high. (D, E) The time courses of the ECFP/FRET emission ratio quantified for the cells in (B, C). (F) The scatter plots (mean ± S.D.) compare the basal and maximal levels of ECFP/FRET ratio in cells with time courses shown in (D) and (E). (**** indicates p < 1.0e-4, n = 21, 25, 20, 19, 22, 23). In cells transfected with Src-tail, Fyn-tail, and EIY CSK-FRET (1.0 μg of DNA on a 24-well plate), the ECFP/FRET ratio values (mean ± S.D.) were 0.297±0.028, 0.373±0.057, or 0.36 ± 0.035 at peak after PDGF stimulation, respectively. For Fyn-tag, Lyn-tag, or KRas-tag CSK-FRET, the ratio values were 0.33 ± 0.031, 0.33 ± 0.033, or 0.44 ± 0.035 at peak, respectively. (G) ECFP/FRET ratiometric images of the Y/F mutant biosensors before and after treatment with 10 μg/mL PDGF. Multiple versions of the biosensor consist of Y/F mutant in different substrate peptides or in the EIY biosensor with submembrane targeted peptides. (H) The scatter plots show quantified FRET changes of the different CSK biosensors along with the negative Y/F mutants in response to 10 μg/mL PDGF stimulation. (**** indicates p < 1.0e-4).
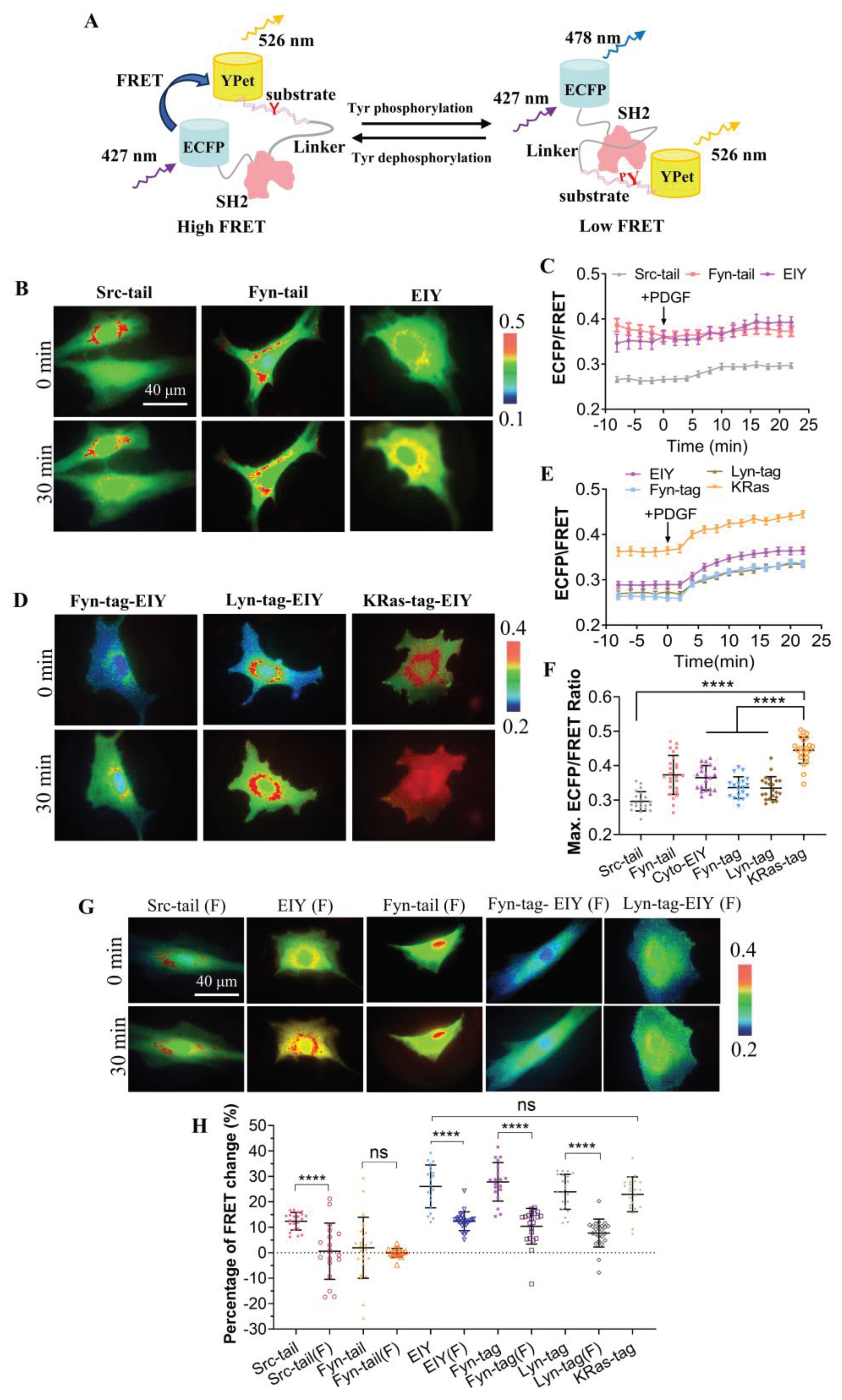
Figure 2.
FRET changes of the biosensor in response to co-transfections of CSK constructs. (A) Representative ECFP/FRET ratiometric images of PDGF-stimulated ASM cells co-transfected with KRas-CSK biosensor (1.0 μg DNA per well in 24-well plate) and vector only or the indicated CSK constructs (1.0 μg DNA each). (B) The time courses of the ECFP/FRET ratio in cells co-expressing the CSK mutants or control vector in (A). (C) The scatter plots compare the basal level and maximal ECFP/FRET ratio in cells with time courses shown in (B). The sample sizes n = 40, 43, 47, 45. In cells co-transfected with control vector, CSK(WT), CSK(ΔSH3), or CSK(R107E), the ECFP/FRET ratio values (mean ± S.D.) were 0.34 ± 0.07, 0.43 ± 0.073, 0.39 ± 0.062, or 0.36 ± 0.046 at the basal level and 0.44 ± 0.078, 0.48 ± 0.059, 0.47 ± 0.061, or 0.42 ± 0.047 at peak after PDGF stimulation, respectively.
Figure 2.
FRET changes of the biosensor in response to co-transfections of CSK constructs. (A) Representative ECFP/FRET ratiometric images of PDGF-stimulated ASM cells co-transfected with KRas-CSK biosensor (1.0 μg DNA per well in 24-well plate) and vector only or the indicated CSK constructs (1.0 μg DNA each). (B) The time courses of the ECFP/FRET ratio in cells co-expressing the CSK mutants or control vector in (A). (C) The scatter plots compare the basal level and maximal ECFP/FRET ratio in cells with time courses shown in (B). The sample sizes n = 40, 43, 47, 45. In cells co-transfected with control vector, CSK(WT), CSK(ΔSH3), or CSK(R107E), the ECFP/FRET ratio values (mean ± S.D.) were 0.34 ± 0.07, 0.43 ± 0.073, 0.39 ± 0.062, or 0.36 ± 0.046 at the basal level and 0.44 ± 0.078, 0.48 ± 0.059, 0.47 ± 0.061, or 0.42 ± 0.047 at peak after PDGF stimulation, respectively.
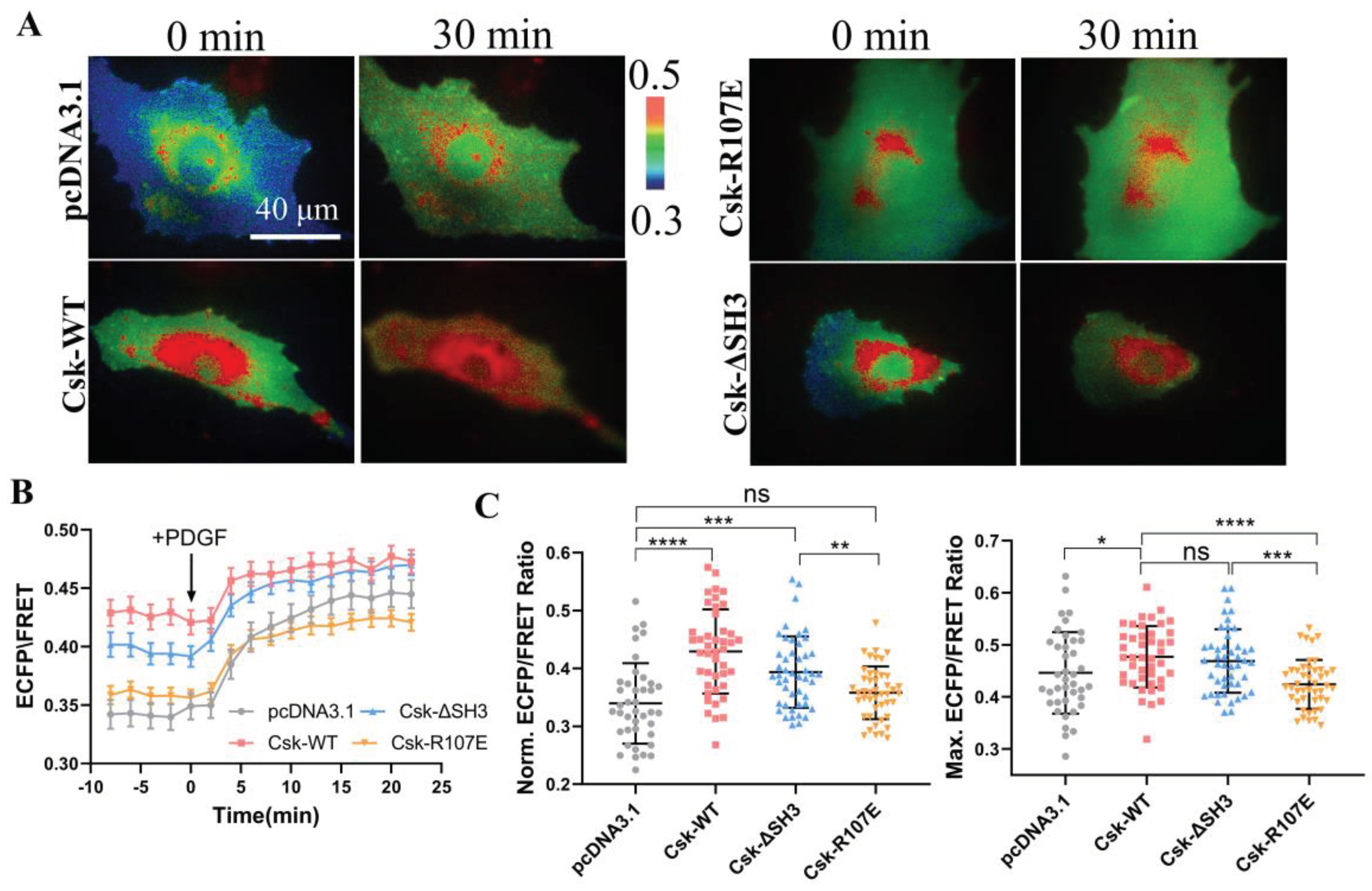
Figure 3.
Specificity of CSK biosensor for CSK kinase. (A) Representative ECFP/FRET ratio images of PDGF-stimulated ASM cells co-transfected with the KRas-CSK biosensor (1.0 μg DNA in 24-well plate) with vector only, Fyn-WT, or Src-WT (0.3 μg DNA each). (B) The average time courses of the ECFP/FRET ratio (mean ± S.E.M.) in cells co-expressing Fyn-WT, Src-WT or control vector in (A) treated with PDGF. (C) The scatter plots (mean ± S.D.) compare the basal level and maximal ECFP/FRET ratio in cells with time courses shown in (B). The sample sizes n=23, 24, 18. (D) Representative ECFP/FRET ratio images of PDGF-stimulated ASM cells co-transfected with the KRas-CSK biosensor (1.0 μg DNA in 24-well plate) and CSK-R107E (0.5 μg DNA each) with vector only, Fyn-WT, or Src-WT (0.5 μg DNA each). (E) The time courses of ECFP/FRET ratio in cells (D) treated with PDGF. (F) The scatter plots compare the basal level and maximal ECFP/FRET ratio in cells with time courses shown in (E). n=18, 16, 19.
Figure 3.
Specificity of CSK biosensor for CSK kinase. (A) Representative ECFP/FRET ratio images of PDGF-stimulated ASM cells co-transfected with the KRas-CSK biosensor (1.0 μg DNA in 24-well plate) with vector only, Fyn-WT, or Src-WT (0.3 μg DNA each). (B) The average time courses of the ECFP/FRET ratio (mean ± S.E.M.) in cells co-expressing Fyn-WT, Src-WT or control vector in (A) treated with PDGF. (C) The scatter plots (mean ± S.D.) compare the basal level and maximal ECFP/FRET ratio in cells with time courses shown in (B). The sample sizes n=23, 24, 18. (D) Representative ECFP/FRET ratio images of PDGF-stimulated ASM cells co-transfected with the KRas-CSK biosensor (1.0 μg DNA in 24-well plate) and CSK-R107E (0.5 μg DNA each) with vector only, Fyn-WT, or Src-WT (0.5 μg DNA each). (E) The time courses of ECFP/FRET ratio in cells (D) treated with PDGF. (F) The scatter plots compare the basal level and maximal ECFP/FRET ratio in cells with time courses shown in (E). n=18, 16, 19.
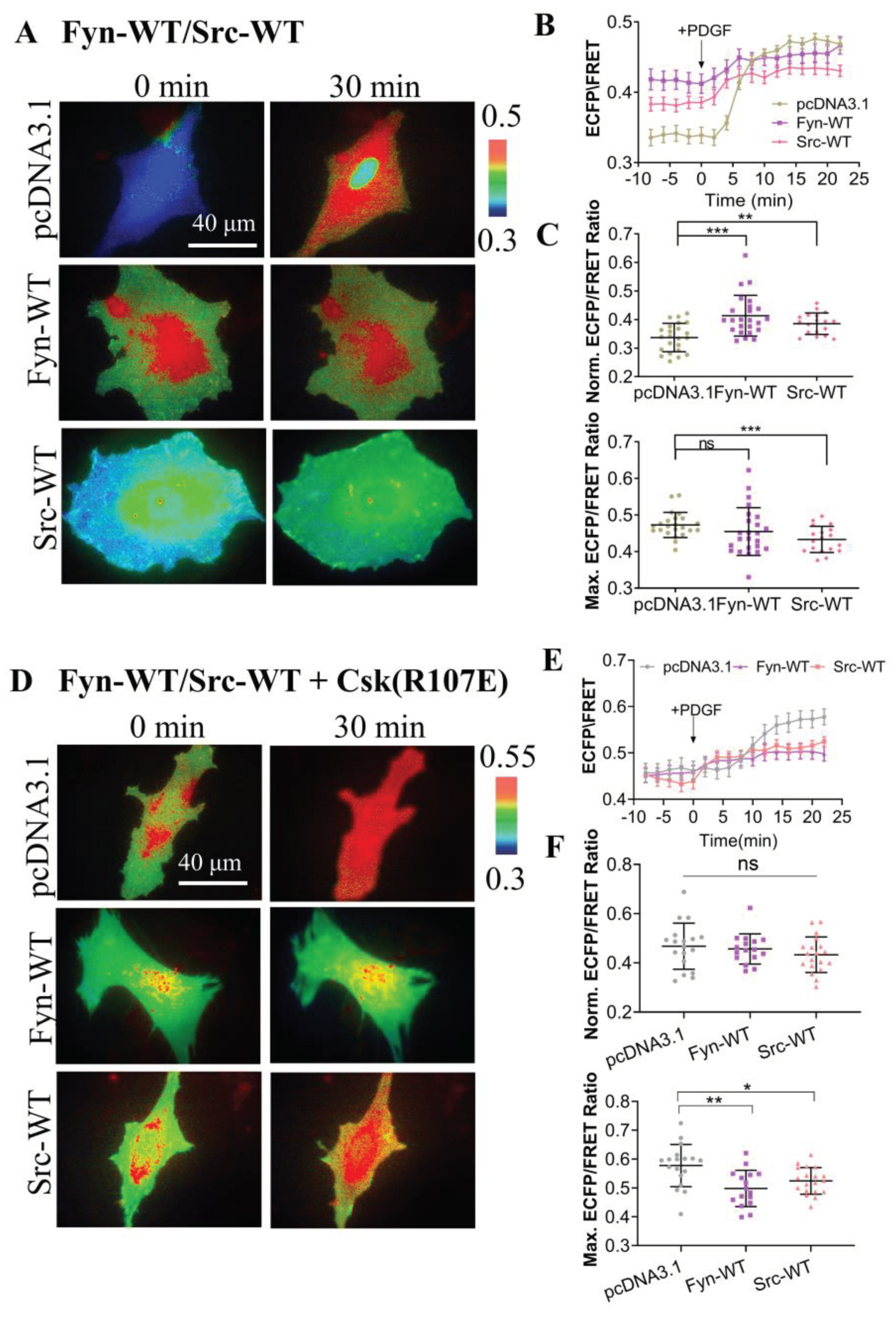
Figure 4.
Comparison of PDGF-induced FRET responses among multiple biosensors in ASM cells. (A) Representative ECFP/FRET ratio images of PDGF-stimulated ASM cells transfected with CSK-FRET, Src-FRET, Fyn-FRET and FAK-FRET biosensors with plasma membrane localizations. (B) The quantified time courses of the ECFP/FRET emission ratio for the cells shown in (A). (C) The normalized time courses of FRET changes in (A). (D) The scatter plots show quantified FRET changes of the biosensor in response to 10 μg/mL PDGF. The sample sizes n = 43, 42 ,42, 33.
Figure 4.
Comparison of PDGF-induced FRET responses among multiple biosensors in ASM cells. (A) Representative ECFP/FRET ratio images of PDGF-stimulated ASM cells transfected with CSK-FRET, Src-FRET, Fyn-FRET and FAK-FRET biosensors with plasma membrane localizations. (B) The quantified time courses of the ECFP/FRET emission ratio for the cells shown in (A). (C) The normalized time courses of FRET changes in (A). (D) The scatter plots show quantified FRET changes of the biosensor in response to 10 μg/mL PDGF. The sample sizes n = 43, 42 ,42, 33.
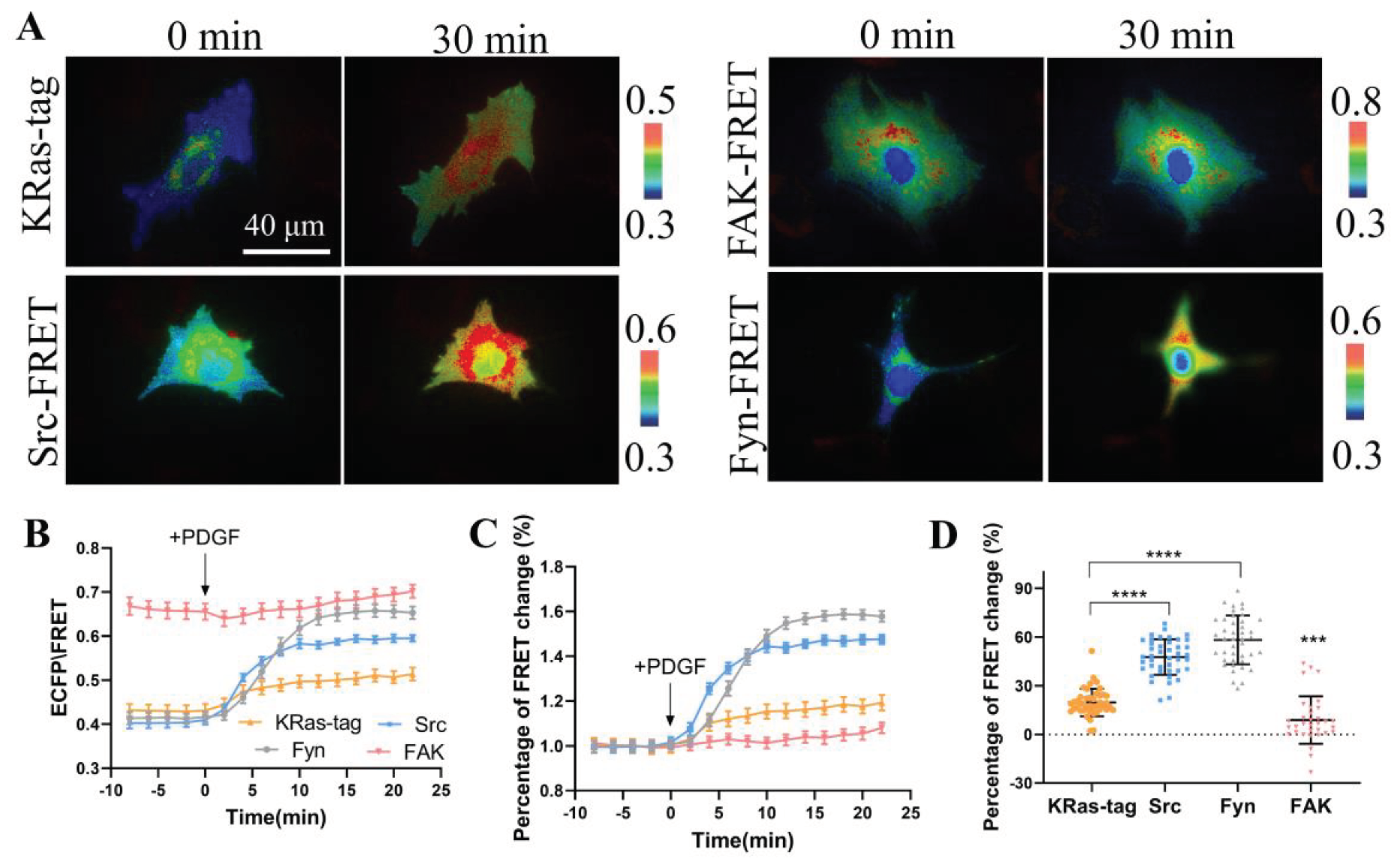
Figure 5.
The regulation of CSK FRET responses by co-transfected PTPα constructs. (A, D) Representative ECFP/FRET ratio images of PDGF-stimulated ASM cells co-transfected with KRas-CSK biosensor (1.0 μg DNA each in 24-well plate) with vector only or the indicated PTPα mutants (1.0 μg DNA each) (A), or co-transfected with KRas-CSK biosensor (1.0 μg DNA each) and CSK-R107E (0.5 μg DNA each) with vector only or the indicated PTPα mutants (0.5 μg DNA each) (D). (B, E) The time courses of the ECFP/FRET ratio (mean ± SEM) in cells co-expressing the PTPα or control vector in (A) and (D) when they are treated with PDGF. (C, F) The scatter plots (mean ± S.D.) compare the basal level and maximal ECFP/FRET ratio in cells with time courses shown in (B) and (E). The sample size n = 55, 61, 66, 68, 67 in (C); n = 58, 62,68, 66, 64 in (F).
Figure 5.
The regulation of CSK FRET responses by co-transfected PTPα constructs. (A, D) Representative ECFP/FRET ratio images of PDGF-stimulated ASM cells co-transfected with KRas-CSK biosensor (1.0 μg DNA each in 24-well plate) with vector only or the indicated PTPα mutants (1.0 μg DNA each) (A), or co-transfected with KRas-CSK biosensor (1.0 μg DNA each) and CSK-R107E (0.5 μg DNA each) with vector only or the indicated PTPα mutants (0.5 μg DNA each) (D). (B, E) The time courses of the ECFP/FRET ratio (mean ± SEM) in cells co-expressing the PTPα or control vector in (A) and (D) when they are treated with PDGF. (C, F) The scatter plots (mean ± S.D.) compare the basal level and maximal ECFP/FRET ratio in cells with time courses shown in (B) and (E). The sample size n = 55, 61, 66, 68, 67 in (C); n = 58, 62,68, 66, 64 in (F).
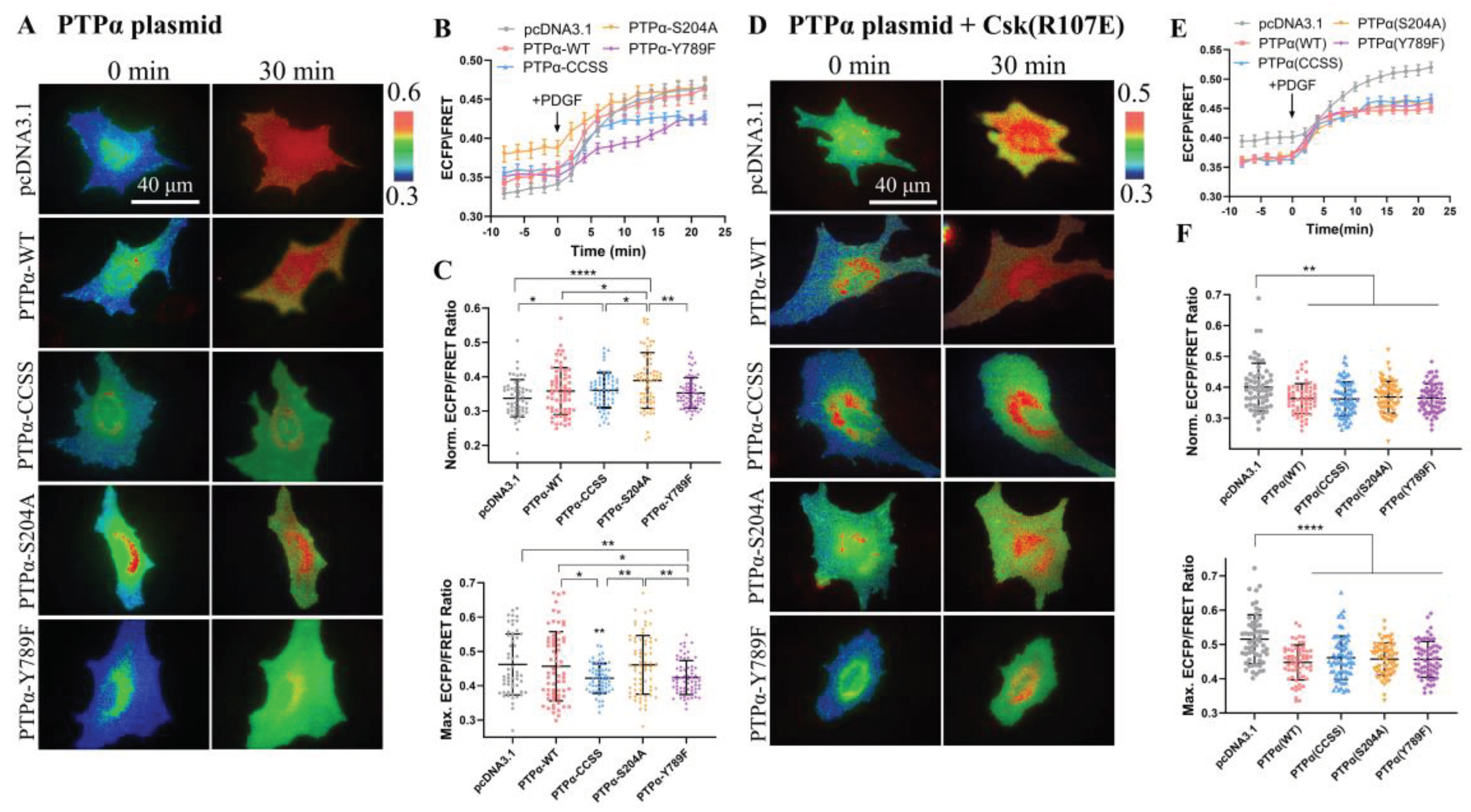
Figure 6.
Detection of CSK FRET levels in tumor cells. (A, D) Representative ECFP/FRET ratio images of EGF-stimulated cells transfected with the KRas-CSK biosensor (1.0 μg DNA per well in 24-well plate). (B, E) The time courses of the ECFP/FRET ratio (mean ± S.E.M.) in cells when they are treated with EGF. (C, F) The scatter plots (mean ± S.D.) compare the maximal ECFP/FRET ratio in cells with time courses shown in (B) and (E).
Figure 6.
Detection of CSK FRET levels in tumor cells. (A, D) Representative ECFP/FRET ratio images of EGF-stimulated cells transfected with the KRas-CSK biosensor (1.0 μg DNA per well in 24-well plate). (B, E) The time courses of the ECFP/FRET ratio (mean ± S.E.M.) in cells when they are treated with EGF. (C, F) The scatter plots (mean ± S.D.) compare the maximal ECFP/FRET ratio in cells with time courses shown in (B) and (E).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



