Submitted:
08 March 2024
Posted:
12 March 2024
You are already at the latest version
Abstract
The BRAFV600E mutation, found in approximately 50% of melanoma cases, is associated with aggressive tumor behavior and poor prognosis. This study aimed to assess its impact on immunogenic cell death (ICD), a pivotal cytotoxic process triggering anti-tumor immune responses. Through comprehensive in silico analysis of The Cancer Genome Atlas data, we explored the association between the BRAFV600E mutation, immune subtype dynamics, and tumor mutation burden (TMB). Our findings revealed that the mutation correlated with a lower TMB, indicating a reduced generation of immunogenic neoantigens. Investigation into immune subtypes reveals an exacerbation of immunosuppression mechanisms in BRAFV600E-mutated tumors. To assess the response to ICD inducers, including doxorubicin and Me-ALA-based photodynamic therapy (PDT), compared to the non-ICD inducer cisplatin, we used distinct melanoma cell lines with wild-type BRAF (SK-MEL-2) and BRAFV600E mutation (SK-MEL-28, A375). We demonstrated a differential response to PDT between the WT and BRAFV600E cell lines. Further transcriptomic analysis revealed upregulation of IFNAR1, IFNAR2, and CXCL10 genes associated with the BRAFV600E mutation, suggesting their involvement in ICD. Using a gene reporter assay, we showed that PDT robustly activated the IFN-1 pathway through cGAS-STING signaling. Collectively, our results underscore the complex interplay between the BRAFV600E mutation and immune responses, emphasizing a putative correlation between tumors carrying the mutation and their responsiveness to therapies inducing the IFN-1 pathway, such as the ICD inducer PDT, possibly mediated by the elevated expression of IFNAR1/2 receptors.
Keywords:
BRAFV600E mutation
; IFN-1 pathway
; melanoma
1. Introduction
Melanoma, a malignant neoplasm originating from melanocytes, represents a significant health concern globally due to its aggressive nature and propensity for metastasis [1]. Among the various genetic alterations implicated in melanoma pathogenesis, the BRAFV600E mutation stands out as one of the most prevalent, occurring in approximately 50% of cases. This mutation, characterized by a valine-to-glutamic acid substitution at codon 600 of the BRAF gene, leads to constitutive activation of the mitogen-activated protein kinase (MAPK) signaling pathway, driving uncontrolled cell proliferation and tumor progression [2,3].
Despite significant advancements in melanoma treatment, including the advent of targeted therapies and immunotherapies, challenges remain in achieving durable responses and overcoming resistance mechanisms. Immunotherapy, particularly immune checkpoint inhibitors (ICIs) targeting programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), has revolutionized the treatment landscape of melanoma. However, a subset of patients does not respond to immunotherapy, while others experience initial responses followed by relapse, highlighting the complexity of immune evasion mechanisms in melanoma [4,5].
Understanding the underlying causes of treatment failure, including intrinsic tumor resistance and acquired resistance mechanisms, is crucial for optimizing treatment strategies and improving patient outcomes. Emerging evidence suggests that resistance to immunotherapy may arise from various factors, including tumor-intrinsic factors such as genetic alterations, tumor heterogeneity, and dysregulated signaling pathways, as well as extrinsic factors involving the tumor microenvironment (TME) and immune escape mechanisms [6,7,8,9].
In this context, immunogenic cell death (ICD) has emerged as a promising therapeutic strategy, triggering an immune response against tumor cells. ICD is characterized by the release of Damage-Associated Molecular Patterns (DAMPs), which activate the immune system [10]. Among various ICD-inducers, photodynamic therapy (PDT) has been investigated, which involves the administration of a photosensitizer (PS) followed by visible light irradiation, leading to the generation of reactive oxygen species (ROS) and localized oxidative stress. In our previous research, we demonstrated that the prodrug Me-ALA induces the production of endogenous PS protoporphyrin IX (PpIX) localized to the endoplasmic reticulum (ER) of murine melanoma cells, triggering ER-stress-mediated apoptotic cell death. PDT-treated melanoma cells also facilitated the maturation of monocyte-derived dendritic cells (DCs), enhancing co-stimulatory signals and chemotaxis towards tumors [11]. Understanding melanoma cell sensitivity to ICD and the underlying molecular mechanisms is crucial for developing effective immunotherapeutic approaches.
Here, our comprehensive analysis, integrating bioinformatics tools and experimental approaches, aimed to investigate the relationship between the BRAFV600E mutation and genomic alterations, immune landscape, and its impact on immunogenic cell death (ICD) in melanoma. Our findings indicate a divergent sensitivity to specific ICD inducers, potentially linked to the BRAF mutation and its modulation of the interferon-1 (IFN-1) pathway. The IFN-1 signaling pathway, mediated by the regulation of interferon-stimulated genes (ISGs) in cancer, plays a crucial role in modulating the TME, regulating the anti-tumor immune response, and influencing therapy sensitivity [12,13]. Overall, our results reveal distinct genomic profiles and immune subtype dynamics associated with the BRAFV600E mutation in melanoma patients. Additionally, we provide insights into the complex interactions among BRAF signaling, ICD, and IFN-1 pathway activation, highlighting potential avenues for therapeutic intervention, immunomodulation, and enhancing responses to ICD in BRAF-mutated melanomas.
2. Materials and Methods
2.1. Cell Cultures
The BRAFV600E human melanoma cell lines SK-MEL-28 and A375 were generously provided by Dr. Pérez Saez (IBYME) and Dr. Álvarez (IMIBIO-SL), respectively. The BRAF WT human melanoma cell line SK-MEL-2 was generously provided by Dr. Álvarez (IMIBIO-SL). All cell lines were cultured according to ATCC protocols. Cells were maintained using DMEM (Modified Eagle medium of Dulbecco) (Gibco™), supplemented with 10% (v/v) fetal bovine serum (FBS) (Internegocios,) and 1% antibiotic-antimycotic (Gibco™) (10,000 units/ml penicillin, 10,000 μg/ml streptomycin and 25 μg/ml antifungal Fungizone) and were grown at 37°C with 5% CO2 in a humidified incubator. The SK-MEL-2-IFN (IFN-1 pathway reporter) cell clone was generated following the subsequent instructions and cultured under identical conditions to the other cell lines.
2.2. Database Analysis
The TCGA PanCancer database, accessible through cBioPortal [14] or Xena [15] was utilized for the in silico analyses (accessed December 2023). Genomic and transcriptomic data were obtained from 363 skin cutaneous melanoma (SKCM) patients via cBioPortal, with evaluation of BRAF gene mutation and mRNA expression of each gene of interest in diploid samples. Patients were categorized into two groups: those with wild-type (WT) BRAF and those with the BRAFV600E mutation. mRNA expression of genes associated with immunogenic cell death (ICD), particularly DAMPs or their modulators, was analyzed in these groups. Additionally, data from 103 patients sourced from the Xena dataset were categorized based on BRAF WT protein form or BRAFV600E mutation, and tumor mutation burden (TMB) was assessed.
2.3. Cell Treatments
2.3.1. Photodynamic Treatment
Cell lines were seeded into a 96-well plate (1x105 cell/mL). The following day, cells were incubated with pro-drug 5-methylaminolevulinic acid (Me-ALA) (Sigma) (0.25, 0.50, 1, and 2 mM) in DMEM 1% FBS for 4 h to allow the endogenous generation of the protoporphyrin IX photosensitizer (PpIX). After incubation, tumor cells were irradiated at room temperature with a light dose of 1 J/cm² (λ: 636 nm). The medium was then replaced with a fresh medium [11].
2.3.2. Chemotherapeutic Treatments
Cell lines were seeded into a 96-well plate (1x105 cell/mL). The following day, cells were treated with doxorubicin (DXR) (Glenmark Life Sciences) (1.5, 3, 6, and 12 μg /ml) or cisplatin (CP) (Deltapharma) (100, 200, 400, and 800 μg/ml) in DMEM 10% FBS for 24 h.
2.3.3. H151 (STING Inhibitor) Treatment and PDT
Cells were seeded into a 6-well plate (1x105 cell/mL). The following day, cells were treated with 1 or 10 𝜇M H151 or DMSO as vehicle in for DMEM 1% FBS for 4 h. A group of cells was simultaneously incubated with 1 mM Me-ALA, followed by the photodynamic treatment described above.
2.4. Cell Viability Assay
Cell viability was determined by the resazurin test using AlamarBlue™ (Invitrogen). According to the manufacturer's protocol, Alamar Blue dye was added to each well and incubated for 6 hours protected from light. Fluorescence was read in Varioskan LUX (Thermo Fisher Scientific) at λem = 570 nm - λex = 590 nm. The lethal dose 50 (LD50), the concentration that induced death in 50% of the treated cells, was determined through the preparation of dose-response curves applying a non-linear regression to the data (GraphPad Prism 8).
2.5. Generation of IFN-1 Pathway Reporter Human Melanoma Cell Line
To generate the reporter cell line, SK-MEL-2 cells were seeded at a density of 3.5x104 cells/mL in a 24-well plate. The following day, cells were transfected with the pMx2-eGFP plasmid containing the IFNα-inducing Mx2 promoter controlling GFP protein expression, using PEI 87 KDa (PolyAr87) in a reagent:DNA ratio of 2:1, as per the manufacturer’s instructions. The pMx2-eGFP plasmid was generously provided by Dr. María de los Milagros Bürgi (UNL) [16]. Stably transfected cells were selected by incubating them with G418 (600 μg/mL) for 21 days with medium refresh every 2 days, followed by limiting dilution cloning. Selected cells were diluted to a final concentration of 5 cells/mL and seeded in 96-well plates with growth medium supplemented with G418. Once wells reached 100% confluence, they were gradually amplified to generate the SK-MEL-2-IFN cell clone.
2.6. Flow Cytometry
After corresponding treatments, cells were trypsinized, resuspended in 1 ml of PBS, and centrifuged (1200 rpm, 4 min, 4°C). The cellular sediment was stained with L/D reagent (Invitrogen™) for 10 min according to the manufacturer's instructions for flow cytometry and then resuspended in FACS buffer (PBS 2% FBS). The samples were analyzed in the flow cytometer (Millipore Guava Easycyte 6 2L) to quantify GFP expression on live cells (L/D negative). For each sample, 10.000 events were acquired in the analyzed region. Data were analyzed with FlowJo vx 0.7 software (Tree Star Inc., Ashland, OR, SA).
2.7. Statistical Analysis
The experimental data are representative of at least three independent experiments and expressed as the means ± SEM and each dot in the graphs represents a biological replicate. Statistical data are informed in the corresponding figure legend. GraphPad Prism 8 (version 8, GraphPad Software, San Diego, CA, USA) software was utilized to carry out the work.
3. Results
3.1. Association of BRAFV600E Mutation with Genomic and Immune Landscape Alterations in Melanoma Patients
In order to investigate the impact of BRAFV600E mutation, known for its prevalence in melanoma [2], on immunogenomic and immune microenvironment in melanoma patients (SKCM), we first conducted an in silico analysis using data from The Cancer Genome Atlas (TCGA) accessed through cBioPortal and Xena platforms. The cBioPortal dataset, comprising TCGA PanCancer data, included 363 patients, with 48.21% exhibiting the wild-type (WT) form of the BRAF protein and 34.44% harboring the BRAFV600E mutation, which was the most prevalent mutation (Figure 1A). We identified that the BRAFV600E mutation in melanoma patients is significantly associated with a lower Tumor Mutation Burden (TMB) (Figure 1B). TMB, reflecting the total number of non-synonymous mutations per megabase of the genome, serves as a metric for genomic instability and potential neoantigen formation [17,18]. Furthermore, analysis of the Xena dataset, also derived from TCGA PanCancer, included 103 patients, among whom 46.60% had the WT BRAF protein form, and 47.67% had the BRAFV600E mutation, confirming this mutation as the most prevalent (Figure 1C). A recent meta-analysis thoroughly examined the immune TME using transcriptomic data, uncovering six distinct immune subtypes across different tumor types [19]. Here, investigation into these immune subtypes unveiled the impact of the BRAFV600E mutation on the profile within the melanoma immune landscape (Figure 1D). Specifically, in BRAFV600E-mutant tumors, there was an abundance of an "Immune C1: Wound healing" profile observed, along with an increased prevalence of other subtypes associated with immune system dysfunction, such as "Immune C4: Lymphocyte Depleted" and "Immune C6: TGF-beta Dominant". Conversely, subtypes indicative of a favorable immune response or infiltration, such as “Immune C2: IFN-gamma Dominant and Immune C3: Inflammatory types”, showed decreased prevalence in melanoma patients carrying the BRAFV600E mutation. Our findings underscore that the presence of the BRAFV600E mutation is linked to a reduced TMB and an overall immunosuppressive TME.
3.2. Impact of BRAFV600E Mutation on Sensitivity to ICD and ICD-Associated Gene Expression Profiles in Melanoma
In our investigation, we aimed to evaluate the impact of the BRAFV600E mutation on sensitivity to ICD, a form of cell death that elicits an immune response against tumor cells [10]. To address this, we conducted in vitro cytotoxicity assays using melanoma cell lines harboring either BRAF wild-type (WT) (SK-MEL-2) or BRAFV600E mutations (SK-MEL-28 and A375). These cell lines were exposed to various ICD inducers, including doxorubicin [20,21,22,23] and Me-ALA-based photodynamic therapy (PDT) [11], as well as a non-ICD inducer, cisplatin [24,25]. Across all treatments, dose-response curves demonstrated that the decrease in cell viability was directly proportional to the concentration of the respective drug used (Figure 2A–C). Notably, substantial distinctions were observed specifically in the context of PDT, as BRAFV600E mutant cell lines showed increased sensitivity compared to BRAF WT SK-MEL-2 (Figure 2A). Conversely, while A375 exhibited the highest resistance to doxorubicin (Figure 2B) and SK-MEL-28 displayed the highest resistance to cisplatin (Figure 2C), no notable distinctions were observed between the remaining cell lines in both instances, with one carrying the BRAF wild-type (WT) and the other bearing the BRAFV600E mutation (Table 1).
To further explore the impact of BRAFV600E mutation on the expression of specific genes associated with ICD, particularly DAMPs, we analyzed data from the cBioPortal database (Figure 1A). Surprisingly, our analysis revealed an upregulation in the expression of genes associated with the IFN-1 pathway [26]: IFNAR1, IFNAR2, and CXCL10 in BRAFV600E-mutated tumors (Figure 2D). Additionally, we observed a decrease in the expression of ectonucleotidase CD39 [27], which plays a role in inhibiting and reversing the pro-inflammatory actions of ATP (Figure 2E). Our findings shed light on the influence of the BRAFV600E mutation on the sensitivity to ICD, particularly highlighting the potential for BRAFV600E-mutated tumors to exhibit increased responsiveness to PDT. These results led the question as to whether this differential sensitivity is linked to the observed distinct profile in the IFN-1 pathway, which is exacerbated in BRAFV600E-mutated tumors.
3.3. Modulation of IFN-1 Pathway Activity in Melanoma Cells by PDT-Induced ICD
The activation of the IFN-1 pathway by ICD was first reported using anthracyclines as an ICD inducer [28]. Additionally, our group observed an upregulation of IFN-α, IFN-β, and certain pro-inflammatory ISGs expression following Me-ALA based PDT in a murine melanoma model linked to dendritic cell maturation [11]. In the present study, we aimed to investigate whether ICD could promote the upregulation of this pathway in a human melanoma model. To address this question, we utilized SK-MEL-2 cells transfected with the MX2-EGFP plasmid, which contains the MX2 promoter, a well-known ISG, controlling the expression of GFP protein. After selection with geneticin, we generated the SK-MEL-2-IFN clone, which serves as a reporter for IFN-1 pathway activity. Subsequently, SK-MEL-2-IFN cells were exposed to cytotoxic doses of various ICD inducers, including doxorubicin and Me-ALA-based photodynamic therapy (PDT), as well as the non-ICD inducer cisplatin. GFP expression, indicative of IFN-1 pathway activity, was evaluated using flow cytometry. PDT emerged as the sole ICD inducer capable of robustly upregulating this pathway, as evidenced by a notable increase in the percentage of GFP+ cells, exhibiting IFN-1 pathway activation (Figure 3A). Unlike previous reports by other researchers [29], treatment with doxorubicin did not show modulation of the IFN-1 pathway in our experimental setup (Figure 3B). In contrast, the non-ICD inducer cisplatin even led to a significant downregulation of the IFN-1 pathway (Figure 3C). These findings highlight the varied impacts of distinct cell death inducers on the IFN-1 pathway, emphasizing the importance of delving deeper into the mechanisms that govern these responses and their subsequent implications. Collectively, our results suggest that the BRAFV600E mutation in melanoma, potentially by upregulating the IFNAR1/2, renders distinct susceptibility to antitumor therapies that induce IFN-1 pathway, such as the ICD-inducer PDT.
3.4. Inhibition of cGAS-STING Signaling Reverses PDT-Induced Upregulation of IFN-1 Pathway Activity
Many anti-tumor therapeutics exert their effects through cytotoxic mechanisms, often involving the destruction of chromosomal DNA [30,31,32,33,34]. The cGAS-STING signaling pathway serves as a crucial cytoplasmic DNA-sensing pathway, playing a pivotal role in regulating immune responses to cancer, infections, and autoimmune diseases by triggering the production of IFN-1 which modulate immune regulation [35]. Building on the preceding findings, we sought to explore whether cGAS-STING signaling might be involved in regulating IFN-1 triggered by PDT. To investigate this hypothesis, we employed the STING inhibitor H-151. Cells were pre-incubated with the inhibitor prior to PDT treatment. At a concentration of 10 µM, the STING inhibitor was capable of reversing the upregulation of the GFP+ cell population indicative of active IFN-1 pathway, as well as the transcriptional activity associated with this pathway. Notably, this reversal effect was observed only under conditions of PDT treatment and not in basal conditions (Figure 4). This suggests a specific interaction between PDT-induced cellular processes and the cGAS-STING signaling pathway, wherein the presence of the STING inhibitor selectively interfered with the downstream activation of IFN-1 pathway triggered by PDT-induced cell death mechanisms. Our study highlights the context-specific nature of cGAS-STING signaling modulation and the intricate interplay between PDT-mediated cellular responses and immune signaling pathways.
4. Discussion
Melanoma represents a paradigmatic example of the intricate interplay between the immune system and cancer progression. Despite being one of the most immunogenic tumors due to its high genomic mutational burden [18], melanoma paradoxically evades immune surveillance through diverse mechanisms [6,7,8,9,36]. This dynamic interaction between melanoma cells and the immune system significantly influences disease progression and treatment outcomes. Thus, comprehending this interplay is crucial for developing effective immunotherapeutic strategies against this aggressive malignancy. Furthermore, The BRAFV600E mutation, found in about 50% of melanomas, complicates the melanoma landscape by driving oncogenic signaling pathways, leading to increased cell proliferation, survival, and metastasis [3,37]. BRAF mutation status guides targeted therapy choices, while immunological features of the TME influence the efficacy of immunotherapeutic approaches in melanoma. Integrating molecular and immune profiling is essential for optimizing treatment outcomes [38,39]. Our study provides comprehensive insights into this complex interplay in melanoma.
Through in silico analyses, we revealed distinct genomic and immune landscape alterations associated with the BRAFV600E mutation in melanoma. Consistent with previous reports [2], we found a heightened frequency of this mutation among melanoma patients. Importantly, this mutation significantly correlated with a decreased TMB, potentially impairing neoantigens formation and hindering immune recognition and response. These findings are in line with studies on other cancers, indicating that probably in the presence of oncogene-driven mutations, including BRAFV600E, the contribution of additional mutations is dispensable in sustaining cancer cell survival and proliferation. A high TMB has been associated with improved clinical outcomes after treatment with immune checkpoint inhibitors (ICIs) [40,41,42,43,44,45,46]. Melanoma, characterized by a high TMB, underscores the intricate relationship between genomic alterations and immune evasion mechanisms [17,18].
It is widely recognized that BRAFV600E in melanoma can act as an immunogenic peptide when presented on MHC-II by CD4+ T-cells. Nonetheless, this mutation is linked to elevated expression of immunosuppressive factors and decreased antigen presentation by MHC-I. The application of BRAF inhibitors has shown promise in reversing tumor-associated immunosuppressive signals [47]. However, it's worth noting that there is limited literature available on this topic, highlighting the need for further research. In a recent metanalysis, significant efforts were made to comprehensively characterize the immune TME across 33 different cancers as analyzed by TCGA. Through this integrated approach, researchers delineated and described six distinct immune subtypes present across various tumor types, highlighting their potential therapeutic and prognostic relevance in cancer management [19]. Notably, our investigation into immune subtypes uncovered substantial alterations within the immune microenvironment of melanoma tumors bearing the BRAFV600E mutation. Specifically, we observed an exacerbation of immune subtype profiles associated with tumor immune evasion, including the “Immune C1: Wound healing profile”, characterized by elevated expression of angiogenic genes and a high proliferation rate. Additionally, we noted an increase in subtypes characterized by loss of immune function, such as “Immune C4: Lymphocyte Depleted” and “Immune C6: TGF-beta Dominant”, which displayed a more prominent macrophage signature and a high TGF-beta signature, respectively. Conversely, subtypes indicative of a favorable immune response, such as “Immune C2: IFN-gamma Dominant” and “Immune C3: Inflammatory”, exhibited decreased prevalence in BRAFV600E-mutated melanoma patients. These findings underscore the immunosuppressive nature of BRAFV600E-mutant tumors compared to BRAF wild-type ones.
In the context of advancing immunotherapies, ICD has emerged as a promising strategy to enhance the immunogenicity of dying cancer cells, potentially boosting the effectiveness of immunotherapeutic interventions [48,49,50]. In this study, our aim was to investigate the relationship between the BRAFV600E mutation and the response to ICD induction, seeking to identify strategies for improving immunostimulant therapies against melanoma. To this end, we explored the responsiveness of BRAFV600E-mutated cells to ICD inducers, including doxorubicin [20,22,23] and PDT using Me-ALA as a prodrug [11]. Indeed, our group was a pioneer in describing Me-ALA-based photodynamic therapy (PDT) as an inducer of ICD [11]. Additionally, we included cisplatin as a control, a conventional chemotherapeutic agent lacking ICD-inducing properties [24,25]. Our findings suggest that tumors harboring the BRAFV600E mutation may exhibit increased susceptibility to PDT-induced cell death. This phenomenon underscores the complex interplay between oncogenic mutations and therapeutic responses in melanoma. The specific molecular alterations driven by the BRAFV600E mutation that likely contribute to the observed differential sensitivity needs to be explored. Our preliminary analysis of transcriptomic data concerning DAMPs revealed intriguing alterations associated with the BRAFV600E mutation, particularly a downregulation in genes related to ATP metabolism (CD39) and upregulation of the type 1 interferon (IFN-1) pathway (IFNAR1, IFNAR2, CXCL10). CD39, along with CD73, converts extracellular ATP (a well-known DAMP) to adenosine, which inhibits T-cell effector functions via the adenosine receptor A2A [51]. The IFN-1 pathway plays a critical role in mediating anti-tumor immunity and is involved in the response to various cancer therapies [52,53,54]. One intriguing aspect highlighted by our results is the potential association between differential sensitivity to ICD and the distinct profile observed in the IFN-1 pathway in BRAFV600E-mutated tumors.
Interferon (IFN)-α2b, as the first approved immunotherapy for melanoma, has historically demonstrated significant benefits in improving both relapse-free survival and overall survival. While no longer a first-line treatment, ongoing research investigates its potential as an adjuvant in combination therapies, highlighting its continued relevance in enhancing the efficacy of other immunotherapies for melanoma patients [55].
The IFN-1 pathway was recently identified as a DAMP involved in ICD [29], which differs from constitutively expressed cDAMPs like calreticulin, ATP, and HMGB1. As an inducible DAMP [56], IFN-1 activation post-ICD serves as a crucial mediator of anti-tumor immunity, facilitating immune effector cell recruitment, antigen presentation enhancement, and durable immune memory generation. Triggered by various stimuli, including viral infections and nucleic ligands, IFN-1 production involves PRRs and cytoplasmic sensors like cGAS, activating transcription factors such as IRFs and NF-κB, culminating in IFN-α and IFN-β production and downstream signaling via IFNAR1/2 receptors [26,28,57,58].
The establishment of the SK-MEL-2-IFN reporter cell line, specifically engineered to express a fluorescent reporter under the control of the IFN-1 pathway [16], played a pivotal role in our investigation into the impact of ICD on the activation of this crucial signaling cascade in melanoma. The establishment of this experimental framework allowed us to monitor and quantify the basal activity of the IFN-1 pathway in SK-MEL-2 melanoma cells. It is important to note the basal activity of the IFN-1 pathway observed in these cells, likely associated the basal activity of IFN-1 exhibited by tumor cells, which can either promote cytotoxicity or confer pro-survival advantages depending on the strength and duration of the response, thereby impacting cancer therapy efficacy [13].
Here, we observed a notable upregulation of the IFN-1 pathway specifically in response to PDT, aligning with our previous findings in a murine melanoma model [11]. Previously, we demonstrated the induction of phosphorylated IRF3 and upregulation of key ISGs, including the cGAS receptor and phosphorylated STAT1, during PDT, suggesting potential autocrine stimulation. Our ongoing investigation aims to elucidate cGAS-STING signaling mechanisms, involving cytoplasmic DNA recognition by cGAS and STING activation [59]. Importantly, inhibiting cGAS-STING with H151 reverses PDT-induced IFN-1 pathway upregulation. Previous studies demonstrated radiotherapy and certain chemotherapeutic drugs induce cytotoxicity in cancer cells, releasing DNA fragments that activate cGAS-STING, prompting IFN-1 production and immune responses [30,31,32,33,34]. Our findings expand understanding of this pathway's role in regulating PDT-induced IFN-1 activity in melanoma, warranting further investigation into the responsible DNA-associated ligand. Emerging studies highlight the synergy between STING agonists and ICIs, enhancing anti-tumor immunity. The cGAS-STING pathway plays a crucial role in innate immune recognition of immunogenic tumors, facilitating APC maturation, cytokine secretion, and CD8+ T cell development targeting tumor-specific antigens. Activation of this pathway reshapes the TME, boosting the anti-tumor immune response and promising new therapeutic approaches for melanoma and other cancers [35]. Given the established curative and synergistic effects observed in preclinical studies with various therapeutic modalities [12], it is plausible that innovative strategies incorporating IFN-1 system activation as part of combination therapies will lead to even better response rates and survival outcomes.
5. Conclusions
Among the ICD inducers evaluated, PDT notably emerged as the sole inducer capable of robustly activating the IFN-1 pathway. The upregulation of IFNAR1/2 associated with the BRAFV600E mutation suggests a potential link to the enhanced susceptibility of tumor cells to PDT, potentially rendering BRAFV600E-mutated cells more responsive to stimuli that activate the IFN-1 pathway. While these findings provide insights into the mechanisms underlying the observed association, further investigation is warranted to validate and elucidate the mechanistic foundations of this phenomenon. Additionally, exploring the broader implications of the IFN-1 pathway in the context of different cell death inducers may offer deeper insights into the complex interplay between oncogenic mutations, immune responses, and therapeutic strategies in melanoma treatment.
Author Contributions
Mentucci FM: Conceptualization, Methodology, Investigation, Data curation, Writing. Romero Nuñez EA: Methodology, Validation, Investigation, Data curation. Ercole A: Methodology, Validation, Investigation, Data curation. Silvetti V: Methodology, Validation, Investigation, Data curation. Dal Col J: Conceptualization, Writing, Review & editing, Supervision. Lamberti MJ: Conceptualization, Validation, Investigation, Data curation, Writing, Funding acquisition.
Funding
This research was funded by the following grants under the supervision of Dr. Lamberti MJ: the Florencio Fiorini para Investigación en Ciencias Biomédicas grant, PICT-2021-GRFTI-00393, PICTO CBA 00034/2022, PIBAA-CONICET, and Jóvenes en Ciencia – MinCyT-Cba. Dr. Lamberti MJ is a member of the Scientific Researcher Career at CONICET. FMM holds a postgraduate fellowship from CONICET. AE holds an undergraduate fellowship from CIN, and EARN holds a fellowship from SeCyT-UNRC.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to acknowledge María de los Milagros Bürgi (UNL) for providing the pMx2-eGFP plasmid, and Dr. Pérez Saez (IBYME) and Dr. Álvarez (IMIBIO-SL) for their assistance with the cell lines. Their contributions were invaluable to the success of this research.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Long, G. V.; Swetter, S.M.; Menzies, A.M.; Gershenwald, J.E.; Scolyer, R.A. Cutaneous Melanoma. Lancet 2023, 402, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The Role of BRAF V600 Mutation in Melanoma. J Transl Med 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- An, Q.; Liu, Z. Comparative Efficacy and Safety of Combination Therapies for Advanced Melanoma: A Network Meta-Analysis. BMC Cancer 2019, 19. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Kartolo, A.; Yeung, C.; Hopman, W.; Baetz, T. Long-Term Toxicities of Immune Checkpoint Inhibitor (ICI) in Melanoma Patients. Current Oncology 2022, Vol. 29, Pages 7953-7963 2022, 29, 7953–7963. [Google Scholar] [CrossRef]
- Mahmoud, F.; Shields, B.; Makhoul, I.; Avaritt, N.; Wong, H.K.; Hutchins, L.F.; Shalin, S.; Tackett, A.J. Immune Surveillance in Melanoma: From Immune Attack to Melanoma Escape and Even Counterattack. 2017. [CrossRef]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.Y.H.; et al. Transcriptional Downregulation of MHC Class I and Melanoma De- Differentiation in Resistance to PD-1 Inhibition. Nature Communications 2020 11:1 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Aiba, S.; Fujimura, T. Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers. Biomolecules 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Failmezger, H.; Muralidhar, S.; Rullan, A.; de Andrea, C.E.; Sahai, E.; Yuan, Y. Topological Tumor Graphs: A Graph-Based Spatial Model to Infer Stromal Recruitment for Immunosuppression in Melanoma Histology a C. Cancer Res 2020, 256, 1199–1209. [Google Scholar] [CrossRef]
- Galluzzi, L.; Agostinis, P.; Vitale, I.; Warren, S.; Adjemian, S.; Coukos, G.; Martinez, A.B.; Chan, T.A.; Edelson, R.L.; Demaria, S.; et al. Consensus Guidelines for the Definition, Detection and Interpretation of Immunogenic Cell Death. J Immunother Cancer 2020, 8:e000337. J Immunother Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Lamberti, M.; Mentucci, F.; Roselli, E.; Araya, P.; Rivarola, V.; Rumie Vittar, N.; Maccioni, M. Photodynamic Modulation of Type 1 Interferon Pathway on Melanoma Cells Promotes Dendritic Cell Activation. Front Immunol. 2019, 10, 2614. [Google Scholar] [CrossRef]
- Borden, E.C. Interferons α and β in Cancer: Therapeutic Opportunities from New Insights. Nat Rev Drug Discov 2019, 18, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.J.; Wang, Y.; Wightman, S.M.; Jackson, M.W.; Stark, G.R. How Cancer Cells Make and Respond to Interferon-I. Trends Cancer 2023, 9, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov 2012, 2, 401. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat Biotechnol 2020, 38, 675. [Google Scholar] [CrossRef]
- Bürgi, M.; Prieto, C.; Oggero, M.; Bollati-Fogolín, M.; Etcheverrigaray, M.; Kratje, R. New Reporter Cell Clones to Determine the Biological Activity of Human Type I Interferons. BMC Proc. 2011, 5, Suppl 8:P4. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What Do All the Mutations Mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med 2017, 9, 1–14. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830e14. [Google Scholar] [CrossRef]
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Immunogenic Cell Death Induction by Anticancer Chemotherapeutics. Oncoimmunology 2017, 6, e1386829. [Google Scholar] [CrossRef]
- Lamberti, M.J.; Montico, B.; Ravo, M.; Nigro, A.; Giurato, G.; Iorio, R.; Tarallo, R.; Weisz, A.; Stellato, C.; Steffan, A.; et al. Integration of MiRNA:MRNA Co-Expression Revealed Crucial Mechanisms Modulated in Immunogenic Cancer Cell Death. Biomedicines 2022, 10, 1896. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. Journal of Experimental Medicine 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, D.; Du, X.; He, X.; Huang, M.; Wang, Y.; Yang, X.; Wang, J. Carrier-Free Nanoassembly of Doxorubicin Prodrug and SiRNA for Combinationally Inducing Immunogenic Cell Death and Reversing Immunosuppression. Nano Today 2020, 35, 100924. [Google Scholar] [CrossRef]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the Immunogenicity of Cisplatin-Induced Cancer Cell Death by Endoplasmic Reticulum Stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, A.; Pirker, C.; Keppler, B.K.; Berger, W. Anticancer Metal Drugs and Immunogenic Cell Death. J Inorg Biochem 2016, 165, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I Interferons in Anticancer Immunity. Nat Rev Immunol 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Lin, Y.S.; Chiang, S.F.; Chen, C.Y.; Hong, W.Z.; Chen, T.W.; Chen, W.T.L.; Ke, T.W.; Yang, P.C.; Liang, J.A.; Shiau, A.C.; et al. Targeting CD73 Increases Therapeutic Response to Immunogenic Chemotherapy by Promoting Dendritic Cell Maturation. Cancer Immunol Immunother 2023, 72, 2283–2297. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Sistigu, A.; Yamazaki, T.; Vitale, I.; Zitvogel, L.; Kroemer, G. Autocrine Signaling of Type 1 Interferons in Successful Anticancer Chemotherapy. Oncoimmunology 2015, 4, e988042. [Google Scholar]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.; Remédios, C.; et al. Cancer Cell-Autonomous Contribution of Type I Interferon Signaling to the Efficacy of Chemotherapy. Nat Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Liu, Y.; Crowe, W.N.; Wang, L.; Lu, Y.; Petty, W.J.; Habib, A.A.; Zhao, D. An Inhalable Nanoparticulate STING Agonist Synergizes with Radiotherapy to Confer Long-Term Control of Lung Metastases. Nat Commun 2019, 10. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur J Pharmacol 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Li, T.; Cheng, H.; Yuan, H.; Xu, Q.; Shu, C.; Zhang, Y.; Xu, P.; Tan, J.; Rui, Y.; Li, P.; et al. Antitumor Activity of CGAMP via Stimulation of CGAS-CGAMP-STING-IRF3 Mediated Innate Immune Response. Sci Rep 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Crowe, W.N.; Wang, L.; Lu, Y.; Petty, W.J.; Habib, A.A.; Zhao, D. An Inhalable Nanoparticulate STING Agonist Synergizes with Radiotherapy to Confer Long-Term Control of Lung Metastases. Nat Commun 2019, 10. [Google Scholar] [CrossRef]
- Luo, M.; Liu, Z.; Zhang, X.; Han, C.; Samandi, L.Z.; Dong, C.; Sumer, B.D.; Lea, J.; Fu, Y.X.; Gao, J. Synergistic STING Activation by PC7A Nanovaccine and Ionizing Radiation Improves Cancer Immunotherapy. J Control Release 2019, 300, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Li, N.; Ye, W.; Gao, H.; Luo, X.; Cheng, B. Activation of Stimulation of Interferon Genes (STING) Signal and Cancer Immunotherapy. Molecules 2022, 27. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Zappasodi, R. A Decade of Checkpoint Blockade Immunotherapy in Melanoma: Understanding the Molecular Basis for Immune Sensitivity and Resistance. Nat Immunol 2022, 23, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Larkin, J.; Ribas, A.; Dréno, B.; Flaherty, K.T.; Ascierto, P.A.; Lewis, K.D.; McKenna, E.; Zhu, Q.; Mun, Y.; et al. Modeled Prognostic Subgroups for Survival and Treatment Outcomes in BRAF V600-Mutated Metastatic Melanoma: Pooled Analysis of 4 Randomized Clinical Trials. JAMA Oncol 2018, 4, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, F.; Crimini, E.; Tarantino, P.; Zagami, P.; Uliano, J.; Corti, C.; Trapani, D.; Curigliano, G.; Ascierto, P.A. First Line Treatment of BRAF Mutated Advanced Melanoma: Does One Size Fit All? Cancer Treat Rev 2021, 99. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, D.C.; Theocharopoulos, C.; Lialios, P.P.; Foteinou, D.; Koumprentziotis, I.A.; Xynos, G.; Gogas, H. Beyond CTLA-4 and PD-1 Inhibition: Novel Immune Checkpoint Molecules for Melanoma Treatment. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949e15. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non–Small Cell Lung Cancer. Science 2015, 348, 124. [Google Scholar] [CrossRef]
- Offin, M.; Rizvi, H.; Tenet, M.; Ni, A.; Sanchez-Vega, F.; Li, B.T.; Drilon, A.; Kris, M.G.; Rudin, C.M.; Schultz, N.; et al. Tumor Mutation Burden and Efficacy of EGFR-Tyrosine Kinase Inhibitors in Patients with EGFR-Mutant Lung Cancers. Clin Cancer Res 2019, 25, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.T.; Wu, Y.L. Combination Chemotherapy Alone Should Be Used in the Treatment of Patients With Stage IV EGFR-Mutant NSCLC Whose Disease Has Progressed on All Available Tyrosine Kinase Inhibitors. J Thorac Oncol 2021, 16, 1627–1631. [Google Scholar] [CrossRef]
- Patel, R.R.; Ramkissoon, S.H.; Ross, J.; Weintraub, L. Tumor Mutational Burden and Driver Mutations: Characterizing the Genomic Landscape of Pediatric Brain Tumors. Pediatr Blood Cancer 2020, 67. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, J.; Schroeder, C.; Martus, P.; Armeanu-Ebinger, S.; Kelemen, O.; Gschwind, A.; Bonzheim, I.; Eigentler, T.; Amaral, T.; Ossowski, S.; et al. TMB and BRAF Mutation Status Are Independent Predictive Factors in High-Risk Melanoma Patients with Adjuvant Anti-PD-1 Therapy. J Cancer Res Clin Oncol 2023, 149, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, K.M.; Correa, I.; Josephs, D.H.; Karagiannis, P.; Egbuniwe, I.U.; Cafferkey, M.J.; Spicer, J.F.; Harries, M.; Nestle, F.O.; Lacy, K.E.; et al. Effects of BRAF Mutations and BRAF Inhibition on Immune Responses to Melanoma. Mol Cancer Ther 2014, 13, 2769–2783. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, M.; Nigro, A.; Mentucci, F.; Rumie Vittar, N.; Casolaro, V.; Dal Col, J. Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity. Pharmaceutics 2020, 256, 3. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Hett, E.; Kroemer, G.; Marincola, F.M. Immunogenic Cell Death in Cancer: Concept and Therapeutic Implications. J Transl Med 2023, 21. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat Rev Immunol 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Zhao, X.; Xiao, J.; Bi, J.; Li, X.Y.; Chen, G.; Lu, L. Review Immune Response of Targeting CD39 in Cancer. Biomark Res 2023, 11. [Google Scholar] [CrossRef]
- Gatti, G.; Nuñez, N.; Nocera, D.; Dejager, L.; Libert, C.; Giraudo, C.; Maccioni, M. Direct Effect of DsRNA Mimetics on Cancer Cells Induces Endogenous IFN-β Production Capable of Improving Dendritic Cell Function. Eur J Immunol 2013, 43, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Núñez, N.; Andreani, V.; Crespo, M.; Nocera, D.; Breser, M.; Morón, G.; Dejager, L.; Libert, C.; Rivero, V.; Maccioni, M. IFNβ Produced by TLR4-Activated Tumor Cells Is Involved in Improving the Antitumoral Immune Response. Cancer Res. 2012, 72, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Nocera, D.; Roselli, E.; Araya, P.; Nuñez, N.; Lienenklaus, S.; Jablonska, J.; Weiss, S.; Gatti, G.; Brinkmann, M.; Kröger, A.; et al. In Vivo Visualizing the IFN-β Response Required for Tumor Growth Control in a Therapeutic Model of Polyadenylic-Polyuridylic Acid Administration. J Immunol. 2016 2016, 196, 2860–2869. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Giubellino, A. The Current State of Treatment and Future Directions in Cutaneous Malignant Melanoma. Biomedicines 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Cullen, S.; Albert, M. Dying Cells Actively Regulate Adaptive Immune Responses. Nat Rev Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Musella, M.; Manic, G.; De Maria, R.; Vitale, I.; Sistigu, A. Type-I-Interferons in Infection and Cancer: Unanticipated Dynamics with Therapeutic Implications. Oncoimmunology 2017, 6, e1314424. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.; Donlin, L. Regulation of Type I Interferon Responses. Nat Rev Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA Sensing by the CGAS-STING Pathway in Health and Disease. Nat Rev Genet 2019, 20, 657–674. [Google Scholar] [CrossRef]
Figure 1.
Genetic and immunological landscape of BRAF mutations in SKCM. a) Distribution of mutation frequencies in BRAF among skin cutaneous melanoma (SKCM) patients, sourced from cBioPortal (TCGA Pan-Cancer Atlas dataset). b) Dot plot showing tumor mutational burden (TMB) comparison between BRAF wild type (WT) (n = 175) and BRAF V600E (n = 125) mutations in SKCM, quantified by the number of proteins carrying non-synonymous mutations. Statistical analysis was conducted using the Student t-test for unmatched samples: * p < 0.05. c) Mutation frequencies distribution in BRAF among SKCM patients, sourced from Xena (TCGA Pan-Cancer Atlas dataset). d) Bar plot showing the classification of immune subtypes in SKCM patients based on BRAF WT (n = 48) and BRAF V600E (n = 49) mutations.
Figure 1.
Genetic and immunological landscape of BRAF mutations in SKCM. a) Distribution of mutation frequencies in BRAF among skin cutaneous melanoma (SKCM) patients, sourced from cBioPortal (TCGA Pan-Cancer Atlas dataset). b) Dot plot showing tumor mutational burden (TMB) comparison between BRAF wild type (WT) (n = 175) and BRAF V600E (n = 125) mutations in SKCM, quantified by the number of proteins carrying non-synonymous mutations. Statistical analysis was conducted using the Student t-test for unmatched samples: * p < 0.05. c) Mutation frequencies distribution in BRAF among SKCM patients, sourced from Xena (TCGA Pan-Cancer Atlas dataset). d) Bar plot showing the classification of immune subtypes in SKCM patients based on BRAF WT (n = 48) and BRAF V600E (n = 49) mutations.
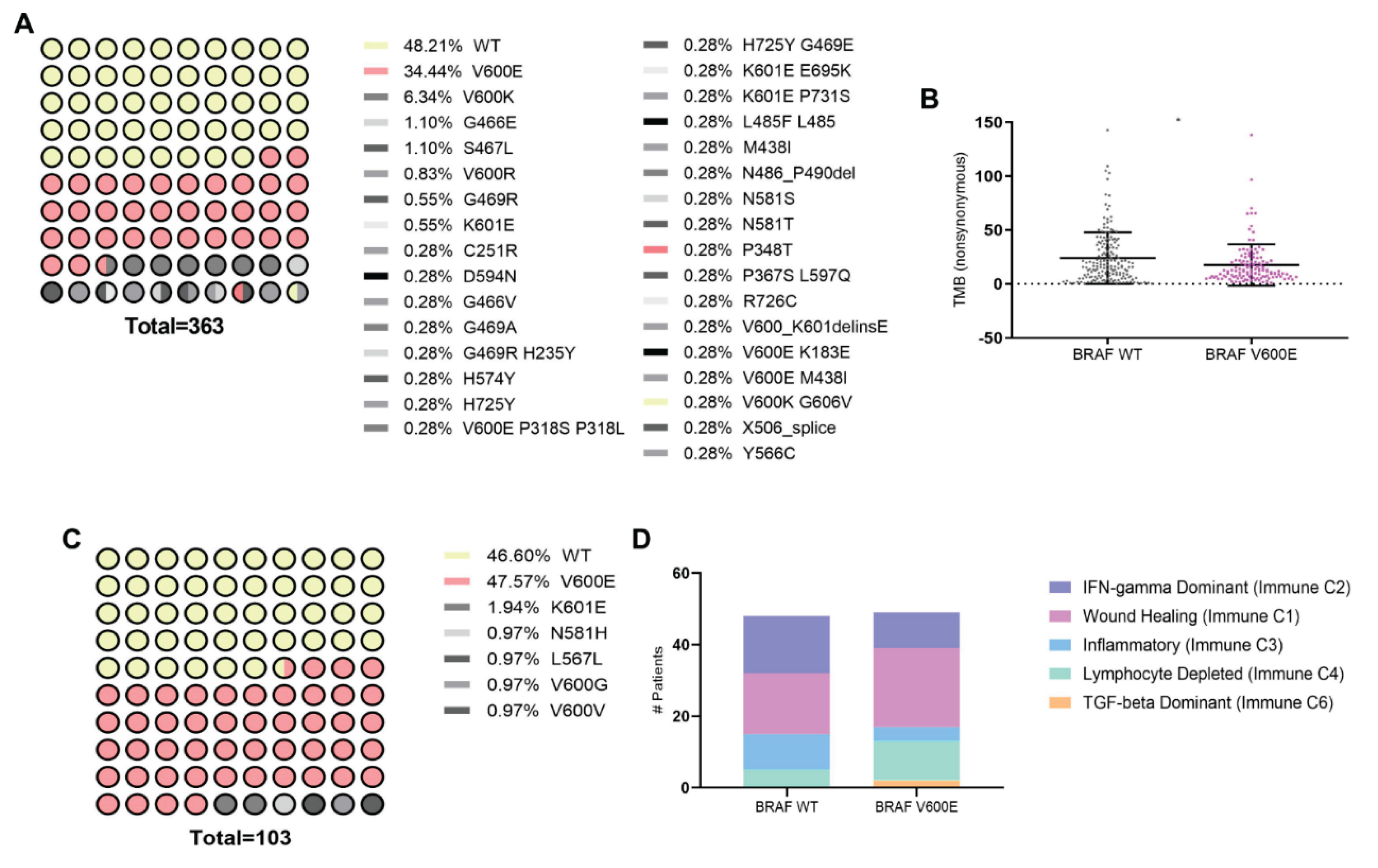
Figure 2.
Response variation of melanoma cell lines to immunogenic cell death inducers and associated gene expression profiles in the context of BRAF mutation. Melanoma cell lines with BRAF wild-type (SK-MEL-2, pink line) or BRAF V600E mutations (SK-MEL-28 and A375, black lines) were exposed to increasing doses of the ICD inducers: a) photodynamic therapy (PDT): cells were first incubated with increasing doses of the pro-drug Me-ALA (0 – 2 mM) for 4 hours, followed by irradiation with a light dose of 1 J/cm2 (λ: 636 nm); b) doxorubicin (DXR): cells were incubated to increasing doses of the chemotherapeutic (0 – 12 µg/ml) for 24 hours; c) cisplatin (CP): cells were incubated to increasing doses of the chemotherapeutic (0 – 800 µg/ml) for 24 hours. Cell viability was assessed 24 hours after treatment using the resazurin assay and expressed as a percentage of non-treated cells (100% represented by the dotted line). Dose-response curves were generated using non-linear regression analysis (GraphPad Prism). d) Heatmap illustrating mRNA expression levels of genes associated with immunogenic cell death (ICD), particularly DAMPs or their modulators, in BRAF wild-type (n = 175) and BRAF V600E (n = 125) melanoma samples sourced from cBioPortal (TCGA Pan-Cancer Atlas dataset). Statistical analysis was conducted using the Student t-test for unmatched samples: *** p < 0.0001. * p < 0.05. e) Dot plots showing mRNA expression levels of genes with statistically significant differences in BRAF V600E samples compared to BRAF wild-type samples. Statistical analysis was conducted using the Student t-test for unmatched samples: *** p < 0.0001. * p < 0.05.
Figure 2.
Response variation of melanoma cell lines to immunogenic cell death inducers and associated gene expression profiles in the context of BRAF mutation. Melanoma cell lines with BRAF wild-type (SK-MEL-2, pink line) or BRAF V600E mutations (SK-MEL-28 and A375, black lines) were exposed to increasing doses of the ICD inducers: a) photodynamic therapy (PDT): cells were first incubated with increasing doses of the pro-drug Me-ALA (0 – 2 mM) for 4 hours, followed by irradiation with a light dose of 1 J/cm2 (λ: 636 nm); b) doxorubicin (DXR): cells were incubated to increasing doses of the chemotherapeutic (0 – 12 µg/ml) for 24 hours; c) cisplatin (CP): cells were incubated to increasing doses of the chemotherapeutic (0 – 800 µg/ml) for 24 hours. Cell viability was assessed 24 hours after treatment using the resazurin assay and expressed as a percentage of non-treated cells (100% represented by the dotted line). Dose-response curves were generated using non-linear regression analysis (GraphPad Prism). d) Heatmap illustrating mRNA expression levels of genes associated with immunogenic cell death (ICD), particularly DAMPs or their modulators, in BRAF wild-type (n = 175) and BRAF V600E (n = 125) melanoma samples sourced from cBioPortal (TCGA Pan-Cancer Atlas dataset). Statistical analysis was conducted using the Student t-test for unmatched samples: *** p < 0.0001. * p < 0.05. e) Dot plots showing mRNA expression levels of genes with statistically significant differences in BRAF V600E samples compared to BRAF wild-type samples. Statistical analysis was conducted using the Student t-test for unmatched samples: *** p < 0.0001. * p < 0.05.
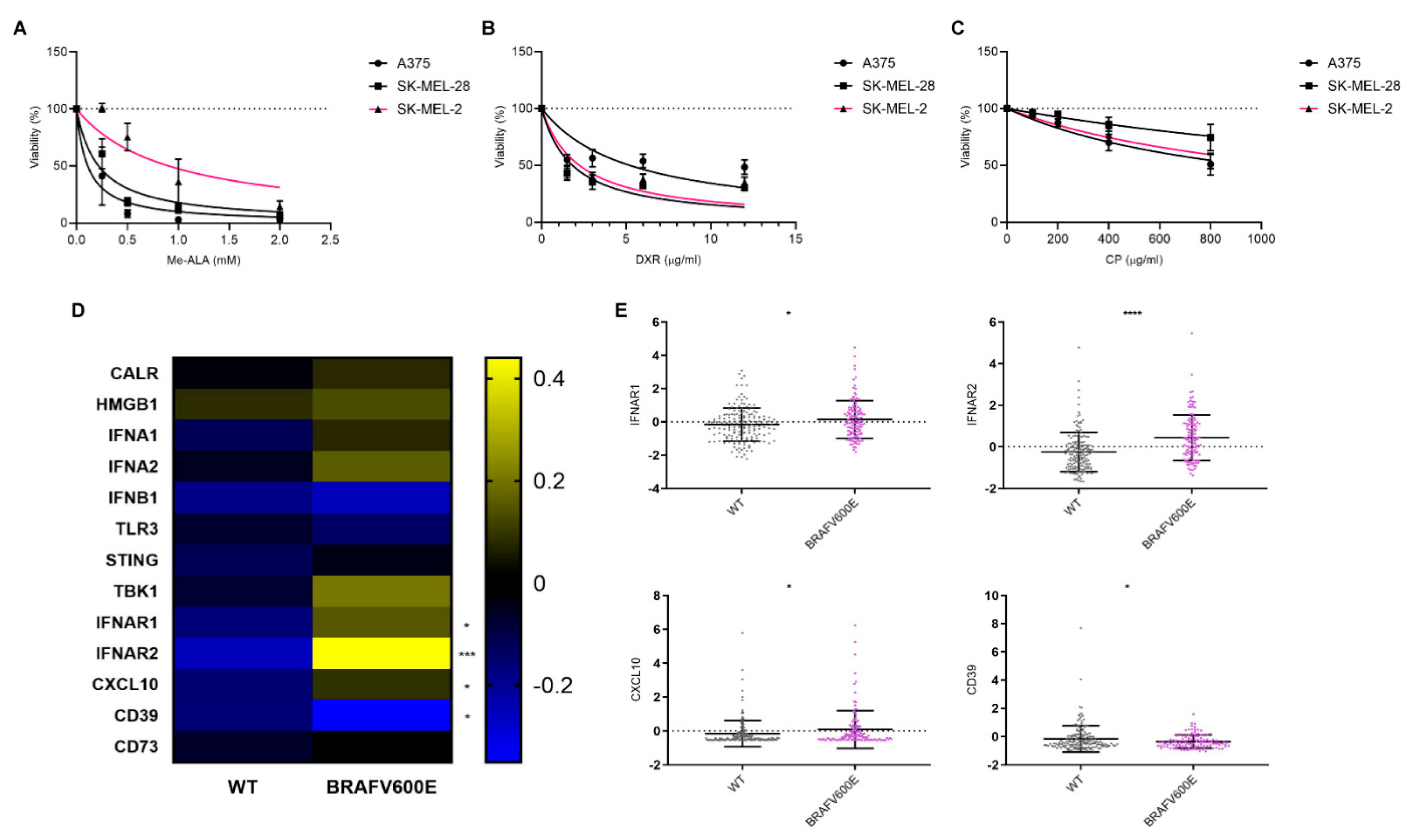
Figure 3.
Assessment of IFN-1 Pathway Activation in Melanoma Cells Exposed to Immunogenic and Non-Immunogenic Cell Death Inducers. a) Gene reporter assay was performed using the IFN-1 pathway reporter cell line SK-MEL-2-IFN exposed to the non-ICD inducer cisplatin (CP) (800 µg/ml) for 24 hours. GFP expression on live cells was quantified from flow cytometry data using FlowJo. Data is presented as the percentage of GFP positive (GFP+) cells representing IFN-1 pathway activation (left), with analysis of the level of IFN-1 activation in GFP+ positive cells performed from GeoMean data (center), all normalized to values corresponding to untreated cells (100% represented by the dotted line). Statistical analysis was conducted using the Student t-test for unmatched samples: * p < 0.05. Representative histograms are shown (right). b) Gene reporter assay was performed using the IFN-1 pathway reporter cell line SK-MEL-2-IFN exposed to the immunogenic cell death (ICD) inducer doxorubicin (DXR) (6 µg/ml) for 24 hours. GFP expression on live cells was quantified from flow cytometry data using FlowJo. Data is presented as the percentage of GFP positive (GFP+) cells representing IFN-1 pathway activation (left), with analysis of the level of IFN-1 activation in GFP+ positive cells performed from GeoMean data (center), all normalized to values corresponding to untreated cells (100% represented by the dotted line). Statistical analysis was conducted using the Student t-test for unmatched samples: absence of asterisks (*) indicates non-significant difference statistically. Representative histograms are shown (right). c) Gene reporter assay was performed using the IFN-1 pathway reporter cell line SK-MEL-2-IFN exposed to the ICD inducer photodynamic therapy (PDT). Cells were first incubated with the pro-drug Me-ALA (1 mM) for 4 hours, followed by irradiation with a light dose of 1 J/cm2 (λ: 636 nm). GFP expression on live cells was quantified from flow cytometry data using FlowJo. Data is presented as the percentage of GFP positive (GFP+) cells representing IFN-1 pathway activation (left), with analysis of the level of IFN-1 activation in GFP+ positive cells performed from GeoMean data (center), all normalized to values corresponding to untreated cells (100% represented by the dotted line). Statistical analysis was conducted using the Student t-test for unmatched samples: ** p < 0.01. Representative histograms are shown (right).
Figure 3.
Assessment of IFN-1 Pathway Activation in Melanoma Cells Exposed to Immunogenic and Non-Immunogenic Cell Death Inducers. a) Gene reporter assay was performed using the IFN-1 pathway reporter cell line SK-MEL-2-IFN exposed to the non-ICD inducer cisplatin (CP) (800 µg/ml) for 24 hours. GFP expression on live cells was quantified from flow cytometry data using FlowJo. Data is presented as the percentage of GFP positive (GFP+) cells representing IFN-1 pathway activation (left), with analysis of the level of IFN-1 activation in GFP+ positive cells performed from GeoMean data (center), all normalized to values corresponding to untreated cells (100% represented by the dotted line). Statistical analysis was conducted using the Student t-test for unmatched samples: * p < 0.05. Representative histograms are shown (right). b) Gene reporter assay was performed using the IFN-1 pathway reporter cell line SK-MEL-2-IFN exposed to the immunogenic cell death (ICD) inducer doxorubicin (DXR) (6 µg/ml) for 24 hours. GFP expression on live cells was quantified from flow cytometry data using FlowJo. Data is presented as the percentage of GFP positive (GFP+) cells representing IFN-1 pathway activation (left), with analysis of the level of IFN-1 activation in GFP+ positive cells performed from GeoMean data (center), all normalized to values corresponding to untreated cells (100% represented by the dotted line). Statistical analysis was conducted using the Student t-test for unmatched samples: absence of asterisks (*) indicates non-significant difference statistically. Representative histograms are shown (right). c) Gene reporter assay was performed using the IFN-1 pathway reporter cell line SK-MEL-2-IFN exposed to the ICD inducer photodynamic therapy (PDT). Cells were first incubated with the pro-drug Me-ALA (1 mM) for 4 hours, followed by irradiation with a light dose of 1 J/cm2 (λ: 636 nm). GFP expression on live cells was quantified from flow cytometry data using FlowJo. Data is presented as the percentage of GFP positive (GFP+) cells representing IFN-1 pathway activation (left), with analysis of the level of IFN-1 activation in GFP+ positive cells performed from GeoMean data (center), all normalized to values corresponding to untreated cells (100% represented by the dotted line). Statistical analysis was conducted using the Student t-test for unmatched samples: ** p < 0.01. Representative histograms are shown (right).
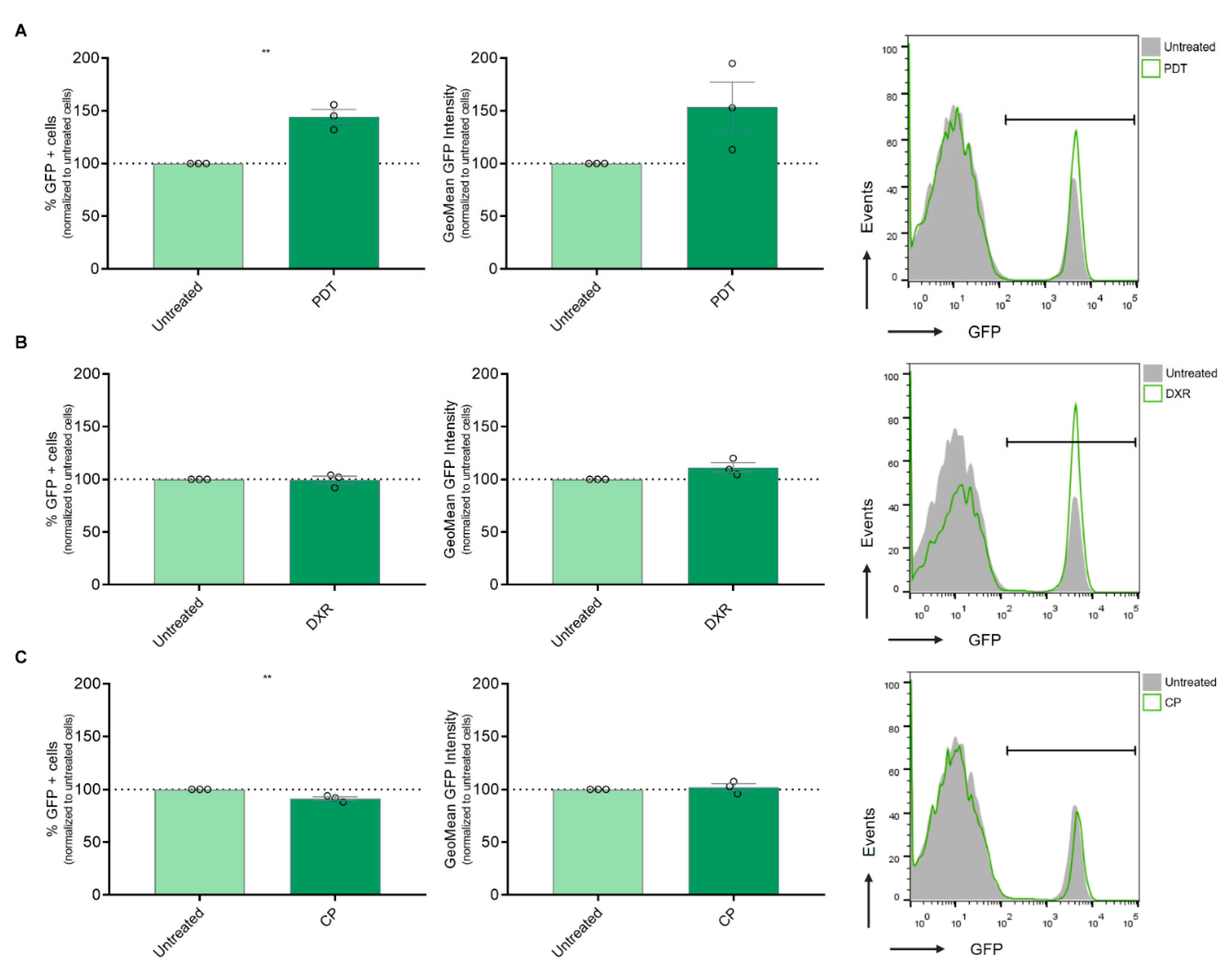
Figure 4.
Modulation of IFN-1 Pathway Activation by PDT mediated by cGAS-STING signaling. Gene reporter assay was performed using the IFN-1 pathway reporter cell line SK-MEL-2-IFN exposed to the immunogenic cell death (ICD) inducer photodynamic therapy (PDT) in the presence or absence of the STING inhibitor H151 (1 mM and 10 mM). Cells were first incubated with the pro-drug Me-ALA (1 mM) for 4 hours with or without H151 or vehicle (DMSO), followed by irradiation with a light dose of 1 J/cm² (λ: 636 nm). GFP expression on live cells (L/D negative), indicative of IFN-1 pathway activation, was quantified from flow cytometry data using FlowJo. The data is presented as the percentage of GFP positive (GFP+) cells representing IFN-1 pathway activation (a), with analysis of the level of IFN-1 activation in GFP+ positive cells performed from GeoMean data (b), all normalized to values corresponding to untreated cells (100% represented by the dotted line). Statistical analysis was conducted using two-way ANOVA with Tukey's post hoc test: **p < 0.01, ***p < 0.001. Representative dot plots are shown (c).
Figure 4.
Modulation of IFN-1 Pathway Activation by PDT mediated by cGAS-STING signaling. Gene reporter assay was performed using the IFN-1 pathway reporter cell line SK-MEL-2-IFN exposed to the immunogenic cell death (ICD) inducer photodynamic therapy (PDT) in the presence or absence of the STING inhibitor H151 (1 mM and 10 mM). Cells were first incubated with the pro-drug Me-ALA (1 mM) for 4 hours with or without H151 or vehicle (DMSO), followed by irradiation with a light dose of 1 J/cm² (λ: 636 nm). GFP expression on live cells (L/D negative), indicative of IFN-1 pathway activation, was quantified from flow cytometry data using FlowJo. The data is presented as the percentage of GFP positive (GFP+) cells representing IFN-1 pathway activation (a), with analysis of the level of IFN-1 activation in GFP+ positive cells performed from GeoMean data (b), all normalized to values corresponding to untreated cells (100% represented by the dotted line). Statistical analysis was conducted using two-way ANOVA with Tukey's post hoc test: **p < 0.01, ***p < 0.001. Representative dot plots are shown (c).
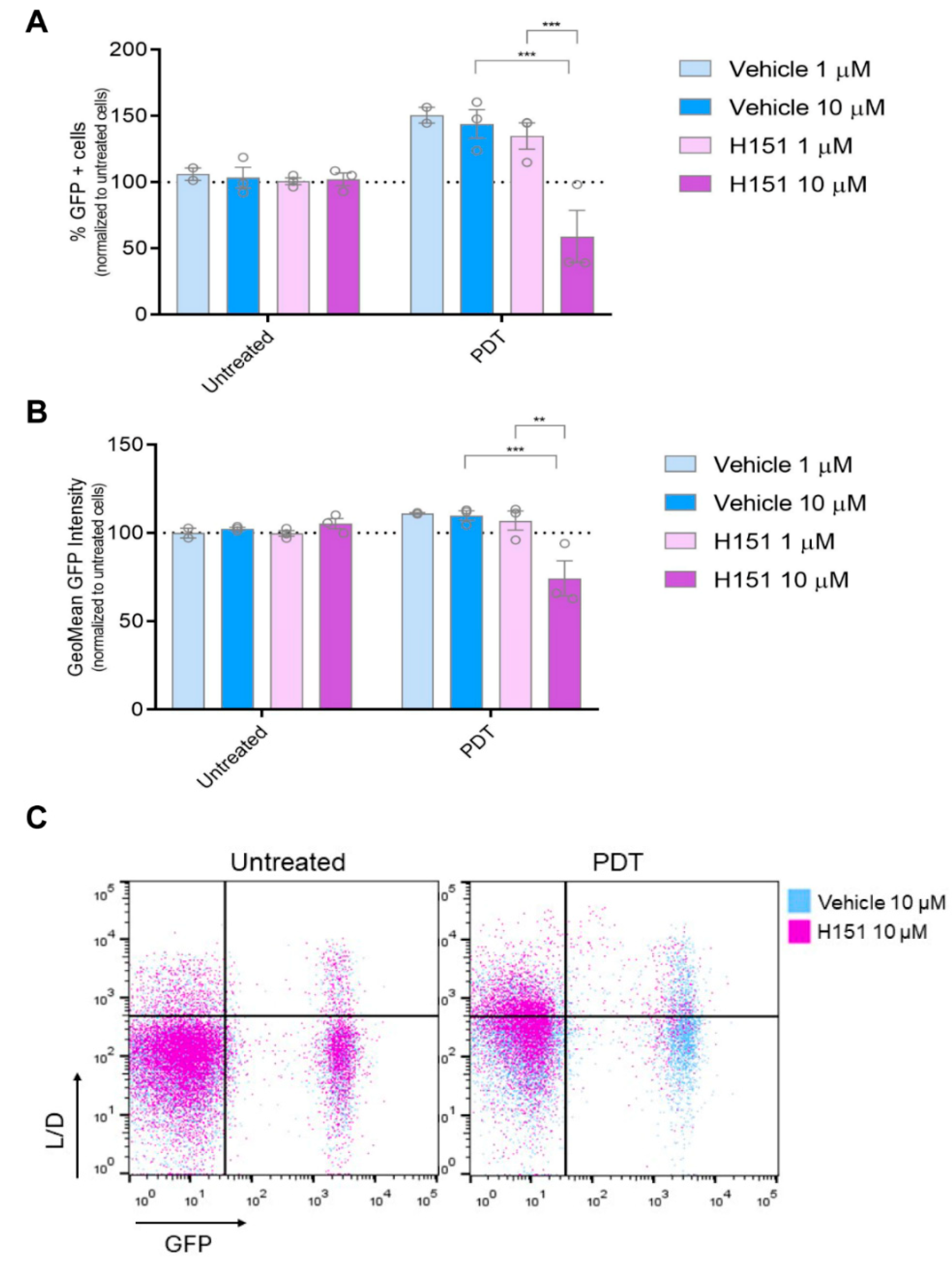

Table 1.
Statistical Comparison of IC50 Values for Melanoma Cell Lines in Response to Different Treatments. Table 1 displays the p values obtained from statistical comparisons of the IC50 (inhibitory concentration 50%) values derived from nonlinear regression analysis of dose-response curves for each pair of cell lines in response to various treatments (PDT: photodynamic therapy, DXR: doxorubicin, CP: cisplatin). Extra sum-of-squares F test was performed to evaluate differences in best-fit parameters (IC50) among cell lines within each treatment (GraphPad Prism). Statistically significant p values (< 0.05) are highlighted in blue, while non-significant ones are highlighted in yellow.
Table 1.
Statistical Comparison of IC50 Values for Melanoma Cell Lines in Response to Different Treatments. Table 1 displays the p values obtained from statistical comparisons of the IC50 (inhibitory concentration 50%) values derived from nonlinear regression analysis of dose-response curves for each pair of cell lines in response to various treatments (PDT: photodynamic therapy, DXR: doxorubicin, CP: cisplatin). Extra sum-of-squares F test was performed to evaluate differences in best-fit parameters (IC50) among cell lines within each treatment (GraphPad Prism). Statistically significant p values (< 0.05) are highlighted in blue, while non-significant ones are highlighted in yellow.
| PDT | |||
| SK-MEL-2 | A375 | SK-MEL-28 | |
| SK-MEL-2 | 0.0001 | 0.0001 | |
| A375 | 0.0001 | 0.87 | |
| SK-MEL-28 | 0.0001 | 0.87 | |
| DXR | |||
| SK-MEL-2 | A375 | SK-MEL-28 | |
| SK-MEL-2 | 0.0072 | 0.4364 | |
| A375 | 0.0072 | 0.005 | |
| SK-MEL-28 | 0.4364 | 0.005 | |
| CP | |||
| SK-MEL-2 | A375 | SK-MEL-28 | |
| SK-MEL-2 | 0.2866 | 0.003 | |
| A375 | 0.2866 | 0.0004 | |
| SK-MEL-28 | 0.003 | 0.0004 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



