
Submitted:
11 March 2024
Posted:
12 March 2024
You are already at the latest version
Abstract
A large diversity of epigenetic factors, such as microRNAs and histones modifications, are known to be capable of regulating gene expression without altering DNA sequence itself. In particular, miR-1 is considered the first essential microRNA in cardiac development. In this study, miR-1 potential role in early cardiac chamber differentiation is analyzed through specific signaling pathways. For this, we performed in chick embryos functional experiments by means of miR-1 microinjections into the posterior cardiac precursors -of both primitive endocardial tubes- committed to sinoatrial region fates. Subsequently, embryos were subjected to whole mount in situ hybridization, immunohistochemistry and RT-qPCR analysis. As a relevant nov-elty, our results revealed that miR-1 increases Tbx5, Gata4 and AMHC1, while this microRNA diminishes Mef2c expression, during early differentiation of cardiac sinoatrial region. Further-more, we observed in this developmental context that miR-1 upregulates CRABPII and RARß, and downregulates CRABPI, which are three crucial factors in retinoic acid signaling pathway. Interestingly, we also noticed that miR-1 directly interacts with HDAC4 and Calmodulin, as well as with Erk2/MAPK1, which are three key factors actively involved in Mef2c regulation. Our study shows, for the first time, a key role of miR-1 as an epigenetic regulator in early differenti-ation of cardiac sinoatrial region through orchestrating opposite actions between retinoic acid and Mef2c, fundamental to properly assign cardiac cells to their respective heart chambers. A better understanding of those molecular mechanisms modulated by miR-1 will definitely help in fields applied to therapy and cardiac regeneration and repair.
Keywords:
miR-1
; epigenetic regulator
; cardiac sinoatrial region
; molecular mechanisms
; HDAC4
; Calmodulin
; pErk2/MAPK1
; Mef2c activity
; retinoic acid signaling
; cardiac development
1. Introduction
During early embryogenesis, the cardiogenic mesoderm organizes -at both sides of the embryo- the primitive endocardial tubes, which fuse to form the primitive heart tube in the midline [1,2,3,4]. Subsequently, the posterior cardiac tube segment differentiates into the cardiac sinoatrial region, including atrium and inflow tract/sinus venosus [5,6]. Concomitantly, the anterior cardiac tube segment contributes to ventricular and outflow tract differentiation [7,8]. Previous studies have revealed that this segmentation process is regulated by several cardiac transcription factors, including Tbx5, Gata4 and Mef2c, among others, which activate the expression of early cardiac structural genes, such as myosin heavy chain (MHC) and myosin light chain (MLC) [9,10,11,12,13]. It is also well known that phenotype of cardiac sinoatrial region is influenced by retinoic acid (RA) signaling, mediating the expression pattern of early molecular markers such as Tbx5, Gata4 and AMHC1 (atrial myosin heavy chain), which together determine the posterior heart tube segment leading to a venous and atrial cell fate [14,15,16]. In fact, exclusion of RA from ventricular precursors is essential for correct specification of the ventricles [17]. Furthermore, RA synthesis is largely controlled by retinaldehyde dehydrogenase 2 (Raldh2/Aldh1a2) [18,19], in such a way that Raldh2−/− knockout mice present reduced Tbx5 and Gata4 expression and severely impaired cardiac sinoatrial region development, while Mef2c expression, which induces ventricular cardiomyocyte differentiation [20], is unaltered [21]. Along with the above, Mef2c−/− knockout mice display ectopic Tbx5 expression, suggesting that the mutant ventricle acquires atrial-specific characteristics and, therefore, Mef2c could act directly on cells and make them nonresponsive to RA inflow differentiation signaling [22].
Several authors have also reported that a large diversity of epigenetic processes, including the expression of non-coding RNAs (especially microRNAs), histones modifications and DNA methylation, are capable of regulating gene expression, by influencing transcription or inhibiting translation through several functional overlapping and cross-talking mechanisms [23,24]. In particular, microRNAs are considered as a post-transcriptional type of epigenetic regulatory processes, since the degree of complementarity microRNA/target mRNA determines its final outcome, either by degradation or translational inhibition [24,25]. Specifically, miR-1 has been the first essential microRNA identified in cardiac development, with a specific expression pattern in cardiac muscle [26,27,28,29,30,31]. Interestingly, experimental overexpression of miR-1 during heart development results in defective ventricular myocyte proliferation, accompanied by hypoplasia of the cardiac ventricular conduction system [32]. To date, the specific effects of miR-1 during early differentiation on cardiac sinoatrial region are unknown. The previous study that analysed the functional role of miR-1, which was performed in murine embryonic stem cells, was focused on the development and function of sinoatrial cells, corresponding specifically to the sinoatrial node, which determines the cardiac pacemaker [33].
Furthermore, it has been proposed that miR-1 modulates cardiomyocyte growth responses by negatively regulating Calmodulin, a miR-1 target gene [34]. In addition, miR-1 regulates human cardiomyocyte progenitor cell differentiation via repression of histone deacetylase 4 (HDAC4), also identified as a miR-1 target gene, altering histone-DNA-binding activity [35]. Nevertheless, HDAC4 can also interact with non-histones by covering the promoter region of a gene, thus inhibiting its expression [36,37,38]. For instance, HDAC4 can interact with Mef2c by repressing its transcriptional activity, which is required for muscle cell differentiation [39]. Additionally, as described in cardiac hypertrophy, it has been reported that the activation of Mef2c transcription is mediated both by Calmodulin and Erk1/2 -extracellular signal regulated kinases 1 and 2, a signal transducer in the family of mitogen activated protein kinase, MAPK- [40,41,42,43,44]. In this sense, it has been recently reported, in a zebrafish model, that Erk1/2 signaling is required to promote ventricular chamber differentiation [45].
Given the complexity of those molecular mechanisms involved in cardiac chamber formation, this experimental study aims to analyse miR-1 potential functions in early differentiation of the cardiac sinoatrial region. By means of gain- and loss-of-function experiments, we show that miR-1 is required for an adequate expression of RA-dependent molecular markers Tbx5, Gata4 and AMHC1, and RA signaling pathway genes RARβ, CRABPII and CRABPI. Significantly, our results reveal that miR-1 suppresses Mef2c by directly repressing Mef2c regulators HDAC4, Calmodulin and Erk2/MAPK1. Additionally, after miR-1 overexpression, we observed morphological alterations, characterized by an enlargement of the heart posterior domain and a reduction of the ventricular region. Altogether, our data support a model in which miR-1 plays a dual role by modulating both Mef2c activity and RA signaling pathway, placed as a key epigenetic regulator of appropriate cardiac cell specification, thus promoting the initial differentiation of the cardiac sinoatrial region.
2. Results
Our results show, as a novel approach, that miR-1 administration enhances the expression of Tbx5, Gata4 and AMHC1, while it diminishes Mef2c expression during early differentiation of the cardiac sinoatrial region. Another original contribution of this work includes the analysis of miR-1 expression pattern, as well as HDAC4, Calmodulin and phospho-Erk2/MAPK1 (pErk2) distributions, from the primitive endocardial tubes to the initial cardiac looping stages. Noticeably, we also observed -for the first time in this developmental context- that miR-1 directly downregulates HDAC4 and Calmodulin, as well as Erk2/MAPK1, which are required for Mef2c expression. Moreover, this microRNA enhances CRABPII and RARß expressions, while it downregulates CRABPI -three essential factors in RA signaling pathway. Supporting the above, after antimiR-1 administration, we obtained the opposite results.
2.1. miR-1 Regulates Specific Gene Expressions during Early Differentiation of Cardiac Sinoatrial Region
In this research work, we analyse the effect of miR-1 on Tbx5, Gata4, AMHC1 and Mef2c expression during early differentiation of cardiac sinoatrial region (Figure 1 and Supplementary Figure S1). For this purpose, we carried out miR-1 gain- and loss-of-function experiments by means of respective microinjections of premiR-1 and antimiR-1 into the posterior cardiac precursors -in primitive endocardial tubes- committed to sinoatrial region fates (Figure 1A). Additionally, in an experimental group, cardiac asa were dissected and subsequently subjected to RNA extraction and RT-qPCR analysis, under each experimental condition (Supplementary Figure S2). Our results show that miR-1 leads to increased Tbx5 (Figure 1B), Gata4 (Figure 1C) and AMHC1 (Figure 1D) expressions, and RT-qPCR analyses support these findings (Figure 1B-D). Interestingly, miR-1 inhibits mRNA and protein levels of Mef2c gene in the cardiac sinoatrial region (Figure 1E). Supporting these data, our miR-1 loss-of-function experiments show an opposite effect (Figure 1, right column, and RT-qPCRs).
Noticeably, in experimental embryos we observed morphological alterations based on an expansion of the posterior domain of the heart after miR-1 administration, concomitantly with a reduced ventricle region (Figure 1 and Supplementary Figure S2A,B).
Additionally, we have observed that microinjections of RA -into the same areas above mentioned- mediate positively the expression of Gata4 (Supplementary Figure S3), and also Tbx5 and AMHC1 as we previously reported [46]. All the above mentioned findings reveal that miR-1 mimics the effects observed with highlighted RA levels.
2.2. miR-1 Modulates RA Signaling Pathway during Early Differentiation of Cardiac Sinoatrial Region
Since miR-1 modulates RA target gene expression during differentiation of cardiac sinoatrial region, through RT-qPCR we analysed RNAs from dissected cardiac asa of embryos microinjected either with premiR-1 or antimiR-1, in order to explore the relationship between miR-1 and those factors involved in the RA signaling pathway (Figure 2).
Our findings reveal that miR-1 does not modify the expression levels of Raldh2 (Figure 2A), a RA synthesis modulator [47]. We also observed that miR-1 is able to increase the endogenous expression levels of CRABPII (Figure 2B), a RA target gene that regulates RA delivery to its nuclear receptors (RARs) [48,49]. In addition, our results show that miR-1 leads to reduced levels of CRABPI (Figure 2C), which functions as a sequester of RA to facilitate its catabolism [50], thus supporting that miR-1 may upregulate RA signaling. In line with the above results, we obtained the opposite effects through antimiR-1 administration (Figure 2).
With respect to the nuclear retinoic acid receptors, it is to note that Rarß is a RA target gene essential for cardiovascular development [51,52,53,54]. In this study we include a detailed analysis of this cardiac gene (Figure 3). By using immunohistochemistry (IMH) in a control group of embryos, we identified Rarß distribution starting in both primitive endocardial tubes, following in the primitive cardiac tube and continuing in the early cardiac looping formation, presenting a location in sinoatrial region (Figure 3A). Our miR-1 gain-of-function experiments led to an increased expression of Rarß and its protein levels (Figure 3B). Supporting these results, we obtained the opposite effect after our miR-1 loss-of-function experiments (Figure 3, right column, and RT-qPCRs). On the other hand, expression modifications of nuclear retinoic acid receptors RARα, RXRα, RARγ and RXRγ were not detected after miR-1 experimental assays (Figure 4).
2.3. miR-1 Modulates HDAC4, Calmodulin and Erk2/MAPK1 during Early Differentiation of Cardiac Sinoatrial Region
Given that our experiments revealed that miR-1 decreases both mRNA and protein levels of Mef2c, which is not a miR-1 target gene, we searched for pivotal miR-1 targets genes as potential Mef2c regulators during differentiation of cardiac sinoatrial region. By using bioinformatics analyses through Target-Scan software, we identified HDAC4, Calmodulin and Erk2/MAPK1 as putative targets of miR-1 (Supplementary Figure S4), suggesting that it might exert control on their post-transcriptional regulation. Our luciferase assays show that luciferase signals from plasmids harbouring 3′UTRs of each gene are reduced with respect to control, proving that miR-1 recognizes and directly binds to HDAC4, Calmodulin and Erk2/MAPK1 3′UTRs, thus triggering their mRNA degradation (Figure 5).
As illustrated in Figure 6, through in situ hybridization (ISH), we report miR-1 expression pattern, starting in both primitive endocardial tubes, following in the primitive cardiac tube and continuing in the early cardiac looping formation. By means of IMH, we observed a similar distribution of HDAC4, Calmodulin, and pErk2 in sinoatrial region during the stages under analyses. Interestingly, our comparative analysis demonstrates a complementary expression pattern among them, suggesting a plausible regulatory link during early posterior cardiac tube differentiation.
With respect to the miR-1 functional role on Mef2c regulators, our gain-of-function experiments show that miR-1 represses both mRNA and protein levels of HDAC4, Calmodulin and Erk2 in the sinoatrial region (Figure 7). In agreement with these results, the opposite effects were observed after antimiR-1 administration (Figure 7, right column, and RT-qPCRs). Therefore, our findings indicate that miR-1 modulates Mef2c expression through the repression of HDAC4, Calmodulin and Erk2 during early differentiation of cardiac sinoatrial region.
3. Discussion
In this work, we have obtained original findings about numerous molecular factors involved in the posterior cardiac tube segment differentiation. In this sense, this is the first time there is clear evidence about the function of miR-1 modulating Mef2c activity and RA signaling pathway during early cardiac chamber differentiation. Our results reveal (Figure 8) that miR-1 administration into the posterior cardiac precursors in the primitive endocardial tubes give rise to high expression levels of Tbx5, Gata4 and AMHC1, which have previously demonstrated to be RA target genes, essential for early differentiation of cardiac sinoatrial region [16,46,55]. Significantly, the expression of Mef2c, a ventricular cardiomyocyte inductor [20,22], was diminished after miR-1 administration. Additionally, morphological alterations were detected after miR-1 overexpression, characterized by an enlarged posterior domain of the heart and a reduced ventricle region. All the above results show that miR-1 mimics the effects previously associated to highlighted RA levels [14,15,16,17,18,19,21], indicating that miR-1 actively interferes with one or more factors involved in RA signaling pathway. In this line, our results reveal that miR-1 upregulates CRABPII and RARß, while it downregulates CRABPI -three crucial factors in RA signaling pathway. Interestingly, this microRNA also mimics the results obtained after deletion of Mef2c [20,22], indicating that miR-1 clearly modulates Mef2c transcription regulators. In addition to the above data, our results show that: i) miR-1 recognizes and directly binds to Erk2/MAPK1, HDAC4 and Calmodulin, the last two also previously reported as miR-1 target genes in culture cardiac cells [34,35]; ii) this microRNA presents a complementary expression pattern with these three target genes during early posterior cardiac tube differentiation; and iii) it represses both mRNA and protein levels of HDAC4, Calmodulin and Erk2/MAPK1 in the sinoatrial region. Our findings support the fact that miR-1 actively interacts with these three potential molecular factors, and also that these factors are involved both in Mef2c regulation and RA signaling pathway during early differentiation of cardiac sinoatrial region (Figure 8 and Figure 9).
3.1. miR-1 Modulates Mef2c Activity
Our results show that miR-1 diminishes both mRNA and protein levels of Mef2c in the posterior domain of the developing heart. It has been proposed that Mef2c promotes outflow-specific differentiation and directs cells in a nonresponsive manner to the inflow-specifying actions of RA [22]. Since Mef2c is not a miR-1 target gene, we analysed the effects of miR-1 on Mef2c modulators and observed that this microRNA represses both mRNA and protein levels of citoplasmatic HDAC4 (cHDAC4), Calmodulin and Erk2/MAPK1 (Figure 9). In this context, it is particularly known that nuclear HDAC4 (nHDAC4) directly binds to Mef2c and blocks its activity [36,39,56]. Through pharmacological inhibition of HDAC4 activity by trichostatin A (TSA) [57], our experimental model (Supplementary Figure S5) revealed that Mef2c expression presented a dramatic increase, proving the importance of histone deacetylation in this transcription factor regulation. It is also known that Calmodulin -through CaMKII induction- leads to phosphorylation of nHDAC4 and its translocation to the cytoplasm, thus promoting Mef2c activation [40,41,42,44,58]. Therefore, in our model (Figure 9), given that low levels of Calmodulin -miR-1 induced- do not allow the export of nHDAC4 from the nucleus to the cytoplasm, miR-1 would increase Mef2c interaction with nHDAC4, which inhibits Mef2c expression. It has also been reported that nHDAC4 together with the nuclear corepressor NCoR1/SMRT synergize to inhibit Mef2c activity [59,60]. Furthermore, NCoR1/SMRT actively translocates cHDAC4 from the cytoplasm into the nucleus and prevents its nuclear export [60,61,62]. The above data support the fact that there are opposite effects of Calmodulin and NCoR1/SMRT on nHDAC4 modulated by miR-1.
On the other hand, it is known that NCoR1/SMRT phosphorylation by Erk2 may destabilize the association between Mef2c and nHDAC4, thus increasing Mef2c expression [60,63,64]. It has also been reported that Mef2c may be enhanced by Erk1/2 through upregulation of coactivator P300/CBP-associated factor (PCAF), which inherently presents histone acetyltransferases (HAT) activity, thus increasing both expression and function of Mef2c [43]. Our results show that miR-1 directly recognizes and represses Erk2/MAPK1 expression (Figure 9), and consequently this microRNA diminishes Mef2c activity, results supported by the above data. In this line, it has been observed during zebrafish development that excessive levels of Erk1/2 in atrial location cause ectopic expression of ventricular specific genes [45], effect that also supports our model, proposing miR-1 as a key regulator of atrial differentiation by repressing Erk1/2 expression.
Based on all the molecular mechanisms mentioned above, we propose in our model (Figure 9) that, in these early stages of cardiac development, miR-1 plays a crucial role to repress Mef2c through Erk2/MAPK1, HDAC4 and Calmodulin modulation.
3.2. miR-1 Modulates RA Signaling Pathway
With respect to miR-1 inductive role on RA signaling pathway during early differentiation of cardiac sinoatrial region (Figure 8), we found that this microRNA increases expression levels of RA target genes Tbx5, Gata4 and AMHC1. It is known that RA establishes an epigenetic switch for histone acetylation, mediated by p300/CBP, which inherently presents histone acetyltransferases (HAT) activity to allow transcription of RARs target genes [65,66,67,68,69,70]. We also observed that miR-1 represses epigenetic repressor HDAC4, which has been reported as a negative regulator of RA target genes [71]. In line with the above, we show in this work that Tbx5, Gata4 and AMHC1 expressions increase with HDAC4 inhibitor TSA (Supplementary Figure S5), confirming that RA regulates these cardiac genes through direct modulation of histone deacetylation. Furthermore, previous studies have reported that nHDAC4 is recruited and retained in the nucleus by NCoR1/SMRT, which is associated to RARs in absence of RA [61,72,73,74]. As a matter of fact, it is known that the presence of RA disrupts the interaction of NCoR1/SMRT with RARs [64,75], thus allowing transcription of RARs target genes (Figure 9). Other studies have described that Erk2/MAPK1 phosphorylates nuclear corepressors N-CoR1/SMRT, reducing the interaction between N-CoR1/SMRT and RARα [63], which would allow us to suggest an increase of RARs target genes transcription. However, our results showed that miR-1 directly supresses Erk2/MAPK1 but it also increases RA target genes. Based on these results, we propose that Erk2/MAPK1 is not sufficient to modulate atrial gene expressions, so that RA signal is necessary to disrupt the interaction between N-CoR1/SMRT and RARs [72]. Keeping all the above in mind, our results show that miR-1 upregulates RA function, thus promoting the expression of its target genes.
Interestingly, we found in our study that miR-1 increases expression levels of CRABPII, a RA target gene -with RA response element (RARE)- involved in delivering RA to its nuclear receptors RARs [48,49,76,77]. In addition, we observed that miR-1 increases both mRNA and proteins of RARβ, a previously defined RA target gene with RARE located in its promoter [51,54,68,75,77]. Moreover, by means of IMH, we carried out a detailed dynamic analysis of RARβ distribution, which showed a specific location in the cardiac sinoatrial region. The above findings, together with the fact that RARβ repression is linked to cardiac abnormalities [52], support miR-1 active role on RA signaling pathway through RARβ. Nevertheless, the levels of nuclear retinoic acid receptors RARα, RXRα, RARγ and RXRγ did not show significant changes after our miR-1 experimental assays, proving that these receptors are not targeted either by miR-1 or RA. It is noteworthy, based on our results, that miR-1 directly suppresses Calmodulin -a protein that also acts as an inhibitor of RARα activity by means of CaMKII induction, which mediates RARα phosphorylation and enhances the interaction between RARα and N-CoR1/SMRT, and subsequently supresses RARα target genes transcription [78]. In our proposed model (Figure 9), miR-1 represses Calmodulin capability to inhibit RARs activity, thus promoting the expression of RA target genes. Supporting these results, previous studies showed that the blockage of RARα function diminishes Gata4 transcripts, specifically interfering with the inflow tract formation [79,80].
Additionally, our results show that miR-1 overexpression does not generate significant changes in the expression level of Raldh2, a RA synthesis modulator [47,74,81] involved in cardiac sinoatrial region development [18,19]. Supporting our results, previous studies [82] have shown that Raldh2 expression is not altered in a nuclear corepressor mutant mouse model –SMRTmRID- which is characterized by enhanced transcription of RARs targets.
Noticeably, we observed that miR-1 diminishes the expression levels of CRABPI, a protein described as a sequester of RA, facilitating its catabolism, limiting RA concentration and regulating the amount of RA that is accessible to nuclear receptors RARs [50,83]. This supports the fact that miR-1 enhances RA activity. Given that CRABPI is not a miR-1 target gene, our model (Figure 9) proposes that miR-1 inhibits TRAP220/MED1 (a miR-1 target gene identified through in silico analysis), which is a CRABPI coactivator associated with RA [84]. Therefore, miR-1 would be able to promote RA signals.
Taking into account all the above, our novel experimental model integrates a network of molecular mechanisms modulated by miR-1, which plays a crucial role during early stages of cardiac chamber formation by promoting atrial differentiation together with complementary suppression of ventricular formation. In conclusion, our study reveals, for the first time, a key role of miR-1 as an epigenetic factor modulating RA and Mef2c in their opposite actions, which are required to properly assign cells to their respective cardiac chambers. Since miR-1 has also been identified in cardiomyopathy processes [85,86,87,88,89,90], further and deeper understanding of this microRNA as a modulator of molecular mechanisms governing specific signaling pathways could be helpful in therapy and cardiac regeneration and repair.
4. Materials and Methods
Experimental protocols with animals were performed in agreement with the Spanish law in application of the EU Guidelines for animal research, and conformed to the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH Publication no.85-23). Approval by the University of Extremadura bioethics board was obtained prior to the initiation of the study.
4.1. Early Chick Whole Embryo Culture
4.2. Embryo Microinjections into the Posterior Cardiac Precursors of Both Primitive Endocardial Tubes
Stage HH 8 (3-4 somites) cultured embryos were microinjected (using an Inject + Matic micro injector system) in both primitive endocardial tubes into the posterior cardiac precursors committed to sinoatrial region fates [19,46].
For gain-of-function experiments, two different groups of embryos were microinjected with premiR-1 and all-trans retinoic acid (RA), respectively. Taking into account that premiR is a precursor miRNA that contains single stem-looped structure to be further processed into mature miRNA, which takes place in the miRNA-induced silencing complex; this, based on the sequence complementary between target mRNA and miRNA, gives rise to translation repression or target RNA degradation [94,95].
Likewise, for loss-of-function experiments, three different group of embryos were microinjected with antimiR-1 (that inhibits the binding of miRNA with its target mRNA); with an inhibitor of RA synthesis, Citral (3,7-dimethyl-2,6-octadienal); and with an inhibitor of HDAC, trichostatin A (TSA). A working solution containing 2.5 mM CFDA (Molecular Probes) and 1/10 volume of 0.5% (wt/vol) fast green FCF was prepared both experimental and control (CFDA) embryos. In the experimental embryos the working solution additionally contained a final concentration of 1μM for microRNAs premiR-1 (Ambion) or antimiR-1 (Ambion); of 10 µg/mL for RA (Sigma), of 10 mmol/L for Citral (Sigma); and as well as of 50 nmol/L for TSA (Sigma).
After 10-12h of additional incubation, embryos were photographed under bright and fluorescent light (Nikon digital, SIGHT DS-U1), and were selected according to the location and extent of the injection. The selected embryos were either fixed in 4% PFA—processed for gene expression (ISH) or immunochemistry (IMH) analysis- or cardiac loops were collected for RNA isolation as previously described [46].
4.3. Whole-Mount In Situ Hybridization (ISH)
Two different ISH-procedures were performed, following our previous procedures [96]. One group of control embryos was processed [97] for LNA-ISH using miR-1 LNA-labelled microRNA probe (miRCURY LNA™ Detection probe 5’-DIG and 3´-DIG labelled, Exiqon), while another group of premiR-1, antimiR-1, RA, Citral and control (CFDA) embryos were processed for ISH following standard procedures [98] using antisense-Tbx5, -Gata4 and -AMHC1 labelled probes [55].
4.4. Whole-Mount Immunohistochemestry (IMH)
Control, experimental and control (CFDA) embryos were subjected to whole mount IMH performed as we previously described [96,99], using polyclonal antibodies rabbit for HDAC4 (1:100, Invitrogen PA5-29103), phospho-Erk1/Erk2 (1:20, Invitrogen 44-680G), RARß (1:100, Invitrogen PA5-33016) and Mef2c (1:50, Proteintech 18290-1-AP), followed by Goat anti-rabbit IgG-HRP antibody (1:1000, Upstate, 12-348) and monoclonal antibody mouse for Calmodulin (1:20, Invitrogen MA3-918), followed by Goat anti-mouse Ig-HRP antibody (1:200, Jackson Immunoresearch Laboratories, West Grove, PA).
4.5. RNA Isolation and RT-qPCR
Cardiac loops isolated from experimental and control (CFDA) embryos (Supplementary Figure S1) were subjected to RT-qPCR analysis following MIQE guidelines [100,101,102]. RNA was extracted and purified by using ReliaPrep RNA Cell Miniprep System Kit (Promega) according to the manufacturer’s instructions. For mRNA expression measurements, 0,5 μg of total RNA was used for retro-transcription with Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Thermo Scientific). Real time PCR experiments were performed with 2μL of cDNA, GoTaq qPCR Master Mix (Promega) and corresponding primer sets (Supplementary Table STI). For miR-1 expression analyses, 20ng of total RNA was used for retro-transcription with Universal cDNA Synthesis Kit II (Exiqon) and the resulting cDNA was diluted 1/80. Real time PCR experiments were performed with 1 μL of diluted cDNA, Go Taq qPCR Master Mix (Promega) as well. All RT-qPCRs were performed using CFX384TM thermocycler (Bio-Rad) following the manufacture’s recommendations. The relative expression of each gene was calculated by using Gusb and Gapdh as internal controls for mRNA expression analyses and 5S and 6U for miR-1 expression analyses, respectively [103]. Each PCR reaction was carried out in triplicate and repeated in at least three distinct biological samples to obtain representative means.
4.6. Analysis In Silico
Target-Scan 8 software (https://www.targetscan.org/vert_80/) was used to perform bioinformatics analysis of the binding sites of miR-1 at 3′UTRs of predicted targets as described before [104].
4.7. Luciferase Assays and 3T3 Transfection
HDAC4, Calmodulin and Erk2 (MAPK1) 3′UTR constructs (Supplementary Table STII) were PCR amplified and cloned into the pMIR_REPORT vector respectively. 3T3 fibroblasts (ATCC) were co-transfected with 100ng of different luciferase-based constructs and 300ng of pcLux vector control for internal normalization, respectively. Luciferase activity (Pierce™ Gaussia Luciferase Flash Assay Kit) was normalized to pcLux vector control (Pierce™ Cypridina Luciferase Flash Assay Kit) and compared to non-transfected controls. Each luciferase assay was carried out in triplicates and repeated in at least three distinct biological samples to obtain representative assays as described previously.
4.8. Statistical Analyses
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
The experimental study was designed and supervised by C.L.-S., A.A., D.F. and V.G.-M., who also wrote the manuscript. C.G.-P., E.L.-V. and V.G.-L. contributed to the conception and design of the study, performed the experiments, and analysed the data. All authors have contributed to the experimental work and analysed and discussed the results. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been financed with research grants IB18123 (to CL-S) and GR21174 (to VG-M, CTS005) from the Junta de Extremadura, with FEDER co-financing, and CTS-446 (to DF and AA) from the Junta de Andalucía Regional Council. C.G.-P. and E.L.-V. have been funded, in part, by a postdoctoral Fellowship of the Junta de Extremadura, with FEDER co-financing.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Laura Ortega Bermejo for her invaluable technical support with embryo handling and sample preparation for in situ hybridisation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Garcia-Martinez, V.; Schoenwolf, G.C. Primitive-streak origin of the cardiovascular system in avian embryos. Dev. Biol. 1993, 159, 706–719. [Google Scholar] [CrossRef]
- Redkar, A.; Montgomery, M.; Litvin, J. Fate map of early avian cardiac progenitor cells. Development 2001, 128, 2269–2279. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Garcia-Masa, N.; Gañan, C.M.; Garcia-Martinez, V. Movement and commitment of primitive streak precardiac cells during cardiogenesis. Int. J. Dev. Biol. 2009, 53, 1445–1455. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Garcia-Lopez, V.; Schoenwolf, G.C.; Garcia-Martinez, V. From epiblast to mesoderm: Elaboration of a fate map for cardiovascular progenitors. In ESC Textbook of Cardiovascular Development; Perez-Pomares, J.M., Kelly, R., Eds.; Oxford University: New York, NY, USA, 2018; pp. 14–22. [Google Scholar]
- Abu-Issa, R.; Kirby, M.L. Patterning of the heart field in the chick. Dev. Biol. 2008, 319, 223–233. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, G.; Abu-Issa, R.; de Boer, B.A.; Hutson, M.R.; de Boer, P.A.; Soufan, A.T.; Ruijter, J.M.; Kirby, M.L.; van den Hoff, M.J.; Moorman, A.F. A caudal proliferating growth center contributes to both poles of the forming heart tube. Circ. Res. 2009, 104, 179–188. [Google Scholar] [CrossRef]
- Waldo, K.L.; Hutson, M.R.; Ward, C.C.; Zdanowicz, M.; Stadt, H.A.; Kumiski, D.; Abu-Issa, R.; Kirby, M.L. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005, 281, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Zaffran, S.; Kelly, R.G. New developments in the second heart field. Differentiation 2012, 84, 17–24. [Google Scholar] [CrossRef]
- Lints, T.J.; Parsons, L.M.; Hartley, L.; Lyons, I.; Harvey, R.P. Nkx-2.5: a novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development 1993, 119, 419–431. [Google Scholar] [CrossRef]
- Kuo, C.T.; Morrisey, E.E.; Anandappa, R.; Sigrist, K.; Lu, M.M.; Parmacek, M.S.; Soudais, C.; Leiden, J.M. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997, 11, 1048–1060. [Google Scholar] [CrossRef]
- Lin, Q.; Schwarz, J.; Bucana, C.; Olson, E.N. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 1997, 276, 1404–1407. [Google Scholar] [CrossRef]
- Bruneau, B.G.; Logan, M.; Davis, N.; Levi, T.; Tabin, C.J.; Seidman, J.G; Seidman, CE. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev. Biol. 1999, 211, 100–108. [Google Scholar] [CrossRef]
- Wolf, M; Basson, C.T. The molecular genetics of congenital heart disease: a review of recent developments. Curr. Opin. Cardiol. 2010, 25, 192–197. [Google Scholar] [CrossRef]
- Yutzey, K.E.; Rhee, J.T.; Bader, D. Expression of the atrial-specific myosin heavy chain AMHC1 and the establishment of anteroposterior polarity in the developing chicken heart. Development 1994, 120, 871–88. [Google Scholar] [CrossRef] [PubMed]
- Kostetskii, I.; Jiang, Y.; Kostetskaia, E.; Yuan, S.; Evans, T.; Zile, M. Retinoid signaling required for normal heart development regulates GATA-4 in a pathway distinct from cardiomyocyte differentiation. Dev. Biol. 1999, 206, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Liberatore, C.M.; Searcy-Schrick, R.D.; Yutzey, K.E. Ventricular expression of tbx5 inhibits normal heart chamber development. Dev. Biol. 2000, 223, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Xavier-Neto, J.; Neville, C.M.; Shapiro, M.D.; Houghton, L.; Wang, G.F.; Nikovits, W., Jr.; Stockdale, F.E.; Rosenthal, N. A retinoic acid-inducible transgenic marker of sino-atrial development in the mouse heart. Development 1999, 126, 2677–2687. [Google Scholar] [CrossRef] [PubMed]
- Xavier-Neto, J.; Shapiro, M.D.; Houghton, L.; Rosenthal, N. Sequential programs of retinoic acid synthesis in the myocardial and epicardial layers of the developing avian heart. Dev. Biol. 2000, 219, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Hochgreb, T.; Linhares, V.L.; Menezes, D.C.; Sampaio, A.C.; Yan, C.Y.; Cardoso,W. V.; Rosenthal, N.; Xavier-Neto, J. A caudorostral wave of raldh2 conveys anteroposterior information to the cardiac field. Development 2003, 130, 5363–5374. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Schwarz, J.; Bucana, C.; Olson, E.N. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 1997, 276, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Vermot, J.; Messaddeq, N.; Schuhbaur, B.; Chambon, P.; Dolle, P. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 2001, 128, 1019–1031. [Google Scholar] [CrossRef]
- Vong, L.; Bi, W.; O’Connor-Halligan, K.E.; Li, C.; Cserjesi, P.; Schwarz, J.J. MEF2C is required for the normal allocation of cells between the ventricular and sinoatrial precursors of the primary heart field. Dev. Dyn. 2006, 235, 1809–1821. [Google Scholar] [CrossRef]
- Vallaster, M.; Vallaster, C.D.; Wu, S.M. Epigenetic mechanisms in cardiac development and disease. Acta Biochim. Biophys. Sin. 2012, 44, 92–102. [Google Scholar] [CrossRef]
- Martinez, S.R.; Gay, M.S.; Zhang, L. Epigenetic mechanisms in heart development and disease. Drug Discov. Today 2015, 20, 799–811. [Google Scholar] [CrossRef]
- Lozano-Velasco, E.; Garcia-Padilla, C.; Del Mar Muñoz-Gallardo, M.; Martinez-Amaro, F.J.; Caño-Carrillo, S.; Castillo-Casas, J.M.; Sanchez-Fernandez, C.; Aranega, A.E.; Franco, D. Post-transcriptional regulation of molecular determinants during cardiogenesis. Int. J. Mol. Sci. 2022, 23, 2839. [Google Scholar] [CrossRef]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Olson, E.N. MicroRNA regulatory networks in cardiovascular development. Dev. Cell 2010, 18, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Wystub, K.; Besser, J.; Bachmann, A.; Boettger, T.; Braun, T. miR-1/133a clusters cooperatively specify the cardiomyogenic lineage by adjustment of myocardin levels during embryonic heart development. PLoS Genet. 2013, 9, e1003793. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yuan, Y.; He, X.; Xia, X.; Mo, X. MicroRNA-1 upregulation promotes myocardiocyte proliferation and suppresses apoptosis during heart development. Mol. Med. Rep. 2017, 15, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- Cianflone, E.; Scalise, M.; Marino, F.; Salerno, L.; Salerno, N.; Urbanek, K.; Torella, D. The negative regulation of gene expression by microRNAs as key driver of inducers and repressors of cardiomyocyte differentiation. Clin. Sci. 2022, 136, 1179–1203. [Google Scholar] [CrossRef]
- Samal, E.; Evangelista, M.; Galang, G.; Srivastava, D.; Zhao, Y.; Vedantham, V. Premature microRNA-1 expression causes hypoplasia of the cardiac ventricular conduction system. Front. Physiol. 2019, 10, 235. [Google Scholar] [CrossRef]
- Benzoni, P.; Nava, L.; Giannetti, F.; Guerini, G.; Gualdoni, A.; Bazzini, C.; Milanesi, R.; Bucchi, A.; Baruscotti, M.; Barbuti, A. Dual role of miR-1 in the development and function of sinoatrial cells. J. Mol. Cell. Cardiol. 2021, 157, 104–112. [Google Scholar] [CrossRef]
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.H.; Ma, Q.; Kang, P.M.; Golub, T.R.; Pu, W.T. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009, 29, 2193–2204. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.; van Mil, A.; van Vliet, P.; Metz, C.H.; Liu, J.; Doevendans, P.A.; Goumans, M.J. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, M.J.; Lo Surdo, P.; Di Giovine, P.; Cirillo, A.; Scarpelli, R.; Ferrigno, F.; Jones, P.; Neddermann, P.; De Francesco, R.; Steinkühler, C.; Gallinari, P.; Carfí, A. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J. Biol. Chem. 2008, 283, 26694–26704. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Sun, L.; Yang, X.J.; Zhu, G.; Wu, Z. Functional characterization of an amino-terminal region of HDAC4 that possesses MEF2 binding and transcriptional repressive activity. J. Biol. Chem. 2003, 278, 23515–23521. [Google Scholar] [CrossRef]
- Wang, Z.; Qin, G.; Zhao, T.C. HDAC4: mechanism of regulation and biological functions. Epigenomics 2014, 6, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol. Cell 2000, 6, 233–244. [Google Scholar] [CrossRef]
- Passier, R.; Zeng, H.; Frey, N.; Naya, F.J.; Nicol, R.L.; McKinsey, T.A.; Overbeek, P.; Richardson, J.A.; Grant, S.R.; Olson, E.N. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Invest. 2000, 105, 1395–1406. [Google Scholar] [CrossRef]
- Youn, H.D.; Grozinger, C.M.; Liu, J.O. Calcium regulates transcriptional repression of myocyte enhancer factor 2 by histone deacetylase 4. J. Biol. Chem. 2000, 275, 22563–22567. [Google Scholar] [CrossRef]
- Backs, J.; Song, K.; Bezprozvannaya, S.; Chang, S.; Olson, E.N. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Invest. 2006, 116, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Wu, S.; Peng, C.; Peng, B.; Luo, X.; Huang, L.; Zhang, H. Interactions between the ERK1/2 signaling pathway and PCAF play a key role in PE induced cardiomyocyte hypertrophy. Mol. Med. Rep. 2021, 24, 636. [Google Scholar] [CrossRef]
- Li, X.Q.; Lu, S.; Xia, L.; Shan, X.L.; Zhao, W.X.; Chen, H.H.; Zhang, C.; Guo, W.; Xu, M.; Lu, R.; Zhao, P. Stachydrine hydrochloride ameliorates cardiac hypertrophy through CaMKII/HDAC4/MEF2C signal pathway. Am. J. Transl. Res. 2022, 14, 3840–3853. [Google Scholar]
- Yao, Y.; Gupta, D.; Yelon, D. The MEK-ERK signaling pathway promotes maintenance of cardiac chamber identity. Development 2024, 151. [Google Scholar] [CrossRef]
- Garcia-Padilla, C.; Garcia-Lopez, V.; Aranega, A.; Franco, D.; Garcia-Martinez, V.; Lopez-Sanchez, C. Inhibition of RhoA and Cdc42 by miR-133a modulates retinoic acid signalling during early development of posterior cardiac tube segment. Int. J. Mol. Sci. 2022, 23, 4179. [Google Scholar] [CrossRef]
- Napoli, J.L. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta 2012, 182, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Durand, B.; Saunders, M.; Leroy, P.; Leid, M.; Chambon, P. All-trans and 9-cis retinoic acid induction of CRABPII transcription is mediated by RAR-RXR heterodimers bound to DR1 and DR2 repeated motifs. Cell 1992, 71, 73–85. [Google Scholar] [CrossRef]
- Dong, D.; Ruuska, S.E.; Levinthal, D.J.; Noy, N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J. Biol. Chem. 1999, 274, 23695–236958. [Google Scholar] [CrossRef] [PubMed]
- Napoli, J.L.; Boerman, M.H.; Chai, X.; Zhai, Y.; Fiorella, P.D. Enzymes and binding proteins affecting retinoic acid concentrations. J. Steroid. Biochem. Mol. Biol. 1995, 53, 497–502. [Google Scholar] [CrossRef]
- de The, H.; Vivanco-Ruiz, M.M.; Tiollais, P.; Stunnenberg, H.; Dejean, A. Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature 1990, 343, 177–180. [Google Scholar] [CrossRef]
- Kostetskii, I.; Linask, K.K.; Zile, M.H. Vitamin A deficiency and the expression of retinoic acid receptors during early cardiogenesis in quail embryo. Rouxs Arch. Dev. Biol. 1996, 205, 260–271. [Google Scholar] [CrossRef]
- Cui, J.; Michaille, J.J.; Jiang, W.; Zile, M.H. Retinoid receptors and vitamin A deficiency: differential patterns of transcription during early avian development and the rapid induction of RARs by retinoic acid. Dev. Biol. 2003, 260, 496–511. [Google Scholar] [CrossRef]
- Bastien, J.; Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004, 328, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanchez, C.; Bartulos, O.; Martinez-Campos, E.; Gañan, C.; Valenciano, A.I.; Garcia-Martinez, V.; De Pablo, F.; Hernandez-Sanchez, C. Tyrosine hydroxylase is expressed during early heart development and is required for cardiac chamber formation. Cardiovasc. Res. 2010, 88, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Miska, E.A.; Karlsson, C.; Langley, E.; Nielsen, S.J.; Pines. ; Kouzarides, T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999, 18, 5099–5107. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.P.; Denicola, M.; Qin, X.; Zhao, Y.; Zhang, L.; Long, X.L.; Zhuang, S.; Liu, P.Y.; Zhao, T.C. HDAC inhibition promotes cardiogenesis and the survival of embryonic stem cells through proteasome-dependent pathway. J. Cell Biochem. 2011, 112, 3246–3255. [Google Scholar] [CrossRef]
- Hou, F.; Wei, W.; Qin, X.; Liang, J.; Han, S.; Han, A.; Kong, Q. The posttranslational modification of HDAC4 in cell biology: Mechanisms and potential targets. J. Cell Biochem. 2020, 121, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.M.; Watson, P.J.; Fairall, L.; Jamieson, A.G.; Schwabe, J.W.R. Insights into the recruitment of class IIa Histone Deacetylases (HDACs) to the SMRT/NCoR transcriptional repression complex. J. Biol. Chem. 2015, 290, 18237–18244. [Google Scholar] [CrossRef] [PubMed]
- Grund, A.; Heineke, J. Targeting cardiac hypertrophy through a nuclear co-repressor. EMBO Mol. Med. 2019, 11, e11297. [Google Scholar] [CrossRef]
- Wu, X.; Li, H.; Park, E.J.; Chen, J.D. SMRTE inhibits MEF2C transcriptional activation by targeting HDAC4 and 5 to nuclear domains. J. Biol. Chem. 2001, 276, 24177–24185. [Google Scholar] [CrossRef]
- Li, C.; Sun, X.N.; Chen, B.Y.; Zeng, M.R.; Du, L.J.; Liu, T.; Gu, H.H.; Liu, Y.; Li, Y.L.; Zhou, L.J.; Zheng, X.J.; Zhang, Y.Y.; Zhang, W.C.; Liu, Y.; Shi, C.; Shao, S.; Shi, X.R.; Yi, Y.; Liu, X.; Wang, J.; Auwerx, J.; Wang, Z.V.; Jia, F.; Li, R.G.; Duan, S.Z. Nuclear receptor corepressor 1 represses cardiac hypertrophy. EMBO Mol. Med. 2019, 11, e9127. [Google Scholar] [CrossRef] [PubMed]
- Jonas, B.A.; Varlakhanova, N.; Hayakawa, F.; Goodson, M.; Privalsky, M.L. Response of SMRT (silencing mediator of retinoic acid and thyroid hormone receptor) and N-CoR (nuclear receptor corepressor) corepressors to mitogen-activated protein kinase kinase kinase cascades is determined by alternative mRNA splicing. Mol. Endocrinol. 2007, 21, 21,1924–1939. [Google Scholar] [CrossRef]
- Mottis, A.; Mouchiroud, L.; Auwerx, J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013, 27, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, V.; Gudas, L.J. Epigenetic regulatory mechanisms distinguish retinoic acid-mediated transcriptional responses in stem cells and fibroblasts. J. Biol. Chem. 2010, 285, 285,14534–14548. [Google Scholar] [CrossRef] [PubMed]
- Linney, E.; Donerly, S.; Mackey, L.; Dobbs-McAuliffe, B. The negative side of retinoic acid receptors. Neurotoxicol. Teratol. 2011, 33, 631–640. [Google Scholar] [CrossRef]
- Laursen, K.B.; Wong, P.M.; Gudas, L.J. Epigenetic regulation by RARalpha maintains ligand-independent transcriptional activity. Nucleic Acids Res. 2012, 40, 40,102–115. [Google Scholar] [CrossRef]
- Urvalek, A.M.; Gudas, LJ. Retinoic acid and histone deacetylases regulate epigenetic changes in embryonic stem cells. J. Biol. Chem. 2014, 289, 19519–19530. [Google Scholar] [CrossRef]
- Berenguer, M.; Meyer, K.F.; Yin, J.; Duester, G. Discovery of genes required for body axis and limb formation by global identification of retinoic acid-regulated epigenetic marks. PLoS Biol. 2020, 18, e3000719. [Google Scholar] [CrossRef]
- Petkovich, M.; Chambon, P. Retinoic acid receptors at 35 years. J. Mol. Endocrinol. 2022, 69, 13–24. [Google Scholar] [CrossRef]
- Clarke, C.J.; Shamseddine, A.A.; Jacob, J.J.; Khalife, G.; Burns, T.A.; Hannun, Y.A. ATRA transcriptionally induces nSMase2 through CBP/p300-mediated histone acetylation. J. Lipid. Res. 2016, 57, 868–881. [Google Scholar] [CrossRef]
- Loinder, K.; Söderström, M. The nuclear receptor corepressor (N-CoR) modulates basal and activated transcription of genes controlled by retinoic acid. J. Steroid. Biochem. Mol. Biol. 2003, 84, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Weston, A.D.; Blumberg, B.; Underhill, T.M. Active repression by unliganded retinoid receptors in development: less is sometimes more. J. Cell Biol. 2003, 161, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Xavier-Neto, J.; Sousa Costa, Â.M.; Figueira, A.C.; Caiaffa, C.D.; Amaral, F.N.; Peres, L.M.; da Silva, B.S.; Santos, L.N.; Moise, A.R.; Castillo, H.A. Signaling through retinoic acid receptors in cardiac development: Doing the right things at the right times. Biochim. Biophys. Acta 2015, 1849, 94–111. [Google Scholar] [CrossRef]
- Kumar, S.; Cunningham, T.J.; Duester, G. Nuclear receptor corepressors Ncor1 and Ncor2 (Smrt) are required for retinoic acid-dependent repression of Fgf8 during somitogenesis. Dev. Biol. 2016, 418, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G. Cellular retinoic acid-binding protein II: a coactivator of the transactivation by the retinoic acid receptor complex RAR-RXR. Nutr. Rev. 2000, 58, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, S.; Zaffran, S. Mechanisms of retinoic acid signaling during cardiogenesis. Mech. Dev. 2017, 143, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Mueller, L.; Collins, S.J. CaMKII regulates retinoic acid receptor transcriptional activity and the differentiation of myeloid leukemia cells. J. Clin. Invest. 2007, 117, 1412–1421. [Google Scholar] [CrossRef]
- Romeih, M.; Cui, J.; Michaille, J.J.; Jiang, W.; Zile, M.H. Function of RARgamma and RARalpha2 at the initiation of retinoid signaling is essential for avian embryo survival and for distinct events in cardiac morphogenesis. Dev. Dyn. 2003, 228, 697–708. [Google Scholar] [CrossRef]
- Zile, M.H. Vitamin A-not for your eyes only: requirement for heart formation begins early in embryogenesis. Nutrients 2010, 2, 532–550. [Google Scholar] [CrossRef]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef]
- Hong, S.H.; Fang, S.; Lu, B.C.; Nofsinger, R.; Kawakami, Y.; Castro, G.L.; Yin, Y.; Lin, C.; Yu, R.T.; Downes, M.; Izpisúa-Belmonte, J.C.; Shilatifard, A.; Evans, R.M. Corepressor SMRT is required to maintain Hox transcriptional memory during somitogenesis. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 10381–10386. [Google Scholar] [CrossRef]
- Won, J.Y.; Nam, E.C.; Yoo, S.J.; Kwon, H.J.; Um, S.J.; Han, H.S.; Kim, S.H.; Byun, Y.; Kim, S.Y. The effect of cellular retinoic acid binding protein-I expression on the CYP26-mediated catabolism of all-trans retinoic acid and cell proliferation in head and neck squamous cell carcinoma. Metabolism 2004, 53, 1007–1012. [Google Scholar] [CrossRef]
- Park, S.W.; Li, G.; Lin, Y.P.; Barrero, M.J.; Ge, K.; Roeder, R.G.; Wei, L.N. Thyroid hormone-induced juxtaposition of regulatory elements/factors and chromatin remodeling of Crabp1 dependent on MED1/TRAP220. Mol. Cell 2005, 19, 643–653. [Google Scholar] [CrossRef]
- Oliveira-Carvalho, V.; da Silva, M.M.; Guimarães, G.V.; Bacal, F.; Bocchi, E.A. MicroRNAs: new players in heart failure. Mol. Biol. Rep. 2013, 40, 2663–2670. [Google Scholar] [CrossRef]
- Duan, L.; Xiong, X.; Liu, Y.; Wang, J. miRNA-1: functional roles and dysregulation in heart disease. Mol. Biosyst. 2014, 10, 2775–82. [Google Scholar] [CrossRef]
- Kura, B.; Kalocayova, B.; Devaux, Y.; Bartekova, M. Potential clinical implications of miR-1 and miR-21 in heart disease and cardioprotection. Int. J. Mol. Sci. 2020, 21, 700. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.; Bahroudi, Z.; Shoorei, H.; Majidpoor, J.; Abak, A.; Taheri, M.; Ghafouri-Fard, S. miR-1: A comprehensive review of its role in normal development and diverse disorders. Biomed. Pharmacother. 2020, 132, 110903. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, E.; Medzikovic, L.; Ruffenach, G.; Eghbali, M. Role of miRNA-1 and miRNA-21 in acute myocardial ischemia-reperfusion injury and their potential as therapeutic strategy. Int. J. Mol. Sci. 2022, 23, 1512. [Google Scholar] [CrossRef] [PubMed]
- Souidi, A.; Nakamori, M.; Zmojdzian, M.; Jagla, T.; Renaud, Y.; Jagla, K. Deregulations of miR-1 and its target Multiplexin promote dilated cardiomyopathy associated with myotonic dystrophy type 1. EMBO Rep. 2023, 24, e56616. [Google Scholar] [CrossRef] [PubMed]
- Hamburger, V.; Hamilton, H.L. A series of normal stages in the development of the chick embryo. J. Morphol. 1951, 88, 49–92. [Google Scholar] [CrossRef] [PubMed]
- Hamburger, V.; Hamilton, H.L. A series of normal stages in the development of the chick embryo. Dev. Dyn. 1992, 195, 231–272. [Google Scholar] [CrossRef]
- Chapman, S.C.; Collignon, J.; Schoenwolf, G.C.; Lumsden, A. Improved method for chick whole-embry oculture using a filter paper carrier. Dev. Dyn. 2001, 220, 284–289. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–24. [Google Scholar] [CrossRef]
- Kalayinia, S.; Arjmand, F.; Maleki, M.; Malakootian, M.; Singh, C.P. MicroRNAs: roles in cardiovascular development and disease. Cardiovasc. Pathol. 2021, 50, 107296. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Franco, D.; Bonet, F.; Garcia-Lopez, V.; Aranega, A.; Garcia-Martinez, V. Negative Fgf8-Bmp2 feed-back is regulated by miR-130 during early cardiac specification. Dev. Biol. 2015, 406, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Darnell, D.K.; Kaur, S.; Stanislaw, S.; Konieczka, J.H.; Yatskievych, T.A.; Antin, P.B. MicroRNA expression during chick embryo development. Dev. Dyn. 2006, 235, 3156–3165. [Google Scholar] [CrossRef] [PubMed]
- Chapman, S.C.; Schubert, F.R.; Schoenwolf, G.C.; Lumsden, A. Analysis of spatial and temporal gene expression patterns in blastula and gastrula stage chick embryos. Dev. Biol. 2002, 245, 187–199. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Garcia-Martinez, V.; Lawson, A.; Chapman, S.C.; Schoenwolf, G.C. Rapid triple-labeling method combining in situ hybridization and double immunocytochemistry. Dev. Dyn. 2004, 230, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; Vandesompele, J.; Wittwer, C.T. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Bonet, F.; Dueñas, Á.; López-Sánchez, C.; García-Martínez, V.; Aránega, A.E.; Franco, D. MiR-23b and miR-199a impair epitelial to mesenchymal transition during atrioventricular endocardial cushion formation. Dev. Dyn. 2015, 244, 1259–1275. [Google Scholar] [CrossRef]
- Lozano-Velasco, E.; Galiano-Torres, J.; Jodar-Garcia, A.; Aranega, A.E.; Franco, D. miR-27 and miR-125 distinctly regulate muscle enriched transcription factors in cardiac and skeletal myocytes. BioMed. Res. Int. 2015, 2015, 391306. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta-Delta C (T)). Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, A.; Expósito, A.; Muñoz, M.D.M.; de Manuel, M.J.; Cámara-Morales, A.; Serrano-Osorio, F.; García-Padilla, C.; Hernández-Torres, F.; Domínguez, J.N.; Aránega, A.; et al. MiR-195 enhances cardiomyogenic differentiation of the proepicardium/ septum transversum by Smurf1 and Foxp1 modulation. Sci. Rep. 2020, 9, 9334. [Google Scholar] [CrossRef] [PubMed]
- Toro, R.; Pérez-Serra, A.; Mangas, A.; Campuzano, O.; Sarquella-Brugada, G.; Quezada-Feijoo, M.; Ramos, M.; Alcalá, M.; Carrera, E.; García-Padilla, C.; Franco, D.; Bonet, F. miR-16-5p suppression protects human cardiomyocytes against endoplasmic reticulum and oxidative stress-induced injury. Int. J. Mol. Sci. 2022, 23, 1036. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Effects of miR-1 gain- and loss-of-function experiments during early differentiation of cardiac sinoatrial region. Embryos microinjected with CFDA (control), premiR-1 or antimiR-1, at the level of the posterior cardiac precursors of both primitive endocardial tubes, and visualization of CFDA (A). Whole-mount ISH for Tbx5 (B) Gata4 (C), AMHC1 (D) and IMH for Mef2c (E). Note the increased and expanded expression of Tbx5, Gata4 and AMHC1 after premiR-1 treatment (blue arrows), accompanied by diminished protein levels of Mef2c (red arrowheads). The posterior diminished expressions of Tbx5, Gata4 and AMHC1 in the heart tube after antimiR-1 treatment is indicated by red arrows, whereas Mef2c is increased (blue arrowheads). Left side illustrates RT-qPCR of RNA from dissected cardiac asa of embryos microinjected either with CFDA, premiR-1 or antimiR-1. High level of miR-1 leads to increased Tbx5, Gata4 and AMHC1 transcripts, whereas miR-1 inhibition leads to Mef2c increased transcripts. Standard deviations are from three independent experiments. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005, **** p < 0.001 with respect to control (CFDA) embryos.
Figure 1.
Effects of miR-1 gain- and loss-of-function experiments during early differentiation of cardiac sinoatrial region. Embryos microinjected with CFDA (control), premiR-1 or antimiR-1, at the level of the posterior cardiac precursors of both primitive endocardial tubes, and visualization of CFDA (A). Whole-mount ISH for Tbx5 (B) Gata4 (C), AMHC1 (D) and IMH for Mef2c (E). Note the increased and expanded expression of Tbx5, Gata4 and AMHC1 after premiR-1 treatment (blue arrows), accompanied by diminished protein levels of Mef2c (red arrowheads). The posterior diminished expressions of Tbx5, Gata4 and AMHC1 in the heart tube after antimiR-1 treatment is indicated by red arrows, whereas Mef2c is increased (blue arrowheads). Left side illustrates RT-qPCR of RNA from dissected cardiac asa of embryos microinjected either with CFDA, premiR-1 or antimiR-1. High level of miR-1 leads to increased Tbx5, Gata4 and AMHC1 transcripts, whereas miR-1 inhibition leads to Mef2c increased transcripts. Standard deviations are from three independent experiments. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005, **** p < 0.001 with respect to control (CFDA) embryos.
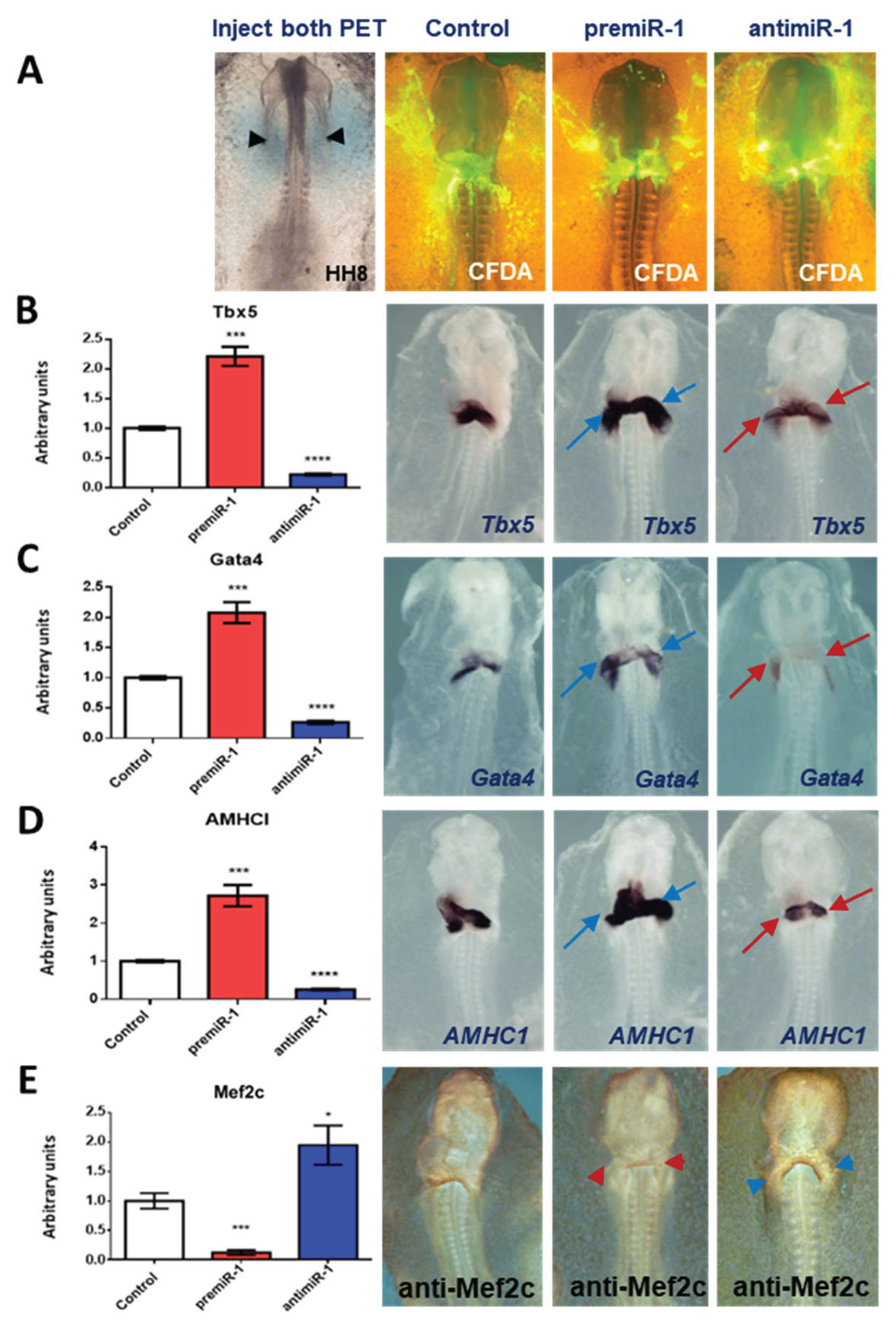
Figure 2.
Effects of miR-1 gain- and loss-of-function experiments on RA pathway during posterior differentiation of cardiac tube. RT-qPCR of RNA from dissected cardiac asa of embryos microinjected either with CFDA, premiR-1 or antimiR-1. High level of miR-1 leads to increased expression of CRABPII and decreased expression of CRABPI. The opposite effect is observed with anti-miR1 treatment. Note that nonsignificant expression of Raldh2 is observed after miR-1 overexpression or miR-1 inhibitor treatment, as compared to control (CFDA). Standard deviations are from three independent experiments. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005, **** p < 0.001 with respect to control (CFDA) embryos.
Figure 2.
Effects of miR-1 gain- and loss-of-function experiments on RA pathway during posterior differentiation of cardiac tube. RT-qPCR of RNA from dissected cardiac asa of embryos microinjected either with CFDA, premiR-1 or antimiR-1. High level of miR-1 leads to increased expression of CRABPII and decreased expression of CRABPI. The opposite effect is observed with anti-miR1 treatment. Note that nonsignificant expression of Raldh2 is observed after miR-1 overexpression or miR-1 inhibitor treatment, as compared to control (CFDA). Standard deviations are from three independent experiments. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005, **** p < 0.001 with respect to control (CFDA) embryos.
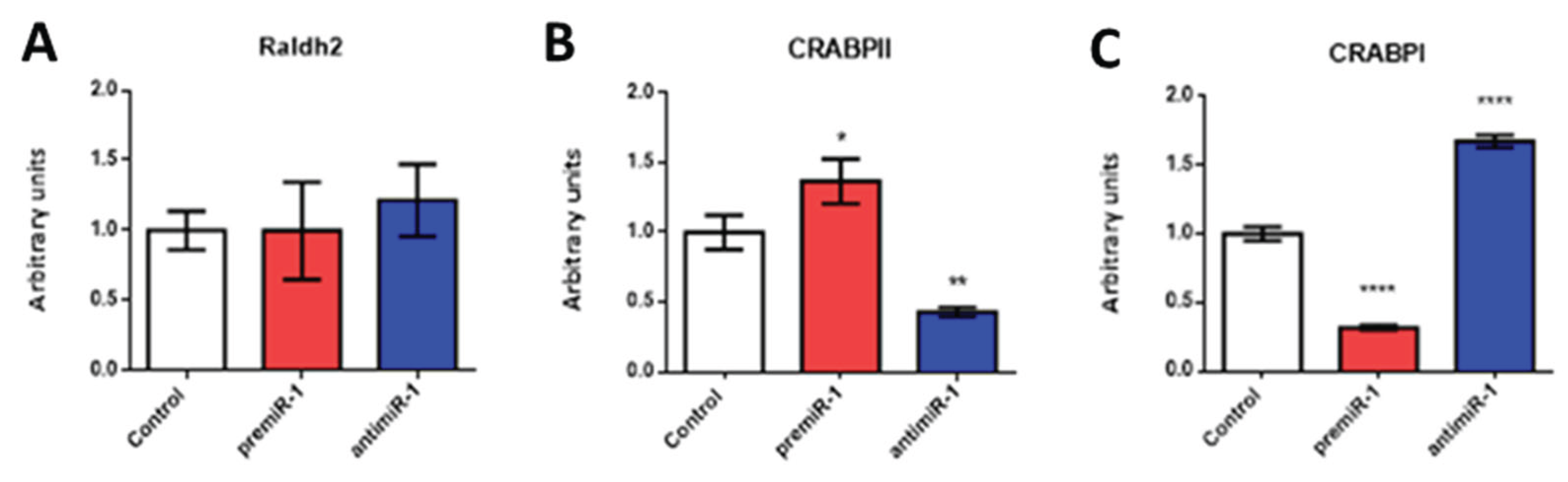
Figure 3.
Embryos subjected to Rarß study. A: Whole-mount IMH for Rarß during early chick cardiac development, from HH8 through HH11, in control embryos. Note Rarß distribution in both primitive endocardial tubes (PET), being observable in sinoatrial (SA) region at later stages. B: Experimental embryos microinjected with CFDA (control), premiR-1 or antimiR-1, at the level of the posterior cardiac precursors of both primitive endocardial tubes (arrowheads), and visualization of CFDA. C: Whole-mount IMH illustrate Rarß markedly increased after miR-1 administration (blue arrows). Note Rarß diminished after antimiR-1 administration (red arrows). RT-qPCR of RNA from dissected cardiac asa (left side) show the levels of Rarß transcripts. Standard deviations are from three independent experiments. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005, **** p < 0.001 with respect to control (CFDA) embryos.
Figure 3.
Embryos subjected to Rarß study. A: Whole-mount IMH for Rarß during early chick cardiac development, from HH8 through HH11, in control embryos. Note Rarß distribution in both primitive endocardial tubes (PET), being observable in sinoatrial (SA) region at later stages. B: Experimental embryos microinjected with CFDA (control), premiR-1 or antimiR-1, at the level of the posterior cardiac precursors of both primitive endocardial tubes (arrowheads), and visualization of CFDA. C: Whole-mount IMH illustrate Rarß markedly increased after miR-1 administration (blue arrows). Note Rarß diminished after antimiR-1 administration (red arrows). RT-qPCR of RNA from dissected cardiac asa (left side) show the levels of Rarß transcripts. Standard deviations are from three independent experiments. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005, **** p < 0.001 with respect to control (CFDA) embryos.
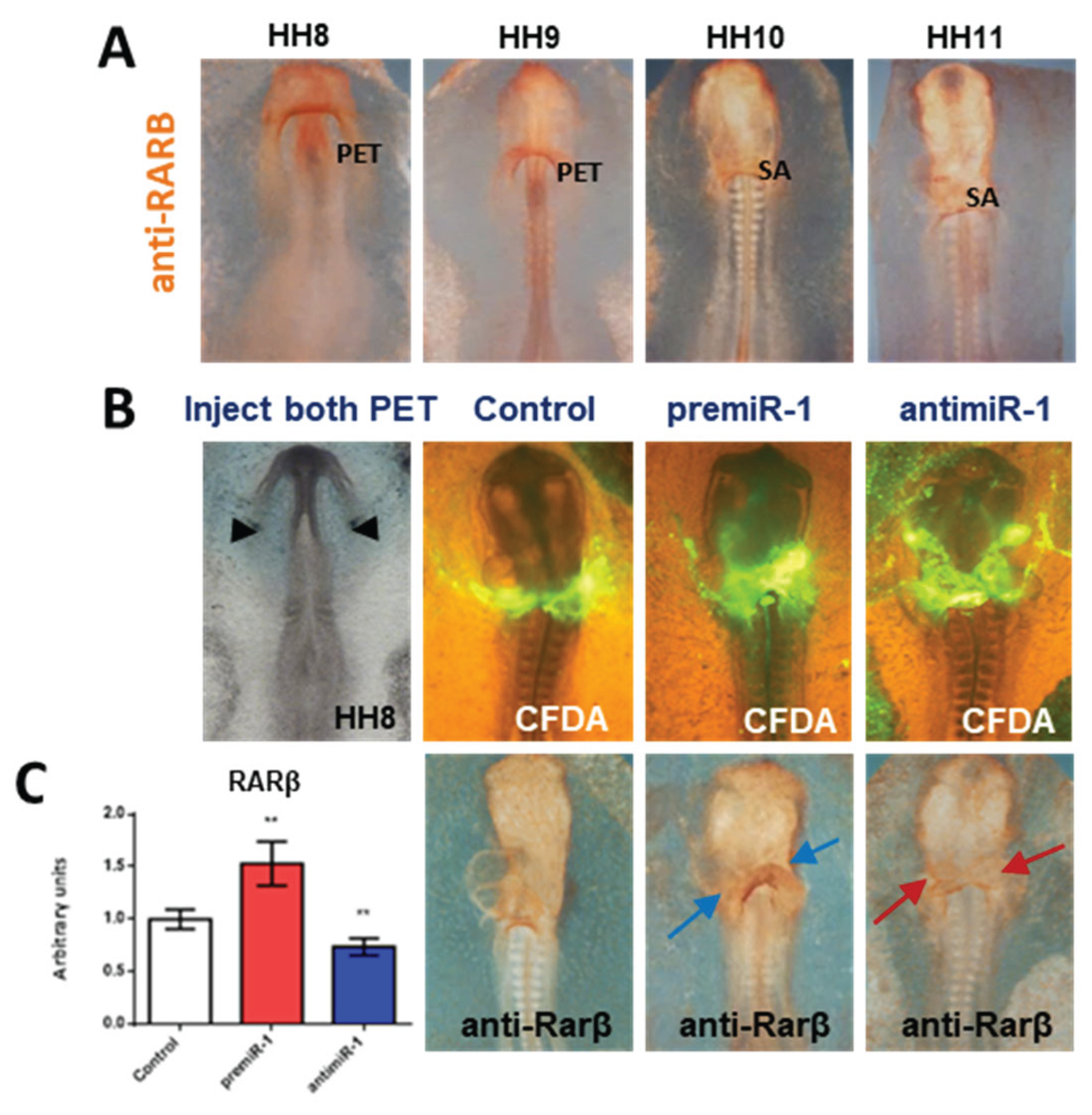
Figure 4.
Effects of miR-1 gain- and loss-of-function experiments on RA pathway during posterior differentiation of cardiac tube. RT-qPCR of RNA from dissected cardiac asa of embryos microinjected either with CFDA, premiR-1 or antimiR-1. Note that nonsignificant expression of RARα, RXRα, RARƳ, RXRƳ are observed after miR-1 overexpression, or miR-1 inhibitor treatment as compared to control (CFDA). Standard deviations are from three independent experiments.
Figure 4.
Effects of miR-1 gain- and loss-of-function experiments on RA pathway during posterior differentiation of cardiac tube. RT-qPCR of RNA from dissected cardiac asa of embryos microinjected either with CFDA, premiR-1 or antimiR-1. Note that nonsignificant expression of RARα, RXRα, RARƳ, RXRƳ are observed after miR-1 overexpression, or miR-1 inhibitor treatment as compared to control (CFDA). Standard deviations are from three independent experiments.
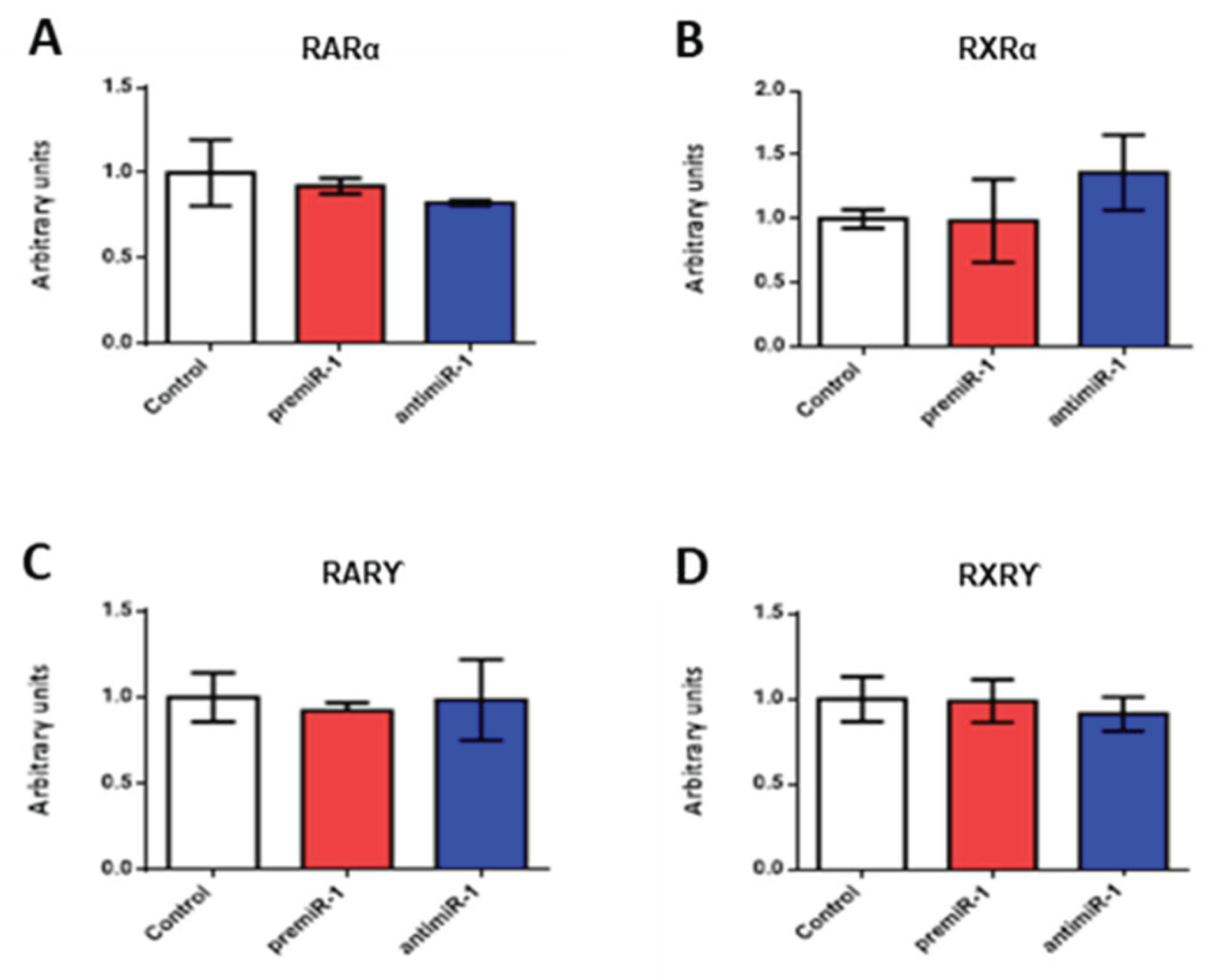
Figure 5.
Representative data of HDAC4 (A1: position 1; A2: position 2), Calmodulin (B) and Erk2/MAPK1 (C) 3′UTR luciferase assays after premiR-1 overexpression in 3T3 fibroblasts. Luciferase activity was compared to non-transfected controls. Each luciferase assay was carried out in triplicate. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005.
Figure 5.
Representative data of HDAC4 (A1: position 1; A2: position 2), Calmodulin (B) and Erk2/MAPK1 (C) 3′UTR luciferase assays after premiR-1 overexpression in 3T3 fibroblasts. Luciferase activity was compared to non-transfected controls. Each luciferase assay was carried out in triplicate. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005.
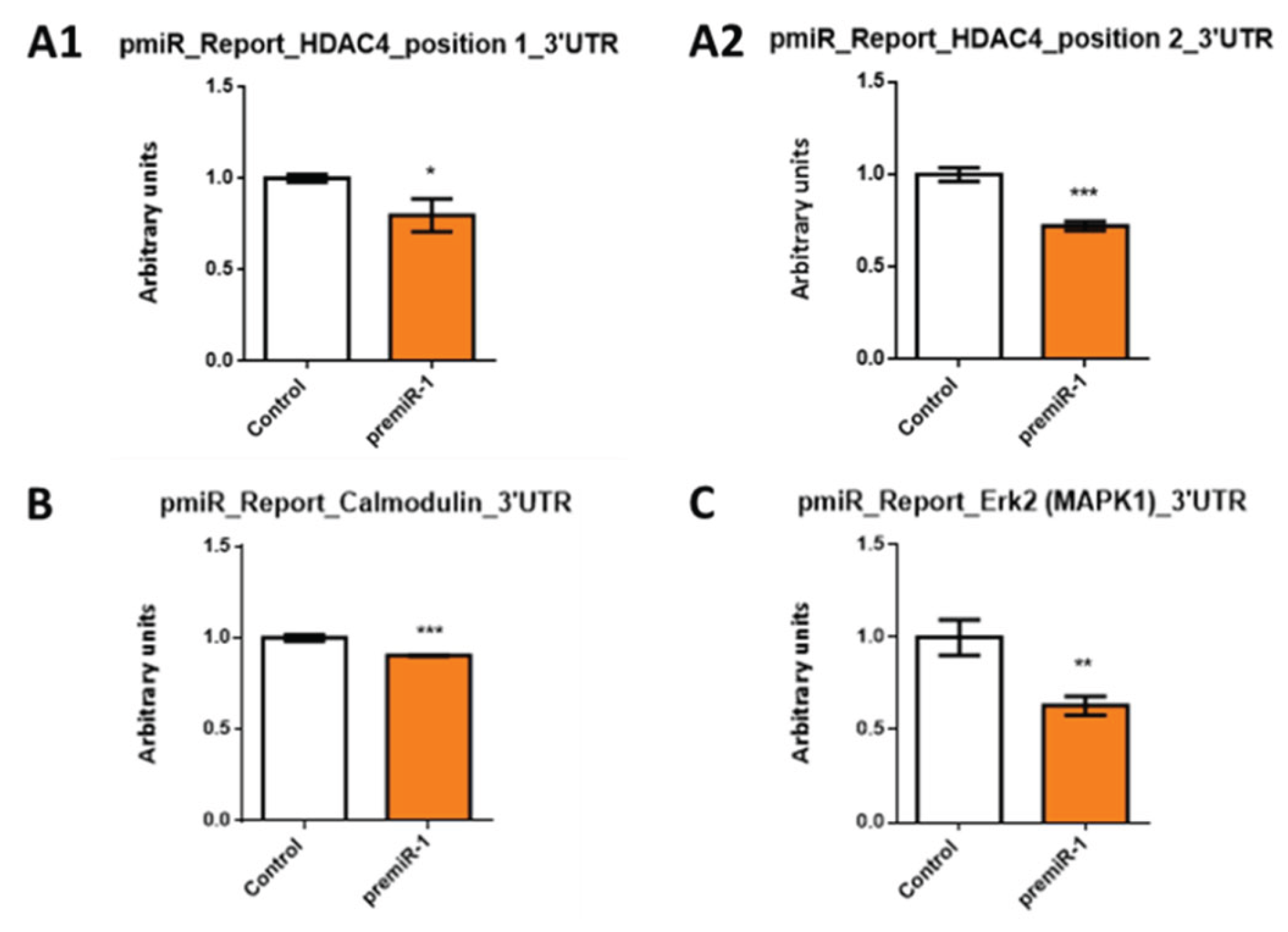
Figure 6.
Whole-mount ISH for miR-1 and whole-mount IMH for HDAC4, Calmodulin and phospho-Erk2 (pErk2) during early chick cardiac development, from HH8 through HH11, in control embryos. Note miR-1 expression pattern in both primitive endocardial tubes (PET), being observable in sinoatrial (SA) region and ventricle (V) at later stages. Note the location of HDAC4, Calmodulin and pErk2 in the PET and, subsequently, in the SA region. Arrows: positive stains.
Figure 6.
Whole-mount ISH for miR-1 and whole-mount IMH for HDAC4, Calmodulin and phospho-Erk2 (pErk2) during early chick cardiac development, from HH8 through HH11, in control embryos. Note miR-1 expression pattern in both primitive endocardial tubes (PET), being observable in sinoatrial (SA) region and ventricle (V) at later stages. Note the location of HDAC4, Calmodulin and pErk2 in the PET and, subsequently, in the SA region. Arrows: positive stains.
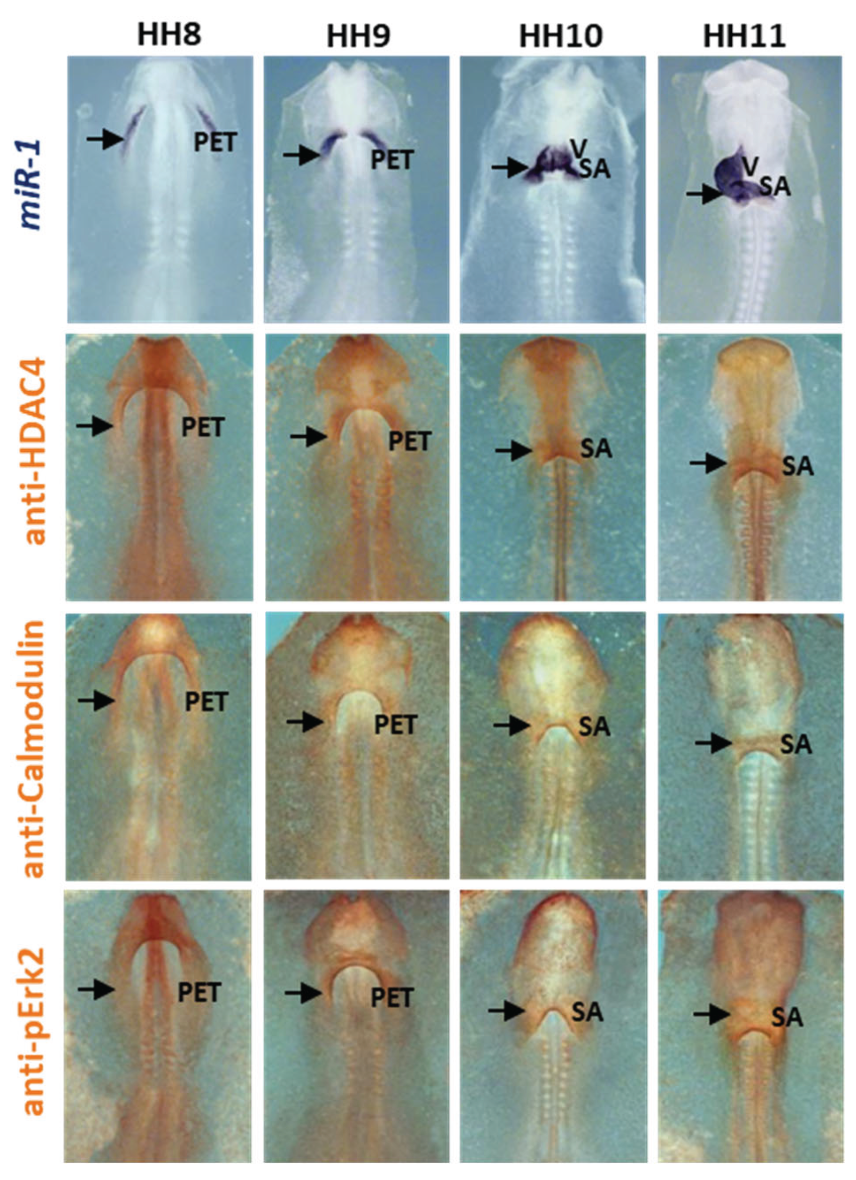
Figure 7.
Embryos microinjected with CFDA (control), premiR-1 or antimiR-1, at the level of the posterior cardiac precursors of both primitive endocardial tubes (arrowheads), and visualization of CFDA (A). Effects of miR-1 gain- and loss-of-function on target genes HDAC4, Calmodulin and phospho-Erk2/MAPK1 (pErk2) during posterior differentiation of cardiac tube (B-D). Whole-mount IMH reveal that HDAC4 (B), Calmodulin (C) and pErk2 (D) are dramatically reduced in the sinoatrial region (red arrows) after premiR-1 administration, whereas they are markedly increased after miR-1 inhibition (blue arrows). RT-qPCR of RNA from dissected cardiac asa (left side) in embryos microinjected either with CFDA, premiR-1 or antimiR-1, show that miR-1 leads to decreased HDAC4, Calmodulin and Erk2/MAPK1 transcripts, whereas miR-1 inhibition leads to increased transcripts. Standard deviations are from three independent experiments. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005, **** p < 0.001 with respect to control (CFDA) embryos.
Figure 7.
Embryos microinjected with CFDA (control), premiR-1 or antimiR-1, at the level of the posterior cardiac precursors of both primitive endocardial tubes (arrowheads), and visualization of CFDA (A). Effects of miR-1 gain- and loss-of-function on target genes HDAC4, Calmodulin and phospho-Erk2/MAPK1 (pErk2) during posterior differentiation of cardiac tube (B-D). Whole-mount IMH reveal that HDAC4 (B), Calmodulin (C) and pErk2 (D) are dramatically reduced in the sinoatrial region (red arrows) after premiR-1 administration, whereas they are markedly increased after miR-1 inhibition (blue arrows). RT-qPCR of RNA from dissected cardiac asa (left side) in embryos microinjected either with CFDA, premiR-1 or antimiR-1, show that miR-1 leads to decreased HDAC4, Calmodulin and Erk2/MAPK1 transcripts, whereas miR-1 inhibition leads to increased transcripts. Standard deviations are from three independent experiments. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005, **** p < 0.001 with respect to control (CFDA) embryos.
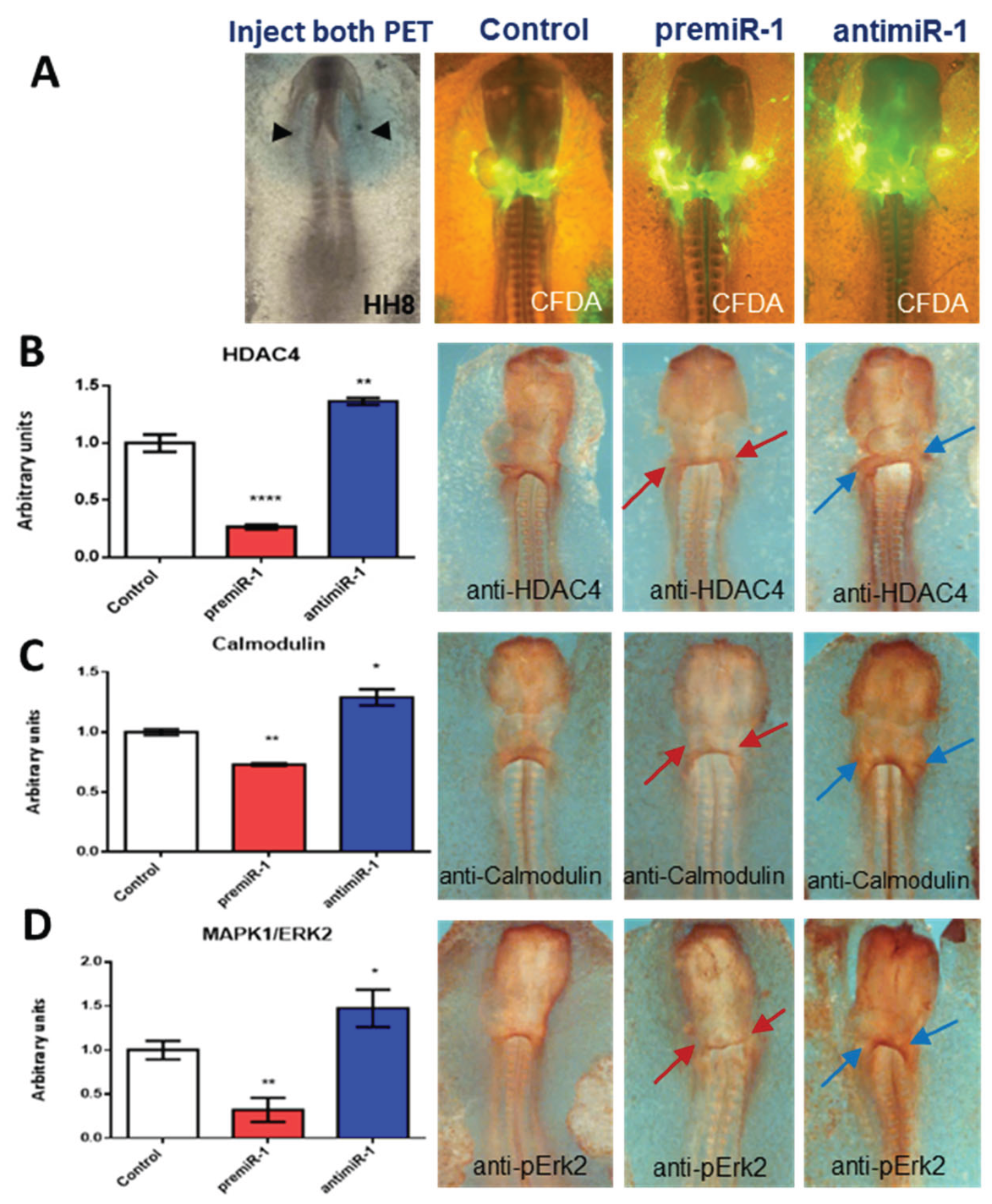
Figure 8.
Schematic drawing that summarizes the results of this work. miR-1 induces Tbx5, Gata4, AMHC1, RARβ and CRABPII, while it suppresses CRABPI, Calmodulin, HDAC4 and Erk2/MAPK1, as well as Mef2c, thus modulating early differentiation of cardiac sinoatrial region.
Figure 8.
Schematic drawing that summarizes the results of this work. miR-1 induces Tbx5, Gata4, AMHC1, RARβ and CRABPII, while it suppresses CRABPI, Calmodulin, HDAC4 and Erk2/MAPK1, as well as Mef2c, thus modulating early differentiation of cardiac sinoatrial region.
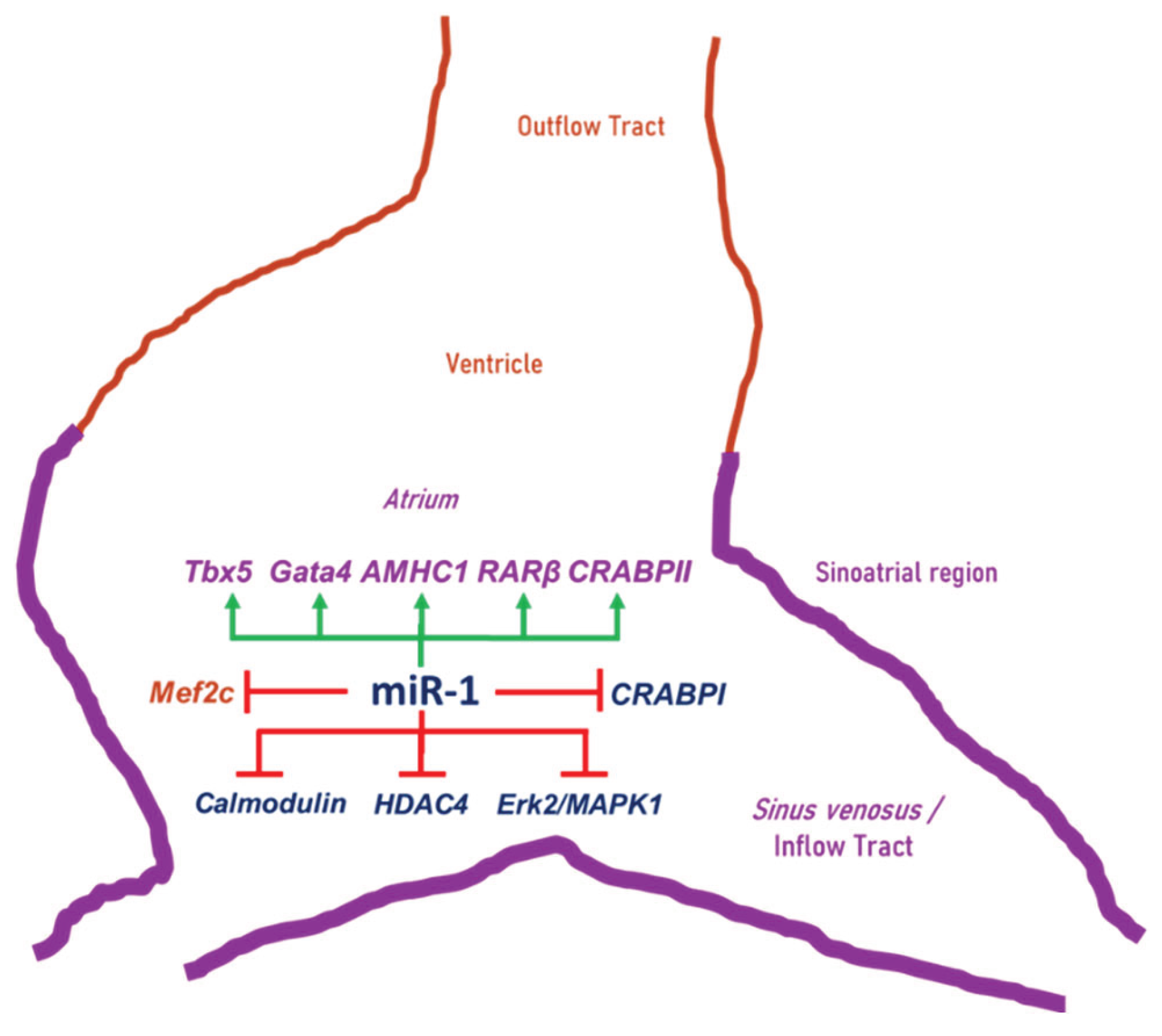
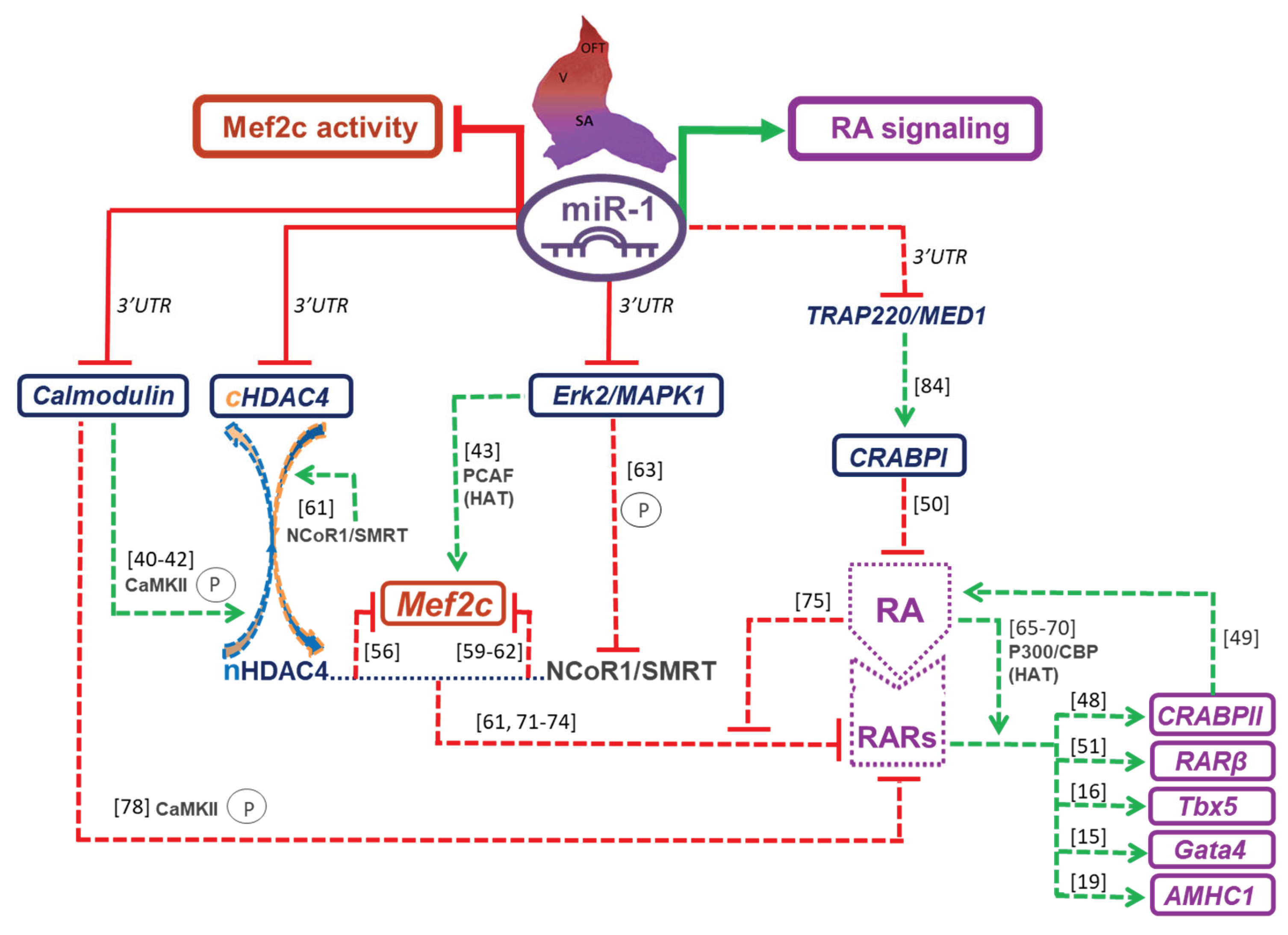
Figure 9.
Proposed model illustrating the network of molecular mechanisms governed by miR-1 during early differentiation of cardiac sinoatrial region. Solid lines indicate our results from this work. Dashed lines correspond to previous studies in different research fields referred in the “Discussion” section. Our model indicates that miR-1 plays a crucial role repressing Mef2c through modulation of HDAC4, Calmodulin and Erk2/MAPK1. In these molecular mechanisms, miR-1 modulates opposite effects between Calmodulin and NCoR1/SMRT on nuclear HDAC4 (nHDAC4), thus increasing Mef2c interaction with nHDAC4, which inhibits Mef2c expression. Also, miR-1 suppresses Erk2/MAPK1 and, consequently, diminishes Mef2c activity. Additionally, our model indicates that RA function is modulated by miR-1, thus promoting the expression of RA target genes Tbx5, Gata4, AMHC1 and RARβ. Moreover, miR-1 increases CRABPII and supresses CRABPI, thus increasing RA activity. The presence of RA disrupts the interaction of nHDAC4-NCoR1/SMRT with RARs, thus allowing transcription of RARs target genes. Also miR-1 represses Calmodulin capability to inhibit RARs activity, thus enhancing the expression of RA target genes. In our model miR-1 modulates the opposite actions between RA and Mef2c, promoting RA signaling pathway and suppressing Mef2c activity to allow properly assign cells to their cardiac chamber. OFT: outflow tract, V: ventricular region, SA: sinoatrial region, P: phosphorylate, [cites listed in the “References” section].
Figure 9.
Proposed model illustrating the network of molecular mechanisms governed by miR-1 during early differentiation of cardiac sinoatrial region. Solid lines indicate our results from this work. Dashed lines correspond to previous studies in different research fields referred in the “Discussion” section. Our model indicates that miR-1 plays a crucial role repressing Mef2c through modulation of HDAC4, Calmodulin and Erk2/MAPK1. In these molecular mechanisms, miR-1 modulates opposite effects between Calmodulin and NCoR1/SMRT on nuclear HDAC4 (nHDAC4), thus increasing Mef2c interaction with nHDAC4, which inhibits Mef2c expression. Also, miR-1 suppresses Erk2/MAPK1 and, consequently, diminishes Mef2c activity. Additionally, our model indicates that RA function is modulated by miR-1, thus promoting the expression of RA target genes Tbx5, Gata4, AMHC1 and RARβ. Moreover, miR-1 increases CRABPII and supresses CRABPI, thus increasing RA activity. The presence of RA disrupts the interaction of nHDAC4-NCoR1/SMRT with RARs, thus allowing transcription of RARs target genes. Also miR-1 represses Calmodulin capability to inhibit RARs activity, thus enhancing the expression of RA target genes. In our model miR-1 modulates the opposite actions between RA and Mef2c, promoting RA signaling pathway and suppressing Mef2c activity to allow properly assign cells to their cardiac chamber. OFT: outflow tract, V: ventricular region, SA: sinoatrial region, P: phosphorylate, [cites listed in the “References” section].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



