
Submitted:
31 March 2024
Posted:
02 April 2024
You are already at the latest version
Abstract
Macro-autophagy (autophagy hereafter) is an evolutionarily conserved cellular process that has long been recognized as an intracellular mechanism for maintaining cellular homeostasis. It involves the formation of a membraned structure called the autophagosome, which carries cargo that includes toxic protein aggregates and dysfunctional organelles to the lysosome for degradation and recycling. Autophagy is primarily considered and studied as a cell-autonomous mechanism. However, recent studies have illuminated an underappreciated facet of autophagy, i.e., non-autonomously regulated autophagy. Non- autonomously regulated autophagy involves the degradation of autophagic components, including organelles, cargo, and signaling molecules, and is induced in neighboring cells by signals from primary adjacent or distant cells/tissues/organs. This review provides insight into the complex molecular mechanisms governing non-autonomously regulated autophagy, highlighting the dynamic interplay between cells within tissue/organ or distinct cell types in different tissues/organs. Emphasis is placed on modes of intercellular communication that include secreted molecules, including microRNAs, and their regulatory roles in orchestrating this phenomenon. Furthermore, we explore the multidimensional roles of non-autonomously regulated autophagy in various physiological contexts, spanning tissue development and aging, as well as its importance in diverse pathological conditions, including cancer and neurodegeneration. By studying the complexities of non-autonomously regulated autophagy, we hope to gain insights into the sophisticated intercellular dynamics within multicellular organisms, including mammals. These studies will uncover novel avenues for therapeutic intervention to modulate intercellular autophagic pathways in altered human physiology.
Keywords:
autophagy
; cell non-autonomous
; disease
; regulation
; cell-autonomous
; intercellular/organ
Introduction
Autophagy is a highly regulated, lysosomal-dependent, intracellular degradative mechanism assisting in the cytoplasmic clearance of deleterious organelles and toxic proteins. It involves cargo sequestration in a double-membrane vesicle called an autophagosome, which then fuses with a lysosome to form an `autolysosome. Within the autolysosome, the cargo gets degraded by lysosomal hydrolases, and the degradation products are recycled back into the cytoplasm. Autophagy at basal levels helps maintain homeostasis within the cells. It is upregulated when the cells, tissues, organs, and organisms are under stress, including nutrient deprivation and oxidative stress. Autophagy is crucial for stem-cell maintenance and differentiation, programmed cell death (PCD), cellular homeostasis, and aging. Altered autophagy has several implications in major human diseases like cancer and neurodegenerative disorders, including Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), and metabolic disorders. Thus, in-depth studies on deciphering mechanisms of autophagy upregulation and suppression are crucial from the therapeutical point of view.
Autophagy is a cell-autonomous mechanism, i.e., occurs within the cell’s cytoplasm. Autophagy is regulated by several growth signaling pathways like mTOR, AMPK, Insulin signaling, etc. [1,2,3,4,5,6,7]. There are three main types of autophagy classified based on the mode of cargo delivery for degradation: microautophagy, chaperon-mediated autophagy (CMA), and macroautophagy [8,9,10,11]. In microautophagy, cargo is directly engulfed by the lysosomal membrane and is the least understood type of autophagy [8]. CMA involves selective degradation of cytoplasmic components, and adaptor proteins called chaperons are important for interaction with the cargo and deliver the cargo to the lysosome directly where the cargo is degraded [9]. Macroautophagy (called autophagy hereafter) is the best understood, most complex form of autophagy that involves the formation of autophagosomes and is a commonly studied type of autophagy regulated by Autophagy-related (Atgs) proteins [10].
Autophagy is mostly considered a cell-autonomous mechanism. However, recently, several reports have shown non-autonomously regulated autophagy wherein autophagy gets altered by neighboring or distant cells/ tissue or even an organ [12]. This review attempts to summarize non-autonomously regulated autophagy and its implications. This review will summarize several reports highlighting the importance of non-autonomously regulated autophagy in several aspects of organismal physiology in wild-type and diseased conditions. We selected the research articles based on three criteria: first, the report should involve at least two neighboring cells/tissues. Second, a biomolecule or factor communicating between cells should be either a secretory biomolecule, a part of the extracellular matrix (ECM), or a receptor on the cell’s membrane. Lastly, biomolecule(s) from one cell/ tissue should alter autophagy in neighboring, adjacent, or distant cells.
Autophagy: An Overview
Autophagy was first described in the 1960s [13]. However, it was only in the 1990s that the Autophagy-related (Atg) genes controlling the process were identified [14,15,16]. For simplicity, autophagy is divided into several steps, i.e., initiation, nucleation, elongation, maturation, fusion with the lysosome, and finally, degradation and release of nutrients in the cytoplasm. Genetic screens have identified more than 40 Atg genes in metazoans that have been shown to control different steps of autophagy [17]. Ulk1 kinase complex (Ulk1/Atg1, Atg13, FIP200, and Atg101) is tightly regulated positively and negatively [18,19,20,21,22]. The initiation of autophagy requires the ULK1 kinase complex, which is tightly regulated by AMPK and mTOR which acts as an activator and inhibitor, respectively [1,5,6,7,23,24,25]. AMPK activates ULK1 through a series of phosphorylation events, while active mTOR inhibits ULK1 through phosphorylation of a different set of serine/threonine residues [1,3,23,26,27]. The activated ULK1 complex, composed of RB1CC1/FIP200, ATG13 and ATG101, can act on the components of the initiation complex as well as class III phosphatidylinositol 3-kinase (PIK3C3) complex, composed of BECN1/Atg6, ATG14L/UVRAG, VPS15 and VPS34, where it phosphorylates both Atg6/Becn1 and Atg14 [26,28,29,30,31,32,33,34,35,36]. Vps34 complex produces a pool of phosphatidylinositol 3-phosphate (PI3P) [35,37]. PI3P formation on the membrane leads to the recruitment of WIPI proteins (WIPI1-4/Atg18) [38,39,40]. WIPI proteins direct ATG2, a lipid transferase, to the phagophore membrane, which transfers phospholipids from the ER (membrane source) in concert with ATG9, which scrambles phospholipids with its scramblase activity [41,42,43,44,45,46,47,48]. WIPI proteins also mediate recruiting of the ATG12–ATG5–ATG16L1 (E3-like) complex in the expanding phagophores [41,42,48,49,50,51,52]. LC3/Atg8 is first cleaved by the ATG4 cysteine proteases to form cytosolic LC3-I, which is then transferred to E1-like activating enzyme ATG7, then subsequently to the E2-like conjugating enzyme ATG3 and finally linked covalently to phosphatidylethanolamine (PE) by E3-like ligase activity of Atg12-Atg5-Atg16 complex [49,50,51,52,53,54,55,56,57,58,59,60,61,62]. The PE-conjugated LC3/Atg8 is referred to as LC3-II/Atg8-PE [10,11,41,49,50,52,61,62]. LC3-II can interact with autophagy receptors such as p62 bound to cargo targeted for degradation via LC3-interacting regions [63]. Fusion of autophagosomes with lysosomes is catalyzed by RAB proteins, SNARE proteins, and HOPS complex proteins to form autolysosomes [10,41,64,65,66,67]. Within the autolysosomes, the cargo is degraded by lysosomal hydrolases (like Cathepsins, phosphatases, nucleases, glycosidases, proteases, peptidases, and lipases), and the degradation products can be reused by the cell after being transported out of the lysosomes (permeases, transporters, etc.) [10]. LC3-II/Atg8-PE bound to the outer membrane of the autolysosomes is cleaved by ATG4 to be reused for a new round of lipidation.
Cell-Autonomous Autophagy
Cell-autonomous autophagy is an extensively studied form of autophagy. This form of autophagy is studied in distinct cell types in vitro and in vivo contexts, including stress. This review focuses on non-autonomously regulated autophagy, and a discussion on cell-autonomous autophagy can be referred to elsewhere [10,11,25,68,69,70,71]. The next sections deal with non-autonomously regulated autophagy in different cell types and contexts in several models and paradigms.
Non-Autonomously Regulated Autophagy
- 1.
- Non-autonomously regulated autophagy in Aging
Aging is best described as a progressive increase in the probability of death for an individual with the passage of time [72,73]. It is well established that there is a gradual decline of autophagy leading to loss of proteostasis with age, and hence, loss of autophagy is often considered one of the causal factors of aging; decreasing proteostasis is systemically controlled among tissues, hence suggesting a cell non-autonomous regulation of autophagy [74,75,76,77,78,79,80].
Studies in D. melanogaster have established that signaling pathways like mTOR, EGFR, and AMPK regulate autophagy [2,3,81]. AMPK signaling in fruitflies has anti-aging effects when neuronally overexpressed, resulting in lifespan extension. Overexpression of AMPK in neurons using Elav-GS led to autophagy induction locally (within the tissue), but amazingly, it also led to a significant autophagy induction non-autonomously regulated in the intestinal cells and muscle cells. Induction of autophagy within the intestine prevented aging-associated intestinal dysfunction. Likewise, autophagy induction in muscles led to improved muscle function during aging. In yet another interesting experiment, AMPK overexpression within the intestine led to autophagy induction in the brain non-autonomously regulated, suggesting that there is crosstalk between these tissues to regulate autophagy [4,82]. Thus, the gut-brain axis is involved in the cross-regulation of autophagy in these tissues. The data further show that slowed intestinal aging led to increased organismal lifespan, and aging in muscle cells was also delayed due to the sequestration and subsequent destruction of damaged proteins and organelles. The researchers demonstrated that the beneficial effects of AMPK/Atg1 overexpression are due to a systemic increase in 4E-BP expression and reduced DILP (Drosophila Insulin-like peptides) levels in the brain [4]. In addition, overexpression of the uppermost hierarchical autophagy kinase Atg1 in the brain distally induces autophagy in midgut enterocytes, which assists in maintaining intestinal homeostasis [4]. The model for this regulation is shown in Figure 3A.
Figure 1.
Autophagy and its regulation. Within the cytoplasmic milieu of eukaryotic cells, induction of autophagy leads to the formation of double-membrane organelles, known as autophagosomes. Autophagosomes carry cytoplasmic cargo of toxic proteins, dysfunctional organelles, and intracellular pathogens such as bacteria and viruses. This orchestrated process, termed autophagy, is initiated by the ULK1 complex, a multimeric assembly comprising ULK/ Atg1, ATG13, FIP200, and ATG101. Vps34 complex consists of Vps34, Vps15, Atg6 and Vps14. WIPI-like proteins, including Atg18, bind phosphoinositides to recruit Atg2 and Atg9. Autophagosome elongation and maturation entail the concerted action of two ubiquitin-like conjugation systems: the LC3/Atg8 (Atg8, Atg4, Atg7, Atg3) and ATG12 (Atg12, Atg5, Atg16, Atg7, Atg10) pathways. Upon closure, autophagosomes undergo irreversible fusion with lysosomes (catalyzed by SNAREs, HOPS complex, Atg14 etc.) culminating in the formation of autolysosomes. Within these acidic compartments, macromolecules undergo enzymatic degradation, yielding amino acids, fatty acids, monosaccharides, and nucleotides, which are subsequently recycled into the cytosol, thus sustaining cellular metabolism and homeostasis.
Figure 1.
Autophagy and its regulation. Within the cytoplasmic milieu of eukaryotic cells, induction of autophagy leads to the formation of double-membrane organelles, known as autophagosomes. Autophagosomes carry cytoplasmic cargo of toxic proteins, dysfunctional organelles, and intracellular pathogens such as bacteria and viruses. This orchestrated process, termed autophagy, is initiated by the ULK1 complex, a multimeric assembly comprising ULK/ Atg1, ATG13, FIP200, and ATG101. Vps34 complex consists of Vps34, Vps15, Atg6 and Vps14. WIPI-like proteins, including Atg18, bind phosphoinositides to recruit Atg2 and Atg9. Autophagosome elongation and maturation entail the concerted action of two ubiquitin-like conjugation systems: the LC3/Atg8 (Atg8, Atg4, Atg7, Atg3) and ATG12 (Atg12, Atg5, Atg16, Atg7, Atg10) pathways. Upon closure, autophagosomes undergo irreversible fusion with lysosomes (catalyzed by SNAREs, HOPS complex, Atg14 etc.) culminating in the formation of autolysosomes. Within these acidic compartments, macromolecules undergo enzymatic degradation, yielding amino acids, fatty acids, monosaccharides, and nucleotides, which are subsequently recycled into the cytosol, thus sustaining cellular metabolism and homeostasis.

Figure 2.
Cell-autonomous and cell non-autonomous regulation of autophagy. Cell-autonomous autophagy occurs within the cytoplasm of the cell upon several stimuli which can culminate on one or more of the proteins shown. AMPK, FOXO and exercise positively regulate autophagy while nutrients, insulin, AKT and mTOR negatively regulates autophagy. Cell non-autonomous autophagy refers to a phenomenon in which the autophagic activity of a cell is influenced by signals or factors originating from neighbouring cells or tissues. This could occur by secreted factors and membrane bound proteins. The induction of cell non-autonomous autophagy can occur in neighbouring cell (green) or a distant cell/tissue (light blue).
Figure 2.
Cell-autonomous and cell non-autonomous regulation of autophagy. Cell-autonomous autophagy occurs within the cytoplasm of the cell upon several stimuli which can culminate on one or more of the proteins shown. AMPK, FOXO and exercise positively regulate autophagy while nutrients, insulin, AKT and mTOR negatively regulates autophagy. Cell non-autonomous autophagy refers to a phenomenon in which the autophagic activity of a cell is influenced by signals or factors originating from neighbouring cells or tissues. This could occur by secreted factors and membrane bound proteins. The induction of cell non-autonomous autophagy can occur in neighbouring cell (green) or a distant cell/tissue (light blue).
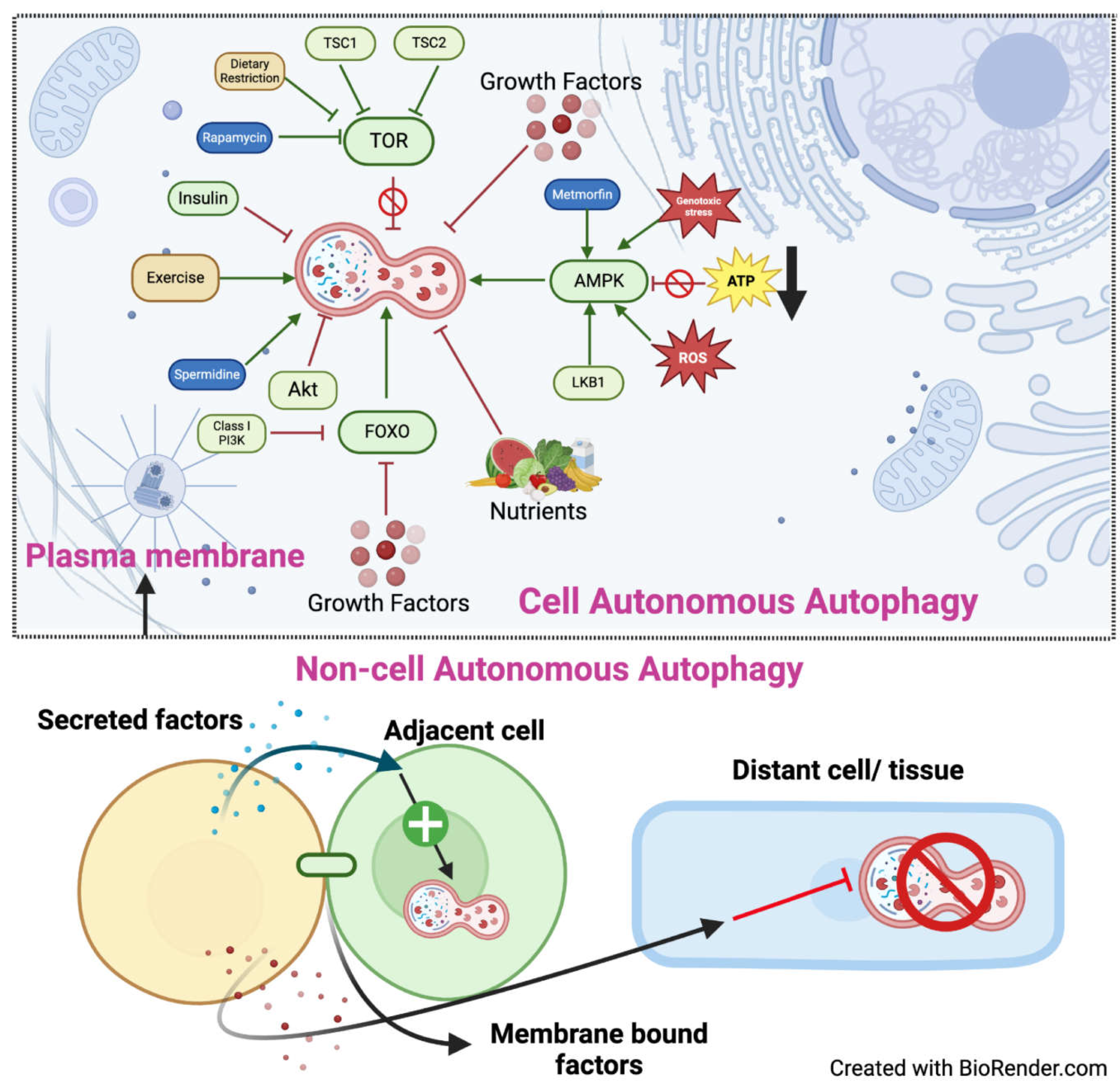
Figure 3.
Cell non-autonomous autophagy in aging. A. Increased levels of AMPK and Atg1 in D. melanogaster adult brain (grey) led to local upregulation of autophagy and reduced DILPs, thus delaying aging locally. Interestingly, this also results in enhanced autophagy cell non-autonomously in intestine and muscles (intestinal cell: yellow, muscle cell: blue) resulting in improved intestinal homeostasis and improved muscle function thus, delaying systemic aging. Further, AMPK overexpression in intestine resulted in autophagy upregulation in brain and muscles, thereby, delaying systemic aging and improved muscle function during aging. B. Aging results in accumulation of miR-83 which inhibits autophagy through CUP-5 and thus, progressing aging. When C. elegans is mutated for miR-83, autophagy is upregulated in BWM (green), which do not express miR-83, suggesting transport of miR-83 from intestine (yellow) to the BWM, thus regulating autophagy cell non-autonomously.
Figure 3.
Cell non-autonomous autophagy in aging. A. Increased levels of AMPK and Atg1 in D. melanogaster adult brain (grey) led to local upregulation of autophagy and reduced DILPs, thus delaying aging locally. Interestingly, this also results in enhanced autophagy cell non-autonomously in intestine and muscles (intestinal cell: yellow, muscle cell: blue) resulting in improved intestinal homeostasis and improved muscle function thus, delaying systemic aging. Further, AMPK overexpression in intestine resulted in autophagy upregulation in brain and muscles, thereby, delaying systemic aging and improved muscle function during aging. B. Aging results in accumulation of miR-83 which inhibits autophagy through CUP-5 and thus, progressing aging. When C. elegans is mutated for miR-83, autophagy is upregulated in BWM (green), which do not express miR-83, suggesting transport of miR-83 from intestine (yellow) to the BWM, thus regulating autophagy cell non-autonomously.
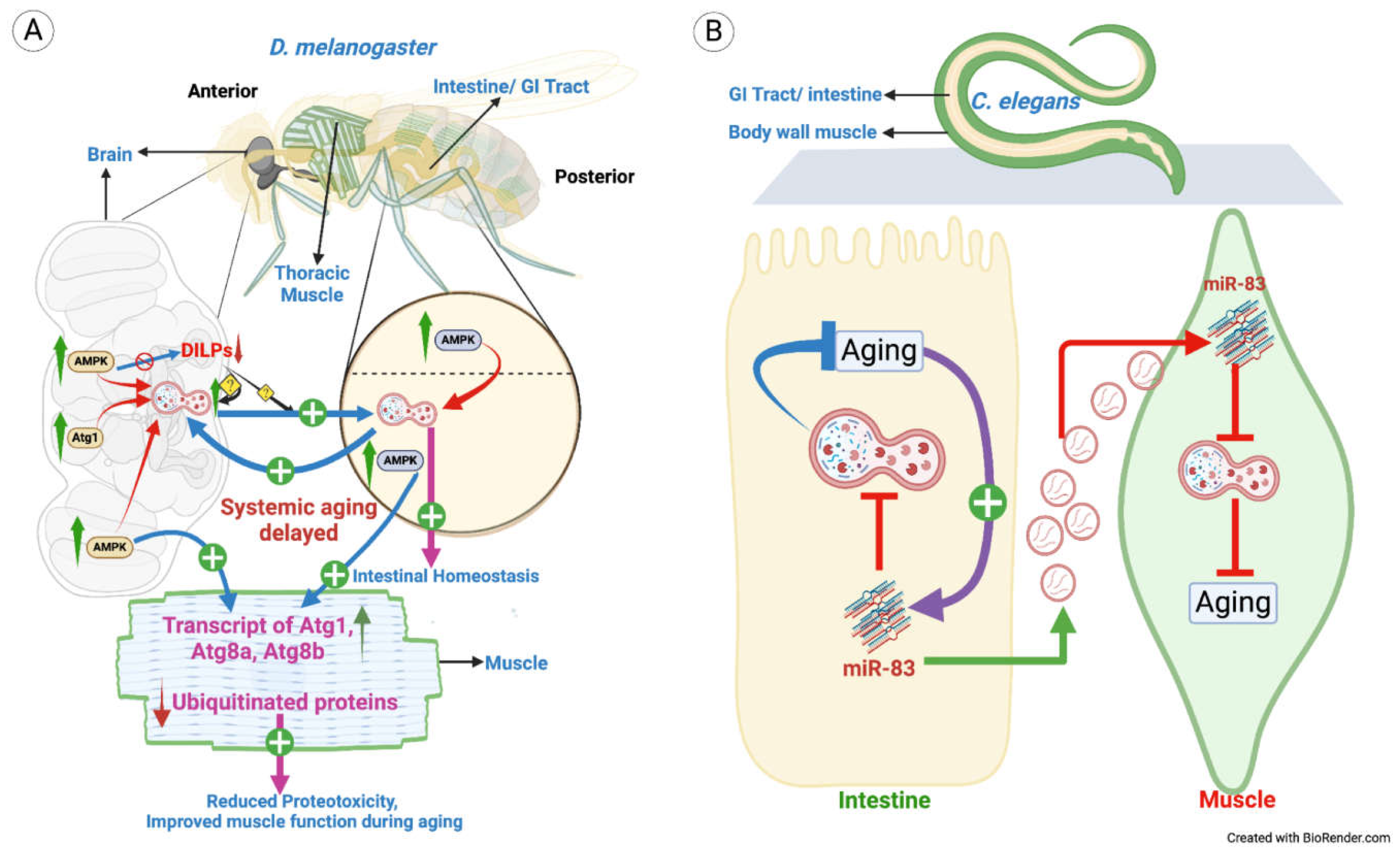
MicroRNAs (miRNAs) are identified as an important regulator of many biological phenomena, including aging and autophagy [83,84]. Interestingly, a few miRNAs have been shown to be secreted out of the source cell into the body fluids [85]. For instance, mir-83 in Caenorhabditis elegans, a homolog of mammalian mir-29, is differentially expressed in neurons and the intestines, and its expression is regulated in an age-dependent manner through heat shock factor-1, the master transcription factor of the heat shock response [86,87]. mir-83 impairs autophagy cell-autonomously by supressing cup-5 in the intestinal cells and it modulates autophagy in the body wall muscles (BWM) non-autonomously regulated also via Cup-5 (Figure 3B). mir-83-/- mutants showed increased autophagosomes and autolysosomes in the BWM, even though BWM does not express mir-83. mir-83 regulates autophagy in BWM non-autonomously through cup-5, as disruption of cup-5 in BWM via RNAi restored autophagy to normal levels [87]. Further, mir-83 transgene, when specifically expressed in the intestine in mir-83-/- mutant animals, resulted in the restoration of autophagy to wildtype levels, thus, suggesting the cell non-autonomous regulation of autophagy, i.e., inter-tissue/organ autophagic regulation [87]. These researchers suggested that transportation of mir-83 across tissue i.e., from intestine to BWM is possibly through extracellular vesicles (EVs).
- 2.
- Cell non-autonomous autophagy in systemic body response
Modulation of signaling molecules within one tissue can lead to systemic upregulation/downregulation of autophagy [4,87,88]. This has been elegantly demonstrated recently in D. melanogaster. D. melanogaster 3rd instar larvae exhibit reduced dTOR activity and a strong autophagy response in their fat body (the mammalian equivalent of the liver) in response to starvation [89]. The authors demonstrate that activated Dmp53 mediates the reduction of dTOR systemically in response to starvation. Dmp53 mediated systemic reduction of dTOR activity led to reduced dILP2 secretion (Drosophila Insulin-like peptide 2) and reduced 4E-BP and InR (Insulin Receptor) expression. In D. melanogaster, dILP2 is secreted from insulin-producing cells (IPCs) located in the brain, and its secretion is stimulated by Upd2 from the body [89]. The researchers demonstrated that Dmp53 affected dILP2 secretion from IPCs via reduced Upd2 production and secretion from the fat body. Finally, the researchers were able to demonstrate that AMPK mediated Dmp53 activation in the fat body in response to starvation. Thus, these data show that AMPK-dependent Dmp53 activation regulates systemic autophagy cell non-autonomously by modulating Dilp2 secretion from IPCs through Upd2 production and secretion from the fat body [89] (Figure 4).
- 3.
- Non-autonomously regulated autophagy in Development
During development, autophagy could be non-autonomously regulated. Tissue and organ morphogenesis in animals depends on the balance between cell survival and death. Developmental programmed cell death is mediated by Caspase-dependent apoptosis and context-specific autophagy-dependent cell death. Steroid hormones are critical regulators of physiology and developmental transitions in mammals and insects [90,91,92,93,94,95].
D. melanogaster larval to pupal transition is regulated by Ecdysone, and during this transition, larval tissues are removed, remodelled, and pave the way for the development of adult tissues [96,97,98,99]. The larval midgut is removed by programmed autophagy. Denton et al. 2019 demonstrated that Dpp (BMP2/4 homolog) signaling needs to be downregulated for midgut clearance by autophagy [100]. The data show that Dpp transcriptionally inhibits several Atgs, including Atg1 and Atg8a, as well as the Ecdysone receptor [100]. In contrast, Ecdysone upregulates several autophagy genes, suggesting a crosstalk between two opposing signaling pathways viz., Dpp signaling (preventing midgut degradation through downregulating Atg genes) and Ecdysone signaling (triggers midgut removal through upregulating Atg genes). Further studies showed that EcR primary response genes, BrC and E74, and several ecdysone biosynthetic genes (spook, phantom, disembodied, shadow, shade) were significantly reduced when the Dpp pathway was upregulated [100]. Additionally, Denton et al. found that a Dpp-expressing midgut prevents the production of ecdysone in the prothoracic glands (PG) [100]. Strong nuclear Mad localization was observed in the PG of animals expressing Dpp. This phenotype was not observed in TkvQ253D, suggesting a signal produced from the midguts, i.e., Dpp, signals to peripheral tissues, including the PG. Thus, Dpp regulates autophagy-dependent midgut removal via cell non-autonomous regulation of ecdysone production, which regulates autophagy cell non-autonomously [100] Figure 5A.
Non-autonomous activation of autophagy by membrane-bound proteins can be observed during D. melanogaster larval salivary gland clearance. Autophagy and Caspases are needed for the degradation of larval salivary glands, but how autophagy regulates the process is poorly understood [101,102,103]. Macroglobulin complement-related (Mcr) is a conserved protein expressed in salivary glands and shows elevated levels after 12 hours of steroid-triggered cell death during puparium formation [104]. Mcr binding to Draper, an immune receptor and a cell-autonomous autophagy regulator, triggers its phosphorylation by Src42, activating Atg1 [82,101,104,105]. When Mcr clones are generated, it blocks autophagy in the Mcr mutant cell, but Mcr+/+ cells neighboring Mcr-/- cells secrete Mcr in the tissue environment, which leads to activation of autophagy in the Mcr-/- [104] Figure 5B. Mcr also mediates an inflammatory response of macrophages, wherein, secreted Mcr from the wounded epithelial cells activates cell non-autonomous autophagy in macrophages migrating towards the wound [104]. This report has shown that Mcr is a cell non-autonomous autophagy regulator during programmed cell death (PCD) of larval salivary gland and during epithelial wound healing, but interestingly, nutrient deprivation-induced autophagy of fat body along with the autophagy of dying midgut is not influenced by Mcr, suggesting its localized function [104].
- 4.
- Cell non-autonomous autophagy in Neurodegeneration and protein aggregation pathologies
Amyotrophic lateral sclerosis (ALS) is a lethal neurodegenerative disease wherein motor neurons are lost. ALS is caused by many mutations, of which SOD1 and C9ORF72 are the most common [106]. It has been reported that piling-up of misfolded ubiquitinated protein aggregates occurs, making this the hallmark of most forms of ALS [107]. The researchers used motor neurons derived from iPSCs generated from an ALS patient (iPS31c8) and healthy control (iPSC1c1) cultured in Astrocyte Conditioned Medium (ACM) obtained from three ALS patients and three healthy controls, wherein they observed significant viability loss of motor neurons cultured in ALS derived ACM, hinting towards a secretory regulation of motor neurons by astrocytes. Interestingly, the phenotype is unique to motor neurons. Also, several studies show that altered autophagy contributes to ALS pathogenesis. C9ORF72, a mutation that is the most common cause of ALS, modulates the ULK1 activity, thereby regulating autophagy initiation [108]. HEK293T cells were cultured in ACM obtained from a patient suffering from ALS, and in ACM derived from healthy patient astrocytes, it was observed that LC3-II levels were significantly lower in ALS derived ACM maintained HEK293T cells, implying decreased autophagy. However, no alterations are seen in BEC-1, pULK1, ATG3, or ATG12 in the same cells. In addition to the LC3-II levels, p62 was significantly increased in ALS-derived ACM-maintained cells, implying decreased autophagy [109]. Thus, it can be inferred that astrocytes of ALS patients secrete factors that target motor neurons wherein these factors impair autophagy in the motor neurons, leading to the accumulation of ALS-related proteins like SOD1 and TDP-43 which otherwise would be degraded, thereby elevating ALS pathogenesis [109]. When autophagy was induced in the ALS models, it reduced the severity of the disease. p62 puncta in cells treated with control ACM and subjected to rapamycin exposure showed no change. However, ALS ACM-induced p62 puncta return to comparable levels to control ACM-induced cells [109]. Therefore, it can be inferred from the data provided that autophagy is regulated from outside of the target cell (motor neuron) by the neighboring astrocytes pointing towards non-autonomous regulation of autophagy in ALS pathogenesis (Figure 6A).
In protein-aggregate pathologies, both cell and cell non-autonomous mechanisms of neurodegeneration are observed, where the latter helps with the propagation of protein aggregates and/or pathologies throughout the nervous system [110,111]. In the transthyretin (TTR) systemic amyloidosis, the healthy liver secretes tetrameric TTR into the bloodstream, where TTR dissociates, misfolds, and aggregates, compromising organ systems such as the heart, autonomic nervous system, and peripheral nervous system [112,113,114]. In C. elegans, TTR toxicity using various mutants of the TTR protein was studied, viz., non-native TTR (NN TTR), WT-TTR, V30M-TTR, and D18G-TTR. In familial amyloid polyneuropathy (FAP) patients, the aggregation of V30M-TTR and its deposition around peripheral and autonomic neurons results in loss of nociception and thermal perception in the extremities [112]. In C. elegans, V30M-TTR expression in BWM led to damage to thermal sensory neurons and loss of thermal sensitivity. This effect could be rescued by RNAi treatment against V30M-TTR in BWM, further supporting cell non-autonomous neuronal proteotoxicity. The authors further provide evidence that TTR tetramers, when secreted into the body-cavity, are then taken up by coelomocytes where TTR is degraded within the lysosomes, i.e., cell non-autonomous degradation of tetramers via autophagosomal-dependent process in coelomocytes [115] (Figure 6B). Compromising coelomocytes or their lysosomal system leads to TTR accumulation. This study reveals that coelomocytes trigger a degradative mechanism, which is autophagosome-dependent or analogous. Hence, modulating autophagy flux could regulate TTR tetramer and oligomer levels and ameliorate NN-TTR-mediated neurotoxicity [115].
- 5.
- Cell non-autonomous autophagy in Cancer and supercompetition
Cancerous cells exhibit several metabolic perks, which gives them a growth advantage in stressful tumor niches [116,117]. Mitochondrial ATP production is depleted in cancerous cells; however, demand for TCA cycle-derived biosynthetic precursors and NADPH is unaltered or elevated [118,119]. Carbon sinking in the TCA cycle comes from glutamine (Gln) to compensate for such conditions. Many cancer cells exhibit higher rates of Gln transport and metabolism due to the high activity of mitochondrial glutaminase [118,119,120,121,122]. Gln is sequentially deaminated to Glutamate (Glu) and eventually to α-ketoglutarate (α-KG). A by-product of glutaminolysis is a potentially toxic compound named ammonia, which is excreted out of the cell by diffusion, by transport, or through incorporation into α-keto-acids [123]. In U2OS cells that are cultured in a medium without replenishment, which are hereafter called cell-conditioned medium (CCM), showed an increase in GFP-LC3 puncta and a decrease in the abundance of p62, both of which are the markers indicative of upregulated autophagy. When the CCM is added to the freshly plated U2OS cells, it induces autophagy [124,125]. Through a series of experiments, the authors deduced that CCM has some metabolites/molecules that diffuse within the cells to induce autophagy. This diffusible metabolite was further characterized and was found to be ammonia [124,125]. This ammonia was present in mM concentrations both in CCM and CCM-derived distillate and was capable of autophagy induction. Cells directly treated with NH4OH induced autophagy with similar kinetics, confirming that ammonia induces autophagy [124,125]. To validate these findings under in-vivo conditions, interstitial fluids were collected from human cancer cell line xenografts, and surprisingly, it was found that this fluid has comparable ammonia concentrations to CCM. Expectedly, Gln-free CCM generated in the presence or absence of α-KG lacked autophagy-stimulating activity. In this Gln-free medium, when Gln is added, even then autophagy induction fails to be restored, implying Gln’s does not induce autophagy but acts as a precursor for the generation of ammonia [124,125]. Interestingly, adding ammonia to Gln-deprived cells partially restored basal autophagy. The authors also demonstrated that ammonia induces autophagy independently of mTOR [124,125]. However, when cells were knocked down for Ulk1 and were subjected to CCM, even ammonia addition failed to induce autophagy, confirming that ammonia-induced autophagy is mTOR-independent but Ulk1-dependent [124,125]. Diffusion of ammonia from tumor cells that utilize glutamine in glutaminolysis provides an autocrine- and paracrine-acting signal that promotes cell-autonomous and non-autonomously regulated autophagy and, in turn, protects cells in different regions of the tumor from stress generated internally as well as environmental stress (Figure 7C).
An interesting study demonstrated that autophagy could be induced non-autonomously within the tumor microenvironment. D. melanogaster eye antennal disc (EAD) RasV12scrib-/- malignant tumor grow and invade the central nervous system and kill the host. Interestingly, as the malignancy progressed, the interaction with the tumor microenvironment triggered autophagy in neighboring non-tumor cells [126]. This phenotype was not observed in benign RasV12 tumors. Interestingly, preventing autophagy within the tumor did not prevent their invasion of the neighboring tissue. However, disruption of autophagy in the microenvironment drastically reduced tumor growth and invasion [126]. In addition to the local cell non-autonomous autophagy activation, systemic cell non-autonomous autophagy activation is observed for distal organs/ tissues like fat body, midgut, and muscles. The induction of non-autonomously regulated autophagy was abolished by overexpressing activated Yorkie, Unpaired 1, and Unpaired 3 [126]. (Figure 7D)
Zinc is a mandatory micronutrient and is involved in a variety of fundamental biological processes [127,128,129,130,131]. A study by Wei et al. has shown Zinc and its transporters have a crucial role in tumorigenesis [132]. The authors used RasV12 and Raf GOF Scrib-/- D. melanogaster as models to mimic the behaviours of various human cancers [133,134,135]. Blocking the Zinc transporter (dZnT7 RNAi) in RasV12 enhanced the growth of cancer, while in RafGOF Scrib-/- led to increased growth as well as invasion. Further, they found that JNK was upregulated, and it, in turn, regulated autophagy both autonomously and non-autonomously [132]. Cell-autonomous autophagy promotes tumor cell growth. Non-cell autonomous autophagy in the surrounding tissues helps the growth of tumors by providing them with vital nutrients [126,132,136]. Chloroquine treatment of RafGOF Scrib-/- reduced tumor growth and invasion, suggesting that inhibition of autophagy could be a therapeutic strategy for cancers [132]. dZnT7 knockdown promoter tumor overgrowth and migration through activation of JNK-mediated cell-autonomous and cell non-autonomous autophagy. (Figure 7D)
The Hippo pathway has been recently shown to be involved in inducing non-autonomously regulated autophagy in eye antennal disc cells in D. melanogaster. Fat is a core component of the Hippo pathway, and fat mutant cells outgrow their neighboring wild-type cells and develop tumors in adult eyes and heads [137]. These fat mutant clones are super competitors (winner), and they induce cell non-autonomous autophagy-mediated cell death in neighboring wild-type loser cells. The loser cells provide nourishment to the winners and aid in the development of the tumor [137,138]. Fat mutant clones activate the cell non-autonomous autophagy-mediated cell death in loser cells in an NF-κB-dependent Hid induction. Interestingly, cell non-autonomous autophagy induction depends on Yki-bantam-mediated TOR signaling activation in winner cells [137]. TOR signaling activation led to upregulation in protein synthesis in supercompetitors [137,138]. It is not clear how autophagy is induced in neighboring cells by fat mutant cells; it may be due to relative differences in protein synthesis levels between winners and losers. A similar phenomenon can be observed even in the competition between Hel25E mutant loser clones when surrounded by wild-type clones [138]. In this scenario, loser cells have compromised protein synthesis due to a mutation in the ribosomal protein Hel25E. Thus, it can be speculated that cells with compromised protein synthesis relative to their respective supercompetitors activate autophagy and eventually lead to cell death (Figure 7A-B). However, how autophagy is induced at the boundary of the winner and loser cells is not understood; but there is a possibility that loser cells respond to some secreted factor form winner cells, which in turn leads to autophagy induction in loser cells [126,137,138]. This data suggests that cell competition can also be autophagy-dependent and may drive tumor expansion during tumorigenesis. This supercompetition is not observed in RasV12scrib-/- malignant eye antennal disc tumor where cell non-autonomous autophagy is induced in wild-type surrounding cells but does not lead to their cell death; however, it is essential for tumor growth and invasion [126]. This difference in mode of action needs to be elucidated further (Figure 7A-D).
- 6.
- Cell non-autonomous autophagy in ECM-mediated regulation and Stem cell maintenance
Extracellular matrix (ECM) forms an intricate network and is essential for the transmission of external signals into the cellular interior. Amongst several ECM proteins heparan sulfate-proteoglycans are found both the cell surface and within the ECM and play indispensable roles in various developmental signaling pathways, including WNT/Wg, Hh, TGF-β, and FGF [139,140,141,142,143]. In Drosophila, Heparan sulfate proteoglycans (HSPGs) coordinate interactions between motor neurons and muscle cells via signaling molecules/pathways at the neuro-muscular junctions (NMJs) [144,145]. Perturbation of HS polymer synthesis or their modifications in muscle cells, led to changes in mitochondrial numbers, and expression of proteins associated with the Golgi and ER. Intracellular changes were also observed in the subsynaptic reticulum (SSR). [146]. The SSR is a highly ordered structure where cytoskeletal, neurotransmitter receptor, and signaling molecules are spatially localized and appears to be sensitive to level of HSPGs [147]. For instance, pancellular or Muscle-specific disruption of HSPG, mediated by ttvRNAi and sflRNAi showed ultrastructural changes at the NMJ of motor-neuron terminals, including expansion of spaces between membrane layers of the SSR. Further, TEM analysis showed presence of mitochondria with altered morphology and increase in number of double-membrane vesicles i.e., autophagosomes [146]. This increase in autophagy in ttvRNAi and sflRNAi was confirm by increased Ref(2)P/p62 accumulation, increased number of GFP-Atg8a puncta and Lysotracker Red-positive vesicles [146]. The authors speculated that HSPG may also be required for autophagy in other tissues/cell types. To test this fatbody specific knockdown of ttv and sfl was performed using RNAi and autophagy was assessed in these cells. The data obtained confirmed the observation from muscle cells i.e., increased in GFP-Atg8a puncta and increase in Lysotracker Red positive structures, indicating that loss of HS biosynthesis leads to autophagy induction. The researchers further addressed if HS biosynthesis is required cell-autonomously or cell non-autonomously in fatbody cells. The authors employed sophisticated inducible FLP-FRT based mitotic recombination system in the fat body to generate mutant cells with compromised HS biosynthesis (ttv-/-; ttv00681/ttv00681) alongside wild-type cells (ttv00681/+) from heterozygous parental cells. ttv00681/ttv00681 mutant cell clones in close proximity to cells with uncompromised HS biosynthesis (Wt cells), did not show increased Lysotracker-Red positive puncta, supporting the notion that HS biosynthesis, mainly mediated by ttv, operates in a non-cell-autonomous manner to regulate autophagy, potentially through modulation of signaling pathways (Figure 8A).
Cellular homeostasis is important to sustain stem cell pools in different tissues, and autophagy plays a critical role in maintaining homeostatic conditions within these cells [148,149]. Germline stem cells are specialized stem cells that produce only eggs or sperm and are crucial for the survival of the species. The authors used D. melanogaster testis as a model to study the role of autophagy in somatic and germline stem cell maintenance and aging. Basal autophagy was active in somatic cells of the testis, and there were fewer mCherryAtg8a puncta in germ cells than in early cyst cells (CCs) and hub cells [88]. When genes involved in autophagosome formation like Atg1, Atg5, and Atg8a were depleted in Cyst Stem Cells (CySCs) and early CCs, it resulted in depletion in CySCs, early CCs, and CCs. It also resulted in a reduction in the GSC number; however, when autophagy is disrupted within GSCs, it did not reduce the GSC number, suggesting that cell-autonomous autophagy is not required in GSCs under homeostatic conditions [88]. Reduced autophagy in CySCs leads to their loss, suggesting that autophagy in cells is autonomously required [88].
Further, the authors discovered that Atg1, Atg5, and Atg8a knockdown resembled EGFRDN mutant phenotype, suggesting a relation between EGFR signaling and autophagy [88,150,151]. Disrupting autophagy in CySCs did not lead to significant changes in dpErk intensity, indicating that autophagy does not act upstream of EGFR signaling [88,152] However, when EGFR is activated in early somatic cells, there is a relative increase in autolysosomes, suggesting EGFR stimulates basal autophagy in somatic cells of the testis. When EGFR signaling is blocked (EGFRDN or fos mutants) in early CCs, a significant decrease in mRNA levels of Atg6 and autolysosome numbers is observed [88]. Furthermore, Demarco et al., 2020 found that high InR/TOR signaling-induced autophagic suppression is required in the CySC daughter cells to promote differentiation [88]. Thus, a model can be drawn out from these genetic interactions that EGFR promotes autophagy in CySCs, which helps in maintaining CySCs and GSCs in opposition to InR/TOR signaling, which suppresses autophagy in early CCs and thereby promoting their differentiation [88] (Figure 8B). Thus, non-autonomously regulated activation of autophagy is crucial for stem cell maintenance in D. melanogaster testis.
A recent report on the non-autonomous regulation of autophagy is described in D. melanogaster male reproductive system. The D. melanogaster testis harbors GSCs, which give rise to sperm and are regulated by their niche cells called the hub cells and CySCs. BMPs (Dpp and Gbb) are produced by both hub cells and CySCs, and upon binding, BMP receptors in GSCs suppress bag-of-marbles (bam), which promotes the differentiation program [153,154,155]. Dpp and Gbb from hub cells and CySCs activate BMP signaling in GSCs and non-autonomously suppress autophagy, which can be detected only at low/basal levels in the GSCs, as seen by reduced Atg5 and Atg8a expression [156]. Autophagy is upregulated, as seen by increased Atg5 expression and increased mCherryAtg8a puncta formation in the differentiated cells where BMP signaling is inactive and Bam is expressed. Further, the authors showed that upregulation of the Dpp suppressed autophagy in GSCs, as seen by reduced lysotracker staining and reduced number of mCherryAtg8a punctate structures. The authors further demonstrated that Atg5 suppression is mediated by Mad-Med binding to the 5th intronic region of the Atg5. However, the suppression of Atg8a by BMPs is not at the transcriptional level. Thus, BMPs signaling suppress autophagy in male GSCs cell non-autonomously and via distinct mechanisms [156].
- 7.
- Cell non-autonomous autophagy in mitochondrial degradation
The mitochondrial quality control system governs processes including mitochondrial biogenesis, mitochondrial dynamics, and their degradation [157,158,159]. Mitophagy, the autophagic degradation of dysfunctional mitochondria, is a cell-autonomous process [157]. Davis et al., identified a phenomenon at the optic nerve head (ONH) of the retinal ganglion cells (RGCs) in wt mice where intact retinal ganglion cell axons shed membrane bound vesicles which were internalized and subsequently degraded in the astrocytes. Using 3D electron microscopic analyses the authors demonstrated that these evulsions in addition to microtubules also contain mitochondria. The authors further provide evidence that these evulsions containing mitochondria are internalized within astrocytes and degraded, as these vesicles colocalized with lysosomes of the astrocytes [160]. Furthermore, expressing mitochondrial specific dual fluorescent reporter MitoEGFPmCherry only in the RGCs the researchers detected a few mCherry-only expressing (acidified mitochondria) mitochondria in astrocytes supporting the fact that mitochondria of the RGCs are taken up in the astrocytes. In addition, by analyzing colocalization of MitoFISH and TUNEL staining the authors demonstrated that these mitochondria now detected in astrocyte cytoplasm are in early stages of degradation [160]. These observations support the notion that although axons represent a significant site for mitochondrial degradation in RGCs, not all axonal mitochondria are degraded autonomously within RGC axons as previously believed, but rather by adjacent astrocytes through what the authors term it as transcellular mitophagy or transmitophagy [160]. Further studies in transmitophagy pathway are essential to understand how this process is controlled at the molecular level.
Concluding Remarks:
The genetic regulation of autophagy was discovered in the early 1990s, and it is much better understood today. However, the understanding of autophagy is limited to the process occurring within the cell. Very few reports have touched upon the regulation of autophagy in non-autonomously regulated fashion. This review is an attempt to describe non-autonomously regulated autophagy in different cellular contexts and types. The cell-autonomous regulation of autophagy has become a prime area of research, and several genetic, biochemical, and cell biology-based screens have been conducted to identify the genes involved.
This review describes cell non-autonomous autophagy during development in healthy and diseased states. It is interesting to know that cell non-autonomous regulation can occur by different mechanisms and is mediated by different kinds of molecules, including microRNAs. Autophagy is a potential target in several diseases, including cancer and neurodegeneration. Hence, understanding cell-autonomous and cell non-autonomous regulation of autophagy is crucial in these diseases. The knowledge obtained by studying cell-autonomous and cell non-autonomous autophagy would aid in designing tailor-made treatment regimens with autophagy inhibitors and inducers. Targeting non-autonomously regulated autophagy would also require designing molecules/drugs that are unique as these are likely to target non-conventional targets such as secreted molecules, ECM components, and even microRNAs.
The study of non-autonomously regulated regulation of autophagy during development would be of enormous significance. How molecules in one region of the tissue/organ regulate autophagy in neighboring cells or distant tissue would fill gaps in our understanding of the morphogenetic process. These studies would be crucial for understanding autophagy regulation in niche-regulated stem cells/progenitor cells in tissues during development and in cancer cells during carcinogenesis. Thus, conducting high-throughput screens based on genetics, biochemistry and cell biology would be important to identify non-autonomously regulated regulation of autophagy in normal physiology, development and several diseases.

Table 1.
Examples of Cell non-autonomous autophagy:.
| Model organism | Source cell/tissue | Target cell/tissue | Molecule | Process affecting | References |
|---|---|---|---|---|---|
| D. melanogaster | Adult brain | Intestine, muscles | DILPs | Aging | [4] |
| C. elegans | Intestine | BWM | miR-83 | Aging | [87] |
| D. melanogaster | Fat body | Brain | Dmp53, Upd2 | Survival during stress | [89] |
| D. melanogaster | Midgut | Prothoracic gland | Dpp, 20E | PCD | [100] |
| D. melanogaster | Salivary gland Mcr+/+ | Salivary gland Mcr-/- | Mcr | PCD | [104] |
| HEK293T cells | ALS-ACM | HEK293T healthy cell | Secreted factors in ACM | Neuronal degradation | [109] |
| C. elegans | BWM | Coelomocyte | NA | Neuronal degradation | [115] |
| D. melanogaster | Hel25EWT cell | Hel25E-/- cell | Relative protein levels | Supercompetition | [138] |
| D. melanogaster | Fat-/- cell | Fat+/+ cell | Relative protein levels | Cancer | [137] |
| U2OS cells | Nutrient-replete cell | Nutrient-deplete cell | Ammonia | Cancer | [124,125] |
| D. melanogaster | Tumor niche | RafGOFScrib-/- | JNK | Cancer | [132] |
| D. melanogaster | Tumor niche | RasV12Scrib-/- | JNK, ROS | Cancer | [126] |
| D. melanogaster | ttv+/+ cell | ttv-/- cell | ECM component | ECM regulation | [146] |
| D. melanogaster | GSCs | CySCs | EGF | GSC maintenance | [88] |
| D. melanogaster | CySCs, Hub cells | GSCs | BMP ligands | GSC maintenance | [156] |
Author Contributions
KS wrote the entire manuscript, tabulated the information, and designed the figures. KS and BVS wrote the manuscript. BVS edited the manuscript.
Funding
This work was supported by grants from SERB-DST CRG/2020/0002716. KS is supported by UGC-JRF fellowship through CSIR-UGC-NET. KS is a registered Ph.D. student affiliated to Dept. of Biotechnology, Savitribai Phule Pune University (SPPU), Pune, India. BVS is affiliated to Savitribai Phule Pune University (SPPU), Pune, India, and is recognized by SPPU as a Ph.D. guide (Biotechnology and Zoology).
Acknowledgments
Thanks to members of the Shravage lab for helpful discussions, especially Kiran Nilangekar, for his comments on the manuscript. We would also like to thank Dr. P. K. Dhakephalkar, Director, Agharkar Research Institute, Pune, for intramural support and the entire ARI fraternity for support and access to facilities.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- S. Alers, A.S. Löffler, S. Wesselborg, B. Stork, Role of AMPK-mTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks, Mol Cell Biol 32 (2012) 2–11. [CrossRef]
- D.M. Gwinn, D.B. Shackelford, D.F. Egan, M.M. Mihaylova, A. Mery, D.S. Vasquez, B.E. Turk, R.J. Shaw, AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint, Mol Cell 30 (2008) 214–226. [CrossRef]
- J. Kim, M. Kundu, B. Viollet, K.L. Guan, AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1, Nat Cell Biol 13 (2011) 132–141. [CrossRef]
- M. Ulgherait, A. Rana, M. Rera, J. Graniel, D.W. Walker, AMPK modulates tissue and organismal aging in a non-cell-autonomous manner, Cell Rep 8 (2014) 1767–1780. [CrossRef]
- T. Noda, Y. Ohsumi, Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast, Journal of Biological Chemistry 273 (1998). [CrossRef]
- D. Meley, C. Bauvy, J.H.P.M. Houben-Weerts, P.F. Dubbelhuis, M.T.J. Helmond, P. Codogno, A.J. Meijer, AMP-activated protein kinase and the regulation of autophagic proteolysis, Journal of Biological Chemistry 281 (2006) 34870–34879. [CrossRef]
- N. Hosokawa, T. Hara, T. Kaizuka, C. Kishi, A. Takamura, Y. Miura, S.I. Iemura, T. Natsume, K. Takehana, N. Yamada, J.L. Guan, N. Oshiro, N. Mizushima, Nutrient-dependent mTORCl association with the ULK1-Atg13-FIP200 complex required for autophagy, Mol Biol Cell 20 (2009). [CrossRef]
- D. Mijaljica, M. Prescott, R.J. Devenish, Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum, Autophagy 7 (2011) 673–682. [CrossRef]
- S. Kaushik, A.M. Cuervo, Chaperone-mediated autophagy: A unique way to enter the lysosome world, Trends Cell Biol 22 (2012) 407–417. [CrossRef]
- N.C. Mulakkal, P. Nagy, S. Takats, R. Tusco, G. Juhász, I.P. Nezis, Autophagy in drosophila: From historical studies to current knowledge, Biomed Res Int 2014 (2014). [CrossRef]
- W. Cao, J. Li, K. Yang, D. Cao, An overview of autophagy: Mechanism, regulation and research progress, Bull Cancer 108 (2021) 304–322. [CrossRef]
- N. Fenouille, A.C. Nascimbeni, J. Botti-Millet, N. Dupont, E. Morel, P. Codogno, To be or not to be cell autonomous? Autophagy says both, Essays Biochem 61 (2017). [CrossRef]
- C. de Duve, R. Wattiaux, Functions of Lysosomes, Annu Rev Physiol 28 (1966) 435–492. [CrossRef]
- M. Tsukada, Y. Ohsumi, Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae, FEBS Lett 333 (1993) 169–174. [CrossRef]
- T.M. Harding, K.A. Morano, S. V. Scott, D.J. Klionsky, Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway, Journal of Cell Biology 131 (1995). [CrossRef]
- M. Thumm, R. Egner, B. Koch, M. Schlumpberger, M. Straub, M. Veenhuis, D.H. Wolf, Isolation of autophagocytosis mutants of Saccharomyces cerevisiae, FEBS Lett 349 (1994). [CrossRef]
- N. Mizushima, T. Yoshimori, Y. Ohsumi, The role of atg proteins in autophagosome formation, Annu Rev Cell Dev Biol 27 (2011). [CrossRef]
- J. Yan, H. Kuroyanagi, A. Kuroiwa, Y.I. Matsuda, H. Tokumitsu, T. Tomoda, T. Shirasawa, M.A. Muramatsu, Identification of mouse ULK1, a novel protein kinase structurally related to C. elegans UNC-51, Biochem Biophys Res Commun 246 (1998). [CrossRef]
- C.A. Mercer, A. Kaliappan, P.B. Dennis, A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy, Autophagy 5 (2009). [CrossRef]
- N. Hosokawa, T. Sasaki, S.I. Iemura, T. Natsume, T. Hara, N. Mizushima, Atg101, a novel mammalian autophagy protein interacting with Atg13, Autophagy 5 (2009). [CrossRef]
- E.Y.W. Chan, A. Longatti, N.C. McKnight, S.A. Tooze, Kinase-Inactivated ULK Proteins Inhibit Autophagy via Their Conserved C-Terminal Domains Using an Atg13-Independent Mechanism, Mol Cell Biol 29 (2009). [CrossRef]
- T. Hara, A. Takamura, C. Kishi, S.I. Iemura, T. Natsume, J.L. Guan, N. Mizushima, FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells, Journal of Cell Biology 181 (2008). [CrossRef]
- Y.C. Kim, K.L. Guan, MTOR: A pharmacologic target for autophagy regulation, Journal of Clinical Investigation 125 (2015) 25–32. [CrossRef]
- I.G. Ganley, D.H. Lam, J. Wang, X. Ding, S. Chen, X. Jiang, ULK1·ATG13·FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy, Journal of Biological Chemistry 284 (2009) 12297–12305. [CrossRef]
- R.C. Scott, O. Schuldiner, T.P. Neufeld, Role and regulation of starvation-induced autophagy in the Drosophila fat body, Dev Cell 7 (2004). [CrossRef]
- P.M. Wong, C. Puente, I.G. Ganley, X. Jiang, The ULK1 complex sensing nutrient signals for autophagy activation, Autophagy 9 (2013) 124–137. [CrossRef]
- F.C. Dorsey, K.L. Rose, S. Coenen, S.M. Prater, V. Cavett, J.L. Cleveland, J. Caldwell-Busby, Mapping the phosphorylation sites of Ulk1., J Proteome Res 8 (2009) 5253–63. [CrossRef]
- H.X. Yuan, R.C. Russell, K.L. Guan, Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy, Autophagy 9 (2013) 1983–1995. [CrossRef]
- J.M. Park, C.H. Jung, M. Seo, N.M. Otto, D. Grunwald, K.H. Kim, B. Moriarity, Y.M. Kim, C. Starker, R.S. Nho, D. Voytas, D.H. Kim, The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14, Autophagy 12 (2016) 547–564. [CrossRef]
- J.M. Park, M. Seo, C.H. Jung, D. Grunwald, M. Stone, N.M. Otto, E. Toso, Y. Ahn, M. Kyba, T.J. Griffin, L.A. Higgins, D.H. Kim, ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction, Autophagy 14 (2018) 584–597. [CrossRef]
- R.C. Russell, Y. Tian, H. Yuan, H.W. Park, Y.-Y. Chang, J. Kim, H. Kim, T.P. Neufeld, A. Dillin, K.-L. Guan, ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase, Nat Cell Biol 15 (2013) 741–750. [CrossRef]
- Z. Yue, S. Jin, C. Yang, A.J. Levine, N. Heintz, Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor, Proc Natl Acad Sci U S A 100 (2003). [CrossRef]
- E. Itakura, N. Mizushima, Atg14 and UVRAG: Mutually exclusive subunits of mammalian Beclin 1-PI3K complexes, Autophagy 5 (2009). [CrossRef]
- E. Itakura, C. Kishi, K. Inoue, N. Mizushima, Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG, Mol Biol Cell 19 (2008). [CrossRef]
- A. Petiot, E. Ogier-Denis, E.F.C. Blommaart, A.J. Meijer, P. Codogno, Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pa7thways that control macroautophagy in HT-29 cells, Journal of Biological Chemistry 275 (2000). [CrossRef]
- K. Lindmo, A. Brech, K.D. Finley, S. Gaumer, D. Contamine, T.E. Rusten, H. Stenmark, The PI 3-kinase regulator Vps15 is required for autophagic clearance of protein aggregates, Autophagy 4 (2008). [CrossRef]
- K. Devereaux, C. Dall’Armi, A. Alcazar-Roman, Y. Ogasawara, X. Zhou, F. Wang, A. Yamamoto, P. de Camilli, G. Di Paolo, Regulation of Mammalian Autophagy by Class II and III PI 3-Kinases through PI3P Synthesis, PLoS One 8 (2013). [CrossRef]
- H.C. Dooley, M. Razi, H.E.J. Polson, S.E. Girardin, M.I. Wilson, S.A. Tooze, WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12-5-16L1, Mol Cell 55 (2014). [CrossRef]
- T. Proikas-Cezanne, S. Waddell, A. Gaugel, T. Frickey, A. Lupas, A. Nordheim, WIPI-1α (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy, Oncogene 23 (2004). [CrossRef]
- Q. Lu, P. Yang, X. Huang, W. Hu, B. Guo, F. Wu, L. Lin, A.L. Kovács, L. Yu, H. Zhang, The WD40 Repeat PtdIns(3)P-Binding Protein EPG-6 Regulates Progression of Omegasomes to Autophagosomes, Dev Cell 21 (2011) 343–357. [CrossRef]
- Y. Ohashi, Activation mechanisms of the vps34 complexes, Cells 10 (2021). [CrossRef]
- S. Chowdhury, C. Otomo, A. Leitner, K. Ohashi, R. Aebersold, G.C. Lander, T. Otomo, Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex, Proc Natl Acad Sci U S A 115 (2018) E9792–E9801. [CrossRef]
- T. Osawa, Y. Ishii, N.N. Noda, Human ATG2B possesses a lipid transfer activity which is accelerated by negatively charged lipids and WIPI4, Genes to Cells 25 (2020) 65–70. [CrossRef]
- A.R.J. Young, E.Y.W. Chan, X.W. Hu, R. Köchl, S.G. Crawshaw, S. High, D.W. Halley, J. Lippincott-Schwartz, S.A. Tooze, Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes, J Cell Sci 119 (2006) 3888–3900. [CrossRef]
- H. Yamamoto, S. Kakuta, T.M. Watanabe, A. Kitamura, T. Sekito, C. Kondo-Kakuta, R. Ichikawa, M. Kinjo, Y. Ohsumi, Atg9 vesicles are an important membrane source during early steps of autophagosome formation, Journal of Cell Biology 198 (2012) 219–233. [CrossRef]
- R. Gómez-Sánchez, J. Rose, R. Guimarães, M. Mari, D. Papinski, E. Rieter, W.J. Geerts, R. Hardenberg, C. Kraft, C. Ungermann, F. Reggiori, Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores, Journal of Cell Biology 217 (2018) 2743–2763. [CrossRef]
- K. Matoba, T. Kotani, A. Tsutsumi, T. Tsuji, T. Mori, D. Noshiro, Y. Sugita, N. Nomura, S. Iwata, Y. Ohsumi, T. Fujimoto, H. Nakatogawa, M. Kikkawa, N.N. Noda, Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion, Nat Struct Mol Biol 27 (2020) 1185–1193. [CrossRef]
- S. Maeda, C. Otomo, T. Otomo, The autophagic membrane tether ATG2A transfers lipids between membranes, Elife 8 (2019). [CrossRef]
- Y. Ohsumi, N. Mizushima, Two ubiquitin-like conjugation systems essential for autophagy, Semin Cell Dev Biol 15 (2004). [CrossRef]
- N. Mizushima, T. Noda, T. Yoshimori, Y. Tanaka, T. Ishii, M.D. George, D.J. Klionsky, M. Ohsumi, Y. Ohsumi, A protein conjugation system essential for autophagy, Nature 395 (1998). [CrossRef]
- N. Mizushima, T. Noda, Y. Ohsumi, Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway, EMBO Journal 18 (1999). [CrossRef]
- N. Fujita, T. Itoh, H. Omori, M. Fukuda, T. Noda, T. Yoshimori, The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy, Mol Biol Cell 19 (2008). [CrossRef]
- E. Hirata, Y. Ohya, K. Suzuki, Atg4 plays an important role in efficient expansion of autophagic isolation membranes by cleaving lipidated Atg8 in Saccharomyces cerevisiae, PLoS One 12 (2017). [CrossRef]
- G. Juhász, B. Érdi, M. Sass, T.P. Neufeld, Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila, Genes Dev 21 (2007) 3061–3066. [CrossRef]
- Y. Ichimura, T. Kirisako, T. Takao, Y. Satomi, Y. Shimonishi, N. Ishihara, N. Mizushima, I. Tanida, E. Kominami, M. Ohsumi, T. Noda, Y. Ohsumi, A ubiquitin-like system mediates protein lipidation, Nature 408 (2000) 488–492. [CrossRef]
- M. Komatsu, S. Waguri, T. Chiba, S. Murata, J.I. Iwata, I. Tanida, T. Ueno, M. Koike, Y. Uchiyama, E. Kominami, K. Tanaka, Loss of autophagy in the central nervous system causes neurodegeneration in mice, Nature 441 (2006). [CrossRef]
- M. Komatsu, S. Waguri, T. Ueno, J. Iwata, S. Murata, I. Tanida, J. Ezaki, N. Mizushima, Y. Ohsumi, Y. Uchiyama, E. Kominami, K. Tanaka, T. Chiba, Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice, Journal of Cell Biology 169 (2005). [CrossRef]
- M. Li, Y. Hou, J. Wang, X. Chen, Z.M. Shao, X.M. Yin, Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates, Journal of Biological Chemistry 286 (2011). [CrossRef]
- Y. Yamada, N.N. Suzuki, Y. Fujioka, Y. Ohsumi, Y. Ichimura, F. Inagaki, Crystallization and preliminary X-ray analysis of Atg3, Acta Crystallogr Sect F Struct Biol Cryst Commun 62 (2006). [CrossRef]
- Y. Yamada, N.N. Suzuki, T. Hanada, Y. Ichimura, H. Kumeta, Y. Fujioka, Y. Ohsumi, F. Inagaki, The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation, Journal of Biological Chemistry 282 (2007). [CrossRef]
- T. Shintani, N. Mizushima, Y. Ogawa, A. Matsuura, T. Noda, Y. Ohsumi, Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast, EMBO Journal 18 (1999). [CrossRef]
- T. Nemoto, I. Tanida, E. Tanida-Miyake, N. Minematsu-Ikeguchi, M. Yokota, M. Ohsumi, T. Ueno, E. Kominami, The mouse APG10 homologue, an E2-like enzyme for Apg12p conjugation, facilitates MAP-LC3 modification, Journal of Biological Chemistry 278 (2003). [CrossRef]
- Y. Ichimura, T. Kumanomidou, Y.S. Sou, T. Mizushima, J. Ezaki, T. Ueno, E. Kominami, T. Yamane, K. Tanaka, M. Komatsu, Structural basis for sorting mechanism of p62 in selective autophagy, Journal of Biological Chemistry 283 (2008). [CrossRef]
- S. Jäger, C. Bucci, I. Tanida, T. Ueno, E. Kominami, P. Saftig, E.L. Eskelinen, Role for Rab7 in maturation of late autophagic vacuoles, J Cell Sci 117 (2004). [CrossRef]
- M.G. Gutierrez, D.B. Munafó, W. Berón, M.I. Colombo, Rab7 is required for the normal progression of the autophagic pathway in mammalian cells, J Cell Sci 117 (2004). [CrossRef]
- P. Jiang, T. Nishimura, Y. Sakamaki, E. Itakura, T. Hatta, T. Natsume, N. Mizushima, The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17, Mol Biol Cell 25 (2014). [CrossRef]
- U. Nair, A. Jotwani, J. Geng, N. Gammoh, D. Richerson, W.L. Yen, J. Griffith, S. Nag, K. Wang, T. Moss, M. Baba, J.A. McNew, X. Jiang, F. Reggiori, T.J. Melia, D.J. Klionsky, SNARE proteins are required for macroautophagy, Cell 146 (2011). [CrossRef]
- A. Kuma, M. Hatano, M. Matsui, A. Yamamoto, H. Nakaya, T. Yoshimori, Y. Ohsumi, T. Tokuhisa, N. Mizushima, The role of autophagy during the early neonatal starvation period, Nature 432 (2004) 1032–1036. [CrossRef]
- M. Rangel, J. Kong, V. Bhatt, K. Khayati, J.Y. Guo, Autophagy and tumorigenesis, FEBS Journal 289 (2022) 7177–7198. [CrossRef]
- D.R. Miller, A. Thorburn, Autophagy and organelle homeostasis in cancer, Dev Cell 56 (2021) 906–918. [CrossRef]
- N. Mizushima, A brief history of autophagy from cell biology to physiology and disease, Nat Cell Biol 20 (2018). [CrossRef]
- A.P.K. Wodrich, A.W. Scott, E. Giniger, What do we mean by “aging”? Questions and perspectives revealed by studies in Drosophila, Mech Ageing Dev 213 (2023). [CrossRef]
- C. López-Otín, M.A. Blasco, L. Partridge, M. Serrano, G. Kroemer, The hallmarks of aging, Cell 153 (2013). [CrossRef]
- H. Koga, S. Kaushik, A.M. Cuervo, Protein homeostasis and aging: The importance of exquisite quality control, Ageing Res Rev 10 (2011) 205–215. [CrossRef]
- M.C. Barbosa, R.A. Grosso, C.M. Fader, Hallmarks of aging: An autophagic perspective, Front Endocrinol (Lausanne) 10 (2019). [CrossRef]
- K.M. Berendzen, J. Durieux, L.W. Shao, Y. Tian, H. eui Kim, S. Wolff, Y. Liu, A. Dillin, Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis, Cell 166 (2016) 1553-1563.e10. [CrossRef]
- D. O’Brien, L.M. Jones, S. Good, J. Miles, M.S. Vijayabaskar, R. Aston, C.E. Smith, D.R. Westhead, P. van Oosten-Hawle, A PQM-1-Mediated Response Triggers Transcellular Chaperone Signaling and Regulates Organismal Proteostasis, Cell Rep 23 (2018) 3905–3919. [CrossRef]
- R.C. Taylor, A. Dillin, XXBP-1 Is a cell-nonautonomous regulator of stress resistance and longevity, Cell 153 (2013) 1435. [CrossRef]
- P. Van Oosten-Hawle, R.S. Porter, R.I. Morimoto, Regulation of organismal proteostasis by transcellular chaperone signaling, Cell 153 (2013) 1366. [CrossRef]
- J.T. Chang, C. Kumsta, A.B. Hellman, L.M. Adams, M. Hansen, Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging, Elife 6 (2017). [CrossRef]
- D.F. Egan, D.B. Shackelford, M.M. Mihaylova, S. Gelino, R.A. Kohnz, W. Mair, D.S. Vasquez, A. Joshi, D.M. Gwinn, R. Taylor, J.M. Asara, J. Fitzpatrick, A. Dillin, B. Viollet, M. Kundu, M. Hansen, R.J. Shaw, Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy, Science (1979) 331 (2011) 456–461. [CrossRef]
- R.C. Scott, G. Juhász, T.P. Neufeld, Direct Induction of Autophagy by Atg1 Inhibits Cell Growth and Induces Apoptotic Cell Death, Current Biology 17 (2007) 1–11. [CrossRef]
- L.R. Lapierre, C. Kumsta, M. Sandri, A. Ballabio, M. Hansen, Transcriptional and epigenetic regulation of autophagy in aging, Autophagy 11 (2015) 867–880. [CrossRef]
- S.V. Thalyana, F.J. Slack, MicroRNAs and their roles in aging, J Cell Sci 125 (2012) 7–17. [CrossRef]
- X. Chen, H. Liang, J. Zhang, K. Zen, C.Y. Zhang, Secreted microRNAs: A new form of intercellular communication, Trends Cell Biol 22 (2012) 125–132. [CrossRef]
- D. Gerlach, E. V. Kriventseva, N. Rahman, C.E. Vejnar, E.M. Zdobnov, miROrtho: Computational survey of microRNA genes, Nucleic Acids Res 37 (2009). [CrossRef]
- Y. Zhou, X. Wang, M. Song, Z. He, G. Cui, G. Peng, C. Dieterich, A. Antebi, N. Jing, Y. Shen, A secreted microRNA disrupts autophagy in distinct tissues of Caenorhabditis elegans upon ageing, Nat Commun 10 (2019). [CrossRef]
- R. Sênos Demarco, B.S. Uyemura, D.L. Jones, EGFR Signaling Stimulates Autophagy to Regulate Stem Cell Maintenance and Lipid Homeostasis in the Drosophila Testis, Cell Rep 30 (2020) 1101-1116.e5. [CrossRef]
- M.C. Ingaramo, J.A. Sánchez, N. Perrimon, A. Dekanty, Fat Body p53 Regulates Systemic Insulin Signaling and Autophagy under Nutrient Stress via Drosophila Upd2 Repression, Cell Rep 33 (2020). [CrossRef]
- Y. Fuchs, H. Steller, Live to die another way: Modes of programmed cell death and the signals emanating from dying cells, Nat Rev Mol Cell Biol 16 (2015) 329–344. [CrossRef]
- S. Arakawa, M. Tsujioka, T. Yoshida, H. Tajima-Sakurai, Y. Nishida, Y. Matsuoka, I. Yoshino, Y. Tsujimoto, S. Shimizu, Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice, Cell Death Differ 24 (2017) 1598–1608. [CrossRef]
- D. Denton, T. Xu, S. Kumar, Autophagy as a pro-death pathway, Immunol Cell Biol 93 (2015) 35–42. [CrossRef]
- L. Galluzzi, I. Vitale, J.M. Abrams, E.S. Alnemri, E.H. Baehrecke, M. V. Blagosklonny, T.M. Dawson, V.L. Dawson, W.S. El-Deiry, S. Fulda, E. Gottlieb, D.R. Green, M.O. Hengartner, O. Kepp, R.A. Knight, S. Kumar, S.A. Lipton, X. Lu, F. Madeo, W. Malorni, P. Mehlen, G. Nũez, M.E. Peter, M. Piacentini, D.C. Rubinsztein, Y. Shi, H.U. Simon, P. Vandenabeele, E. White, J. Yuan, B. Zhivotovsky, G. Melino, G. Kroemer, Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012, Cell Death Differ 19 (2012) 107–120. [CrossRef]
- J. Yuan, G. Kroemer, Alternative cell death mechanisms in development and beyond, Genes Dev 24 (2010) 2592–2602. [CrossRef]
- K. King-Jones, C.S. Thummel, Nuclear receptors - A perspective from Drosophila, Nat Rev Genet 6 (2005) 311–323. [CrossRef]
- D. Denton, M.T. Aung-Htut, S. Kumar, Developmentally programmed cell death in Drosophila, Biochim Biophys Acta Mol Cell Res 1833 (2013) 3499–3506. [CrossRef]
- S. Kumar, D. Cakouros, Transcriptional control of the core cell-death machinery, Trends Biochem Sci 29 (2004) 193–199. [CrossRef]
- S. Nicolson, D. Denton, S. Kumar, Ecdysone-mediated programmed cell death in Drosophila, International Journal of Developmental Biology 59 (2015) 23–32. [CrossRef]
- E.H. Baechrecke, Steroid regulation of programmed cell death during Drosophila development, Cell Death Differ 7 (2000). [CrossRef]
- D. Denton, T. Xu, S. Dayan, S. Nicolson, S. Kumar, Dpp regulates autophagy-dependent midgut removal and signals to block ecdysone production, Cell Death Differ 26 (2019) 763–778. [CrossRef]
- D.L. Berry, E.H. Baehrecke, Growth Arrest and Autophagy Are Required for Salivary Gland Cell Degradation in Drosophila, Cell 131 (2007) 1137–1148. [CrossRef]
- P.E. Joubert, G. Meiffren, I.P. Grégoire, G. Pontini, C. Richetta, M. Flacher, O. Azocar, P.O. Vidalain, M. Vidal, V. Lotteau, P. Codogno, C. Rabourdin-Combe, M. Faure, Autophagy Induction by the Pathogen Receptor CD46, Cell Host Microbe 6 (2009) 354–366. [CrossRef]
- M.A. Sanjuan, C.P. Dillon, S.W.G. Tait, S. Moshiach, F. Dorsey, S. Connell, M. Komatsu, K. Tanaka, J.L. Cleveland, S. Withoff, D.R. Green, Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis, Nature 450 (2007) 1253–1257. [CrossRef]
- L. Lin, F.S.L.M. Rodrigues, C. Kary, A. Contet, M. Logan, R.H.G. Baxter, W. Wood, E.H. Baehrecke, Complement-Related Regulates Autophagy in Neighboring Cells, Cell 170 (2017) 158-171.e8. [CrossRef]
- T.K. Chang, B. V. Shravage, S.D. Hayes, C.M. Powers, R.T. Simin, J. Wade Harper, E.H. Baehrecke, Uba1 functions in Atg7- and Atg3-independent autophagy, Nat Cell Biol 15 (2013) 1067–1078. [CrossRef]
- A.M. Blokhuis, E.J.N. Groen, M. Koppers, L.H. Van Den Berg, R.J. Pasterkamp, Protein aggregation in amyotrophic lateral sclerosis, Acta Neuropathol 125 (2013) 777–794. [CrossRef]
- A. Bodansky, J.M.H. Kim, L. Tempest, A. Velagapudi, R. Libby, J. Ravits, TDP-43 and ubiquitinated cytoplasmic aggregates in sporadic ALS are low frequency and widely distributed in the lower motor neuron columns independent of disease spread, Amyotrophic Lateral Sclerosis 11 (2010) 321–327. [CrossRef]
- M. Yang, L. Chen, K. Swaminathan, S. Herrlinger, F. Lai, R. Shiekhattar, J.F. Chen, A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy, Sci Adv 2 (2016). [CrossRef]
- M. Madill, K. McDonagh, J. Ma, A. Vajda, P. McLoughlin, T. O’Brien, O. Hardiman, S. Shen, Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms, Mol Brain 10 (2017). [CrossRef]
- M. Goedert, B. Falcon, F. Clavaguera, M. Tolnay, Prion-like mechanisms in the pathogenesis of tauopathies and synucleinopathies, Curr Neurol Neurosci Rep 14 (2014) 1–11. [CrossRef]
- C.I. Nussbaum-Krammer, R.I. Morimoto, Caenorhabditis elegans as a model system for studying non-cellautonomous mechanisms in protein-misfolding diseases, DMM Disease Models and Mechanisms 7 (2014) 31–39. [CrossRef]
- T. Coelho, M.S. Maurer, O.B. Suhr, THAOS-The Transthyretin Amyloidosis Outcomes Survey: Initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis, Curr Med Res Opin 29 (2013) 63–76. [CrossRef]
- S.M. Johnson, S. Connelly, C. Fearns, E.T. Powers, J.W. Kelly, The transthyretin amyloidoses: From delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug, J Mol Biol 421 (2012) 185–203. [CrossRef]
- J.D. Schonhoft, C. Monteiro, L. Plate, Y.S. Eisele, J.M. Kelly, D. Boland, C.G. Parker, B.F. Cravatt, S. Teruya, S. Helmke, M. Maurer, J. Berk, Y. Sekijima, M. Novais, T. Coelho, E.T. Powers, J.W. Kelly, Peptide probes detect misfolded transthyretin oligomers in plasma of hereditary amyloidosis patients, Sci Transl Med 9 (2017). [CrossRef]
- K. Madhivanan, E.R. Greiner, M. Alves-Ferreira, D. Soriano-Castell, N. Rouzbeh, C.A. Aguirre, J.F. Paulsson, J. Chapman, X. Jiang, F.K. Ooi, C. Lemos, A. Dillin, V. Prahlad, J.W. Kelly, S.E. Encalada, Cellular clearance of circulating transthyretin decreases cell-nonautonomous proteotoxicity in Caenorhabditis elegans, Proc Natl Acad Sci U S A 115 (2018) E7710–E7719. [CrossRef]
- M.G.V. Heiden, L.C. Cantley, C.B. Thompson, Understanding the warburg effect: The metabolic requirements of cell proliferation, Science (1979) 324 (2009) 1029–1033. [CrossRef]
- Warburg 1956, (n.d.).
- R.J. DeBerardinis, J.J. Lum, G. Hatzivassiliou, C.B. Thompson, The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation, Cell Metab 7 (2008) 11–20. [CrossRef]
- R.J. DeBerardinis, A. Mancuso, E. Daikhin, I. Nissim, M. Yudkoff, S. Wehrli, C.B. Thompson, Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis, Proc Natl Acad Sci U S A 104 (2007). [CrossRef]
- P. Gao, I. Tchernyshyov, T.C. Chang, Y.S. Lee, K. Kita, T. Ochi, K.I. Zeller, A.M. De Marzo, J.E. Van Eyk, J.T. Mendell, C. V. Dang, C-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism, Nature 458 (2009) 762–765. [CrossRef]
- M. Szeliga, M. Obara-Michlewska, Glutamine in neoplastic cells: Focus on the expression and roles of glutaminases, Neurochem Int 55 (2009) 71–75. [CrossRef]
- B.P. Bode, B.C. Fuchs, B.P. Hurley, J.L. Conroy, J.E. Suetterlin, K.K. Tanabe, D.B. Rhoads, S.F. Abcouwer, W.W. Souba, Molecular and functional analysis of glutamine uptake in human hepatoma and liver-derived cells, Am J Physiol Gastrointest Liver Physiol 283 (2002). [CrossRef]
- R.J. Deberardinis, T. Cheng, Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer, Oncogene 29 (2010) 313–324. [CrossRef]
- C.H. Eng, K. Yu, J. Lucas, E. White, R.T. Abraham, Ammonia derived from glutaminolysis is a diffusible regulator of autophagy, Sci Signal 3 (2010). [CrossRef]
- C.H. Eng, R.T. Abraham, Glutaminolysis yields a metabolic by-product that stimulates autophagy, Autophagy 6 (2010). [CrossRef]
- N.S. Katheder, R. Khezri, F. O’Farrell, S.W. Schultz, A. Jain, M.K.O. Schink, T.A. Theodossiou, T. Johansen, G. Juhász, D. Bilder, A. Brech, H. Stenmark, T.E. Rusten, Microenvironmental autophagy promotes tumour growth, Nature 541 (2017) 417–420. [CrossRef]
- M. Hershfinkel, W.F. Silverman, I. Sekler, The zinc sensing receptor, a link between zinc and cell signaling, in: Molecular Medicine, 2007: pp. 331–336. [CrossRef]
- R.S. Macdonald, Zinc and Health: Current Status and Future Directions The Role of Zinc in Growth and Cell Proliferation 1,2, 2000. https://academic.oup.com/jn/article-abstract/130/5/1500S/4686427.
- M. Yan, Y. Song, C.P. Wong, K. Hardin, E. Ho, Zinc Deficiency Alters DNA Damage Response Genes in Normal Human Prostate Epithelial Cells 1,2,3, 2008.
- T. Kambe, T. Tsuji, A. Hashimoto, N. Itsumura, The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism, Physiol Rev 95 (2015). [CrossRef]
- K.A. Mccall, C.-C. Huang, C.A. Fierke, Zinc and Health: Current Status and Future Directions Function and Mechanism of Zinc Metalloenzymes 1, J. Nutr 130 (2000).
- T. Wei, X. Ji, Y. Gao, X. Zhu, G. Xiao, ZnT7 RNAi favors RafGOFscrib−/−-induced tumor growth and invasion in Drosophila through JNK signaling pathway, Oncogene 40 (2021) 2217–2229. [CrossRef]
- K. Doggett, F.A. Grusche, H.E. Richardson, A.M. Brumby, Loss of the Drosophila cell polarity regulator Scribbled promotes epithelial tissue overgrowth and cooperation with oncogenic Ras-Raf through impaired Hippo pathway signaling, BMC Dev Biol 11 (2011). [CrossRef]
- J.A. McCubrey, L.S. Steelman, W.H. Chappell, S.L. Abrams, E.W.T. Wong, F. Chang, B. Lehmann, D.M. Terrian, M. Milella, A. Tafuri, F. Stivala, M. Libra, J. Basecke, C. Evangelisti, A.M. Martelli, R.A. Franklin, Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance, Biochim Biophys Acta Mol Cell Res 1773 (2007) 1263–1284. [CrossRef]
- M. Uhlirova, H. Jasper, D. Bohmann, Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model, Proc Natl Acad Sci U S A 102 (2005). [CrossRef]
- L. Poillet-Perez, E. White, Role of tumor and host autophagy in cancer metabolism, Genes Dev 33 (2019). [CrossRef]
- R. Nagata, N. Akai, S. Kondo, K. Saito, S. Ohsawa, T. Igaki, Yorkie drives supercompetition by non-autonomous induction of autophagy via bantam microRNA in Drosophila, Current Biology 32 (2022) 1064-1076.e4. [CrossRef]
- R. Nagata, M. Nakamura, Y. Sanaki, T. Igaki, Cell Competition Is Driven by Autophagy, Dev Cell 51 (2019) 99-112.e4. [CrossRef]
- J.R. Bishop, M. Schuksz, J.D. Esko, Heparan sulphate proteoglycans fine-tune mammalian physiology, Nature 446 (2007) 1030–1037. [CrossRef]
- U. Häcker, K. Nybakken, N. Perrimon, Heparan sulphate proteoglycans: The sweet side of development, Nat Rev Mol Cell Biol 6 (2005) 530–541. [CrossRef]
- C.A. Kirkpatrick, S.B. Selleck, Heparan sulfate proteoglycans at a glance, J Cell Sci 120 (2007) 1829–1832. [CrossRef]
- X. Lin, Functions of heparan sulfate proteoglycans in cell signaling during development, Development 131 (2004) 6009–6021. [CrossRef]
- A.D. Lander, S.B. Selleck, The elusive functions of proteoglycans: In vivo veritas, Journal of Cell Biology 148 (2000). [CrossRef]
- K.G. Johnson, A.P. Tenney, A. Ghose, A.M. Duckworth, M.E. Higashi, K. Parfitt, O. Marcu, T.R. Heslip, J.L. Marsh, T.L. Schwarz, J.G. Flanagan, D. Van Vactor, The HSPGs Syndecan and dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development, Neuron 49 (2006) 517–531. [CrossRef]
- E. Stryker, K.G. Johnson, LAR, liprin α and the regulation of active zone morphogenesis, J Cell Sci 120 (2007) 3723–3728. [CrossRef]
- C.E. Reynolds-Peterson, N. Zhao, J. Xu, T.M. Serman, J. Xu, S.B. Selleck, Heparan sulfate proteoglycans regulate autophagy in Drosophila, Autophagy 13 (2017) 1262–1279. [CrossRef]
- Y. Ren, C.A. Kirkpatrick, J.M. Rawson, M. Sun, S.B. Selleck, Cell type-specific requirements for heparan sulfate biosynthesis at the Drosophila neuromuscular junction: Effects on synapse function, membrane trafficking, and mitochondrial localization, Journal of Neuroscience 29 (2009) 8539–8550. [CrossRef]
- D. Drummond-Barbosa, Stem cells, their niches and the systemic environment: An aging network, Genetics 180 (2008) 1787–1797. [CrossRef]
- B. Levine, D.J. Klionsky, Development by self-digestion: Molecular mechanisms and biological functions of autophagy, Dev Cell 6 (2004). [CrossRef]
- A.G. Hudson, B.B. Parrott, Y. Qian, C. Schulz, A Temporal Signature of Epidermal Growth Factor Signaling Regulates the Differentiation of Germline Cells in Testes of Drosophila melanogaster, PLoS One 8 (2013). [CrossRef]
- L.A. Perkins, M.R. Johnson, M.B. Melnick, N. Perrimon, The nonreceptor protein tyrosine phosphatase Corkscrew functions in multiple receptor tyrosine kinase pathways in Drosophila, Dev Biol 180 (1996). [CrossRef]
- Y. Yung, Y. Dolginov, Z. Yao, H. Rubinfeld, D. Michael, T. Hanoch, E. Roubini, Z. Lando, D. Zharhary, R. Seger, Detection of ERK activation by a novel monoclonal antibody, FEBS Lett 408 (1997). [CrossRef]
- M. de Cuevas, E.L. Matunis, The stem cell niche: Lessons from the Drosophila testis, Development 138 (2011) 2861–2869. [CrossRef]
- A.A. Shivdasani, P.W. Ingham, Regulation of Stem Cell Maintenance and Transit Amplifying Cell Proliferation by TGF-β Signaling in Drosophila Spermatogenesis, Current Biology 13 (2003) 2065–2072. [CrossRef]
- E. Kawase, M.D. Wong, B.C. Ding, T. Xie, Gbb/Bmp signaling is essential for maintaining germline stem cells and for repressing bam transcription in the Drosophila testis, Development 131 (2004) 1365–1375. [CrossRef]
- V.B. Varga, D. Schuller, F. Szikszai, J. Szinyákovics, G. Puska, T. Vellai, T. Kovács, Autophagy is required for spermatogonial differentiation in the Drosophila testis, Biol Futur 73 (2022) 187–204. [CrossRef]
- G. Ashrafi, T.L. Schwarz, The pathways of mitophagy for quality control and clearance of mitochondria, Cell Death Differ 20 (2013) 31–42. [CrossRef]
- R.C. Scarpulla, R.B. Vega, D.P. Kelly, Transcriptional integration of mitochondrial biogenesis, Trends in Endocrinology and Metabolism 23 (2012) 459–466. [CrossRef]
- R.J. Youle, A.M. Van Der Bliek, Mitochondrial fission, fusion, and stress, Science (1979) 337 (2012) 1062–1065. [CrossRef]
- C.H.O. Davis, K.Y. Kim, E.A. Bushong, E.A. Mills, D. Boassa, T. Shih, M. Kinebuchi, S. Phan, Y. Zhou, N.A. Bihlmeyer, J. V. Nguyen, Y. Jin, M.H. Ellisman, N. Marsh-Armstrong, Transcellular degradation of axonal mitochondria, Proc Natl Acad Sci U S A 111 (2014) 9633–9638. [CrossRef]
Figure 4.
Cell non-autonomous autophagy in systemic response to metabolic stress. Nutrient stress results in activation of AMPK (a positive regulator of autophagy) in the fat body (shown in yellow), results in upregulation of Dmp53 which inhibits Upd2. In absence of Upd2, DILP2 levels decreased in brain (dark yellow) thus, upregulating autophagy throughout the body, resulting in adaptation to stress followed by prolonged survival.
Figure 4.
Cell non-autonomous autophagy in systemic response to metabolic stress. Nutrient stress results in activation of AMPK (a positive regulator of autophagy) in the fat body (shown in yellow), results in upregulation of Dmp53 which inhibits Upd2. In absence of Upd2, DILP2 levels decreased in brain (dark yellow) thus, upregulating autophagy throughout the body, resulting in adaptation to stress followed by prolonged survival.
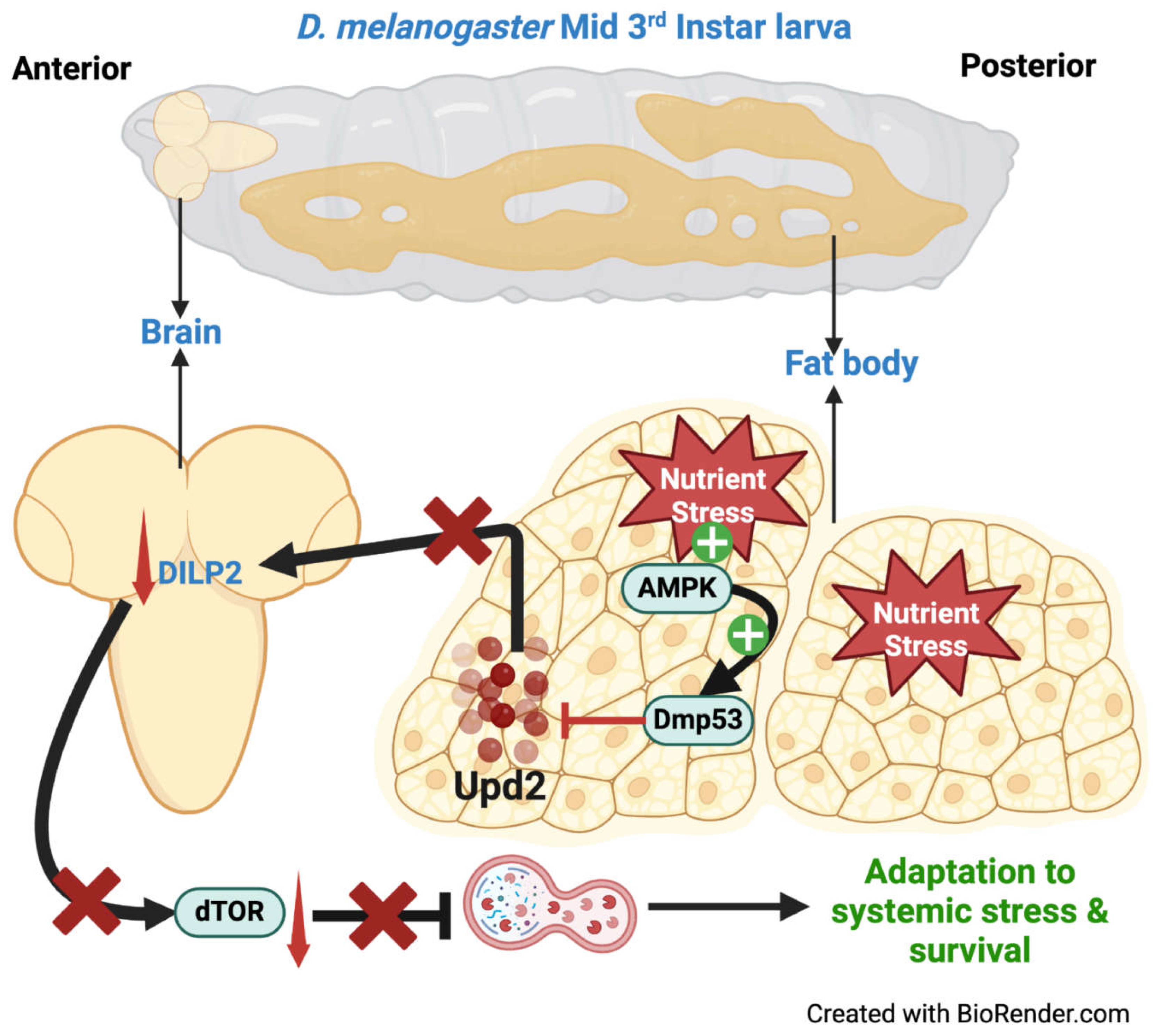
Figure 5.
Cell non-autonomous autophagy in developmental programmed cell death. A. During the D. melanogaster larval midgut degradation (LL3) Dpp prevents the precocious activation of midgut removal by suppressing autophagy. 20-hydroxyecdysone, a hormone which is responsible for activating autophagy-dependent cell death of the midgut is released from the prothoracic gland, is negatively regulated by Dpp signaling initiated from the midgut. Thus, Dpp cell non-autonomously acts on ecdysone production to suppress autophagy in the midgut. B. D. melanogaster larval salivary gland degradation is an autophagy-dependent process. Mcr binds to Draper which through Src42A triggers autophagy which is followed by PCD. When salivary gland is subjected to mosaic analysis, and the Mcr-/- cell (yellow) is in contact with Mcr+/+ (wildtype) (green), autophagy is restored in the Mcr-/- cell as well. This clearly shows that, Mcr cell non-autonomously regulates the autophagy.
Figure 5.
Cell non-autonomous autophagy in developmental programmed cell death. A. During the D. melanogaster larval midgut degradation (LL3) Dpp prevents the precocious activation of midgut removal by suppressing autophagy. 20-hydroxyecdysone, a hormone which is responsible for activating autophagy-dependent cell death of the midgut is released from the prothoracic gland, is negatively regulated by Dpp signaling initiated from the midgut. Thus, Dpp cell non-autonomously acts on ecdysone production to suppress autophagy in the midgut. B. D. melanogaster larval salivary gland degradation is an autophagy-dependent process. Mcr binds to Draper which through Src42A triggers autophagy which is followed by PCD. When salivary gland is subjected to mosaic analysis, and the Mcr-/- cell (yellow) is in contact with Mcr+/+ (wildtype) (green), autophagy is restored in the Mcr-/- cell as well. This clearly shows that, Mcr cell non-autonomously regulates the autophagy.
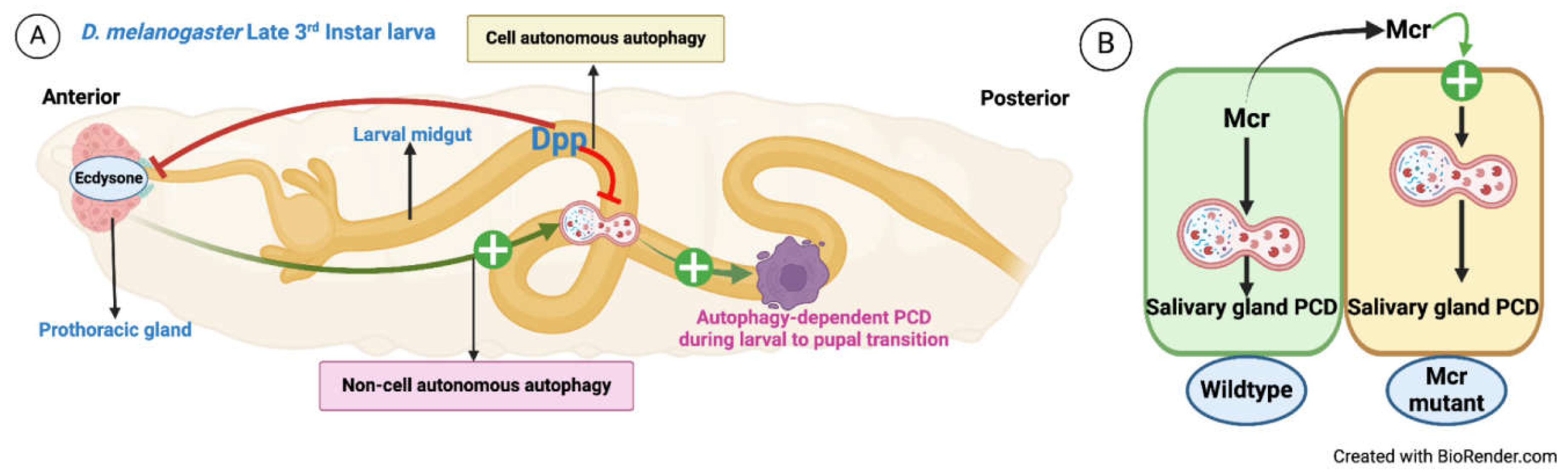
Figure 6.
Cell non-autonomous autophagy in neurodegeneration and protein aggregation pathologies. A. Astrocytes from healthy patients grown on a Petri dish. These astrocytes secrete factors into the growth medium termed as healthy astrocyte-conditioned medium (ACM). HEK293T cells grown in an astrocyte-conditioned medium have normal levels of basal autophagy. Astrocytes isolated from patients suffering from Amyotrophic lateral sclerosis (ALS) grown on a Petri dish. These astrocytes secrete factors into the growth medium termed an ALS astrocyte-conditioned medium (ACM). HEK293T cells grown in an ALS astrocyte-conditioned medium have reduced levels of basal autophagy leading to accumulation of p62 and reduced formation of autophagosomes as judged by LC3-II levels. B. In C. elegans when transthyretin (TTR) is released from body wall muscle (BWM) into the body cavity and forms TTR tetramers, it gets carried towards the neuron leading to neuronal degradation if not degraded. When these TTR tetramers being taken-up by coelomocyte (autophagosomal degradation) and degraded, neuronal degradation is delayed. Thus, coelomocyte degrades TTR tetramers cell non-autonomously by coelomocyte.
Figure 6.
Cell non-autonomous autophagy in neurodegeneration and protein aggregation pathologies. A. Astrocytes from healthy patients grown on a Petri dish. These astrocytes secrete factors into the growth medium termed as healthy astrocyte-conditioned medium (ACM). HEK293T cells grown in an astrocyte-conditioned medium have normal levels of basal autophagy. Astrocytes isolated from patients suffering from Amyotrophic lateral sclerosis (ALS) grown on a Petri dish. These astrocytes secrete factors into the growth medium termed an ALS astrocyte-conditioned medium (ACM). HEK293T cells grown in an ALS astrocyte-conditioned medium have reduced levels of basal autophagy leading to accumulation of p62 and reduced formation of autophagosomes as judged by LC3-II levels. B. In C. elegans when transthyretin (TTR) is released from body wall muscle (BWM) into the body cavity and forms TTR tetramers, it gets carried towards the neuron leading to neuronal degradation if not degraded. When these TTR tetramers being taken-up by coelomocyte (autophagosomal degradation) and degraded, neuronal degradation is delayed. Thus, coelomocyte degrades TTR tetramers cell non-autonomously by coelomocyte.
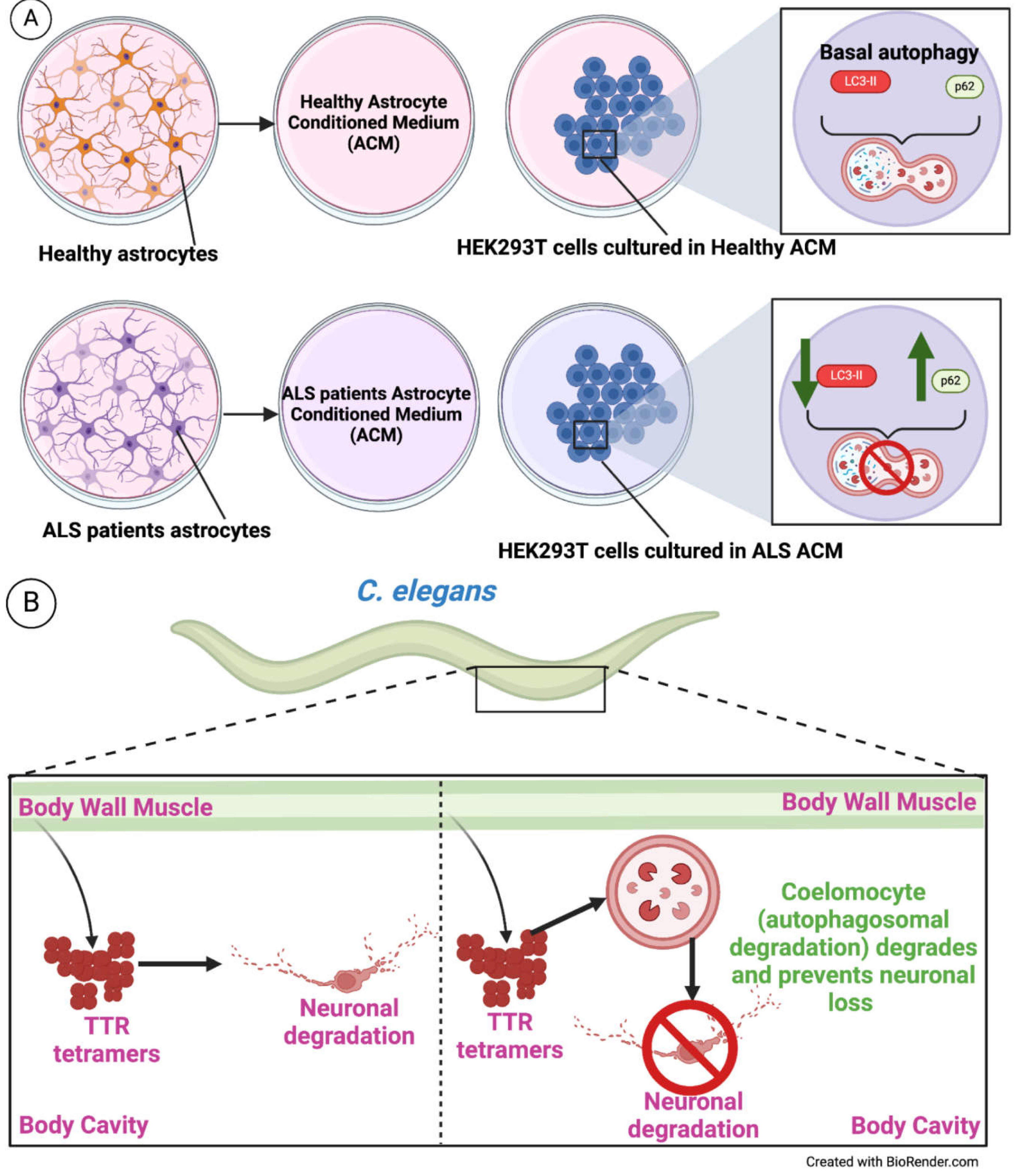
Figure 7.
Cell non-autonomous autophagy in supercompetition and cancer. A. Hel25E-/- mutant (orange) cell has reduced protein synthesis and thus is less fit to survive and readily gets eliminated and replaced by the neighboring Hel25EWT (yellow) winner cell in an autophagy mediated apoptosis. B. Fat-/- mutant (yellow) cell is a hippo pathway mutant cancer cell with a Yorkie dependent increased protein synthesis levels and the neighboring Fat+/+ (orange) cell (wildtype) is with normal protein synthesis levels, in this condition is less fit and readily gets eliminated by the Fat-/- mutant cell in an autophagy-dependent cell death further aiding in the growth of Fat-/- mutant cell, thereby increasing the cancer phenotype. Note: A. and B. shows that relative global protein levels in the cells from the tissue leads to supercompetition. Orange cells are losers and have been eliminated by autophagy-mediated cell death triggered by neighboring yellow, winner cells. C. A cancer is a condition where all cells within the tumor do not get the same level of access to the nutrients, and thus cancer cell in nutrient-replete (yellow) state supports the cell which is in nutrient-deplete (orange) condition by providing NH3+ via action of Glutaminase. This diffused NH3+ triggers autophagy in the nutrient-deplete cancer cell, which further helps in sustaining them and avoiding TNF-α induced cell death. D. Malignant tumor (RafGOF Scrib-/- and RasV12 Scrib-/-) triggers ER stress or oxidative stress. These stresses activate autophagy in tumor microenvironment (wildtype) which provides amino acids and nutrition to the tumor and thereby supporting the proliferation and malignancy of tumor.
Figure 7.
Cell non-autonomous autophagy in supercompetition and cancer. A. Hel25E-/- mutant (orange) cell has reduced protein synthesis and thus is less fit to survive and readily gets eliminated and replaced by the neighboring Hel25EWT (yellow) winner cell in an autophagy mediated apoptosis. B. Fat-/- mutant (yellow) cell is a hippo pathway mutant cancer cell with a Yorkie dependent increased protein synthesis levels and the neighboring Fat+/+ (orange) cell (wildtype) is with normal protein synthesis levels, in this condition is less fit and readily gets eliminated by the Fat-/- mutant cell in an autophagy-dependent cell death further aiding in the growth of Fat-/- mutant cell, thereby increasing the cancer phenotype. Note: A. and B. shows that relative global protein levels in the cells from the tissue leads to supercompetition. Orange cells are losers and have been eliminated by autophagy-mediated cell death triggered by neighboring yellow, winner cells. C. A cancer is a condition where all cells within the tumor do not get the same level of access to the nutrients, and thus cancer cell in nutrient-replete (yellow) state supports the cell which is in nutrient-deplete (orange) condition by providing NH3+ via action of Glutaminase. This diffused NH3+ triggers autophagy in the nutrient-deplete cancer cell, which further helps in sustaining them and avoiding TNF-α induced cell death. D. Malignant tumor (RafGOF Scrib-/- and RasV12 Scrib-/-) triggers ER stress or oxidative stress. These stresses activate autophagy in tumor microenvironment (wildtype) which provides amino acids and nutrition to the tumor and thereby supporting the proliferation and malignancy of tumor.
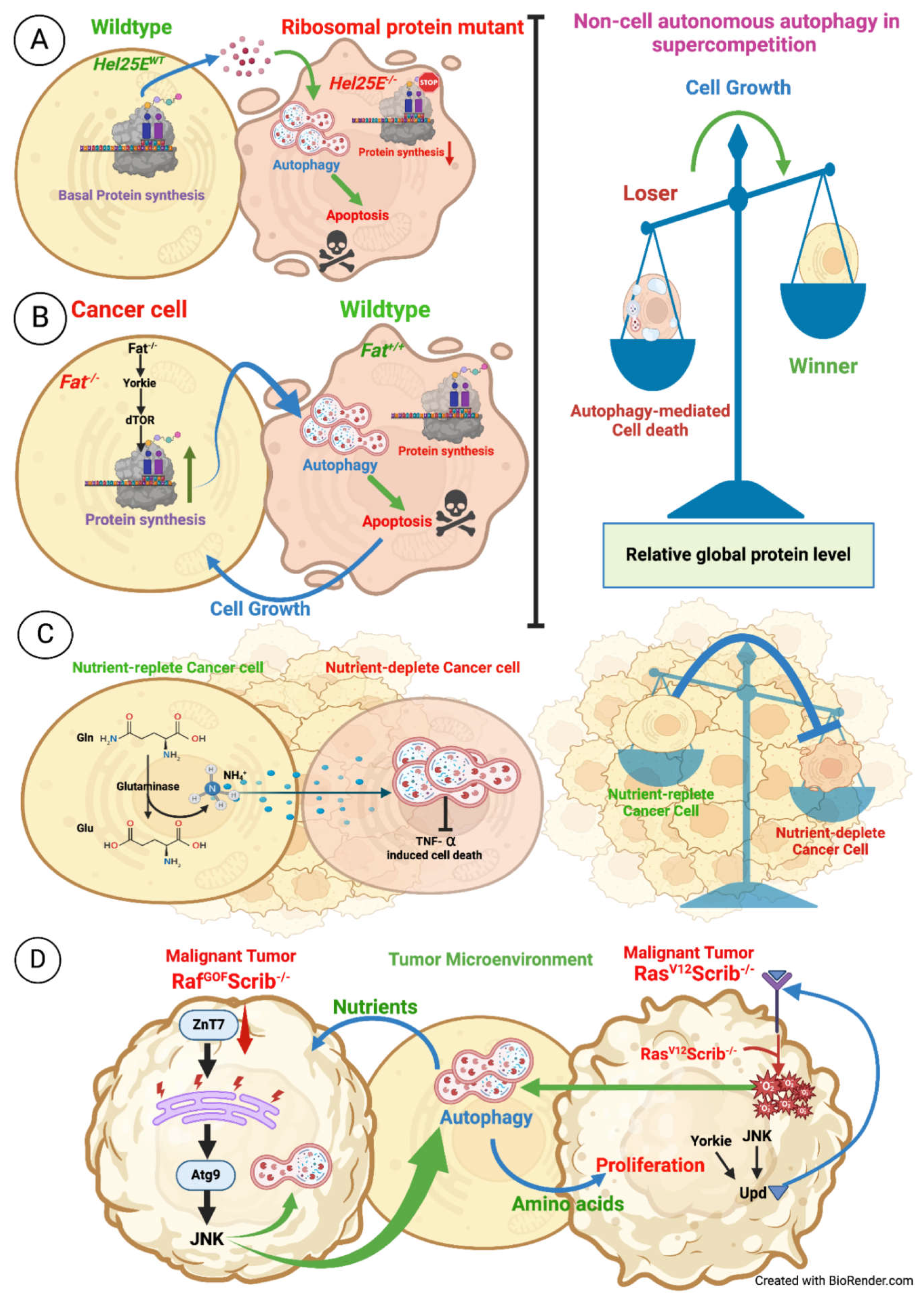
Figure 8.
Cell non-autonomous autophagy during ECM regulation and Stem cell maintenance. A. Heparan-sulfate (HS) proteoglycan synthesis is important part for the ECM which helps in canonical signaling regulation. When fat body is subjected to mosaic-clone technique, it generated two types of cells; HS-producing (yellow) and HS-mutant (blue) cell. HSPGs knocked-down or mutant cells resulted in autophagy upregulation. However, when HS-mutant clone (blue) is in contact with HS-producing (yellow) cell, upregulated autophagy is rescued to basal levels. This shows that HS and ECM regulate autophagy, in this case, negatively. B. Stem cell maintenance: EGF secreted from D. melanogaster male GSCs leads to activation of EGFR signaling in Cyst stem cell (CySC) leads to autophagy induction, thus maintaining CySC maintenance which in turn maintains GSCs. However, when DILPs activates insulin signaling in CySC, it inhibits autophagy via mTOR pathway, leading to CySC differentiation in early cyst cell, which is entailed by GSCs/ germline differentiation.
Figure 8.
Cell non-autonomous autophagy during ECM regulation and Stem cell maintenance. A. Heparan-sulfate (HS) proteoglycan synthesis is important part for the ECM which helps in canonical signaling regulation. When fat body is subjected to mosaic-clone technique, it generated two types of cells; HS-producing (yellow) and HS-mutant (blue) cell. HSPGs knocked-down or mutant cells resulted in autophagy upregulation. However, when HS-mutant clone (blue) is in contact with HS-producing (yellow) cell, upregulated autophagy is rescued to basal levels. This shows that HS and ECM regulate autophagy, in this case, negatively. B. Stem cell maintenance: EGF secreted from D. melanogaster male GSCs leads to activation of EGFR signaling in Cyst stem cell (CySC) leads to autophagy induction, thus maintaining CySC maintenance which in turn maintains GSCs. However, when DILPs activates insulin signaling in CySC, it inhibits autophagy via mTOR pathway, leading to CySC differentiation in early cyst cell, which is entailed by GSCs/ germline differentiation.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



