
Submitted:
01 April 2024
Posted:
02 April 2024
You are already at the latest version
Abstract
Research indicates that brain region-specific synapse loss and dysfunction are early hallmarks and stronger neurobiological correlates of cognitive decline in Alzheimer’s disease (AD) than counts of amyloid plaques, neurofibrillary tangles, and neuronal loss. Even though the precise mechanisms underlying increased synaptic pruning in AD are still unknown, it has been confirmed that dysregulation of the balance between complement activation and inhibition is a crucial driver of pathology. The complement includes three distinct activation mechanisms, with activation products C3a and C5a, potent inflammatory effectors, and a membrane attack complex (MAC), leading to cell lysis. Besides pro-inflammatory cytokines, dysregulated complement proteins released by activated microglia bind to amyloid β at synaptic regions and cause microglia to engulf synapses. Additionally, research indicating that microglia-removed synapses are not always degenerating, and that suppression of synaptic engulfment can repair cognitive deficits point to an essential opportunity for intervention that can prevent the loss of intact synapses. In this study, we focus on the latest research on the role and mechanisms of complement-mediated microglial synaptic pruning at different stages of AD to find the right targets that could interfere with complement dysregulation and be relevant for therapeutic intervention at the early stages of the disease.
Keywords:
complement proteins
; cognitive decline
; synapse loss
; microglia
; neurodegeneration
Introduction
Alzheimer's disease (AD) is a neurodegenerative disease that causes large amounts of amyloid plaques and neurofibrillary tangles to build up in the brain. It is one of the leading causes of illness and death around the world. Given the absence of effective therapeutic methods and the mostly unexplained etiology of AD, scientists have been pushed to look deeper into molecular mechanisms and pathways to comprehend the disease's pathophysiology. It has been established that innate immune responses play a significant role in AD development and progression. The Genome-wide Association Studies (GWAS) revealed many AD risk genes that converge on microglial cell phagocytic pathways (Hansen et al., 2018a) (Lambert et al., 2009). Among risk variations are single nucleotide polymorphisms (SNPs) in complement receptor 1 (CR1), clusterin (CLU) (Lambert et al., 2009), and C1S (Bellenguez et al., 2022).
It is commonly known that the complement system is a key component of the innate immune response and a strong inducer of neuroinflammation in AD. Increasing evidence shows that the Aβ protein stimulates complement activity (Tenner, 2020) (Z. Li et al., 2023), which causes localized chronic inflammation and activation of the glial cells in the vicinity (Shah et al., 2021). By co-localizing with amyloid β in the capillaries of the brain complement proteins can cause cerebral amyloid angiopathy (Matsuo et al., 2017). On the contrary, the levels of complement regulators significantly decrease in AD brains, which emphasizes potentially significant disturbances in the regulation of inflammatory reactions. The continuous production of inflammatory mediators and complement proteins by microglia exaggerates the amyloid pathology. It leads to a self-supported neuroinflammation loop between overactivated microglia, the complement system, and Aβ plaques, worsening AD pathology.
Complement-dependent glial-mediated synaptic pruning has been shown among the primary contributors to the loss of synapses at the early stages of AD (Bohlson & Tenner, 2023). The glial cells isolated from AD brains had higher concentrations of synaptic proteins than did microglia and astrocytes from non-AD individuals (Taddei et al., 2023) (Tzioras et al., 2023). Additionally, the aberrant tau oligomers in synapses as well as the presence of amyloid pathology enhance glial-mediated synapse removal in the brains of AD patients (Taddei et al., 2023) (Tzioras et al., 2023). In culture, synapses obtained from AD patients were more easily phagocytosed by primary mouse and human microglia and astrocytes than synapses derived from control brains. Research also showed that inhibiting the opsonophagocytic mechanism can regulate glial synapse ingestion (Tzioras et al., 2023). Additionally, recent research has shown that the excessive synaptic loss associated with several neurodegenerative diseases can be reduced by the deletion of not only C1q but also C3 and C4 complement proteins (Hong et al., 2016a) (Shi et al., 2017a). Taken together, these findings imply that the opsonophagocytic mechanism, which involves the activation of complement components is crucial in the internalization of synapses by astrocytes and microglia in human AD brains and mouse models.
However, the facts indicating the role of complement activation in the acceleration of AD progression, as well as evidence that suppressing the complement system is not without risk, raise the idea that targeting the complement, especially in the brain, requires closer attention. Therefore, important issues about complement-mediated synapse loss in AD, still need to be resolved. Moreover, it is unclear what physiological processes in AD make synapses more susceptible to being "tagged." In this article, we go over how the microglia-complement interactions affect brain functions while also playing a role in synaptic loss. Also, we present an overview of recent findings on complement-mediated synaptic decline in AD and provide clarification regarding the underlying mechanism.
Complement Proteins
Complement is a crucial element of an innate immune system that functions in the protection of the body from pathogens or injured cells (Nonaka & Kimura, 2006) (J. D. Lee et al., 2019). It promotes inflammatory reactions through the generation of anaphylatoxins (Yanamadala & Friedlander, 2010). The complement includes an enzymatic cascade with approximately 50 proteins and membrane receptors (Yanamadala & Friedlander, 2010) (Shah et al., 2021). Classical, lectin and alternative pathways are well-known mechanisms for complement activation (Ricklin et al., 2016).
The classical pathway is activated when C1 (C1qrs) interacts with immune complexes, microbes, as well as specific proteins (such as Aβ or α-synuclein protein). All three pathways join to form a C3 convertase, which then cuts C3 to release C3a and C3b. The latter then cleaves C5 into C5a and C5b. Being anaphylatoxins that provide immune cell recruitment and inflammation, C3a and C5a (Ricklin et al., 2016) act via binding to their respective receptors. The C3a receptor (C3aR) and the C5a receptor (C5aR1), are present on the membrane of neutrophils and macrophages. The second C5a-like receptors for binding with C5a are C5L2 and C5aR2. Binding of C3b to C3R on the surface of phagocytes leads to an increase in their phagocytic activity (Wu et al., 2019). In contrast, activation of C5aR1 initiates proinflammatory responses via stimulation of cytokine or chemokine production and activation of phagocytes. Nevertheless, C3aR signaling is mainly immunomodulatory and, depending on the context, may exert pro- or anti-inflammatory effects(Coulthard & Woodruff, 2015) (Shinjyo et al., 2021). Ricklin et al. (2016) found that C5b encourages the formation of the membrane-attack-complex (MAC), which breaks down the integrity of the cellular plasma membrane followed by death and lysis of cells.
Different regulators or controllers of complement activation (RCA), such as factor H (FH), factor I, C1-inhibitor, clusterin, etc., function to limit complement activation both in time and place and prevent autologous tissue damage. When an immune response takes place against a pathogen the complement system is activated and triggers an inflammatory response that helps immune cells combat infection by increasing or "complementing" the capacity of antibodies and phagocytic cells to eliminate microbes and injured cells.
Despite the well-known functions of the complement system in peripheral organs, its roles in the central nervous system (CNS) are still obscure and under investigation. In the CNS complement proteins, as well as their regulatory proteins and receptors, are secreted primarily by glial cells and neurons (Gasque et al., 1996). The CNS uses the complement for several crucial processes, including synaptic plasticity throughout life, synapse elimination, neurogenesis, apoptosis, and neuronal plasticity. In the process of synaptic pruning during development complement proteins contribute to the elimination of inactive synapses to allow the strengthening and maturing of more healthy connections (Schafer et al., 2012).
Components of classical complement cascade, representing a group of secreted ‘eat me’ signals mediate recognition and phagocytosis of weak synapses by microglia (Figure 1). This happens when synapses are tagged by C1q, opsonized by C3b, and then taken up by microglia through CR3 activation (Sekar et al., 2022)(Schafer et al., 2012). The quantity of phagocytic microglia and the degree of early synapse loss are greatly decreased when C1q, C3, or the complement receptor CR3 on the microglia are suppressed (Schafer et al., 2012) (Hong et al., 2016a). Although CNS complement is mostly produced by microglia and astrocytes, significant levels of C1q and CR3 have been demonstrated to be secreted by microglia. In microglial-specific conditional C1q knockout mice, even though the blood C1q levels remained stable, in brain tissue C1q was not detected, indicating that microglia are the primary CNS source of C1q (Fonseca et al., 2017).
Disturbances in the regulation of the complement system in the CNS contribute to various neurological disorders, including neurodevelopmental disorders such as autism and schizophrenia (Brown, 2012) (Sekar et al., 2022) (Yilmaz et al., 2021), neurodegenerative diseases, diseases associated with disturbances in blood-brain barrier (BBB) functions, etc. Additionally, recent research has demonstrated the critical role of complement cascade in the development of neurological complications of SARS-CoV-2 infection (Jaffry et al., 2022) (M. H. Lee et al., 2022).
Microglia in Health and Disease
Microglia are macrophage-like cells providing immunological surveillance of the central nervous system (CNS). They derive in the yolk sac and in mice colonize the neuroepithelium at embryonic day 10.5 (Ginhoux et al., 2010) (Gomez Perdiguero et al., 2015). Reaching the brain, they propagate, ramify, and scatter all over the CNS (Ginhoux et al., 2010). During a lifetime, microglial homeostasis maintains the balance between their proliferation and apoptosis (Askew et al., 2017). The proliferation of microglial cells is regulated by IL-34, IFN regulatory factor 8, the transcription factor PU.1, and macrophage colony-stimulating factor 1 receptor (Askew et al., 2017).
Microglial depletion studies showed that the source of replenishment of the depleted microglial population is exclusively brain-resident cells and takes place via an IL-1α-dependent pathway (Bruttger et al., 2015). However, research has confirmed that in diseased conditions associated with BBB breakdowns, such as brain ischemia, autoimmune encephalomyelitis, and whole-body radiotherapy 2 (Bruttger et al., 2015), the peripheral monocytes can penetrate the brain parenchyma and populate the microglial niche. These findings suggest that circulating monocytes may contribute to the pathophysiology of AD, where BBB breakdown is apparent (Sweeney et al., 2018).
As classic tissue-resident macrophages, microglia play vital roles in CNS tissue maintenance, homeostasis, and health (Bartels et al., 2020). They dynamically survey the environment and provide brain protection, response to tissue injury, and repair by actively removing phagocytic cellular debris (Hansen et al., 2018b). To morpho-functionally adapt to their environment, microglia use a whole arsenal of resources, including plasticity of elements of the cytoskeleton, a system of intracellular proteins, and microglial receptors, that recognize pathogen-associated molecular patterns and damage-associated molecular patterns (Franco-Bocanegra et al., 2019). Additionally, they can phagocytose and produce a variety of substances that are involved in tissue maintenance and immunological defense, including chemokines, cytokines, trophic factors, and nitric oxide.
Recent studies on developing and adult brains showed that microglia are not just immune cells; they are involved in adult neurogenesis (Kettenmann et al., 2013) (Frost & Schafer, 2016), formation of brain architecture and wiring neural circuits, and vasculature development (Dudvarski Stankovic et al., 2016). Importantly, there is strong evidence supporting the role of microglia in the regulation of synaptic plasticity, a crucial cellular mechanism, involved in learning and memory (Bailey et al., 2015). These cells engulf synapses and reshape synaptic connections during normal brain development as well as its decline. This process varies with developmental stages, brain regions, as well as stages of the disease.
The homeostatic state of microglia in a healthy brain is supported by neuronal–microglial crosstalk that includes the signaling pathways involving CD200–CD200R and CX3CL1–CX3CR1 (Simon et al., 2019). However, in the brains of patients with AD, the expression of CD200, as well as receptors CD200R and CX3CR1 are diminished, leading to a reduced physiological regulation of microglial behavior (Holtman et al., 2015). Hence, in response to persistent or chronic stimulation, microglia adopt a significantly different signature from the homeostatic one and undergo changes in morphology, proteomic markers, and behavior. This significantly depends on the region affected and stage of the disease and the severity of the pathogenic environment in brain tissue.
In the context of AD, microglial priming is increasingly being proposed as a prerequisite process when chronic low-level stimuli (such as systemic inflammation and aging) cause naive microglia to assume an altered state, resulting in an enhanced or inappropriate inflammatory reaction in response to repeated pathological stimulation (Perry & Holmes, 2014). Alterations of microglial phenotype in AD lead to activation of inflammasome signaling, and the release of excessive inflammatory cytokines, which in turn promotes neurotoxicity and excessive microglial synapse elimination (Hong et al., 2016b) (Venegas et al., 2017) (Batista et al., 2024). Findings confirm that neuroinflammation is an ongoing process that does not resolve on its own and is regarded as a critical driver of AD (Heneka et al., 2015). Moreover, the growing body of evidence suggests that caspase-1 activation and proinflammatory cytokine (such as IL-1β) production occur before AD pathology, showing that activation of the microglial NLRP3 inflammasome represents an early pathogenic event in AD (Hanslik & Ulland, 2020).
On the other hand, individuals with disrupted microglial functioning (which could be acquired or hereditary susceptibility) experience cognitive loss at an earlier clinical stage of AD (Jack et al., 2013) than those with intact microglial function. Thus, microglial behavior partially determines the vulnerability of people to AD concerning more severe clinical manifestations at a particular stage of the disease. Additionally, in late-stage AD, ineffective Aβ clearance and tau fibrillation negatively impact the microglial defense activities and induce a persistent harmful microglial activation, contributing to neurodegeneration (Butovsky & Weiner, 2018).
Activation of Complement Proteins in AD
Although the mechanisms, involved in the contribution of complement cascade as well as complement-related genes to AD pathogenesis are under investigation, findings on human brain tissue and animal models demonstrate that complement regulatory dysfunction plays a significant role in the development of AD (Batista et al., 2024). Post-mortem studies of AD brains demonstrated a dramatic increase in the activation of all complement components (Reichwald et al., 2009) (Maier et al., 2008). Indeed, higher levels of C1q, C3, and C4 have been found in different brain regions, including temporal cortex, especially near Aβ plaques and tau aggregates (Shen et al., 2001) (Thambisetty et al., 2013). Furthermore, distinct disease stages can be distinguished by elevated quantities of complement proteins and activating components in the bloodstream and cerebrospinal fluid (CSF). In this regard, products of complement and immune system dysregulation might serve as biomarkers for early diagnosis of AD and as predictors of disease progression (Morgan, 2018) (Carpanini et al., 2022).
Triggers of pathological C1q upregulation in surrounding microglia and complement cascade activation are oligomeric/fibrillar Aβ and hyperphosphorylated tau (Shen et al., 2001) (Morgan, 2018) (Doens & Fernández, 2014). Additionally, a variety of other complementary components have been linked to AD. Among those are complement protein 3a (C3a) and its receptor C3aR, complement protein 5a (C5a) and its receptor C5aR1, complex C5b-C9, complement component 9, factor B and factor D (Litvinchuk et al., 2018) (Goetzl et al., 2018) (Goetzl et al., 2018) (Wyss-Coray & Rogers, 2012) (Kretzschmar et al., 2021).
Initiator of nuclear factor-κB (NFκB) in astroglia, complement C3 was shown to act via the neuronal C3aR (Lian et al., 2015). Studies show that C3 is an astroglia-specific NFκB target, sufficient to cause neuronal damage via C3aR and intraneuronal calcium signaling (Lian et al., 2015). Notably, several neurological disorders, including AD, have impaired synaptic function as a primary result of calcium dysregulation (Mattson, 2007). Thus, activation of neuronal C3aR leads to intraneuronal calcium increase, which in turn, enhances synaptic excitation and impairs dendritic morphology, both of which contribute to neuronal dysfunction. Furthermore, C3 produced by astrocytes also interacts with C3aR on microglia exacerbating Aβ pathology. Since dendritic and synaptic loss in neurons is caused by NFκB/C3/C3aR signaling, it seems reasonable that cognition is improved in APP/PS1 mice administered a C3aR antagonist (Lian et al., 2015) (Batista et al., 2024). These studies provide evidence that unique neuron-glia signaling pathways link Aβ pathology with synaptic abnormalities and neuronal hyperexcitability.
Moreover, the identification of terminal membrane attack complex (MAC), the final product of activation of complement pathways, the near fibrillar Aβ plaques, and on synapses highlighted the activation of not only classical but all three complement pathways in AD (Fonseca et al., 2017). In particular, APP knock-in (KI) mouse models of AD demonstrated elevated levels of MAC, C1q, and C3 in brain synaptosomes (Carpanini et al., 2022). The application of MAC-blocking antibodies, as well as the depletion of MAC component C6, confirmed the detrimental role of the MAC in synaptic loss (Carpanini et al., 2022), though the core mechanisms need further clarification.
Human studies have also confirmed the contribution of the MAC to synaptic pruning in AD. In the hippocampus and frontal cortex of AD patients, there was a significant reduction in both the mRNA and levels of complement defense 59 (CD59) protein, which prevents complement membrane attack complex (MAC) assembly (L. B. Yang et al., 2000). However, levels of complement component 9 (C9), the last component needed for the formation of MAC, showed a significant increase. Furthermore, a strong association was discovered between the decline in CD59 and synaptophysin (L. B. Yang et al., 2000), suggesting that deficiencies in CD59 expression could lead to greater MAC generation and the destruction of synapses in AD.
Extensive reports highlight the important role the complement proteins play in the inflammatory response in AD (Lian et al., 2016) (Xie et al., 2023) (El Gaamouch et al., 2020). It has been hypothesized that complement dysfunction may cause neuroinflammation in an AD patient decades before clinical symptoms appear (Shah et al., 2021). The facts demonstrating that complement activation may speed up the development of AD leads to the idea that inhibiting complements could be a possible therapeutic strategy (B. P. Morgan, 2018).
Is Complement Activation Beneficial or Detrimental in AD?
Determining whether complement activation is advantageous or harmful for AD is still highly debatable. In some experiments, complement deficiency has been shown to protect against Alzheimer's disease in some, yet to have no effect, or possibly worsen the condition, in others (Maier et al., 2008) (Shi et al., 2017a) (M. Li et al., 2024). Some authors claim that C1q deletion did not affect amyloid load, implying that C1q functions downstream of Aβ (Fonseca et al., 2017). Furthermore, when the C1q complement component is inhibited, microglia are unable to remove glutamate-containing vesicular blebs produced by injured neurons and apoptotic cells (Fraser et al., 2010). Increased neurodegeneration has been shown in the 3xTg mouse model deficient for C1q (Benoit et al., 2013).
Other studies show that complement inhibition or deficiency causes accelerated amyloid pathology (Maier et al., 2008) (Wyss-Coray & Rogers, 2012), emphasizing the role of C3 and CR3 in Aβ phagocytosis (Maier et al., 2008). Indeed, as fibrillar amyloid plaque is accessible to complement components, the activated complement components opsonize and facilitate its elimination (Maier et al., 2008). Genetical studies provided evidence that mutations in complement receptor 1 (C1R) may contribute to the progression of AD through modulation of Aβ accumulation (Zhu et al., 2020). A study carried out using 12- and 17-month-old amyloid precursor protein (APP)/C3−/− mice demonstrated the beneficial role of C3 in plaque clearance and neuronal health (Maier et al., 2008). Damaged neurons have not been also cleared and eliminated in C3-deficient animals. Likewise, in studies that used APP-transgenic mice and other models, C3 inhibition accelerated Aβ plaque deposition (Maier et al., 2008) (Shi et al., 2015) (Kinney et al., 2018).
APPswe/PS1ΔE9 transgenic mice administered with the C3aR1 antagonist SB290157 showed decreased microgliosis and Aβ pathology (Lian et al., 2016), in contrast to other findings that demonstrated decreased microgliosis and amyloid load via treatment of C3aR1 agonist (El Gaamouch et al., 2020). Remarkably, it has been documented that when SB290157 is administered at a concentration of 10 μM, it functions as an agonist for C3aR1 instead of an antagonist (Woodruff & Tenner, 2015) (Mathieu et al., 2005), thus demonstrating the beneficial role of C3aR1 activation in AD. Furthermore, the scientists showed that complement synthesis and C3aR1 activation via C3a control the expression of homeostatic genes during development, limiting the alterations in the transcriptome of microglia (El Gaamouch et al., 2020).
Low levels of CSF complement proteins have been positively correlated with faster cognitive deterioration and acceleration in the progression of AD according to recent studies (M. Li et al., 2024). Reduction in levels of complement proteins has been partially linked to their capture in amyloid plaques. Additionally, decreased C1q levels were strongly correlated with lower mental status and cognitive performance, according to the study. Similar findings have been reported by another cohort study using the Luminex assay, linking lower levels of CSF C3 to accelerated decline in cognition (Toledo et al., 2014).
Nevertheless, other animal models show that the complement plays a detrimental role in AD. Increasing evidence shows that along with diminishing the number of phagocytic microglia the inhibition of complement components C1q, C3, or CR3 also reduces the extent of synapse loss (Hemonnot et al., 2019). In an AD mouse model, C1q deletion (either by use of C1q neutralizing antibodies) or reducing C3 enhanced the number of synapses and improved cognitive performance (Berg et al., 2012). In other studies, C3 deficiency prevented the elimination of synapses from the damaged neurons compared to the control (Brucato & Benjamin, 2020).
It is well established that the most vulnerable regions of the brain for synapse loss are the hippocampus and frontal cortex. Interestingly, familial AD-mutant hAPP ("J20") transgenic mice showed more pronounced C1q elevation and enhanced levels of microglial lysosomal protein CD68 in those areas (Hong et al., 2016). Moreover, the authors reported that C1q synaptic localization was increased even before plaques were formed (Hong et al., 2016). Quantification of colocalized pre- and postsynaptic markers (synaptophysin and PSD95, synaptotagmin, and homer) in those animals revealed a significant loss of synapses at 3–4 months old, an age that precedes plaque deposition. Furthermore, the microglia of wild animals that have been challenged with intracerebroventricular injection of soluble Aβ had much higher levels of CD68 immunoreactivity and more pronounced microglial pruning of synapses and synaptic loss. However, C1q, C3, or C3 receptor knockout mice as well as those receiving pharmacological treatment with an anti-C1qa-blocking antibody, rescued synapse loss and did not show increased CD68 immunoreactivity in response to Aβ (Hong et al., 2016). Additionally, the absence of C1q in the transgenic hAPP Tg2576 mouse model prevented the generation of the downstream products C3a and C5a of the classical complement pathway (Fonseca et al., 2004). It also kept synaptophysin and MAP2 from being lost in the CA3 area of the hippocampus. These data provide evidence that synapse loss in AD is mostly caused by complement-dependent microglial clearance of synapses.
On the other side, the activation of the complement system exacerbated tau pathology (Hansen et al., 2018). In a separate study, tau-P301S transgenic mice were used for unbiased proteomic analysis of postsynaptic densities (PSDs) to uncover tau-dependent synaptic changes before explicit neurodegeneration. Besides disturbances in other proteins and pathways, the authors observed the depletion of a set of GTPase-regulatory proteins involved in the rearrangement of the actin cytoskeleton, which eventually led to the loss of dendritic spines. In this study, complement C1q accumulation in the PSDs of tau-P301S mice and AD patients has been correlated to the amount of phospho-tau. The accumulation of complement C1q in the PSDs in those models resulted in augmented engulfment of synapses by microglia and a reduction of synapse density. Additionally, a C1q-blocking antibody reduced microglial-induced synapse loss and restored synapse density in tau-P301S mice as well as in cultured neurons, suggesting that synaptic pruning initiated by tau pathology depends on C1q (Dejanovic et al., 2018) (Brucato & Benjamin, 2020).
Additionally, C3 inhibition or knockout provides neuroprotection by inhibiting all complement activation pathways (Shi et al., 2015) (Shi et al., 2017) (Hong et al., 2016). In comparison to aged wild-type mice, aged C3 knockout mice showed increased long-term potentiation, implying increased synaptic activity and connectivity in the hippocampus and, as a consequence, improvement in learning and memory. Accordingly, animals deficient in C3 did not show typical age-related hippocampus degeneration (Shi et al., 2015). A similar effect was observed in hippocampal CA3 region APP/PS1 mice, which demonstrated protective effects of C3 deficiency against synaptic and neuronal loss (Shi et al., 2017). The consequence is clear: complement may interfere with synaptic health in old age and AD (Brucato & Benjamin, 2020).
Furthermore, the microgliosis and astrogliosis in the hippocampus of the animal AD models were alleviated and brain levels of pro-inflammatory cytokines were reduced by C3 loss in APP/PS1 mice. These aged APP/PS1; C3 KO mice exhibited altered glial cell morphology and position around plaques, demonstrating that a C3 deficiency may influence the glial cell response to plaques (Shi et al., 2017b). These findings suggest that C3 may be a key factor in neuroinflammation that promotes synaptic dysfunction.
Other controversies debate on effect of C3a and its C3a receptor on tau pathology. Recently, tau hyperphosphorylation induced by okadaic acid (OA) as well as the activity of glycogen synthase kinase 3β (GSK3β) has been inhibited by antagonist of C3a receptor SB290157 (J. Hu et al., 2019). The findings suggest that the C3a receptor has a unique role in controlling the phosphorylation of tau protein via GSK3 signaling pathways (J. Hu et al., 2019). In addition to the decrease of plaque-associated synapse loss in PS2APP animals, complement inhibition via deleting C3 also reduced neuron loss and brain atrophy in tauP301S mice. The authors stated that enhanced levels of C3 in AD patients' CSF correlate with tau accumulation and that diminishing C3 function may be helpful in other types of tauopathies (Wu et al., 2019).
The mechanism by which microglia and astrocytes ingest excitatory and inhibitory synapses, respectively has been demonstrated in the PS19 tau animal model (Dejanovic et al., 2022). It's interesting to note that excitatory synapses have been found in lysosomes of astrocytes, while microglial lysosomes contain inhibitory synapses. Furthermore, the lack of C1q ameliorated synaptic elimination in the PS19 mouse model and reduced pruning of synapses by microglia and astrocytes.
It should be highlighted, nonetheless, that not all research has discovered a strong relationship between changes in CLU and CR on the other side as well as CSF Aβ and tau on the other (Schjeide et al., 2011). The reason for these contradicting results might be that the majority of mice models used to replicate early-onset AD only contain single-gene alterations as opposed to the prevalent multi-gene mutations observed in late-onset AD. Therefore, it's possible that the conclusions drawn from these mice models are limited in scope and cannot be applied to other cases.
On the other hand, controverting conclusions coming from the published data can be explained by the varied experimental models (acute or chronic) utilized, different brain areas, and the different stages of disease studied. Moreover, in mouse models of amyloid or tau neuropathology, the developmental timing of C3aR1 ablation and the duration of activation or inhibition of the C3aR1 pathway may be crucial factors. For instance, in a study by Lian et al. (2016), short-term (1 h) treatment of microglia with C3 or C3a enhanced phagocytosis, while longer treatment (24 h) reduced it (Lian et al., 2016). Additionally, beneficial effects of C3 for Aβ clearance have been shown in APP and hAPP mice older than 10 months with sustained Aβ plaque pathology (Maier et al., 2008). However, studies demonstrating the deleterious effects of C1q, C3, and CR3 on synaptic homeostasis investigated the pre-plague stages of the disease in J20 (Hong et al., 2016) and APP animals (Czirr et al., 2017).
Complement Components in Different Stages of AD
There are still many significant unanswered concerns regarding the molecular mechanisms that underlie the complement cascade's regulation and impact on neuronal function and dysfunction in the AD brain. It has been revealed that minor memory loss at the initial stage of AD is directly related to synapse loss and neuron dysfunction and is caused by the classical complement cascade (Hong et al., 2016). Studies show that, initially, the Aβ peptides attach to a collagen-like domain (CLF) within C1q (Figure 2). Because the binding of Aβ 1-42 to C1q is more effective than Aβ 1-40, this pathway has more impact on Aβ 1-42, resulting in its assembly into aggregates and fibrils. This interaction leads to the activation of the classical complement pathway and results in synapse loss. Indeed, C1q, as well as TNF-α and interleukin-1α (IL-1α), released from activated microglia promote astrocytes to change into their reactive pattern, namely A1 astrocytes, which are main producers of complement protein C3 (Liddelow et al., 2017). Complement C3 released from A1-reactive astrocytes accumulates on amyloid plaques, NFTs, and weakened synapses (Batista et al., 2024).
The question is whether synaptic pruning mechanism is helpful initially but subsequently gets dysregulated in the long-term chronic way, impairing the neurons it was trying to conserve. The steps in the activation of the complement cascade depending on the stage and progression of AD has been reported in many studies (Tenner, 2020). (Z. Li et al., 2023). It has been shown that in the early stages of the disease, C1q expression is increased by tissue injury, amyloid oligomers, apoptotic neurons, and neuronal blebs. The binding of C1q to those structures initiates phagocytosis by microglia. In the absence of other pathogens or damage-associated molecular patterns, it provides anti-inflammatory activity.
The accumulation of fibrillary Aβ and local damage, however, leads to further C1r, C1s, C4, and C3 synthesis and chronic activation of the complement cascade. Newly generated C3b/iC3b is covalently bound to fibrillary Aβ and may lead to phagocytosis via CR3. Nevertheless, in the more advanced stages, in addition to 2b, 3b, and 4b bound to fibrillary Aβ there is also a generation of C5a and C5b. C5a induces chemotaxis in microglia, providing their recruitment to the amyloid plaque. C5a diffuses from the plaque, binds to the anaphylatoxin receptor C5aR1 on microglia, and induces a chronic inflammatory state by acting on MAPKs and activating the production of pro-inflammatory mediators (Tenner, 2020). According to Carvalho et al. (Carvalho et al., 2022b) the primary way that C5a-C5aR1 signaling works in AD is via stimulating pathways that activate microglia, leading to disease progression. As was mentioned, the fibrillary Aβ also binds to microglial TLR receptors. Interestingly, C5a–C5aR1 signaling was found to synergize with TLR2 and TLR4 (Schartz & Tenner, 2020). Acting in concert, they enhance proinflammatory cytokine responses and reactive oxygen species production, leading to a neurotoxic environment. Thus, while the binding of C5a and C3a recruits phagocytic cells to the plaque, the intracellular signaling of C5a and C3a on microglia induces a chronic inflammatory state (J. Yang et al., 2020). AD patients had higher serum levels of C5a, TNF-α, IL-1β, IL-6, and CRP than non-AD subjects did (Z. Li et al., 2023). Furthermore, more severe AD patients demonstrated higher levels of C5a compared to patients with mild and moderate AD. These levels had a positive correlation with plasma pro-inflammatory factor levels and a negative correlation with cognitive function (Z. Li et al., 2023). Moreover, C5a initiates apoptosis in neurons (Carvalho et al., 2022b), and C5a/C5aR1 signaling increases vascular permeability by causing endothelial cell apoptosis further exacerbating brain pathology (Mahajan et al., 2016).
Consequently, in large quantities, fibrillary Aβ plaques cannot be properly removed from the brain tissue, and a chronic inflammatory environment develops, contributing to more fibrillary Aβ production, greater neuronal damage, and death.
Complement-Modulating Approaches in AD Treatment
Increasing evidence shows that selective genetic or antibody-based complement inhibitory approaches that protect synapses and memory loss can be beneficial in slowing down the progression of AD (Morgan, 2018) (Carpanini et al., 2022). For instance, overexpression of CD55, an inhibitor of complement pathways produced in neurons at inflammation, resulted in the alleviation of synaptic loss in the dentate gyrus of the hippocampus (Wang et al., 2020). That also led to fewer synaptic dissociations in the CA1 area of the rodent hippocampus and less remote memory loss (Wang et al., 2020).
Blocking the complement receptors C3aR and C5a-C5aR1 signaling reduces synaptic pruning, the amyloid burden, slows the appearance of plaque-associated dystrophic neurites, and avoids microglial polarization toward a more detrimental disease-associated inflammatory phenotype, allowing for inhibition of inflammation in AD mouse models. Even though all the upstream complement components involved in synaptic pruning were still there, cognition was better and AD pathology was less severe when the C5a-C5aR1 interaction was blocked with drugs or deleted genetically (Schartz & Tenner, 2020). Aβ-induced inflammatory response initiated by C5a was inhibited by C5a-targeting vaccines as well as by PMX205, a C5aR antagonist in two mouse models of AD (Tg2576 and 3xTg) (Landlinger et al., 2015). Animals showed improved contextual memory and reduced cerebral amyloid plaque. Since JAK/STAT3 signaling is involved in the neuroinflammatory responses to Aβ and C5a, inhibiting this route with an antagonist, AG490, prevented the generation of proinflammatory cytokines as well (An et al., 2018). In other studies, PMX205 treatment reduced microglial and astroglia activation besides diminishing fibrillar amyloid accumulation and cognitive loss (Fonseca et al., 2009). In the 3xTg model that gets tangles, PMX205 treatment caused hyperphosphorylated tau to drop by 70%. Moreover, the cytotoxic and inflammatory effects of Aβ42 on microglial BV-2 that were initiated by C5a have also been blocked by the C5aR antagonist, PMX205 (An et al., 2018).
Furthermore, the neuronal damage was prevented by the C5aR1 antagonist PMX53, a near analog of PMX205. Primary neurons obtained from C5aR1 null mice showed reduced Aβ42-induced damage after being exposed to the maximum C5a dose examined. The authors speculate that the C5aR1 antagonist's positive benefits in AD mice models may be because it protects neurons from C5a's harmful effects. (Hernandez et al., 2017).
The beneficial effects of PMX205 and PMX53 on the loss of neurons have been also demonstrated in other neurodegenerative diseases and culture models (Tenner, 2020) (Hernandez et al., 2017). In hSOD1G93A mice, a model of amyotrophic lateral sclerosis (ALS), Lee et al. (2017) used PMX205 as a C5aR antagonist (J. D. Lee et al., 2017). According to the study, oral administration of PMX205 increased the animals' grip strength, retarded the progression of the disease, and increased their chances of survival by penetrating the brain at a pharmacologically effective concentration (Lee et al., 2017). In Tg2576 animals, the antagonist prevented the loss of pre-synaptic markers and decreased dystrophic neurites and Aβ levels (Gomez-Arboledas et al., 2022). Additionally, it partially restored microglial homeostatic genes and alleviated memory loss associated with AD (Gomez-Arboledas et al., 2022).
Another experiment was conducted on a transgenic AD model that contains a third "Arctic" mutation in the human APP transgene with high production of fibrillar Aβ plaques. Deletion of the C5aR1 gene rescued the loss of neuronal complexity in the CA1 region of the hippocampus and behavioral deficits compared to the C5aR1-sufficient Arctic mice at 10 months of age without any change in plaque accumulation (Hernandez et al., 2017).
Carvalho et al. (2022) demonstrated that in AD models, C5a-C5aR1 signaling mostly affects disease progression through accelerating microglial activation pathways (Carvalho et al., 2022a). Microglial activation and astrogliosis have been reduced in C5a-deficient mice (Gomez-Arboledas et al., 2022). Whereas interaction of C5a with C5aR2 demonstrates neuroprotective function, binding of C5a to C5aR1 results in disease progression. In Arctic mice with C5a overexpression, the authors showed that specific pharmacological inhibition of C5aR1 could be a potential treatment strategy for AD (Carvalho et al., 2022b). In a different study, which used the AD amyloidosis mouse model (5xFAD model), it was demonstrated that EP67, a modified C5a receptor agonist, increased clearance of both fibrillar and non-fibrillar Aβ by microglia, therefore mitigating synaptic degeneration and ameliorating cognitive impairment (Panayiotou et al., 2019).
The deletion of C5AR1 and TYROBP in the other mice with modified inflammatory processes showed a restoration of gene expression circuits and cognitive decline. However, there was no decrease in amyloid plaque deposition (Haure-Mirande et al., 2019). These findings are similar to reports of cognitively normal people with significant plaque pathologic state at autopsy (Elobeid et al., 2014), implying that it is the induced response to plaque rather than the plaques themselves that are harmful in AD. Moreover, C5aR1 deletion restricts the polarization of microglia to more harmful disease-associated inflammatory cells in animal models of AD, allowing for continuing phagocytosis and degradation with fewer inflammatory side effects (Schartz & Tenner, 2020). Overall, the data from mice and people suggest that blocking C5aR1 is protective and may reduce inflammation and improve homeostasis in AD.
Finally, recent research found that C8γ, being one of the three subunits of complement protein C8 (α, β, γ), which makes up MAC can be a promising therapeutic target for AD and other neurological conditions (Kim et al., 2021). The authors identified the inhibitory effects of C8γ on glial hyperactivation, neuroinflammation, and cognitive loss.
Nevertheless, additional investigation into the mechanisms governing complement-mediated synaptic loss is essential to identify potential biomarker candidates that aid in the early diagnosis of complement dysregulation, slow the disease's progression, and potentially enhance cognitive and memory performances for AD patients in the future.
Conclusions, Challenges in Targeting Complement and Future Perspectives
Along with neuroprotective properties, relying on the degree of activation and the initial targets, complement components and microglial cells can also be neurotoxic, causing synaptic loss, neuroinflammation, and ultimately progressive neurodegeneration. Activation of the terminal complement pathway is implicated in driving synaptic loss in Alzheimer's, making it a potential therapeutic target. Additionally, anti-inflammatory treatments may protect against Alzheimer's by mitigating beta-amyloid-mediated complement system activation. Insights from synapse pruning in normal brain development offer promising drug targets to preserve cognition in AD before plaque formation. However, the complement system is intricate, with multiple components and regulatory mechanisms. Understanding and precisely manipulating this complexity is challenging. Therefore, targeting complement to prevent synaptic loss in Alzheimer's disease faces several troubles and should be addressed in future investigations.
References
- Askew, K., Li, K., Olmos-Alonso, A., Garcia-Moreno, F., Liang, Y., Richardson, P., Tipton, T., Chapman, M. A., Riecken, K., Beccari, S., Sierra, A., Molnár, Z., Cragg, M. S., Garaschuk, O., Perry, V. H., & Gomez-Nicola, D. (2017). Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Reports, 18(2), 391–405. [CrossRef]
- Bailey, C. H., Kandel, E. R., & Harris, K. M. (2015). Structural Components of Synaptic Plasticity and Memory Consolidation. Cold Spring Harbor Perspectives in Biology, 7(7), a021758. [CrossRef]
- Bartels, T., De Schepper, S., & Hong, S. (2020). Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science (New York, N.Y.), 370(6512), 66–69. [CrossRef]
- Batista, A. F., Khan, K. A., Papavergi, M.-T., & Lemere, C. A. (2024). The Importance of Complement-Mediated Immune Signaling in Alzheimer’s Disease Pathogenesis. International Journal of Molecular Sciences, 25(2), 817. [CrossRef]
- Bellenguez, C., Küçükali, F., Jansen, I. E., Kleineidam, L., Moreno-Grau, S., Amin, N., Naj, A. C., Campos-Martin, R., Grenier-Boley, B., Andrade, V., Holmans, P. A., Boland, A., Damotte, V., van der Lee, S. J., Costa, M. R., Kuulasmaa, T., Yang, Q., de Rojas, I., Bis, J. C., … Lambert, J.-C. (2022). New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nature Genetics, 54(4), 412–436. [CrossRef]
- Benoit, M. E., Hernandez, M. X., Dinh, M. L., Benavente, F., Vasquez, O., & Tenner, A. J. (2013). C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity. The Journal of Biological Chemistry, 288(1), 654–665. [CrossRef]
- Berg, A., Zelano, J., Stephan, A., Thams, S., Barres, B. A., Pekny, M., Pekna, M., & Cullheim, S. (2012). Reduced removal of synaptic terminals from axotomized spinal motoneurons in the absence of complement C3. Experimental Neurology, 237(1), 8–17. [CrossRef]
- Bohlson, S. S., & Tenner, A. J. (2023). Complement in the Brain: Contributions to Neuroprotection, Neuronal Plasticity, and Neuroinflammation. Annual Review of Immunology, 41(1), 431–452. [CrossRef]
- Brown, A. S. (2012). Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Developmental Neurobiology, 72(10), 1272–1276. [CrossRef]
- Brucato, F. H., & Benjamin, D. E. (2020). Synaptic Pruning in Alzheimer’s Disease: Role of the Complement System. Global Journal of Medical Research, 20(6). [CrossRef]
- Bruttger, J., Karram, K., Wörtge, S., Regen, T., Marini, F., Hoppmann, N., Klein, M., Blank, T., Yona, S., Wolf, Y., Mack, M., Pinteaux, E., Müller, W., Zipp, F., Binder, H., Bopp, T., Prinz, M., Jung, S., & Waisman, A. (2015). Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity, 43(1), 92–106. [CrossRef]
- Butovsky, O., & Weiner, H. L. (2018). Microglial signatures and their role in health and disease. Nature Reviews Neuroscience, 19(10), 622–635. [CrossRef]
- Carpanini, S. M., Torvell, M., Bevan, R. J., Byrne, R. A. J., Daskoulidou, N., Saito, T., Saido, T. C., Taylor, P. R., Hughes, T. R., Zelek, W. M., & Morgan, B. P. (2022). Terminal complement pathway activation drives synaptic loss in Alzheimer’s disease models. Acta Neuropathologica Communications, 10(1), 99. [CrossRef]
- Carvalho, K., Schartz, N. D., Balderrama-Gutierrez, G., Liang, H. Y., Chu, S.-H., Selvan, P., Gomez-Arboledas, A., Petrisko, T. J., Fonseca, M. I., Mortazavi, A., & Tenner, A. J. (2022a). Modulation of C5a–C5aR1 signaling alters the dynamics of AD progression. Journal of Neuroinflammation, 19(1), 178. [CrossRef]
- Carvalho, K., Schartz, N. D., Balderrama-Gutierrez, G., Liang, H. Y., Chu, S.-H., Selvan, P., Gomez-Arboledas, A., Petrisko, T. J., Fonseca, M. I., Mortazavi, A., & Tenner, A. J. (2022b). Modulation of C5a-C5aR1 signaling alters the dynamics of AD progression. Journal of Neuroinflammation, 19(1), 178. [CrossRef]
- Coulthard, L. G., & Woodruff, T. M. (2015). Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. Journal of Immunology (Baltimore, Md.: 1950), 194(8), 3542–3548. [CrossRef]
- Czirr, E., Castello, N. A., Mosher, K. I., Castellano, J. M., Hinkson, I. V., Lucin, K. M., Baeza-Raja, B., Ryu, J. K., Li, L., Farina, S. N., Belichenko, N. P., Longo, F. M., Akassoglou, K., Britschgi, M., Cirrito, J. R., & Wyss-Coray, T. (2017). Microglial complement receptor 3 regulates brain Aβ levels through secreted proteolytic activity. Journal of Experimental Medicine, 214(4), 1081–1092. [CrossRef]
- Dejanovic, B., Huntley, M. A., De Mazière, A., Meilandt, W. J., Wu, T., Srinivasan, K., Jiang, Z., Gandham, V., Friedman, B. A., Ngu, H., Foreman, O., Carano, R. A. D., Chih, B., Klumperman, J., Bakalarski, C., Hanson, J. E., & Sheng, M. (2018). Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron, 100(6), 1322-1336.e7. [CrossRef]
- Dejanovic, B., Wu, T., Tsai, M.-C., Graykowski, D., Gandham, V. D., Rose, C. M., Bakalarski, C. E., Ngu, H., Wang, Y., Pandey, S., Rezzonico, M. G., Friedman, B. A., Edmonds, R., De Mazière, A., Rakosi-Schmidt, R., Singh, T., Klumperman, J., Foreman, O., Chang, M. C., … Hanson, J. E. (2022). Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nature Aging, 2(9), 837–850. [CrossRef]
- Doens, D., & Fernández, P. L. (2014). Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. Journal of Neuroinflammation, 11(1), 1–14. [CrossRef]
- Dudvarski Stankovic, N., Teodorczyk, M., Ploen, R., Zipp, F., & Schmidt, M. H. H. (2016). Microglia–blood vessel interactions: A double-edged sword in brain pathologies. Acta Neuropathologica, 131(3), 347–363. [CrossRef]
- El Gaamouch, F., Audrain, M., Lin, W.-J., Beckmann, N., Jiang, C., Hariharan, S., Heeger, P. S., Schadt, E. E., Gandy, S., Ehrlich, M. E., & Salton, S. R. (2020). VGF-derived peptide TLQP-21 modulates microglial function through C3aR1 signaling pathways and reduces neuropathology in 5xFAD mice. Molecular Neurodegeneration, 15(1), 4. [CrossRef]
- Elobeid, A., Rantakömi, S., Soininen, H., & Alafuzoff, I. (2014). Alzheimer’s disease-related plaques in nondemented subjects. Alzheimer’s & Dementia, 10(5), 522–529. [CrossRef]
- Fonseca, M. I., Ager, R. R., Chu, S.-H., Yazan, O., Sanderson, S. D., LaFerla, F. M., Taylor, S. M., Woodruff, T. M., & Tenner, A. J. (2009). Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. The Journal of Immunology, 183(2), 1375–1383. [CrossRef]
- Fonseca, M. I., Chu, S.-H., Hernandez, M. X., Fang, M. J., Modarresi, L., Selvan, P., MacGregor, G. R., & Tenner, A. J. (2017). Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. Journal of Neuroinflammation, 14(1), 48. [CrossRef]
- Franco-Bocanegra, D. K., McAuley, C., Nicoll, J. A. R., & Boche, D. (2019). Molecular Mechanisms of Microglial Motility: Changes in Ageing and Alzheimer’s Disease. Cells, 8(6), E639. [CrossRef]
- Fraser, D. A., Pisalyaput, K., & Tenner, A. J. (2010). C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. Journal of Neurochemistry, 112(3), 733–743. [CrossRef]
- Frost, J. L., & Schafer, D. P. (2016). Microglia: Architects of the Developing Nervous System. Trends in Cell Biology, 26(8), 587–597. [CrossRef]
- Gasque, P., Chan, P., Mauger, C., Schouft, M. T., Singhrao, S., Dierich, M. P., Morgan, B. P., & Fontaine, M. (1996). Identification and characterization of complement C3 receptors on human astrocytes. Journal of Immunology (Baltimore, Md.: 1950), 156(6), 2247–2255.
- Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., Mehler, M. F., Conway, S. J., Ng, L. G., Stanley, E. R., Samokhvalov, I. M., & Merad, M. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (New York, N.Y.), 330(6005), 841–845. [CrossRef]
- Goetzl, E. J., Schwartz, J. B., Abner, E. L., Jicha, G. A., & Kapogiannis, D. (2018). High complement levels in astrocyte-derived exosomes of Alzheimer disease: ADE Complement Proteins in AD. Annals of Neurology, 83(3), 544–552. [CrossRef]
- Gomez Perdiguero, E., Klapproth, K., Schulz, C., Busch, K., Azzoni, E., Crozet, L., Garner, H., Trouillet, C., de Bruijn, M. F., Geissmann, F., & Rodewald, H.-R. (2015). Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature, 518(7540), 547–551. [CrossRef]
- Gomez-Arboledas, A., Carvalho, K., Balderrama-Gutierrez, G., Chu, S.-H., Liang, H. Y., Schartz, N. D., Selvan, P., Petrisko, T. J., Pan, M. A., Mortazavi, A., & Tenner, A. J. (2022). C5aR1 antagonism alters microglial polarization and mitigates disease progression in a mouse model of Alzheimer’s disease. Acta Neuropathologica Communications, 10(1), 116. [CrossRef]
- Hansen, D. V., Hanson, J. E., & Sheng, M. (2018a). Microglia in Alzheimer’s disease. The Journal of Cell Biology, 217(2), 459–472. [CrossRef]
- Hansen, D. V., Hanson, J. E., & Sheng, M. (2018b). Microglia in Alzheimer’s disease. Journal of Cell Biology, 217(2), 459–472. [CrossRef]
- Hanslik, K. L., & Ulland, T. K. (2020). The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Frontiers in Neurology, 11, 570711. [CrossRef]
- Haure-Mirande, J.-V., Wang, M., Audrain, M., Fanutza, T., Kim, S. H., Heja, S., Readhead, B., Dudley, J. T., Blitzer, R. D., Schadt, E. E., Zhang, B., Gandy, S., & Ehrlich, M. E. (2019). Correction: Integrative approach to sporadic Alzheimer’s disease: deficiency of TYROBP in cerebral Aβ amyloidosis mouse normalizes clinical phenotype and complement subnetwork molecular pathology without reducing Aβ burden. Molecular Psychiatry, 24(3), 472. [CrossRef]
- Hemonnot, A.-L., Hua, J., Ulmann, L., & Hirbec, H. (2019). Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Frontiers in Aging Neuroscience, 11, 233. [CrossRef]
- Heneka, M. T., Carson, M. J., Khoury, J. E., Landreth, G. E., Brosseron, F., Feinstein, D. L., Jacobs, A. H., Wyss-Coray, T., Vitorica, J., Ransohoff, R. M., Herrup, K., Frautschy, S. A., Finsen, B., Brown, G. C., Verkhratsky, A., Yamanaka, K., Koistinaho, J., Latz, E., Halle, A., … Kummer, M. P. (2015). Neuroinflammation in Alzheimer’s disease. The Lancet Neurology, 14(4), 388–405. [CrossRef]
- Hernandez, M. X., Namiranian, P., Nguyen, E., Fonseca, M. I., & Tenner, A. J. (2017). C5a increases the injury to primary neurons elicited by fibrillar amyloid beta. ASN Neuro, 9(1), 1759091416687871. [CrossRef]
- Holtman, I. R., Raj, D. D., Miller, J. A., Schaafsma, W., Yin, Z., Brouwer, N., Wes, P. D., Möller, T., Orre, M., Kamphuis, W., Hol, E. M., Boddeke, E. W. G. M., & Eggen, B. J. L. (2015). Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathologica Communications, 3, 31. [CrossRef]
- Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., Merry, K. M., Shi, Q., Rosenthal, A., Barres, B. A., Lemere, C. A., Selkoe, D. J., & Stevens, B. (2016a). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (New York, N.Y.), 352(6286), 712–716. [CrossRef]
- Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., Merry, K. M., Shi, Q., Rosenthal, A., Barres, B. A., Lemere, C. A., Selkoe, D. J., & Stevens, B. (2016b). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (New York, N.Y.), 352(6286), 712–716. [CrossRef]
- Jack, C. R., Knopman, D. S., Jagust, W. J., Petersen, R. C., Weiner, M. W., Aisen, P. S., Shaw, L. M., Vemuri, P., Wiste, H. J., Weigand, S. D., Lesnick, T. G., Pankratz, V. S., Donohue, M. C., & Trojanowski, J. Q. (2013). Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. The Lancet. Neurology, 12(2), 207–216. [CrossRef]
- Jaffry, M., Department of Neurology, Rutgers New Jersey Medical School, Newark, NJ, USA, Faiz, I., Department of Neurology, Rutgers New Jersey Medical School, Newark, NJ, USA, Jaffry, K., Department of Neurology, Rutgers New Jersey Medical School, Newark, NJ, USA, Souayah, N., & Department of Neurology, Rutgers New Jersey Medical School, Newark, NJ, USA. (2022). Neurological Manifestations of SARS-CoV-2 Infection and the Role of Complement Activation. US Neurology, 18(2), 86. [CrossRef]
- Kettenmann, H., Kirchhoff, F., & Verkhratsky, A. (2013). Microglia: New Roles for the Synaptic Stripper. Neuron, 77(1), 10–18. [CrossRef]
- Kim, J.-H., Afridi, R., Han, J., Jung, H.-G., Kim, S.-C., Hwang, E. M., Shim, H. S., Ryu, H., Choe, Y., Hoe, H.-S., & Suk, K. (2021). Gamma subunit of complement component 8 is a neuroinflammation inhibitor. Brain, 144(2), 528–552. [CrossRef]
- Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., & Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s & Dementia (New York, N. Y.), 4, 575–590. [CrossRef]
- Kretzschmar, G. C., Bumiller-Bini, V., Gasparetto Filho, M. A., Zonta, Y. R., Yu, K. S. T., de Souza, R. L. R., Dias-Melicio, L. A., & Boldt, A. B. W. (2021). Neutrophil Extracellular Traps: A Perspective of Neuroinflammation and Complement Activation in Alzheimer’s Disease. Frontiers in Molecular Biosciences, 8, 630869. [CrossRef]
- Lambert, J.-C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., Combarros, O., Zelenika, D., Bullido, M. J., & Tavernier, B. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genetics, 41(10), 1094–1099. [CrossRef]
- Landlinger, C., Oberleitner, L., Gruber, P., Noiges, B., Yatsyk, K., Santic, R., Mandler, M., & Staffler, G. (2015). Active immunization against complement factor C5a: A new therapeutic approach for Alzheimer’s disease. Journal of Neuroinflammation, 12(1), 1–13. [CrossRef]
- Lee, J. D., Coulthard, L. G., & Woodruff, T. M. (2019). Complement dysregulation in the central nervous system during development and disease. Seminars in Immunology, 45, 101340. [CrossRef]
- Lee, J. D., Kumar, V., Fung, J. N., Ruitenberg, M. J., Noakes, P. G., & Woodruff, T. M. (2017). Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. British Journal of Pharmacology, 174(8), 689–699. [CrossRef]
- Lee, M. H., Perl, D. P., Steiner, J., Pasternack, N., Li, W., Maric, D., Safavi, F., Horkayne-Szakaly, I., Jones, R., Stram, M. N., Moncur, J. T., Hefti, M., Folkerth, R. D., & Nath, A. (2022). Neurovascular injury with complement activation and inflammation in COVID-19. Brain: A Journal of Neurology, 145(7), 2555–2568. [CrossRef]
- Li, M., Ma, Y.-H., Guo, Y., Liu, J.-Y., Tan, L., & Alzheimer’s Disease Neuroimaging Initiative. (2024). Associations of cerebrospinal fluid complement proteins with Alzheimer’s pathology, cognition, and brain structure in non-dementia elderly. Alzheimer’s Research & Therapy, 16(1), 12. [CrossRef]
- Li, Z., Wu, H., Luo, Y., & Tan, X. (2023). Correlation of serum complement factor 5a level with inflammatory response and cognitive function in patients with Alzheimer’s disease of different severity. BMC Neurology, 23(1), 319. [CrossRef]
- Lian, H., Litvinchuk, A., Chiang, A. C.-A., Aithmitti, N., Jankowsky, J. L., & Zheng, H. (2016). Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. The Journal of Neuroscience, 36(2), 577–589. [CrossRef]
- Lian, H., Yang, L., Cole, A., Sun, L., Chiang, A. C.-A., Fowler, S. W., Shim, D. J., Rodriguez-Rivera, J., Taglialatela, G., Jankowsky, J. L., Lu, H.-C., & Zheng, H. (2015). NFκB-Activated Astroglial Release of Complement C3 Compromises Neuronal Morphology and Function Associated with Alzheimer’s Disease. Neuron, 85(1), 101–115. [CrossRef]
- Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., Bennett, M. L., Münch, A. E., Chung, W.-S., Peterson, T. C., Wilton, D. K., Frouin, A., Napier, B. A., Panicker, N., Kumar, M., Buckwalter, M. S., Rowitch, D. H., Dawson, V. L., Dawson, T. M., … Barres, B. A. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541(7638), 481–487. [CrossRef]
- Litvinchuk, A., Wan, Y.-W., Swartzlander, D. B., Chen, F., Cole, A., Propson, N. E., Wang, Q., Zhang, B., Liu, Z., & Zheng, H. (2018). Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron, 100(6), 1337-1353.e5. [CrossRef]
- Mahajan, S. D., Tutino, V. M., Redae, Y., Meng, H., Siddiqui, A., Woodruff, T. M., Jarvis, J. N., Hennon, T., Schwartz, S., Quigg, R. J., & Alexander, J. J. (2016). C5a induces caspase-dependent apoptosis in brain vascular endothelial cells in experimental lupus. Immunology, 148(4), 407–419. [CrossRef]
- Maier, M., Peng, Y., Jiang, L., Seabrook, T. J., Carroll, M. C., & Lemere, C. A. (2008). Complement C3 deficiency leads to accelerated amyloid β plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. Journal of Neuroscience, 28(25), 6333–6341.
- Mathieu, M.-C., Sawyer, N., Greig, G. M., Hamel, M., Kargman, S., Ducharme, Y., Lau, C. K., Friesen, R. W., O’Neill, G. P., Gervais, F. G., & Therien, A. G. (2005). The C3a receptor antagonist SB 290157 has agonist activity. Immunology Letters, 100(2), 139–145. [CrossRef]
- Matsuo, K., Shindo, A., Niwa, A., Tabei, K.-I., Akatsu, H., Hashizume, Y., Akiyama, H., Ayaki, T., Maki, T., Sawamoto, N., Takahashi, R., Oikawa, S., & Tomimoto, H. (2017). Complement Activation in Capillary Cerebral Amyloid Angiopathy. Dementia and Geriatric Cognitive Disorders, 44(5–6), 343–353. [CrossRef]
- Mattson, M. P. (2007). Calcium and neurodegeneration. Aging Cell, 6(3), 337–350. [CrossRef]
- Morgan, B. P. (2018). Complement in the pathogenesis of Alzheimer’s disease. Seminars in Immunopathology, 40(1), 113–124. [CrossRef]
- Nonaka, M., & Kimura, A. (2006). Genomic view of the evolution of the complement system. Immunogenetics, 58(9), 701–713. [CrossRef]
- Panayiotou, E., Fella, E., Andreou, S., Papacharalambous, R., Gerasimou, P., Costeas, P., Angeli, S., Kousiappa, I., Papacostas, S., & Kyriakides, T. (2019). C5aR agonist enhances phagocytosis of fibrillar and non-fibrillar Aβ amyloid and preserves memory in a mouse model of familial Alzheimer’s disease. PLOS ONE, 14(12), e0225417. [CrossRef]
- Perry, V. H., & Holmes, C. (2014). Microglial priming in neurodegenerative disease. Nature Reviews. Neurology, 10(4), 217–224. [CrossRef]
- Reichwald, J., Danner, S., Wiederhold, K.-H., & Staufenbiel, M. (2009). Expression of complement system components during aging and amyloid deposition in APP transgenic mice. Journal of Neuroinflammation, 6(1), 35. [CrossRef]
- Ricklin, D., Reis, E. S., & Lambris, J. D. (2016). Complement in disease: A defence system turning offensive. Nature Reviews Nephrology, 12(7), 383–401. [CrossRef]
- Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., Ransohoff, R. M., Greenberg, M. E., Barres, B. A., & Stevens, B. (2012). Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron, 74(4), 691–705. [CrossRef]
- Schartz, N. D., & Tenner, A. J. (2020). The good, the bad, and the opportunities of the complement system in neurodegenerative disease. Journal of Neuroinflammation, 17(1), 354. [CrossRef]
- Schjeide, B.-M. M., Schnack, C., Lambert, J.-C., Lill, C. M., Kirchheiner, J., Tumani, H., Otto, M., Tanzi, R. E., Lehrach, H., Amouyel, P., Von Arnim, C. A. F., & Bertram, L. (2011). The Role of Clusterin, Complement Receptor 1, and Phosphatidylinositol Binding Clathrin Assembly Protein in Alzheimer Disease Risk and Cerebrospinal Fluid Biomarker Levels. Archives of General Psychiatry, 68(2), 207. [CrossRef]
- Sekar, A., Bialas, A. R., de Rivera, H., Davis, A., Hammond, T. R., Kamitaki, N., Tooley, K., Presumey, J., Baum, M., Van Doren, V., Genovese, G., Rose, S. A., Handsaker, R. E., Schizophrenia Working Group of the Psychiatric Genomics Consortium, Daly, M. J., Carroll, M. C., Stevens, B., & McCarroll, S. A. (2022). Author Correction: Schizophrenia risk from complex variation of complement component 4. Nature, 601(7892), E4–E5. [CrossRef]
- Shah, A., Kishore, U., & Shastri, A. (2021). Complement System in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(24), 13647. [CrossRef]
- Shen, Y., Lue, L.-F., Yang, L.-B., Roher, A., Kuo, Y.-M., Strohmeyer, R., Goux, W. J., Lee, V., Johnson, G. V., & Webster, S. D. (2001). Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neuroscience Letters, 305(3), 165–168.
- Shi, Q., Chowdhury, S., Ma, R., Le, K. X., Hong, S., Caldarone, B. J., Stevens, B., & Lemere, C. A. (2017a). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Science Translational Medicine, 9(392), eaaf6295. [CrossRef]
- Shi, Q., Chowdhury, S., Ma, R., Le, K. X., Hong, S., Caldarone, B. J., Stevens, B., & Lemere, C. A. (2017b). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Science Translational Medicine, 9(392), eaaf6295. [CrossRef]
- Shi, Q., Colodner, K. J., Matousek, S. B., Merry, K., Hong, S., Kenison, J. E., Frost, J. L., Le, K. X., Li, S., Dodart, J.-C., Caldarone, B. J., Stevens, B., & Lemere, C. A. (2015). Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 35(38), 13029–13042. [CrossRef]
- Shinjyo, N., Kagaya, W., & Pekna, M. (2021). Interaction Between the Complement System and Infectious Agents – A Potential Mechanistic Link to Neurodegeneration and Dementia. Frontiers in Cellular Neuroscience, 15, 710390. [CrossRef]
- Simon, E., Obst, J., & Gomez-Nicola, D. (2019). The Evolving Dialogue of Microglia and Neurons in Alzheimer’s Disease: Microglia as Necessary Transducers of Pathology. Neuroscience, 405, 24–34. [CrossRef]
- Sweeney, M. D., Sagare, A. P., & Zlokovic, B. V. (2018). Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nature Reviews Neurology, 14(3), 133–150. [CrossRef]
- Taddei, R. N., Perbet, R., Mate de Gerando, A., Wiedmer, A. E., Sanchez-Mico, M., Connors Stewart, T., Gaona, A., Melloni, A., Amaral, A. C., Duff, K., Frosch, M. P., & Gómez-Isla, T. (2023). Tau Oligomer-Containing Synapse Elimination by Microglia and Astrocytes in Alzheimer Disease. JAMA Neurology, 80(11), 1209–1221. [CrossRef]
- Tenner, A. J. (2020). Complement-Mediated Events in Alzheimer’s Disease: Mechanisms and Potential Therapeutic Targets. The Journal of Immunology, 204(2), 306–315. [CrossRef]
- Thambisetty, M., An, Y., Nalls, M., Sojkova, J., Swaminathan, S., Zhou, Y., Singleton, A. B., Wong, D. F., Ferrucci, L., Saykin, A. J., Resnick, S. M., & Baltimore Longitudinal Study of Aging and the Alzheimer’s Disease Neuroimaging Initiative. (2013). Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biological Psychiatry, 73(5), 422–428. [CrossRef]
- Toledo, J. B., Korff, A., Shaw, L. M., Trojanowski, J. Q., Zhang, J., & the Alzheimer’s Disease Neuroimaging Initiative. (2014). Low levels of cerebrospinal fluid complement 3 and factor H predict faster cognitive decline in mild cognitive impairment. Alzheimer’s Research & Therapy, 6(3), 36. [CrossRef]
- Tzioras, M., Daniels, M. J. D., Davies, C., Baxter, P., King, D., McKay, S., Varga, B., Popovic, K., Hernandez, M., Stevenson, A. J., Barrington, J., Drinkwater, E., Borella, J., Holloway, R. K., Tulloch, J., Moss, J., Latta, C., Kandasamy, J., Sokol, D., … Spires-Jones, T. L. (2023). Human astrocytes and microglia show augmented ingestion of synapses in Alzheimer’s disease via MFG-E8. Cell Reports. Medicine, 4(9), 101175. [CrossRef]
- Venegas, C., Kumar, S., Franklin, B. S., Dierkes, T., Brinkschulte, R., Tejera, D., Vieira-Saecker, A., Schwartz, S., Santarelli, F., Kummer, M. P., Griep, A., Gelpi, E., Beilharz, M., Riedel, D., Golenbock, D. T., Geyer, M., Walter, J., Latz, E., & Heneka, M. T. (2017). Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature, 552(7685), 355–361. [CrossRef]
- Wang, C., Yue, H., Hu, Z., Shen, Y., Ma, J., Li, J., Wang, X.-D., Wang, L., Sun, B., Shi, P., Wang, L., & Gu, Y. (2020). Microglia mediate forgetting via complement-dependent synaptic elimination. Science, 367(6478), 688–694. [CrossRef]
- Woodruff, T. M., & Tenner, A. J. (2015). A Commentary On: “NFκB-Activated Astroglial Release of Complement C3 Compromises Neuronal Morphology and Function Associated with Alzheimer’s Disease”. A cautionary note regarding C3aR. Frontiers in Immunology, 6, 220. [CrossRef]
- Wu, T., Dejanovic, B., Gandham, V. D., Gogineni, A., Edmonds, R., Schauer, S., Srinivasan, K., Huntley, M. A., Wang, Y., Wang, T.-M., Hedehus, M., Barck, K. H., Stark, M., Ngu, H., Foreman, O., Meilandt, W. J., Elstrott, J., Chang, M. C., Hansen, D. V., … Hanson, J. E. (2019). Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Reports, 28(8), 2111-2123.e6. [CrossRef]
- Wyss-Coray, T., & Rogers, J. (2012). Inflammation in Alzheimer Disease—A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harbor Perspectives in Medicine, 2(1), a006346–a006346. [CrossRef]
- Xie, J., Cools, L., Van Imschoot, G., Van Wonterghem, E., Pauwels, M. J., Vlaeminck, I., De Witte, C., El Andaloussi, S., Wierda, K., De Groef, L., Haesebrouck, F., Van Hoecke, L., & Vandenbroucke, R. E. (2023). Helicobacter pylori-derived outer membrane vesicles contribute to Alzheimer’s disease pathogenesis via C3-C3aR signalling. Journal of Extracellular Vesicles, 12(2), e12306. [CrossRef]
- Yanamadala, V., & Friedlander, R. M. (2010). Complement in neuroprotection and neurodegeneration. Trends in Molecular Medicine, 16(2), 69–76. [CrossRef]
- Yang, J., Wise, L., & Fukuchi, K. (2020). TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Frontiers in Immunology, 11, 724. [CrossRef]
- Yang, L. B., Li, R., Meri, S., Rogers, J., & Shen, Y. (2000). Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer’s disease. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 20(20), 7505–7509. [CrossRef]
- Yilmaz, M., Yalcin, E., Presumey, J., Aw, E., Ma, M., Whelan, C. W., Stevens, B., McCarroll, S. A., & Carroll, M. C. (2021). Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nature Neuroscience, 24(2), 214–224. [CrossRef]
- Zhu, X., Dai, W., & Ma, T. (2020). Impacts of CR1 genetic variants on cerebrospinal fluid and neuroimaging biomarkers in alzheimer’s disease. BMC Medical Genetics, 21(1), 181. [CrossRef]
Figure 1.
Complement-mediated synapse elimination. The initial step in synapse pruning involves the production of complement component C1q by glial cells. This is followed by cleavage of C4 and C2, activation of C3 convertase, and production of C3a and C3b. Then the opsonin iC3b, which formed from C3b is recognized by the CR3 receptor on microglia. Subsequent microglial activation leads to synapse phagocytosis.
Figure 1.
Complement-mediated synapse elimination. The initial step in synapse pruning involves the production of complement component C1q by glial cells. This is followed by cleavage of C4 and C2, activation of C3 convertase, and production of C3a and C3b. Then the opsonin iC3b, which formed from C3b is recognized by the CR3 receptor on microglia. Subsequent microglial activation leads to synapse phagocytosis.
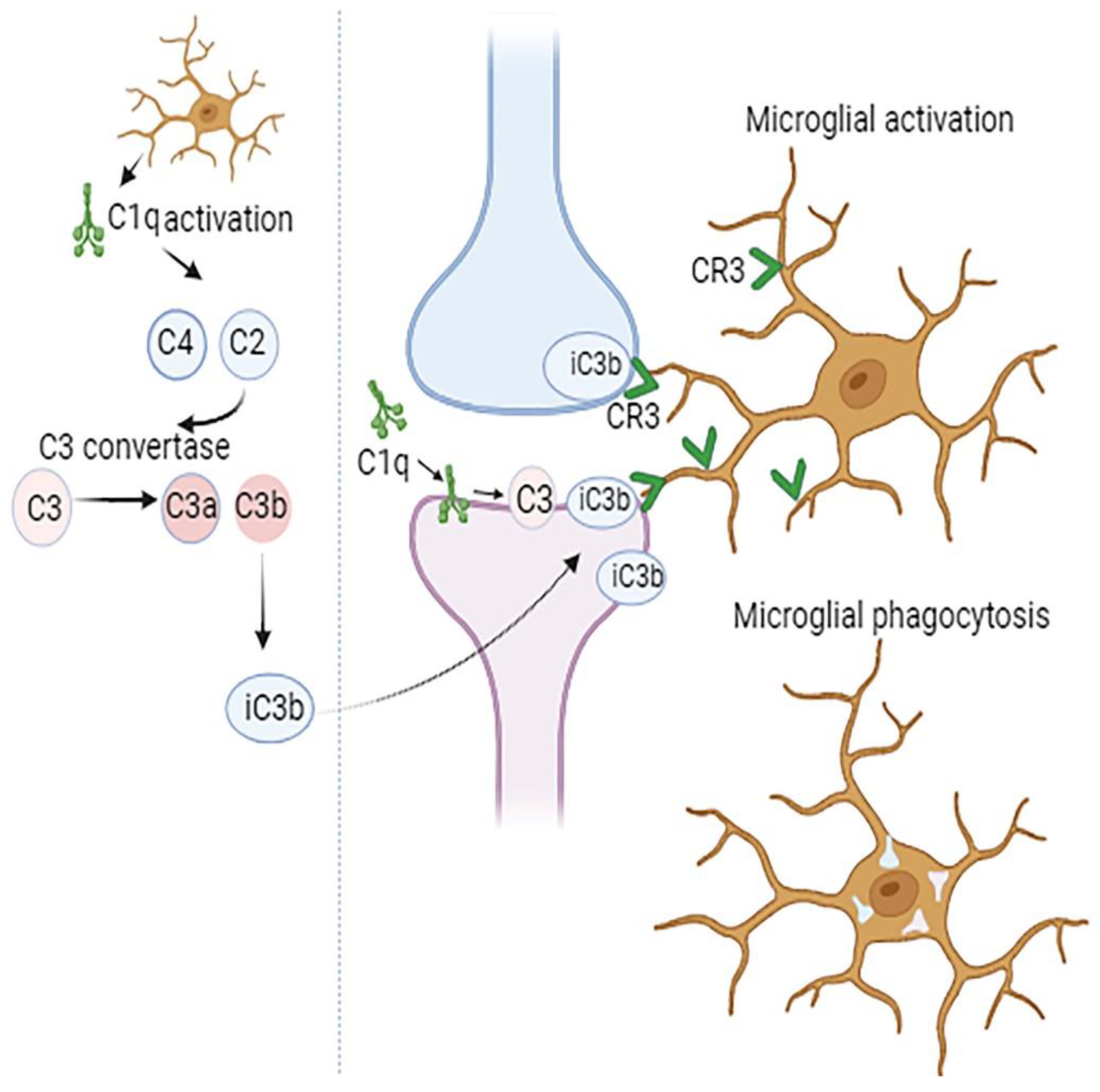
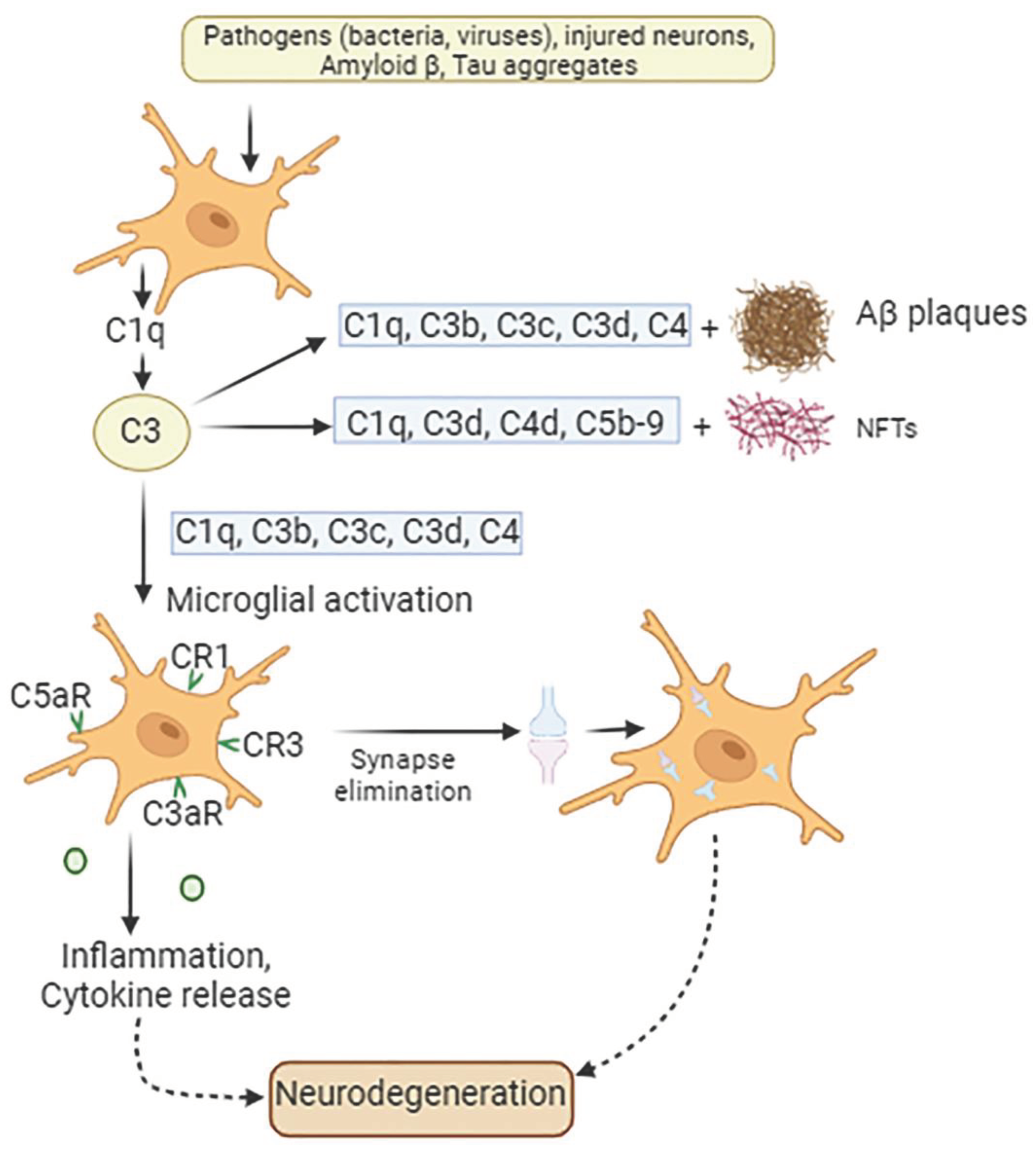
Figure 2.
Involvement of complement proteins in AD pathogenesis. In the presence of pathogens such as bacteria or viruses in the CNS, signals released by injured tissue, apoptotic neurons, Aβ protein, and tau aggregates microglia produce C1q along with other molecules. This is followed by the release from astrocytes of C3 complement protein, which co-localizes with NFTs, weakened synapses, and amyloid plaques. Complement components C1q, C3b, C3c, and C3d accumulate on amyloid plaques and dystrophic neurites, while complement proteins C1q, C3d, C4d, and C5b-9 deposit on neurofibrillary tangles (NFTs). Both amyloid plaques and NFT initiate microglial activation. Additionally, microglia possess complement receptors CR1, CR3, C3aR, and C5aR1, which allow these cells to bind various complement opsonin’s, such as C1q, C3b, iC3b, C3d, C4b, C3a and C5a. These interactions result in microglial activation, followed by synaptic elimination, chronic inflammation, and exacerbation of neurodegeneration.
Figure 2.
Involvement of complement proteins in AD pathogenesis. In the presence of pathogens such as bacteria or viruses in the CNS, signals released by injured tissue, apoptotic neurons, Aβ protein, and tau aggregates microglia produce C1q along with other molecules. This is followed by the release from astrocytes of C3 complement protein, which co-localizes with NFTs, weakened synapses, and amyloid plaques. Complement components C1q, C3b, C3c, and C3d accumulate on amyloid plaques and dystrophic neurites, while complement proteins C1q, C3d, C4d, and C5b-9 deposit on neurofibrillary tangles (NFTs). Both amyloid plaques and NFT initiate microglial activation. Additionally, microglia possess complement receptors CR1, CR3, C3aR, and C5aR1, which allow these cells to bind various complement opsonin’s, such as C1q, C3b, iC3b, C3d, C4b, C3a and C5a. These interactions result in microglial activation, followed by synaptic elimination, chronic inflammation, and exacerbation of neurodegeneration.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



