
Submitted:
14 March 2024
Posted:
18 March 2024
You are already at the latest version
Abstract
Background. Gene rearrangements affecting KMT2A are frequent in acute myeloid leukemia (AML) and are often associated with a poor prognosis. KMT2A gene fusions are often detected by chromosome banding analysis and confirmed by fluorescence in-situ hybridization. However, small intragenic insertions, termed KMT2A partial tandem duplication (PTD), are particularly challenging to detect using standard molecular and cytogenetic approaches. Methods. We have validated the use of a custom hybrid capture based next-generation sequencing (NGS) panel for comprehensive profiling of AML patients seen at our institution. This NGS panel targets the entire consensus coding DNA sequence of KMT2A; to deduce the presence of a PTD we used the relative ratio of KMT2A exons coverage. We sought to corroborate the KMT2A-PTD NGS results using (1) multiplex-ligation probe amplification (MLPA) and (2) optical genome mapping (OGM). Results. We analyzed 932 AML cases and identified 41 individuals harboring a KMT2A-PTD. MLPA, NGS and OGM confirmed the presence of a KMT2A-PTD in 22 of the cases analyzed where orthogonal testing was possible. Two false positive PTD calls by NGS could be explained by the presence of cryptic structural variants impacting KMT2A and interfering with PTD analysis. OGM revealed the nature of these previously undetected gene rearrangements in KMT2A, while MLPA yielded in-conclusive results. MLPA analysis for KMT2A-PTD is limited to exon 4, whereas NGS and OGM resolved KMT2A-PTD sizes and copy number levels. Conclusion. KMT2A-PTDs are complex gene rearrangements that cannot be fully ascertained using a single genomic platform. MLPA, NGS panels and OGM are complementary technologies applied in standard of care testing for AML patients. MLPA and NGS panels are designed for targeted copy number analysis; however, our results show that integration of concurrent genomic alterations is needed for accurate KMT2A-PTD identification. Unbalanced chromosomal rearrangements overlapping with KMT2A can interfere with the diagnostic sensitivity and specificity of copy number based KMT2A-PTD detection methodologies.
Keywords:
acute leukemia
; KMT2A patial tandem duplication (KMT2A-PTD)
; structural variation
; optical genome mapping
; next generation sequecing
; multiplex-ligation probe amplication
; OGM
; NGS
; MLPA
1. Introduction
Structural variants (SVs) affecting the Lysine (K)-Specific Methyltransferase 2A (KMT2A) gene, formerly known as MLL (myeloid/lymphoid or mixed-lineage leukemia) on chromosome band 11q23.3 are recurrently encountered in acute myeloid leukemia (AML) and are often indicative of early relapse and an overall poor prognosis[1,2,3,4,5,6]. These KMT2A gene rearrangements can occur in the context of fusion with other gene partners or result from partial tandem duplication within KMT2A (i.e., KMT2A-PTD). KMT2A fusions have been reported in 3% of primary pediatric and adult leukemia as well as 10% of secondary leukemia occurring following treatment with DNA topoisomerase II inhibitors [7,8,9,10,11,12,13]. KMT2A-PTDs are identified in 3-10% of AML cases, particularly in up to 25% of patients with a concurrent trisomy of chromosome 11[1,4].
KMT2A-PTDs are small intragenic in-frame duplications within the N-terminal end of KMT2A. The breakpoints of these cryptic SVs often occur in flanking intronic sequences of exons 2 to 10 and are mediated by Alu elements [14,15,16,17,18]. Typically, KMT2A-PTDs, such as the recurrent duplication spanning exons 1 to 10 of KMT2A, occur below the limit of detection of classical cytogenetics karyotyping or FISH techniques. KMT2A-PTDs were traditionally investigated by classical molecular approaches such as reverse transcriptase PCR (RT-PCR) and Southern blot [6,19,20,21,22,23,24]. However, next-generation sequencing (NGS) panels have become part of the current standard of care testing for AML, as they can enable comprehensive molecular profiling and risk prognostication for sequence variants in addition to identifying KMT2A-PTDs [16,25,26,27,28,29,30].
Identification of KMT2A-PTDs is crucial to the clinical management of AML patients; however, the detection of the PTD is not straightforward. First, these cryptic SVs occur below the resolution of G-banding and FISH [24,31,32]. Second, these intragenic duplications exceed the amplification capacity of traditional PCR strategies. For comparison, the canonical KMT2A exons 1 to 10 PTDs are hundreds of times larger than FLT3-ITD (i.e., internal tandem duplications in the FLT3 gene), which are notoriously difficult to amplify using amplicon based NGS. Third, analyzing the PTD by NGS requires uniform KMT2A sequence coverage, which is difficult to achieve using amplicon-based NGS assays. Lastly, it is challenging to detect SVs such as KMT2A-PTD from amplicon or hybrid capture based on short reads NGS. Here, we present our experience with KMT2A-PTD detection in 932 adult AML patients using a custom hybrid-capture NGS panel. To validate the use of NGS data for KMT2A-PTD calling, we confirmed the presence of KMT2A-PTD using two orthogonal methodologies for the detection of copy number alterations: multiplex-ligation probe amplification (MLPA) and optical genome mapping (OGM).
2. Materials and Methods
Study cohort. This consisted of 932 unselected AML patients diagnosed at the Princess Margaret Cancer Centre, Toronto, Canada. DNA was extracted from peripheral blood (PB) or bone marrow (BM) samples of AML cases studied herein. The study was approved by the University Health Network Research Ethics Board.
Conventional cytogenetics. G-banding analyses were conducted on all cases with analyzable metaphases. Where there was a suspicion of an 11q23.3 chromosomal rearrangement, KMT2A break-apart FISH (Abbott Molecular, Intermedico, Markham, Canada) was conducted to investigate the presence of KMT2A rearrangements.
DNA target enrichment and sequencing. NGS was conducted using a custom hybrid-capture NGS panel (heme-NGS) with probes from OGT (Oxford Gene Technology, United Kingdom) targeting clinically relevant myeloid genes regions such as the entire consensus DNA sequence (CCDS) of KMT2A. Data analysis used a custom bioinformatics analysis pipeline following GATK best practices for data pre-processing, where reads were aligned to GRCh37/hg19 human genome reference, Burrows-Wheeler Aligner v 0.7.12; marking duplicating reads, Picard v1.130; and correcting base quality scores, GATK v3.3.0 Base Quality Score Recalibration algorithm. Variant calling was performed using Varscan v2.3.8 and mean depth of coverage of KMT2A exon interval calculated using Picard.
KMT2A-PTD detection by NGS. Only samples with depth of KMT2A exon coverage >100x were considered for PTD analysis. For PTD detection, we utilized the ratio of the mean depth of coverage for each of the PTD specific KMT2A exons in N-ter (i.e., exons 1 to 10) relative to reference KMT2A exons in C-ter (i.e., exons 27 and 36). Using both exons 27 and 36 safeguards against intragenic structural rearrangements in one or the other exon (Figure 1).
KMT2A-PTD detection by MLPA. Where adequate sample was available, suspicious KMT2A-PTDs identified by NGS were confirmed using the multiplex ligation-dependent probe amplification (MLPA) P414 probe mix from MRC-Holland (Amsterdam, The Netherlands). Here, PTD calling is derived from a PTD specific probe in KMT2A exon 4 and a reference probe in KMT2A exon 36 (Figure 1). Copy number ratios of exon 4 to exon 36 >1.3 but <1.65 are in keeping with a KMT2A-PTD.
KMT2A-PTD detection by OGM. Where adequate sample was available, the presence of a KMT2A-PTD was further investigated using optical genome mapping (OGM). Unlike NGS and MLPA, OGM is an agnostic KMT2A-PTD detection method that does not require the use of gene specific probes (Figure 1). Ultra-high molecular weight DNA was extracted from patient samples and labeled with the Bionano DLS labeling kit. Labelled molecules were run on nanochannel flow cells and imaged by Bionano Saphyr (Bionano Inc, San Diego, USA). Assembly was performed using the Rare Variant Assembly (Solve 3.7, Access 1.7) and visualized using Bionano Access. Analysis of SVs was performed as described by Levy et al., 2024[33].
3. Results
KMT2A exon z-score by NGS. The presence of a PTD was inferred from the coverage of the PTD specific KMT2A exons (i.e., exons 1 to 10) relative to a reference KMT2A exon (i.e., exons 27 or 36). The depth of exon coverage can vary based on several factors such as sample loading quantity, exon length, number of NGS probes, and GC content (Figure 2A). To account for this variability, we first sought to normalize the KMT2A exon coverage by applying heme-NGS to a baseline cohort of 100 cases with no detectable SVs on chromosome 11q23.3 by classical cytogenetics. Within the baseline cohort, we determined the relative mean depth of each PTD specific KMT2A exons to the mean depth of a reference KMT2A exon. We then derived the corresponding standard deviation and z-score of these exon ratios (Figure 2B) using the following formula:
z-score =[ (PTD exon mean depth / reference exon mean depth) – baselinemean ] / baseline ratiosd
KMT2A-PTD detection threshold by NGS. AML patients often harbor concurrent numerical and/or SVs involving chromosome 11 that can interfere with KMT2A-PTD detection. We applied heme-NGS to a test cohort of 25 samples with no evidence of PTD, but harboring numerical and/or SVs involving KMT2A. Within this test cohort, the KMT2A exon z-score were kept within 2 standard deviation (Figure 2C). We thus determined that cases with KMT2A exon z-score >2 to be suspicious for the presence of a KMT2A-PTD (Figure 2D).
KMT2A-PTD detection by NGS. Intragenic duplications can occur in any gene region. However, unlike internal duplications seen in other myeloid genes (e.g., FLT3-ITD), KMT2A-PTDs often span over multiple exons and are located in the N-ter portion from exons 1 to 10 of KMT2A (Figure 1, Figure 2 and Figure 3). KMT2A-PTDs are hereby defined as intragenic duplications involving solitary (e.g., Figure 3, case 41) or multiple (e.g., Figure 3, case 1) consecutive exons in the N-ter of KMT2A (i.e., exons 1 to 10). Suspicious KMT2A-PTD cases by NGS have a KMT2A exon z-score >2 for multiple consecutive N-ter exons (Figure 2D). KMT2A z-scores were derived from the ratio of each PTD specific exon (i.e., 1 to 10) over a reference exon (27 or 36). Determining the z-score for both exon 27 and 36 increases the likelihood of a PTD. Typically, we require that the KMT2A z-score is >2 for both exons 27 and 36. Exceptionally, samples showing KMT2A z-score <2 for only one of these reference exons were considered as PTD positive provided that one or multiple flanking exons also had a KMT2A z-score >2 for both exon 27 and exon 36 (e.g., Figure 3, cases 2 and 3). In applying this scheme to a cohort of 932 retrospective AML cases seen at our institution, we identified 41 samples suspicious for a KMT2A-PTD.
KMT2A-PTD detection by MLPA. We sought to confirm the KMT2A-PTD calls made by heme-NGS for 21 patients, for which sufficient DNA could be obtained for gene dosage by MLPA. MLPA also detected the presence of a KMT2A-PTD (i.e., KMT2A exon 4 to exon 36 ratio >1.3) in 17 cases (Figure 3). All of these cases had a duplication involving KMT2A exon 4. MLPA yielded inconclusive results in 3 individuals (cases 2, 3, and 35) showing a normal dosage (i.e., copy number ratio: 0.99- 1.14) of the PTD specific probe (exon 4), thus making a PTD of KMT2A exon 4 unlikely. Instead, cases 2, 3, and 35 showed an apparent deletion (i.e., copy number: 0.51 – 0.65) of the PTD reference probe (exon 36) suggesting a C-ter KMT2A deletion. However, heme-NGS exon coverage across the entire KMT2A coding sequence (i.e., exons 1 to 36) did not suggest a deletion involving the C-ter KMT2A exons in any of these 3 samples. In fact, KMT2A z-scores values obtained for both exons 27 and 36 (Figure 2D and Figure 3) were in keeping with a KMT2A-PTD in all cases (i.e., case 2: PTD of exons 1 to 10, case 3: PTD of exons 1 to 9, case 35: PTD of exon 2). MLPA did not detect a PTD in case 32 showing a normal dosage for the PTD specific and reference probes in KMT2A exon 4 and 36, respectively (Figure 3).
KMT2A-PTD detection by OGM. OGM was performed on 15 AML cases for which concurrent heme-NGS results were also available. OGM and heme-NGS supported the presence of a KMT2A-PTD in 11 of the samples analyzed. These findings were also validated by concordant MLPA results obtained in 6 samples (Figure 3). Discordant calls between OGM and heme-NGS were seen for cases 3 and 35 with suspicious false positive calls by heme-NGS. In case 3, OGM detected the presence of two SVs overlapping KMT2A: (1) a cryptic insertion of approximately 100 kb of 5′ KMT2A sequence into chromosome 10 within the MLLT10 gene resulting in a cryptic KMT2A::MLLT10 fusion and (2) a 0.82 Mb deletion on the presumed derivative chromosome 11 deleting both proximal (5′) and distal (3′) sequence in the KMT2A gene region (Figure 4). It should be noted that this deletion is below the resolution of conventional karyotyping and is likely on the derivative 11 as KMT2A::MLLT10 fusions are known to often come about from multiple rearrangements. In case 35, OGM detected the presence of a translocation involving chromosomes 11 and 19 juxtaposing the genes KMT2A and ELL (Figure 5). Conventional karyotyping failed for case 35; however, OGM showed a very complex karyotype with multiple copy numbers and SVs. KMT2A break-apart FISH also confirmed the presence of a KMT2A rearrangement and evidence of higher ploidy. Also seen in Figure 5 is the copy number imbalance at both the KMT2A and ELL breakpoints indicating an unbalanced translocation. The imbalance of the translocation results in loss of 3′ KMT2A sequences distal to the translocation breakpoint which explains both the false positive NGS PTD call and the inconclusive MLPA result. A discordant call between OGM and MLPA was seen for case 32, where the MLPA PTD specific probe in KMT2A exon 4 could not detect the presence of a KMT2A-PTD. Instead, both OGM and heme-NGS revealed the presence of a KMT2A-PTD in this individual suggesting a false negative result by MLPA (Figure 2D and Figure 3).
4. Discussion
Detecting SVs by NGS requires both good coverage of all targeted gene regions and uniformity of sequences covered across all areas targeted. However, these requirements are not often met by many commercial NGS heme panels. Here, we used a hybrid-capture based NGS assay to overcome the challenges with KMT2A-PTD analysis. Relying on uniform KMT2A exonic sequence coverage, we utilized the relative coverage of N-ter (i.e., exon 1 to 10) exons over C-ter exons (i.e., exons 27 or 36) of KMT2A to deduce the presence of an intragenic duplication in N-ter (Figure 2). Co-occurring SVs on chromosome 11q23.3, where KMT2A is located, may interfere with KMT2A-PTD analysis. To account for these PTD ‘look-alikes’, we established a threshold for KMT2A exon coverage (i.e., KMT2A exon z-score >2) after normalizing KMT2A exon coverage against a cohort of 100 baseline samples as well as a cohort of 25 test samples as explained above (Figure 2).
We analyzed 932 AML cases and used this PTD analysis scheme to identify 41 individuals with a KMT2A-PTD (Figure 3). These include patients with multi-exon (e.g., cases 1-32, 40) as well as single exon PTD (cases 33-39, 41). The heme-NGS findings of a KMT2A-PTD were also concordant with MLPA (N = 17/21, 81%) and OGM (N= 11/13, 85%) results, where a sample could be obtained for analysis. In total, we confirmed the heme-NGS PTD results to be true positive calls in 22 samples based on supportive MLPA and/or OGM results. Of interest, none of these 22 samples had another known SV on chromosome 11q23.3, other than the KMT2A-PTD identified. Of the 22 true positive KMT2A-PTDs detected by NGS, cases 7, 15 and 20 also had trisomy of chromosome 11 (Table 1), indicating that the heme-NGS PTD detection algorithm is able to identify intragenic KMT2A duplications in the context of chromosome 11 aneuploidy. Trisomy of chromosome 11 is seen in up to 25% of AML cases with KMT2A-PTD and can interfere with PTD analysis, given that there is presence of an intragenic gene duplication in the background of an additional copy of chromosome 11. Due to the uniform gene coverage achieved using heme-NGS, chromosome 11 aneuploidy impacts KMT2A exons coverage at similar levels across the entire coding region. As a result, the relative ratio of the KMT2A N-ter (i.e., exon 1 to 10) to C-ter (i.e., exons 27 or 36) does not vary, whether in presence of gain or loss of chromosome 11 copies. Thus, PTD detection by heme-NGS is not affected by the co-occurring aneuploidies in cases 7, 15 and 20.
Unlike aneuploidies, however, unbalanced gene rearrangements on chromosome 11q23.3 result in differences in exon coverage within the KMT2A gene with heme-NGS. When such SVs are present, detecting a KMT2A-PTD is more challenging. For instance, a C-terminal deletion combined with an N-ter with normal copy number appears to be a relative gain of the N-ter region. Calculating the KMT2A exon z-score from two reference exons in C-ter safeguards against some intragenic KMT2A C-ter deletions, but not all SVs overlapping with KMT2A. In this study, heme-NGS detected the presence of a KMT2A-PTD (Figure 2D) in case 2 (PTD exon 1 to 10), 3 (PTD exon 1 to 9) and 35 (PTD exon 2); however, concurrent MLPA and OGM results obtained were not in keeping with the presence of an intragenic KMT2A duplication. Indeed, MLPA showed a normal copy number for KMT2A exon 4 in all 3 cases (Figure 3). MLPA results instead suggested these individuals had relative copy number loss of KMT2A exon 27, suggesting that a different KMT2A SV was causing abnormal results in these 3 patients. Of note, G-banded karyotyping did not identify any abnormality on chromosome 11q23 in cases 2 and 3 and was unsuccessful for patient 35. However, OGM showed that case 3 and 35 had other (non-PTD) KMT2A rearrangements: a cryptic KMT2A::MLLT10 gene fusion and a KMT2A deletion (case 3, Figure 4); a KMT2A::ELL fusion in the context of a complex genome with 3′ KMT2A copy number loss (case 35, Figure 5). Sufficient material to conduct additional investigations was not available for case 2; however, similar to cases 3 and 35, there is also a possibility that a KMT2A SV led to a false positive PTD call in this patient.
NGS panels can combine DNA point mutations and limited analysis of SVs (including copy number analysis) on a single platform, and are therefore extremely useful for standard of care testing in AML. However, accurate assessment of SVs using NGS panels is challenging due to the targeted nature of those panels, not to mention the challenges with SV detection using short-read sequencing technologies[34]. Alternative KMT2A-PTD detection approaches should be considered in conjunction to NGS, particularly in patients with overlapping SVs on chromosome 11q23.3. Note that some KMT2A rearrangements including the KMT2A-PTD and cryptic insertions, for example, occur below the resolution of karyotyping (Table 1). Therefore, methodologies such as OGM, that detect genome-wide SVs at a higher resolution than karyotyping are more suitable for KMT2A-PTD investigations. In the absence of OGM, and since FISH is often integrated into the diagnostic workup of AML cases, reflex FISH testing (with KMT2A break-apart probe) of positive KMT2A-PTD calls made by NGS may assist in some cases to rule out potential PTD mimics. For instance, the signal pattern of the KMT2A FISH break-apart probes in case 35 is in keeping with a KMT2A rearrangement and will thus invalidate the heme-NGS PTD results of a KMT2A-PTD. Conversely, FISH did not show a KMT2A rearrangement in cases 6-8, 10, 16-18, 22, 28, 30, 34, 37 and 38; here FISH supports the KMT2A-PTD heme-NGS calls in these 14 patients (Table 1, Figure 2 and Figure 3). However, unlike OGM, cryptic gene rearrangements may not be seen by FISH and still yield false negative results.
KMT2A gene rearrangements resulting in fusions with multiple partner genes are more amenable to classical molecular and cytogenetic techniques and thus have been investigated more thoroughly. In comparison, several questions remain unanswered with respect to KMT2A-PTD, beginning with the very definition of a PTD. For instance, it is unclear whether the following should be considered as KMT2A-PTD: (1) single exon intragenic duplications in N-ter of KMT2A or, (2) intragenic duplications not involving KMT2A exons 1 to 10. Currently, it is unclear whether single exon KMT2A-PTD should be interpreted as variant of uncertain significance or clinically actionable alterations in the context of AML. KMT2A-PTD have traditionally been assayed using classical molecular genetics techniques that provide little information on the composition of these variable genetic lesions. As such, it has not been determined if single exon PTDs are also indicative of a poorer AML outcome as shown for multi-exons KMT2A-PTD.
Higher resolution technologies, such as NGS and OGM, are elucidating the variable composition in size and copy number of intragenic duplications within KMT2A. For instance, a KMT2A exon z-score >2 by heme-NGS suggests a duplication (Figure 2). However, KMT2A exon z-score values ranged from 2.02 to 8.52, indicating that more than two copies (i.e., duplication) of the “PTD region” are present in some KMT2A-PTD positive patients (Figure 3). MLPA (i.e., exon triplication ratio: 1.75-2.15) and OGM results also showed similar results. Using OGM, KMT2A-PTDs could be visualized directly. OGM provided the most accurate resolution of KMT2A-PTD composition in size and copy numbers, as shown for individuals harboring two (cases 15, 20 Figure 6A-B), three (cases 36, Figure 6C) or four (cases 9, 32 Figure 6D-E) PTDs or variable length within KMT2A. In comparison, heme-NGS did not allow a clear distinction of cases with two or more PTD copies. For instance, case 36 (2.05-2.14) has a lower KMT2A z-score than case 15 (3.03-3.09), yet OGM showed that case 36 had three PTD copies versus two PTD copies for case 15 (Figure 3). PTD copy number estimates by MLPA was not consistent, as shown for case 9 (ratio: 1.62 - quadruplication), case 15 (ratio 1.57- duplication) and case 20 (ratio of 1.34; duplication). Comparing OGM, NGS and MLPA for KMT2A-PTD copy number estimation it is important to consider the impact on cancer cell fraction on MLPA and NGS calculations. Higher cancer cell fraction (also with possible presence of LOH) may increase copy number estimate using MLPA or NGS. Conversely, since OGM uses long and intact DNA molecules, the size and label pattern for each molecule in the region can be visualized to more accurately determine the composition of the PTD (Figure 6). Recent studies suggest higher levels of PTD copy number to be positively associated with relapse and risk of disease transformation[35]. However, at this time, PTD copy number and complexity are not yet taken into consideration in the clinical management of AML patients as data on large cohorts with high resolution PTD analysis are currently unavailable.

Table 1.
Characteristics of KMT2A-PTD cases identified by the heme-NGS assay. INC. Inconclusive. N/T. Not Tested. For the OGM nomenclature, values in square brackets (e.g., variant allele frequency/VAF for structural variants and fractional copy number/fCN for copy number changes) are reported as proportion of the sample as per the ISCN 2024 recommendations [36].
Table 1.
Characteristics of KMT2A-PTD cases identified by the heme-NGS assay. INC. Inconclusive. N/T. Not Tested. For the OGM nomenclature, values in square brackets (e.g., variant allele frequency/VAF for structural variants and fractional copy number/fCN for copy number changes) are reported as proportion of the sample as per the ISCN 2024 recommendations [36].
| # | G-banding Optical Genome Mapping |
KMT2A FISH |
KMT2A-PTD | ||
|---|---|---|---|---|---|
| NGS | MLPA | OGM | |||
| 1 | N/T | N/T | Yes | N/T | N/T |
| 2 | 46,XY,-20,+21[8]/46,idem,der(3)inv(3)(p23q27)inv(3)(q?21q26.2)[12] | N/T | Yes | INC | N/T |
| 3 | 48,XY,+8,+19[20] ogm[GRCh38] (8)x3[0.96],del(11)(q23.3)(117,817,690_118,650,394)[0.99], t(10;11)(p12.31;q23.3)(21,642,030;118,493,942)MLLT10::KMT2A [0.98],(19)x3[0.66] |
N/T | Yes | INC | No |
| 4 | N/T | N/T | Yes | N/T | N/T |
| 5 | 45,XX,-7[5]/49,XX,+8,+13,+22[1]/46,XX[17] ogm[GRCh38] (8)x3[0.27],ins(11;?)(q23.3;?)(118,470,405_118,479,068)[0.90],(13)x3[0.28] |
N/T | Yes | Yes | Yes |
| 6 | 46,XY,del(11)(p15p11.2)[19]/46,XY[1] ogm[GRCh38] del(11)(p14.3p11.2)(24,233,253_45,484,059)[0.36], ins(11;?)(q23.3;?)(118,470,405_118,479,068)[0.85] |
No | Yes | Yes | Yes |
| 7 | 47,XY,del(11)(p15p11.2),+del(11)[13]/48,XY,+11,+13[6]/46,XY[2] | No | Yes | Yes | N/T |
| 8 | Inconclusive | No | Yes | N/T | N/T |
| 9 | 46,XY[20] ogm[GRCh38] ins(11;?)(q23.3;?)(118,470,405_118,479,068)[0.81] |
N/T | Yes | Yes | Yes |
| 10 | 46,XY,inv(7)(q11.2q22)?c[22] | No | Yes | Yes | N/T |
| 11 | 46,XY,del(7)(q22q32)[17]/46,XY[3] | N/T | Yes | Yes | N/T |
| 12 | 46,XY,add(7)(q32)[20] | N/T | Yes | N/T | N/T |
| 13 | 46,XY,add(2)(p13),add(14)(q24)[16]/46,idem,add(7)(q22)[4] | N/T | Yes | N/T | N/T |
| 14 | 46,XY[20] | N/T | Yes | Yes | N/T |
| 15 | 47,XY,del(9)(q13q22),+11[10] ogm[GRCh38] del(9)(q21.11q22.31)(67,717,842_92,504,226)[0.90],(11)x3[0.91], ins(11;?)(q23.3;?)(118,470,405_118,479,068)[0.84] |
N/T | Yes | Yes | Yes |
| 16 | 46,XX,del(12)(p12p13)[22] | No | Yes | Yes | N/T |
| 17 | 46,XY[20] | No | Yes | Yes | N/T |
| 18 | Inconclusive | No | Yes | Yes | N/T |
| 19 | 46,XX[20] ogm[GRCh38] ins(11;?)(q23.3;?)(118,470,405_118,479,068)[0.91] |
N/T | Yes | Yes | Yes |
| 20 | 47,XY,+11[19]/46,XY[1] ogm[GRCh38] (11)x3[0.90],dup(11)(q23.3q23.3)(118,452,293_118,479,068)[0.85] |
N/T | Yes | Yes | Yes |
| 21 | 46,XY[20] | N/T | Yes | Yes | N/T |
| 22 | Inconclusive | No | Yes | Yes | N/T |
| 23 | 46,XX[21] | N/T | Yes | Yes | N/T |
| 24 | ogm[GRCh38] del(4)(q24)(105061991_105450148)x1[0.5], ins(11;?)(q23.3;?)(118,470,405_118,479,068)[0.86] | N/T | Yes | N/T | Yes |
| 25 | N/T | N/T | Yes | N/T | N/T |
| 26 | 46,XX[21] | N/T | Yes | N/T | N/T |
| 27 | 45,XY,-7[9]/46,idem,+mar[9]/46,XY[3] ogm[GRCh38] (7)x1[0.63],ins(11;?)(q23.3;?)(118,470,405_118,479,068)[0.74] |
N/T | Yes | N/T | Yes |
| 28 | 46,XY[21] | No | Yes | N/T | N/T |
| 29 | 46,XY,+1,der(1;14)(q10;q10)[15]/46,XY[5] | N/T | Yes | Yes | N/T |
| 30 | 46,XX[20] | No | Yes | N/T | N/T |
| 31 | N/T | N/T | Yes | N/T | N/T |
| 32 | ogm[GRCh38] ins(11;?)(q23.3;?)(118,470,101_118,477,357)[0.84] | N/T | Yes | Yes | Yes |
| 33 | 46,XY[20] | N/T | Yes | N/T | N/T |
| 34 | Inconclusive | No | Yes | N/T | N/T |
| 35 | ogm[GRCh38] (3,4,7,8,11,12,15,19,20)cx, del(5)(q21.3q32)(108917351_146240776)[0.54], t(11;19)(q23.3;p13.11)(118,493,942 ;18,499,964)KMT2A::ELL[0.54] |
Yes | Yes | INC | No |
| 36 | 47,XY,+8[12]/46,XY[11] ogm[GRCh38] (8)x3[0.42],ins(11;?)(q23.3;?)(118,461,867_118,479,068)[0.62] |
N/T | Yes | N/T | Yes |
| 37 | 46,XY[21] ogm[GRCh38] ins(11;?)(q23.3;?)(118,470,405_118,479,068)[0.87] |
No | Yes | N/T | Yes |
| 38 | 46,XY[24] | No | Yes | N/T | N/T |
| 39 | 46,XX[20] | N/T | Yes | N/T | N/T |
| 40 | 46,XX,der(6)t(1;6)(q12;p23),del(12)(p11.2p13)[4]/46,XX,del(12)(p11.2p13),der(19)t(1;19)(q12;p13)[3]/46,XX[6] | N/T | Yes | N/T | N/T |
| 41 | 44,XY,der(3)add(3)(p22-24),del(5)(q13q33),-7,-8,add(11)(p15),-12, add(12)(p13),add(13)(q10),add(14)(q32),+mar[9]/46,XY[1] |
No | Yes | N/T | N/T |
KMT2A-PTD are intragenic variants that pose challenges to molecular and cytogenetic diagnostic approaches as they are difficult to detect with NGS panels due to their size but far too small to ascertain by conventional cytogenetic approaches. Here, we used MLPA, NGS and OGM to explore KMT2A-PTD detection. In our experience, none of these approaches were able to fully characterize the complex nature of KMT2A-PTDs. KMT2A SV analysis by MLPA is limited to a single PTD specific probe in KMT2A exon 4 (Figure 1). Indeed, all the KMT2A-PTD positive patients identified by MLPA had a PTD involving KMT2A exon 4. Non-canonical PTD that do not involve KMT2A exon 4 will thus fail detection by MLPA. For instance, case 32 with a PTD spanning over KMT2A exons 2 and 3 by heme-NGS (Figure 2D), was not determined to be KMT2A-PTD positive using MLPA (Figure 3). Inconclusive MLPA results such as the ones seen in cases 2, 3, and 35 can be attributed to various factors (e.g., DNA quantity or quality) that are not related to KMT2A-PTD. These inconclusive MLPA results do not provide more information on the KMT2A genotype. Thus, additional investigations are required to resolve the MLPA findings.
The heme-NGS panel covers the entire KMT2A coding region and thus allows the analysis of canonical (e.g., exon 1 to 10) as well as atypical KMT2A-PTD (e.g., single exon PTD). Here, we focused our analyses on KMT2A-PTD encompassing exons 1 to 10; however other researchers have detected larger PTD extending further downstream of KMT2A exon 10 using NGS[35]. Heme-NGS provided a better exon level resolution of KMT2A-PTD than MLPA and/or OGM. Indeed, MLPA (single probe in exon 4) and OGM (4 informative labels) only rely on a few indicators for KMT2A-PTD analysis (Figure 1). For example, OGM could not determine the exact breakpoints of the KMT2A-PTD in case 32. In fact, OGM estimated the PTD to span over KMT2A exons 3 and 4 (Figure 6E), whereas heme-NGS did not indicate a duplication of KMT2A exon 4 (Figure 2D). Absence of a PTD involving exon 4 as suggested by heme-NGS is also in keeping with the false negative KMT2A-PTD results by MLPA (Figure 3). In general, OGM could not inform on the extent of KMT2A-PTD beyond exon 5 (Figure 6), given that there are only two informative labels in KMT2A between exons 5 and 16 (Figure 1). Therefore, approximate KMT2A-PTD length is derived from the size of the duplicated segment on OGM. Overall, the estimated KMT2A-PTDs size obtained by OGM were comparable to the theoretical Hg38 values (Figure 3) and were also in agreement with the corresponding heme-NGS KMT2A-PTDs size estimate.
Heme-NGS identified 8 patients with a single exon KMT2A-PTD. Note that due to a single PTD-specific probe in KMT2A exon 4, MLPA will yield false negative results in 7 of these patients (e.g., cases 33-35: exon 2 KMT2A-PTD, cases 36-37: exon 3 KMT2A-PTD, case 39: exon 8 KMT2A-PTD, and case 41: exon 9 KMT2A-PTD. OGM was applied to cases 36 and 37 and determined that these individuals harbor multi-exons KMT2A-PTDs (i.e., case 36: exons 3 to 5 KMT2A-PTD, Figure 6C , case 37: exons 3 to 4 KMT2A-PTD) . Misidentification of multi-exons KMT2A-PTD as a single exon KMT2A-PTD may impact clinical management of AML. Currently, such single exons KMT2A-PTD will be considered of uncertain significance, whereas multi-exons KMT2A-PTD are clinically actionable and often associated with a poorer AML outcome. Note that the KMT2A z-score for exon 4 is close to the PTD threshold in case 37, thus corroborating the OGM findings of multi-exons KMT2A-PTD involving exons 3 to 4 in this patient. As seen with case 37, seemingly single exon KMT2A-PTDs by heme-NGS may be true multi-exons KMT2A-PTDs. Low cancer fraction and variable depth of KMT2A exons coverage can mislead KMT2A-PTD detection by NGS from failure to detect one or multiple KMT2A exons involved in a PTD. Indeed, similar to cases 36 and 37, lower KMT2A z-score (i.e., z-score <2) were observed for N-ter exons of several other patients (e.g., cases 2, 10 and 12) and prevented accurate KMT2A-PTD length assessment. Unlike short read NGS, OGM enables a high-resolution scan of ultra-long DNA molecules and therefore provides a more comprehensive interrogation of KMT2A. Only OGM was able to clearly distinguish true KMT2A-PTD from “PTD copy number mimics” in a single assay as evidenced by cases 3 and 35 (Figure 3, Figure 4 and Figure 5). As shown in this study, assay design can have substantial outcomes on analytical sensitivity and specificity, especially for SVs such as KMT2A-PTD greater than 1kb but smaller than cytogenetic resolution (~10 Mb).
5. Conclusions
KMT2A-PTD are intragenic gene rearrangements of clinical importance to the management of myeloid neoplasms. These alterations have been assayed using a wide range of cytogenetics and molecular approaches; yet, the biological consequences of the varied sizes and composition of KMT2A-PTDs remain poorly understood. Novel high throughput DNA sequencing and/or mapping methodologies are further elucidating the complex nature of KMT2A-PTDs. NGS panels are widely established as standard of care testing in AML. While myeloid NGS assays have mainly focused on DNA point mutations detection, using NGS data to also detect larger SVs (e.g., KMT2A-PTD detection) adds further utility in many disease contexts. Here, we utilized the relative coverage of KMT2A exons to derive the presence of a KMT2A-PTD. Although, this approach led to the successful identification of patients with KMT2A-PTD, short read NGS (or other coverage-based copy number approach) can yield false positive results (e.g., cases 2, 3 and 35) and thus warrants caution when interpreting. We have identified the presence of confounding SVs on chromosome 11q23.3 to be a key limiting factor to KMT2A-PTD detection by NGS. Additional testing (e.g., KMT2A FISH break-apart) can help distinguish true positive KMT2A-PTD calls from other KMT2A-PTD mimickers. In our experience, other technologies, such as OGM, provide better resolution and identification of SVs on chromosome 11q23.3, including the KMT2A-PTD. OGM and NGS are complementary approaches that can be used to better characterize and interpret KMT2A-PTD in a clinical setting. Several questions on the clinical consequences of the size and composition of KMT2A-PTD are left unanswered and need to be addressed on larger cohorts and likely by additional long-range sequencing characterization and with functional assays.
Author Contributions
Conceptualization, ACS. and JC.; methodology, AS, GD, OK, SS, AB, KC, GS, NB, MDM, ACS. and JC software, AS and GD; validation, AS, GD, OK, SS, AB, ACS. and JC.; formal analysis, AS, GD, OK, SS, AB, KC, GS, NB, MDM, ACS. and JC.; investigation, ACS. and JC.; resources, MDM, ACS and JC.; data curation, AS, GD, OK, SS, AB, ACS. and JC.; writing—original draft preparation, AS, GD, ACS and JC.; writing—review and editing, AS, GD, OK, SS, AB, MDM, ACS. and JC. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study was approved by the University Health Network Research Ethics Board (CAPCR# 20-6121).
Informed Consent Statement
Not applicable for quality innovation studies on retrospective cohort of patients not involving humans. A consent waiver was granted by the University Health Network Research Ethics Board.
Data Availability Statement
Data is unavailable due to privacy or ethical restrictions.
Acknowledgments
We acknowledge the contribution of patients studied herein as well as genetics staffs from the Department of Laboratory Medicine and Pathobiology, University of Toronto.
Conflicts of Interest
ACS and AB declare stock ownership in Bionano Inc. All other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Döhner, K.; Tobis, K.; Ulrich, R.; Fröhling, S.; Benner, A.; Schlenk, R.F.; Döhner, H. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2002, 20, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- Krauter, J.; Wagner, K.; Schäfer, I.; Marschalek, R.; Meyer, C.; Heil, G.; Schaich, M.; Ehninger, G.; Niederwieser, D.; Krahl, R.; et al. Prognostic factors in adult patients up to 60 years old with acute myeloid leukemia and translocations of chromosome band 11q23: individual patient data-based meta-analysis of the German Acute Myeloid Leukemia Intergroup. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2009, 27, 3000–3006. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. The New England journal of medicine 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Kinkelin, U.; Schoch, C.; Heinecke, A.; Haase, D.; Haferlach, T.; Büchner, T.; Wörmann, B.; Hiddemann, W.; Griesinger, F. Screening for MLL tandem duplication in 387 unselected patients with AML identify a prognostically unfavorable subset of AML. Leukemia 2000, 14, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Larghero, P.; Almeida Lopes, B.; Burmeister, T.; Gröger, D.; Sutton, R.; Venn, N.C.; Cazzaniga, G.; Corral Abascal, L.; Tsaur, G.; et al. The KMT2A recombinome of acute leukemias in 2023. Leukemia 2023, 37, 988–1005. [Google Scholar] [CrossRef] [PubMed]
- Bera, R.; Chiu, M.-C.; Huang, Y.-J.; Huang, G.; Lee, Y.-S.; Shih, L.-Y. DNMT3A mutants provide proliferating advantage with augmentation of self-renewal activity in the pathogenesis of AML in KMT2A-PTD-positive leukemic cells. Oncogenesis 2020, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO classification of tumours of haematopoietic and lymphoid tissues. International agency for research on cancer Lyon, 2008; Volume 2. [Google Scholar]
- Schoch, C.; Schnittger, S.; Klaus, M.; Kern, W.; Hiddemann, W.; Haferlach, T. AML with 11q23/MLL abnormalities as defined by the WHO classification: incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood 2003, 102, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Satake, N.; Maseki, N.; Nishiyama, M.; Kobayashi, H.; Sakurai, M.; Inaba, H.; Katano, N.; Horikoshi, Y.; Eguchi, H.; Miyake, M.; et al. Chromosome abnormalities and MLL rearrangements in acute myeloid leukemia of infants. Leukemia 1999, 13, 1013–1017. [Google Scholar] [CrossRef]
- Cox, M.C.; Panetta, P.; Lo-Coco, F.; Del Poeta, G.; Venditti, A.; Maurillo, L.; Del Principe, M.I.; Mauriello, A.; Anemona, L.; Bruno, A.; et al. Chromosomal aberration of the 11q23 locus in acute leukemia and frequency of MLL gene translocation: results in 378 adult patients. American journal of clinical pathology 2004, 122, 298–306. [Google Scholar] [CrossRef]
- Stock, W.; Thirman, M.J.; Dodge, R.K.; Rowley, J.D.; Diaz, M.O.; Wurster-Hill, D.; Sobol, R.E.; Davey, F.R.; Larson, R.A.; Westbrook, C.A.; et al. Detection of MLL gene rearrangements in adult acute lymphoblastic leukemia. A Cancer and Leukemia Group B study. Leukemia 1994, 8, 1918–1922. [Google Scholar]
- Super, H.J.; McCabe, N.R.; Thirman, M.J.; Larson, R.A.; Le Beau, M.M.; Pedersen-Bjergaard, J.; Philip, P.; Diaz, M.O.; Rowley, J.D. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood 1993, 82, 3705–3711. [Google Scholar] [CrossRef] [PubMed]
- Górecki, M.; Kozioł, I.; Kopystecka, A.; Budzyńska, J.; Zawitkowska, J.; Lejman, M. Updates in KMT2A Gene Rearrangement in Pediatric Acute Lymphoblastic Leukemia. Biomedicines 2023, 11, 821. [Google Scholar] [CrossRef] [PubMed]
- Basecke, J.; Whelan, J.T.; Griesinger, F.; Bertrand, F.E. The MLL partial tandem duplication in acute myeloid leukaemia. British journal of haematology 2006, 135, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Nilson, I.; Löchner, K.; Siegler, G.; Greil, J.; Beck, J.D.; Fey, G.H.; Marschalek, R. Exon/intron structure of the human ALL-1 (MLL) gene involved in translocations to chromosomal region 11q23 and acute leukaemias. British journal of haematology 1996, 93, 966–972. [Google Scholar] [CrossRef]
- Audemard, E.O.; Gendron, P.; Feghaly, A.; Lavallée, V.-P.; Hébert, J.; Sauvageau, G.; Lemieux, S. Targeted variant detection using unaligned RNA-Seq reads. Life Science Alliance 2019, 2, e201900336. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Hoshii, T.; Armstrong, S.A. Mixed-Lineage Leukemia Fusions and Chromatin in Leukemia. Cold Spring Harbor Perspectives in Medicine 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Vetro, C.; Haferlach, T.; Meggendorfer, M.; Stengel, A.; Jeromin, S.; Kern, W.; Haferlach, C. Cytogenetic and molecular genetic characterization of <em>KMT2A</em>-PTD positive acute myeloid leukemia in comparison to <em>KMT2A</em>-Rearranged acute myeloid leukemia. Cancer Genetics 2020, 240, 15–22. [Google Scholar] [CrossRef]
- Jamal, R.; Taketani, T.; Taki, T.; Bessho, F.; Hongo, T.; Hamaguchi, H.; Horiike, S.; Taniwaki, M.; Hanada, R.; Nakamura, H.; et al. Coduplication of the MLL and FLT3 genes in patients with acute myeloid leukemia. Genes, chromosomes & cancer 2001, 31, 187–190. [Google Scholar] [CrossRef]
- Shih, L.Y.; Liang, D.C.; Fu, J.F.; Wu, J.H.; Wang, P.N.; Lin, T.L.; Dunn, P.; Kuo, M.C.; Tang, T.C.; Lin, T.H.; et al. Characterization of fusion partner genes in 114 patients with de novo acute myeloid leukemia and MLL rearrangement. Leukemia 2006, 20, 218–223. [Google Scholar] [CrossRef]
- Steudel, C.; Wermke, M.; Schaich, M.; Schäkel, U.; Illmer, T.; Ehninger, G.; Thiede, C. Comparative analysis of MLL partial tandem duplication and FLT3 internal tandem duplication mutations in 956 adult patients with acute myeloid leukemia. Genes, chromosomes & cancer 2003, 37, 237–251. [Google Scholar] [CrossRef]
- Whitman, S.P.; Liu, S.; Vukosavljevic, T.; Rush, L.J.; Yu, L.; Liu, C.; Klisovic, M.I.; Maharry, K.; Guimond, M.; Strout, M.P.; et al. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood 2005, 106, 345–352. [Google Scholar] [CrossRef]
- Burmeister, T.; Meyer, C.; Gröger, D.; Hofmann, J.; Marschalek, R. Evidence-based RT-PCR methods for the detection of the 8 most common MLL aberrations in acute leukemias. Leukemia Research 2015, 39, 242–247. [Google Scholar] [CrossRef]
- Li, Q.; Xing, S.; Zhang, H.; Mao, X.; Xiao, M.; Wang, Y. FISH improves risk stratification in acute leukemia by identifying KMT2A abnormal copy number and rearrangements. Scientific Reports 2022, 12, 9585. [Google Scholar] [CrossRef] [PubMed]
- Ross, L.L.; Peter, J.M.V. Next-generation sequencing in the diagnosis and minimal residual disease assessment of acute myeloid leukemia. Haematologica 2019, 104, 868–871. [Google Scholar] [CrossRef]
- Dai, B.; Yu, H.; Ma, T.; Lei, Y.; Wang, J.; Zhang, Y.; Lu, J.; Yan, H.; Jiang, L.; Chen, B. The Application of Targeted RNA Sequencing for KMT2A–Partial Tandem Duplication Identification and Integrated Analysis of Molecular Characterization in Acute Myeloid Leukemia. The Journal of Molecular Diagnostics 2021, 23, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Afrin, S.; Zhang, C.R.C.; Meyer, C.; Stinson, C.L.; Pham, T.; Bruxner, T.J.C.; Venn, N.C.; Trahair, T.N.; Sutton, R.; Marschalek, R.; et al. Targeted Next-Generation Sequencing for Detecting MLL Gene Fusions in Leukemia. Molecular Cancer Research 2018, 16, 279–285. [Google Scholar] [CrossRef] [PubMed]
- McKerrell, T.; Moreno, T.; Ponstingl, H.; Bolli, N.; Dias, J.M.L.; Tischler, G.; Colonna, V.; Manasse, B.; Bench, A.; Bloxham, D.; et al. Development and validation of a comprehensive genomic diagnostic tool for myeloid malignancies. Blood 2016, 128, e1–e9. [Google Scholar] [CrossRef]
- Engvall, M.; Cahill, N.; Jonsson, B.-I.; Höglund, M.; Hallböök, H.; Cavelier, L. Detection of leukemia gene fusions by targeted RNA-sequencing in routine diagnostics. BMC Medical Genomics 2020, 13, 106. [Google Scholar] [CrossRef]
- Hinai, A.S.A.A.; Pratcorona, M.; Grob, T.; Kavelaars, F.G.; Bussaglia, E.; Sanders, M.A.; Nomdedeu, J.; Valk, P.J.M. The Landscape of KMT2A-PTD AML: Concurrent Mutations, Gene Expression Signatures, and Clinical Outcome. HemaSphere 2019, 3, e181. [Google Scholar] [CrossRef]
- Creutzig, U.; van den Heuvel-Eibrink, M.M.; Gibson, B.; Dworzak, M.N.; Adachi, S.; de Bont, E.; Harbott, J.; Hasle, H.; Johnston, D.; Kinoshita, A.; et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 2012, 120, 3187–3205. [Google Scholar] [CrossRef]
- Smith, A.C.; Neveling, K.; Kanagal-Shamanna, R. Optical genome mapping for structural variation analysis in hematologic malignancies. American Journal of Hematology 2022, 97, 975–982. [Google Scholar] [CrossRef]
- Levy, B.; Kanagal-Shamanna, R.; Sahajpal, N.S.; Neveling, K.; Rack, K.; Dewaele, B.; Olde Weghuis, D.; Stevens-Kroef, M.; Puiggros, A.; Mallo, M.; et al. A framework for the clinical implementation of optical genome mapping in hematologic malignancies. American Journal of Hematology 2024, n/a, 1–20. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nature Communications 2019, 10, 1784. [Google Scholar] [CrossRef]
- Tsai, H.K.; Gibson, C.J.; Murdock, H.M.; Davineni, P.; Harris, M.H.; Wang, E.S.; Gondek, L.P.; Kim, A.S.; Nardi, V.; Lindsley, R.C. Allelic complexity of KMT2A partial tandem duplications in acute myeloid leukemia and myelodysplastic syndromes. Blood Advances 2022, 6, 4236–4240. [Google Scholar] [CrossRef]
- Moore, S., McGowan-Jordan, J., Smith, A. C., Rack, K., Koehler, U., Stevens-Kroeg, M., Barseghyan, H., Kanagal-Shamanna, R., & Hastings, R. Genome Mapping Nomenclature. Cytogenetic and Genome Research. 2023. [CrossRef]
Figure 1.
Schematic representation of the 11q23.3 region and the KMT2A gene. An ideogram of chromosome 11 is shown (top) and the 11q23.3 chromosome band is indicated. Detailed view of the KMT2A gene structure on band 11q23.3 is shown with exons indicated as rectangles with relevant exons numbered. Below the KMT2A gene map the coverage of each of the techniques used is shown for comparison. Top line shows the chromosome 11 reference map for the KMT2A region for Optical Genome Mapping (OGM). Vertical lines on the reference represent a label used to detect structural variants by OGM. DNA is labelled wherever in the genome the sequence CTTAAG is present and usually occurs with an average spacing of approximately 6kb. As seen by the reference map the labels are not uniformly spaced. However it should be noted that the rare variant assembly can detect not only changes in label patterns but also changes in distance between labels enabling the detection of deletions, duplications and insertions down to 5kb. Below the OGM reference the regions are the KMT2A gene regions captured for sequencing by the heme-NGS assay. These includes the consensus coding DNA sequences (e.g., exons 1 to 36) of KMT2A. KMT2A-PTD analysis using MLPA uses a PTD specific probe in exon 4 as well as a reference probe in exon 36.
Figure 1.
Schematic representation of the 11q23.3 region and the KMT2A gene. An ideogram of chromosome 11 is shown (top) and the 11q23.3 chromosome band is indicated. Detailed view of the KMT2A gene structure on band 11q23.3 is shown with exons indicated as rectangles with relevant exons numbered. Below the KMT2A gene map the coverage of each of the techniques used is shown for comparison. Top line shows the chromosome 11 reference map for the KMT2A region for Optical Genome Mapping (OGM). Vertical lines on the reference represent a label used to detect structural variants by OGM. DNA is labelled wherever in the genome the sequence CTTAAG is present and usually occurs with an average spacing of approximately 6kb. As seen by the reference map the labels are not uniformly spaced. However it should be noted that the rare variant assembly can detect not only changes in label patterns but also changes in distance between labels enabling the detection of deletions, duplications and insertions down to 5kb. Below the OGM reference the regions are the KMT2A gene regions captured for sequencing by the heme-NGS assay. These includes the consensus coding DNA sequences (e.g., exons 1 to 36) of KMT2A. KMT2A-PTD analysis using MLPA uses a PTD specific probe in exon 4 as well as a reference probe in exon 36.
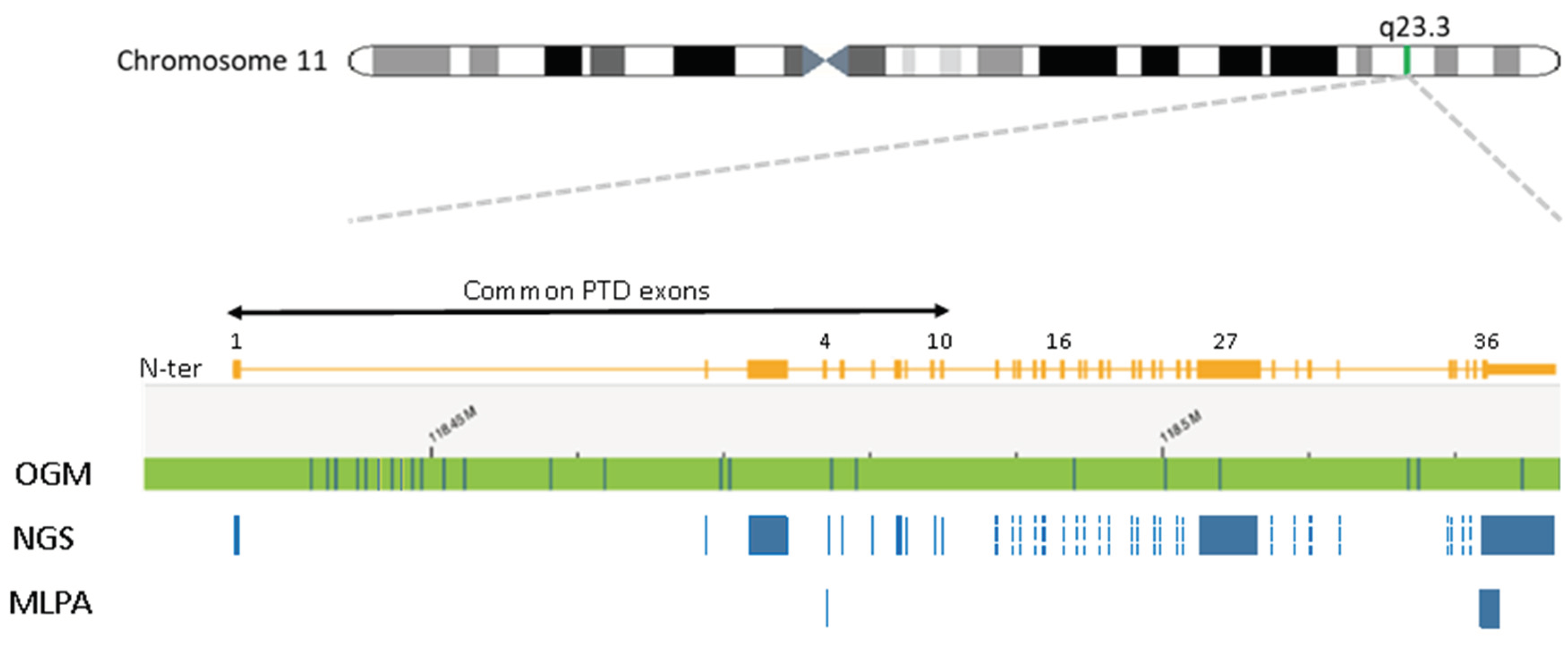
Figure 2.
KMT2A-PTD detection using heme-NGS. A. KMT2A-PTD specific exon 1 to 10 coverage in a cohort of baseline samples with no detectable numerical or structural variant on chromosome 11q23.3, B. KMT2A z-score in a cohort of baseline samples, using KMT2A exon 27 as a reference exon. C. KMT2A z-score in a cohort of patients with numerical (i.e., trisomy 11) or structural variants impacting KMT2A, using KMT2A exon 27 as a reference exon. D. KMT2A z-score ratio in the KMT2A-PTD postive cases detected by NGS, using KMT2A exon 27 as a reference exon.
Figure 2.
KMT2A-PTD detection using heme-NGS. A. KMT2A-PTD specific exon 1 to 10 coverage in a cohort of baseline samples with no detectable numerical or structural variant on chromosome 11q23.3, B. KMT2A z-score in a cohort of baseline samples, using KMT2A exon 27 as a reference exon. C. KMT2A z-score in a cohort of patients with numerical (i.e., trisomy 11) or structural variants impacting KMT2A, using KMT2A exon 27 as a reference exon. D. KMT2A z-score ratio in the KMT2A-PTD postive cases detected by NGS, using KMT2A exon 27 as a reference exon.
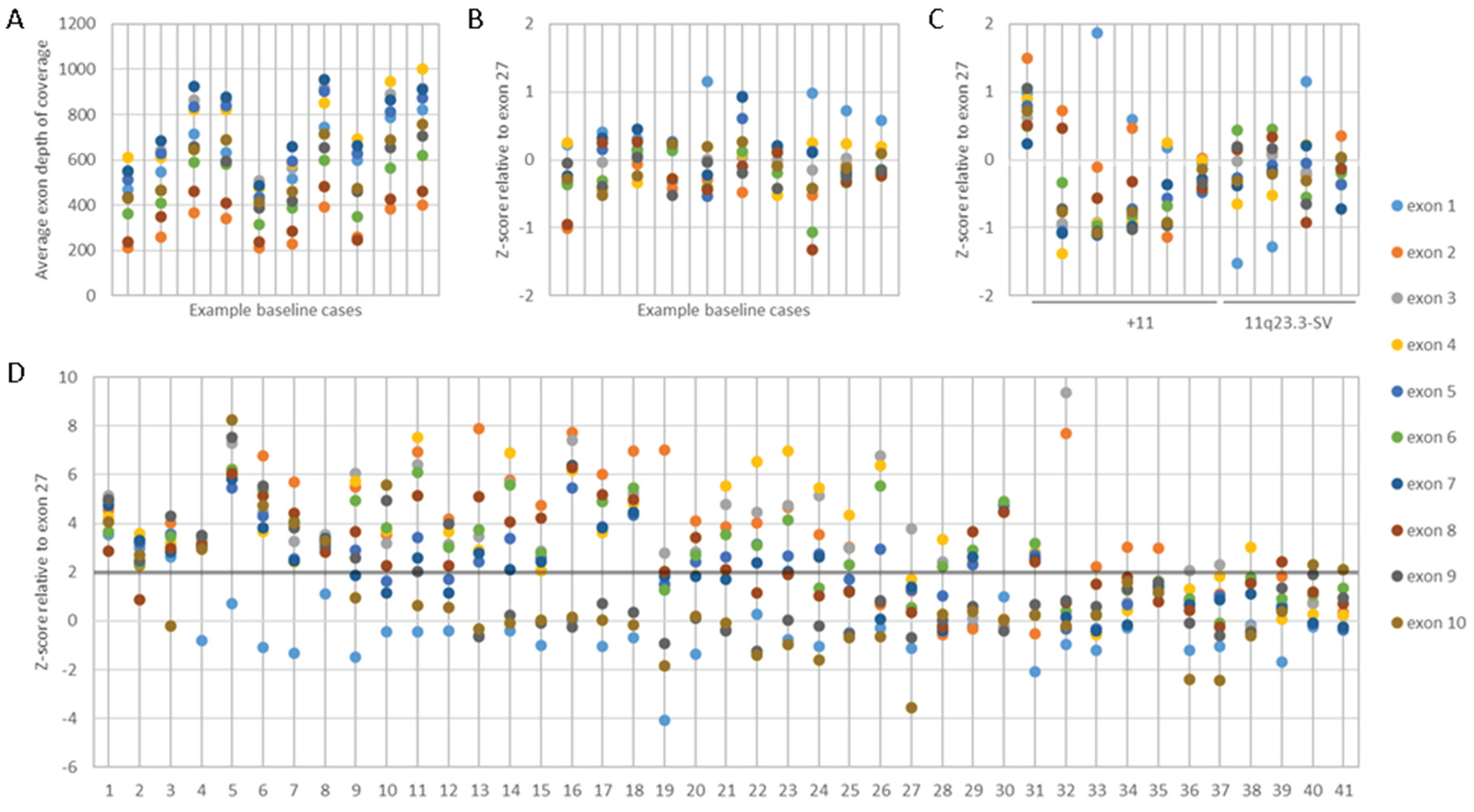
Figure 3.
Comparative analyses of NGS, MLPA and OGM for KMT2A-PTD detection. PTD. Partial tandem duplication. Inc. Inconclusive MLPA results. Y. Yes – KMT2A-PTD detected. N. No KMT2A-PTD detected. N/T. Not tested. Ratio 4/36. MLPA copy number ratio for the exon 4 KMT2A-PTD specific over the exon 36 KMT2A reference probe. OGM KMT2A-PTD size were obtained from the Access analysis software and compared to appromixate sizes derived from Human reference genome Hg38 build. Boxed. Cases showing discordant results by NGS, MLPA or OGM.
Figure 3.
Comparative analyses of NGS, MLPA and OGM for KMT2A-PTD detection. PTD. Partial tandem duplication. Inc. Inconclusive MLPA results. Y. Yes – KMT2A-PTD detected. N. No KMT2A-PTD detected. N/T. Not tested. Ratio 4/36. MLPA copy number ratio for the exon 4 KMT2A-PTD specific over the exon 36 KMT2A reference probe. OGM KMT2A-PTD size were obtained from the Access analysis software and compared to appromixate sizes derived from Human reference genome Hg38 build. Boxed. Cases showing discordant results by NGS, MLPA or OGM.
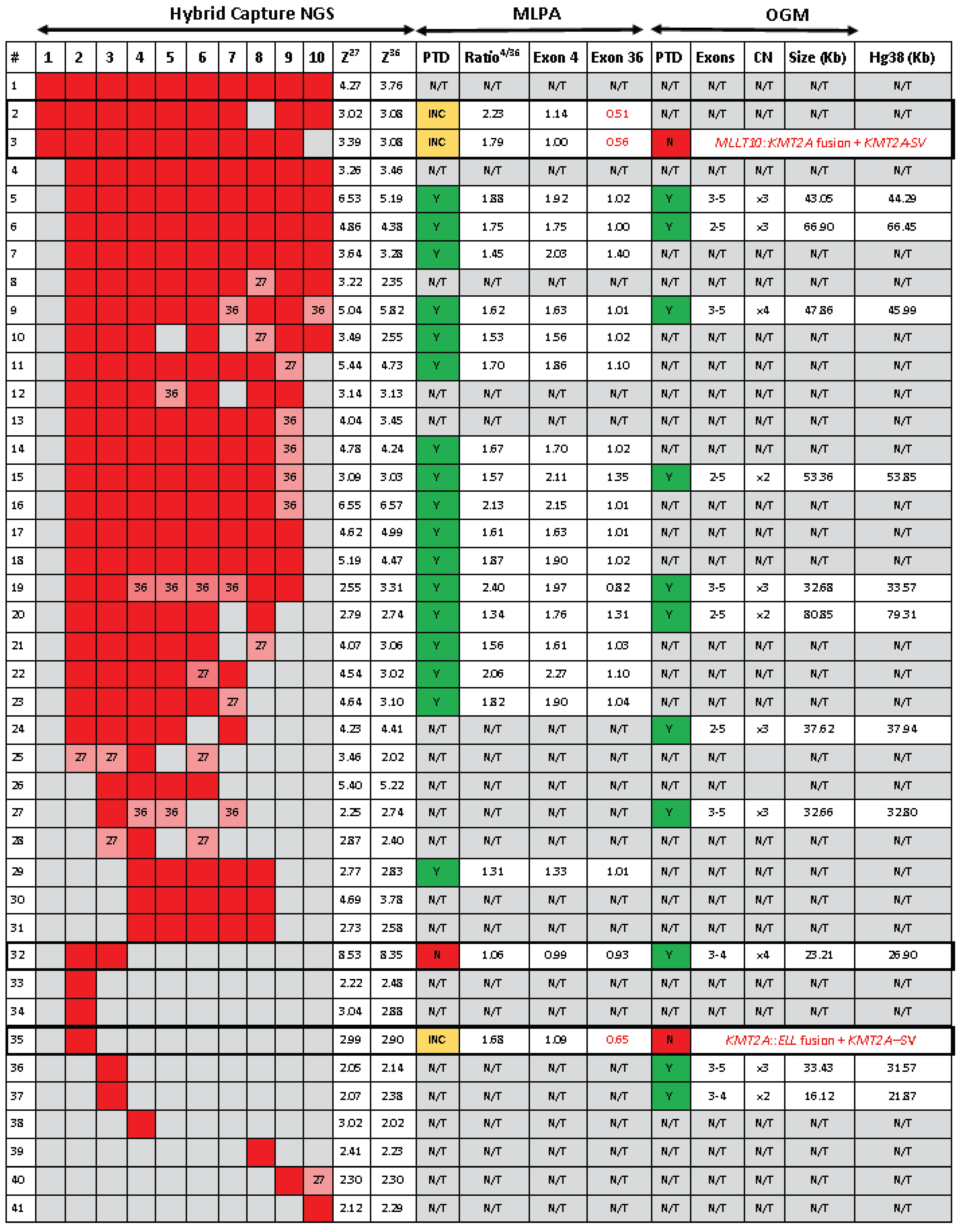
Figure 4.
Cytogenetic analyses for Case 3. A. G-banded karyotype showing a karyotype of 48,XY,+8,+19. B. Optical Genome Mapping circos plot showing trisomy for chromosomes 8 and 19, in addition a rearrangement between chromsomes 10p and 11q is observed (magenta line). Circos plot elements from outer ring to inner ring: chromosome number, ideogram, intrachromosomal SV (<5Mb), copy number, aneuploidy bar, intrachromosomal SV (>5 Mb) or interchromosomal SV. C. Genome view of the rearrangement involving chromosomes 10 and 11. Reference chromosome 10 is the top bar and reference chromosome 11 is the bottom bar. A hybrid map (blue bar, middle) shows the alignment of this cryptic insertion to both chromsomes 10 and 11. The matchlines (grey) show alignment of labels to the specific segment on the reference sequence and it can be observed that sequence from chromosome 11 (KMT2A gene) is flanked by sequence from chromosome 10 indicating that the mechanism of rearrangement is an insertion. D. KMT2A break-apart FISH shows a signal pattern consistent with a rearrangement. One set of signals is fused (red + green fusion) and represents the normal chromosome 11. The remaining signals are separated indicating a rearrangement. Note that the residual separated signals appear diminished compared to the normal chromosome 11 signals – consistent with the deletion of sequence overlapping the probe region.
Figure 4.
Cytogenetic analyses for Case 3. A. G-banded karyotype showing a karyotype of 48,XY,+8,+19. B. Optical Genome Mapping circos plot showing trisomy for chromosomes 8 and 19, in addition a rearrangement between chromsomes 10p and 11q is observed (magenta line). Circos plot elements from outer ring to inner ring: chromosome number, ideogram, intrachromosomal SV (<5Mb), copy number, aneuploidy bar, intrachromosomal SV (>5 Mb) or interchromosomal SV. C. Genome view of the rearrangement involving chromosomes 10 and 11. Reference chromosome 10 is the top bar and reference chromosome 11 is the bottom bar. A hybrid map (blue bar, middle) shows the alignment of this cryptic insertion to both chromsomes 10 and 11. The matchlines (grey) show alignment of labels to the specific segment on the reference sequence and it can be observed that sequence from chromosome 11 (KMT2A gene) is flanked by sequence from chromosome 10 indicating that the mechanism of rearrangement is an insertion. D. KMT2A break-apart FISH shows a signal pattern consistent with a rearrangement. One set of signals is fused (red + green fusion) and represents the normal chromosome 11. The remaining signals are separated indicating a rearrangement. Note that the residual separated signals appear diminished compared to the normal chromosome 11 signals – consistent with the deletion of sequence overlapping the probe region.
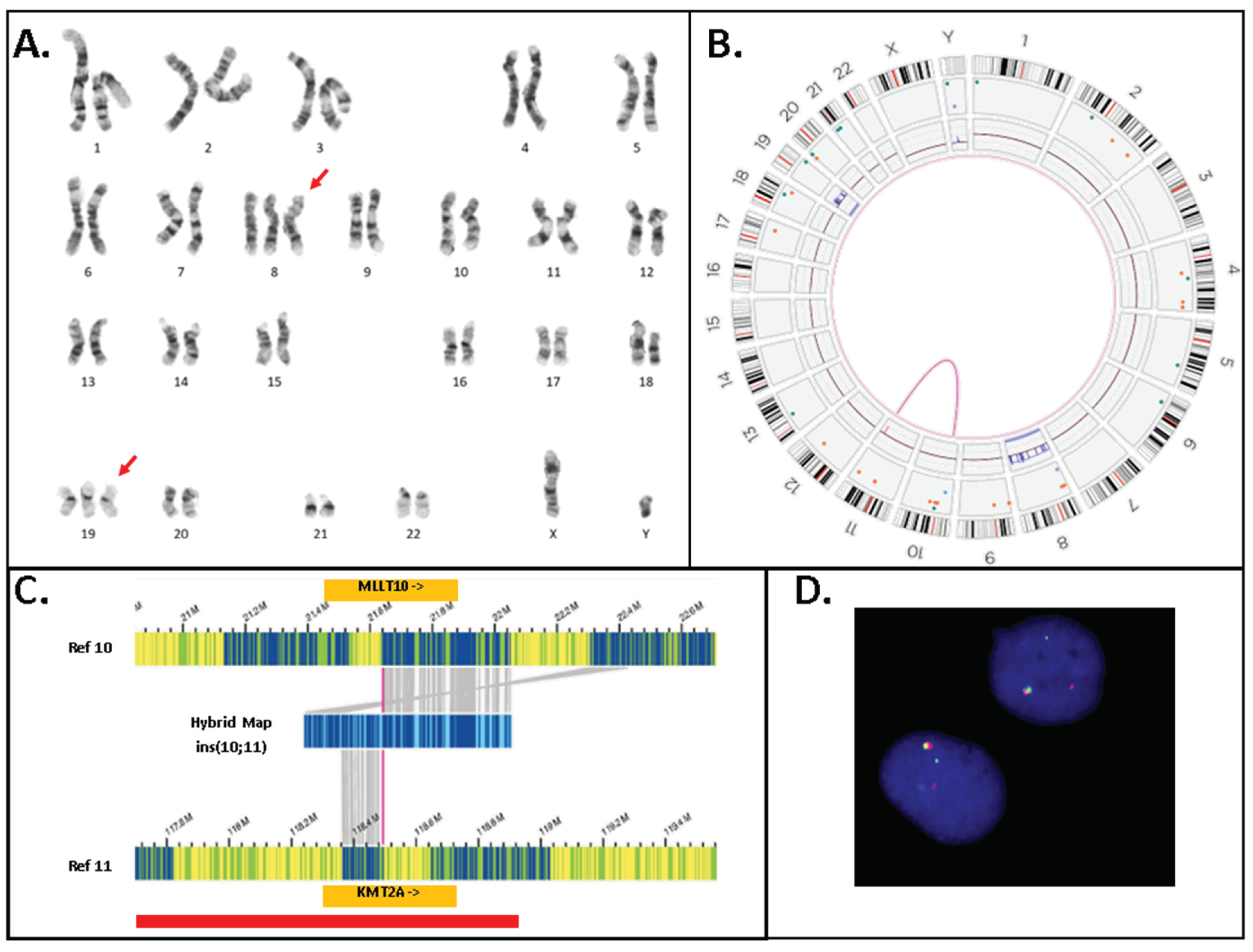
Figure 5.
Cytogenetic analyses for Case 35. A. Optical Genome Mapping circos plot showing a complex genome with multiple intra- and interchromosomal SVs (magenta lines), copy number changes (red and blue boxes on the copy number track of the circos plot) and aneuploidies (orange or blue line spanning the full chromosome) . Circos plot elements from outer ring to inner ring: chromosome number, ideogram, intrachromosomal SV (<5Mb), copy number, aneuploidy bar, intrachromosomal SV (>5 Mb) or interchromosomal SV. A KMT2A::ELL translocation was identified and is highlighted among the other interchromosomal SVs by a dotted blue line extending from chromosome 11q to chromosome 19p. B. KMT2A break-apart FISH shows multiple signals for the 5′ and 3′ probes for KMT2A. There are multiple signals for both the 5′ and 3′ probes indicating a polyploid karyotype. Both fused and separated 5′ and 3′ signals are observed consistent with a rearrangement of KMT2A. C. Genome view of the rearrangement involving chromosomes 11 and 19. Reference chromosome 11 is the top green bar and reference chromosome 19 is the bottom green bar. A hybrid map (blue bar, middle) shows the alignment of this unbalnced translocation to both chromsomes 11 and 19. The matchlines (grey) show alignment of labels to the specific segment on the reference genome and it can be observed that sequence from chromosome 11 in the 5′ KMT2A gene region breaks and is joined to the region containing the ELL gene on chromsome 19. Copy number track for chromosome 11 appears above the reference (green bar) and shows loss of 3′ KMT2A after the breakpoint. Conversely, the copy number plot for chromosome 19 shows a gain indicating that this rearrangement is unbalanced.
Figure 5.
Cytogenetic analyses for Case 35. A. Optical Genome Mapping circos plot showing a complex genome with multiple intra- and interchromosomal SVs (magenta lines), copy number changes (red and blue boxes on the copy number track of the circos plot) and aneuploidies (orange or blue line spanning the full chromosome) . Circos plot elements from outer ring to inner ring: chromosome number, ideogram, intrachromosomal SV (<5Mb), copy number, aneuploidy bar, intrachromosomal SV (>5 Mb) or interchromosomal SV. A KMT2A::ELL translocation was identified and is highlighted among the other interchromosomal SVs by a dotted blue line extending from chromosome 11q to chromosome 19p. B. KMT2A break-apart FISH shows multiple signals for the 5′ and 3′ probes for KMT2A. There are multiple signals for both the 5′ and 3′ probes indicating a polyploid karyotype. Both fused and separated 5′ and 3′ signals are observed consistent with a rearrangement of KMT2A. C. Genome view of the rearrangement involving chromosomes 11 and 19. Reference chromosome 11 is the top green bar and reference chromosome 19 is the bottom green bar. A hybrid map (blue bar, middle) shows the alignment of this unbalnced translocation to both chromsomes 11 and 19. The matchlines (grey) show alignment of labels to the specific segment on the reference genome and it can be observed that sequence from chromosome 11 in the 5′ KMT2A gene region breaks and is joined to the region containing the ELL gene on chromsome 19. Copy number track for chromosome 11 appears above the reference (green bar) and shows loss of 3′ KMT2A after the breakpoint. Conversely, the copy number plot for chromosome 19 shows a gain indicating that this rearrangement is unbalanced.

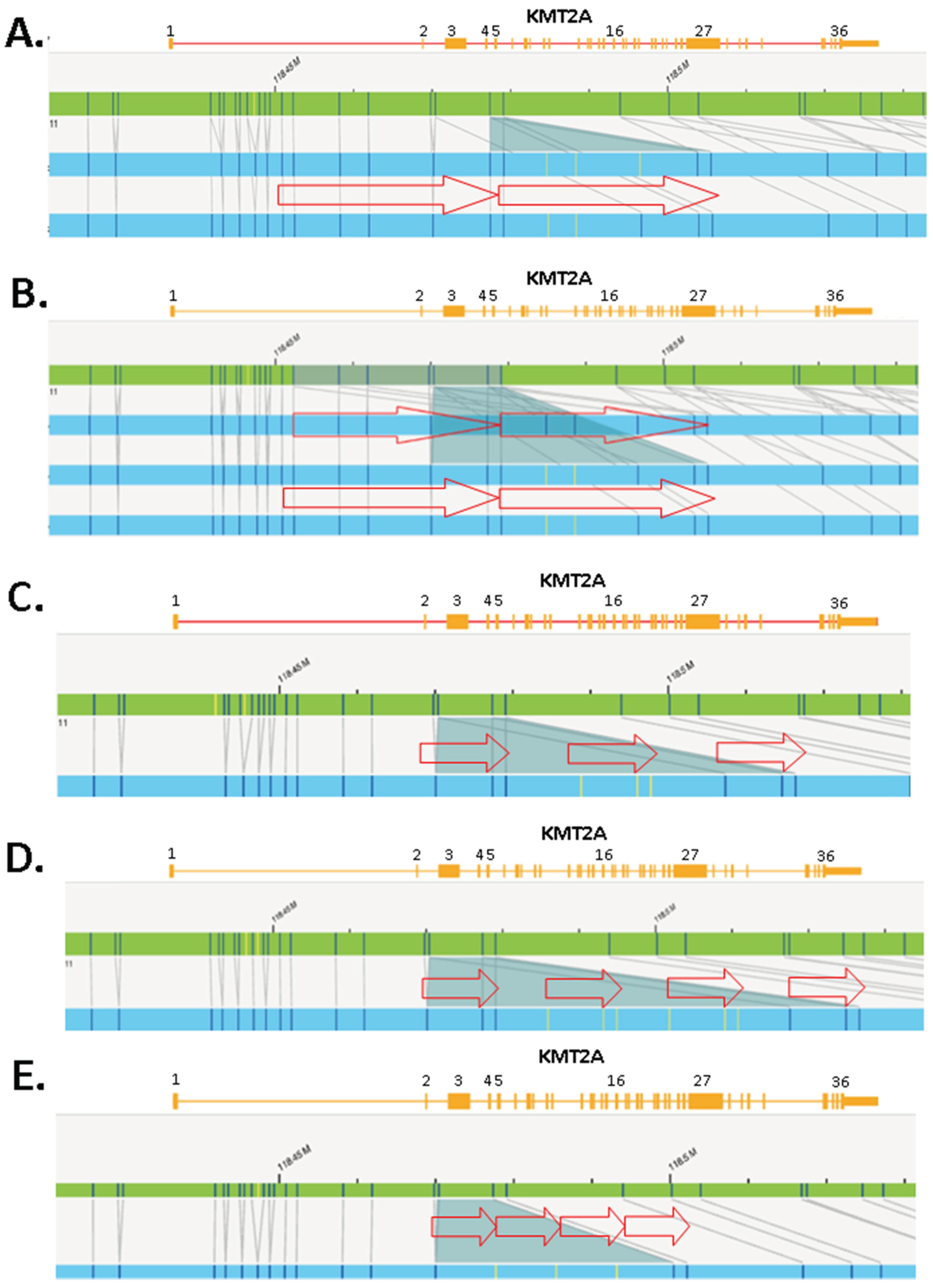
Figure 6.
Examples of KMT2A-PTD cases showing size and composition differences detected by heme-NGS and confirmed by OGM. An ideogram showing the KMT2A gene region from exons 1 to 36 is depicted. Green bar - Reference genome. Blue bar- Hybrid map . Red arrows. Duplicated segment. A. Genome view for the KMT2A-PTD in case 15 . OGM estimates an SV of 53kb with two PTD copies encompassing KMT2A exons 2 to 5. Note a 5′ PTD breakpoint deep in KMT2A intron 1. Note the presence of multiple SV hybrid maps. Top hybrid and bottom hybrid maps highlighted as KMT2A intragenic insertions by the Bionano Access software. Smaller intragenic duplications (i.e., here an SV of 27kb) may be identified as insertions of unknown materiall; ins(11;?) when insufficient labels are present to definitevely align an SV back to the genome reference. B. Genome view for PTD in case 20. OGM estimates an SV of 80kb with two PTD copies encompassing KMT2A exons 2 to 5. Note a 5′ PTD breakpoint deep in KMT2A intron 1. Note the difference in KMT2A-PTD size of 27kb between cases 15 and 20, although the corresponding PTDs in these patients are of approximate lengths. An additional SV hybrid map is listed in this patient. Similar to case 15, the middle and bottom hybrid maps are highlighted as KMT2A intragenic insertions. The top hybrid map shows a duplication of overlapping KMT2A gene regions. Although not confirmed by OGM, the presence of KMT2A-PTDs in more than one 11q23.3 homologue is likely. Case 20 was shown to harbor a trisomy 11 by OGM and G-banded karyotype. The PTD size difference between case 15 (i.e., 53 kb) and 20 (i.e., 80 kb), the VAF of the KMT2A-PTDs (>50%) as well as the label patterns and breakpoints for each of the hybrid maps are additional supportive evidence for KMT2A-PTDs on more than one homologue. C. Genome view for PTD in case 36. OGM estimates an SV of 33kb with three PTD copies encompassing KMT2A exons 2 to 5. The 5′ breakpoint of the PTD is estimated closer to exon 2 than in cases 15 and 20. Note that gaps between the duplicated KMT2A segments. It is clear from the spacing between the duplications that the breakpoint is at 3′ of exon 5 due to the amount of sequence without a label, however, the exact breakpoints cannot be determined by OGM. D. Genome view for the PTD in case 9. OGM estimates an SV of 48kb with four PTD copies encompassing KMT2A exons 3 to 5. Note that similar to case 36 above, it is likey that the PTD extend farther 3′ of KMT2A exon 5. The 5′ breakpoint of the PTD is presumed to be even closer than in cases 15, 20 and 9. E. Genome view for the PTD in case 32. OGM estimates an SV of 23kb with four PTD copies encompassing KMT2A exons 3 to 4. Note that from the labels pattern of case 32 (two labels) compared to cases 36 and 9 (three labels), the involvement of KMT2A exon 5 in the PTD is unlikely.
Figure 6.
Examples of KMT2A-PTD cases showing size and composition differences detected by heme-NGS and confirmed by OGM. An ideogram showing the KMT2A gene region from exons 1 to 36 is depicted. Green bar - Reference genome. Blue bar- Hybrid map . Red arrows. Duplicated segment. A. Genome view for the KMT2A-PTD in case 15 . OGM estimates an SV of 53kb with two PTD copies encompassing KMT2A exons 2 to 5. Note a 5′ PTD breakpoint deep in KMT2A intron 1. Note the presence of multiple SV hybrid maps. Top hybrid and bottom hybrid maps highlighted as KMT2A intragenic insertions by the Bionano Access software. Smaller intragenic duplications (i.e., here an SV of 27kb) may be identified as insertions of unknown materiall; ins(11;?) when insufficient labels are present to definitevely align an SV back to the genome reference. B. Genome view for PTD in case 20. OGM estimates an SV of 80kb with two PTD copies encompassing KMT2A exons 2 to 5. Note a 5′ PTD breakpoint deep in KMT2A intron 1. Note the difference in KMT2A-PTD size of 27kb between cases 15 and 20, although the corresponding PTDs in these patients are of approximate lengths. An additional SV hybrid map is listed in this patient. Similar to case 15, the middle and bottom hybrid maps are highlighted as KMT2A intragenic insertions. The top hybrid map shows a duplication of overlapping KMT2A gene regions. Although not confirmed by OGM, the presence of KMT2A-PTDs in more than one 11q23.3 homologue is likely. Case 20 was shown to harbor a trisomy 11 by OGM and G-banded karyotype. The PTD size difference between case 15 (i.e., 53 kb) and 20 (i.e., 80 kb), the VAF of the KMT2A-PTDs (>50%) as well as the label patterns and breakpoints for each of the hybrid maps are additional supportive evidence for KMT2A-PTDs on more than one homologue. C. Genome view for PTD in case 36. OGM estimates an SV of 33kb with three PTD copies encompassing KMT2A exons 2 to 5. The 5′ breakpoint of the PTD is estimated closer to exon 2 than in cases 15 and 20. Note that gaps between the duplicated KMT2A segments. It is clear from the spacing between the duplications that the breakpoint is at 3′ of exon 5 due to the amount of sequence without a label, however, the exact breakpoints cannot be determined by OGM. D. Genome view for the PTD in case 9. OGM estimates an SV of 48kb with four PTD copies encompassing KMT2A exons 3 to 5. Note that similar to case 36 above, it is likey that the PTD extend farther 3′ of KMT2A exon 5. The 5′ breakpoint of the PTD is presumed to be even closer than in cases 15, 20 and 9. E. Genome view for the PTD in case 32. OGM estimates an SV of 23kb with four PTD copies encompassing KMT2A exons 3 to 4. Note that from the labels pattern of case 32 (two labels) compared to cases 36 and 9 (three labels), the involvement of KMT2A exon 5 in the PTD is unlikely.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



