
Submitted:
15 March 2024
Posted:
18 March 2024
You are already at the latest version
Abstract
Vimentin has been reported to play diverse roles in cell processes such as spreading, migration, and cell-matrix adhesion. Here, we use vimentin knock out cells (Vim KO) to assess how vimentin impacts cell spreading, morphology, and myofibroblast transformation of human corneal fibro-blasts. Overall, loss of vimentin did not significantly impact corneal fibroblast spreading, as indicated by Vim KO morphology and mechanical activity (traction force). However, we found that Vim KO cells had reduced elongation in response to PDGF as compared to controls. Furthermore, absence of vimentin did not completely block TGFβ induced myofibroblast transformation, since αSMA protein expression was detected in Vim KO cells. Nonetheless, the degree of transformation and amount of αSMA protein was reduced as compared to control cells. Proteomics showed that Vim KO cells cultured in TGFβ had a similar pattern of protein expression as controls. One exception included periostin, an ECM protein associated with wound healing and fibrosis in other cell types, which was highly expressed only in Vim KO cells. We also demonstrate for the first time that LRRC15, a protein previously associated with myofibroblast transformation of cancer-associated fibroblasts, is also expressed by corneal myofibroblasts. Overall, our data shows that vimentin has a limited, yet measurable impact on corneal fibroblast spreading and myofibroblast transformation. We also identified novel proteins that may regulate corneal myofibroblast transformation in the presence and/or absence of vimentin.
Keywords:
cornea
; vimentin
; cytoskeleton
; fibroblasts
; myofibroblasts
; wound healing
; TGFb
; PDGF
1. Introduction
The cytoskeleton is composed of three filamentous proteins: F-actin, microtubules, and intermediate filaments (IFs). These cytoskeletal filament proteins form highly structured and dynamic networks. Cytoskeletal filament proteins reorganize quickly in response to external and internal signals, allowing cells to adapt their shape in response to environmental or chemical cues [1,2]. These changes in cell shape are important in cell biological processes such as cell division, migration, adhesion, and the response to external forces [3,4,5,6,7]. The three cytoskeletal filament proteins differ not only in their chemical and physical structure but also in the types of filaments and networks they form [1]. While the role of F-actin and microtubules in the spreading and morphology of cells is well established, questions remain regarding the functional roles of IFs. The family of proteins forming IFs is vast, they are differentially expressed, and the form and structure of the network often depends on IF type, cell type as well as differentiation status [8].
Vimentin is a type III IF protein expressed mainly in mesenchymal cell types [9]. Fibroblasts are cells of mesenchymal origin that are broadly found in many tissues and organs [10]. Vimentin intermediate filaments, VIFs, are generally distributed homogenously throughout the cytoplasm of fibroblasts [11]. Recent studies have shown VIFs in fibroblasts play diverse roles in cell mechanical processes, such as modulating cell extensions [12], regulating migration and movement [13], contributing to cell growth and development of cell-matrix adhesions [14], and maintaining cellular integrity [9]. VIFs also provide the cells with mechanical support to stabilize them and allow them to withstand large amounts of stress without damage [8,15]; VIFs are elastic polymers that can withstand deformations of up to 300% of their initial length [15]. Nonetheless, the precise mechanisms of action through which VIFs function have not been fully elucidated.
The cornea is an optically transparent tissue at the front of the eye that is responsible for two thirds of its refractive power. The corneal stroma, which makes up 90% of the corneal thickness, is a highly ordered structure consisting of lamellae of aligned collagen fibrils that endow the tissue with its mechanical stability and optical transparency. Corneal keratocytes are sandwiched between these collagen lamellae and interact with the extracellular matrix (ECM) to regulate fundamental biological processes such as developmental morphogenesis and wound healing. Dysregulated wound healing can result in corneal fibrosis and scarring, which is a leading cause of blindness worldwide. [16,17]. Stromal haze is caused by activation of keratocytes into migratory fibroblasts which repopulate the wound and transform into myofibroblasts, which secrete a disorganized fibrotic ECM [17,18].
Recent studies have shown that a decrease in the steady state levels of vimentin or inhibition of vimentin phosphorylation inhibit myofibroblast differentiation of corneal keratocytes, resulting in decreased fibrosis following corneal injury in a mouse model [19,20]. In this study, we explored the role of VIFs in modulating both spreading and myofibroblast transformation of cultured human corneal fibroblasts. First, we developed vimentin knock out (Vim KO) and knock down (Vim KD) cells and analyzed their spreading and morphology on 2D substrates and in 3D collagen matrices. Subsequently, we analyzed the effect of vimentin depletion on myofibroblast transformation of corneal fibroblasts, and conducted proteomics analysis to compare the patterns of protein expression between vimentin KO cells and wild type (WT) cells.
2. Materials and Methods
2.1. Materials
Dulbecco's modified Eagle medium (DMEM) and transforming growth factor beta (TGFβ) were obtained from Sigma-Aldrich (St. Louis, MO). OptiMEM was obtained from Gibco (Grand Island, NY). Fibroblast growth media and fibroblast growth kit-low serum were obtained from ATCC (Gaithersburg, MD). 0.25% trypsin/EDTA solution and RNAiMAX transfection reagent were purchased from Invitrogen (Gaithersburg, MD). Platelet-derived growth factor BB isotype (PDGF) was obtained from Upstate Biotechnology, Inc. (Lake Placid, NY). Fetal bovine serum (FBS), fatty acid-free and fraction V bovine serum albumin (BSA), penicillin, streptomycin, and amphotericin B were obtained from Lonza (Walkersville, MD). Vimentin siRNA was purchased from ThermoFisher (Waltham, MA). Non-targeting siRNA and siTOX transfection control were obtained from Horizon Discovery Biosciences (Cambridge, UK). Cas9/tracRNA/crRNA RNP and vimentin primers were obtained from Integrated DNA Technologies (Coralville, IA). Pierce RIPA buffer and the Protease and Phosphatase Inhibitor cocktail were obtained from Thermo Scientific (Waltham, MA). Mini-protean TGX gels, PVDF membrane, and HRP-conjugated goat anti-mouse antibody were obtained from Bio-Rad (Hercules CA). Type I rat tail collagen high concentration was purchased from BD Biosciences (Bedford, MA). Alexa Fluor 488, 546, 633, and propidium iodide (PI) were obtained from Molecular Probes, Inc. (Eugene, OR). RNase (DNase free) was purchased from Roche (Indianapolis, IN). Vimentin antibody, vimentin conjugated 488 and 546, and GAPDH antibody were obtained from Santacruz (Dallas, TX). FITC goat-anti-rabbit conjugated and goat-anti-mouse were obtained from Jackson ImmunoResearch (West Grove, PA).
2.2. Cell Culture
We used an established cell line (HTK) in which the catalytic subunit of human telomerase (h-TERT) was used to extend the lifespan of corneal fibroblasts [21]. HTK cells were maintained in tissue culture flasks at 37°C in a 5% CO2 humidified incubator using DMEM containing 10% FBS and supplemented with 1% penicillin, 1% streptomycin, and 1% amphotericin B.
All experiments were carried out with serum free basal media (SF), composed of DMEM and supplemented with essential amino acids, ascorbic acid, vitamins and 5 mg/ml BSA. PDGF-BB (50 ng/ml) or TGFβ1 (5 ng/ml) was added to media to study the cell response to growth factors. For cell spreading experiments, media containing PDGF was used and incubations were carried out for 4 and 24 hours. SF media was used as control. For myofibroblast transformation, cells were incubated for 6 days with media containing TGFβ and SF media was used as control. Collection of protein for western blotting was carried out after the 6th day of incubation with TGFβ1.
2.3. Vimentin Knockdown
For siRNA vimentin reverse transfection, a vimentin siRNA master mix solution was prepared by combining transfection reagent RNAiMAX at a ratio of 1:100 with OptiMEM. Vimentin siRNA was added at final concentration of 100 nM. Vimentin siRNA solution was poured into a well plate. An equal amount of solution containing trypsinized cells (200,000 cells) was poured on top of the same well and mixed with siRNA solution. For control experiments, vimentin siRNA was replaced with non-targeting siRNA or siTOX. After 24 hours of incubation, media was changed to DMEM. Following 72 hours of incubation, a second reverse transfection was carried out in the same way as described above. Media was changed to DMEM after 24 hours of incubation, and after 72 hours, cells were collected for experiments.
2.4. Vimentin Knockout
For vimentin knock-out experiments, HTK cells were electroporated with Cas9/tracRNA/crRNA RNP (1350 V, 30 ms, single pulse). The crRNA used was Hs.Cas9.Vim.1.AB (Seq: GGACTCGGTGGACTTCTCGC, PAM: TGG). Electroporated cells were screened using VIM exon 2 primers to confirm editing. Heterogeneous cell populations were sorted by flow cytometry into 96 well plates. Single cells were grown in fibroblast basal medium supplemented with the low serum growth kit. The fastest growing clones were scaled up and analyzed for Vim KO via qPCR, EnGen, and western blotting. Clones were then expanded and cultured for experiments.
2.5. 3D Collagen Matrix Model
A neutralized collagen solution was prepared by mixing high concentration rat tail type I collagen with 0.1 N NaOH, 10XMEM, H2O and DMEM containing cells at a density of 5X104 cells/ml to achieve a final collagen concentration of 2 mg/ml. 150 µl solution was poured on Mattek glass dishes. Samples were then placed into a humidified incubator (37°C, 5% CO2) for 60 minutes for polymerization. After samples polymerized, media containing PDGF was added to stimulate cell spreading.
2.6. Collagen Matrix Contraction
Collagen matrices were prepared as described above. Immediately after samples polymerized and media was added, matrix height was measured. Samples were returned into the humidified incubator and incubated for 24 hours. Matrix height was measured again. Height is measured by focusing on the top and bottom of each matrix. Since the bottom of the matrix remains attached to the dish, the change in height is a measure of the matrix reorganization produced by the cells, expressed percentage of matrix contraction. Duplicate samples were analyzed for each condition in each experiment.
2.7. Immunostaining
Samples were fixed using 3% paraformaldehyde in phosphate buffered saline (PBS) for 10 minutes, and permeabilized with 0.5% Triton X-100 in PBS for 15 minutes. Samples were then washed with PBS and blocked with 1% BSA fraction V in PBS for 1 hour. For vimentin labeling, samples were incubated with a Vimentin monoclonal antibody for 120 minutes, washed for 60 minutes and then incubated with a FITC conjugated goat anti-mouse secondary antibody. Samples were washed for 60 minutes and then incubated with Alexa-fluor 546 Phalloidin (1:150 ratio) to label F-actin. After 60 minutes of washing, samples were incubated for 30 minutes with DAPI (1:100 ratio) in PBS containing 1:100 RNase-DNase free to label cell nuclei. All staining processes were carried out on the same plates used for the experiments to avoid cell or matrix distortion.
Fluorescence Images were collected with a laser confocal microscope (Leica SP8, Heidelberg). An Argon laser (488 nm) was used to visualize vimentin, a GreNe laser (543nm) was used for F-actin, and a UV laser (405 nm) was used to image nuclei. Images were acquired sequentially to avoid cross talk between the channels. A stack of optical sections was acquired by changing the position of the focal plane in the z-direction with a step size of 1 or 2 µm, using a 63X oil immersion objective or 25X water immersion objective, respectively.
2.8. Immunoblotting
Protein was extracted with a lysis buffer containing Pierce RIPA buffer and the Protease and Phosphatase Inhibitor cocktail at 100:1 ratio. Lysates were clarified at 10000g at 4°C for 10 minutes. Protein was subjected to SDS-PAGE electrophoresis using a Bio-Rad electrophoresis chamber and mini-protean TGX gels. Transfer to PVDF membranes was carried out with a trans-blot turbo transfer system from Bio-Rad. Blots were probed with mouse anti-alpha smooth muscle actin, anti-GAPDH, and anti-Vimentin antibodies followed by HRP-conjugated goat anti-mouse antibodies. ImageQuant TL software was used to quantify protein expression, which was normalized to GAPDH.
2.9. Image Processing and Analysis
Imaging processing of collected images was carried out using MetaMorph software version 7.7 (Molecular Devices, Inc). Z-stacks images from each channel were integrated into single maximum intensity projection images and combined using the color overlay function. For morphometric measurements, Image J software was used to outline cells (manually) and calculate cell area, length, and aspect ratio.
2.10. Statistical Analysis
All statistical analysis was performed using GraphPad Prism 10. Two-way ANOVA was used to compare group means, and post hoc multiple comparisons tests were performed using the Sidak or Tukey methods. A p-value of less than 0.05 was considered statistically significant.
2.11. Proteomics
Protein was extracted as for western blotting as described previously. Protein was subjected to SDS-PAGE electrophoresis in mini-protean TGX gels for 20 minutes to cluster protein. Gels were stained with Coomassie dye for 1 hour and then washed twice with de-stain solution for 30 minutes. Gel bands were diced into 1 mm³ cubes and placed in Eppendorf tubes. Samples were digested overnight with trypsin (Pierce) following reduction and alkylation with DTT and iodoacetamide (Sigma–Aldrich). The samples then underwent solid-phase extraction cleanup with an Oasis HLB plate (Waters) and the resulting samples were injected onto a QExactive HF mass spectrometer coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system. Samples were injected onto a 75 um i.d., 15-cm long EasySpray column (Thermo) and eluted with a gradient from 0-28% buffer B over 90 minutes with a flow rate of 250 nL/min. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer operated in positive ion mode with a source voltage of 2.5 kV and an ion transfer tube temperature of 275 °C. MS scans were acquired at 120,000 resolution in the Orbitrap and up to 20 MS/MS spectra were obtained for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2-8. Dynamic exclusion was set for 20 s after an ion was selected for fragmentation.
Raw MS data files were analyzed using Proteome Discoverer v.3.0 (Thermo), with peptide identification performed using Sequest HT searching against the human reviewed protein database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.02 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification and oxidation of Met was set as a variable modification. The false-discovery rate (FDR) cutoff was 1% for all peptides.
3. Results
3.1. Knockout of Vimentin alters PDGF-Induced Elongation of Corneal Fibroblasts
3.1.1. Corneal Fibroblast Spreading on 2D Substrates
Studies have shown that vimentin intermediate filaments (VIFs) can modulate cellular extensions and contribute to cell movement and migration in other cell types [12,13]. To study the role of VIFs in the spreading of corneal fibroblasts, we proceeded to develop vimentin knock out (Vim KO) cells, culture them, and evaluate their spreading. We verified that vimentin is not present in Vim KO cells using PCR (not shown) and western blotting (Figure 1).
Our first set of experiments was carried out on 2D substrates with media containing PDGF or serum-free (SF) basal media. Cells were incubated for 4 and 24 hours. After just 4 hours of incubation, wild type cells, editing control cells, and Vim KO cells were all able to spread normally in SF media (Figure 2A). Although the cell area for the editing control cells was significantly higher than the cell area of wild type and Vim KO cells (Figure 2B). The addition of PDGF-BB induced an increase in cell length and/or the degree of elongation (aspect ratio) in wild type and control cells, consistent with previous studies. However, Vim KO cells did not show significant elongation in response to PDGF.
3.1.2. Corneal Fibroblast Spreading in 3D Collagen Matrices
We next evaluated the spreading of Vim KO cells in 3D collagen matrices. After 4 hours of incubation, wild type cells, editing control cells, and Vim KO cells were all able to spread within 3D matrices (Figure S1A). In SF media, the cell area and length were similar for all three cell types. However, PDGF-induced elongation was higher in wild type cells as compared to both editing control and Vim KO cells (Figure S1B). Incubation in 3D collagen matrices for 24 hours showed that all cell types continued to spread and elongate (Figure 4A). The addition of PDGF induced an increase in cell length and the degree of elongation (aspect ratio) in wild type and control cells. However, Vim KO cells did not show significant elongation in response to PDGF (Figure 4B).
Previous studies in our lab have shown that cell polarization and elongation is a common response induced by PDGF [22]. The results of both the 2D and 3D cell spreading experiments in the current study suggest while the ability of cells to spread is generally maintained in the absence of vimentin, the elongation response is somewhat mitigated in Vim KO cells.
3.2. Vim KO and Wild-Type Cells Produce Similar Amounts of Matrix Reorganization
Cells exert tractional forces on the matrix during spreading. To test if Vim KO cells exert similar amounts of tractional force as wild type cells, we used a collagen matrix contraction assay. In this assay, the height of an attached collagen matrix is reduced over time due to cell-induced matrix reorganization. Interestingly, wild type and KO cells induced similar amounts of matrix contraction in SF media, and a significant increase was observed in response to PDGF in both cell types (Figure 5).
3.3. Knockdown of Vimentin Does not Alter Corneal Fibroblast Spreading
To assess whether the ability of Vim KO cells to continue to spread in the absence of the protein was due to compensatory mechanisms developed over time, we evaluated the effect of transient vimentin knockdown on corneal fibroblast spreading (Figure 6). Vim KD cells, non-targeting control cell (NT) and wild-type cells were incubated in 3D collagen matrices as described above and labeled for F-actin. Interestingly, no obvious changes in the spreading of Vim KD cells and their response to PDGF were observed (Figure 7), consistent with our results showing only subtle changes in cell spreading in Vim KO cells.
3.4. Vimentin Is not Requred for Myofibroblast Transformation of Corneal Keratocytes
Recent evidence has shown that VIFs may play a role in the development of corneal fibrosis by inhibiting myofibroblast transformation in a mouse model [19,20]. In order to determine whether vimentin is required for myofibroblast transformation of human corneal fibroblasts, Vim KO and wild type cells were cultured on collagen coated dishes in the presence of TGFβ (a promoter of myofibroblasts transformation) and in basal SF media. Myofibroblasts are reparative cells that synthetize, secrete, and reorganize the ECM. Not all myofibroblast are alike, however they are characterized by the universal expression of αsmooth muscle actin (αSMA) [23]. Interestingly, corneal fibroblasts were able to transform into myofibroblasts in response to TGFβ in the absence of vimentin, as indicated by the development of prominent F-actin stress fibers and positive αSMA labeling (Figure 8A). However, the amount of positive αSMA labeling appeared to be reduced as compared to WT or control cells.
To quantify αSMA protein expression we conducted Western blot analysis. Westen blot images confirmed that αSMA protein was expressed in Vim KO cells (Figure 8B). Quantitative analysis indicated that the amount of αSMA expression induced by TGFβ was reduced in Vim KO cells, consistent with our imaging results.
3.5. Proteomics Shows Similar Expression Profiles for KO and Wild Type Cells
As shown above, vim KO cells expressed αSMA protein when cultured with TGFβ, albeit at lower levels than controls. Next, we wanted to investigate whether vim KO cells express other proteins associated with myofibroblast transformation of wild type cells. To answer this question, we conducted proteomics analysis of Vim KO, control, and wild type cells following culture in TGFβ. TGFβ is a growth factor known to induce myofibroblast transformation in vivo, and is produced by macrophages, epithelial cells, and fibroblasts in many organs and tissues [24]. TGFβ is also known to promote expression of extracellular matrix (ECM) proteins such as collagen 1 and V, fibronectin and the proteoglycans biglycan and versican, all of which are involved in wound repair and remodeling [24,25,26]. In addition, TGFβ is known to up-regulate tropomyosins, proteins which are responsible for incorporating αSMA into stress fibers [27]. TGFβ also increases the induction of connective tissue growth factor, a potent stimulator of myofibroblast differentiation and ECM production [28].
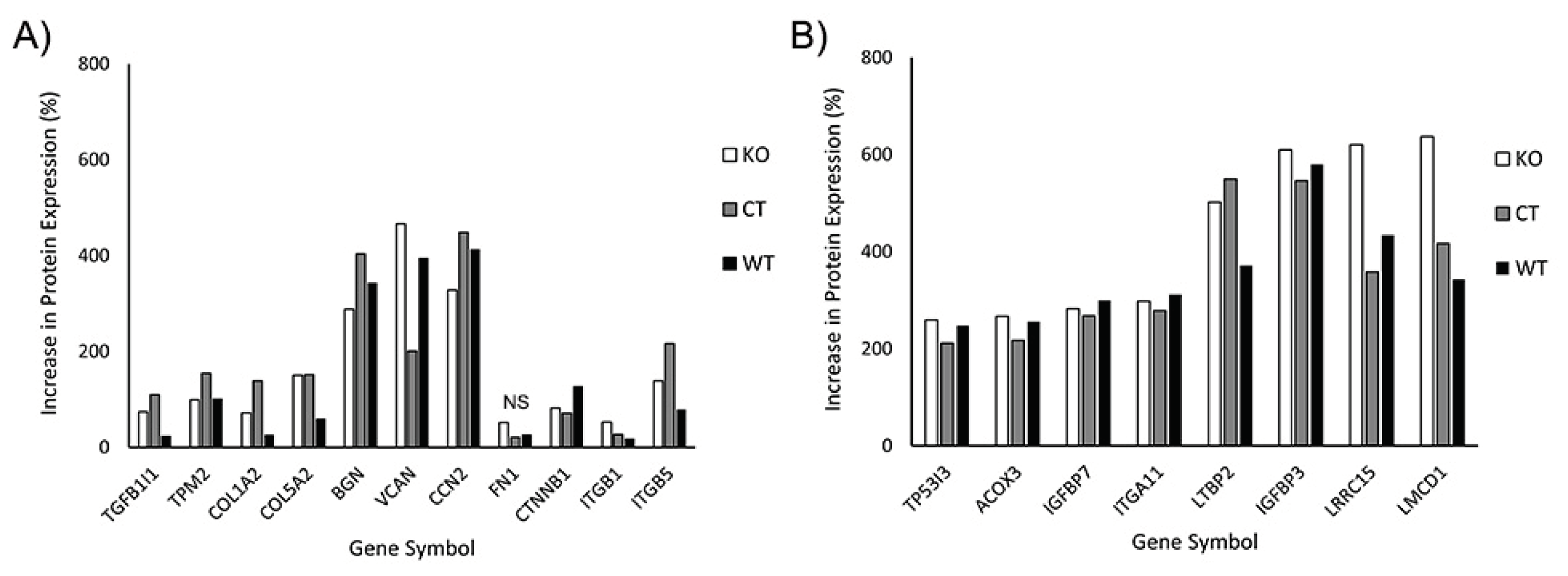
In our initial analysis, we evaluated the effects of TGFβ1 on expression of proteins known to be associated with myofibroblast transformation. This included transforming growth factor beta 1 induced transcript 1 (TGFB1I1) which regulates the TGFβ signaling pathway; Tropomyosin 2 (TPM2), collagen 1 and V (COL1A2, COL5A2), fibronectin (FN1), the proteoglycans biglycan and versican (BGN, VCAN), and connective tissue growth factor (CCN2). We also included additional proteins reported to be upregulated in corneal myofibroblasts, such as β-catenin (CTNNB1), and integrin β1 and β5 (ITGB1, ITGB5) [24,29]. Figure 9A shows the percentage increase in the above-mentioned proteins induced by culture in TGFβ1. Note that similar increases in protein expression were observed in WT, control and Vim KO cells, suggesting that Vim KO cells follow a similar differentiation pathway as control and wild type cells during myofibroblast transformation.
In our proteomics analysis, we found 8 additional proteins that had at least a two-fold increase in expression following culture in TGFβ1 (Figure 9B). Except for acyl-CoA oxidase 3 (ACOX3) protein, all other 7 proteins have been reported to play a potential role in myofibroblast behavior and/or fibrosis in other cell types. Specifically, Insulin like growth factor is often found embedded in the ECM where it interacts with matrix proteins, and insulin like growth factor binding proteins such as IGFBP7 play an essential role in this process [30]. Integrins such as ITGA11 are essential for strong cell-matrix attachments to resist highly contractile forces [31,32]. Tumor protein P53 inducible protein 3 (TP53I3) provides apoptotic resistance to myofibroblasts during fibrosis [33]. LMCD1 has been shown to be highly up-regulated by myofibroblasts in order to increase expression of ECM proteins and maintain high levels of αsma [34]. Latent transforming growth factor binding proteins (LTBP2) are essential for securing TGFβ to the ECM through the latency-associated pro-peptide, LAP [35,36]. Interestingly, LRRC15, a protein previously identified in cancer myofibroblasts [37], was highly expressed in corneal myofibroblasts, both with and without vimentin.
In general, the overall pattern of protein expression in response to TGFβ was similar in all three cell types. One protein that is found in fibrotic tissue and facilitates myofibroblast differentiation is periostin, POSTN [29,38]. Interestingly periostin expression was upregulated only in Vim KO cells, with an increase of 282%; no significant increase was observed in control or wild type cells.
4. Discussion
Vimentin has been associated with myofibroblast transformation of corneal keratocytes and the development of fibrosis in vivo [19,20,39,40], and studies in cell types suggest it can play a central role in regulating key aspects of cell mechanical behavior, including mechanosensing, polarization, and directional migration [12,15,20,41,42,43,44]. In this study, we explored the role of vimentin intermediate filaments in the spreading of corneal fibroblasts in 2D and 3D environments, and also investigated whether vimentin was required for their transition into myofibroblasts. To achieve our goal, we developed vimentin knock out corneal fibroblasts and did comparative experiments with wild type and control cells.
During cell spreading, there is a complex interactive process between the dynamics of the cytoskeletal filament proteins and the formation of focal adhesion complexes that attach cells to the substrate. Previous studies in other cell types suggest that vimentin null cells retain the ability to spread and exert tractional forces [45] but that vimentin plays a role in regulating cell polarization, as well as the orientation of these forces [42]. In our studies, the loss of vimentin intermediate filaments in corneal fibroblasts did not significantly influence their ability to spread on 2D collagen coated glass or inside 3D collage matrices. However, consistent with previous studies, decreased elongation (i.e., polarization) of Vim KO cells was observed in response to PDGF. It should be noted that the role of vimentin in regulating cell morphology is dependent on the stiffness of the substrate. [15]. Specifically, on compliant substrates where less actomyosin contractility is required, vimentin intermediate filaments play a more significant role in cell spreading. Interestingly, our results were similar on both rigid 2D substrates, and compliant 3D collagen matrices. One possible reason is that in addition to mechanical stiffness, matrix porosity may also modulate cell spreading and morphology in 3D collagen matrices [46]. In addition, it has been suggested that in 3D environments, cell density and matrix composition can also impact the spreading and contractility of vimentin null cells [15].
Our previous studies have shown that corneal fibroblasts develop focal adhesions and apply traction forces to the extracellular matrix to spread and/or migrate [47]. This process is recognized to involve the actomyosin machinery and the dynamics of vimentin in other cell types [48,49]. Our studies using the collagen matrix contraction model show that Vim KO cells and wild type cells produced a similar amount of matrix reorganization, suggesting that similar amounts of tractional force are generated by the cells. However, this model does not allow for the assessment of the pattern of tractional force generation in individual cells, or the dynamics of that process. Thus, there could still be differences in the subcellular pattern of tractional force generation between Vim KO and wild type cells. Given that the pattern of force generation generally correlates with the F-actin cytoskeletal organization, it is unlikely that large differences are present under the conditions used in this study.
Fibrosis in the cornea produces haze that can be observed at the site of the damaged tissue and impairs corneal transparency [16,17]. Haze is caused in part by transformation of corneal fibroblast to myofibroblasts, which secrete a disorganized, fibrotic ECM [17,18]. Studies with vimentin knockout mice have shown that fibroblast migration, wound contraction, and appearance of myofibroblasts are all delayed during the healing of incisional wounds in mouse embryos [50]. Vimentin has been reported to be overexpressed by myofibroblasts at the site of corneal alkali injury or laser injury in the rabbit [40,51]. The use of Withaferrin A (WTA), which blocks polymerization of vimentin, has also been shown to inhibit corneal fibrosis and improve corneal clarity during in vivo wound healing [20]. These and other studies suggest that vimentin could be used as a target to inhibit the development of corneal fibrosis [19,52]. However, one challenge with in vivo studies is that knockout models or WFA treatment can impact multiple cell types in the cornea which are known to interact during healing, and may also inhibit corneal fibroblast migration into the wound. Thus, the specific role of vimentin in myofibroblast transformation of corneal keratocytes is difficult to assess in vivo. In our study, absence of vimentin did not block myofibroblast transformation, since αSMA protein expression was detected by both western blot and fluorescent staining in Vim KO cells. Nonetheless, the degree of transformation and amount of αSMA protein was reduced as compared to control cells, consistent with previous studies indicating a role for vimentin in this process.
In our initial proteomics analysis, we evaluated the effects of TGFβ1 on expression of proteins known to be associated with myofibroblast transformation. This included transforming growth factor beta 1 induced transcript 1 (TGFB1I1) which regulates the TGFβ signaling pathway; Tropomyosin 2 (TPM2), collagen 1 and V (COL1A2, COL5A2), fibronectin (FN1), the proteoglycans biglycan and versican (BGN, VCAN), connective tissue growth factor (CCN2), β-catenin (CTNNB1), and integrin β1 and β5 (ITGB1, ITGB5) [24,29]. Similar increases in expression of these proteins were observed in WT, control and Vim KO cells, suggesting that Vim KO cells follow a similar differentiation pathway as control and wild type cells during myofibroblast transformation.
Our proteomics study also identified 8 additional proteins whose expression was highly upregulated in response to TGFβ in both Vim KO and control cells. These include Insulin like growth factor binding proteins 3 and 7 (IGFBP3 and IGFBP7). Insulin like growth factor (IGF) contributes to fibrosis by stimulating collagen production, and changes in IGF expression have been found in fibrotic tissue. IGF binding proteins (IGFBPs) are deposited in the ECM and they regulate, at least in part, the biological activity of IGF by mediating access of IGF to its receptor and potentiating its effect in tissues [53]. We found high levels of IGFBP 3 and 7, which have been reported in fibrosis of the lung and liver [54,55]. TGFβ has been shown to increase IGFBP3 production [54,56], which is essential for fibroblast to myofibroblast differentiation [57].
Another protein highly expressed in myofibroblasts was integrin α11. Integrin α11β1 is a collagen receptor, and has been reported to be involved in myofibroblast differentiation of other cell types [32,58]. Decreases in the levels of integrin α11 can reduce expression of αSMA, which is essential for the myofibroblast phenotype [58]. The latent transforming growth factor beta binding protein 2, LTBP2, was also highly expressed in both Vim KO and control cells. TGFβ is secreted with a latency-associated pro-peptide, LAP, which in this state is unable to bind its receptors and therefore remains latent. Latent TGFβ-binding proteins bind this complex and secure TGFβ to the ECM [35,36]. LTBP2 is overexpressed in myofibroblasts in fibrotic tissue, and studies have shown that silencing LTBP2 suppresses fibroblast-myofibroblast differentiation, whereas overexpression of LTBP2 promotes pulmonary fibroblast-myofibroblast differentiation even in the absence of TGFβ [59].
Another protein highly expressed in Vim KO and control cells is the tumor protein P53 inducible protein 3 (TP53I3), which is a potent transcription factor responsible for the induction of cell-cycle arrest, apoptosis, and senescence [60]. During pathological wound healing, myofibroblasts have been shown to adopt an apoptotic-resistant phenotype to perpetuate fibrosis [33]. Myofibroblast persistence leads to more ECM deposition, remodeling, tissue contraction, and formation of pathological scars. Increased expression of TP53I3 may help prevent persistent fibrosis through a negative feedback loop.
Recently, myofibroblast populations identified in cancer have been characterized by the expression of the leucine rich repeat containing protein 15, LRRC15, and are known as LRRC15 myofibroblasts [61,62]. Interestingly, LRRC15 was highly expressed by both Vim KO and control cells in our study. In cancer, the differentiation of fibroblasts into LRRC15 myofibroblasts is carried out through TGFβ signaling [37]. It is remarkable to note the cells which transformed into myofibroblasts in our study were not derived from any cancer lineage, suggesting for the first time that LRRC15 is more universal and not just restricted to cancer fibroblast lineage.
Another protein that was highly expressed in both Vim KO and control cells was the LIM and cysteine rich domains 1, LMCD1, which is a transcription factor. Evidence suggests that LMCD1 may facilitate profibrotic gene expression and in tissue fibrosis, it is found to be consistently elevated in pulmonary and renal fibrosis [34,63]. Depletion of LMCD1 expression has been shown to reduce the expression of ECM proteins and αSMA. On the other hand, high levels of LMCD1 correlate with high levels of ECM and αSMA protein, features of tissue fibrosis [34].
In summary, our proteomics study shows that many proteins associated with fibrosis were highly expressed in both Vim KO and control cells, suggesting that the VIFs were not essential for corneal fibroblasts to be in this state. Interestingly, our proteomics analysis also found that Periostin, a protein that has been associated with fibrosis and myofibroblast differentiation in other organ systems [29,38] was highly expressed in Vim KO cells, but not control cells. Additional studies are needed to determine the functional role of this protein in corneal myofibroblasts and its relationship to vimentin. Furthermore, the identification of LRRC15 as a potential marker and/or regulator of corneal myofibroblast transformation warrants further investigation.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Vim KO cell spreading in 3D collagen matrices for 4 hours in the presence of PDGF and SF media.
Author Contributions
Conceptualization, Miguel Miron-Mendoza, John Hulleman and and W. Matthew Petroll; Data curation, Sophie DiCesare, Emi Nakahari and Meet Bhatt; Formal analysis, Kara Poole, Meet Bhatt and and W. Matthew Petroll; Funding acquisition, and W. Matthew Petroll; Investigation, Miguel Miron-Mendoza, Sophie DiCesare and Emi Nakahari; Methodology, Miguel Miron-Mendoza, Sophie DiCesare, Emi Nakahari, John Hulleman and and W. Matthew Petroll; Project administration, and W. Matthew Petroll; Supervision, John Hulleman and and W. Matthew Petroll; Writing – original draft, Miguel Miron-Mendoza and Kara Poole; Writing – review & editing, Miguel Miron-Mendoza, Kara Poole and and W. Matthew Petroll.
Funding
This research was funded by NIH grants R01 EY013322 and P30 EY030413, and a Challenge Grant from Research to Prevent Blindness, Inc.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is available upon request.
Acknowledgments
The authors wish to thank the UT Southwestern Proteomics Core and its director Prof. Andrew Lemoff for the invaluable help in processing and analyzing our proteomics data.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hohmann, T.; Dehghani, F. The Cytoskeleton-A Complex Interacting Meshwork. Cells 2019, 8. [Google Scholar] [CrossRef]
- Ingber, D. E. Tensegrity and mechanotransduction. J Bodyw Mov Ther 2008, 12, 198–200. [Google Scholar] [CrossRef]
- Bear, J. E.; Haugh, J. M. Directed migration of mesenchymal cells: where signaling and the cytoskeleton meet. Curr Opin Cell Biol 2014, 30, 74–82. [Google Scholar] [CrossRef]
- McIntosh, J. R. Mitosis. Cold Spring Harb Perspect Biol 2016, 8. [Google Scholar] [CrossRef]
- Seetharaman, S.; Etienne-Manneville, S. Cytoskeletal Crosstalk in Cell Migration. Trends Cell Biol 2020, 30, 720–735. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Horwitz, A. R. Adhesion dynamics at a glance. J Cell Sci 2011, 124 Pt 23, 3923–3927. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K. A.; Donato, D. M.; Balcioglu, H. E.; Schmidt, T.; Danen, E. H.; Koenderink, G. H. A guide to mechanobiology: Where biology and physics meet. Biochim Biophys Acta 2015, 1853, 3043–3052. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S. Cytoplasmic Intermediate Filaments in Cell Biology. Annu Rev Cell Dev Biol 2018, 34, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Ridge, K. M.; Shumaker, D.; Robert, A.; Hookway, C.; Gelfand, V. I.; Janmey, P. A.; Lowery, J.; Guo, M.; Weitz, D. A.; Kuczmarski, E.; et al. Methods for Determining the Cellular Functions of Vimentin Intermediate Filaments. Methods Enzymol 2016, 568, 389–426. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M. V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S. M.; Herzog, E. L.; Driskell, R. R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Lin, J. J.; Feramisco, J. R. Disruption of the in vivo distribution of the intermediate filaments in fibroblasts through the microinjection of a specific monoclonal antibody. Cell 1981, 24, 185–193. [Google Scholar] [CrossRef]
- Ding, I.; Ostrowska-Podhorodecka, Z.; Lee, W.; Liu, R. S. C.; Carneiro, K.; Janmey, P. A.; McCulloch, C. A. Cooperative roles of PAK1 and filamin A in regulation of vimentin assembly and cell extension formation. Biochim Biophys Acta Mol Cell Res 2020, 1867. [Google Scholar] [CrossRef]
- Karoii, D. H.; Azizi, H.; Amirian, M. Signaling Pathways and Protein-Protein Interaction of Vimentin in Invasive and Migration Cells: A Review. Cell Reprogram 2022, 24, 165–174. [Google Scholar] [CrossRef]
- Ostrowska-Podhorodecka, Z.; McCulloch, C. A. Vimentin regulates the assembly and function of matrix adhesions. Wound Repair Regen 2021, 29, 602–612. [Google Scholar] [CrossRef]
- Mendez, M. G.; Restle, D.; Janmey, P. A. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys J 2014, 107, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Kivanany, P. B.; Grose, K. C.; Tippani, M.; Su, S.; Petroll, W. M. Assessment of Corneal Stromal Remodeling and Regeneration after Photorefractive Keratectomy. Sci Rep 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Netto, M. V.; Mohan, R. R.; Sinha, S.; Sharma, A.; Dupps, W.; Wilson, S. E. Stromal haze, myofibroblasts, and surface irregularity after PRK. Exp Eye Res 2006, 82, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Kivanany, P. B.; Grose, K. C.; Petroll, W. M. Temporal and spatial analysis of stromal cell and extracellular matrix patterning following lamellar keratectomy. Exp Eye Res 2016, 153, 56–64. [Google Scholar] [CrossRef]
- Das, S. K.; Gupta, I.; Cho, Y. K.; Zhang, X.; Uehara, H.; Muddana, S. K.; Bernhisel, A. A.; Archer, B.; Ambati, B. K. Vimentin knockdown decreases corneal opacity. Invest Ophthalmol Vis Sci 2014, 55, 4030–4040. [Google Scholar] [CrossRef]
- Bargagna-Mohan, P.; Lei, L.; Thompson, A.; Shaw, C.; Kasahara, K.; Inagaki, M.; Mohan, R. Vimentin Phosphorylation Underlies Myofibroblast Sensitivity to Withaferin A In Vitro and during Corneal Fibrosis. PLoS One 2015, 10, e0133399. [Google Scholar] [CrossRef]
- Jester, J. V.; Huang, J.; Fisher, S.; Spiekerman, J.; Chang, J. H.; Wright, W. E.; Shay, J. W. Myofibroblast differentiation of normal human keratocytes and hTERT, extended-life human corneal fibroblasts. Invest Ophthalmol Vis Sci 2003, 44, 1850–1858. [Google Scholar] [CrossRef]
- Miron-Mendoza, M.; Lin, X.; Ma, L.; Ririe, P.; Petroll, W. M. Individual versus collective fibroblast spreading and migration: regulation by matrix composition in 3D culture. Exp Eye Res 2012, 99, 36–44. [Google Scholar] [CrossRef]
- Younesi, F. S.; Son, D. O.; Firmino, J.; Hinz, B. Myofibroblast Markers and Microscopy Detection Methods in Cell Culture and Histology. Methods Mol Biol 2021, 2299, 17–47. [Google Scholar] [CrossRef] [PubMed]
- Kim, K. K.; Sheppard, D.; Chapman, H. A. TGF-beta1 Signaling and Tissue Fibrosis. Cold Spring Harb Perspect Biol 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Malmstrom, J.; Lindberg, H.; Lindberg, C.; Bratt, C.; Wieslander, E.; Delander, E. L.; Sarnstrand, B.; Burns, J. S.; Mose-Larsen, P.; Fey, S.; et al. Transforming growth factor-beta 1 specifically induce proteins involved in the myofibroblast contractile apparatus. Mol Cell Proteomics 2004, 3, 466–477. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-beta Signaling. Biomolecules 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Prunotto, M.; Bruschi, M.; Gunning, P.; Gabbiani, G.; Weibel, F.; Ghiggeri, G. M.; Petretto, A.; Scaloni, A.; Bonello, T.; Schevzov, G.; et al. Stable incorporation of alpha-smooth muscle actin into stress fibers is dependent on specific tropomyosin isoforms. Cytoskeleton (Hoboken) 2015, 72, 257–267. [Google Scholar] [CrossRef]
- Yanagihara, T.; Tsubouchi, K.; Gholiof, M.; Chong, S. G.; Lipson, K. E.; Zhou, Q.; Scallan, C.; Upagupta, C.; Tikkanen, J.; Keshavjee, S.; et al. Connective-Tissue Growth Factor Contributes to TGF-beta1-induced Lung Fibrosis. Am J Respir Cell Mol Biol 2022, 66, 260–270. [Google Scholar] [CrossRef]
- Layton, T. B.; Williams, L.; McCann, F.; Zhang, M.; Fritzsche, M.; Colin-York, H.; Cabrita, M.; Ng, M. T. H.; Feldmann, M.; Sansom, S. N.; et al. Cellular census of human fibrosis defines functionally distinct stromal cell types and states. Nat Commun 2020, 11, 2768. [Google Scholar] [CrossRef]
- Taipale, J.; Keski-Oja, J. Growth factors in the extracellular matrix. FASEB J 1997, 11, 51–59. [Google Scholar] [CrossRef]
- Tschumperlin, D. J. Matrix, mesenchyme, and mechanotransduction. Ann Am Thorac Soc 2015, 1 (Suppl 1), S24–29. [Google Scholar] [CrossRef]
- Carracedo, S.; Lu, N.; Popova, S. N.; Jonsson, R.; Eckes, B.; Gullberg, D. The fibroblast integrin alpha11beta1 is induced in a mechanosensitive manner involving activin A and regulates myofibroblast differentiation. J Biol Chem 2010, 285, 10434–10445. [Google Scholar] [CrossRef]
- McElhinney, K.; Irnaten, M.; O'Brien, C. p53 and Myofibroblast Apoptosis in Organ Fibrosis. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Bogatkevich, G. S.; Atanelishvili, I.; Bogatkevich, A. M.; Silver, R. M. Critical Role of LMCD1 in Promoting Profibrotic Characteristics of Lung Myofibroblasts in Experimental and Scleroderma-Associated Lung Fibrosis. Arthritis Rheumatol 2023, 75, 438–448. [Google Scholar] [CrossRef]
- Roman, J. Fibroblasts-Warriors at the Intersection of Wound Healing and Disrepair. Biomolecules 2023, 13. [Google Scholar] [CrossRef]
- Munger, J. S.; Huang, X.; Kawakatsu, H.; Griffiths, M. J.; Dalton, S. L.; Wu, J.; Pittet, J. F.; Kaminski, N.; Garat, C.; Matthay, M. A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Krishnamurty, A. T.; Shyer, J. A.; Thai, M.; Gandham, V.; Buechler, M. B.; Yang, Y. A.; Pradhan, R. N.; Wang, A. W.; Sanchez, P. L.; Qu, Y.; et al. LRRC15(+) myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature 2022, 611, 148–154. [Google Scholar] [CrossRef]
- Nikoloudaki, G.; Snider, P.; Simmons, O.; Conway, S. J.; Hamilton, D. W. Periostin and matrix stiffness combine to regulate myofibroblast differentiation and fibronectin synthesis during palatal healing. Matrix Biol 2020, 94, 31–56. [Google Scholar] [CrossRef] [PubMed]
- Bargagna-Mohan, P.; Paranthan, R. R.; Hamza, A.; Zhan, C. G.; Lee, D. M.; Kim, K. B.; Lau, D. L.; Srinivasan, C.; Nakayama, K.; Nakayama, K. I.; et al. Corneal antifibrotic switch identified in genetic and pharmacological deficiency of vimentin. J Biol Chem 2012, 287, 989–1006. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, S. S.; Kaur, H.; de Medeiros, F. W.; Smith, S. D.; Wilson, S. E. Dynamics of the expression of intermediate filaments vimentin and desmin during myofibroblast differentiation after corneal injury. Exp Eye Res 2009, 89, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Ding, L.; Burckhardt, C. J.; Lowery, J.; Zaritsky, A.; Sitterley, K.; Mota, A.; Costigliola, N.; Starker, C. G.; Voytas, D. F.; et al. Vimentin Intermediate Filaments Template Microtubule Networks to Enhance Persistence in Cell Polarity and Directed Migration. Cell Syst 2016, 3, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Costigliola, N.; Ding, L.; Burckhardt, C. J.; Han, S. J.; Gutierrez, E.; Mota, A.; Groisman, A.; Mitchison, T. J.; Danuser, G. Vimentin fibers orient traction stress. Proc Natl Acad Sci U S A 2017, 114, 5195–5200. [Google Scholar] [CrossRef] [PubMed]
- Murray, M. E.; Mendez, M. G.; Janmey, P. A. Substrate stiffness regulates solubility of cellular vimentin. Mol Biol Cell 2014, 25, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Patteson, A. E.; Pogoda, K.; Byfield, F. J.; Mandal, K.; Ostrowska-Podhorodecka, Z.; Charrier, E. E.; Galie, P. A.; Deptula, P.; Bucki, R.; McCulloch, C. A.; et al. Loss of Vimentin Enhances Cell Motility through Small Confining Spaces. Small 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- van Loosdregt, I.; Weissenberger, G.; van Maris, M.; Oomens, C. W. J.; Loerakker, S.; Stassen, O.; Bouten, C. V. C. The Mechanical Contribution of Vimentin to Cellular Stress Generation. J Biomech Eng 2018, 140. [Google Scholar] [CrossRef]
- Miron-Mendoza, M.; Seemann, J.; Grinnell, F. The differential regulation of cell motile activity through matrix stiffness and porosity in three dimensional collagen matrices. Biomaterials 2010, 31, 6425–6435. [Google Scholar] [CrossRef] [PubMed]
- Miron-Mendoza, M.; Vazquez, D.; Garcia-Ramila, N.; Ikebe, H. R.; Petroll, W. M. Coupling of Fibrin Reorganization and Fibronectin Patterning by Corneal Fibroblasts in Response to PDGF BB and TGFbeta1. Bioengineering (Basel) 2020, 7. [Google Scholar] [CrossRef]
- Ndiaye, A. B.; Koenderink, G. H.; Shemesh, M. Intermediate Filaments in Cellular Mechanoresponsiveness: Mediating Cytoskeletal Crosstalk From Membrane to Nucleus and Back. Front Cell Dev Biol 2022, 10, 882037. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Gonzalez, A. M.; Debiase, P. J.; Trejo, H. E.; Goldman, R. D.; Flitney, F. W.; Jones, J. C. Recruitment of vimentin to the cell surface by beta3 integrin and plectin mediates adhesion strength. J Cell Sci 2009, 122 Pt 9, 1390–1400. [Google Scholar] [CrossRef]
- Eckes, B.; Colucci-Guyon, E.; Smola, H.; Nodder, S.; Babinet, C.; Krieg, T.; Martin, P. Impaired wound healing in embryonic and adult mice lacking vimentin. J Cell Sci 2000, 113 ( Pt 13) Pt 13, 2455–2462. [Google Scholar] [CrossRef]
- Ishizaki, M.; Zhu, G.; Haseba, T.; Shafer, S. S.; Kao, W. W. Expression of collagen I, smooth muscle alpha-actin, and vimentin during the healing of alkali-burned and lacerated corneas. Invest Ophthalmol Vis Sci 1993, 34, 3320–3328. [Google Scholar]
- Mohan, R.; Bargagna-Mohan, P. The Use of Withaferin A to Study Intermediate Filaments. Methods Enzymol 2016, 568, 187–218. [Google Scholar] [CrossRef] [PubMed]
- Hwa, V.; Oh, Y.; Rosenfeld, R. G. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr Rev 1999, 20, 761–787. [Google Scholar] [CrossRef]
- Pilewski, J. M.; Liu, L.; Henry, A. C.; Knauer, A. V.; Feghali-Bostwick, C. A. Insulin-like growth factor binding proteins 3 and 5 are overexpressed in idiopathic pulmonary fibrosis and contribute to extracellular matrix deposition. Am J Pathol 2005, 166, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Castillo, M.; Rosique-Oramas, D.; Medina-Avila, Z.; Perez-Hernandez, J. L.; Higuera-De la Tijera, F.; Santana-Vargas, D.; Montalvo-Jave, E. E.; Sanchez-Avila, F.; Torre, A.; Kershenobich, D.; et al. Differential production of insulin-like growth factor-binding proteins in liver fibrosis progression. Mol Cell Biochem 2020, 469, (1–2). [Google Scholar] [CrossRef]
- Izumi, K.; Kurosaka, D.; Iwata, T.; Oguchi, Y.; Tanaka, Y.; Mashima, Y.; Tsubota, K. Involvement of insulin-like growth factor-I and insulin-like growth factor binding protein-3 in corneal fibroblasts during corneal wound healing. Invest Ophthalmol Vis Sci 2006, 47, 591–598. [Google Scholar] [CrossRef]
- Sampson, N.; Zenzmaier, C.; Heitz, M.; Hermann, M.; Plas, E.; Schafer, G.; Klocker, H.; Berger, P. Stromal insulin-like growth factor binding protein 3 (IGFBP3) is elevated in the diseased human prostate and promotes ex vivo fibroblast-to-myofibroblast differentiation. Endocrinology 2013, 154, 2586–2599. [Google Scholar] [CrossRef]
- Talior-Volodarsky, I.; Connelly, K. A.; Arora, P. D.; Gullberg, D.; McCulloch, C. A. alpha11 integrin stimulates myofibroblast differentiation in diabetic cardiomyopathy. Cardiovasc Res 2012, 96, 265–275. [Google Scholar] [CrossRef]
- Zou, M.; Zou, J.; Hu, X.; Zheng, W.; Zhang, M.; Cheng, Z. Latent Transforming Growth Factor-beta Binding Protein-2 Regulates Lung Fibroblast-to-Myofibroblast Differentiation in Pulmonary Fibrosis via NF-kappaB Signaling. Front Pharmacol 2021, 12, 788714. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K. T.; Mello, S. S.; Attardi, L. D. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lovrot, J.; Larsson, C.; Sommarin, M.; Madsen, C. D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun 2018, 9, 5150. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C. X.; Muller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T. W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov 2020, 10, 232–253. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Tian, M.; He, P.; Chen, J.; Zhao, Z.; Zhang, Y.; Zhang, B. Suppression of LMCD1 ameliorates renal fibrosis by blocking the activation of ERK pathway. Biochim Biophys Acta Mol Cell Res 2022, 1869, 119200. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Western blot showing lack of vimentin protein expression in Vimentin knockout (KO) cells as compared to wild type (WT) and controls.
Figure 1.
Western blot showing lack of vimentin protein expression in Vimentin knockout (KO) cells as compared to wild type (WT) and controls.
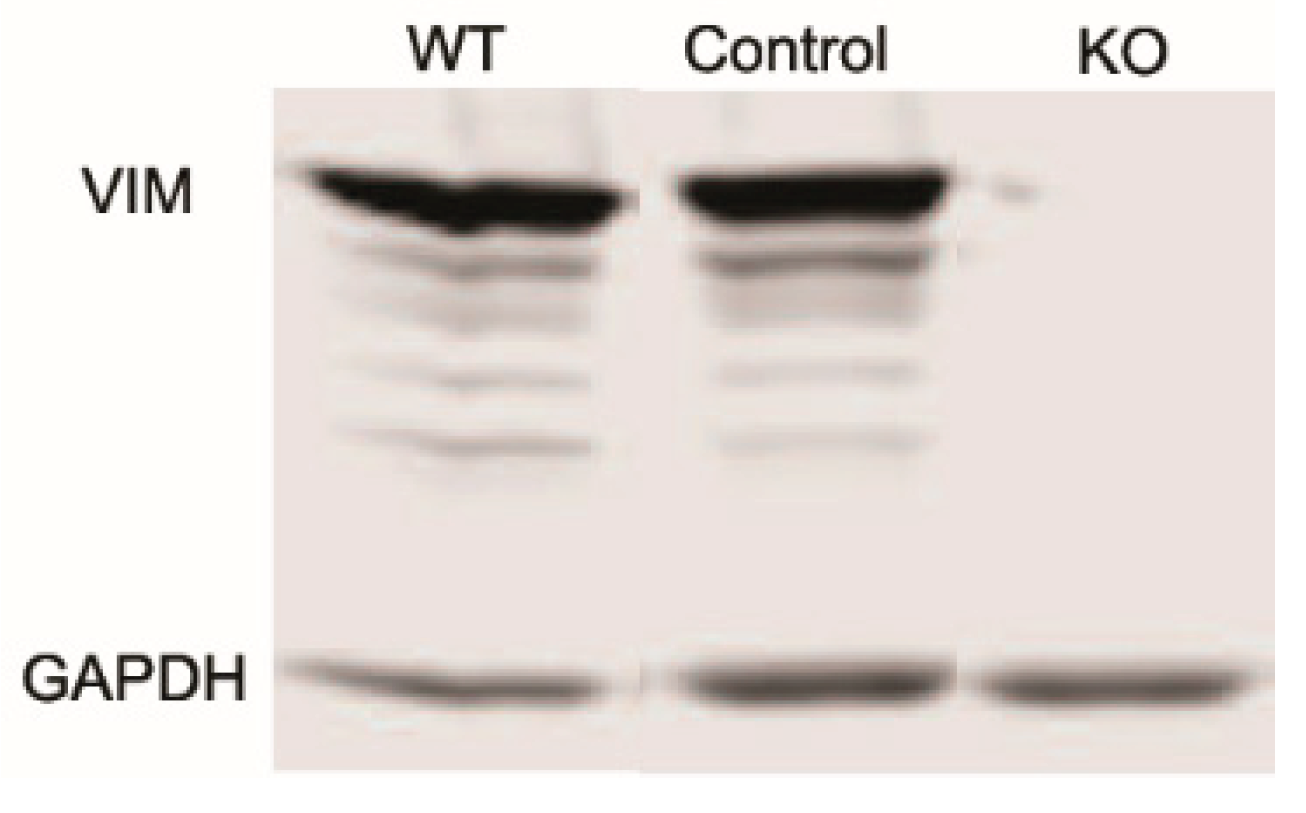
Figure 2.
The effect of Vim KO on corneal fibroblasts spreading on 2D substrates following culture for 4 hours in the presence of serum-free (SF) media with or without the addition of PDGF. A) Representative pictures of samples that were fixed and labeled for F-actin (red) and nuclei (blue). Scale Bar is 50 μm. B) Graphs showing morphological data from 3 independent experiments. Error bars show standard deviations.
Figure 2.
The effect of Vim KO on corneal fibroblasts spreading on 2D substrates following culture for 4 hours in the presence of serum-free (SF) media with or without the addition of PDGF. A) Representative pictures of samples that were fixed and labeled for F-actin (red) and nuclei (blue). Scale Bar is 50 μm. B) Graphs showing morphological data from 3 independent experiments. Error bars show standard deviations.
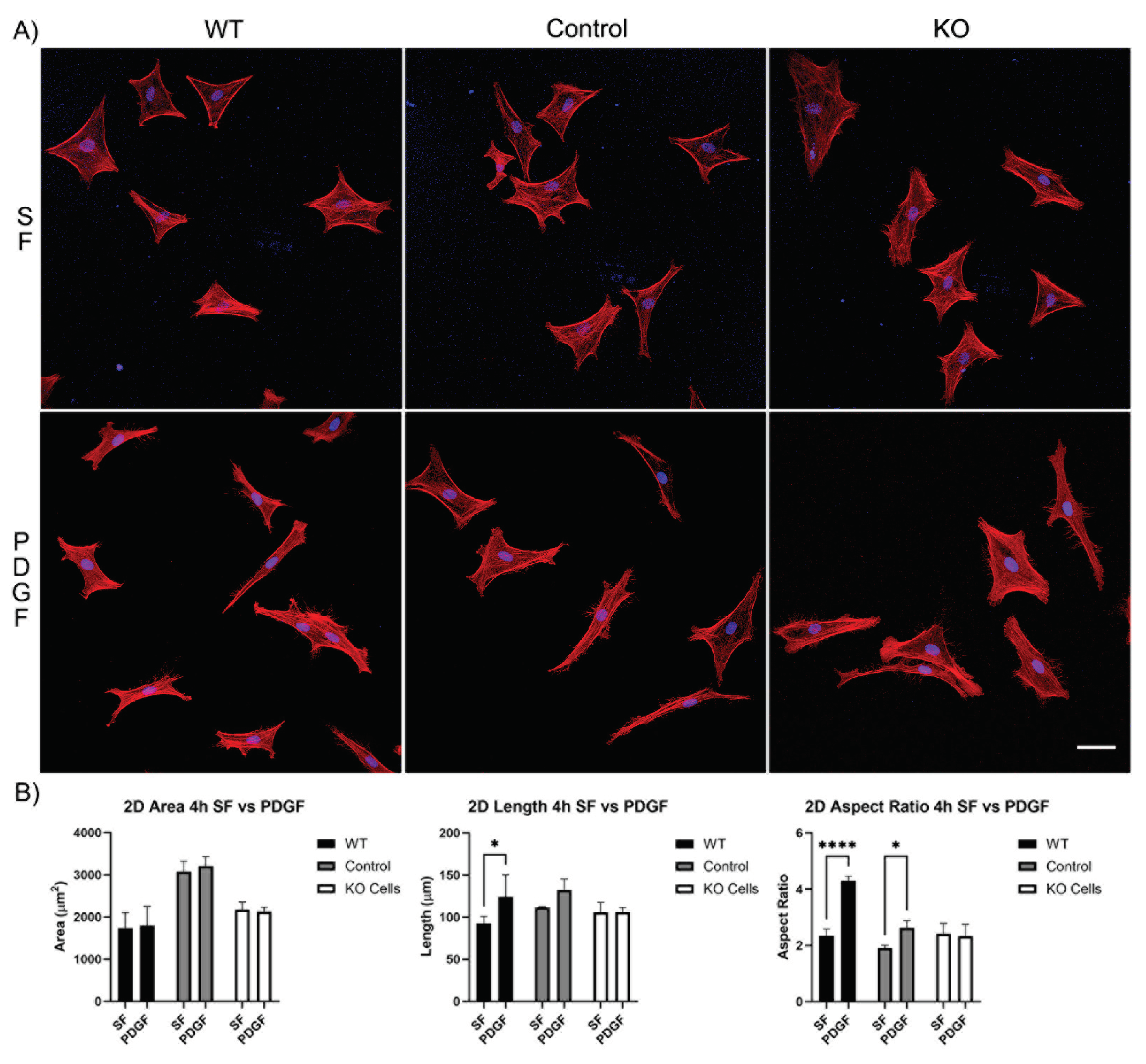
Figure 3.
The effect of Vim KO on corneal fibroblasts spreading on 2D substrates following culture for 24 hours in the presence of serum-free (SF) media with or without the addition of PDGF. A) Representative pictures of samples that were fixed and labeled for F-actin (red). Scale Bar is 50 μm. B) Graphs showing morphological data from 3 independent experiments. Error bars show standard deviations.
Figure 3.
The effect of Vim KO on corneal fibroblasts spreading on 2D substrates following culture for 24 hours in the presence of serum-free (SF) media with or without the addition of PDGF. A) Representative pictures of samples that were fixed and labeled for F-actin (red). Scale Bar is 50 μm. B) Graphs showing morphological data from 3 independent experiments. Error bars show standard deviations.
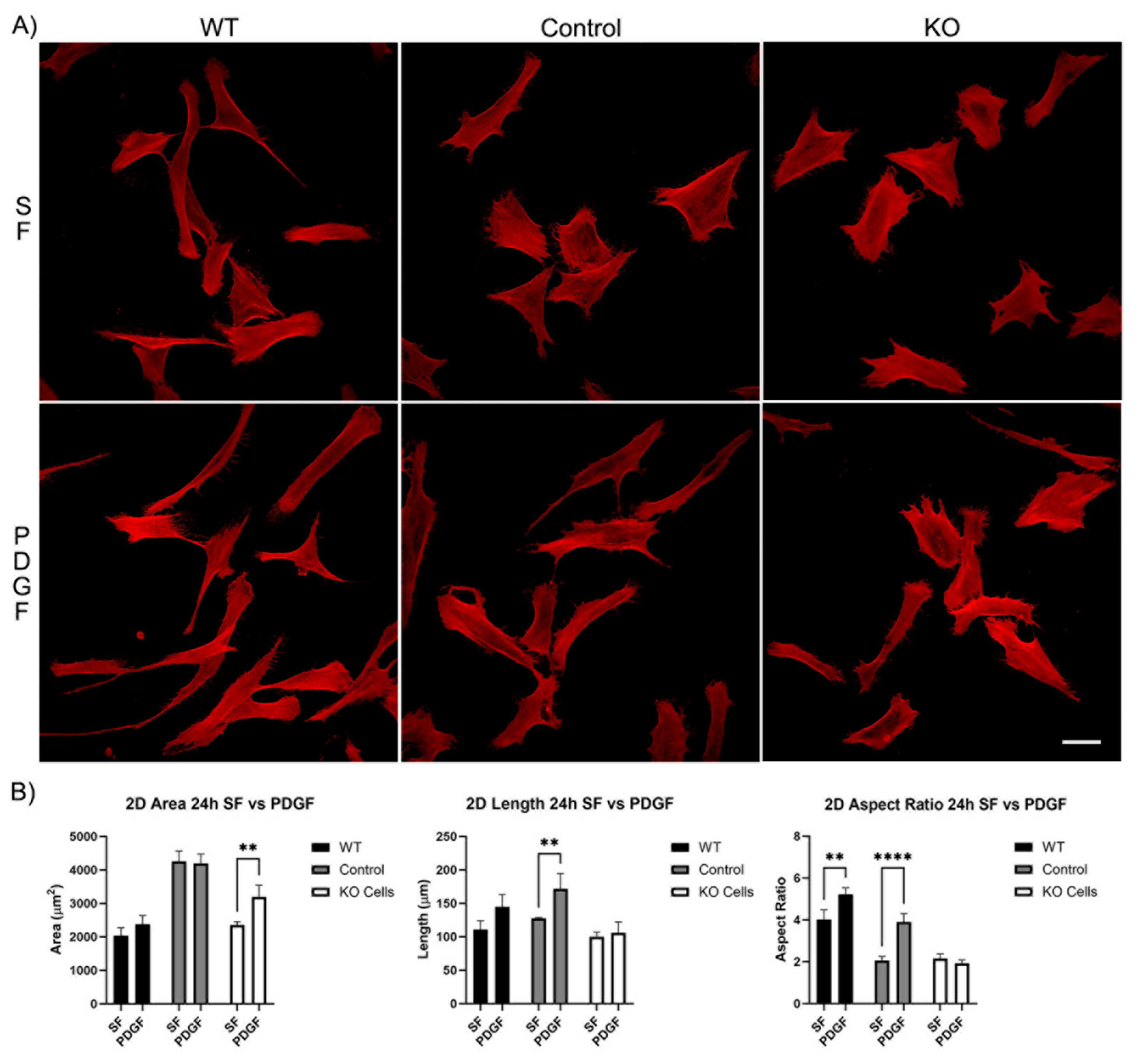
Figure 4.
Vim KO cell spreading in 3D collagen matrices for 24 hours in the presence of PDGF and SF media. A) Representative pictures of samples that were fixed and labeled for F-actin. Scale Bar is 50 μm. B) Graphs are morphological data from 3 different experiments.
Figure 4.
Vim KO cell spreading in 3D collagen matrices for 24 hours in the presence of PDGF and SF media. A) Representative pictures of samples that were fixed and labeled for F-actin. Scale Bar is 50 μm. B) Graphs are morphological data from 3 different experiments.
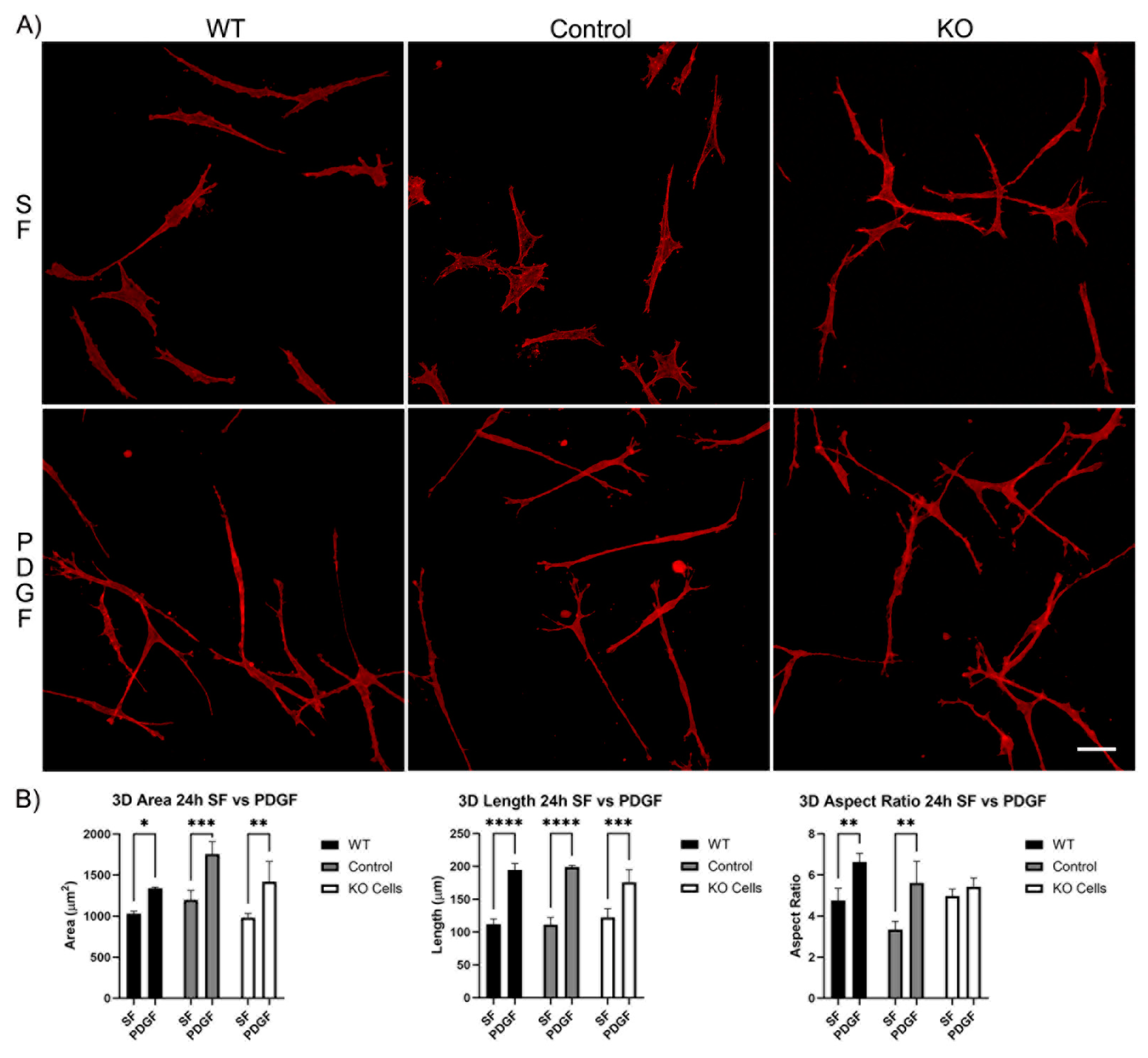
Figure 5.
Collagen matrix contraction induced by wild type (WT) and Vim KO cells. Graphs are data from 3 different experiments.
Figure 5.
Collagen matrix contraction induced by wild type (WT) and Vim KO cells. Graphs are data from 3 different experiments.
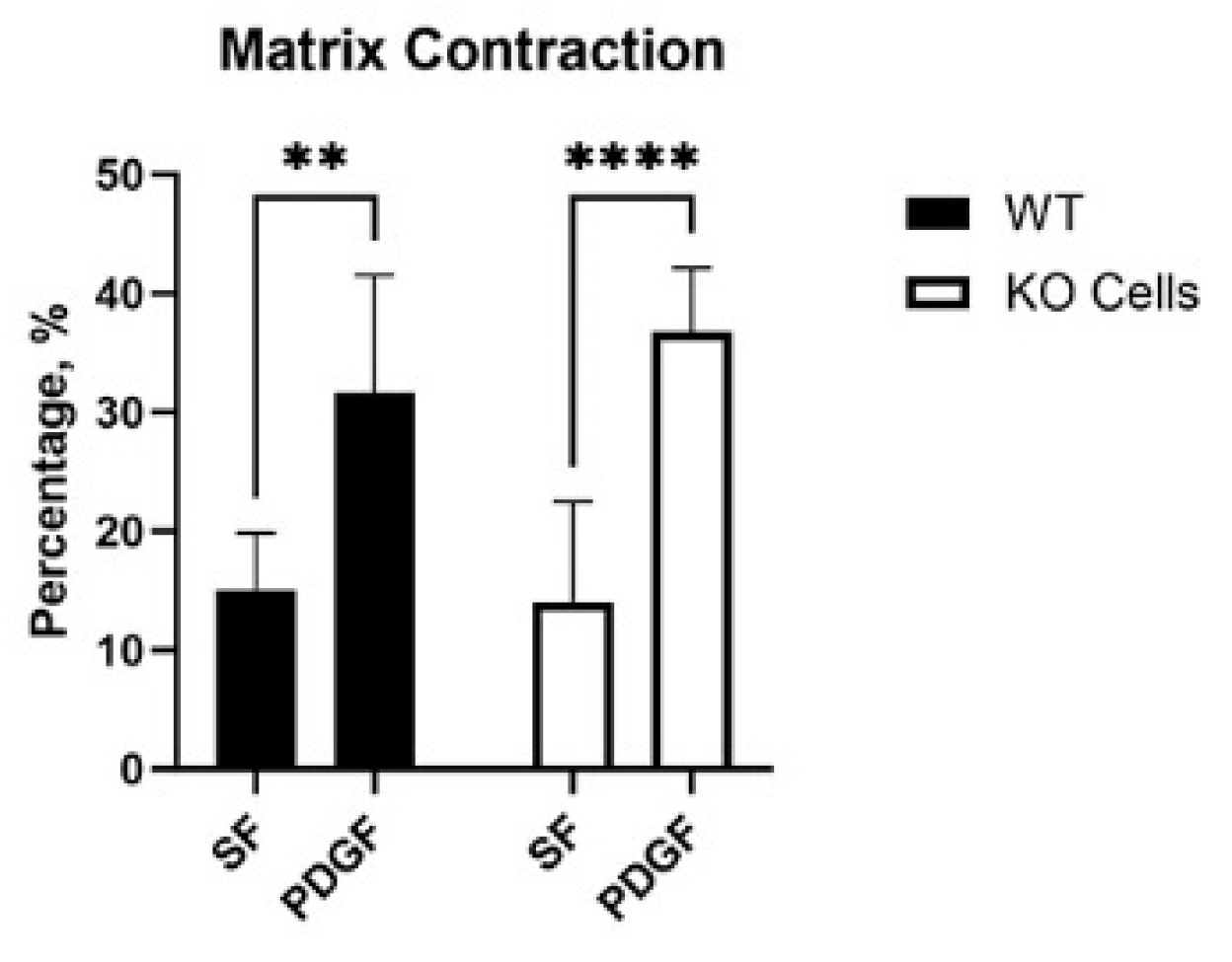
Figure 6.
Western blot showing the effect of double siRNA reverse transfection of vimentin in human corneal fibroblasts. Transfection reduced vimentin protein expression by 82.2%.
Figure 6.
Western blot showing the effect of double siRNA reverse transfection of vimentin in human corneal fibroblasts. Transfection reduced vimentin protein expression by 82.2%.
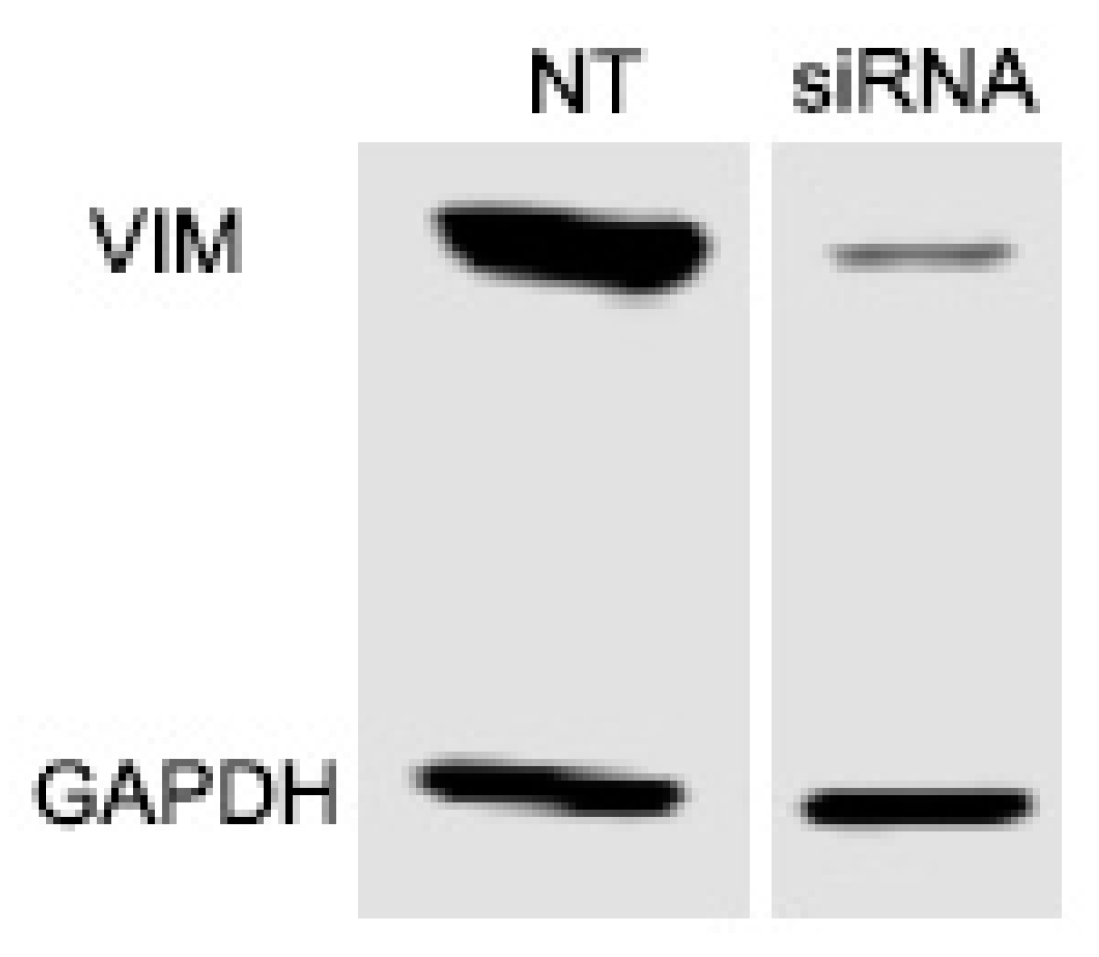
Figure 7.
Vim KD cell spreading in 3D collagen matrices for 24 hours in the presence of PDGF and SF media. A) Representative pictures of cells that were fixed and labeled for F-actin. Scale bar is 50 μm.
Figure 7.
Vim KD cell spreading in 3D collagen matrices for 24 hours in the presence of PDGF and SF media. A) Representative pictures of cells that were fixed and labeled for F-actin. Scale bar is 50 μm.
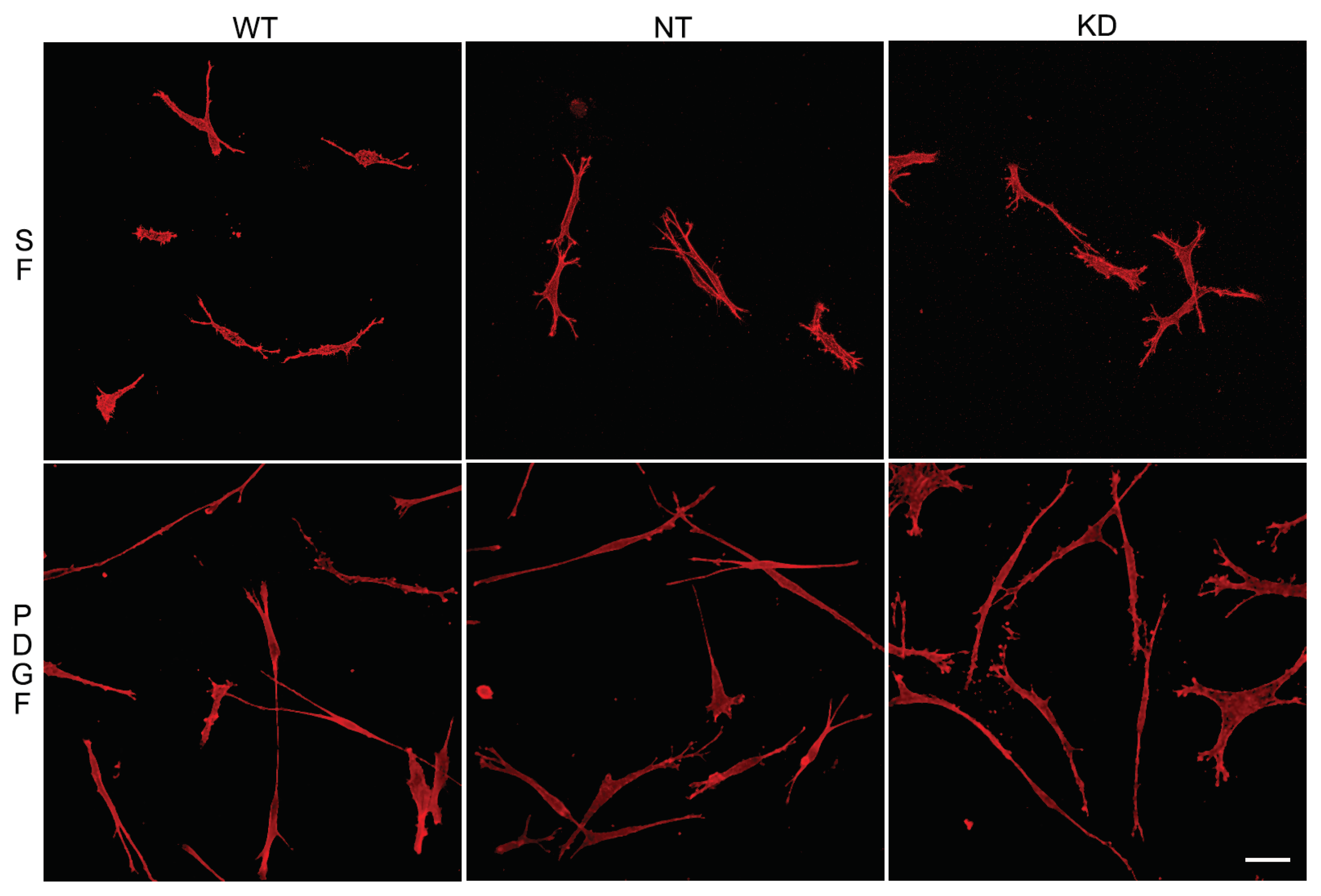
Figure 8.
Vim KO cells transform into myofibroblasts. A) Representative pictures of corneal fibroblasts cultured on 2D substrates for 6 days in serum-free media (SF) with or without TGFβ1. After incubation, cells were fixed and labeled for F-actin (red in top panel), αSMA (green in top panel), vimentin (cyan in bottom panel), and nuclei (red in bottom panel). Bar is 100 μm. B) Western blot shows αsma protein expression following incubation in TGFβ for all cell types. C) Quantification of protein expression from western blots. Data from 3 independent experiments.
Figure 8.
Vim KO cells transform into myofibroblasts. A) Representative pictures of corneal fibroblasts cultured on 2D substrates for 6 days in serum-free media (SF) with or without TGFβ1. After incubation, cells were fixed and labeled for F-actin (red in top panel), αSMA (green in top panel), vimentin (cyan in bottom panel), and nuclei (red in bottom panel). Bar is 100 μm. B) Western blot shows αsma protein expression following incubation in TGFβ for all cell types. C) Quantification of protein expression from western blots. Data from 3 independent experiments.
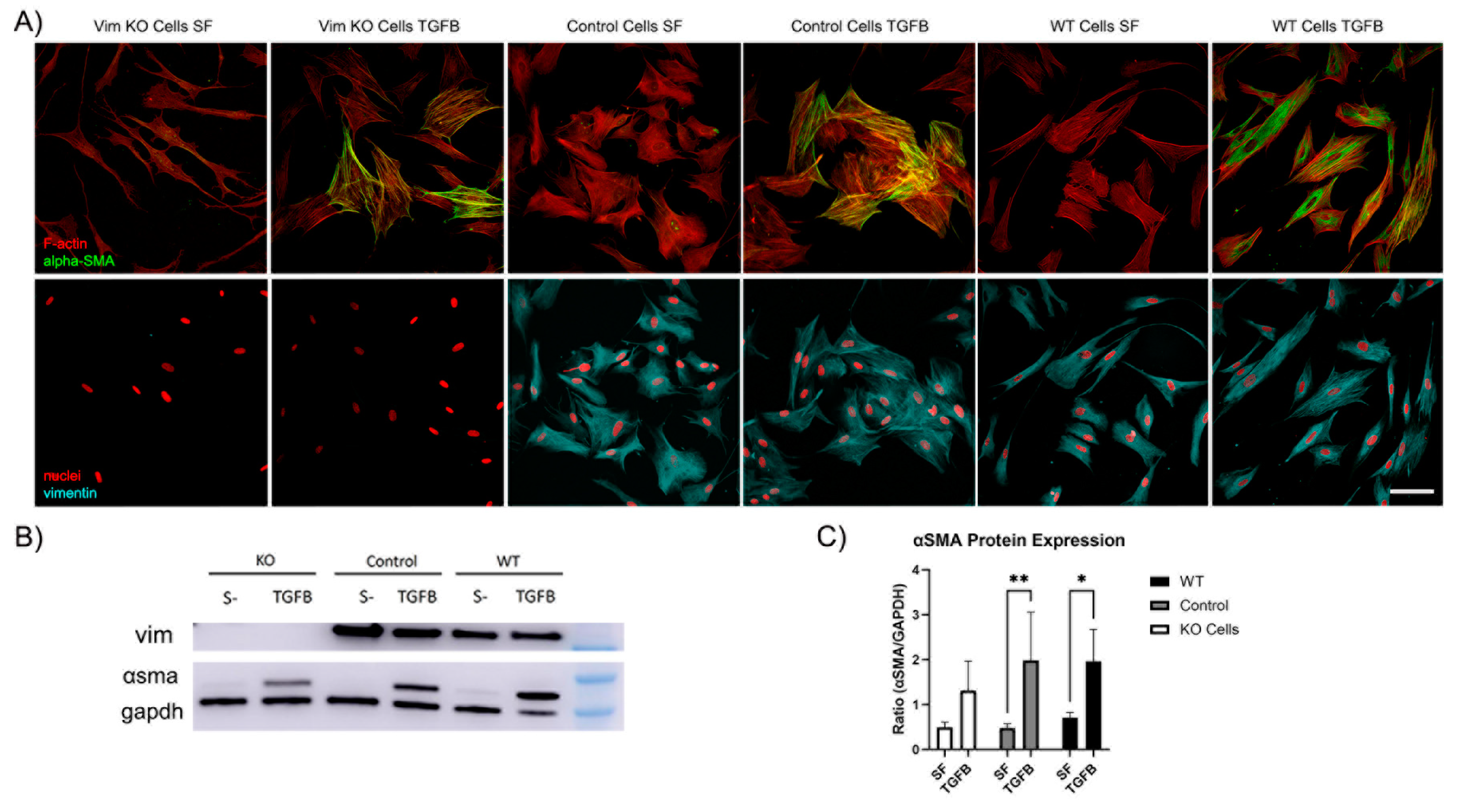
Figure 9.
Increase in protein expression induced by TGFβ1 (as compared to SF media). All 3 cell types were incubated for 6 days with media containing TGFβ or SF basal media. Subsequently protein was extracted and processed for mass spectrometry. A) Increase in expression of known proteins associated with corneal myofibroblasts. B) Additional proteins that were found to have at least two-fold increases in all 3 cell types. .
Figure 9.
Increase in protein expression induced by TGFβ1 (as compared to SF media). All 3 cell types were incubated for 6 days with media containing TGFβ or SF basal media. Subsequently protein was extracted and processed for mass spectrometry. A) Increase in expression of known proteins associated with corneal myofibroblasts. B) Additional proteins that were found to have at least two-fold increases in all 3 cell types. .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



