
Submitted:
18 March 2024
Posted:
19 March 2024
You are already at the latest version
Abstract
Glioblastoma (GBM) is the most aggressive brain cancer. To model GBM in research, orthotopic brain tumor models, including syngeneic models like GL261 and genetically engineered mouse models like TRP, are used. In longitudinal studies, tumor growth and treatment response are typ-ically tracked with in vivo imaging, including bioluminescence imaging (BLI), which is quick, cost-effective, and easily quantifiable. However, BLI requires luciferase-tagged cells, and recent studies indicate that the luciferase gene can elicit an immune response, leading to tumor rejection and experimental variation. We sought to optimize the engraftment of two luciferase-expressing GBM models, GL261 Red-FLuc and TRP-mCherry-FLuc, showing differences in tumor take, with GL261 Red-FLuc cells requiring immunocompromised mice for 100% engraftment. Immuno-histochemistry and MRI revealed distinct tumor characteristics: GL261 Red-FLuc tumors were well-demarcated with densely packed cells, high mitotic activity, and vascularization. In con-trast, TRP-mCherry-FLuc tumors were large, invasive, and necrotic, with perivascular invasion. Quantifying tumor volume using the HALO® AI analysis platform yielded results comparable to manual measurements, providing a standardized and efficient approach for reliable high-throughput analysis of luciferase-expressing tumors. Our study highlights the importance of considering tumor engraftment when using luciferase-expressing GBM models, providing in-sights for preclinical research design.
Keywords:
blood-brain barrier
; glioblastoma
; luciferase
; bioluminescence imaging
; preclinical
; GL261 Red-FLuc
; TRP-mCherry-FLuc
; AI
1. Introduction
Glioblastoma (GBM) is one of the most aggressive forms of brain cancer, with a 5-year survival rate of only 5.5% [1]. Despite the use of the standard of care (resection, radiation, and chemotherapy with the alkylating agent temozolomide), recurrence is nearly inevitable due to the invasion of remnant GBM cells into the surrounding brain parenchyma [2,3].
In pursuing effective treatments for improving patient survival, researchers use preclinical mouse GBM models to study disease characteristics and test novel therapies. These models are broadly divided into four categories: cell-line xenografts, patient-derived xenografts, syngeneic mouse models, and genetically engineered mouse models (GEMMs) [4]. Tumors of GEMMs can be dissociated for subsequent implantation to create models known as homografts [5]. Among these, syngeneic models and homografts allow implanting GBM cells of murine origin into mice of a similar genetic background, resulting in tumors with highly consistent growth rates and survival. Furthermore, the parental GEMM tumors develop de novo and are derived from specific mutations shared with human GBM tumors. Examples of syngeneic models and GEMMs include mouse glioma 261 (GL261) and TRP murine GBM, respectively.
GL261 is a widely used syngeneic GBM model established through intracranial implantation of carcinogenic 3-methylcholanthrene pellets, followed by tumor harvesting [6]. Subsequent tumor maintenance techniques involved serial fragment transplantation into both C57BL/6 mouse brain and flank [7]. Over time, in vitro cultures were established, enabling long-term propagation and widespread use as a preclinical GBM model. GL261 tumors share many pathological and molecular features with human GBM, including anaplasia, pleomorphic cells with atypical nuclei, hypoxia, angiogenesis, and increased mitotic rate [8]. Furthermore, GL261 tumors are radio- and chemo-sensitive and harbor mutations in tumor suppressor protein 53 (p53) and Kristen rat sarcoma viral oncogene (KRAS) [9,10,11,12]. The TRP model was derived from a GEMM harboring inactive retinoblastoma protein (RB), constitutively active KRAS, and phosphatase and tensin homolog (PTEN) deletion, which results in activation of the receptor tyrosine kinase (RTK)/phosphoinositide 3-kinase (PI3K) pathway and p53 missense mutations [13]. Notably, RB, RTK/PI3K and p53 pathway dysregulation is present in about 78%, 88%, and 87% of GBM tumors, respectively, with 74% harboring alterations in all three pathways [14]. These mutations lead to tumors with pathological features resembling patient GBM samples, including pseudopalisading necrosis, vessel co-option, and invasion [13]. These features collectively make GL261 and TRP valuable models for preclinical research.
When using preclinical intracranial GBM models, monitoring treatment response is typically limited to traditional analyses, such as histopathology or MRI, which have limitations. Histopathology is a labor-intensive process that requires sacrificing animals, which limits downstream endpoint analyses. Contrast-enhanced MRI involves time-consuming animal preparation with tail vein catheter placement and variable scan times and sequences. The nature of these procedures does not make them feasible for high-throughput studies.
To avoid these limitations, cancer cell lines are often genetically modified to express reporter genes, such as the firefly luciferase enzyme. Luciferase expression allows for in vivo bioluminescent imaging of tumors in live animals, which yields real-time information on relative tumor size and treatment response [15]. The most common luciferase reporters include Luc (original), Luc2 (next-generation codon-modified), and Red-FLuc (latest generation red-shifted, highest intensity, suitable for deep tissue imaging) [16,17]. However, cancer cells expressing reporter proteins, including luciferase, can be immunogenic, which is associated with spontaneous tumor regression [18,19]. Sanchez et al. recently demonstrated that more than half of C57BL/6 mice implanted with GL261 Red-FLuc cells rejected their tumors when injected with cell numbers ranging from 50,000 to 300,000 cells, suggesting that further characterization and optimization of luciferase-expressing tumor models is needed [20].
For the present study, we compared the tumor take and growth characteristics of luciferase-expressing GL261 Red-FLuc and TRP-mCherry-FLuc (TRP-mCF) tumors in immunocompetent (albino B6) mice. Due to the low tumor take of GL261 Red-FLuc tumors in immunocompetent mice, we switched to immunocompromised (J: NU) mice for further tumor characterization. We then characterized the histopathology and imaging features of GL261 Red-FLuc and TRP-mCF tumors in immunocompromised and immunocompetent hosts, respectively. Implantation of luciferase-expressing GL261 Red-FLuc cells into J: NU mice increased tumor take to 100% compared to 38% in albino B6 mice while maintaining histopathological features of this tumor. In contrast, TRP-mCherry-FLuc tumor take was 100% in 4 out of the 5 injected cell numbers. We also report a method of detecting luciferase-positive tumor cells using artificial intelligence to quantify tumor volume and high throughput analysis of histopathology samples. These studies directly compare two luciferase-expressing mouse GBM models, emphasizing the impact of luciferase expression on tumor engraftment, a critical aspect often overlooked in preclinical model development that is largely underreported in existing literature.
2. Materials and Methods
2.1. Cell Line and Culture Conditions
The parental GL261 cell line was purchased from the Division of Cancer Treatment and Diagnosis Tumor Repository (National Cancer Institute, NIH, Bethesda, MD), and the Bioware® Brite GL261 Red-FLuc cell line was purchased from PerkinElmer (BW134246; Waltham, MA, USA). Both cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 1,000 mg/L glucose, 584 mg/L L-glutamine, and 3.7 g/L sodium bicarbonate (D6046; MilliporeSigma, Saint Louis, MO, USA), and 10% fetal bovine serum (FBS; 89510-186, VWR, Radnor, PA, USA). GL261 Red-FLuc medium was supplemented with 2 µg/ml puromycin (1861; BioVision, Waltham, MA, USA) for maintaining luciferase expression. TRP-mCherry-FLuc (TRP-mCF) cells were kindly provided by Dr. Shawn Hingtgen (University of North Carolina, Chapel Hill, NC, USA) and maintained in DMEM containing 4500 mg/L glucose, 584 mg/L L-glutamine, 3.7 g/L sodium bicarbonate (Corning, Corning, NY, USA), 10% heat-inactivated fetal bovine serum, and 1x penicillin-streptomycin (MP Biomedicals, Solon, OH, USA).
Cells were incubated in a Heraeus HERAcell 150 CO2 incubator (Thermo Fisher Scientific, Waltham, PA, USA) at 37°C and 5% CO2. Cell morphology, proliferation, and confluence were assessed at 100-fold magnification with a TELAVAL 31 inverted transmitted light microscope (Zeiss, White Plains, NY, USA). Once cells were 80-90% confluent, they were treated with 0.05% trypsin-EDTA (25-053-Cl; Corning, Corning, NY, USA) in phosphate-buffered saline (PBS with 1.05 mM KH2PO4, 154 mM NaCl, 5.6 mM Na2HPO4; SH30256.01; HyClone Laboratories, Logan, UT, USA) for 3 min at 37°C. Trypsinization was stopped with cell culture medium twice the added 0.05% trypsin-EDTA volume. Cells were centrifuged (200 g, 5 min, RT) and resuspended in cell culture medium. We used a Scepter 2.0 automated cell counter to count cells (MilliporeSigma, Saint Louis, MO, USA). Cells were regularly tested for mycoplasma using either the PCR Mycoplasma Test Kit I/C (PK-CA91-1096; PromoCell GmbH, Heidelberg, DE) or the MycoStrip™ Mycoplasma Detection Kit (InvivoGen, San Diego, CA, USA).
2.2. Mice
All animal experiments were approved by the University of Kentucky Institutional Animal Care and Use Committee (IACUC #2018-2947; PI: Bauer). The University of Kentucky Division of Laboratory Animal Resources is an AAALAC-accredited institution, and experiments were carried out per the US Department of Agriculture Animal Welfare Act and by the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Eight-week-old female homozygous J: NU (immunocompromised; strain number 007850) and albino B6 (B6(Cg)-Tyrc-2J/J; immunocompetent; strain number 000058) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Mice were group-housed in cages connected to an EcoFlo ventilation system (Allentown Inc., Allentown, NJ, USA) in an AAALAC-accredited temperature- and humidity-controlled facility at the University of Kentucky (21-22°C, 30-70% humidity, 14:10-h light:dark cycle). Mice received water and standard chow ad libitum (Envigo Teklad Chow 2918, Envigo, Indianapolis, IN, USA).
2.3. Stereotaxic Intracranial Tumor Cell Implantations
GBM cell implantations were based on previously published protocols from Carlson et al. [21] and El Meskini et al. [22]. On the day before tumor cell implantations, the heads of albino B6 mice were shaved with a cordless hair trimmer under 2% isoflurane anesthesia. On the morning of the procedure, all mice were injected with Buprenorphine ER-LAB (1 mg/kg, s.c.; ZooPharm, Laramie, WY, USA). Cells were collected as described above, resuspended in PBS from 2,500 to 25,000 cells/µL depending on the total number of cells injected, and kept on ice throughout the surgeries. Isoflurane anesthesia (induction: 2.5%, room air: 21% O2) was delivered with a SomnoSuite® low-flow anesthesia system connected to an induction chamber (Kent Scientific, Torrington, CT, USA). Once anesthetized, mice were transferred to a platform with an infrared warming pad controlled by a RightTemp® temperature monitoring and homeothermic control module (Kent Scientific, Torrington, CT, USA) and positioned into a stereotaxic head frame and anesthesia mask (David Kopf Instruments, Tujunga, CA, USA). We lubricated the eyes with OptixCare® eye lube (Covetrus, Portland, ME, USA), and maintenance anesthesia was set to 1-2% isoflurane for the remainder of the procedure. The shaved scalp was disinfected with 2% chlorhexidine solution (Covetrus, Portland, ME, USA) and sterile saline (Covetrus, Portland, ME, USA) that were applied alternately using sterile cotton-tipped wood applicators (3 times, 1 min each). Following disinfection of the surgical area, a 1 cm midline incision was made using a 22-blade sterile disposable scalpel (Sklar, West Chester, PA, USA). To remove the remaining periosteum and to visualize bregma, the skull was swabbed with 3% H2O2 (Ward’s Science, Rochester, NY, USA). A 0.9 mm burr hole was created using an MH-170 rotary handpiece (Foredom Electric Company, Bethel, CT, USA) 2 mm mediolateral and -2 mm anteroposterior from bregma. Once the burr hole was created, cells were gently resuspended and pulled into a 5 µL Hamilton syringe with a 22sG needle (Hamilton Company, Reno, NV, USA). The exterior of the needle was cleaned with an alcohol prep pad.
For the GL261 Red-FLuc model, the needle was slowly inserted over 10 s into the burr hole to a depth of 4 mm, then removed 1 mm to create a pocket for the cells. The GL261 Red-FLuc cell dilution was injected over 2 min (2 µL; 1 µL/min) using an UltraMicroPump 3 with SMARTouch™ Controller (David Kopf Instruments, Tujunga, CA, USA). The needle remained for 1 min, then was slowly removed over 10 s. Any leakage/blood at the injection site was removed with a cotton-tipped applicator, followed by gentle scrubbing with an EtOH-soaked cotton-tipped applicator to remove any cells that might have made it onto the skull.
The TRP-mCF injection protocol was adapted from El Meskini et al. [22]. The needle was incrementally inserted into the burr hole at a rate of 1 mm/min to a depth of 4 mm, then retracted 1 mm to create a pocket for the cells. The TRP-mCF cell dilution was injected over 6 min (2 µL; 0.33 µL/min) using an UltraMicroPump 3 with SMARTouch™ Controller (David Kopf Instruments, Tujunga, CA, USA). The needle remained for 1 minute, then was removed incrementally at a rate of 1 mm/min. Any leakage/blood at the injection site was removed with a cotton-tipped applicator, followed by gentle scrubbing with an EtOH-soaked cotton-tipped applicator to remove any cells that might have made it onto the skull. A piece of bone wax (Covetrus, Portland, ME, USA) was shaped into a cone (~1 mm) and placed into the burr hole to prevent any extracranial growth.
For both models, the burr hole was sealed by heating standard pattern forceps (Fine Science Tools, Foster City, CA, USA) with a Germinator 500 Glass Bead Sterilizer (CellPoint Scientific, Gaithersburg, MD, USA) to melt bone wax (Covetrus, Portland, ME, USA), which was applied over the injection site. The skin was closed with wound clips (Fine Science Tools, Foster City, CA, USA), and the mouse was transferred to a clean cage on a heating pad (Stryker, Kalamazoo, MI, USA). Mice were monitored for at least 3 h post-op until they returned to normal behavior (e.g., movement, eating, drinking, cleaning). On the days following the injection, mice were observed at least once daily until they reached a humane endpoint (25% bodyweight loss or other adverse clinical signs, such as altered behavior, imbalance, head tilt, or altered respiration as approved per the IACUC protocol [23,24]).
2.4. In Vitro Bioluminescence Imaging
Luciferase expression of GL261 Red-FLuc and TRP-mCF cells was verified using a modified protocol from previously described [17]. Briefly, 5,000, 10,000, 15,000, and 20,000 cells/well were seeded in black clear-bottom 96-well plates (Corning, Corning, NY, USA) and incubated overnight (37°C, 5% CO2). After 24 h, media was aspirated, and 100 µL of phenol red-free DMEM (Thermo Fisher Scientific, Waltham, PA, USA) was added to each well. XenoLight® RediJect™ D-luciferin (PerkinElmer, Waltham, MA, USA) was diluted 1:100 in phenol red-free DMEM, and 100 µL of the luciferin dilution (0.15 µg/well) was added to each well. The plate was promptly transferred to an Ami HT optical imaging system (Spectral Instruments Imaging, Tucson, AZ, USA). Bioluminescence was determined by 2D imaging (FOV: 15 cm, exposure time: 1 s, f-stop: 1.2, binning: 4) at the following time points: 1-5, 10, 15, 20, 25, and 30 min and quantified using Aura 4.0.7 imaging software (Spectral Instruments Imaging, Tucson, AZ, USA).
2.5. In Vivo Bioluminescence Imaging
Bioluminescence imaging of mice harboring luciferase-expressing tumors was conducted weekly to verify tumor take and monitor tumor growth. Mice received 150 mg/kg (5 µL/g; i.p.) of XenoLight® RediJect™ D-luciferin and were anesthetized with 2% isoflurane. Eight min post-luciferin injection, immunocompetent (B6(Cg)-Tyrc-2J/J) and immunocompromised (J: NU) mice were relocated to the heated imaging stage of either an IVIS® Spectrum in vivo imaging system or a Lago in vivo optical imaging system (Spectral Instruments Imaging, Tucson, AZ, USA), respectively. Ten min post-luciferin injection, tumor bioluminescence was determined by 2D imaging (IVIS - FOV: 21.6 cm, f-stop: 8, binning: 4 or Lago – FOV: 25 cm, f-stop: 2, binning: 2), and images were analyzed using Aura 4.0.7 imaging software (Spectral Instruments Imaging, Tucson, AZ, USA). Tumor doubling times were calculated using a log-linked model, "Biofluorescence ~ ln(Cells) + Week", where ln(Cells) was the natural logarithm of cell dose.
2.6. Magnetic Resonance Imaging
MRI was conducted at the University of Kentucky Magnetic Resonance Imaging and Spectroscopy Center. Before handling mice, tail vein catheters were prepared by fitting a 29G needle into one side of a Tygon catheter tubing (50” long; 0.03” od x 0.01” id; Braintree Scientific Inc., Braintree, MA, USA) and inserting a 1 mL TB syringe with 30G needle onto the other end. Sterile saline was loaded into the syringe and perfused through the tubing until saline was expelled from the 29G needle. Mice were anesthetized with 1.5-2% isoflurane, and a tail vein catheter was placed to administer the contrast agent. Once the tail vein catheter was secured, mice were transferred to the platform of a 7T Bruker BioSpec, small animal MRI scanner (Bruker BioSpin, Billerica, MA, USA). We lubricated the eyes with OptixCare® eye lube (Covetrus, Portland, ME, USA), and a respiration pad transducer and rodent rectal temperature probe were positioned to monitor and document each animal’s vital signs. Pre-contrast T1-weighted (repetition time (TR) = 4000 ms, time to echo (TE) = 2.2 ms, field of view (FOV) = 20 x 20 x 8 mm) and T2-weighted scans (TR = 4000 ms, TE = 33 ms, FOV = 20 x 20 x 8) were acquired. Following the acquisition of pre-contrast scans, gadolinium (1 mmol/mL; Gadavist® (gadobutrol), Bayer AG, Whippany, NJ, USA) was diluted 1:10 in sterile saline and administered at a dose of 0.6 mmol/kg through the tail vein catheter. The catheter was flushed with 70 µL sterile saline to ensure the administration of the full dose of gadolinium (dead volume of 50” of tubing with a 0.01” inner diameter ≈ 64 µL). Post-contrast T1-weighted images (same settings as above) were acquired 10 min after gadolinium injection. Acquired images were analyzed using syngo.via software (Siemens Medical Solutions USA, Inc., Malvern, PA, USA). A conventional T2-RARE sequence was used to identify anatomical features, in conjunction with a standard T1 sequence with administration of a contrast agent to assess contrast enhancement and calculate tumor volume. Enhancing tumor area was determined using the “Freehand ROI” function, and total tumor area was calculated using the equation:
where distance between slices = 400 µm.
2.7. Histopathology
Histopathology samples were processed as previously described [17]. At week 3 post-implantation, mice were anesthetized with sodium pentobarbital (150 mg/kg, i.p.), transcardially perfused with PBS (100 mL, 10 mL/min), followed by perfusion with 10% formalin (50 mL, 10 mL/min; MilliporeSigma, Saint Louis, MO, USA). Mice were decapitated, brains were removed and placed in 5 mL 10% formalin and stored at RT overnight. The following day, brains were transferred to 70% ethanol and stored at 4°C until further processing. Samples were dehydrated in increasing concentrations of ethanol (70 – 100%), defatted with xylene, and embedded in paraffin. Brains were sectioned into two consecutive 4 µm coronal sections at 0.2 mm intervals: one for immunohistochemistry with anti-luciferase antibody and the other for hematoxylin and eosin (H&E) staining. Decreasing concentrations of ethanol (100 – 70%) and xylene were used to deparaffinize and rehydrate the tissue, respectively. Anti-luciferase immunohistochemistry was conducted using the Discovery Ultra Research staining system (Ventana Co., Tucson, AZ, USA). Immunohistochemistry antigen retrieval was performed using an EDTA-based heat-induced antigen retrieval (CC1) method at 91°C for 64 min. Slides were stained with anti-luciferase antibody (ab181640, Abcam, Cambridge, MA, USA) at a 1:250 dilution (4 µg/mL) for 4 h at RT. The antigen-antibody complex was detected using the OmniMap anti-goat multimer RUO detection system and DAB detection kit (Ventana Co., Tucson, AZ, USA). All slides were counterstained with hematoxylin. Brain slides were imaged with an Aperio ScanScope XT (Leica Biosystems, Buffalo Grove, IL, USA) at 20X magnification. Tumor area in each slice was determined manually by outlining the tumor with the Annotations feature of the Aperio ImageScope software. Tumor area was also determined using the Deep Learning Classifier Add-On of the HALO® AI analysis platform (Indica Labs Inc., Albuquerque, NM, USA). Tumor area was calculated using the equation:
where slice thickness + distance between slices = 204 µm.
2.8. Data Analysis & Statistics
Unless specified otherwise, all data were analyzed by generalized linear mixed-level models (glmer) to account for correlated measurements by individual mice [25]. C57BL/6 mice were modeled with Biofluorescence ~ Cells + Response + Cells×Response, where "Response" was acceptance or rejection of tumor. J: NU mice were modeled with Biofluorescence ~ Cells + Week + Cells×Week. Tumor volume vs. analytical method was modeled with Volume ~ Timepoint + Method + Timepoint×Method. Quantitative data were scaled by standard deviation, and predictors also centered by mean values. Coefficients are, therefore, standardized values. Error families (i.e., Gaussian, gamma, or inverse Gaussian) and link functions (identity or log) were compared by second-order Akaike Information Criterion (AIC) [26]. Specific links are indicated in the results. Survival was modeled with Cox proportional hazards modeling [27] and parametric survival regression [28], using the Akaike Information Criterion with a correction for small sample sizes (AICc) to compare effects of cell counts as quantities and as levels of a factor under assumptions of proportionality or a specific parametric distribution. For tumor volume assessment analyses, models were built to determine how methods of volume assessment differed in overall estimates and how well the AI histology and MRI methods agreed with manual assessment of histology. Specifically, for level estimates, we used a mixed-level generalized linear model "Cell type + Method + Cell Type × Method", with individual animal as a random intercept. To determine agreement between AI histology or MIR with manual histology, we built parallel linear models of each alternate method vs. manual histology for each cell type. We then used R2 to quantify percent linear agreement and compared the square roots of R2 by the Fisher z transformation. Modeling was followed by analysis of deviance (ANOVA) and pairwise comparisons of selected estimated marginal means for level estimates [29]. Coefficients of determination (R2) were Nagelkerke's for levels [30] and traditional R2 for agreement. Partial R2 for each effect in level estimate modeling were estimated by calculating R2 for nested sub-models lacking an effect and subtracting this from the overall model R2. Omnibus (overall) ANOVAs for level estimates were χ2 tests of models vs. intercept-only or intercept and animal random effects only. The R environment [31] was used to perform analyses, using the lme4 [32] and emmeans [33] packages.
3. Results
3.1. In Vitro Verification of Luciferase Activity
We confirmed luciferase activity of the luciferase-expressing GL261 Red-FLuc and TRP-mCherry-FLuc cell lines through in vitro bioluminescence imaging (Figure 1A and 1B). Bioluminescence of GL261 Red-FLuc cells peaked 2 min after adding luciferin, slightly decreased through 5 min, then increased and stabilized from 5 to 30 min, depending on cell number. Bioluminescence of TRP-mCF cells was highest 1 min after adding luciferin, decreased through 5 min, then increased slightly and stabilized from 5 to 30 min. After 30 minutes, stable bioluminescence was observed, showing a linear correlation between cell number and bioluminescence for both cell lines (GL261 Red-FLuc: R2 = 0.985, p < 0.0001; TRP-mCF: R2 = 0.932, p < 0.0001) (Figures 1C and 1D). This suggests that as cell numbers increase, bioluminescence production rises consistently and without plateau, which is important for tracking tumor growth in vivo. These data verify that both GL261 Red-FLuc and TRP-mCF express active luciferase.
3.2. GL261 Red-FLuc Tumor Take and Survival in Immunocompetent (Albino B6) Mice
To establish luciferase-expressing GL261 Red-FLuc tumors in immunocompetent hosts, we intracranially implanted 5x104 GL261 Red-FLuc cells into 8-week-old female albino C57BL/6 mice. We verified the successful implantation of cells into mouse brains one week after injection and tracked tumor growth weekly through bioluminescence imaging using the IVIS® Spectrum in vivo imaging system. Figures 2A and 2B show representative images and quantified bioluminescence for albino C57BL/6 mice 1-4 weeks post-implantation. Mice with successful tumor take throughout the end of the study (40 days post-implantation) are labeled as “accepted”, and mice with no evidence of tumor by the end of the study are labeled as “rejected”. Only 22 of 51 (43.1%) mice showed evidence of tumor two weeks after implantation, which decreased to 20 of 51 (39.2%) by week 3. The doubling time of accepted tumors was 1.6 ± 0.1 days (Figure 2B). By the end of the study, mortality was 38% ± 15/12% (Figure 2C; SE estimated by Cox model of survival vs. intercept). In contrast, tumors were rejected in 31 of 51 (61%) of mice, which is consistent with previous reports using GL261 Red-FLuc cells in immunocompetent mice [20] (Figures 2A-C). Median survival of mice that accepted tumors and reached endpoint was 27 days (Figure 2C). In summary, most GL261 Red-FLuc tumors are rejected in immunocompromised mice, making this a suboptimal model for GBM research.
3.3. GL261 Red-FLuc Tumor Take and Survival in Immunocompromised (J: NU) Mice
Given the high rate of tumor regression in albino B6 mice, we sought to increase tumor engraftment by implanting GL261 Red-FLuc cells into immunocompromised, female athymic nude (J: NU) mice. The injection procedure of luciferase-expressing cells in J: NU mice was identical to C57BL/6 mice. To test survival, the number of intracranially implanted cells ranged from 5,000 to 50,000. Representative bioluminescence images and average bioluminescence quantifications are shown in Figure 3A and 3B, respectively. No representative images are shown for mice implanted with 15,000, 25,000, or 50,000 cells at week 4 since most mice had already reached endpoint.
Tumor growth, measured by bioluminescence, appears to have been in the log phase in weeks 1-3 (Figures 3B,C). Therefore, explicit modeling was restricted to these time points. We used a log-linked model, “Biofluorescence ~ ln(Cells) + Week”, where ln(Cells) was the natural logarithm of the number of cells implanted. This model was significant for both variables (cells and week; Figure 3B); however, there was no significant interaction between cell number and time, suggesting that the tumor doubling times for each injected cell number cannot be presumed to differ significantly. With this model, we estimated the average tumor doubling time as 2.13 ± 0.1 days (Figure 3C and Table 1).
When we compared different survival models, AICc favored a parametric survival vs. cell dose model with a logistic distribution (Figure 3D). Specifically, as we increased the number of injected GL261 cells, survival decreased as expected. Unlike in the albino C57BL/6 mice, all starting cell numbers yielded 100% tumor take in J: NU mice. Due to this 100% tumor take and the longest survival of the starting cell numbers, we injected J: NU mice with 5x103 GL261 Red-FLuc cells in all remaining studies.
3.4. TRP-mCherry-FLuc Tumor Take and Survival in Immunocompetent (Albino B6) Mice
Since tumor engraftment of GL261 Red-FLuc tumors in immunocompetent mice was low, we aimed to assess the tumor take of another luciferase-expressing cell line, TRP-mCF. Notably, TRP-mCF cells behave more aggressively than GL261 Red-FLuc in vitro, demonstrating shorter doubling times when seeded at cell densities up to 4,000 cells (Figure S1; Table S1). Therefore, we hypothesized that these more proliferative cells could overcome the immunoreactive microenvironment of immunocompetent mice.
Analogous to GL261 Red-FLuc cells, we intracranially implanted 5x103 to 5x104 TRP-mCF cells into 8-week-old female albino C57BL/6 mice. We verified the successful cell implantation into mouse brains one week after injection and tracked tumor growth weekly through bioluminescence imaging using the IVIS® Spectrum in vivo imaging system. Representative weekly bioluminescence images and average quantified bioluminescence are shown in Figures 4A and 4B, respectively. Most mice reached the prespecified endpoint during week 3, with tumor bioluminescence no longer detectable in one of the five mice injected with 10,000 cells by the second week post-implantation. As measured by bioluminescence, tumor growth appears to have been in the log phase from weeks 1 to 3 (Figures 4A–C). A log-linked model, "Biofluorescence ~ Cells + Week", was significant for both variables, suggesting that cell number did affect weekly tumor size (Figure 4B). A potential model without interaction of cell count and time was more likely by AICc comparison than one with interaction, suggesting that the impact of cell number on bioluminescence (tumor size) is consistent across time points and vice versa. This model was also used to estimate doubling time as 2.42 ± 0.28/0.22 days (Figure 4C and Table 2).
When different survival models were compared, a parametric model of survival vs. cell dose, with a lognormal distribution (Figure 4D), was favored by AICc. Specifically, as cell dose increased, survival decreased in a rational fashion. Overall, these data highlight that TRP-mCherry-FLuc tumors can overcome the immunosuppressive microenvironment of albino B6 mice, yielding 100% tumor take with most cell numbers injected.
3.5. MRI Features and Tumor Volume Determination
We imaged J: NU and albino B6 mice injected with 5x103 GL261 Red-FLuc and TRP-mCF cells using MRI three weeks post-implantation. Representative MR images for two animals of each model are shown in Figure 5.
GL261 Red-FLuc tumors were evident on pre-contrast T1-weighted, post-contrast T1-weighted, and T2-weighted images, and the average tumor volume was 28.9 ± 18.8 mm3. On pre-contrast T1-weighted images, tumors appeared as well-demarcated hypodense masses in the right hemisphere (Figure 5A). Following i.v. injection of a gadolinium (Gd)-based contrast agent, tumors displayed heterogenous contrast enhancement, signifying blood-brain barrier disruption (Figure 5B). T2-weighted images showed well-demarcated masses with light (red arrow) and dark (yellow arrow) regions, suggesting the presence of fluid and hematoma, respectively (Figure 5C). An appreciable mass effect (white arrows) was seen in all mice, with involvement of the contralateral hemisphere in larger tumors. Similarly, TRP-mCF tumors were apparent on pre-contrast T1-weighted, post-contrast T1-weighted, and T2-weighted images, with an average tumor volume of 109.5 ± 38.9 mm3. Tumors were less demarcated than GL261 Red-FLuc tumors on pre-contrast T1-weighted images, with cloudy tumor borders, suggesting tumor infiltration and/or peritumoral changes (Figure 5D). Moderate contrast enhancement was observed on post-contrast T1-weighted images, with some tumors exhibiting diminished enhancement in their central regions (yellow arrow), suggesting necrosis, hemorrhage, increased cell density, or increased interstitial pressure in these areas (Figure 5E). On T2-weighted images, tumor borders were less discernable, and there was a larger mass effect (white arrows) compared to GL261 Red-FLuc (Figure 5F). Some tumors contained dark regions (yellow arrow), suggesting the presence of hematoma.
These data show that GL261 Red-FLuc tumors manifest as circular, well-demarcated masses, while TRP-mCF tumors exhibit increased signs of invasion, along with necrotic and/or densely compact cores.
3.6. Immunohistochemical Features of GL261 Red-FLuc and TRP-mCherry-FLuc Tumors
To verify the characteristics observed on MRI with increased resolution, we transcardially perfused and fixed the brains of nude and albino B6 mice injected with 5x103 GL261 Red-FLuc or TRP-mCherry-FLuc cells, respectively, at three weeks post-implantation. For GL261 Red-FLuc tumors, histological morphology assessed using H&E staining demonstrated densely packed cells with high mitotic activity, nuclear pleomorphisms, and vascularization (Figures 6A and 6B). Similar to what we observed by MRI, tumors were well-circumscribed with minimal invasive cells, similar to characterization of parental GL261 tumors in immunocompetent C57BL/6 mice [34,35,36]. IHC revealed strongly stained luciferase-positive cells with a well-demarcated border (Figure 6C). At the same time point post-implantation, TRP-mCF tumors occupied most of the right hemisphere, causing a significant mass effect, with many tumors invading the contralateral hemisphere (Figure 6D). H&E staining displayed areas of necrosis, perivascular tumor invasion, and increased mitotic activity (Figure 6E). Unlike GL261 Red-FLuc tumors, invasive cell nests were seen around the main tumor border, which was verified on anti-luciferase IHC (Figure 6F).
3.7. High Throughput Determination of GL261 Red-FLuc and TRP-mCF Tumor Volume and Comparison to MRI
We also calculated tumor volumes from histopathology to verify volumes determined by MRI. The tumor area of each slice was calculated manually using Aperio ImageScope software and HALO®’s AI Deep Learning Classifier Add-On. Examples of manually analyzed anti-luciferase IHC slices are shown in Figure 7A and 7C, with tumors outlined in green. Examples of slices analyzed using AI are shown in Figures 7B and 7D, with red, green, and yellow corresponding to tumor, non-tumor, and glass/background, respectively. Total tumor volume was calculated by multiplying the sum of the tumor area by the distance between the slices for each mouse. Average GL261 Red-FLuc and TRP-mCF tumor volumes, as determined from histopathology slices, are listed in Table 3. Paired tumor volumes, determined manually and with AI from histopathology samples, are compared to MRI in Figure 8.
Mean tumor volumes were similar by both manual and AI interpretation of histopathology (Figures 8A and 8C). There was some significant (p = 0.017) but small elevation of estimation for GL261 Red-FLuc tumors. These observed disparities may be attributed to the detection of minor artifacts and darker staining regions (e.g., hippocampus). On the other hand, tumor volume estimates were noticeably higher overall for MRI (Figures 8A and 8C), particularly for TRP-mCF. There were two potential visual outliers in the GL261 Red-FLuc readings, but when we examined normalized Pearson residuals, no points deviated sufficiently from model predictions to consider rejection. The difference between MRI and histopathology was further underlined when comparing the linear agreement between AI and manual assessments of histopathology and between MRI and manual assessments of histopathology (Figures 8B and 8D), arbitrarily choosing the manual assessment as a "standard". For both GL261 Red-FLuc and TRP-mCF tumors, agreement (as R2) was > 99%. On the other hand, agreement between manual histopathology and MRI was only 71% for GL261 and 67% for TRP. When we performed a z test on Fisher-transformed square roots of R2, both differences were significant (GL261 z = 2.959, p = 0.002; TRP z = 3.846, p < 0.001). ANOVAs for the models were significant (p ≤ 0.05), but this was trivial since relationships were mostly monotonic.
4. Discussion
Preclinical models that accurately replicate key features of human GBM serve as a valuable platform for investigating tumor biology, testing novel therapies, and predicting clinical responses. These models play a critical role in bridging the gap between preclinical studies and human trials, contributing to the development of more precise and effective interventions compared to the standard of care for GBM.
Among the syngeneic models used in GBM research, the GL261 model stands out as the most used due to its high reproducibility and shared pathological and molecular features with human GBM [9,10]. However, this model has limitations, as it lacks other features of high-grade gliomas, including invasive cells, vessel co-option, and pseudopalisading necrosis. In contrast, the TRP-mCF model exemplifies a GEMM that reproduces these additional traits accurately. Regardless of the model, luciferase-tagged GBM cells are used to monitor tumor growth and preclinical treatment response in animals, allowing non-invasive in vivo imaging. However, recent reports have highlighted inconsistent tumor engraftment of luciferase-tagged cells in immunocompetent mice, rendering these models unreliable for high-throughput studies. Consequently, further optimization and validation of these models are imperative to ensure their reliability and suitability for research purposes [20,37].
Here, we validate earlier findings of GL261 Red-FLuc immunoreactivity in immunocompetent mice, present an alternative approach to improve tumor take, and characterize GL261 Red-FLuc tumor growth rate and survival. We found that GL261 Red-FLuc tumors spontaneously regressed in more than 60% of immunocompetent (albino B6) mice injected with 5x104 GL261 Red-FLuc cells. These data are consistent with previous work by Sanchez et al. that demonstrates spontaneous regression and long-term survival (>100 days) in 60% of C57BL/6 mice [20]. This phenomenon is most likely due to the immunogenicity of the Red-FLuc tag, which induces a proinflammatory microenvironment with increased macrophage and T-cell infiltration compared to parental, untagged GL261 cells, leading to GBM implant rejection.
Variable tumor growth and spontaneous tumor regression are not suitable characteristics of a reliable preclinical model due to the inability to distinguish between treatment effects from immune-mediated tumor regression. Therefore, we aimed to generate a luciferase-expressing GL261 model with consistent tumor growth. Using immunocompromised mice (J:NU nude), we achieved a 100% tumor take rate following the implantation of 5,000-50,000 GL261 Red-FLuc cells. Increasing cell numbers led to a cell number-dependent decrease in median survival, ranging from 19 to 27 days. Szatmári et al. [9], who used the parental GL261 model in immunocompetent C57BL/6 mice, revealed similar survival rates, with median survivals of 27 and 25 days for mice implanted with 1x104 and 1x105 cells, respectively. On the other hand, we show that cells with a more aggressive phenotype, TRP-mCF, can overcome the immunoreactivity of firefly luciferase to achieve high tumor engraftment in immunocompetent mice.
Data from our MRI and histopathological analyses of GL261 Red-FLuc tumors in immunocompromised mice are consistent with features of parental GL261 tumors in immunocompetent mice [8,36,38]. On day 21 post-implantation, GL261 Red-FLuc tumors exhibited well-demarcated borders with a small degree of edema and demonstrated heterogeneous contrast enhancement on post-Gd T1-weighted images. Tumors showed vascular proliferation, high cellularity, nuclear pleomorphism, high mitotic activity, and distinct borders, which is a characteristic feature of carcinogen-induced mouse gliomas [39]. Relative to GL261 Red-FLuc tumors, TRP-mCF tumors were much larger by day 21 and displayed increased evidence of necrosis, edema, and contralateral hemisphere involvement on MRI. On histopathology, TRP-mCF tumors were invasive and exhibited necrosis, vessel co-option, and high mitotic activity.
Alongside descriptive histopathology, we also introduce an AI-based approach for automating tumor volume analysis. The automatization of histopathological analysis has advantages, including increased efficiency, consistency, standardization, reduced subjectivity, and the ability to handle large datasets. We trained HALO’s AI™ Deep Learning Classifier Add-On to classify regions as glass (background), non-tumor, or tumor tissue on slides stained for firefly luciferase. On day 21 post-implantation, tumor volume was assessed manually, with noticeable differences observed when employing AI. However, these differences were minimal (GL261 Red-FLuc: 17.7±16.3 mm3 vs. 19.0±17.0 mm3; TRP-mCF: 64.4±19.0 mm3 vs. 64.4±19.1 mm3 for manual and AI calculation, respectively) and could be attributed to factors such as the small sample size (n=7-8 tumors) or limitations of AI in accurately discerning necrotic regions, which may resemble glassy backgrounds or dark brain regions with high cellular density resembling tumor tissue. Furthermore, calculated tumor volumes differed significantly when determined by histopathology and MRI; however, this discrepancy is consistent with previous studies comparing brain tumor volumes using these methods [17,40,41]. These differences could be due to various factors, including resolution differences, tissue processing with either shrinkage artifacts or delicate tumor fragments prone to detachment, tumor edema, or imaging artifacts. Regardless, these differences suggest that tumor volume quantification via histopathology and MRI are not interchangeable and should be consistent within a study.
The use of the GL261 Red-FLuc tumor model in immunocompromised mice is limited, i.e., the lack of immune response, which is why syngeneic models are commonly used in preclinical research. However, the level of immunodeficiency in J: NU mice is not as severe as in other immunocompromised strains, such as NCG and NOD SCID. J: NU mice lack T cells, but B cells, dendritic cells, macrophages, natural killer cells, and hemolytic complement are present. Furthermore, our characterization of the GL261 Red-FLuc tumors in J: NU mice shows that the tumors maintain histopathological features of parental GL261 tumors in C57BL/6 mice reported in the literature [8,36,38]. The GL261 Red-FLuc model in J: NU mice could be valuable in studies requiring large animal numbers and high tumor take and in tumor growth or direct cytotoxic studies without the confounding factor of an intact immune response. The use of both immunocompetent and immunocompromised mice would be valuable in studies characterizing the role of the immune system in treatment response [42,43]. Overall, our study demonstrates the challenges and opportunities associated with optimizing and validating luciferase-expressing GBM models, emphasizing the importance of selecting the most appropriate model for specific research questions and experimental objectives.
5. Conclusions
By characterizing the GL261 Red-FLuc model in J: NU mice, we have addressed the limitations of inconsistent tumor take observed in C57BL/6 mice, providing a model useful for studies either focusing on direct cytotoxic effects of therapies or without the confounding effects of the immune system. We also demonstrate that other luciferase-expressing models, like TRP-mCF, can yield high tumor engraftment in immunocompetent hosts. In addition, while acknowledging the challenges in AI-based tumor volume analysis, we use these models to demonstrate their potential to complement traditional methods of tumor volume analysis, yielding values similar to those of manual analysis while saving time and effort. In conclusion, we demonstrate how to achieve high GL261 Red-FLuc and TRP-mCF tumor take to track tumor growth and treatment response reliably in GBM research without experimental variability. Since each model has strengths and weaknesses, selecting the appropriate preclinical model should be based on the project goals.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Comparison of GL261 Red-FLuc and TRP-mCF in vitro growth characteristics; Table S1: In Vitro GL261 Red-FLuc and TRP-mCF Growth Characteristics.
Author Contributions
Conceptualization, L.T.R, J.A.S, A.M.S.H, and B.B.; methodology, L.T.R. and J.A.S.; software, L.T.R and B.M.; validation, L.T.R. and J.A.S.; formal analysis, L.T.R. and B.M.; investigation, L.T.R., J.A.S., and B.B.; resources, A.M.S.H. and B.B.; data curation, B.M.; writing – original draft preparation, L.T.R., B.M., and B.B.; writing – review and editing, L.T.R., J.A.S., B.M., A.M.S.H., and B.B.; visualization, L.T.R. and B.M.; supervision, A.M.S.H. and B.B.; project administration, B.B.; funding acquisition, L.T.R. and B.B. All authors have read and agreed to the published version of the manuscript.
Funding
Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health, grant number R01NS107548 (BB); the National Center for Advancing Translational Sciences of the National Institutes of Health, grant number TL1TR001997 (LTR); and the Northern Kentucky/Greater Cincinnati UK Alumni Club Fellowship (LTR). The content is solely the authors’ responsibility and does not necessarily represent the official views of the NINDS or the NIH.
Institutional Review Board Statement
The animal study protocol was approved by the University of Kentucky Institutional Animal Care and Use Committee (IACUC #2018-2947; PI: Bauer).
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Acknowledgments
We thank all Bauer and Hartz research team members for proofreading the manuscript. Special thanks to Yuma Tega and Yingying Gu for technical assistance with various procedures. This research was supported by the Biospecimen Procurement and Translational Pathology Shared Resource Facility of the University of Kentucky Markey Cancer Center (P30CA177558) and the University of Kentucky Magnetic Resonance Imaging and Spectroscopy Center. The Aperio ScanScope XT is supported by the University of Kentucky Alzheimer’s Disease Research Center grant P30 AG072946.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016-2020. Neuro Oncol 2023, 25, iv1–iv99. [Google Scholar] [CrossRef]
- Robin, A.M.; Pawloski, J.A.; Snyder, J.M.; Walbert, T.; Rogers, L.; Mikkelsen, T.; Noushmehr, H.; Lee, I.; Rock, J.; Kalkanis, S.N.; Rosenblum, M.L. Neurosurgery's Impact on Neuro-Oncology-"Can We Do Better?"-Lessons Learned Over 50 Years. Neurosurgery 2022, 68, 17–26. [Google Scholar] [CrossRef]
- Seker-Polat, F.; Pinarbasi Degirmenci, N.; Solaroglu, I.; Bagci-Onder, T. Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Haddad, A.F.; Young, J.S.; Amara, D.; Berger, M.S.; Raleigh, D.R.; Aghi, M.K.; Butowski, N.A. Mouse models of glioblastoma for the evaluation of novel therapeutic strategies. Neurooncol Adv 2021, 3, vdab100. [Google Scholar] [CrossRef] [PubMed]
- Bourré, L.u. Tumor Homograft Model Generation: Dissociated Tumor Cells vs Tumor Fragments - Crown Bioscience. Available online: https://blog.crownbio.com/tumor-homograft-models-dtc-tumor-fragments (accessed on.
- Seligman, A.M.S., M. J.; Alexander, L. Studies in Carcinogenesis: VIII. Experimental Production of Brain Tumors in Mice with Methylcholanthrene. The American Journal of Cancer 1939, 37, 364–395. [Google Scholar] [CrossRef]
- Ausman, J.I.; Shapiro, W.R.; Rall, D.P. Studies on the chemotherapy of experimental brain tumors: development of an experimental model. Cancer Res 1970, 30, 2394–2400. [Google Scholar] [PubMed]
- Cha, S.; Johnson, G.; Wadghiri, Y.Z.; Jin, O.; Babb, J.; Zagzag, D.; Turnbull, D.H. Dynamic, contrast-enhanced perfusion MRI in mouse gliomas: correlation with histopathology. Magn Reson Med 2003, 49, 848–855. [Google Scholar] [CrossRef]
- Szatmari, T.; Lumniczky, K.; Desaknai, S.; Trajcevski, S.; Hidvegi, E.J.; Hamada, H.; Safrany, G. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci 2006, 97, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, E.W.Z., D. The Murine GL261 Glioma Experimental Model to Assess Novel Brain Tumor Treatments. In CNS Cancer, Meir, E., Ed.; Cancer Drug Discovery and Development; 2009.
- Delgado-Goni, T.; Julia-Sape, M.; Candiota, A.P.; Pumarola, M.; Arus, C. Molecular imaging coupled to pattern recognition distinguishes response to temozolomide in preclinical glioblastoma. NMR Biomed. 2014, 27, 1333–1345. [Google Scholar] [CrossRef]
- Candolfi, M.; Yagiz, K.; Wibowo, M.; Ahlzadeh, G.E.; Puntel, M.; Ghiasi, H.; Kamran, N.; Paran, C.; Lowenstein, P.R.; Castro, M.G. Temozolomide does not impair gene therapy-mediated antitumor immunity in syngeneic brain tumor models. Clin Cancer Res 2014, 20, 1555–1565. [Google Scholar] [CrossRef]
- El Meskini, R.; Iacovelli, A.J.; Kulaga, A.; Gumprecht, M.; Martin, P.L.; Baran, M.; Householder, D.B.; Van Dyke, T.; Weaver Ohler, Z. A preclinical orthotopic model for glioblastoma recapitulates key features of human tumors and demonstrates sensitivity to a combination of MEK and PI3K pathway inhibitors. Dis Model Mech 2015, 8, 45–56. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Dinca, E.B.; Sarkaria, J.N.; Schroeder, M.A.; Carlson, B.L.; Voicu, R.; Gupta, N.; Berger, M.S.; James, C.D. Bioluminescence monitoring of intracranial glioblastoma xenograft: response to primary and salvage temozolomide therapy. J Neurosurg 2007, 107, 610–616. [Google Scholar] [CrossRef]
- Liu, S.; Su, Y.; Lin, M.Z.; Ronald, J.A. Brightening up Biology: Advances in Luciferase Systems for in Vivo Imaging. ACS Chem. Biol. 2021, 16, 2707–2718. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.A.; Rodgers, L.T.; Kryscio, R.J.; Hartz, A.M.S.; Bauer, B. Characterization and comparison of human glioblastoma models. BMC Cancer 2022, 22, 844. [Google Scholar] [CrossRef] [PubMed]
- Baklaushev, V.P.; Grinenko, N.F.; Yusubalieva, G.M.; Abakumov, M.A.; Gubskii, I.L.; Cherepanov, S.A.; Kashparov, I.A.; Burenkov, M.S.; Rabinovich, E.Z.; Ivanova, N.V.; et al. Modeling and integral X-ray, optical, and MRI visualization of multiorgan metastases of orthotopic 4T1 breast carcinoma in BALB/c mice. Bull. Exp. Biol. Med. 2015, 158, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Baklaushev, V.P.; Kilpelainen, A.; Petkov, S.; Abakumov, M.A.; Grinenko, N.F.; Yusubalieva, G.M.; Latanova, A.A.; Gubskiy, I.L.; Zabozlaev, F.G.; Starodubova, E.S.; et al. Luciferase Expression Allows Bioluminescence Imaging But Imposes Limitations on the Orthotopic Mouse (4T1) Model of Breast Cancer. Sci Rep 2017, 7, 7715. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, V.E.; Lynes, J.P.; Walbridge, S.; Wang, X.; Edwards, N.A.; Nwankwo, A.K.; Sur, H.P.; Dominah, G.A.; Obungu, A.; Adamstein, N.; et al. GL261 luciferase-expressing cells elicit an anti-tumor immune response: an evaluation of murine glioma models. Sci Rep 2020, 10, 11003. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.L.; Pokorny, J.L.; Schroeder, M.A.; Sarkaria, J.N. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. In Curr Protoc Pharmacol, 2011/07/12 ed.; James Wiley & Sons: Hoboken NJ, 2011; pp. 1–14. [Google Scholar]
- El Meskini, R.; Atkinson, D.; Weaver Ohler, Z. Translational Orthotopic Models of Glioblastoma Multiforme. J Vis Exp 2023. [Google Scholar] [CrossRef] [PubMed]
- Toth, L.A. Defining the Moribound Condition as an Experimental Endpoint for Animal Research. ILAR Journal 2000, 41, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J. Humane endpoints and cancer research. ILAR J 2000, 41, 87–93. [Google Scholar] [CrossRef]
- Detry, M.A.; Ma, Y. Analyzing Repeated Measurements Using Mixed Models. JAMA 2016, 315, 407–408. [Google Scholar] [CrossRef]
- Burnham, K.P.; Anderson, D.R. Model Selection and Multimodel Inference: A practical information-theoretic approac, 2 ed.; Spring New York: New York, 2002; p. 488.
- Andersen, P.K.; Gill, R.D. Cox's Regression Model for Counting Processes: A Large Sample Study. The Annals of Statistics 1982, 10, 1100–1120, 1127. [Google Scholar] [CrossRef]
- Kalbfleisch, J.D.; Prentice, R.L. The Statistical Analysis of Failure Time Data, 2nd ed.; John Wiley & SOns: New York, 2002. [Google Scholar]
- Searle, S.R., Speed, F. M., Milliken, G. A. Population marginal means in the linear model: An alternative to least squares means. The American Statistician 1980, 34, 216-221.
- Nagelkerke, N.J.D. A Note on a General Definition of the Coefficient of Determination. Biometrika 1991, 78, 691–692. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. Journal of Computational and Graphical Statistics 1996, 5, 299–314. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Statistical Software 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Lenth, R.V.; Buerkner, P.; Gine-Vazquez, I.; Herve, M.; Jung, M.; Love, J.; Miguiz, F.; Riebl, H.; Singmann, H. emmeans: Estimated Marginal Means, aka Least-Squares Means. Available online: https://cran.r-project.org/web/packages/emmeans/.
- Renner, D.N.; Malo, C.S.; Jin, F.; Parney, I.F.; Pavelko, K.D.; Johnson, A.J. Improved Treatment Efficacy of Antiangiogenic Therapy when Combined with Picornavirus Vaccination in the GL261 Glioma Model. Neurotherapeutics 2016, 13, 226–236. [Google Scholar] [CrossRef]
- Fayzullin, A.; Tuvnes, F.A.; Skjellegrind, H.K.; Behnan, J.; Mughal, A.A.; Langmoen, I.A.; Vik-Mo, E.O. Time-lapse phenotyping of invasive glioma cells ex vivo reveals subtype-specific movement patterns guided by tumor core signaling. Exp. Cell Res. 2016, 349, 199–213. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Hudson, A.L.; Prasanna Kumar, R.; Wilmott, J.S.; Attrill, G.H.; Long, G.V.; Scolyer, R.A.; Clarke, S.J.; Wheeler, H.R.; Diakos, C.I.; Howell, V.M. Temporal and spatial modulation of the tumor and systemic immune response in the murine Gl261 glioma model. PLoS One 2020, 15, e0226444. [Google Scholar] [CrossRef]
- Bausart, M.; Bozzato, E.; Joudiou, N.; Koutsoumpou, X.; Manshian, B.; Preat, V.; Gallez, B. Mismatch between Bioluminescence Imaging (BLI) and MRI When Evaluating Glioblastoma Growth: Lessons from a Study Where BLI Suggested "Regression" while MRI Showed "Progression". Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Leten, C.; Struys, T.; Dresselaers, T.; Himmelreich, U. In vivo and ex vivo assessment of the blood brain barrier integrity in different glioblastoma animal models. J Neurooncol 2014, 119, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Yeo, A.T.; Charest, A. Immune Checkpoint Blockade Biology in Mouse Models of Glioblastoma. J Cell Biochem 2017, 118, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, S.; Murle, B.; Felix, M.; Arns, A.; Groden, C.; Wenz, F.; Hug, A.; Glatting, G.; Kramer, M.; Giordano, F.A.; Brockmann, M.A. Imaging of Orthotopic Glioblastoma Xenografts in Mice Using a Clinical CT Scanner: Comparison with Micro-CT and Histology. PLoS One 2016, 11, e0165994. [Google Scholar] [CrossRef]
- Bouckaert, C.; Christiaen, E.; Verhoeven, J.; Descamps, B.; De Meulenaere, V.; Boon, P.; Carrette, E.; Vonck, K.; Vanhove, C.; Raedt, R. Comparison of In Vivo and Ex Vivo Magnetic Resonance Imaging in a Rat Model for Glioblastoma-Associated Epilepsy. Diagnostics 2021, 11. [Google Scholar] [CrossRef]
- Ruotsalainen, J.; Martikainen, M.; Niittykoski, M.; Huhtala, T.; Aaltonen, T.; Heikkilä, J.; Bell, J.; Vähä-Koskela, M.; Hinkkanen, A. Interferon-Beta Sensitivity of Tumor Cells Correlates with Poor Response to VA7 Virotherapy in Mouse Glioma Models. Molecular Therapy 2012, 20, 1529–1539. [Google Scholar] [CrossRef]
- Kober, C.; Weibel, S.; Rohn, S.; Kirscher, L.; Szalay, A.A. Intratumoral INF-γ triggers an antiviral state in GL261 tumor cells: a major hurdle to overcome for oncolytic vaccinia virus therapy of cancer. Molecular Therapy Oncolytics 2015, 2. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |
Figure 1.
In vitro verification of luciferase activity. (A) Quantified in vitro bioluminescence of 5,000; 10,000; 15,000; and 20,000 GL261 Red-FLuc cells one to 30 min following the addition of D-luciferin (0.5 µg/well) (n = 3, 3 technical replicates; data points represent mean ± SEM); (B) Quantified in vitro bioluminescence of 5,000; 10,000; 15,000; and 20,000 TRP-mCF cells one to 30 min following the addition of D-luciferin (0.5 µg/well) (n = 3, 3 technical replicates; data points represent mean ± SEM); (C) Representative image of in vitro bioluminescence of GL261 Red-FLuc (G) and TRP-mCF (T) cells 30 min following the addition of D-luciferin; (D) Linear correlation between seeded cell number and bioluminescence for GL261 Red-FLuc and TRP-mCF cells (GL261 Red-FLuc: Y = 7366*X - 10058920, R2 = 0.985, p < 0.0001; TRP-mCF: Y = 7268*X + 6461663, R2 = 0.932, p < 0.0001). Statistics: Simple linear regression.
Figure 1.
In vitro verification of luciferase activity. (A) Quantified in vitro bioluminescence of 5,000; 10,000; 15,000; and 20,000 GL261 Red-FLuc cells one to 30 min following the addition of D-luciferin (0.5 µg/well) (n = 3, 3 technical replicates; data points represent mean ± SEM); (B) Quantified in vitro bioluminescence of 5,000; 10,000; 15,000; and 20,000 TRP-mCF cells one to 30 min following the addition of D-luciferin (0.5 µg/well) (n = 3, 3 technical replicates; data points represent mean ± SEM); (C) Representative image of in vitro bioluminescence of GL261 Red-FLuc (G) and TRP-mCF (T) cells 30 min following the addition of D-luciferin; (D) Linear correlation between seeded cell number and bioluminescence for GL261 Red-FLuc and TRP-mCF cells (GL261 Red-FLuc: Y = 7366*X - 10058920, R2 = 0.985, p < 0.0001; TRP-mCF: Y = 7268*X + 6461663, R2 = 0.932, p < 0.0001). Statistics: Simple linear regression.
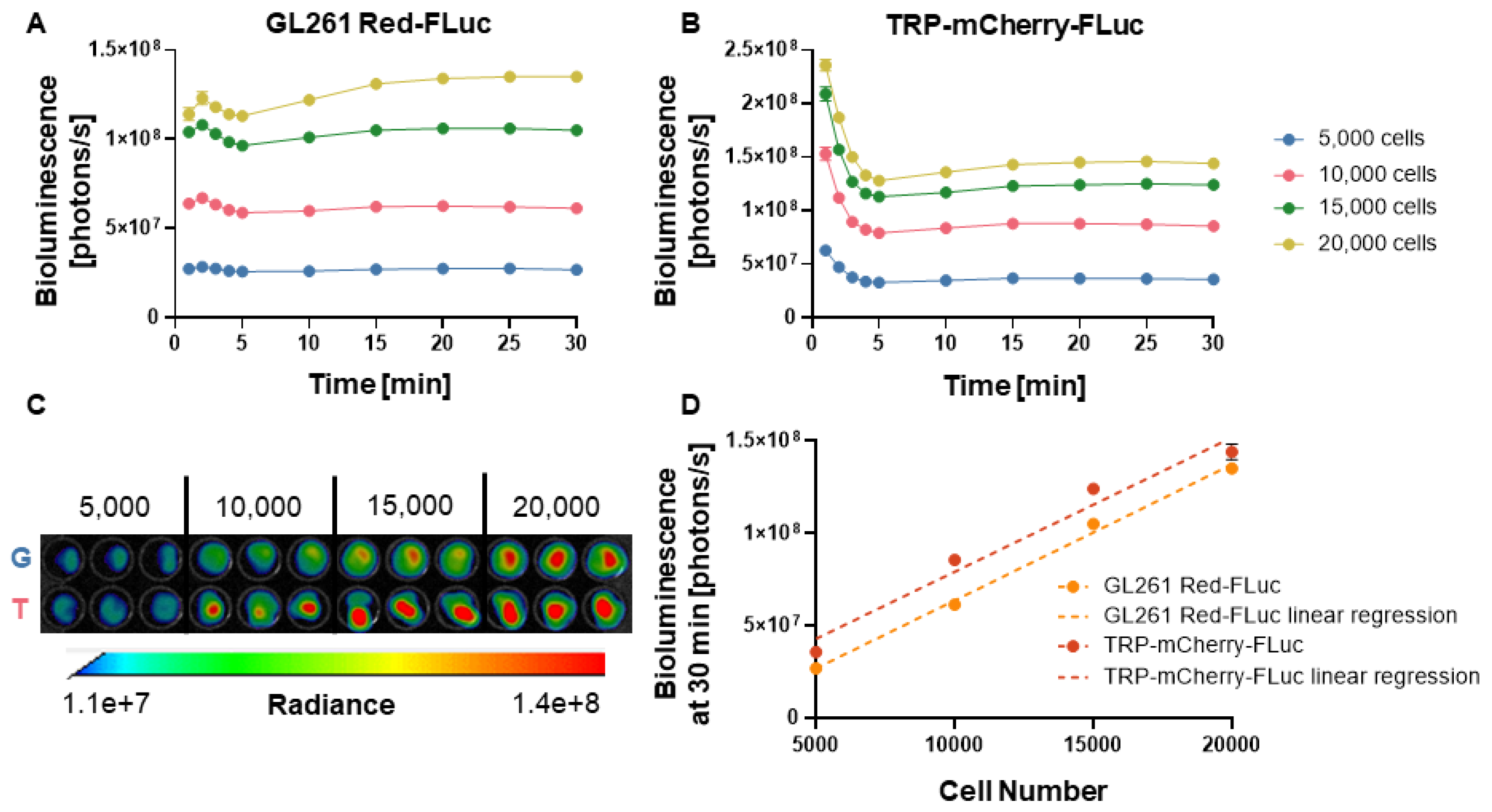
Figure 2.
GL261 Red-FLuc tumor take and survival in immunocompetent (albino B6) mice. (A) Representative in vivo bioluminescence images in albino B6 mice intracranially injected with 50,000 GL261 Red-FLuc cells. Total emission range: 1*106 to 2*108 photons/s. (B) Quantified bioluminescence of accepting and rejecting mice over 4 weeks post-implantation (above) with descriptive statistics of modeled data (below). Coefficient estimates are on log link. R2 is for fixed effects. (C) Survival curve of mice with accepted or rejected intracranial GL261 Red-FLuc tumors. Data points represent mean ± SEM (n = 51).
Figure 2.
GL261 Red-FLuc tumor take and survival in immunocompetent (albino B6) mice. (A) Representative in vivo bioluminescence images in albino B6 mice intracranially injected with 50,000 GL261 Red-FLuc cells. Total emission range: 1*106 to 2*108 photons/s. (B) Quantified bioluminescence of accepting and rejecting mice over 4 weeks post-implantation (above) with descriptive statistics of modeled data (below). Coefficient estimates are on log link. R2 is for fixed effects. (C) Survival curve of mice with accepted or rejected intracranial GL261 Red-FLuc tumors. Data points represent mean ± SEM (n = 51).
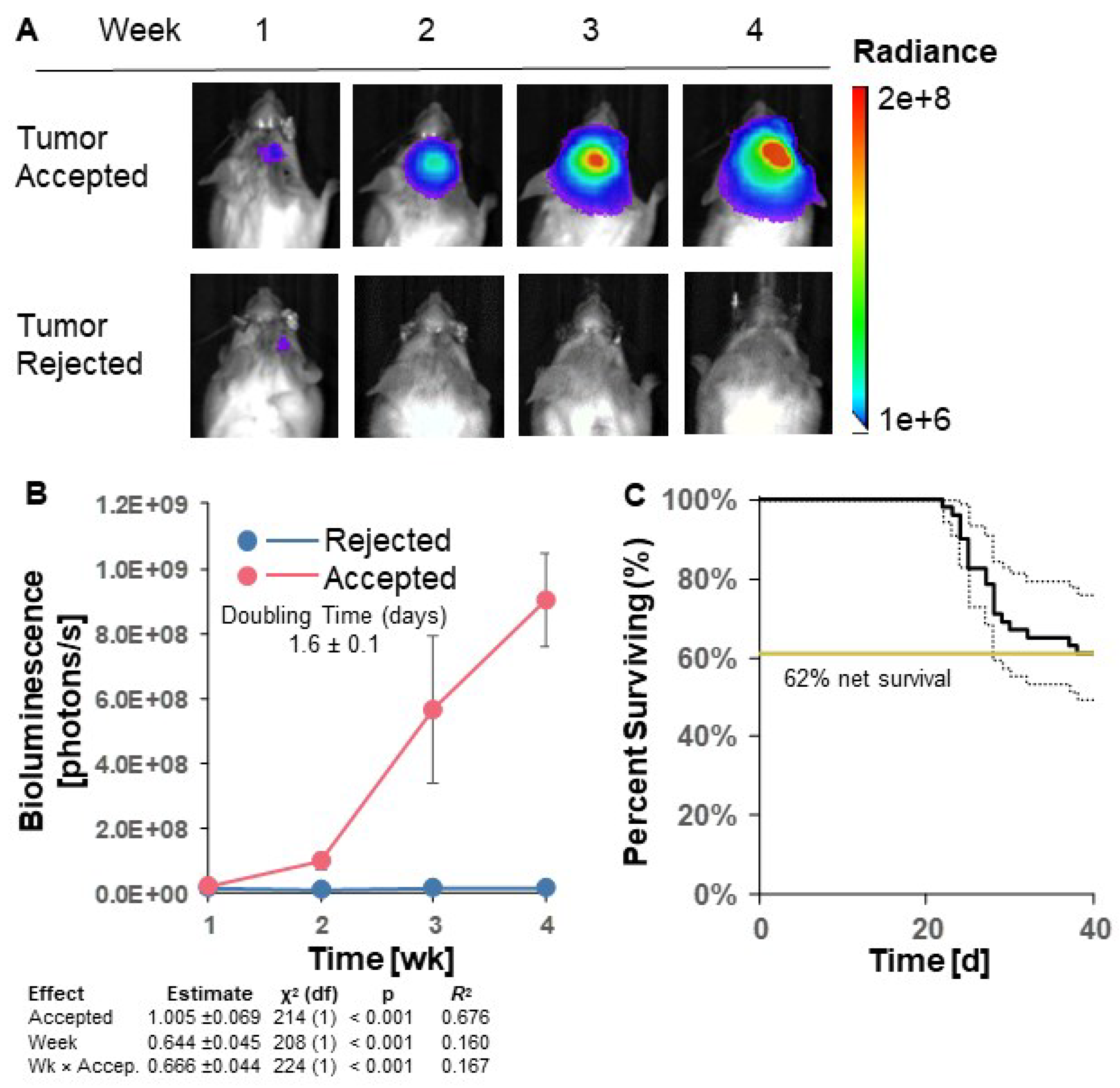
Figure 3.
GL261 Red-FLuc tumor take and survival in immunocompromised (J: NU) mice. (A) Representative images of tumor bioluminescence in J: NU mice intracranially injected with GL261 Red-FLuc cell doses ranging from 5,000 to 50,000 cells. Total emission range: 1x107 to 3x109 photons/s. (B) Quantified bioluminescence of mice by dose and week post-implantation (above) and descriptive statistics of modeled data (below). Coefficients estimated on log link. (C) Log-response of quantified bioluminescence by dose and week post-implantation. (D) Survival modeling of mice by dose and days post-implantation (above) and descriptive statistics of modeled data (below). Solid lines show the predicted effects of cell dose (logistic link). Points joined by dashed lines are Kaplan-Meier curves segregated by dose. Statistics (χ2, p, R2) are from the parametric survival model. Data points represent mean ± SEM. For group numbers, see Table 1.
Figure 3.
GL261 Red-FLuc tumor take and survival in immunocompromised (J: NU) mice. (A) Representative images of tumor bioluminescence in J: NU mice intracranially injected with GL261 Red-FLuc cell doses ranging from 5,000 to 50,000 cells. Total emission range: 1x107 to 3x109 photons/s. (B) Quantified bioluminescence of mice by dose and week post-implantation (above) and descriptive statistics of modeled data (below). Coefficients estimated on log link. (C) Log-response of quantified bioluminescence by dose and week post-implantation. (D) Survival modeling of mice by dose and days post-implantation (above) and descriptive statistics of modeled data (below). Solid lines show the predicted effects of cell dose (logistic link). Points joined by dashed lines are Kaplan-Meier curves segregated by dose. Statistics (χ2, p, R2) are from the parametric survival model. Data points represent mean ± SEM. For group numbers, see Table 1.
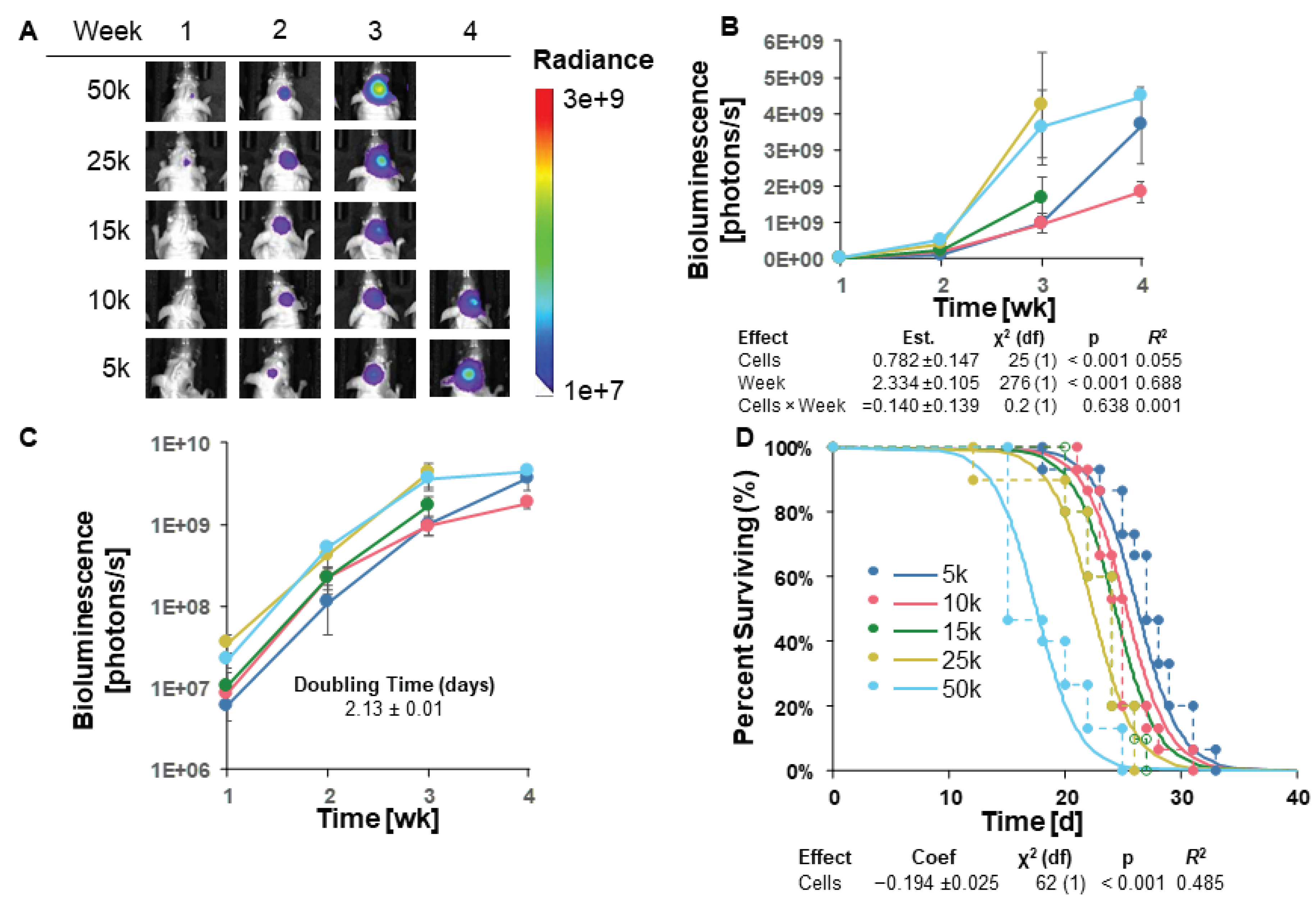
Figure 4.
TRP-mCherry-FLuc tumor take and survival in immunocompetent (albino B6) mice. (A) Representative images of tumor bioluminescence in albino B6 mice intracranially injected with TRP-mCF cell doses ranging from 5,000 to 50,000 cells. Total emission range: 1x107 to 3x109 photons/s. (B) Quantified bioluminescence of mice by dose and week post-implantation (above) and descriptive statistics of modeled data (below). Coefficients estimated on log link. (C) Log-response of quantified bioluminescence by dose and week post-implantation. (D) Survival modeling of mice by dose and days post-implantation (above) and descriptive statistics of modeled data (below). Solid lines show the predicted effects of cell dose (logistic link). Points joined by dashed lines are Kaplan-Meier curves segregated by dose. Statistics (χ2, p, R2) are from the parametric survival model. Data points represent mean ± SEM (n = 5/group).
Figure 4.
TRP-mCherry-FLuc tumor take and survival in immunocompetent (albino B6) mice. (A) Representative images of tumor bioluminescence in albino B6 mice intracranially injected with TRP-mCF cell doses ranging from 5,000 to 50,000 cells. Total emission range: 1x107 to 3x109 photons/s. (B) Quantified bioluminescence of mice by dose and week post-implantation (above) and descriptive statistics of modeled data (below). Coefficients estimated on log link. (C) Log-response of quantified bioluminescence by dose and week post-implantation. (D) Survival modeling of mice by dose and days post-implantation (above) and descriptive statistics of modeled data (below). Solid lines show the predicted effects of cell dose (logistic link). Points joined by dashed lines are Kaplan-Meier curves segregated by dose. Statistics (χ2, p, R2) are from the parametric survival model. Data points represent mean ± SEM (n = 5/group).
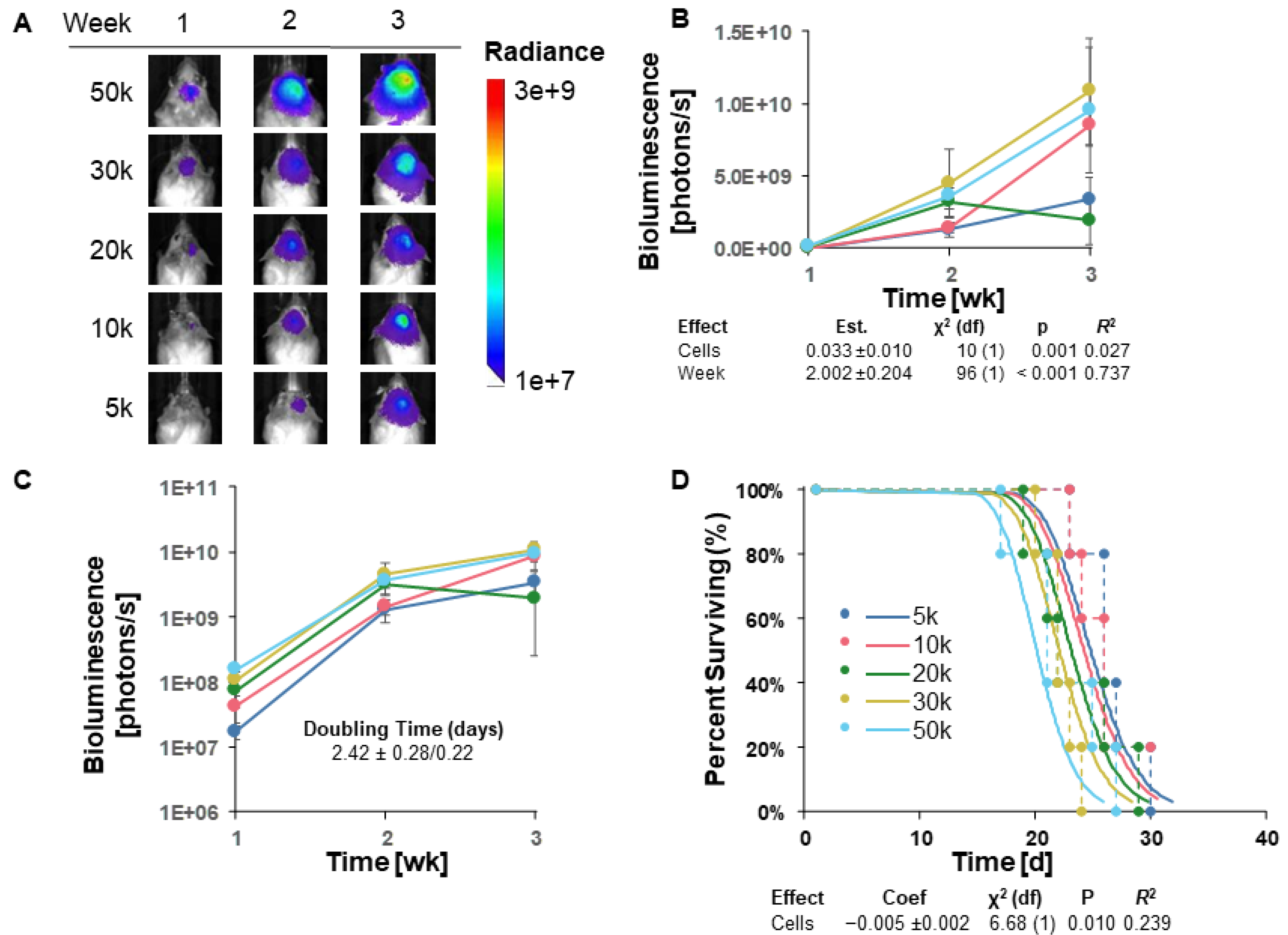
Figure 5.
MRI features and tumor volume determination. Top: Representative (A) pre-contrast T1-weighted, (B) post-contrast T1-weighted, and (C) T2-weighted images of J: NU mice injected with 5,000 GL261 Red-FLuc cells. Bottom: Representative (D) pre-contrast T1-weighted, (E) post-contrast T1-weighted, and (f) T2-weighted images of albino B6 mice injected with 5,000 TRP-mCF cells. Images were taken three weeks post-intracranial injection. Red arrow: light region (fluid); yellow arrow: dark region (hematoma) or low Gd contrast enhancement; white arrow: mass effect. Size bar = 2 mm.
Figure 5.
MRI features and tumor volume determination. Top: Representative (A) pre-contrast T1-weighted, (B) post-contrast T1-weighted, and (C) T2-weighted images of J: NU mice injected with 5,000 GL261 Red-FLuc cells. Bottom: Representative (D) pre-contrast T1-weighted, (E) post-contrast T1-weighted, and (f) T2-weighted images of albino B6 mice injected with 5,000 TRP-mCF cells. Images were taken three weeks post-intracranial injection. Red arrow: light region (fluid); yellow arrow: dark region (hematoma) or low Gd contrast enhancement; white arrow: mass effect. Size bar = 2 mm.
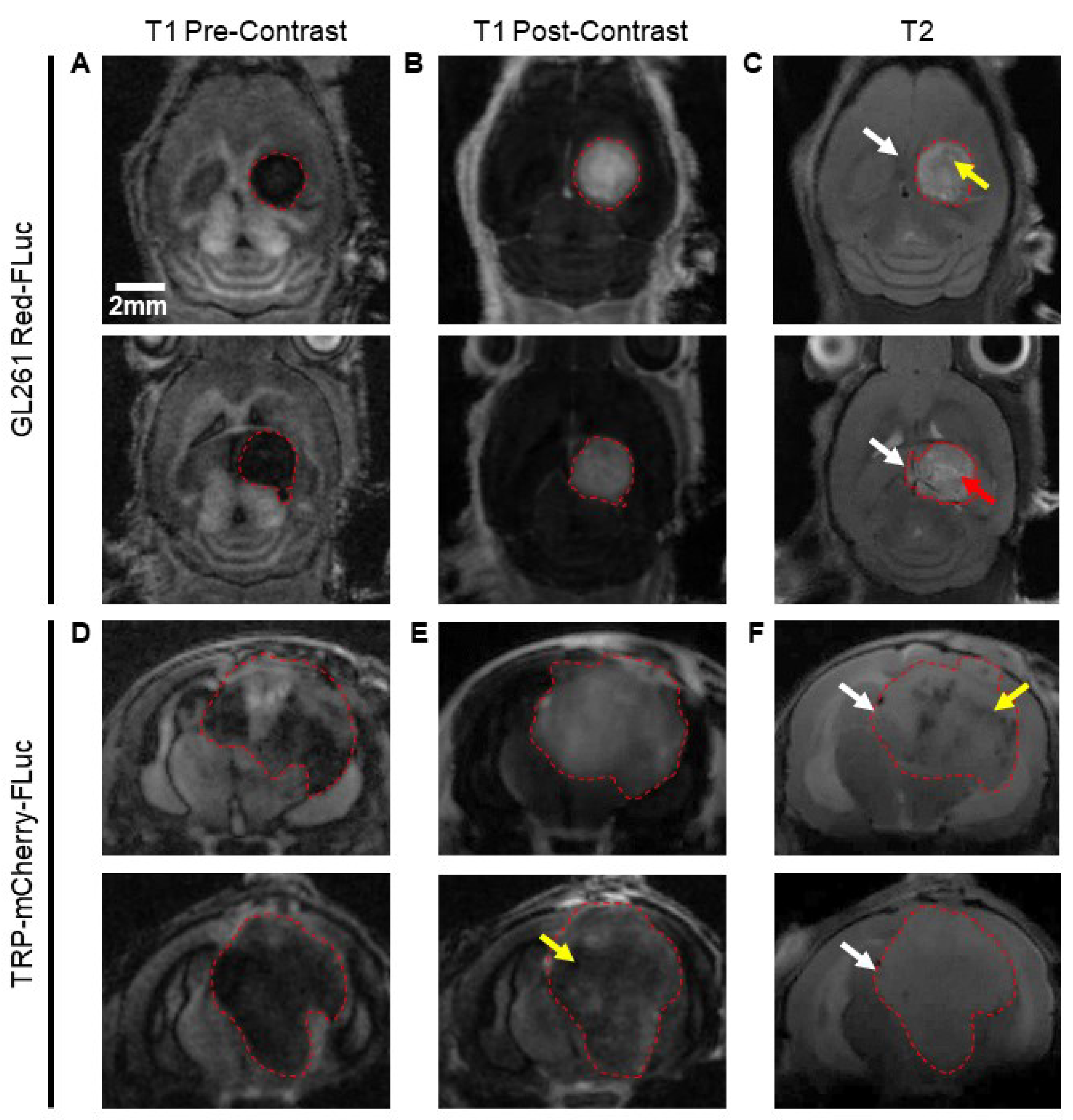
Figure 6.
Immunohistochemical features of GL261 Red-FLuc and TRP-mCherry-FLuc tumors. (A) Representative H&E-stained section of GL261 Red-FLuc tumor from J: NU mouse. Size bar = 2 mm (full slice) and 200 µm (11x zoom). (B) Close-up images of GL261 Red-FLuc H&E showing vascularization, pleomorphisms, and enhanced mitotic activity. Size bar = 10 µm. (C) Representative anti-firefly luciferase IHC of GL261 Red-FLuc tumor from J: NU mouse. Size bar = 2 mm (full slice) and 200 µm (11x zoom). (D) Representative H&E-stained section of TRP-mCF tumor from albino B6 mouse. Size bar = 2 mm (full slice) and 200 µm (11x zoom). (E) Close-up images of TRP-mCF H&E showing necrosis, perivascular tumor invasion, and enhanced mitotic activity. Size bar = 100 µm (left) and 10 µm (middle and right). (F) Representative anti-firefly luciferase IHC of TRP-mCF tumor from albino B6 mouse. Size bar = 2 mm (full slice) and 200 µm (11x zoom).
Figure 6.
Immunohistochemical features of GL261 Red-FLuc and TRP-mCherry-FLuc tumors. (A) Representative H&E-stained section of GL261 Red-FLuc tumor from J: NU mouse. Size bar = 2 mm (full slice) and 200 µm (11x zoom). (B) Close-up images of GL261 Red-FLuc H&E showing vascularization, pleomorphisms, and enhanced mitotic activity. Size bar = 10 µm. (C) Representative anti-firefly luciferase IHC of GL261 Red-FLuc tumor from J: NU mouse. Size bar = 2 mm (full slice) and 200 µm (11x zoom). (D) Representative H&E-stained section of TRP-mCF tumor from albino B6 mouse. Size bar = 2 mm (full slice) and 200 µm (11x zoom). (E) Close-up images of TRP-mCF H&E showing necrosis, perivascular tumor invasion, and enhanced mitotic activity. Size bar = 100 µm (left) and 10 µm (middle and right). (F) Representative anti-firefly luciferase IHC of TRP-mCF tumor from albino B6 mouse. Size bar = 2 mm (full slice) and 200 µm (11x zoom).
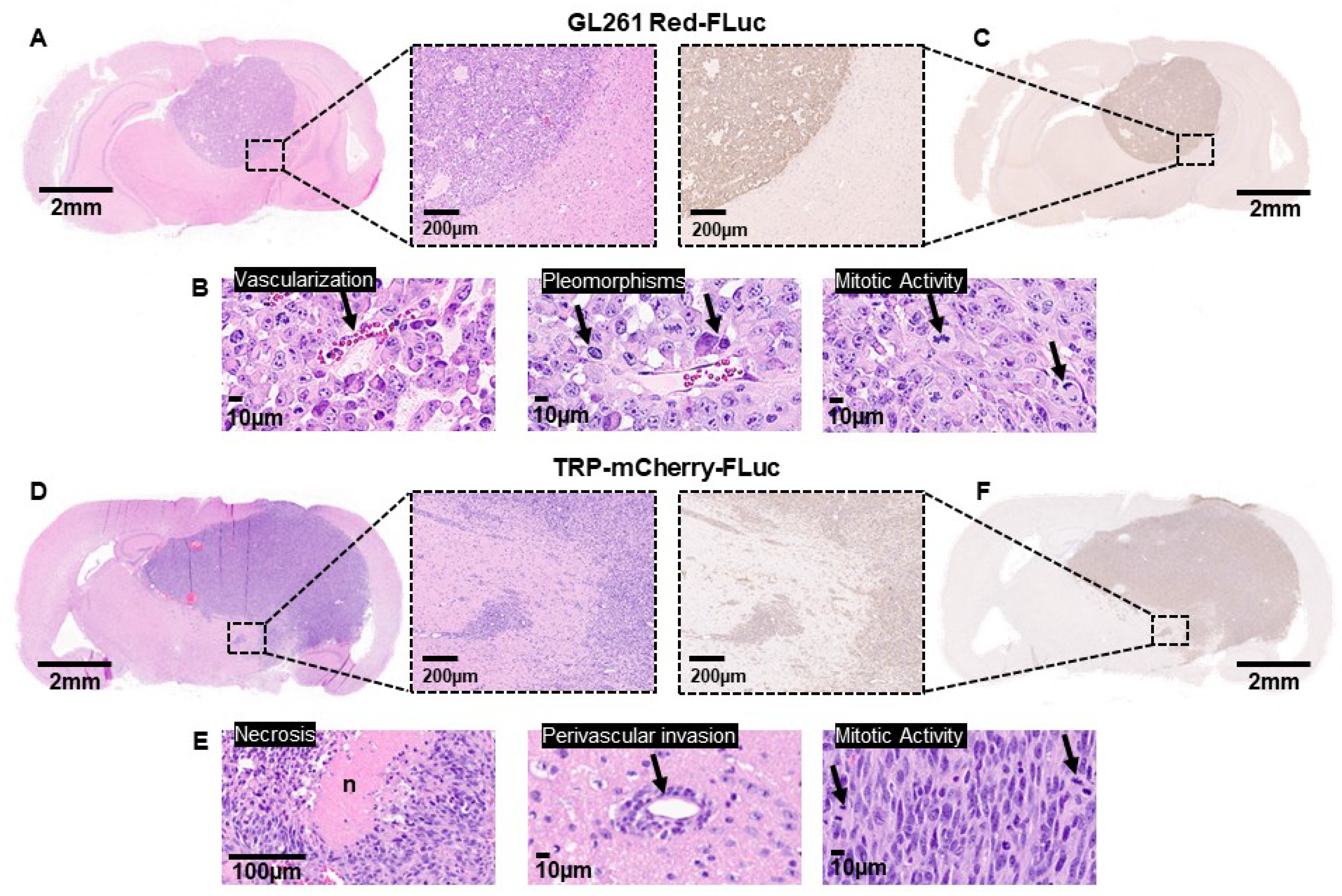
Figure 7.
High throughput determination of GL261 Red-FLuc and TRP-mCF tumor volume. (A) Representative anti-firefly luciferase immunohistochemistry of GL261 Red-FLuc tumors from J: NU mice injected with 5,000 cells three weeks post-implantation. (B) Analysis of tumor area determined by HALO AI™ Deep Learning Classifier Add-On. Red = tumor; green = non-tumor; yellow = glass/background. (C) Representative anti-firefly luciferase immunohistochemistry of TRP-mCF tumors from albino B6 mice injected with 5,000 cells three weeks post-implantation. (D) Analysis of tumor area determined by HALO AI™ Deep Learning Classifier Add-On. Red = tumor; green = non-tumor; yellow = glass/background.
Figure 7.
High throughput determination of GL261 Red-FLuc and TRP-mCF tumor volume. (A) Representative anti-firefly luciferase immunohistochemistry of GL261 Red-FLuc tumors from J: NU mice injected with 5,000 cells three weeks post-implantation. (B) Analysis of tumor area determined by HALO AI™ Deep Learning Classifier Add-On. Red = tumor; green = non-tumor; yellow = glass/background. (C) Representative anti-firefly luciferase immunohistochemistry of TRP-mCF tumors from albino B6 mice injected with 5,000 cells three weeks post-implantation. (D) Analysis of tumor area determined by HALO AI™ Deep Learning Classifier Add-On. Red = tumor; green = non-tumor; yellow = glass/background.
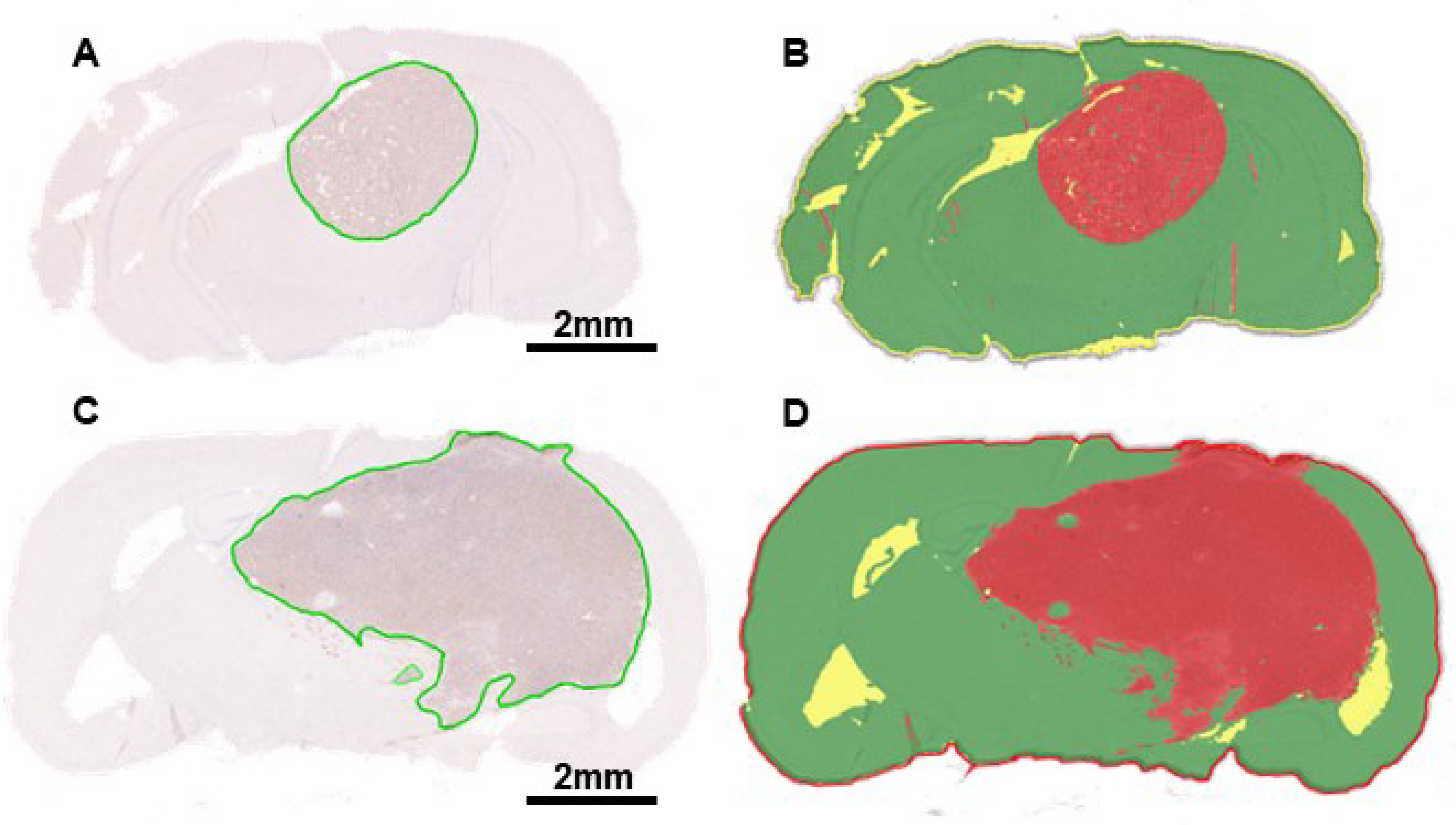
Figure 8.
Comparison of tumor volume estimation methods. (A, B) GL261 Red-FLuc and (C, D) TRP-mCF tumor volumes were measured by manual and AI quantification of histopathology samples and by MRI. We compared (A, C) differences in overall estimated levels and (B, D) agreement between methods. Rose-colored squares = mean ± SEM.
Figure 8.
Comparison of tumor volume estimation methods. (A, B) GL261 Red-FLuc and (C, D) TRP-mCF tumor volumes were measured by manual and AI quantification of histopathology samples and by MRI. We compared (A, C) differences in overall estimated levels and (B, D) agreement between methods. Rose-colored squares = mean ± SEM.


Table 1.
In vivo tumor growth characteristics of mice injected with decreasing numbers of GL261 Red-FLuc cells.
Table 1.
In vivo tumor growth characteristics of mice injected with decreasing numbers of GL261 Red-FLuc cells.
| Cell Number | Doubling Time (d) 1 | Median Survival (d) | N |
|---|---|---|---|
| 50k | 2.7 ± 0.5/0.7 | 19 | 17 |
| 25k | 2.4 ± 0.2/0.2 | 24 | 10 |
| 15k | 2.3 ± 0.1/0.1 | 24 | 10 |
| 10k | 2.2 ± 0.1/0.1 | 25 | 15 |
| 5k | 2.2 ± 0.2/0.1 | 27 | 15 |
1Tumor doubling times were calculated using a log-linked model, “Biofluorescence ~ ln(Cells) + Week", where ln(Cells) was the natural logarithm of cell dose. Values are presented as mean ± SD.
Table 2.
In vivo tumor growth characteristics of mice injected with decreasing numbers of TRP-mCherry-FLuc cells.
Table 2.
In vivo tumor growth characteristics of mice injected with decreasing numbers of TRP-mCherry-FLuc cells.
| Cell Number | Doubling Time (d) 1 | Median Survival (d) | N |
|---|---|---|---|
| 50k | 2.4 ± 0.4/0.7 | 20 | 5 |
| 30k | 2.4 ± 0.3/0.3 | 21 | 5 |
| 20k | 2.4 ± 0.2/0.3 | 21 | 5 |
| 10k | 2.4 ± 0.3/0.4 | 25 | 5 |
| 5k | 2.4 ± 0.3/0.4 | 25 | 5 |
1Tumor doubling times were calculated using a log-linked model, “Biofluorescence ~ ln(Cells) + Week", where ln(Cells) was the natural logarithm of cell dose. Values are presented as mean ± SD.
Table 3.
GL261 Red-FLuc and TRP-mCF Tumor Volume as Determined from Histopathology and MRI.
| Model | Tumor Volume (mm3) as Determined by: | N | ||
|---|---|---|---|---|
| Manual | AI | MRI | ||
| GL261 Red-FLuc | 17.7 ± 16.3 | 19.0 ± 17.0 | 28.9 ± 18.8 | 8 |
| TRP-mCF | 64.4 ± 19.0 | 64.4 ± 19.1 | 109.5 ± 38.9 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



