
Submitted:
18 March 2024
Posted:
22 March 2024
You are already at the latest version
Abstract
Gene regulation is vital for cellular function and homeostasis, involving diverse mechanisms controlling specific gene products and contributing to tissue-specific gene expression. Dysregulation leads to diseases, emphasizing the need for understanding these mechanisms. Non-coding regions, notably enhancers and super-enhancers (SEs), act as crucial gene regulators, orchestrating transcriptional activity via transcription factors (TFs). SEs, with high activity, are pivotal in cell identity and disease progression. Current computational approaches focus on individual regulators like TFs and miRNAs, neglecting SE interactions. We emphasize incorporating SEs into models to enhance understanding. Experimental studies have recently linked miRNAs and TFs to SEs. Chromatin immunoprecipitation followed by sequencing (ChIP-Seq) provides valuable SE information. In this review, we categorize computational methods leveraging TF and miRNA data into three areas and explore challenges integrating enhancers/SEs. These include unraveling indirect regulatory networks, identifying network motifs, and enriching pathway identification by dissecting gene regulators, including SEs. We hypothesize that addressing these challenges will enhance our understanding of gene regulation, aiding in identifying therapeutic targets and disease biomarkers. We believe that understanding the role of ChIP-Seq data in predicting the effect of miRNA on gene regulation is crucial to construct statistical/computational model that would tackle these challenges.
Keywords:
Super-enhancer
; miRNA
; ChIP-Seq
; Gene regulatory network
; Transcription factor
; Multi-omics
1. Introduction
Gene regulation represents a collection of mechanisms by which cells increase or decrease the production of specific gene products. Cells from different tissues turn on specific genes and turn off others thus contributing to tissue-related differences in the expressions of the whole repertoire of genes. Aberration in gene regulation may promote deviation from normal pathway activity, sometimes leading to complex diseases. Expression of any gene may be regulated due to DNA modification, transcriptional regulation, epigenetic gene regulation, regulation by RNA etc. While only a little over genome code for protein [1], the remaining genome that was once thought of as junk has emerged as a crucial hub of gene regulators.
These gene regulatory elements comprise non-coding DNA sequences that provide binding sites for some proteins, called transcription factors (TFs). These TFs play an important role in controlling the transcription rate of genetic material from DNA to RNA. TFs either alone or with other proteins form complexes to promote (called activators) or block (called repressors) the recruitment of RNA polymerase to specific genes. TFs participate in gene regulation by binding themselves to certain DNA regions such as enhancers and promoters often located near the genes. Enhancers are non-coding DNA sequences that contain multiple activator and repressor binding sites. In contrast to enhancers, silencers are regions on DNA that, when bound by specific TFs, repress gene expression. Marked by distinct chromatin features, enhancers contribute to the repertoire of epigenetic mechanisms responsible for cellular memory and cell type-specific gene expression [2]. In addition, enhancers play a vital role in spatiotemporal gene expression which is tissue-specific activation of genes at specific times during development. To regulate gene expression, active enhancers come in contact with closely located promoter regions of genes or distant promoter regions of genes through DNA looping [3]. While an active enhancer interacts with multiple promoters [2], an active promoter also interacts with multiple enhancers. Genetic mutations within enhancer have been implicated with tumors and various common diseases [4,5]. Clusters of regular enhancers in close genomic proximity that are associated with high levels of transcriptional co-activators (e.g. mediators), bear active chromatin marks and contain sequence motifs corresponding to cell-type specific TFs more than typical enhancers, are called super-enhancers, abbreviated as SEs [6]. The enhancers comprising an SE might be responsive to different signals, thus allowing multiple signaling pathways to regulate the transcription of a single gene [7]. Many GWAS SNPs are shown to be significantly enriched in SEs [8,9,10].
Role of ChIP-Seq technology: Chromatin immunoprecipitation followed by sequencing (ChIP-Seq) is a standard assay for mapping genome-wide protein–DNA interactions viz. characterization of binding sites for TFs and other DNA-binding proteins, enables identification of sequence motifs etc. [11]. ChIP-Seq also contributes to the identification of histone marks that act as major modulators of chromatin structure which regulates gene expression epigenetically. Apart from bioinformatic approaches [12], algorithms such as Hypergeometric Optimization of Motif EnRichment (HOMER) [13], and Rank-Ordering of Super-Enhancers (ROSE) [6,14] identify SEs from ChIP-Seq data. Since gene regulation is closely correlated with the 3D conformation of chromatin, other experimental assays such as Hi-C (high-throughput chromosome conformation capture) and Hi-C derivative technologies viz. ChIA-PET (chromatin interaction analysis based on paired-end-tag sequencing), HiChIP (in situ Hi-C followed by chromatin immunoprecipitation) etc. are also studied for enhancer identification. These technologies explore the chromatin structure to identify enhancer-target gene maps [15] that are also well validated by experimental methods [16]. Several databases using these technologies unravel information on enhancer-target gene, enhancer-disease, enhancer- target TF, enhancer-cancer type etc. along with enhancer and SE identification. Based on ChIP-Seq experiments, SEs from cancer or normal tissue/cell lines are identified by databases such as SEdb 2.0 [17], SEanalysis [18], CenhANCER [19], ENdb [20], EnhancerAtlas 2.0 [21] etc.
Role of miRNA in gene regulation: Like non-coding DNA, non-coding RNA molecules such as microRNA (miRNA) are also involved in regulation of gene expression. These approximately 21 nucleotide long miRNAs form are a class of small RNA regulators participating in RNA silencing and controlling gene expression post-transcriptionally [22]. miRNAs bind to target messenger RNA (mRNA) transcripts of protein-coding genes and regulate gene translation. Individual miRNAs often target hundreds of mRNAs to modulate expression of genes in various critical pathways such as developmental, metabolic etc. or completely switch on or off mRNA translation [22]. Animal and plant miRNAs are respectively complementary to 3′ UTR and coding regions of target mRNAs. For partial complementary miRNAs to recognize their targets, 2-7 nucleotides (called seed region) of the miRNA must be perfectly complementary to the target mRNA. Oftentimes, animal miRNAs form imprecise base pairing causing translational repression while the near-perfect base pairing in plants results in target degradation. Experimental identification of miRNA genes and their targets was challenging and involved laborious techniques leading to the development of computational algorithms [23] such as base-pairing patterns, thermodynamic stability of miRNA-mRNA hybrid etc. [24]. Using these algorithms, several computational target prediction methods viz. TargetScan [25], TargetScanS [26], PicTar [27], miRanda [28] etc. have been implemented. Many databases such as miRBase Targets [29], TarBase5.0 [30] etc. have been built for integrating functional annotations with the predicted miRNA sequences and their targets [24]. These databases are either based on literature mining or computational algorithms that use the aforementioned target prediction methods. However, depending on the algorithms used, computational methods incur varying amounts of false positive results in identifying target mRNAs. Thus, the overlap of information across different databases is often low [24]. Some databases like GOMir [31], miRecords [32] etc. integrates information from multiple prediction algorithms to get accurate results but it often worsens the inference [33]. This necessitates experimental validation to check the interaction between miRNA and its target mRNA directly [24] or computational methods that would learn from the true positive results of the existing large databases [34]. However, recent databases such as mirDIP 4.1 [35] claims to provide more accurate miRNA-target predictions by combining predictions from 30 independent resources using integrative score. Other methods such as MiRror [36], miRGate [37] also provides integrated target predictions.
Joint effect of miRNAs and TFs in gene regulation: Apart from regulating one another, sometimes miRNAs and TFs co-regulate ’target hubs’ of genes related to specific pathways [38]. Experimental methods showed that the miR-200 family and miR-205 together regulate epithelial-to-mesenchymal transition (EMT), by targeting E-cadherin transcriptional repressors ZEB1 and SIP1, which are previously implicated in EMT and tumour metastasis. Since EMT facilitates tissue remodelling during embryonic development and is viewed as an essential early step in tumour metastasis, these miRNAs are implicated in tumour progression via TFs [39]. Other experimental studies also illustrate the role of miRNA-TF co-regulation in playing a vital role in gene regulation [40]. The interaction between miRNAs and TFs involving auto-regulatory feedback loops is a common regulatory mechanism for controlling gene expression [41]. Many computational methods and databases identify networks to predict miRNA-TF, miRNA-mRNA, miRNA-TF-mRNA [42], and miRNA-disease regulatory relations [43]. Some of these methods are based on the networks that consider feed-forward, feed-backward etc. loops that controls gene regulation [44].
Evidence of gene regulation jointly via miRNAs, TFs and SEs: On the other hand, collective evidence illustrate the potential of SEs in unravelling gene regulatory network to a great extent. Recent experimental validation of the involvement of SEs in miRNA production, miRNA-associated disease, and TFs [45,46] provides an insight into its potential in unravelling gene regulation. For example, over-expression of miR-1301 induced by the disruption of SE KLF6 leads to a significant inhibition of cell proliferation [47]. Although enhancers and SEs are found to be associated with disease genes experimentally, they have not been computationally explored much to identify their connection in gene regulation network. Although some databases [48,49,50] connecting SE/enhancer with target genes, miRNAs and TFs are available but still there is a lack of computational method that might systematically explore the regulation network. Besides, the databases are constructed based on the overlaps between SEs and (a) the neighborhood of promoter regions of target genes, (b) TF binding sites or genes encoding for TFs and (c) the neighborhood of the transcription start site of miRNAs [49,50]. SEs are usually identified using ROSE or HOMER algorithm from different publicly available ChIP-Seq data [48] and integrated with other information based on the above approaches. Although these databases provide information within the transcriptional regulatory regions but are unable to explore the joint effect of these regulators on gene expression. As these databases are built on multiple databases gathering specific information, computational methods might leverage insight from them to develop robust methods and/or validate the known findings.
In this review, we first summarize our current understanding of the computational method unravelling gene regulation network in connection to miRNAs and TFs obtained from ChIP-based experiments. We also outline the necessity of incorporating SEs, another ChIP-Seq derived information, into the network and formulate open problems that highlights computationally motivating research areas. We broadly divide the research areas in three categories: (1) Unravelling indirect network of gene regulation, (2) Identification of network motifs to increase the precision target genes, and (3) Enriched pathway identification by jointly dissecting genes regulators. In the first category, we review computational methods or pipelines that explore effect of miRNAs on gene regulation through indirect networks. Some methods [51] accumulate information from databases on TFs associated target genes and miRNAs and combine them to predict genes that miRNAs target via TFs, while others explore the agreement between different data types and iteratively refine predictions [52]. We highlight that similar idea can be explored in the realm of SEs as well because they interact with miRNAs to deregulate genes, and multiple databases are available to provide specific information about SEs. In the second category, we explore methods identifying network motifs through which miRNAs impact target genes. Most of the work in this area deals with finding auto-regulatory loops involving TFs. Although experimentally SEs have been identified to form similar loops, but such connections remain untapped computationally. Moreover, presence of interactions identified in auto-regulatory loop-specific databases [50] in connection to SEs emphasize the importance of systemic identification of such network motifs. The third category highlights leveraging SEs to identify important pathways. We speculate that this will provide substantial insight in tissue-specific functions. Although this research area is well established in connection to TFs, miRNAs and target genes, SEs remained neglected largely despite of its potential in gene regulation network. Although experimental studies and databases have identified association of SEs in gene expression network, these methods are either very specific (as in experimental studies) or too generalized (as identified in databases) often measured across multiple studies/tissues/cell lines. But appropriate computational methods for systematic identification of the regulators in gene regulatory network is crucial. However, existing context-specific databases (summarized in Table 1) might be leveraged for developing such methods, validation of the findings through experimental studies would be ideal. But validation from existing publications also provides evidence of the efficacy of the developed methods.
In the next sections, we provide three computationally motivating and challenging areas of research related to unravelling some of the well-known players (viz. miRNAs, TFs) of gene regulatory networks, along with some recently popular ones like SEs (in Figure 1). With the existing methods available in each area (summarized in Table 2), we illustrate the potential insights and challenges that would be gained by incorporating SEs and provide motivational open problems for understanding gene regulation network a little better. The emerging experimental studies illustrating gene regulation in context of SE association (summarized in Table 3) provides a compelling reason to dissect SEs via computational methods to have improved understanding of gene regulation network.
2. Unravelling Indirect Network of Gene Regulation
2.1. Status
Identifying the mechanism orchestrating gene regulation is quite complex as it involves an intricate network of multiple cellular functions. Although experimental validations are required to confirm any cellular function, computational methods help in zeroing in on the plausible major players. To understand the role of miRNA in gene regulation, many challenges are encountered viz., identification of miRNAs, prediction of target mRNA for the corresponding miRNA directly or indirectly via TFs or SEs etc. Computational approaches for identifying miRNAs and predicting direct targets (with physical binding sites) of miRNAs have been vividly explored. However, the smaller overlap of miRNA targets across databases illustrates the need for higher accuracy in miRNA-mRNA mapping. To obtain this, it is imperative to include more information. Since it is known that gene expression is modulated by, TFs and/or SEs at the transcriptional level and miRNAs at the post-transcriptional level, with both expression regulators having the ability to regulate each other, dissecting their interplay might aid in understanding gene regulation [53]. Computational methods for identifying miRNA-TF-target gene is not straightforward. Recently, Sayed et al. developed miRinGO [51] to predict gene ontology (GO) annotations indirectly targeted by miRNAs via TFs. Interestingly, this approach integrates (1) predicted TF targets of miRNAs using the TargetScan database [25], with (2) computationally predicted tissue-specific TF associated with genes [54] using the PANDA [55] algorithm, to find indirect mRNA/gene targets of the corresponding miRNAs. The method PUMA [52] also uses PANDA to identify regulatory networks between miRNA and target genes. PUMA extracts information from TargetScan and miRanda databases [28] to construct the network. However, this method does not consider the simultaneous regulatory effect of TFs on target genes.
PANDA is a widely used approach for exploring agreement between different data types and iteratively refine predictions. Explicitly, this algorithm explores the co-regulatory effect between networks and efficiently circulates information among the networks. Originally, the PANDA algorithm combined information from gene expression, protein-protein interaction, and sequence motif data (or TF regulatory data) to reconstruct genome-wide gene regulatory networks. The network in PANDA consists of two types of nodes viz. effectors and affected, and three types of edges. Agents that control the behavior of subsequent ‘affected’’ targets (called affected nodes) are called effector nodes. The three types of edges either between pairs of effector nodes, from an effector to an affected node, or between pairs of affected nodes, represent three sources of information. PANDA uses Tanimoto similarity to obtain normalized z-scores between two sets of network edge weights. This allows PANDA to have a high similarity value when the edges either exist or do not exist in both networks. Initially, three networks viz., protein-protein, regulatory (from motif data), and gene co-expression are constructed respectively based on (1) whether the motif of TF i (say) is present in the control region of gene j (say), (2) whether TF k (say) interacts with TF (say), and (3) Pearson’s correlation. For final network validation, gold-standard experimental results (eg., ChIP-Seq analysis) based on stringent p-value cutoff are used to identify (1) whether TF i (say) significantly binds with the control region of gene j (say), (2) whether TF i (say) significantly bind TF j, and (3) whether any TF significantly binds to two genes, respectively. All the networks are then estimated and updated based on the weights of the edges. Messages are passed between networks using the concepts of ’responsibility’ and ’availability’, which are simultaneously estimated and updated along with other regulatory networks. Since multiple TFs can target a set of genes, the ’responsibility’ (defined by Tanimoto similarity) of an edge from TF i to gene j in the regulatory network determines the level of agreement between the TFs that target gene j and TFs that cooperate with TF i via physical protein complex etc. On the other hand, since genes that are targeted by the same TF are co-regulated, the ’availability’ (also defined by Tanimoto similarity) of an edge between TF i and gene j in the regulatory network, determines the level of agreement between the regulatory targets of TF i and the set of genes with which gene j is co-regulated. Since it is intuitive that regulation requires (1) a TF responsible for the regulatory status of its target gene, and (2) the target gene is available to be regulated by the TF, the network is updated till convergence of the average of responsibility and availability. Thus, PANDA explores the agreement between different data types and iteratively refine predictions.
2.2. Open Problem
Although computational approaches for combining TFs with miRNAs to identify target mRNAs are available (such as miRinGO), SEs have not been explored much in this context even when experimental methods have implicated SEs with miRNA production and miRNA-associated disease [45]. SEs together with histone marks influence tissue-specific evolutionarily conserved atlas of miRNA expression and function. The role of SE constituents in boosting cell-specific miRNA production has been validated by CRISPR/Cas9 assay [45]. SEs have been implicated along with key oncogenes in multiple cancer types [10,14]. The discovery of cancer hallmarks obtained from dissecting cancer-related miRNAs that are associated with SE alterations has paved the way for the identification of SE-miRNA biomarkers [45,56]. Although experimental methods implicate SEs for miRNA processing, SE-miRNA regulatory relations have not been computationally explored much. On the other hand, the SE-TF regulatory network that plays a crucial role in the carcinogenesis of malignant tumours [57] has not been explored in connection to target gene prediction of miRNAs. Using ChIP-Seq data, a few databases identifying enhancers [50], and SEs [17] have become available in recent times, which might be explored to better understand the interplay between genes and their regulators. So, ChIP-Seq data has great potential to dissect the indirect regulatory role of miRNAs on the target genes.
3. Identification of Network Motifs to Increase the Precision Target Genes
3.1. Status
Transcription networks regulate responses of living cells through different biochemical wiring patterns called network motifs. One of these motifs is feed-forward loop (FFL). In a regulatory network of genes altered by an miRNA and a TFs (or two miRNAs) via FFL, a target gene is regulated by both the miRNA and TF (or the miRNAs). In an FFL with miRNA-TF co-regulators, the TF often appears to regulate the miRNA or gets regulated by the miRNA or both. Computational methods identified these FFLs and hub of target genes each potentially targeted by multiple miRNAs and TFs [38]. Based on statistical ranks of the computationally predicted FFLs, dChip-GemiNI [58] identifies differential gene and miRNA expression between two biological conditions such as normal and cancer. Other methods [43,59] also explore crosstalk between the two regulators and their targets. Moreover, to achieve increased precision of gene regulatory network, it might be important to have a detailed understanding of the direction of FFLs. These could either speed up the response time of the target gene expression following stimulus steps in one direction but not in the other direction. These FFL are called incoherent FFLs. Another type of FFL called coherent FFL act as sign-sensitive delays. Since FFL has three nodes, each edge can possibly have two kinds of interactions (activator or repressor) that gives rise to eight different structural configurations of FFL. Identification of the FFL structure [60] might provide some insight into the architecture of the gene regulatory network.
On the other hand, enhancers are regulatory elements that regulates expression of protein-coding genes or miRNAs by recruiting TFs in a tissue-specific manner. To activate adjacent genes, enhancers tend to loop to them [2]. However, many enhancers map to target gene promoters located large distances away, by forming loops in the three-dimensional nuclear space to achieve close spatial proximity to distant genes [61]. Given that promoter interactomes are highly cell-type specific, enhancers have been linked with active promoter of genes [62]. Besides, SEs that are often associated with high signals of active histone marks from ChIP-Seq experiments, plays an important role in the miRNA production. In cancer cells, miRNAs with SE gain are found to be more associated with oncogenic roles while those with SE loss correlated with tumor-suppressive behavior [45]. Experimental studies [6] identified large enhancer domains that are densely occupied by TFs and Mediators and are associated with genes encoding key regulators of cell identity. In pluripotent embryonic stem cells (ESCs), these domains based on their sizes, transcription factor density and content, ability to activate transcription, and sensitivity to perturbation, have been identified as SEs. In ESCs, alteration in the TF or Mediator causes preferential loss of expression of SE-associated genes compared to other genes. In more differentiated cells, SEs containing cell-type-specific master TFs are also found to be associated with genes that define cell identity. It has been observed that genes encoding ESC master TFs are themselves driven by SEs, forming feedback loops where key TFs regulate their own expression [6]. IKAROS, prominently associated with leukemia, collaborates with TFs and SEs via FFL, and triggers aberrant gene expression program in a B-cell epithelial transition [63]. Experimental studies conducted by a scientific group over a decade apart identified involvement of deregulated miRNAs, SEs, and TFs in natural killer/T-cell lymphoma (NKTL) pathways [64,65]. This provides a clear motivation that SEs or enhancers should be involved in identifying network motifs to predict target genes with increased precision. But computational methods for identifying FFL or other network motifs involving SEs, TFs and miRNAs in transcription regulation are not yet explored in detail.
3.2. Open Problem
Even though SEs have been implicated for influencing tissue-specific miRNAs, it has not been studied to explore the network motifs in relation to transcription regulation. Database EnhFFL [49] provides FFL and other network motifs in relation with SE and typical enhancer with TFs, miRNAs and genes based on deterministic connections. But networks might come with intrinsic and/ or experimental uncertainties. This necessitates exploring of its stochastic properties. Such methods would make allowances for incorporating intrinsic uncertainties of the network building blocks into the model. It is important to note that, functionally related network motifs are not identical to topologically related motifs. Topological networks that are often identified by deterministic approaches might be contaminated with noise from experimental assays or databases. Stochastic networks allow for the circumventing the effect of such noisy observations through statistical or probabilistic modeling. Although some methods are available in conjunction with protein-protein interaction or gene regulation [66,67,68] but methods dealing with miRNAs, enhancers or SEs and TFs in connection to gene regulation have not been explored.
4. Enriched Pathways Identification by Jointly Dissecting Gene Regulators
4.1. Status
Gene regulation network is complex phenomenon that involves not only regulation of a particular gene by its multiple regulators such as TFs, miRNAs etc., but regulation of the expression of a bunch of regulators as well. Computationally identifying associations between individual TF or miRNA with genes might not be enough because the corresponding target gene might be targeted by other TFs and/or miRNAs. On the other hand, any TF or miRNA often targets multiple genes. Studies have shown that these genes are co-expressed at varying degrees depending on the gene regulators [69]. For example, compared to miRNAs, genes targeted by the same TFs tend to be much more co-regulated at mRNA and protein level. However, genes sharing common TFs or miRNAs have been associated with same disease. The complexity of gene regulation network increases because some genes also regulate expression of multiple miRNAs and TFs. Experimental studies provide important evidence that confirms the role of proto-oncogene c-Myc in regulating expression of TF E2F1 by altering expression of a cluster of miRNAs [70]. Conversely, E2F1 regulates c-Myc, thus revealing a putative positive feedback circuit [71]. Both c-Myc and E2F1 regulates cell growth and death by inducing transcription and modulating signal transduction [71]. It has been found that miRNAs regulate and gets regulated by E2F1 creating an auto-regulatory loop [72]. Collectively, these experimental studies reflect the complex interplay of various gene regulators in important pathways. Other experimental studies confirmed promotion of oncogenic MYC expression via SEs in Wnt signaling pathway [73]. Besides, SEs associated with TFs and other complexes plays an important role in gene regulation [63,65]. Thus, it is imperative to develop computational method to systematically dissect information on the gene regulators along with the target genes to understand context-specific gene regulation. This might lead to identification of functionally enriched pathways that would in turn encourage experimental studies to explore in detail specific players of gene regulation identified in a given context. While some method [74] leveraged miRNA-TF co-regulatory networks to identify pathways under miRNA control, and significantly enriched the proportion of true miRNA-target interactions, others constructed tools for identifying enrichment analysis for miRNAs [75,76]. Availability of such methods for miRNAs underscores the importance of developing similar methods for SEs.
4.2. Open Problem
Apart from playing a prominent role in miRNA biogenesis [45], SEs have been implicated in altering expression of genes key to cell identity in normal and diseased cells. In particular, experimental studies [7] show that cancer cells more often acquire SEs at genes that foster tumorigenesis. In addition, these genes are sensitive to perturbation of oncogenic signaling pathways. Furthermore, the enhancers constituting an SE may individually respond to various signals, enabling the regulation of a single gene’s transcription by multiple signaling pathways. Multiple studies have unraveled the potential involvement of cancer-specific SEs in the dysregulation of signaling pathway [77]. Coupled with these observations, the role of SEs in driving the biogenesis of miRNAs crucial for cell identity via enhancement of both transcription and Drosha/DGCR8-mediated primary miRNA processing [45], highlights the importance of identifying the enriched pathways by jointly dissecting gene regulators. Other studies also implicate miRNAs driven by SEs are associated with diseases or pathways. For example, miRNAs driven by SEs positively regulate Hippo pathway during liver development [78], deletion of KLF6 SE inhibits cell proliferation in HepG2 cells via miR-1301 over-expression [47], and in diseases such as black-fever [79] miRNAs driven by SEs mediates immune-suppression. Efforts have been made to relate SE with miRNA-associated gene regulation in pan-cancer analysis. Contrary, to the common inverse proportionality relation in expression between miRNA and mRNAs, studies [80] showed top-ranked positively correlated miRNA/gene sites are more likely to form SEs in major human cancers. Although such studies highlight the potential of decoding the relationship between SEs and associated miRNA in understanding gene regulation, computational methods to systematically explore such relations is still lacking. Plethora of associations identified by SEanalysis 2.0 [18] database connecting SEs, pathways, TFs, and genes. Existence of such database provides important evidence for identifying inter-relationship gene regulators in functionally relevant pathways. Although computational method for identifying pathways have been explored in context of miRNAs and TFs [74], methods involving SEs are still unavailable.

Table 1.
SE related databases and softwares.
| Database/Softwares | Feature |
|---|---|
| ROSE [6,14] | Pipeline identifying SEs from ChIP-Seq data; separates SEs and typical |
| enhancers | |
| HOMER [13] | Software for motif discovery and ChIP-Seq analysis; identifies enhancers |
| and SEs | |
| SEdb 2.0 [17] | Database for SE resource and annotate the potential roles in gene |
| transcription | |
| SEanalysis 2.0 [18] | Web server for identifying association connecting SEs, pathways, TFs, |
| and genes | |
| CenhANCER [19] | Database for cancer enhancers from primary tissues and cell lines |
| ENdb [20] | Manually curated database of experimentally supported enhancers |
| EnhancerAtlas 2.0 [21] | Database with enhancer annotation across nine species |
| TRmir [48] | Database for miRNA related transcriptional regulation especially typical |
| enhancer and SE | |
| EnhFFL [49] | Database for enhancer related FFLs based on deterministic connections |
| EnhancerDB [50] | Database for enhancer related transcriptional regulatory associations |
Table 2.
Computational methods related to miRNA, mRNA, TF, and sequence motif to understand gene regulation network.
Table 2.
Computational methods related to miRNA, mRNA, TF, and sequence motif to understand gene regulation network.
| Methods | Feature |
|---|---|
| miRinGO [51] | Accumulate information from databases on TFs associated target genes |
| and miRNAs; then combine them to predict genes that miRNAs target | |
| via TFs | |
| PANDA [55] | A message-passing model integrating protein-protein interaction, gene |
| expression, and sequence motif data to predict regulatory relationships | |
| PUMA [52] | Identify gene regulatory networks under miRNA control using PANDA |
| and target genes | |
| Sonawane et al. [54] | Computationally predict tissue-specific TF associated with genes |
| using PANDA | |
| dChip-GemiNI [58] | Statistically ranks computationally predicted FFLs to account for |
| differential gene and miRNA expression between two biological | |
| conditions | |
| FFLtool [59] | A web based tool for detecting FFL of TF-miRNA-target regulation in |
| human | |
| Mangan and Alon [60] | Theoretically analyze the functions of all possible structural types of |
| FFLs | |
| Jiang et al. [66] | Identify network motif using stochastic networks |
| Yeger-Lotem et al. [67] | Developed algorithms for detecting networks motifs with two or |
| more types of interactions | |
| Kashtan et al. [68] | Algorithms for detecting network motif generalizations |
| Prompsy et al. [74] | Leveraged miRNA-TF co-regulatory networks to identify pathways |
| under miRNA control, and significantly enriched the proportion of true | |
| miRNA-target interactions | |
| MiEAA [75] | A web-based application for miRNA set enrichment analysis and |
| annotation | |
| miRFA [76] | Pipeline for biomarker discovery involving mature miRNAs |
| Shalgi et al. [38] | Identifies miRNA-TF regulatory network |
Table 3.
Experimental studies illustrating gene regulation in context of SE association.
| Studies | Feature |
|---|---|
| Whyte et al. [6] | SEs play key roles in the control of mammalian cell identity; formation of |
| SE driven feedback loops; regulation of SE-associated gene expression | |
| via master TFs | |
| Hnisz et al. [7] | SEs are occupied more frequently by terminal TFs of the Wnt, TGF-b, and |
| LIF signaling pathways in ESCs/cancer cells; and SE-driven genes respond | |
| to manipulation of these pathways compared to typical enhancers | |
| Hnisz et al. [10] | Cancer cells generate SE at oncogenes and other genes related to tumor |
| pathogenesis | |
| Lovén et al. [14] | SEs are associated with critical oncogenic drivers in cancer cells |
| Suzuki et al. [45,56] | SEs potentially drive the biogenesis of miRNAs crucial for cell identity via |
| enhancement of both transcription and Drosha/DGCR8-mediated primary | |
| miRNA processing | |
| Ri et al. [47] | Over-expression of miR-1301 induced by deletion of KLF6 SE inhibits cell |
| proliferation in HepG2 cells | |
| Liang et al. [57] | SE-TF regulatory network plays a crucial role in the carcinogenesis of |
| malignant tumour | |
| Javierre et al. [62] | Promoter interactions are highly cell-type specific and enriched for |
| association between active promoters and epigenetically marked enhancers | |
| Hu et al. [63] | IKAROS, prominently associated with leukemia, collaborates with TFs and |
| SEs via FFL, and triggers aberrant gene expression program in a B-cell | |
| epithelial transition | |
| Zhou et al. [65] | SE-driven TF gene mediates oncogenesis in Natural Killer/T Cell |
| Lymphoma | |
| Scholz et al. [73] | WNT signaling activates MYC expression via SE in cancer cells |
| Zhang et al. [78] | miRNAs driven by SEs positively regulate Hippo pathway during liver |
| development | |
| Das et al. [79] | miRNAs driven by SEs mediates immune-suppression |
| Tan et al. [80] | miRNAs/genes with positive correlations tend to form super-enhancer-like |
| regions | |
| Turunen et al. [81] | Synergistic role of miRNAs and TFs on SEs associated with Hippo |
| signaling pathway |
5. Discussion
miRNAs and TFs have been extensively studied in connection to gene regulation. Aberration in these gene regulators have been implicated in various diseases. In this review, we highlight the role of ChIP-Seq data that potentially generates much more information other than TF, to unravel gene regulation network under miRNA control. Recent experimental studies have illustrated the synergistic role of miRNAs and TFs on enhancers and SEs in gene regulation network, particularly in cancer [81]. SEs are not only observed near genes with cell-type specific functions, but they are also considered sensitive to alteration in chromatin-based mechanism of gene regulation [10]. For example, in Burkitt’s lymphoma, strong enhancers come in close proximity of oncogenes via genomic rearrangements such as translocation. But compared to normal enhancers, SEs are enriched in sequence motifs corresponding to cell-type specific master TFs. SEs have been implicated to drive the biogenesis of miRNAs that are crucial for cell identity via enhancement of both transcription and Drosha/DGCR8-mediated primary miRNA (pri-miRNA) processing. CRISPR/Cas9 genomics revealed that SEs facilitate Drosha/DGCR8 recruitment and pri-miRNA processing that boosts cell-specific miRNA production [45]. Tissue-specific and evolutionarily conserved atlas of miRNA expression and function are observed to be largely shaped by SEs and broad H3K4me3 domains identified by ChIP-Seq. Both well studied cancer-related miRNAs [82] and other miRNAs with SE alterations have been associated with cancer hallmarks. Moreover, targeting SE components such as disruption of SE structure or inhibition of SE cofactors have attracted therapeutic options across various cancers [77].
It is well known that a single miRNA can target multiple genes and mediate their post-translational regulations. But accurately identifying these target genes experimentally and computationally is still a challenge. Numerous databases are constructed based on curated experimental gene targets and/or computationally predicted targets. But the little overlap between these databases indicates presence of false positive predictions. The reason behind this discrepancy might be due to the lack of appropriate method available to combine more players involved in the regulatory network. With the advent of NGS technology, we have access to a huge collection of high-throughput data. But it is still challenging to understand the complexity in the gene regulation. Computationally TFs and miRNAs are extensively studied in relation to gene expression regulation because of their better biological understanding. But SEs have not been explored much in parallel to TFs and miRNAs, despite of experimental validation of its involvement in gene regulation network. Although, SEs could be identified from the Chip-Seq experiments like TFs, identification of SEs is not straightforward [83]. Oftentimes, putative enhancers in close genomic proximity are termed as SEs. Although the definition of SEs is not very clear, a few databases such as HOMER [13], ROSE [6,14], EnhancerDB [50] etc. are now available that provides information on SEs and enhancers.
Here, we aim to provide the status of the existing computational method that unfold the gene regulatory network in connection to SE, TF and miRNA jointly. We also bring forth the gap in the knowledge in three different areas as open problems for the scientific community to consider. In connection to gene expression regulation, we first explore networks that dissects the role of miRNA, TFs and gene expressions collected from different databases using integrative computational approaches. A number of these methods are constructed based on iteratively updating information of one data source using different data sources. Other methods use multiple databases and integrate partial information to get an overall picture of predicted associations. But none of these methods have explored SEs in this connection, despite of being experimentally implicated in aberration of gene expression. Secondly, different network motifs or biochemical wiring patterns have been studied relating to genes and its regulators such as, TFs and miRNAs. Computationally identifying these patterns might provide a greater insight in the gene regulatory network. Motifs such as FFL and others aids in understanding the actual direction of the flow of information. But identifying motifs in connection with SEs or enhancers have been largely overlooked. But presence of databases such as EnhFFL, that curate motifs in connection to enhancers/SEs emphasizes the importance of such information to scientific community. But these FFLs are created based on physical overlaps between either enhancer-TF-miRNA, enhancer-TF-gene, enhancer-TF, or enhancer-miRNA-gene across several tissue/cell lines in human and mouse. Lastly, we discuss about the importance of identifying pathways through enrichment analysis. Since gene regulatory network is quite complex with multiple genes being target of one or more miRNAs and vice-versa, it is imperative to focus collectively on genes having similar functions and their regulators. Moreover, since chromatin folding plays a huge role in gene regulation we emphasize in dissecting information from ChIP-Seq experiments to better understand the effect of gene regulators in close-knit networks or pathways. The availability of SE databases such as SEanalysis 2.0 curated from multiple Chip-Seq data could mark an important piece of information. Although, computational methods [74] for identifying pathways associated with miRNAs and TFs exists, we are yet to develop similar approaches involving SEs that might provide substantial insight for gene regulation network.
In conclusion, while significant progress has been made in understanding gene regulation networks involving TFs and miRNAs, the integration of SEs into computational analyses remains an open research area. Developing computational methods to incorporate SEs into regulatory network predictions will enhance our understanding of gene regulation mechanisms and facilitate the identification of functionally relevant pathways in health and disease.
Author Contributions
Conceptualization, S.D. and S.R.; writing—original draft preparation, S.D.; writing—review and editing, S.R.; visualization, S.D.; supervision, S.R. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
Dr. Shesh Rai was generously supported by UC Cancer Center; Dr. William Barrett, Co-Director for UC Cancer Center, Charles M. Barrett M.D. Endowed Chair, & Professor and Dr. Syed Ahmad, Chief of Section of Surgical Oncology, Vice Chair for Faculty Development, Hayden Family Endowed Chair for Cancer Research.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ponting, C.P.; Hardison, R.C. What fraction of the human genome is functional? Genome Res 2011, 21, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.T.; Corces, V.G. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 2011, 12, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Levine, M. Transcriptional enhancers in animal development and evolution. Curr Biol 2010, 20, R754–R763. [Google Scholar] [CrossRef] [PubMed]
- Murakawa, Y.; Yoshihara, M.; Kawaji, H.; Nishikawa, M.; Zayed, H.; Suzuki, H.; Hayashizaki, Y.; Consortium, F.; others. Enhanced identification of transcriptional enhancers provides mechanistic insights into diseases. Trends in Genetics 2016, 32, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Karnuta, J.M.; Scacheri, P.C. Enhancers: bridging the gap between gene control and human disease. Human molecular genetics 2018, 27, R219–R227. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Oldridge, D.A.; Wood, A.C.; Weichert-Leahey, N.; Crimmins, I.; Sussman, R.; Winter, C.; McDaniel, L.D.; Diamond, M.; Hart, L.S.; Zhu, S.; Durbin, A.D.; Abraham, B.J.; Anders, L.; Tian, L.; Zhang, S.; Wei, J.S.; Khan, J.; Bramlett, K.; Rahman, N.; Capasso, M.; Iolascon, A.; Gerhard, D.S.; Guidry Auvil, J.M.; Young, R.A.; Hakonarson, H.; Diskin, S.J.; Look, A.T.; Maris, J.M. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 2015, 528, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Nakayamada, S.; Tanaka, Y. Critical roles of super-enhancers in the pathogenesis of autoimmune diseases. Inflamm Regen 2020, 40, 16. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Park, P.J. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 2009, 10, 669–680. [Google Scholar] [CrossRef]
- Kleftogiannis, D.; Kalnis, P.; Bajic, V.B. Progress and challenges in bioinformatics approaches for enhancer identification. Brief Bioinform 2016, 17, 967–979. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Zhang, Y.; Wong, C.H.; Birnbaum, R.Y.; Li, G.; Favaro, R.; Ngan, C.Y.; Lim, J.; Tai, E.; Poh, H.M.; Wong, E.; others. Chromatin connectivity maps reveal dynamic promoter–enhancer long-range associations. Nature 2013, 504, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.R.; Satpathy, A.T.; Boyle, E.A.; Dai, C.; Gowen, B.G.; Cho, S.W.; Nguyen, M.L.; Rubin, A.J.; Granja, J.M.; Kazane, K.R.; others. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nature genetics 2017, 49, 1602–1612. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, J.; Liu, Z.; Duan, Y.; Li, C. Integrative approaches based on genomic techniques in the functional studies on enhancers. Brief Bioinform 2023, 25. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.C.; Zhou, L.W.; Li, Y.Y.; Yu, Z.M.; Li, L.D.; Wang, Y.Z.; Xu, M.C.; Wang, Q.Y.; Li, C.Q. SEanalysis 2.0: a comprehensive super-enhancer regulatory network analysis tool for human and mouse. Nucleic Acids Res 2023, 51, W520–W527. [Google Scholar] [CrossRef]
- Luo, Z.H.; Shi, M.W.; Zhang, Y.; Wang, D.Y.; Tong, Y.B.; Pan, X.L.; Cheng, S. CenhANCER: a comprehensive cancer enhancer database for primary tissues and cell lines. Database (Oxford) 2023, 2023. [Google Scholar] [CrossRef]
- Bai, X.; Shi, S.; Ai, B.; Jiang, Y.; Liu, Y.; Han, X.; Xu, M.; Pan, Q.; Wang, F.; Wang, Q.; others. ENdb: a manually curated database of experimentally supported enhancers for human and mouse. Nucleic acids research 2020, 48, D51–D57. [Google Scholar] [CrossRef]
- Gao, T.; Qian, J. EnhancerAtlas 2.0: an updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic acids research 2020, 48, D58–D64. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Nazarov, P.V.; Kreis, S. Integrative approaches for analysis of mRNA and microRNA high-throughput data. Comput Struct Biotechnol J 2021, 19, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Yoon, S. Got target? Computational methods for microRNA target prediction and their extension. Exp Mol Med 2010, 42, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.h.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; Rajewsky, N. Combinatorial microRNA target predictions. Nat Genet 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol 2003, 5, R1. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Papadopoulos, G.L.; Reczko, M.; Simossis, V.A.; Sethupathy, P.; Hatzigeorgiou, A.G. The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res 2009, 37, D155–D158. [Google Scholar] [CrossRef]
- Zotos, P.; Papachristoudis, G.; Roubelakis, M.G.; Michalopoulos, I.; Pappa, K.I.; Anagnou, N.P.; Kossida, S. GOmir: a stand-alone application for human microRNA target analysis and gene ontology clustering. 2008 8th IEEE International Conference on BioInformatics and BioEngineering. IEEE, 2008, pp. 1–6.
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res 2009, 37, D105–D110. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, P.; Maragkakis, M.; Papadopoulos, G.L.; Reczko, M.; Hatzigeorgiou, A.G. Lost in translation: an assessment and perspective for computational microRNA target identification. Bioinformatics 2009, 25, 3049–3055. [Google Scholar] [CrossRef] [PubMed]
- Cihan, M.; Andrade-Navarro, M.A. Detection of features predictive of microRNA targets by integration of network data. PLoS One 2022, 17, e0269731. [Google Scholar] [CrossRef] [PubMed]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.C.; Tsay, M.; Lu, R.; Jurisica, I. mirDIP 4.1-integrative database of human microRNA target predictions. Nucleic Acids Res 2018, 46, D360–D370. [Google Scholar] [CrossRef] [PubMed]
- Friedman, Y.; Naamati, G.; Linial, M. MiRror: a combinatorial analysis web tool for ensembles of microRNAs and their targets. Bioinformatics 2010, 26, 1920–1921. [Google Scholar] [CrossRef] [PubMed]
- Andres-Leon, E.; González Peña, D.; Gomez-Lopez, G.; Pisano, D.G. miRGate: a curated database of human, mouse and rat miRNA–mRNA targets. Database 2015, 2015, bav035. [Google Scholar] [CrossRef] [PubMed]
- Shalgi, R.; Lieber, D.; Oren, M.; Pilpel, Y. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol 2007, 3, e131. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 2008, 455, 1124–1128. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Nersisyan, S.; Galatenko, A.; Galatenko, V.; Shkurnikov, M.; Tonevitsky, A. miRGTF-net: Integrative miRNA-gene-TF network analysis reveals key drivers of breast cancer recurrence. PLoS One 2021, 16, e0249424. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, P.H.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P.; Cannataro, M. Analysis of miRNA, mRNA, and TF interactions through network-based methods. EURASIP Journal on Bioinformatics and Systems Biology 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Arora, S.; Rana, R.; Chhabra, A.; Jaiswal, A.; Rani, V. miRNA-transcription factor interactions: a combinatorial regulation of gene expression. Mol Genet Genomics 2013, 288, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Young, R.A.; Sharp, P.A. Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell 2017, 168, 1000–1014. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, H.; Suzuki, H.I. Systems and Synthetic microRNA Biology: From Biogenesis to Disease Pathogenesis. Int J Mol Sci 2019, 21. [Google Scholar] [CrossRef] [PubMed]
- Ri, K.; Kim, C.; Pak, C.; Ri, P.; Om, H. The KLF6 Super Enhancer Modulates Cell Proliferation via MiR-1301 in Human Hepatoma Cells. Microrna 2020, 9, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, C.; Zhang, Y.; Song, C.; Chen, J.; Li, Y.; Wei, L.; Qian, F.; Ai, B.; Liu, Y.; Zhu, J.; Su, X.; Li, C.; Wang, Q. TRmir: A Comprehensive Resource for Human Transcriptional Regulatory Information of MiRNAs. Front Genet 2022, 13, 808950. [Google Scholar] [CrossRef]
- Kang, R.; Tan, Z.; Lang, M.; Jin, L.; Zhang, Y.; Zhang, Y.; Guo, T.; Guo, Z. EnhFFL: A database of enhancer mediated feed-forward loops for human and mouse. Precis Clin Med 2021, 4, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zhang, Y.; Huang, Q.; Meng, J.; Ding, R.; Chang, Y.; Xiong, L.; Guo, Z. EnhancerDB: a resource of transcriptional regulation in the context of enhancers. Database (Oxford) 2019, 2019. [Google Scholar] [CrossRef]
- Sayed, M.; Park, J.W. miRinGO: Prediction of biological processes indirectly targeted by human microRNAs. Non-coding RNA 2023, 9, 11. [Google Scholar] [CrossRef]
- Kuijjer, M.L.; Fagny, M.; Marin, A.; Quackenbush, J.; Glass, K. PUMA: PANDA Using MicroRNA Associations. Bioinformatics 2020, 36, 4765–4773. [Google Scholar] [CrossRef] [PubMed]
- Mitsis, T.; Efthimiadou, A.; Bacopoulou, F.; Vlachakis, D.; Chrousos, G.P.; Eliopoulos, E. Transcription factors and evolution: An integral part of gene expression. World Academy of Sciences Journal 2020, 2, 3–8. [Google Scholar] [CrossRef]
- Sonawane, A.R.; Platig, J.; Fagny, M.; Chen, C.Y.; Paulson, J.N.; Lopes-Ramos, C.M.; DeMeo, D.L.; Quackenbush, J.; Glass, K.; Kuijjer, M.L. Understanding Tissue-Specific Gene Regulation. Cell Rep 2017, 21, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Glass, K.; Huttenhower, C.; Quackenbush, J.; Yuan, G.C. Passing messages between biological networks to refine predicted interactions. PLoS One 2013, 8, e64832. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Katsura, A.; Matsuyama, H.; Miyazono, K. MicroRNA regulons in tumor microenvironment. Oncogene 2015, 34, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Li, L.; Xin, T.; Li, B.; Zhang, D. Superenhancer–transcription factor regulatory network in malignant tumors. Open Medicine 2021, 16, 1564–1582. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Shah, P.K.; Amin, S.B.; Samur, M.K.; Huang, N.; Wang, X.; Misra, V.; Ji, H.; Gabuzda, D.; Li, C. Integrative analysis of gene and miRNA expression profiles with transcription factor-miRNA feed-forward loops identifies regulators in human cancers. Nucleic Acids Res 2012, 40, e135. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.Y.; Xia, M.; Miao, Y.R.; Luo, M.; Zhang, Q.; Guo, A.Y. FFLtool: a web server for transcription factor and miRNA feed forward loop analysis in human. Bioinformatics 2020, 36, 2605–2607. [Google Scholar] [CrossRef] [PubMed]
- Mangan, S.; Alon, U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A 2003, 100, 11980–11985. [Google Scholar] [CrossRef]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113. [Google Scholar] [CrossRef]
- Javierre, B.M.; Burren, O.S.; Wilder, S.P.; Kreuzhuber, R.; Hill, S.M.; Sewitz, S.; Cairns, J.; Wingett, S.W.; Várnai, C.; Thiecke, M.J.; Burden, F.; Farrow, S.; Cutler, A.J.; Rehnström, K.; Downes, K.; Grassi, L.; Kostadima, M.; Freire-Pritchett, P.; Wang, F.; Stunnenberg, H.G.; Todd, J.A.; Zerbino, D.R.; Stegle, O.; Ouwehand, W.H.; Frontini, M.; Wallace, C.; Spivakov, M.; Fraser, P.; BLUEPRINT Consortium. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 2016, 167, 1369–1384.e19. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, Z.; Kashiwagi, M.; Yoshida, T.; Joshi, I.; Jena, N.; Somasundaram, R.; Emmanuel, A.O.; Sigvardsson, M.; Fitamant, J.; El-Bardeesy, N.; Gounari, F.; Van Etten, R.A.; Georgopoulos, K. Superenhancer reprogramming drives a B-cell-epithelial transition and high-risk leukemia. Genes Dev 2016, 30, 1971–1990. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Yan, J.; Huang, G.; Selvarajan, V.; Tay, J.L.S.; Lin, B.; Bi, C.; Tan, J.; Kwong, Y.L.; Shimizu, N.; Aozasa, K.; Chng, W.J. Dysregulated microRNAs affect pathways and targets of biologic relevance in nasal-type natural killer/T-cell lymphoma. Blood 2011, 118, 4919–4929. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Toh, S.H.M.; Tan, T.K.; Balan, K.; Lim, J.Q.; Tan, T.Z.; Xiong, S.; Jia, Y.; Ng, S.B.; Peng, Y.; Jeyasekharan, A.D.; Fan, S.; Lim, S.T.; Ong, C.A.J.; Ong, C.K.; Sanda, T.; Chng, W.J. Super-enhancer-driven TOX2 mediates oncogenesis in Natural Killer/T Cell Lymphoma. Mol Cancer 2023, 22, 69. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Tu, Z.; Chen, T.; Sun, F. Network motif identification in stochastic networks. Proc Natl Acad Sci U S A 2006, 103, 9404–9409. [Google Scholar] [CrossRef] [PubMed]
- Yeger-Lotem, E.; Sattath, S.; Kashtan, N.; Itzkovitz, S.; Milo, R.; Pinter, R.Y.; Alon, U.; Margalit, H. Network motifs in integrated cellular networks of transcription-regulation and protein-protein interaction. Proc Natl Acad Sci U S A 2004, 101, 5934–5939. [Google Scholar] [CrossRef]
- Kashtan, N.; Itzkovitz, S.; Milo, R.; Alon, U. Topological generalizations of network motifs. Phys Rev E Stat Nonlin Soft Matter Phys 2004, 70, 031909. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Alexander, K.; Clark, A.G.; Grimson, A.; Yu, H. Integrated network analysis reveals distinct regulatory roles of transcription factors and microRNAs. RNA 2016, 22, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef]
- Matsumura, I.; Tanaka, H.; Kanakura, Y. E2F1 and c-Myc in cell growth and death. Cell Cycle 2003, 2, 333–338. [Google Scholar] [CrossRef]
- Lee, K.H.; Chen, Y.L.; Yeh, S.D.; Hsiao, M.; Lin, J.T.; Goan, Y.G.; Lu, P.J. MicroRNA-330 acts as tumor suppressor and induces apoptosis of prostate cancer cells through E2F1-mediated suppression of Akt phosphorylation. Oncogene 2009, 28, 3360–3370. [Google Scholar] [CrossRef] [PubMed]
- Scholz, B.A.; Sumida, N.; de Lima, C.D.M.; Chachoua, I.; Martino, M.; Tzelepis, I.; Nikoshkov, A.; Zhao, H.; Mehmood, R.; Sifakis, E.G.; Bhartiya, D.; Göndör, A.; Ohlsson, R. WNT signaling and AHCTF1 promote oncogenic MYC expression through super-enhancer-mediated gene gating. Nat Genet 2019, 51, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Prompsy, P.B.; Toubia, J.; Gearing, L.J.; Knight, R.L.; Forster, S.C.; Bracken, C.P.; Gantier, M.P. Making use of transcription factor enrichment to identify functional microRNA-regulons. Comput Struct Biotechnol J 2021, 19, 4896–4903. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Khaleeq, Q.T.; Meese, E.; Keller, A. miEAA: microRNA enrichment analysis and annotation. Nucleic Acids Res 2016, 44, W110–W116. [Google Scholar] [CrossRef]
- Borgmästars, E.; de Weerd, H.A.; Lubovac-Pilav, Z.; Sund, M. miRFA: an automated pipeline for microRNA functional analysis with correlation support from TCGA and TCPA expression data in pancreatic cancer. BMC Bioinformatics 2019, 20, 393. [Google Scholar] [CrossRef]
- Tang, F.; Yang, Z.; Tan, Y.; Li, Y. Super-enhancer function and its application in cancer targeted therapy. NPJ Precis Oncol 2020, 4, 2. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, Y.Y.; Chen, P.P.; Xu, H.; Xie, S.J.; Xu, S.J.; Li, B.; Li, J.H.; Liu, S.; Yang, J.H.; Zhou, H.; Qu, L.H. Genome-wide identification of microRNA targets reveals positive regulation of the Hippo pathway by miR-122 during liver development. Cell Death Dis 2021, 12, 1161. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mukherjee, S.; Ali, N. Super enhancer-mediated transcription of miR146a-5p drives M2 polarization during Leishmania donovani infection. PLoS Pathog 2021, 17, e1009343. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Huang, S.; Zhang, Z.; Qian, X.; Sun, P.; Zhou, X. Pan-cancer analysis on microRNA-associated gene activation. EBioMedicine 2019, 43, 82–97. [Google Scholar] [CrossRef]
- Turunen, T.; Hernández de Sande, A.; Pölönen, P.; Heinäniemi, M. Genome-wide analysis of primary microRNA expression using H3K36me3 ChIP-seq data. Comput Struct Biotechnol J 2021, 19, 1944–1955. [Google Scholar] [CrossRef]
- Wang, D.; Gu, J.; Wang, T.; Ding, Z. OncomiRDB: a database for the experimentally verified oncogenic and tumor-suppressive microRNAs. Bioinformatics 2014, 30, 2237–8. [Google Scholar] [CrossRef] [PubMed]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat Genet 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
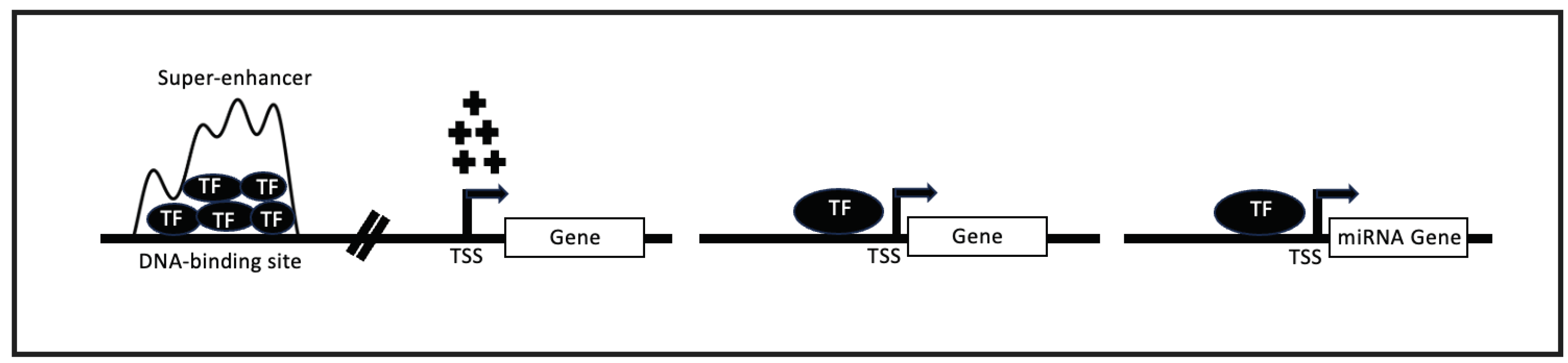
Figure 1.
Graphical representation of gene regulation in context of Super-enhancer (SE), transcription factor(TF), miRNA genes, and target genes. SE located away (denoted by breaks on the DNA) from the transcription start site (TSS) of target genes regulates expression of the corresponding genes via TFs and other protein complexes; TFs bind to promoter regions of miRNA and other genes to regulate expression of the corresponding genes but sometimes complex mechanisms occur between them in form of feed-forward and other loops (not shown here).
Figure 1.
Graphical representation of gene regulation in context of Super-enhancer (SE), transcription factor(TF), miRNA genes, and target genes. SE located away (denoted by breaks on the DNA) from the transcription start site (TSS) of target genes regulates expression of the corresponding genes via TFs and other protein complexes; TFs bind to promoter regions of miRNA and other genes to regulate expression of the corresponding genes but sometimes complex mechanisms occur between them in form of feed-forward and other loops (not shown here).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



