
Submitted:
17 October 2024
Posted:
17 October 2024
You are already at the latest version
Abstract
C-C motif chemokine receptor-8 (CCR8) belongs to class A of G protein-coupled receptors (GPCRs). CCR8 interacts with the specific chemokine ligand CCL1/I-309 in humans, which is produced by various cells, including tumor-associated macrophages and regulatory T cells (Treg). CCR8 is highly expressed on Treg and T-helper 2 (TH2) cells recruited to the inflammation site and is implicated in allergy, asthma, and cancer progression. The CCR8+Treg has been suggested to be an important regulator in the immunosuppressive tumor microenvironment (TME); therefore, it has been desired to develop sensitive monoclonal antibodies (mAbs) for CCR8. This study developed a specific mAb for human CCR8 (hCCR8), which is useful for flow cytometry by employing the Cell-Based Immunization and Screening (CBIS) method. The established anti-hCCR8 mAb, C8Mab-21, (mouse IgM, kappa) reacted with hCCR8-overexpressed Chinese hamster ovary-K1 (CHO/hCCR8) cells, TALL-1 (acute T lymphoblastic leukemia), CCRF-HSB2 (human T-lymphoblastic leukemia), and natural killer cells, which express endogenous hCCR8 by flow cytometry. Furthermore, C8Mab-21 demonstrated a moderate binding affinity for CHO/hCCR8 and TALL-1 with a dissociation constant of 6.5×10-8 M and 2.0×10-8 M, respectively. C8Mab-21, which was established by the CBIS method, could be a useful tool for analyzing the hCCR8-related biological response using flow cytometry.
Keywords:
CCR8
; CBIS method
; monoclonal antibody
; flow cytometry
1. Introduction
Targeting of immune checkpoint has become effective and powerful strategy for cancer therapy.[1] In particular, the development of antibody drugs targeting immune checkpoint molecules, such as programmed-cell death-1 (PD-1), cytotoxic T lymphocyte antigen 4 (CTLA-4), and PD-1 ligand 1 (PD-L1), has achieved remarkable therapeutic results.[2,3,4] PD-1 inhibits the excessive activation of conventional T cells by suppressing costimulatory signaling and renders them dysfunctional or exhausted.[5] PD-1 and CTLA-4 are also expressed in regulatory T cells (Treg), one of the immunosuppressors in the tumor microenvironment (TME). The inhibition could potentiate the activation and immunosuppressive function of Treg cells.[6]
Treg is defined as a CD4+T cell with CD25 and Foxp3 and maintains self-tolerance to prevent excessive immune responses and autoimmune diseases.[7] Treg suppresses the effector functions of T cells through the secretion of immunosuppressive cytokines, such as interleukin-10, transforming growth factor-β, and cytotoxic granzyme/perforin.[7,8,9,10,11] Intratumoral Treg suppresses antitumor T cell responses and thus resists the effects of immune checkpoint inhibitor therapy.[12,13] Antibodies against T cell immunoreceptors with Ig and ITIM domains (TIGIT), one of the immune checkpoint molecules, improve the effectiveness of PD-L1 antibodies by suppressing Treg.[14] Therefore, the development of immunotherapy targeting Treg is expected.[6,15]
Intratumoral Treg expresses a high level of C-C motif chemokine receptor-8 (CCR8). Additionally, the CCR8-expressing Treg has also increased the expression of CD25 and Foxp3 compared with CCR8-negative Treg.[16] Therefore, CCR8-expressing Treg is considered to have potent immunosuppressive functions. The CCR8-expressing Treg is known to be correlated with poor prognosis in some cancer patients.[17,18] CCR8 is attractive as a target molecule for the next cancer immunotherapy.[19] Anti-CCR8 drugs, including S-531011,[20] IPG7236,[21] and SRF114[22] are undergoing clinical trials.
CCR8 is one of the seven transmembrane-spanning G protein-coupled receptors (GPCRs). Human CCR8 is known to bind to five C-C chemokine ligands (CCLs): CCL1/I-309, CCL4, CCL16, CCL17, and CCL18.[23] CCR8 is upregulated in not only Treg, but also various cancers, including breast, non-small cell lung (NSCLC), bladder, and colorectal cancer.[17,18] CCR8 mediates bladder cancer cell migration, invasion, and epithelial-mesenchymal transition by interacting with CCL18.[24] CCR8 and its specific ligand CCL1/I-309 regulate the immune system, which mediates the progression of diseases such as cancers. CCR8-CCL1/I-309 axis promotes migration and inhibits apoptosis in Treg and lymphomas.[25,26] Therefore, CCR8-targeting antibodies will contribute to the elucidation of pathological mechanisms, diagnosis, and therapy.
Using the Cell-Based Immunization and Screening (CBIS) method, we previously developed numerous monoclonal antibodies (mAbs) against chemokine receptors, including mouse CCR3,[27,28] mouse CCR8,[29,30,31] human CCR9,[32] and mouse CXCR4.[33] In this study, we have successfully developed an anti-human CCR8 (hCCR8) mAb using the CBIS method, which is applicable to flow cytometry.
2. Materials and Methods
2.1. Preparation of Cell Lines
LN229, Chinese hamster ovary (CHO)-K1, and P3X63Ag8U.1 (P3U1) cells were obtained from the American Type Culture Collection (Manassas, VA). TALL-1 and CCRF-HSB2 cells were obtained from the Japanese Collection of Research Bioresources (JCRB) Cell Bank (Osaka, Japan). The human natural killer (NK) cells (donor lot. 4022602, purity > 70%) were purchased from Takara Bio (Shiga, Japan). pCMV6neo-myc-DDK vector with hCCR8 (Accession No.: NM_005201) was purchased from OriGene Technologies, Inc. (Rockville, MD). The plasmid was transfected into the cell lines using a Neon transfection system (Thermo Fisher Scientific, Inc., Waltham, MA). Subsequently, LN229 and CHO-K1, which stably overexpressed hCCR8 with C-terminal myc-DDK tags (hereinafter described as LN229/hCCR8 and CHO/hCCR8, respectively) were established using a cell sorter (SH800; Sony Corp., Tokyo, Japan), following cultivation in a medium containing 0.5 mg/mL G418 (Nacalai Tesque, Inc., Kyoto, Japan).
2.2. Production of Hybridomas
For developing anti-hCCR8 mAbs, two female 6-week-old BALB/c mice were immunized intraperitoneally with 1 × 108 cells of LN229/hCCR8. The immunogen was harvested after brief exposure to 1 mM ethylenediaminetetraacetic acid (EDTA; Nacalai Tesque, Inc.). We added Imject Alum Adjuvant (Thermo Fisher Scientific Inc.) as an adjuvant in the first immunization. Three additional injections of 1 × 108 cells of LN229/hCCR8 were performed without an adjuvant every week. We performed a final booster immunization of 1 × 108 cells of LN229/hCCR8 intraperitoneally two days before harvesting splenocytes from mice. We conducted cell-fusion of the harvested splenocytes with P3U1 cells using polyethylene glycol 1500 (PEG1500; Roche Diagnostics, Indianapolis, IN).
2.3. Flow Cytometric Analyses
CHO-K1 and CHO/hCCR8 cells were harvested after brief exposure to 1 mM EDTA. CHO-K1, CHO/hCCR8, TALL-1, and CCRF-HSB2 cells were washed with 0.1% bovine serum albumin in phosphate-buffered saline and treated with primary mAbs for 30 min at 4°C. Afterward, cells were treated with Alexa Fluor 488-conjugated anti-mouse IgG (1:1000) following the collection of fluorescence data, using the SA3800 Cell Analyzer (Sony Corp.). The anti-human CD198 (CCR8) mAbs (clones S19017D and L263G8) were purchased from BioLegend (San Diego, CA). The Alexa Fluor 488-conjugated anti-mouse IgG was purchased from Cell Signaling Technology, Inc. (Danvers, MA).
2.4. Determination of the EC50 by Flow Cytometry
CHO/hCCR8 and TALL-1 were suspended in 100 μL serially diluted of C8Mab-21 (100 µg/mL to 0.006 µg/mL), S19017D (10 µg/mL to 0.0006 µg/mL for CHO/hCCR8, 10 µg/mL, 2.5 µg/mL to 0.0006 µg/mL for TALL-1), or L263G8 (10 µg/mL to 0.0006 µg/mL for CHO/hCCR8, 0.625 µg/mL to 0.0006 µg/mL for TALL-1), after which Alexa Fluor 488-conjugated anti-mouse IgG (1:200) was added. Fluorescence data were subsequently collected, using the BD FACSLyric (BD Biosciences, Franklin Lakes, NJ), following the calculation of EC50 by fitting the binding isotherms into the built-in; one-site binding model in GraphPad PRISM 6 (GraphPad Software, Inc., La Jolla, CA).
3. Results
3.1. Establishment of Anti-hCCR8 mAbs by the CBIS Method
To develop anti-hCCR8 mAbs, we employed the CBIS method using hCCR8-overexpressed cells. Anti-hCCR8 mAbs-producing hybridoma screening was conducted using flow cytometry (Figure 1). Two mice were intraperitoneally immunized with LN229/hCCR8 weekly for a total of 5 times. Subsequently, hybridomas were seeded into 96-well plates, after which flow cytometric analysis was used to select CHO/hCCR8-reactive and CHO-K1-nonreactive supernatants of hybridomas. Afterward, we obtained CHO/hCCR8-reactive supernatant in only 1 of 956 wells (0.10%). We finally established clone C8Mab-21 (mouse IgM, kappa) by limiting dilution and additional screening.
3.2. Flow Cytometric Analysis
Flow cytometric analysis was conducted using purified C8Mab-21 (Supplementary Figure S1) and commercially available anti-human CD198 (CCR8) mAbs (clone S19017D and L263G8) against CHO-K1, CHO/hCCR8, TALL-1, CCRF-HSB2, and NK cells. Results showed that C8Mab-21, S19017D, and L263G8 recognize CHO/hCCR8 dose-dependently (Figure 2A). C8Mab-21, and L263G8 did not react with parental CHO-K1 cells even at 20 µg/mL of mAbs. S19017D slightly reacted to CHO-K1 cells at 20 µg/mL and also 2 µg/mL (Figure 2B). Regarding endogenously hCCR8-expressing cells in Figure 3, C8Mab-21 recognized TALL-1, CCRF-HSB2, and NK cells at a concentration of 2 µg/mL or 20 µg/mL (Figure 3). S19017D reacted to TALL-1 and CCRF-HSB2 dose-dependently even at a concentration of 0.02 μg/mL. However, NK cells were not recognized by S19017D even at a concentration of 20 µg/mL (Figure 3). L263G8 reacted to TALL-1 and CCRF-HSB2 even at 0.02 μg/mL, and also NK cells at a concentration of 2 μg/mL or higher of mAb (Figure 3).To conclude, C8Mab-21 could detect exogenously and endogenously expressed hCCR8 in native conformation using flow cytometry.
3.3. Titration of C8Mab-21 and Anti-CCR-8 Control Antibodies on hCCR8 Overexpressed and Endogenously hCCR8-Expressing Cell Lines
The titration of C8Mab-21, S19017D, and L263G8 was assessed with exogenously hCCR8-expressed CHO/hCCR8 using flow cytometry. Results showed that the EC50 values of C8Mab-21, S19017D, and L263G8 for CHO/hCCR8 are 6.5×10-8 M, 2.6×10-9 M, and 1.2×10-9 M, respectively (Figure 4). These results indicate that C8Mab-21 possesses a moderate affinity for exogenously overexpressed hCCR8 in CHO-K1 cells.
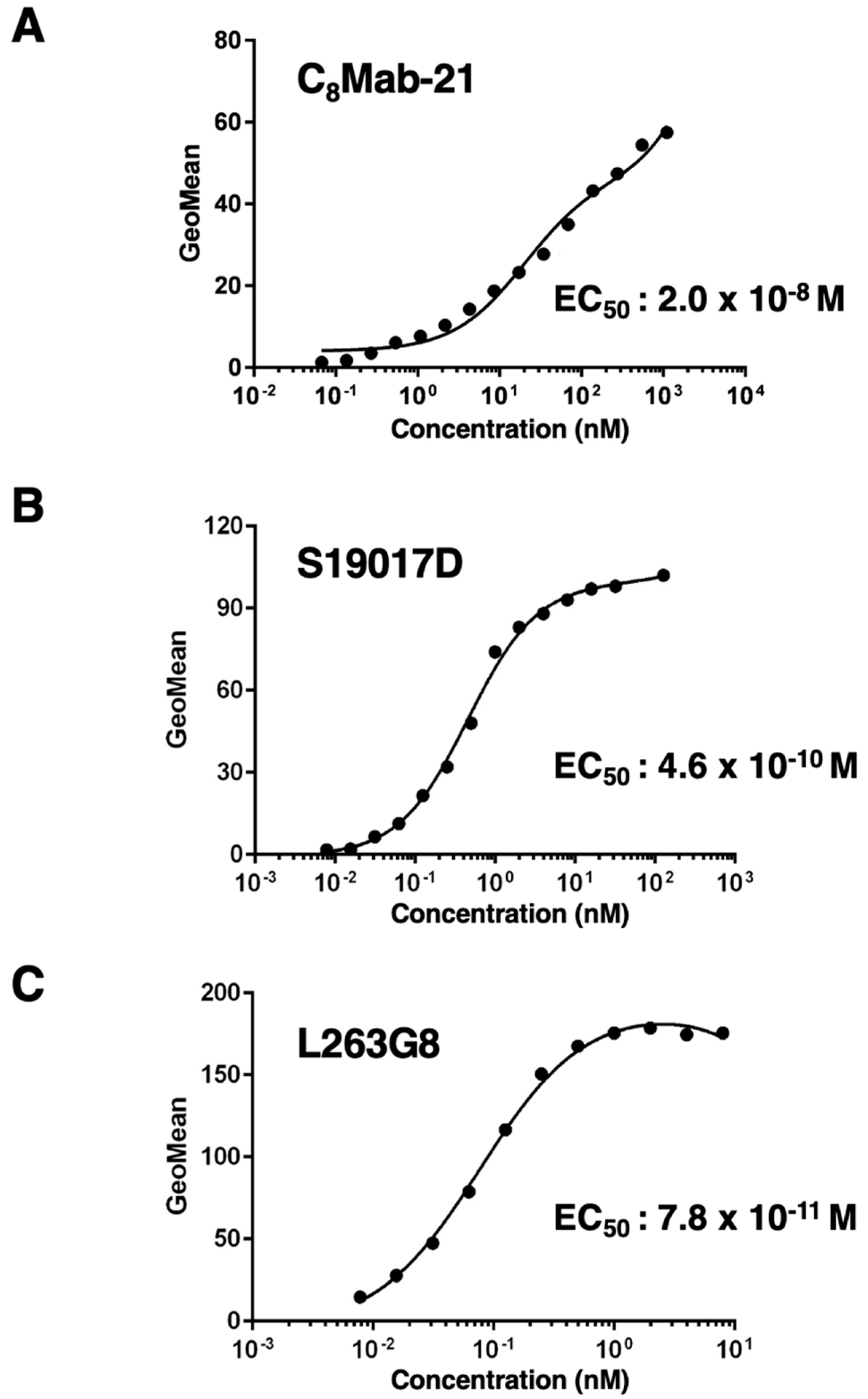
The titration of C8Mab-21, S19017D, and L263G8 was analyzed with endogenously hCCR8-expressing TALL-1 using flow cytometry. Results indicated that the EC50 values of C8Mab-21, S19017D, and L263G8 for TALL-1 are 2.0×10-8 M, 4.6×10-10 M, and 7.8×10-11 M, respectively (Figure 5). These results showed that C8Mab-21 possesses a moderate affinity for endogenously expressing hCCR8 in TALL-1 leukemia cells.
4. Discussion
GPCRs, including chemokine receptors, are focused as targets for many diseases, including inflammatory disorders and cancers.[34,35] GPCRs transmit signals to intracellular molecules about extracellular conditions and govern broad cell dynamics, such as proliferation, homeostasis, migration, and motility of the cells.[34,35] Therapeutic drugs, including mAbs targeting GPCRs, have been developed to date; however, the complexity of the structure, the small area of epitope regions, and the difficulty of purifying the protein as an immunogen pose high hurdles.[36,37]
Unlike protein purifications, the preparation of antigens is not complicated in the CBIS method. Furthermore, it is possible to retain the antigen structure and modification such as glycosylation and folding using the CBIS method. We have successfully developed multiple mAbs by the CBIS method against human epidermal growth factor receptor 1 (HER1; EGFR),[38] HER3,[39] trophoblast cell surface antigen 2,[40] epithelial cell adhesion molecule,[41] CD44,[42,43,44] and podoplanin.[45] Furthermore, some of the developed mAbs by the CBIS method exhibited cancer specificity by recognizing unique cancer-specific epitopes.[46,47,48,49,50,51] Therefore, the CBIS method is an efficient and useful tactic for generating diverse antibodies targeting membrane proteins.
In the immunosuppressive TME, CD8+T cells are exhausted along with the induction of CCR8+Treg. CCR8+Treg infiltration is confirmed to be associated with high TOX, an exhaustion marker of T cells, expression in CD8+T cells in some types of cancer patients.[52,53] Elimination of CCR8+Treg using antibodies is expected to advance the treatment of these cancer patients. Targeting CCR8 might be more specific in antitumor activity than other approaches aimed at Treg removal.[53,54] In mice, CCR8+T cell depletion therapy using anti-CCR8 mAbs induces tumor-specific immune responses without triggering autoimmune responses or immune reactions in TME.[55] Since C8Mab-21 could recognize cell surface hCCR8, we plan to investigate the function of C8Mab-21 against Treg, such as detection and interfering effects in future studies. Furthermore, we have previously enhanced antibody-dependent cellular cytotoxicity (ADCC) activity and complement-dependent cytotoxicity (CDC) by modifying isotypes and defucosylation in mAbs.[56,57] C8Mab-21 is mouse IgM, which has no ADCC activity, Although the property of IgM such as crosslinking activity is lost, it will be converted into a mouse IgG2a version to evaluate the effect of antitumor activities in xenograft models. We successfully cloned the cDNA of VH and VL region of C8Mab-21 (Supplementary Figure S2).
Interestingly, a correlation between cancer-associated fibroblasts (CAFs) and CCR8 has been found from the results of omics analysis.[22] CCR8 is suggested to be involved in the pathogenesis of various cancer types. CAFs are one of the tumor suppressor factors, the same as Treg, that are known to interfere with the function of tumor immune cells by promoting fibrosis and constructing an extracellular matrix in the TME.[58,59] CAFs with a myofibroblastic-like phenotype transfer large amounts of proteins to the surrounding endothelial cells through matrix-bound vesicles, which might contribute to cancer progression and treatment resistance.[60,61] Recently, the phenotypes of CAF have been reported to be associated with better or worse outcomes in NSCLC patients.[62] Although further functional analysis of CCR8-expressing CAFs is required, targeting CCR8 might suppress Treg and CAFs, and lead to synergistic antitumor immunotherapy results. In that case, it is well worth evaluating the impact of C8Mab-21 on CAFs as well in future studies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
T.T. and G.L. performed the experiments. M.K.K. and Y.K. designed the experiments. T.T. analyzed the data. T.T., H.S., and Y.K. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported, in part, by Japan Agency for Medical Research and Development (AMED) under Grant Nos.: JP23ama121008 (to Y.K.), JP23am0401013 (to Y.K.), JP23bm1123027 (to Y.K.), and JP23ck0106730 (to Y.K.), and by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (KAKENHI) Grant Nos.: 21K20789 (to T.T.), 22K06995 (to H.S.), 21K07168 (to M.K.K.), and 22K07224 (to Y.K.).
Institutional Review Board Statement
The animal study protocol was approved by the Animal Care and Use Committee of Tohoku University (Permit number: 2022MdA-001) for studies involving animals.
Conflicts of Interest
The authors have no conflict of interest.
References
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- van den Bulk, J.; Verdegaal, E.M.; de Miranda, N.F. Cancer immunotherapy: broadening the scope of targetable tumours. Open Biol 2018;8(6).
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Forde, P.M.; Hammers, H.J.; et al. Antagonists of PD-1 and PD-L1 in Cancer Treatment. Semin Oncol 2015, 42, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer 2020, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell 2000, 101, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Brunkow, M.E.; Jeffery, E.W.; Hjerrild, K.A.; et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 2001, 27, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kuniyasu, Y.; Toda, M.; et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol 1998, 10, 1969–1980. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.; Tanaka, A.; Sakaguchi, S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell 2023, 41, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K. Selective targeting or reprogramming of intra-tumoral Tregs. Med Oncol 2024, 41, 71. [Google Scholar] [CrossRef] [PubMed]
- Wakiyama, H.; Kato, T.; Furusawa, A.; et al. Treg-Dominant Tumor Microenvironment Is Responsible for Hyperprogressive Disease after PD-1 Blockade Therapy. Cancer Immunol Res 2022, 10, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hu, R.; Choi, Y.; et al. Anti-TIGIT antibody improves PD-L1 blockade through myeloid and T(reg) cells. Nature 2024.
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Wang, L.; Simons, D.L.; Lu, X.; et al. Connecting blood and intratumoral Treg cell activity in predicting future relapse in breast cancer. Nat Immunol 2019, 20, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- De Simone, M.; Arrigoni, A.; Rossetti, G.; et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity 2016;45(5): 1135-1147.
- Plitas, G.; Konopacki, C.; Wu, K.; et al. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016;45(5): 1122-1134.
- Barsheshet, Y.; Wildbaum, G.; Levy, E.; et al. CCR8(+)FOXp3(+) Treg cells as master drivers of immune regulation. Proc Natl Acad Sci U S A 2017;114(23): 6086-6091.
- Nagira, Y.; Nagira, M.; Nagai, R.; et al. S-531011, a Novel Anti-Human CCR8 Antibody, Induces Potent Antitumor Responses through Depletion of Tumor-Infiltrating CCR8-Expressing Regulatory T Cells. Mol Cancer Ther 2023;22(9): 1063-1072.
- Wu, Y.; Xi, J.; Li, Y.; et al. Discovery of a Potent and Selective CCR8 Small Molecular Antagonist IPG7236 for the Treatment of Cancer. J Med Chem 2023;66(7): 4548-4564.
- Kim, N.; Kim, M.H.; Pyo, J.; et al. CCR8 as a Therapeutic Novel Target: Omics-Integrated Comprehensive Analysis for Systematically Prioritizing Indications. Biomedicines 2023;11(11).
- Märkl, F.; Huynh, D.; Endres, S.; Kobold, S. Utilizing chemokines in cancer immunotherapy. Trends Cancer 2022;8(8): 670-682.
- Liu, X.; Xu, X.; Deng, W.; et al. CCL18 enhances migration, invasion and EMT by binding CCR8 in bladder cancer cells. Mol Med Rep 2019;19(3): 1678-1686.
- Spinetti, G.; Bernardini, G.; Camarda, G.; et al. The chemokine receptor CCR8 mediates rescue from dexamethasone-induced apoptosis via an ERK-dependent pathway. J Leukoc Biol 2003;73(1): 201-207.
- Denis, C.; Deiteren, K.; Mortier, A.; et al. C-terminal clipping of chemokine CCL1/I-309 enhances CCR8-mediated intracellular calcium release and anti-apoptotic activity. PLoS One 2012;7(3): e34199.
- Saito, M.; Harigae, Y.; Li, G.; et al. C(3)Mab-2: An Anti-Mouse CCR3 Monoclonal Antibody for Immunocytochemistry. Monoclon Antib Immunodiagn Immunother 2022;41(1): 45-49.
- Asano, T.; Suzuki, H.; Tanaka, T.; et al. C(3)Mab-3: A Monoclonal Antibody for Mouse CC Chemokine Receptor 3 for Flow Cytometry. Monoclon Antib Immunodiagn Immunother 2022;41(2): 74-79.
- Saito, M.; Suzuki, H.; Tanaka, T.; et al. Development of an Anti-Mouse CCR8 Monoclonal Antibody (C(8)Mab-1) for Flow Cytometry and Immunocytochemistry. Monoclon Antib Immunodiagn Immunother 2022;41(6): 333-338.
- Tanaka, T.; Nanamiya, R.; Takei, J.; et al. Development of Anti-Mouse CC Chemokine Receptor 8 Monoclonal Antibodies for Flow Cytometry. Monoclon Antib Immunodiagn Immunother 2021;40(2): 65-70.
- Suzuki, H.; Saito, M.; Asano, T.; et al. C(8)Mab-3: An Anti-Mouse CCR8 Monoclonal Antibody for Immunocytochemistry. Monoclon Antib Immunodiagn Immunother 2022;41(2): 110-114.
- Nanamiya, R.; Takei, J.; Asano, T.; et al. Development of Anti-Human CC Chemokine Receptor 9 Monoclonal Antibodies for Flow Cytometry. Monoclon Antib Immunodiagn Immunother 2021;40(3): 101-106.
- Ouchida, T.; Suzuki, H.; Tanaka, T.; Kaneko, M.K.; Kato, Y. Cx(4)Mab-1: A Novel Anti-Mouse CXCR4 Monoclonal Antibody for Flow Cytometry. Monoclon Antib Immunodiagn Immunother 2024;43(1): 10-16.
- Proudfoot, A.E. Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol 2002;2(2): 106-115.
- Schöneberg, T.; Liebscher, I. Mutations in G Protein-Coupled Receptors: Mechanisms, Pathophysiology and Potential Therapeutic Approaches. Pharmacol Rev 2021;73(1): 89-119.
- Jo, M.; Jung, S.T. Engineering therapeutic antibodies targeting G-protein-coupled receptors. Exp Mol Med 2016;48(2): e207.
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 2017;16(12): 829-842.
- Itai, S.; Yamada, S.; Kaneko, M.K.; et al. Establishment of EMab-134, a Sensitive and Specific Anti-Epidermal Growth Factor Receptor Monoclonal Antibody for Detecting Squamous Cell Carcinoma Cells of the Oral Cavity. Monoclon Antib Immunodiagn Immunother 2017;36(6): 272-281.
- Asano, T.; Ohishi, T.; Takei, J.; et al. Anti-HER3 monoclonal antibody exerts antitumor activity in a mouse model of colorectal adenocarcinoma. Oncol Rep 2021;46(2).
- Tanaka, T.; Ohishi, T.; Asano, T.; et al. An anti-TROP2 monoclonal antibody TrMab-6 exerts antitumor activity in breast cancer mouse xenograft models. Oncol Rep 2021;46(1).
- Kaneko, M.K.; Ohishi, T.; Takei, J.; et al. Anti-EpCAM monoclonal antibody exerts antitumor activity against oral squamous cell carcinomas. Oncol Rep 2020;44(6): 2517-2526.
- Kudo, Y.; Suzuki, H.; Tanaka, T.; Kaneko, M.K.; Kato, Y. Development of a Novel Anti-CD44 Variant 5 Monoclonal Antibody C(44)Mab-3 for Multiple Applications against Pancreatic Carcinomas. Antibodies (Basel) 2023;12(2).
- Suzuki, H.; Ozawa, K.; Tanaka, T.; Kaneko, M.K.; Kato, Y. Development of a Novel Anti-CD44 Variant 7/8 Monoclonal Antibody, C(44)Mab-34, for Multiple Applications against Oral Carcinomas. Biomedicines 2023;11(4).
- Ejima, R.; Suzuki, H.; Tanaka, T.; et al. Development of a Novel Anti-CD44 Variant 6 Monoclonal Antibody C(44)Mab-9 for Multiple Applications against Colorectal Carcinomas. Int J Mol Sci 2023;24(4).
- Suzuki, H.; Kaneko, M.K.; Kato, Y. Roles of Podoplanin in Malignant Progression of Tumor. Cells 2022;11(3).
- Kaneko, M.K.; Suzuki, H.; Ohishi, T.; et al. A Cancer-Specific Monoclonal Antibody against HER2 Exerts Antitumor Activities in Human Breast Cancer Xenograft Models. Int J Mol Sci 2024;25(3).
- Kaneko, M.K.; Suzuki, H.; Kato, Y. Establishment of a Novel Cancer-specific Anti-HER2 Monoclonal Antibody H2Mab-250/H2CasMab-2 for breast cancers. Monoclon Antib Immunodiagn Immunother 2024;in press.
- Arimori, T.; Mihara, E.; Suzuki, H.; et al. Locally misfolded HER2 expressed on cancer cells is a promising target for development of cancer-specific antibodies. Structure 2024. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Ohishi, T.; Tanaka, T.; Kaneko, M.K.; Kato, Y. A Cancer-Specific Monoclonal Antibody against Podocalyxin Exerted Antitumor Activities in Pancreatic Cancer Xenografts. Int J Mol Sci 2023;25(1).
- Suzuki, H.; Ohishi, T.; Kaneko, M.K.; Kato, Y. A Humanized and Defucosylated Antibody against Podoplanin (humLpMab-23-f) Exerts Antitumor Activities in Human Lung Cancer and Glioblastoma Xenograft Models. Cancers (Basel) 2023;15(20).
- Kato, Y.; Kaneko, M.K. A cancer-specific monoclonal antibody recognizes the aberrantly glycosylated podoplanin. Sci Rep 2014, 4, 5924. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, A.; Nogami, W.; Nashiki, K.; et al. Immunotherapy Targeting CCR8+ Regulatory T Cells Induces Antitumor Effects via Dramatic Changes to the Intratumor CD8+ T Cell Profile. J Immunol 2023;211(4): 673-682.
- Kidani, Y.; Nogami, W.; Yasumizu, Y.; et al. CCR8-targeted specific depletion of clonally expanded Treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc Natl Acad Sci U S A 2022;119(7).
- Zheng, D.; Wang, X.; Cheng, L.; et al. The Chemokine Receptor CCR8 Is a Target of Chimeric Antigen T Cells for Treating T Cell Malignancies. Front Immunol 2022;13: 808347.
- Campbell, J.R.; McDonald, B.R.; Mesko, P.B.; et al. Fc-Optimized Anti-CCR8 Antibody Depletes Regulatory T Cells in Human Tumor Models. Cancer Res 2021;81(11): 2983-2994.
- Li, G.; Suzuki, H.; Ohishi, T.; et al. Antitumor activities of a defucosylated anti-EpCAM monoclonal antibody in colorectal carcinoma xenograft models. Int J Mol Med 2023;51(2).
- Tanaka, T.; Suzuki, H.; Ohishi, T.; Kaneko, M.K.; Kato, Y. Antitumor activities against breast cancers by an afucosylated anti-HER2 monoclonal antibody H(2) Mab-77-mG(2a) -f. Cancer Sci.
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat Rev Cancer 2016;16(9): 582-598.
- Santi, A.; Kay, E.J.; Neilson, L.J.; et al. Cancer-associated fibroblasts produce matrix-bound vesicles that influence endothelial cell function. Sci Signal 2024;17(827): eade0580.
- Eskandari-Malayeri, F.; Rezaei, M. Immune checkpoint inhibitors as mediators for immunosuppression by cancer-associated fibroblasts: A comprehensive review. Front Immunol 2022;13: 996145.
- Cords, L.; Engler, S.; Haberecker, M.; et al. Cancer-associated fibroblast phenotypes are associated with patient outcome in non-small cell lung cancer. Cancer Cell 2024, 42, 396–412.e395. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A schematic procedure of anti-hCCR8 mAbs production. The procedure of the Cell-Based Immunization and Screening (CBIS) method for antibody development. (A) LN229/hCCR8 cells were immunized into two mice using intraperitoneal injection. (B) The spleen cells from mice were fused with P3U1 myeloma cells. (C) The culture supernatants of hybridoma were screened by flow cytometry using CHO-K1 and CHO/hCCR8. (D) After limiting dilution of hybridomas and additional analysis, C8Mab-21 was finally established.
Figure 1.
A schematic procedure of anti-hCCR8 mAbs production. The procedure of the Cell-Based Immunization and Screening (CBIS) method for antibody development. (A) LN229/hCCR8 cells were immunized into two mice using intraperitoneal injection. (B) The spleen cells from mice were fused with P3U1 myeloma cells. (C) The culture supernatants of hybridoma were screened by flow cytometry using CHO-K1 and CHO/hCCR8. (D) After limiting dilution of hybridomas and additional analysis, C8Mab-21 was finally established.
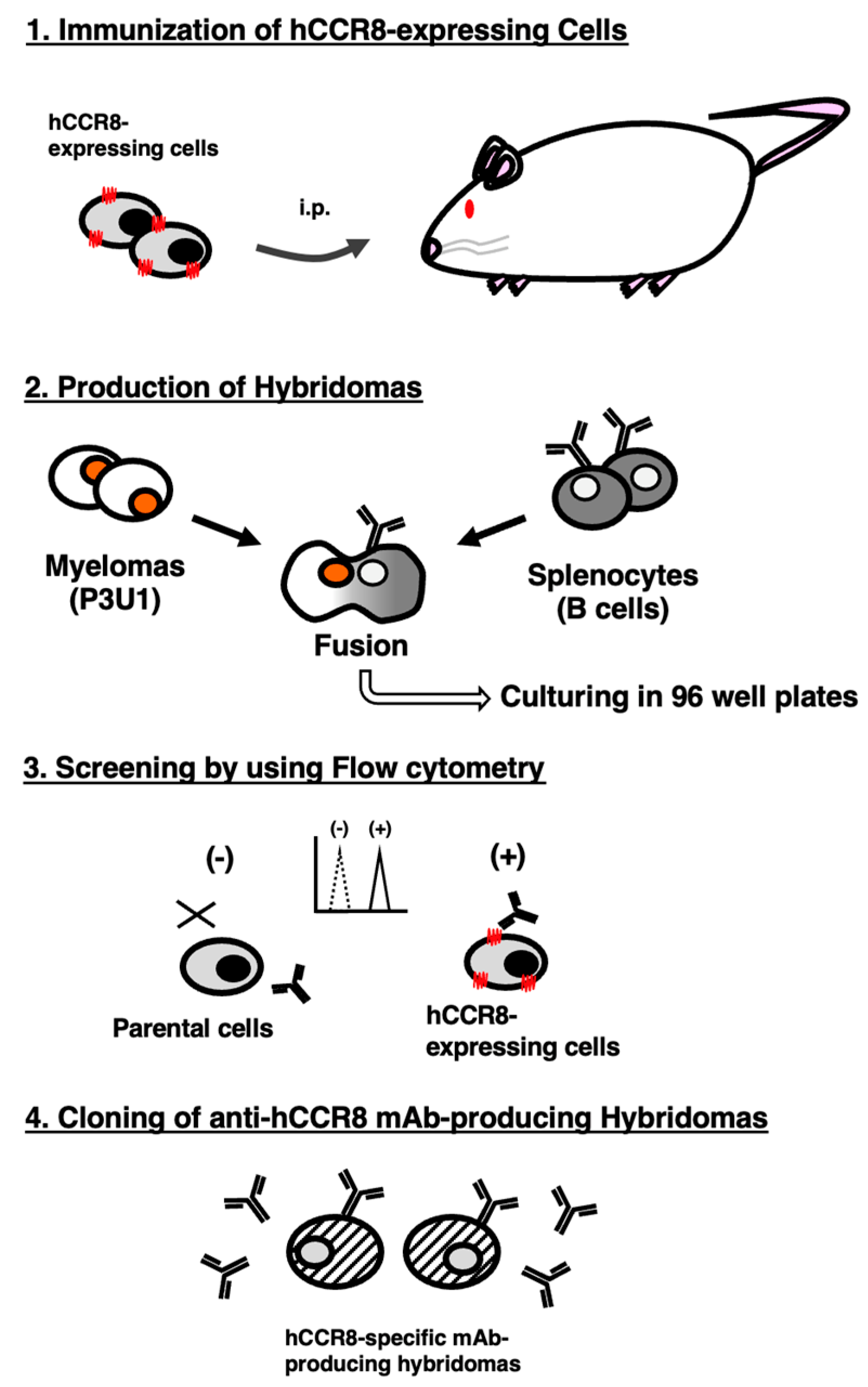
Figure 2.
Flow cytometric analysis of anti-hCCR8 mAbs against CHO/hCCR8 and CHO-K1. CHO/hCCR8 (A) and CHO-K1 cells (B) were treated with 0.02–20 µg/mL of C8Mab-21, S19017D, and L263G8 (red line), followed by treatment with Alexa Fluor 488-conjugated anti-mouse IgG. Fluorescence data were collected using the SA3800 Cell Analyzer. Black line, control (no primary antibody treatment).
Figure 2.
Flow cytometric analysis of anti-hCCR8 mAbs against CHO/hCCR8 and CHO-K1. CHO/hCCR8 (A) and CHO-K1 cells (B) were treated with 0.02–20 µg/mL of C8Mab-21, S19017D, and L263G8 (red line), followed by treatment with Alexa Fluor 488-conjugated anti-mouse IgG. Fluorescence data were collected using the SA3800 Cell Analyzer. Black line, control (no primary antibody treatment).
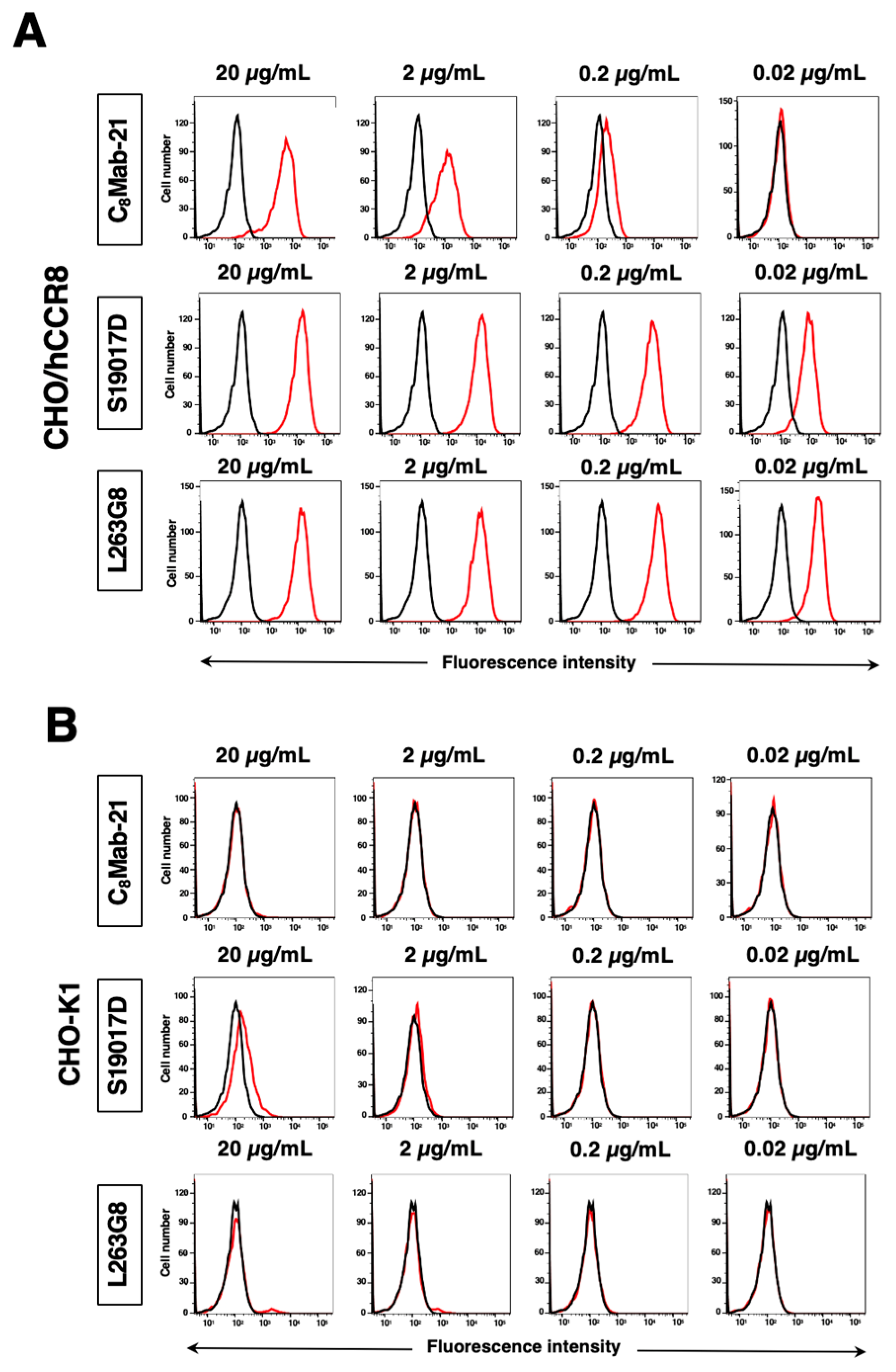
Figure 3.
Flow cytometric analysis of anti-hCCR8 mAbs against endogenously hCCR8-expressing cells. TALL-1 (A), CCRF-HSB2 (B), and NK cells (C) were treated with 0.02–20 µg/mL of C8Mab-21, S19017D, and L263G8 (red line), followed by treatment with Alexa Fluor 488-conjugated anti-mouse IgG. Fluorescence data were collected using the SA3800 Cell Analyzer. Black line, control (no primary antibody treatment).
Figure 3.
Flow cytometric analysis of anti-hCCR8 mAbs against endogenously hCCR8-expressing cells. TALL-1 (A), CCRF-HSB2 (B), and NK cells (C) were treated with 0.02–20 µg/mL of C8Mab-21, S19017D, and L263G8 (red line), followed by treatment with Alexa Fluor 488-conjugated anti-mouse IgG. Fluorescence data were collected using the SA3800 Cell Analyzer. Black line, control (no primary antibody treatment).
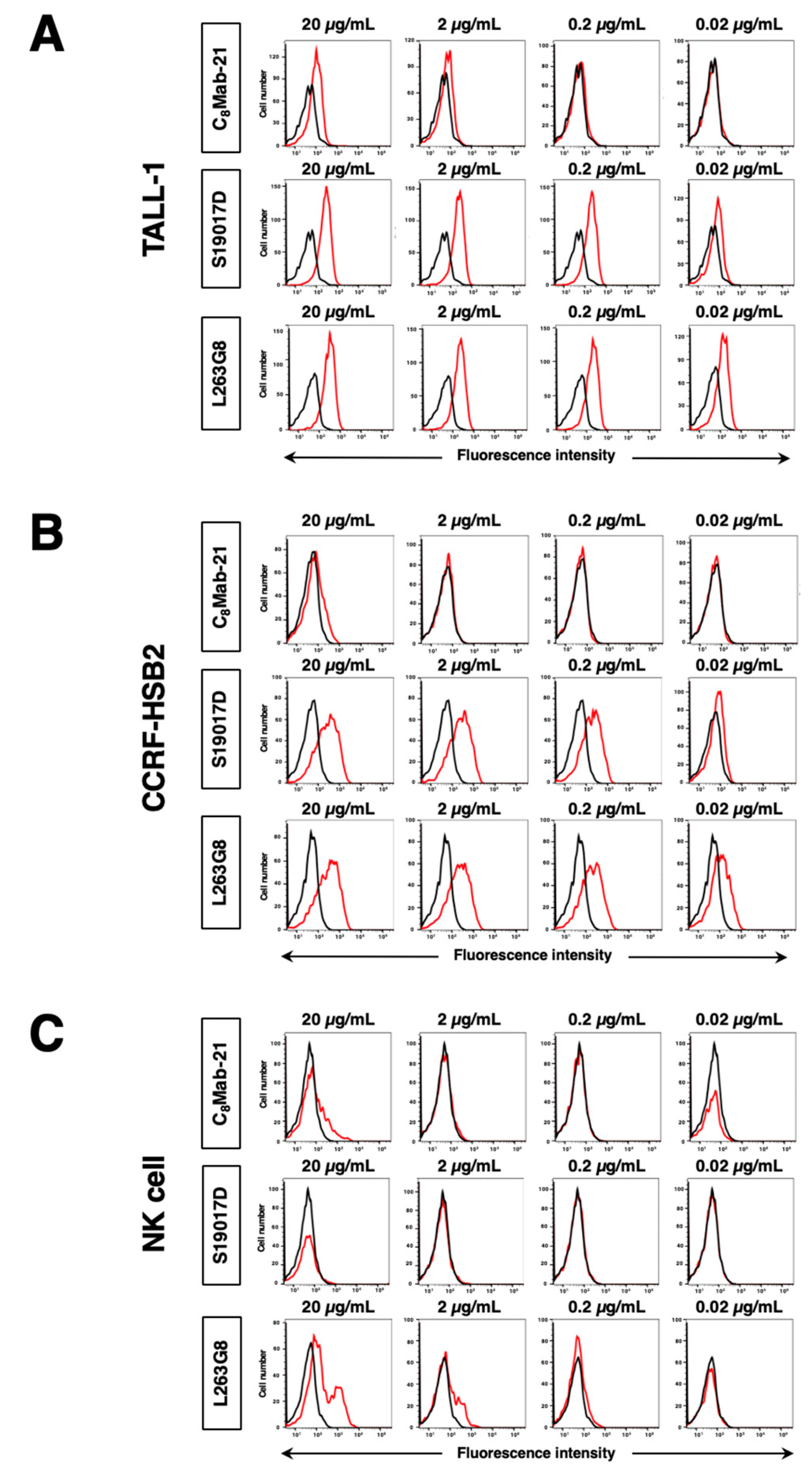
Figure 4.
The analysis of the binding affinity of anti-hCCR8 mAbs for CHO/hCCR8. CHO/hCCR8 cells were suspended in 100 µL of serially diluted C8Mab-21 (100 µg/mL to 0.006 µg/mL) (A), S19017D (10 µg/mL to 0.0006 µg/mL) (B), or L263G8 (10 µg/mL to 0.0006 µg/mL) (C). Then, cells were treated with Alexa Fluor 488-conjugated anti-mouse IgG. Fluorescence data were subsequently collected using the BD FACSLyric, following the calculation of EC50 by GraphPad PRISM 6.
Figure 4.
The analysis of the binding affinity of anti-hCCR8 mAbs for CHO/hCCR8. CHO/hCCR8 cells were suspended in 100 µL of serially diluted C8Mab-21 (100 µg/mL to 0.006 µg/mL) (A), S19017D (10 µg/mL to 0.0006 µg/mL) (B), or L263G8 (10 µg/mL to 0.0006 µg/mL) (C). Then, cells were treated with Alexa Fluor 488-conjugated anti-mouse IgG. Fluorescence data were subsequently collected using the BD FACSLyric, following the calculation of EC50 by GraphPad PRISM 6.
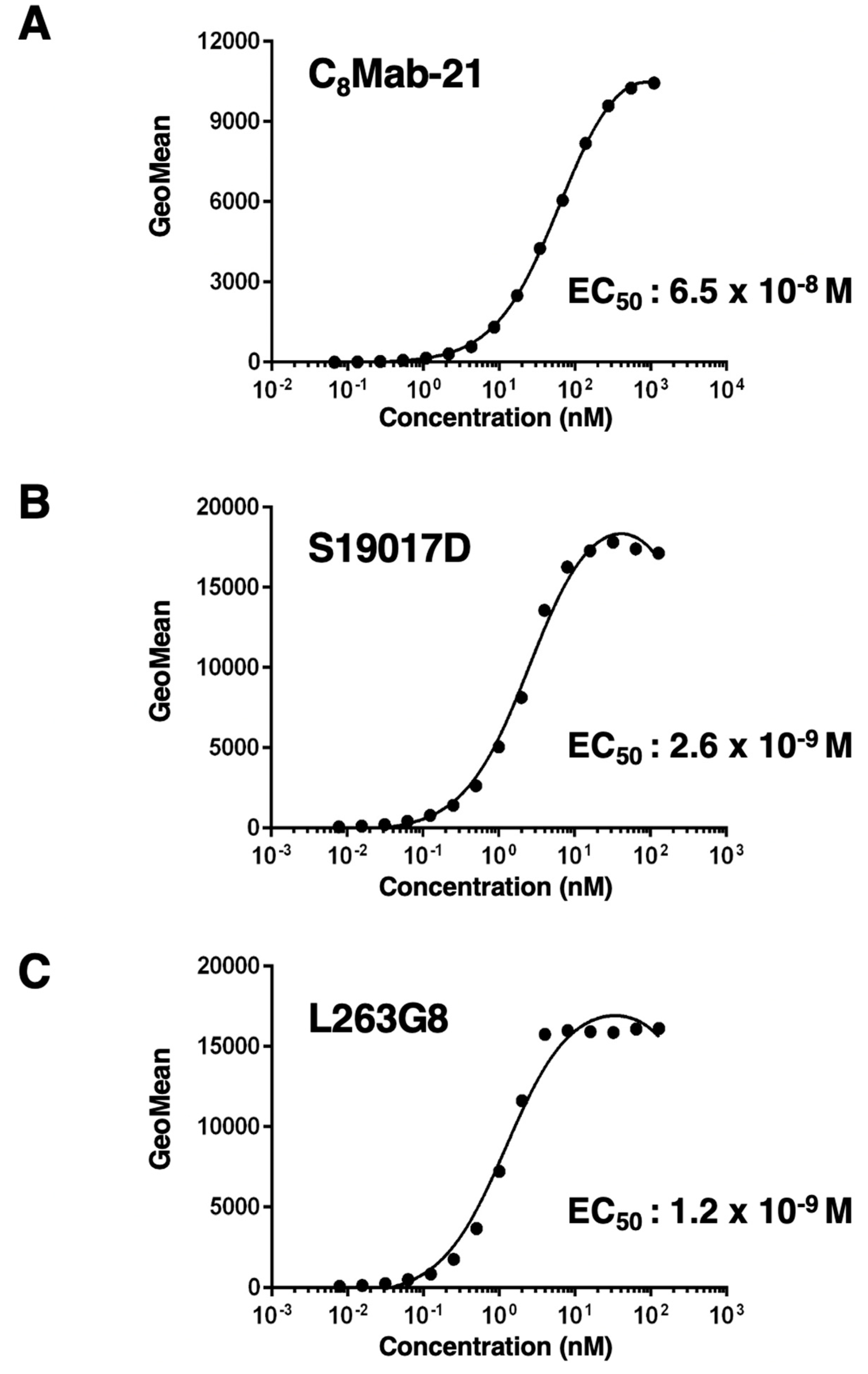
Figure 5.
The analysis of the binding affinity of anti-hCCR8 mAbs for TALL-1. TALL-1 cells were suspended in 100 µL of serially diluted C8Mab-21 (100 µg/mL to 0.006 µg/mL) (A), S19017D (10 µg/mL, 2.5 µg/mL to 0.0006 µg/mL) (B), or L263G8 (0.625 µg/mL to 0.0006 µg/mL) (C). Then, cells were treated with Alexa Fluor 488-conjugated anti-mouse IgG. Fluorescence data were subsequently collected using the BD FACSLyric, following the calculation of the EC50 by GraphPad PRISM 6.
Figure 5.
The analysis of the binding affinity of anti-hCCR8 mAbs for TALL-1. TALL-1 cells were suspended in 100 µL of serially diluted C8Mab-21 (100 µg/mL to 0.006 µg/mL) (A), S19017D (10 µg/mL, 2.5 µg/mL to 0.0006 µg/mL) (B), or L263G8 (0.625 µg/mL to 0.0006 µg/mL) (C). Then, cells were treated with Alexa Fluor 488-conjugated anti-mouse IgG. Fluorescence data were subsequently collected using the BD FACSLyric, following the calculation of the EC50 by GraphPad PRISM 6.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



