
Submitted:
20 March 2024
Posted:
20 March 2024
You are already at the latest version
Abstract
Protein tyrosine phosphatase receptor type E (PTPRE) is a member of the “classical” protein tyrosine phosphatase subfamily and regulates a variety of cellular processes in a tissue-specific manner by antagonizing the function of protein tyrosine kinases. PTPRE plays a tumorigenic role in different human cancer cells, but its role in retinoblastoma (RB), the most common malignant eye cancer in children, remains to be elucidated. Etoposide resistant RB cell lines and RB patients display significant higher PTPRE expression levels compared to chemosensitive counterparts and the healthy human retina, respectively. PTPRE promotor methylation analyses revealed that PTPRE expression in RB is not regulated via this mechanism. Lentiviral PTPRE knockdown (KD) induced a significant decrease in growth kinetics, cell viability, and anchorage independent growth of etoposide resistant Y79 and WERI RB cells. Caspase-dependent apoptosis rates were significantly increased and a re-sensitization for etoposide could be observed after PTPRE depletion. In vivo chicken chorioallantoic membrane (CAM) assays revealed decreased tumor formation capacity as well as reduced tumor size and weight following PTPRE KD. Expression levels of miR631 were significantly downregulated in etoposide resistant RB cells and patients. Transient miR631 overexpression resulted in significantly decreased PTPRE levels and concomitantly decreased proliferation and increased apoptosis levels in etoposide resistant RB cells, impacts mirroring PTPRE KD effects and indicating PTPRE regulation via this miR. Additionally, PTPRE KD led to altered phosphorylation of protein kinase SGK3 and - dependent on the cell line - also AKT and ERK1/2, suggesting potential PTPRE downstream signaling pathways. In summary, these results indicate an oncogenic role of PTPRE in chemoresistant retinoblastoma.
Keywords:
PTPRE
; miR631
; retinoblastoma
; CAM assay
; etoposide
; chemoresistance
; tumorigenesis
1. Introduction
Retinoblastoma (RB) is a malignant tumor that arises in the developing retina of children, affecting approximately 1 in 16,000 live births [1,2,3]. In more than 95% of the cases, the tumor initially develops through biallelic loss of the tumor suppressor gene RB1 and further progresses after additional genetic/epigenetic changes (for review: [1]). RB is curable, but tumors become advanced if treatment is delayed, hindering vision and globe salvages with a risk of metastasis [4]. VEC (vincristine, etoposide, carboplatin) combination chemotherapy is commonly used for globe sparing with different routes of drug delivery [3,5]. However, RB treatment is often limited, side effects of the chemotherapeutics and development of drug resistances leading to relapses or secondary carcinomas [6]. Besides, similar to other cancers, therapeutics that selectively target the tumor while sparing the normal retina are desired.
Protein tyrosine phosphatases (PTPs) working in concert with protein tyrosine kinases (PTK) to control cellular homeostasis are novel targets for cancer drug discovery. Cell signaling is tightly controlled by a balance between pathway activators like PTKs, and inactivators like PTPs [7,8]. Aberrant regulation of protein phosphorylation on tyrosine residues is a well-established cause of cancer [9,10]. Receptor-type protein tyrosine phosphatases (PTPRs) are a subgroup of PTPs comprising 21 members that share a transmembrane domain (for review: [7]). Receptor-type protein tyrosine phosphatase epsilon (PTPRE), a member of the PTPR family, has two isoforms, a membrane type PTPRE (memPTPRE) that anchors in the cell membrane and is highly expressed in brain, testes, lymph node and lung and a cytosol type PTPRE (cytPTPRE), which is mainly localized in cytosol and is found in spleen, lung and thymus [10]. Alterations in expression of these phosphatases, e.g. by promoter methylation, cause aberrations in the activation of crucial cellular pathways like proliferation, apoptosis, survival, adhesion, migration, and invasion, suggesting that PTPRs are critical components in malignant transformation and tumorigenesis [7,10]. In general, PTPs have been controversially discussed as either tumor suppressors or oncogenes [7,11,12]. Upregulated levels of PTPRE were found in several cancer entities, including acute myeloid leukemia and renal cell carcinoma [13,14]. It has been shown that promoter methylation is an important mechanism to regulate PTPR expression (for review; [7]) and aberrant PTPRE promoter methylation has been observed in various cancers [15,16]. Furthermore, PTPRE was described as a downstream target of miR631 in hepatocellular carcinoma [17] and an up-regulation and activation of PTPRE was observed in MCF-7 cells and MDA-MB-231 upon stimulation with fibroblast growth factor (FGF) [18]. At the downstream signaling level, receptor-type PTPRE dephosphorylates and thereby activates Src in murine mammary tumor cells, which in turn contributes to oncogenesis [9,10,19,20]. Besides, PTPRE was found to contribute to ERK1/2 and AKT activation in human breast cancer cells [18] and it has been shown that in mice PTPRE antagonizes the activation of voltage-gated potassium channels by tyrosine kinases like Src [21].
Studies of PTPRE signaling in cancer in general and in RB in particular will help to develop novel RB treatment strategies targeting this tyrosine phosphatase or up- and downstream signaling components. Thus, in the study presented, we set out to unravel the expression profile of PTPRE in chemosensitive and chemoresistant RB cell lines and patient tumors and its potential regulation by promotor methylation, miR631 targeting, and FGF signaling. Besides, the effects of a lentiviral PTPRE knockdown on survival, growth and re-sensitization as well as PTPRE downstream signaling of etoposide resistant RB cells were investigated in vitro and in vivo.
2. Results
2.1. PTPRE is Differentially Expressed in Retinoblastoma Cell Lines and Patient Tumors
We analyzed the expression of PTPRE in the parental, chemosensitive RB suspension cell lines Y79, WERI, Rbl-13, Rbl30, RB247 and RB383 as well as in the adherent cell line RB355. Compared to the healthy human retina, PTPRE was differentially expressed with significantly higher mRNA levels in WERI, RB355, Rbl-13, Rbl30 and RB247 cells and significantly decreased expression in Y79 and RB383 cells (Figure 1a). Western Blot analysis confirmed this expression pattern at PTPRE protein level (Figure 1a). In addition, compared to chemosensitive counterparts, we found an increase in PTPRE expression in the etoposide resistant RB cell lines Y79_Etop and WERI_Etop, however, only reaching full significance in the latter cell line (Figure 1b). Western Blot analysis confirmed this expression pattern at PTPRE protein level (Figure 1b). Moreover, significantly increased PTPRE mRNA and protein expression levels were detected in RB patient tumors compared to the healthy human retina (Figure 1c).
The PTPRE expression pattern in a RB patient tumor was revealed by immunohistochemical staining and is shown in Figure 2.
2.2. PTPRE Expression Is Not Regulated by Promotor Methylation
Since an epigenetic regulation of PTPRE via promotor methylation has been described for pituitary adenomas and papillary thyroid carcinoma [15,16], we performed PTPRE promotor methylation analyses for the etoposide resistant RB cell lines Y79 and WERI and their chemosensitive counterparts. Bisulfite conversion and subsequent sequencing revealed that only 1-12% of the 7 CpG islands within the PTPRE promotor region are methylated and no differences in methylation status were discernible when comparing parental, chemosensitive and etoposide resistant Y79 and WERI cells (Figure 3). Thus, PTPRE expression in RB cells is not regulated by promotor methylation.
2.3. Involvement of miR631 in the Regulation of PTPRE Expression
PTPRE has been suggested as a direct downstream target of miR631 in hepatocellular carcinoma cells [17]. Attempting to decipher the mechanisms regulating the expression of PTPRE in etoposide resistant RB cells, expression patterns of miR631 was analyzed in Y79_Etop and WERI_Etop RB cell lines and RB patient tumors. Compared to the healthy human retina, miR631 levels were significantly downregulated in RB patient tumors and in both etoposide resistant RB cell lines (Figure 4 a,b). Thus, miR631 and PTPRE display opposing expression patterns and we set out to test for a hypothesized regulation of PTPRE expression by this respective miR. Overexpression of miR631 resulted in significantly decreased PTPRE protein levels (Figure 4c) as well as significantly decreased proliferation rates (Figure 4d) with concomitantly upregulated apoptosis levels (Figure 4e). All effects seen mirrored those obtained following PTPRE knockdown, strongly indicating that PTPRE expression in etoposide resistant RB is regulated by miR361.
2.4. Involvement of FGFb Signaling in the Regulation of PTPRE Expression
In a proteome profiler human oncology array we previously found FGFb to be upregulated in etoposide resistant RB cells (data not shown). As it has been shown that expression of PTPRE is induced by FGF [18] and RB cells were reported to express all four known FGF receptors (FGFR; [22]), we hypothesized that inhibition of the FGFR, prohibiting binding of FGFb, potentially results in decreased PTPRE expression, whereas treatment with recombinant FGF (rFGF) possibly increases PTPRE expression. Thus, we performed FGFR inhibitor studies or treated the cells with rFGF. Inhibition of the FGFR with Pemigatinib resulted in significantly decreased cell viability (Figure 5a) and slightly, yet not significantly increased apoptosis levels (Figure 5b) of etoposide resistant Y79 and WERI cells, effects also seen upon PTPRE knockdown (see below). However, PTPRE protein expression was not clearly altered upon FGFR inhibition and administration of rFGF did not induce any changes in PTPRE levels independent of the treatment duration and the concentration used (data not shown), indicating that PTPRE expression in etoposide resistant RB cells is most likely not regulated via the FGF signaling axis.
2.5. PTPRE Knockdown Influences Cell Viability, Proliferation and GROWth of Etoposide Resistant Y79 and WERI Retinoblastoma Cell Lines
We performed lentiviral PTPRE knockdown experiments in the etoposide resistant RB cell lines Y79 and WERI, both exhibiting high endogenous PTPRE expression levels (Figure 1b). Efficient PTPRE knockdown levels were confirmed by quantitative Real-time PCR (Figure 6a) and Western blot analysis (Figure 6b).
As revealed by WST-1 assays and BrdU cell counts, both etoposide resistant RB cell lines investigated exhibited significantly lower cell viabilities (Figure 7a) and decreased proliferation rates (Figure 7b) following PTPRE knockdown. In both RB cell lines investigated additional growth curve analyses (Figure 7c,d) after PTPRE knockdown confirmed the effects on cell proliferation shown above.
2.6. PTPRE Knockdown Induces Caspase Dependent Apoptosis in Etoposide Resistant RB Cell Lines
PTPRE knockdown resulted in a significant increase in apoptosis levels of both etoposide resistant RB cell lines investigated (Figure 8a). To analyze whether PTPRE induced apoptosis is caspase dependent, etoposide resistant, PTPRE depleted RB cells were treated with the broad spectrum caspase inhibitor Boc-D-Fmk. Compared to cell death levels after PTPRE KD alone, apoptosis was significantly reduced after Boc-D-Fmk treatment in both cell lines, indicating the involvement of caspases in PTPRE mediated apoptosis (Figure 8a). Additional caspase assays and immunocytochemical stains against caspase 3 revealed significantly increased caspase 3/7 activity (Figure 8b) as well as significantly increased numbers of cleaved and thereby activated caspase 3 positive cells (Figure 8 c,d), confirming that PTPRE induced apoptosis is caspase-3/7 mediated in both etoposide resistant RB cell lines investigated.
2.7. PTPRE Knockdown Reduces Anchorage Independent Growth of Etoposide Resistant RB Cell Lines
Compared to their parental counterparts, PTPRE depleted WERI_Etop cells showed a significantly reduced colony formation capacity (Figure 9a), displaying significantly smaller colonies in soft agarose assays testing for changes in anchorage-independent growth capability (Figure 9 b,c). Y79_Etop cells likewise displayed significantly smaller colonies following PTPRE knockdown (Figure 9 b,c), colony formation capacity did, however, not significantly change (Figure 9a).
2.8. PTPRE Knockdown Decreases Tumorigenicity and Migration Potential of Etoposide Resistant RB Cells In Vivo
To investigate whether the effects of a PTPRE knockdown on etoposide resistant RB cell growth in vitro likewise influence their tumor growth and migration potential in vivo, we used the chicken in ovo chorioallantoic membrane (CAM) assay as a model system. PTPRE depleted Y79_Etop or WERI_Etop cells and control cells were inoculated onto the CAM of embryonic developmental day 10 (EDD10) chicken embryos. Photo-documentation of CAM tumors developing from inoculated RB cells (Figure 10a) and quantification of tumor weight (Figure 10b) and size (Figure 10c) revealed that PTPRE depleted, etoposide resistant RB cells develop significantly smaller tumors (Figure 10 a,c) than control cells, with in the case of Y79_Etop cells also significantly lower tumor weight (Figure 10b).
To exemplarily test for changes in migratory potential following PTPRE knockdown, GFP-labeled etoposide resistant WERI-Rb1 RB cells were injected into a CAM vein. PTPRE depleted WERI_Etop RB cells showed no reduced extravasation from the CAM vasculature into the surrounding tissue and did not displayed significant changes in migration rate compared to their respective controls as revealed by human GAPDH real-time PCR analyses of lower CAM punches (data not shown).
2.9. Re-Sensitization of Resistant RB Cells towards Etoposide after PTPRE Knockdown
To determine whether PTPRE depletion contributes to a re-sensitization of etoposide resistant RB cells, we again knocked down PTPRE in etoposide resistant Y79_Etop and WERI_Etop cell lines. Treatment of PTPRE depleted etoposide resistant RB cell lines with etoposide did indeed result in significantly decreased cell viability (Figure 11) compared to the PTPRE knockdown alone, indicating that depletion of PTPRE increases the susceptibility of etoposide resistant RB cells for this chemotherapeutic drug.
2.10. PTPRE Downstream Signaling
It has been shown that many cancer entities are connected to aberrant activation of tyrosine kinases, including PTPRE. PTPRE regulates phosphorylation processes with the involvement of different other kinases like SRC [10,20,36], ERK1/2 [23], glucocorticoid kinase 3 (SGK3) and AKT downstream signaling [24,25]. As all above mentioned signaling molecules mediate effects related to cell proliferation, growth, viability, tumor initiation and tumor cell migration [26,27,28], effects likewise seen to be influenced following PTPRE silencing, we investigated ERK/p-ERK, SRC/pSRC, AKT/p-AKT, and SGK3/pSGK3 expression after PTPRE knockdown in order to reveal possible PTPRE downstream signaling mechanisms in etoposide resistant RB cells.
Western blot analyses revealed no changes in SRC expression or phosphorylation upon PTPRE KD in both etoposide resistant RB lines investigated (Figure 12a,b). By contrast, in PTPRE depleted Y79_Etop RB cells significantly increased ERK1/2 as well as pERK1/2 protein levels (Figure 12a,c) and slightly to significantly decreased pAKT (Figure 12a,d) and pSGK3 levels were observed (Figure 12 a+e). In WERI-Etop RB cells, however, changes in ERK1/2 levels were not significant and no pERK expression was detectable (Figure 12a,c). PTPRE depletion in WERI-Etop RB cells significantly increased AKT levels, but did not significantly change pAKT levels (Figure 12a,d). Phospho SGK3 levels, by contrast, were likewise reduced in WERI-Etop cells (Figure 12a,e).
3. Discussion
Protein tyrosine phosphatases (PTPs) are enzymes that remove phosphate groups from proteins. Dysregulation of dephosphorylation can lead to uncontrolled cell growth and proliferation, apoptosis arrest and ultimately cancer development [10,12]. Initially, PTPs have been described as tumor suppressors as they terminate signaling by desphosphorylation of oncogenic kinases. However, it became clear that PTPs are overexpressed in several cancers and in this setting do not suppress tumor growth but rather promote tumor development [12]. Thus, PTPs and receptor-type protein tyrosine phosphatases (PTPRs), a subgroup of PTPs sharing a transmembrane domain [7], can act as tumor suppressors and oncogenes [7,11,12].
Our present study aimed to explore the involvement of receptor type protein tyrosine phosphatase epsilon (PTPRE) in retinoblastoma progression in general and chemotherapy resistance in particular. PTPRE has been studied e.g. in osteoclasts, nerve cells, and cancer cells, and it exerts different functions among various tissues (for review see: [10]). Expression of PTPRE was found to be significantly higher than average in acute myeloid leukemia (AML). PTPRE expression did, however, not display a correlation with overall AML patients’ survival [13]. No significant difference was observed in PTPRE expression in different sexes of patients, but expression displayed a weak correlation with patient age [13]. In lung adenocarcinoma PTPRE was suggested as one of the genes potentially helpful to categorize different prognostic and therapeutic subgroups [29]. Overexpression of PTPRE was likewise reported for all stages of renal cell carcinoma [14]. Besides, PTPRE is highly expressed in thyroid cancer clinical samples and cell lines and overexpression correlates with TNM stage [30]. Fittingly, in the study presented, we likewise observed increased PTPRE expression levels in etoposide resistant compared to chemosensitive RB cell lines as well as in RB patient tumors compared to the healthy human retina.
To unravel molecular mechanisms potentially regulating PTPRE expression, we analyzed (i) promotor methylation of PTPRE in RB, (ii) miR631 as a putative PTPRE targeting miRand (iii) the potential involvement of FGF receptor (FGFR) signaling.
Promoter methylation is an important mechanism for PTPR inactivation in cancer (for review see: [7]). It has been shown, that the promoter methylation status of PTPRE is strongly associated with lymph node metastasis (LNM) of papillary thyroid carcinoma (PTC) and high expression correlates with a significantly better survival [15]. Thus, PTPRE promoter methylation status has been suggested as a useful predictive biomarker of LNM in PTC [15]. Promoter hypermethylation and decreased expression level of PTPRE were described for nonfunctioning pituitary adenomas (NFPAS), whereby invasive and non-invasive NFPAs displayed only slight differences in their methylation profiles [16]. In our study presented, PTPRE promotor methylation analyses revealed no differences in methylation status when comparing chemosensitive and etoposide resistant RB cell lines. Therefore, PTPRE expression in RB cells is not regulated by promotor methylation.
PTPRE was described as a downstream target of miR631 in hepatocellular carcinoma (HCC) [17]. MiR631 targets genes, including PTPRE, are involved in different cellular processes including proliferation, apoptosis, invasion, and drug resistance and sensitivity, therefore playing an important role in tumor progression (for review see: [33]). MiR631 levels were found to be significantly down-regulated in hepatocellular carcinoma (HCC) [17]. In prostate cancer, one of the most common cancers, the expression of miR631 is likewise low. Increasing expression levels of this miR inhibits metastasis and invasion of prostate cancer cells by inhibiting Zeta-chain-associated protein kinase 70 (ZAP70) [31]. In addition, miR631 expression is low in non-small-cell lung cancer (NSCLC) cancer and its induction leads to an inhibition of E2F2, which regulates PI3K/AKT signaling, in turn reducing malignancy [32]. Fittingly, in the study presented miR631 levels were likewise significantly downregulated in RB patient tumors and etoposide resistant RB cell lines when compared to the healthy human retina. Thereby miR631 and PTPRE displayed opposing expression patterns suggesting a regulation of PTPRE expression by this miR. Overexpression of miR631 resulted in the exact same effects seen upon PTPRE knockdown, strongly indicating that PTPRE expression in etoposide resistant RB is regulated by miR361.
RB cells were reported to express all four known FGF receptors (FGFR; [22]) and an up-regulation and activation of PTPRE was observed upon FGF stimulation of human breast cancer cells [18]. As we previously found FGFb to be upregulated in a proteom profiler oncology array of etoposide resistant RB cells (data not shown), we hypothesized that inhibition of the FGFR might result in decreased PTPRE expression, whereas treatment with recombinant FGF (rFGF) possibly increases PTPRE expression. FGFR inhibition with Pemigatinib resulted indeed in significantly decreased viability of etoposide resistant RB cells, effects also seen upon PTPRE knockdown. FGFR inhibition did, however, not significantly decrease PTPRE expression levels. Treatment of WERI_Etop and Y79_Etop cells with recombinant FGF likewise did not induce any changes in PTPRE levels, indicating that PTPRE expression in etoposide resistant RB cells is unlikely to be regulated via the FGF-FGFR signaling axis.
As PTPs in general and PTPRE in particular have been controversially discussed as tumor suppressors or oncogenes [7,11,12,19], in the study presented we set out to investigate the functional effects of a PTPRE knockdown in etoposide resistant RB cell lines. Upregulated PTPRE has been shown to promote cell proliferation, whereas PTPRE KD resulted in the opposite effect indicating an oncogenic function in thyroid carcinoma [30]. Moreover, cells isolated from mammary tumors induced by Neu in mice genetically lacking PTPRE proliferated less than corresponding mammary tumor cells isolated from mice expressing the phosphatase [9]. Further along this line, reduced PTPRE levels decreased the viability and anchorage-independent growth of human breast cancer cells [18]. These findings are in good accordance with data from our study presented, showing significantly reduced cell viability, growth and proliferation upon PTPRE KD in etoposide resistant RB cell lines, indicating an oncogenic role of PTPRE in retinoblastoma. In addition, the PTPRE depletion also led to a re-sensitization of the resistant RB cell lines towards etoposide, underlining its potential role in cancer cell transformation processes [10].
It has been shown that PTPRE expression in murine mammary glands leads to massive hyperplasia and associated tumorigenesis [9] and high PTPRE expression in thyroid cancer correlates with tumor size [30]. In accordance with these findings, in our study presented, PTPRE depleted, etoposide resistant RB cells developed significantly smaller tumors in ovo than control cells, supporting the notion of PTPRE being an oncogene rather than a tumor suppressor in RB. PTPRE depleted etoposide resistant WERI-Rb1 cells did, however, not display significant changes in migration rates in ovo, contradicting findings that PTPRE promotes migration in macrophages [34] and acts as a metastatic promoter in hepatocellular carcinoma [35].
In order to investigate putative downstream signaling pathways, we set out to analyze the protein expression and phosphorylation status of the four known downstream target of PTPRE SRC, ERK1/2, AKT and SGK3. It has been reported that at the molecular level, PTPRE dephosphorylates and thereby activates SRC [9,19,20]. Activation of SRC has been shown to activate downstream pathways like e.g. the PI3K-AKT pathway, playing an essential role in cell proliferation, survival, growth, tumor initiation, metastasis, and drug resistance (for review see: [26]). The activity of SRC is significantly reduced in tumor cells lacking PTPRE and restoring SRC activity revealed that it is only its reduced activity that caused aberrant proliferation rates of tumor cells lacking PTPRE [9]. Further along this line, lack of PTPRE increased SRC phosphorylation at its C-terminal inhibitory Tyr527 sites by 51% in Neu-induced Ptpre-/- murine mammary carcinoma tumor cells [19]. By contrast, our study revealed that although PTPRE KD slightly increased pSRC levels in etoposide resistant WERI-Rb1 cells, effects did not reach significance, indicating that the anti-tumorigenic effects of a PTPRE KD in etoposide resistant RB cells are not mediated via this downstream mediator.
Previous studies suggested an involvement of ERK1/2 in PTPRE downstream signaling [18,23,30]. The serotonin-tyrosine kinase ERK, phosphorylated and thereby activated by MEK, primarily plays a role in promoting cell proliferation and anti-apoptotic effects (for review see: [27]). Unexpectedly, silencing of PTPRE expression in human breast cancer cells has been shown to abolish ERK1/2 activation, leading to significantly diminished pERK levels [18] and siPTPRE transfected thyroid cancer cells likewise exhibited decreased pERK1/2 levels [30]. In line with these findings, ectopic expression of PTPRE in MCF-7 cell lines resulted in activation of ERK1/2 [18] and pERK levels were clearly increased in thyroid cancer cells upon PTPRE upregulation [30]. By contrast, phosphorylation and thereby activation of ERK1/2 was reduced after PTPRE transfection of non-carcinogenic HEK293T cells [23]. Fittingly, in our study presented significantly increased pERK levels were observed upon PTPRE KD in Y79_Etop RB cells. Upregulated pERK1/2 levels do, however, not fit the anti-proliferative and anti-survival effects seen after PTPRE KD in these cells. Besides, in WERI-Etop RB cells, no pERK expression was detectable. Thus, the ERK/pERK signaling is most likely not involved in mediating PRPRE effects in RB cells.
Retinoblastoma cells have been described to activate the AKT pathway [36]. Phosphorylated and thereby activated AKT is known to be implicated in regulation of cell survival, growth and apoptosis (for review see: [28]). As PTPRE was found to contribute to AKT activation [18], we hypothesizes that PTPRE might signal via the AKT axis in RB cells as well. In the study presented, PTPRE KD did not change pAKT levels in etoposide resistant WERI-Rb1 cells, but resulted in slightly yet not significantly decreased pAKT levels in etoposide resistant Y79 RB cells. Fittingly, PTPRE silencing in MCF-7 and MDA-MB-231 cells abolished AKT activation by significantly diminishing pAKT levels [18] and siPTPRE transfected thyroid cancer cells likewise exhibited decreased pAKT levels [30]. However, as PTPRE is a phosphatase, dephosphorylation of its targets should be abolished or at least diminished upon its silencing, resulting in increased rather than decreased pAKT levels. Thus, although reduced pAKT levels would fit reduced cell proliferation and growth seen in PTPRE-depleted etoposide resistant RB cells, these effects cannot be directly mediated via PTPRE.
The PI3K-dependent serum and glucocorticoid inducible protein kinase 3 (SGK3) shares a substrate specificity to AKT and has been suggested to play a critical role in AKT-independent oncogenic signaling (for review see: [37]). It has been shown that in tumor cells with minimal AKT activation, SGK3 conferred increased cell viability [37]. Fittingly, in the study presented the effects of PTPRE depletion on pAKT levels did not reach significance, whereas pSGK3 levels were significantly decreased in etoposide resistant Y79 RB cells and likewise reduced in WERI_Etop cells. However, as already discussed above, PTPRE is a phosphatase, and thus, one would expect an increase rather than a decrease in pSGK3 levels upon its depletion. Nevertheless, PTPRE downstream effects might be mediated indirectly via the AKT-independent SGK3 axis rather than the AKT pathway.
Summarizing, one can state that PTPRE acts as an oncogene in RB and is most likely involved in chemotherapy resistance mechanisms by enhancing cell proliferation and tumor growth and inhibiting apoptosis. Although molecular signaling pathways remains to be elucidated in more detail in the future, miR631 seems to be a key player in the regulation of PTPRE in RB. As small molecules and antibodies inhibiting the activity of tyrosine kinases are effective tools in cancer treatment, regulators of tyrosine kinase activity like the tyrosine phosphatase PTPRE hold the potential of new future RB therapy targets.
4. Materials and Methods
4.1. Human Retina and Retinoblastoma Samples
Postmortem healthy human retina (hRet) and patient RB samples were used for comparative expression studies. The study methodologies conformed to the standards set by the Declaration of Helsinki. The Ethics Committee of the Medical Faculty of the University of Duisburg-Essen approved the use of human retina (approval # 06-30214) and RB samples (approval # 14-5836-BO) for research conducted in the course of the study presented, and written informed consent has been obtained from patients’ relatives or parents.
4.2. Cell Lines and Culture
The human retinoblastoma (RB) cell lines RB355, Rbl-13, Rbl30, RB247, R383 [38], Y79 [39] and WERI-Rb1 (WERI) [40] as well as the corresponding etoposide resistant RB cell lines Y79_Etop and WERI_Etop were kindly provided by Dr H. Stephan. RB cell lines were cultivated as described previously [41]. Human embryonic kidney cells (HEK293T) were grown as adherent cell culture in DMEM (PAN-Biotech, Aidenbach, Germany) with 10% FBS (PAN-Biotech, Aidenbach, Germany), 4 mM L-glutamine (Gibco, Karlsruhe, Germany), 100 U penicillin/ml, and 100 µg streptomycin/ml (Gibco, Karlsruhe, Germany) at 37°C, 5% CO2 and 95% humidity. No approval from an ethics committee was required for work with the human cell lines.
4.3. Lentiviral PTPRE Knockdown
In order to generate lentiviral particles, 6x106 human embryonic kidney cells (HEK293T) were transfected with 6 µg of each of the following plasmid DNAs: (I) packaging vectors pczVSV-G[42] and pCD NL-BH [42], (II) shPTPRE clone#198 (clone TRCN0000315198) for transduction of Y79_Etop cells, (III) shPTPRE clone #895 (clone TRCN0000002895) for transduction of WERI_Etop Rb1 cells, (V) pPRIME-CMV-Neo-FF3 (p234) as a negative control for all knockdown experiments or (VI) GFP expression vector (pCL7EGwo) each in the presence of 45 µg polyethyleneimine (PEI, branched, Sigma-Aldrich, St. Louis, Missouri, USA) in DMEM medium. After 24 h the medium was changed to Iscove´s Modified Dulbecco´s medium (IMDM, Pan-Biotech, Aidenbach, Germany) with 10% FBS and 1% penicillin/streptomycin and 72h after transfection viral supernatants were harvested, filtered (0.45 µm filter) and cryoconserved.
For stable transduction, 1.25x106 RB cells were seeded in DMEM medium. After 24h the medium was removed and the cells were transducted with PTPRE virus particles or control virus particles each in the presence of polybrene (5 µl per ml lentivirus; H9268, Sigma-Aldrich, München, Germany). After 24h DMEM medium with supplements (twice the volume of the virus particles) was added and 48h later the medium was completely changed and the cells were incubated for another 72h.
4.4. PTPRE Promotor Analysis
Genomic DNA from all RB cell lines was isolated using the DNeasy Blood and Tissue Kit (Qiagen) and bisulfite-converted with the EZ DNA Methylation-Gold Kit (Zymo Research) to convert all cytosines to uracil, while the methylated cytosines remain unmodified. PTPRE promoter sequences were amplified from bisulfite-converted DNA by PCR using the following protocol: 5 min at 95 °C, 50 cycles of 1 min 95 °C, 2 min 53/54 °C, 1 min 72 °C followed by final 10 min 72 °C step. PCR forward and reverse primer sequences were as follows: PTPRE (for) 5´-GTGAGTGTTTGTTATATTTTGATTTTT-3´ and PTPRE (rev) 5`- CTATCCTCAACCTAACAAAAAATTTA-3´. Afterwards, the PCR product was purified from agarose gels by Illustra GFX PCR DNA and Gel Band Purification Kit (VWR Lifescience) and subcloned into the pCR®4-TOPO® vector using a TOPO TA Cloning Kit for Sequencing (Invitrogen). Plasmid isolation was performed with the GeneJET Plasmid Miniprep Kit (Thermo Scientific) and individual clones were sequenced by Microsynth using the M13 forward and reverse primer. Sequences of at least 5 clones were analyzed using the Clone Manager 9 software to identify methylated cytosine residues.
4.5. Plasmids and miR631 Overexpression
To generate a miR631 expression vector (pSG5-miR631), the human miR631 sequence was amplified from cDNA of HEK293T cells by RT-PCR using the forward primer 5´-CGGAATTCAACAGGCAGAGATCAGAGGG -3´ and the reverse primer 5´-CGGGATCCTGTCGGGATTACAGGTGTGG-3´ containing EcoRI and BamHI restriction sites. The PCR product was cloned into the pCRII-TOPO vector (Invitrogen, Karlsruhe, Germany), excised by digestion with EcoRI and BamHI and ligated into the pSG5 vector (#216201, Stratagene, La Jolla, USA) digested with the same restriction enzymes. The empty pSG5 vector was used as control and transfection was done as described previously [43]. 24 h after transfection the cells were harvested and used for protein and RNA isolation.
4.6. FGF Receptor Inhibitor Studies
For FGF receptor inhibition studies, 7.5x106 cells were seeded in 25 ml medium and incubated with 100 µM FGFR inhibitor (Pemigatinib INCB054828; MedChemExpress, Monmouth Junction, USA) for 120 h. DMSO (Sigma-Aldrich, München, Germany) treated cells served as a solvent control.
4.7. Re-Sensitization Studies
To study re-sensitizing effects of PTPRE depletion on the chemosensitivity of etoposide resistant RB cells, 1x106 Y79_Etop and WERI_Etop cells were seeded in 2 ml DMEM with supplements with or without etoposide (3 µmol/ml or 5 µmol/ml for Y79_Etop and WERI_Etop, respectively) in a 6 well plate and cultivated at 37°C. 1 mL DMEM with supplements was added after 96 h of cultivation. After 7 days, 4x104 cells were seeded in a 96 well plate in 100 µl DMEM with supplements with or without etoposide (3 µmol/ml or 5 µmol/ml for Y79_Etop and WERI_Etop, respectively) in triplicates. 10 µl of a water-soluble tetrazolium (WST-1) salt solution (Sigma-Aldrich, München, Germany) were added to each well and cells were incubated for a designated period at 37°C. Formazan dye produced by viable cells was quantified by measuring the absorbance in a microplate reader at 450 nm (Agilent BioTek, Santa Clara, CA, USA).
4.8. RNA Extraction and Quantitative Real-Time PCR
RNA isolations from RB cells and CAM tissue were performed using the NucleoSpin® RNA II Kit (Macherey & Nagel, Düren, Germany) and the miRNeasy Kit (Qiagen, Hilden, Germany), respectively. For quantitative Real-time PCR analyses, cDNA was synthesized with the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) according to the manufacturer´s protocol. For analysis of PTPRE expression a SYBRTM green PCR assay (Applied Biosystem, Darmstadt, Germany) was used with specific primers for PTPRE 5’-CTGAGCCAACTGGATGGAAT-3’ (forward) and 5’-TTGGGACCTTGAGCTGCTAT-3’ (reverse) as well as 5’-ACCCACTCCTCCACCTTTGA-3’ (forward) and 5’-CTGTTGCTGTAGCCAAATTCGT-3’ (reverse) for human GAPDH (hGAPDH) as an endogenous control. Real-time PCR reactions were conducted in triplicates in 20 µl of SYBRTM green PCR Mastermix (Applied Biosystem, Darmstadt, Germany) performing 40 cycles of the following program: 95°C for 15 min; 94°C for 15 sec., 55°C for 30 sec. and 70°C for 34 sec. For micro-RNA expression analyses a miScript PCR Starter Kit (# 2181193; Qiagen, Hilden, Germany) was used, following the manufacturer´s protocol. The designated miScript HiSpec Buffer (Qiagen, Hilden, Germany) for quantification of mature miRNA was used along with specific primers for miR631 5’-AGACCTGGCCCAGACCTCAGC-3’ and 5.8S RNA (5’-CTACGCCTGTCT GAGCGTCGCTT-3’) as an endogenous control. The reactions were performed in duplicates using a QuantStudioTM 3 real-time PCR system (Thermo Fisher, Darmstadt, Germany), with the following program: 95°C for 15 min; 94°C for 15 sec., 55°C for 30 sec. and 70°C for 34 sec. and 40 cycles. RNA isolation from chicken chorioallantoic membrane (CAM) tissue punches (see below) was performed as described previously [44]. Quantitative real-time PCR analyses were performed and quantified following the protocol published previously [44].
4.9. Western Blotting
For Western blot analyses, cells were washed in phosphate buffered saline (PBS, PAN-Biotech GmbH, Aidenbach, Germany) and lysed in radioimmunoprecipitation assay (RIPA) buffer plus supplements [45] for 30 min at 4°C on a shaker and centrifuged at 10,000 rpm at 4°C for 30 min. Afterwards, protein concentrations were measured by bicinchoninic acid assay (BCA; Thermo-Scientific, Oberhausen, Germany) according to the manufacturer’s protocol. Equal amounts of protein extracts were separated on a 10% SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were incubated with primary antibodies at 4°C overnight (Table 1).
HRP-conjugated species-specific secondary antibodies (goat-anti-rabbit; P0448 and rabbit-anti-mouse; P0260; DAKO, Glostrup, Denmark) were used in dilutions of 1:10,000 at room temperature for 1 hour on a tabletop shaker. The signals were developed by adding Western Bright Chemiluminescence Reagent (Cytiva, Buckinghamshire, UK).
4.10. Cell Viability Assays
For cell viability assays, 4 x 104 cells in 100 µl medium were seeded in a 96-well plate in two triplicates. After different incubation times depending on the experimental settings, 10 µl of a water-soluble tetrazolium (WST-1) salt solution (Sigma-Aldrich, München, Germany) were added to each well and cells were incubated at 37°C for a designated period. Quantification of the formazan dye produced by viable cells was performed by measuring the absorbance at 450 nm in a microplate reader (Agilent BioTek, Santa Clara, CA, USA).
4.11. Growth Kinetic
To determine growth kinetics in a 24-well plate format, 3 x 105 cells were seeded in 500 µl DMEM medium (PAN-Biotech, Aidenbach, Germany) with supplements in triplicates and the number of vital cells was counted manually every 24 hours (5 time points: 0 h, 24 h, 48 h, 72 h and 96 h) in a Neubauer chamber using the trypan blue exclusion method.
4.12. Cell Proliferation and Apoptosis Detection
Cell proliferation was determined by 5-Bromo-2´-deoxyuridine (BrdU; Sigma, Hamburg, Germany) incorporation. For BrdU immunochemistry, 4 h prior to PFA fixation 5 µM BrdU was added to the cells. Afterwards, cells were permeabilized and incubated with a rat anti-BrdU antibody (1:1,000; ab6326; Abcam, Cambridge, UK) and the BrdU signal was visualized using a goat anti-rat secondary antibody labelled with Alexa Fluor 594 (1:1,000; Molecular Probes, Eugene, USA). For each experiment, five coverslips were stained and the percentages of proliferating cells were calculated by setting BrdU-positive cells in relation to the total amount of DAPI-positive cells.
4.13. Caspase Dependent Apoptosis
In order to inhibit endogenous caspase activity, the broad spectrum caspase inhibitor Boc-D-Fmk (Calbiochem, Dramstadt, Germnay) was used. Cells were seeded on poly-D-lysin coated coverslips as described before and were cultured in the presence of 50 µl Boc-D-Fmk for 24 h. Afterwards the cells were fixed with 4% PFA and DAPI stained to determine the number of pycnotic nuclei. To analyze caspase 3 and 7 cleavage activity after PTPRE knockdown a caspase-Glo 3/7 assay (Promega, Madison, WI USA) was used. Therefore, 1.5 x 105 PTPRE depleted and control cells were seeded in 500 µl growth medium supplemented with 2% FBS in a 24-well plate format and incubated overnight. After 24 h 60 µl of each cell suspension were mixed with 60 µl of caspase-3/7 reagent and seeded in a white 96-well plate for 2 h at room temperature protected from light. Afterwards, luminescence was measured with an Orion II microplate luminometer (Berthold Detection Systems, Pforzheim, Germany) following the manufacturer´s instructions. Measurements were performed three times in five replicates. Cleaved, activated caspase 3 was detected immunocytochemically using a rabbit monoclonal cleaved caspase 3 antibody (#9664; 5A1E; Cell Signalling) diluted 1:400 in PBS with 0.1% Triton™ X-100, 4% BSA and 1% NGS at 4°C overnight in a humidified chamber after 1 h PFA fixation and methanol permeabilisation. A goat anti-rabbit antibody, labelled with Alexa-fluor®594 (Molecular Probes, Eugene, USA), diluted 1:1000 in PBS with 1% BSA, was used to visualize cleaved caspase 3 positive cells via fluorescent microscopy with a Nikon Eclipse E600 microscope (Nikon, Düsseldorf, Germany).
4.14. Colony Formation and Soft Agarose Assay
Soft agarose assays were performed as described previously [43]. 5000 cells were seeded in 2 mL soft agarose in a six-well dish in triplicates and cultivated for 3 weeks. Colony formation capacity (%) was calculated by counting the number of colony-forming cells and viable single cells in six visual fields (10x magnification) in triplicates per assay. Colony size was measured by capturing images using a Nikon Eclipse TS2 microscope equipped with a digital camera and IC MEASURE 1.0 software (Nikon, Düsseldorf, Germany). For the determination of colony size, eight colonies per well were measured.
4.15. CAM Assays
In order to study the effects of PTPRE knockdown on tumor formation and migration capacity in vivo, PTPRE depleted etoposide resistant RB and control cells were inoculated on the extraembryonic chorioallantoic membrane (CAM) of chicken embryos on EDD10 mainly following the protocols published by Zijlstra and Palmer [46,47]. Ten eggs were inoculated with 1x106 cells suspended in 50 µl PBS in at least three independent experiments. Seven days after grafting, at EDD17, grown tumors were excised, measured, weighted and photographed as described previously [44].
Intravenous injection of GFP-labeled Y79_Etop and WERI_Etop control and PTPRE knockdown cells was carried out on EDD12 as described previously by our group [44]. Five days after injection (EDD17) the chicken embryos were sacrificed and six punches of the ventral CAM, opposing the injection site, were collected and processed as described previously [48] [47]. Successful migration after injection was monitored by the identification of the GFP-labelled cells via fluorescence microscopy of the CAM punches. RNA isolations and quantification of hGAPDH of the pooled CAM punches tissue were performed as described previously [44].
Wholemount immunofluorescent staining of CAM vessels was carried out as described previously [49]. The first antibody detecting chicken desmin (D33; ab8470, Abcam, Cambridge, UK) was diluted 1:20 in PBS with 3% BSA and incubated in a humidified chamber at 4°C overnight. Alexa-fluor®594 goat anti mouse IgG (Molecular Probes, Eugene, USA) was diluted 1:1000 in 300 µl of PBS and CAM punches were incubated in a 24-well plate format on a shaker overnight at 4°C. Subsequent fluorescence microscopy was carried out with a Nikon ECLIPSE E600 microscope and NIS Elements Imaging 5.20.02 software (Nikon, Düsseldorf, Germany).
4.16. Immunohistochemistry
For PTPRE immunohistochemical localization in formalin fixed, paraffin embedded retinoblastoma and to immunohistochemically localize GFP labeled cells in fixed and embedded CAM tumors, sample sections were deparaffinized and steamed for 1h in EDTA buffer (pH 9 for PTPRE and GFP) to improve antigen retrieval. Sections were analyzed as previously described [50] following the manufacturers description of the Vectastain Elite ABC kit (Biozol, Eching, Germany). Immunostaining was performed using a rabbit monoclonal antibody against PTPRE (#PA5-82543; Invitrogen, Thermofisher Scientific) and GFP (2955S; Cell Signaling Technology, Danvers, USA) at a dilution of 1:200 (PTPRE and GFP) in antibody dilution buffer provided in the Vectastain kit (Biozol, Eching, Germany) at 4°C overnight in a humidified chamber. As controls, in all cases, antibody dilution buffer was substituted for the primary antisera to test for nonspecific labeling. No specific cellular staining was observed when the primary antiserum was omitted. Images were acquired by a slide scanner (Leica, Wetzlar, Germany) and viewed using the Aperio Image Scope Software (Leica).
4.17. Statistical Analysis
All assays were performed at least in triplicates. Statistical analyses were performed using GraphPad Prism 9. Data represent means ± SEM of three independent experiments from independent RB cell cultures. Results were analyzed by a Student`s t-test and considered significantly different if p-value <0.05 (*), p-value <0.01 (**), p-value <0.001 (***) or p-value <0.0001 (****). Statistics on the growth curves was performed using a free web interface http://bioinf.wehi.edu.au/software/compareCurves/, which uses the “compare growth curves” function from a statistical modeling package called statmod, available from the “R Project for Statistical Computing”: http://www.r-project.org, previously described elsewhere [51].
Author Contributions
Conceptualization, MAB, ND; Methodology LM, AD, NM, OD, MAB; Formal Analysis LM, AD, NM, MAB; Investigation, LM, AD, NM, OD; Data Curation, MAB and ND; Writing—Original Draft Preparation, LM, AD, ND, MAB; Writing—Review and Editing, ND and MAB; Visualization, LM, AD, MAB; Supervision, ND and MAB; Project Administration, ND.
Funding
The research was funded by the ELAN Promotionskolleg of the Medical Faculty of the University Duisburg-Essen and Stiftung Universitätsmedizin Essen. We also acknowledge the support by the Open Access Publication Fund of the University of Duisburg-Essen.
Institutional Review Board Statement
The use of retinoblastoma samples (ethic approval # 14-5836-BO) and human retina (ethic approval # 06-30214) for research conducted in the course of the study presented was approved of by the Ethics Committee of the Medical Faculty of the University of Duisburg-Essen.
Informed Consent Statement
Written informed consent has been obtained from patients` relatives or parents.
Acknowledgments
The authors would like to thank Dr. H. Hanenberg for the lentiviral vectors and Dr. H. Stephan for providing the RB cell lines and the Institute for Pathology for valuable support. We also would like to thank A. Bollmeier for excellent technical assistance and D. Gioè for valuable help with the cell counts.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kaewkhaw, R.; Rojanaporn, D. Retinoblastoma: Etiology, Modeling, and Treatment. Cancers 2020, 12. [Google Scholar] [CrossRef]
- Bornfeld, N.; Lohmann, D.; Bechrakis, N.E.; Biewald, E. Retinoblastom. Der Ophthalmologe : Zeitschrift der Deutschen Ophthalmologischen Gesellschaft 2020, 117, 389–402. [Google Scholar] [CrossRef]
- Munier, F.L.; Beck-Popovic, M.; Chantada, G.L.; Cobrinik, D.; Kivelä, T.T.; Lohmann, D.; Maeder, P.; Moll, A.C.; Carcaboso, A.M.; Moulin, A.; et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. "Alive, with good vision and no comorbidity". Progress in retinal and eye research 2019, 73, 100764. [Google Scholar] [CrossRef]
- Berry, J.L.; Kogachi, K.; Aziz, H.A.; McGovern, K.; Zolfaghari, E.; Murphree, A.L.; Jubran, R.; Kim, J.W. Risk of metastasis and orbital recurrence in advanced retinoblastoma eyes treated with systemic chemoreduction versus primary enucleation. Pediatric blood & cancer 2017, 64. [Google Scholar]
- Shields, C.L.; Lally, S.E.; Leahey, A.M.; Jabbour, P.M.; Caywood, E.H.; Schwendeman, R.; Shields, J.A. Targeted retinoblastoma management: when to use intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Current opinion in ophthalmology 2014, 25, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Temming, P.; Arendt, M.; Viehmann, A.; Eisele, L.; Le Guin, C.H.D.; Schündeln, M.M.; Biewald, E.; Astrahantseff, K.; Wieland, R.; Bornfeld, N.; et al. Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatric blood & cancer 2017, 64, 71–80. [Google Scholar]
- Du, Y.; Grandis, J.R. Receptor-type protein tyrosine phosphatases in cancer. Chinese journal of cancer 2015, 34, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Hale, A.J.; Steege, E. ter; Hertog, J. den. Recent advances in understanding the role of protein-tyrosine phosphatases in development and disease. Developmental biology 2017, 428, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Berman-Golan, D.; Granot-Attas, S.; Elson, A. Protein tyrosine phosphatase epsilon and Neu-induced mammary tumorigenesis. Cancer metastasis reviews 2008, 27, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shi, J.; Wang, N.; Zhao, H.; Sun, J. Tuning the Protein Phosphorylation by Receptor Type Protein Tyrosine Phosphatase Epsilon (PTPRE) in Normal and Cancer Cells. Journal of Cancer 2019, 10, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Frankson, R.; Yu, Z.-H.; Bai, Y.; Li, Q.; Zhang, R.-Y.; Zhang, Z.-Y. Therapeutic Targeting of Oncogenic Tyrosine Phosphatases. Cancer research 2017, 77, 5701–5705. [Google Scholar] [CrossRef] [PubMed]
- Bollu, L.R.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2017, 23, 2136–2142. [Google Scholar] [CrossRef]
- Kabir, N.N.; Rönnstrand, L.; Kazi, J.U. Deregulation of protein phosphatase expression in acute myeloid leukemia. Medical oncology (Northwood, London, England) 2013, 30, 517. [Google Scholar] [CrossRef] [PubMed]
- LACZMANSKA, I.; LACZMANSKI, L.; SASIADEK, M.M. Expression Analysis of Tyrosine Phosphatase Genes at Different Stages of Renal Cell Carcinoma. Anticancer Res 2020, 40, 5667–5671. [Google Scholar] [CrossRef]
- Gan, S.; Ye, R.; Ha, Y.; Xiong, Y.; Li, R.; Di, X.; Zou, Z.; Sun, Y.; Zhang, Z. Prediction Biomarkers Associated with Lymph Node Metastasis and Prognosis were Identified in Papillary Thyroid Carcinoma via Integrated Bioinformatics Analysis. Combinatorial chemistry & high throughput screening 2021, 24, 1395–1409. [Google Scholar]
- Kober, P.; Boresowicz, J.; Rusetska, N.; Maksymowicz, M.; Goryca, K.; Kunicki, J.; Bonicki, W.; Siedlecki, J.A.; Bujko, M. DNA methylation profiling in nonfunctioning pituitary adenomas. Molecular and cellular endocrinology 2018, 473, 194–204. [Google Scholar] [CrossRef]
- Chen, B.; Liao, Z.; Qi, Y.; Zhang, H.; Su, C.; Liang, H.; Zhang, B.; Chen, X. miR-631 Inhibits Intrahepatic Metastasis of Hepatocellular Carcinoma by Targeting PTPRE. Frontiers in oncology 2020, 10, 565266. [Google Scholar] [CrossRef]
- Nunes-Xavier, C.E.; Elson, A.; Pulido, R. Epidermal growth factor receptor (EGFR)-mediated positive feedback of protein-tyrosine phosphatase epsilon (PTPepsilon) on ERK1/2 and AKT protein pathways is required for survival of human breast cancer cells. The Journal of biological chemistry 2012, 287, 3433–3444. [Google Scholar] [CrossRef]
- Gil-Henn, H.; Elson, A. Tyrosine phosphatase-epsilon activates Src and supports the transformed phenotype of Neu-induced mammary tumor cells. The Journal of biological chemistry 2003, 278, 15579–15586. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Yamada, N.; Shimizu, H.; Shiota, M.; Tamura, M.; Kim-Mitsuyama, S.; Miyazaki, H. Tyrosine phosphatase epsilonM stimulates migration and survival of porcine aortic endothelial cells by activating c-Src. Biochemical and biophysical research communications 2004, 325, 314–319. [Google Scholar] [CrossRef]
- Peretz, A.; Gil-Henn, H.; Sobko, A.; Shinder, V.; Attali, B.; Elson, A. Hypomyelination and increased activity of voltage-gated K(+) channels in mice lacking protein tyrosine phosphatase epsilon. The EMBO journal 2000, 19, 4036–4045. [Google Scholar] [CrossRef]
- Siffroi-Fernandez, S. Acidic Fibroblast Growth Factor (FGF-1) and FGF Receptor 1 Signaling in Human Y79 Retinoblastoma. Archives of ophthalmology (Chicago, Ill. : 1960) 2005, 123, 368. [Google Scholar] [CrossRef] [PubMed]
- Wabakken, T.; Hauge, H.; Finne, E.F.; Wiedlocha, A.; Aasheim, H. Expression of human protein tyrosine phosphatase epsilon in leucocytes: a potential ERK pathway-regulating phosphatase. Scandinavian journal of immunology 2002, 56, 195–203. [Google Scholar] [CrossRef]
- Bruhn, M.A.; Pearson, R.B.; Hannan, R.D.; Sheppard, K.E. Second AKT: the rise of SGK in cancer signalling. Growth factors (Chur, Switzerland) 2010, 28, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Xu, Z.; Wang, J.; Cigliano, A.; Pilo, M.G.; Ribback, S.; Zhang, S.; Qiao, Y.; Che, L.; Pascale, R.M.; et al. Functional role of SGK3 in PI3K/Pten driven liver tumor development. BMC cancer 2019, 19, 343. [Google Scholar] [CrossRef] [PubMed]
- Raji, L.; Tetteh, A. ; Amin, A R M Ruhul. Role of c-Src in Carcinogenesis and Drug Resistance. Cancers 2023, 16. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal transduction and targeted therapy 2023, 8, 455. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Molecular cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Song, C.; Lu, Z.; Lai, K.; Li, D.; Hao, B.; Xu, C.; Pan, S.; Li, N.; Geng, Q. Identification of an inflammatory response signature associated with prognostic stratification and drug sensitivity in lung adenocarcinoma. Scientific reports 2022, 12, 10110. [Google Scholar] [CrossRef]
- Peng, C.; Zhang, C.; Yu, W.; Le Li; Zhang, Z. ; Liu, T.; Zhang, Y.; Fan, G.; Huangfu, H. Receptor Type Protein Tyrosine Phosphatase Epsilon (PTPRE) Plays an Oncogenic Role in Thyroid Carcinoma by Activating the AKT and ERK1/2 Signaling Pathway. Current cancer drug targets 2023, 23, 471–481. [Google Scholar] [CrossRef]
- Fu, D.; Liu, B.; Zang, L.E.; Jiang, H. MiR-631/ZAP70: A novel axis in the migration and invasion of prostate cancer cells. Biochemical and biophysical research communications 2016, 469, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Yu, J.; Zhang, H.; Qian, X.; Wang, Q.; Lu, B.; Sun, Y. MicroRNA-631 deriving from bone marrow mesenchymal stem cell exosomes facilitates the malignant behavior of non-small cell lung cancer via modulating the E2F family of transcription factor 2/phosphatidylinositol 3-kinase/Akt signaling pathway. Bioengineered 2022, 13, 8382–8395. [Google Scholar] [CrossRef] [PubMed]
- Najafi, F.; Kelaye, S.K.; Kazemi, B.; Foruzandeh, Z.; Allahverdizadeh, F.; Vakili, S.; Rad, K.K.; Derakhshani, M.; Solali, S.; Alivand, M.R. The role of miRNA-424 and miR-631 in various cancers: Focusing on drug resistance and sensitivity. Pathology, research and practice 2022, 239, 154130. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, F.; Turcotte, S.; Roy, J.; Bissonnette, E.; Rola-Pleszczynski, M.; Stankova, J. RPTPε promotes M2-polarized macrophage migration through ROCK2 signaling and podosome formation. Journal of cell science 2020, 133. [Google Scholar] [CrossRef]
- Liao, Z.; Chen, L.; Zhang, X.; Zhang, H.; Tan, X.; Dong, K.; Lu, X.; Zhu, H.; Liu, Q.; Zhang, Z.; et al. PTPRε Acts as a Metastatic Promoter in Hepatocellular Carcinoma by Facilitating Recruitment of SMAD3 to TGF-β Receptor 1. Hepatology (Baltimore, Md.) 2020, 72, 997–1012. [Google Scholar] [CrossRef]
- Xie, C.; Freeman, M.J.; Lu, H.; Wang, X.; Forster, C.L.; Sarver, A.L.; Hallstrom, T.C. Retinoblastoma cells activate the AKT pathway and are vulnerable to the PI3K/mTOR inhibitor NVP-BEZ235. Oncotarget 2017, 8, 38084–38098. [Google Scholar] [CrossRef]
- Bruhn, M.A.; Pearson, R.B.; Hannan, R.D.; Sheppard, K.E. AKT-independent PI3-K signaling in cancer - emerging role for SGK3. Cancer management and research 2013, 5, 281–292. [Google Scholar] [PubMed]
- Griegel, S.; Hong, C.; Frötschl, R.; Hülser, D.F.; Greger, V.; Horsthemke, B.; Rajewsky, M.F. Newly established human retinoblastoma cell lines exhibit an "immortalized" but not an invasive phenotype in vitro. International journal of cancer 1990, 46, 125–132. [Google Scholar] [CrossRef]
- Reid TW, Albert DM, Rabson AS, Russell P, Craft J, Chu EW, Tralka TS, Wilcox JL. Characteristics of an established cell line of retinoblastoma. Characteristics of an established cell line of retinoblastoma. J Nat Cancer Inst 1974, 347–360. [Google Scholar]
- McFall, R.C.; Sery, T.W.; Makadon, M. Characterization of a new continuous cell line derived from a human retinoblastoma. Cancer research 1977, 37, 1003–1010. [Google Scholar]
- Busch, M.; Papior, D.; Stephan, H.; D�nker, N. Characterization of etoposide- and cisplatin-chemoresistant retinoblastoma cell lines. Oncol Rep 2017. [CrossRef]
- Hartmann, L.; Neveling, K.; Borkens, S.; Schneider, H.; Freund, M.; Grassman, E.; Theiss, S.; Wawer, A.; Burdach, S.; Auerbach, A.D.; et al. Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. American journal of human genetics 2010, 87, 480–493. [Google Scholar] [CrossRef]
- Große-Kreul, J.; Busch, M.; Winter, C.; Pikos, S.; Stephan, H.; Dünker, N. Forced Trefoil Factor Family Peptide 3 (TFF3) Expression Reduces Growth, Viability, and Tumorigenicity of Human Retinoblastoma Cell Lines. PloS one 2016, 11, e0163025. [Google Scholar] [CrossRef]
- Busch, M.; Philippeit, C.; Weise, A.; Dünker, N. Re-characterization of established human retinoblastoma cell lines. Histochemistry and cell biology 2015, 143, 325–338. [Google Scholar] [CrossRef]
- Weise, A.; Dünker, N. High trefoil factor 1 (TFF1) expression in human retinoblastoma cells correlates with low growth kinetics, increased cyclin-dependent kinase (CDK) inhibitor levels and a selective down-regulation of CDK6. Histochemistry and cell biology 2013, 139, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, A.; Mellor, R.; Panzarella, G.; Aimes, R.T.; Hooper, J.D.; Marchenko, N.D.; Quigley, J.P. A quantitative analysis of rate-limiting steps in the metastatic cascade using human-specific real-time polymerase chain reaction. Cancer research 2002, 62, 7083–7092. [Google Scholar] [PubMed]
- Palmer, T.D.; Lewis, J.; Zijlstra, A. Quantitative analysis of cancer metastasis using an avian embryo model. Journal of visualized experiments : JoVE 2011. [Google Scholar] [CrossRef]
- Kim, J.; Yu, W.; Kovalski, K.; Ossowski, L. Requirement for Specific Proteases in Cancer Cell Intravasation as Revealed by a Novel Semiquantitative PCR-Based Assay. Cell 1998, 94, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Dräger, O.; Metz, K.; Busch, M.; Dünker, N. Role of L1CAM in retinoblastoma tumorigenesis: identification of novel therapeutic targets. Molecular oncology 2022, 16, 957–981. [Google Scholar] [CrossRef] [PubMed]
- van Meenen, D.; Doege, A.; Alefeld, E.; Haase, A.; Beier, M.; Kiefer, T.; Biewald, E.; Metz, K.; Dräger, O.; Busch, M.A.; et al. ADAM10 and ADAM17-Novel Players in Retinoblastoma Carcinogenesis. International journal of molecular sciences 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Elso, C.M.; Roberts, L.J.; Smyth, G.K.; Thomson, R.J.; Baldwin, T.M.; Foote, S.J.; Handman, E. Leishmaniasis host response loci (lmr1-3) modify disease severity through a Th1/Th2-independent pathway. Genes and immunity 2004, 5, 93–100. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Endogenous PTPRE expression in chemosensitive (a) and etoposide resistant (Etop) retinoblastoma cell lines (b) and RB patient tumor specimen (c) as revealed by Western blot (a, b) analyses and quantitative Real-time PCR (c). ctr: chemosensitive control cells. Indicated intensity ratios relative to ß-actin, used as a loading control (a, b), were calculated using Micro Manager 1.4 software. (c) PTPRE expression levels in enucleated RB patient eyes as compared to the healthy human retina (hRet). Values are means of at least three independent experiments ± SEM. ns p>0.05; *p<0.05; ***p<0.001; ****p<0.0001 statistical differences compared to the control group calculated by Student´s t-test.
Figure 1.
Endogenous PTPRE expression in chemosensitive (a) and etoposide resistant (Etop) retinoblastoma cell lines (b) and RB patient tumor specimen (c) as revealed by Western blot (a, b) analyses and quantitative Real-time PCR (c). ctr: chemosensitive control cells. Indicated intensity ratios relative to ß-actin, used as a loading control (a, b), were calculated using Micro Manager 1.4 software. (c) PTPRE expression levels in enucleated RB patient eyes as compared to the healthy human retina (hRet). Values are means of at least three independent experiments ± SEM. ns p>0.05; *p<0.05; ***p<0.001; ****p<0.0001 statistical differences compared to the control group calculated by Student´s t-test.
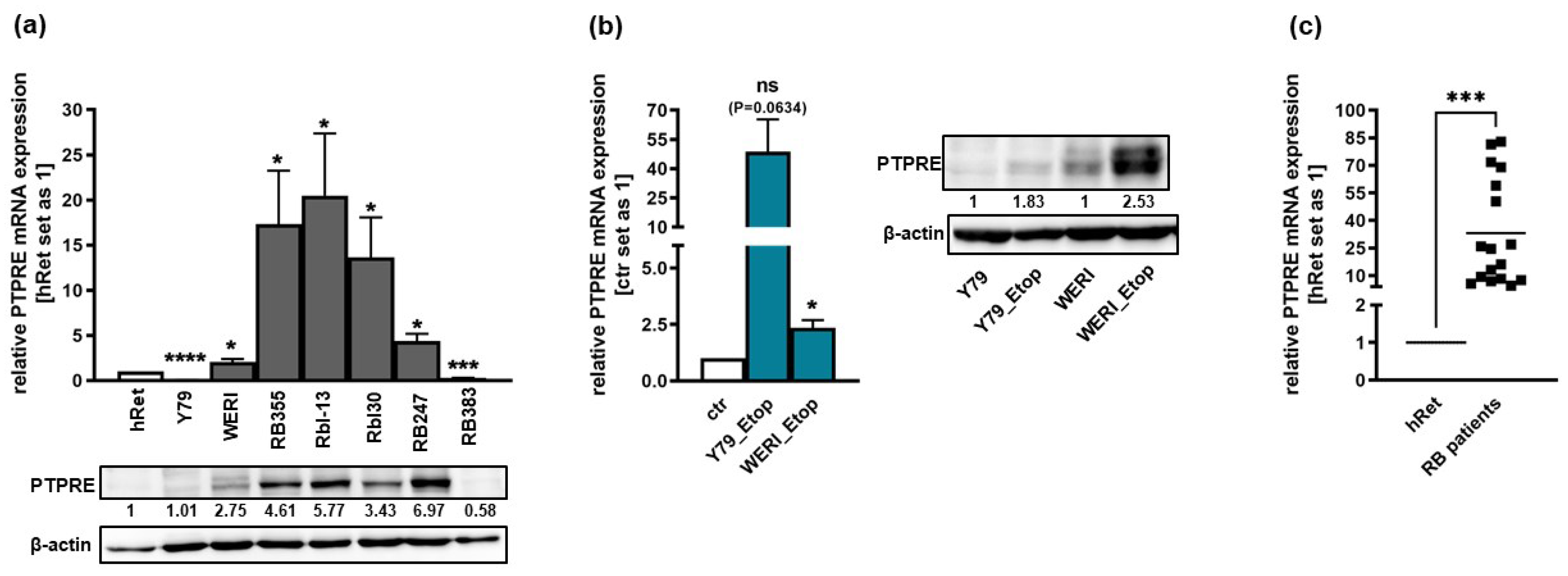
Figure 2.
PTPRE expression in a primary RB patient tumor. (a) Immunohistochemical analysis of PTPRE expression (brown signal) in an exemplary paraffin section of a human RB patient tumor counterstained with hematoxylin (blue nuclei staining). (b) Corresponding hematoxylin and eosine (H&E) stain. Scale bars, 2mm and 300 µm (magnified insets).
Figure 2.
PTPRE expression in a primary RB patient tumor. (a) Immunohistochemical analysis of PTPRE expression (brown signal) in an exemplary paraffin section of a human RB patient tumor counterstained with hematoxylin (blue nuclei staining). (b) Corresponding hematoxylin and eosine (H&E) stain. Scale bars, 2mm and 300 µm (magnified insets).
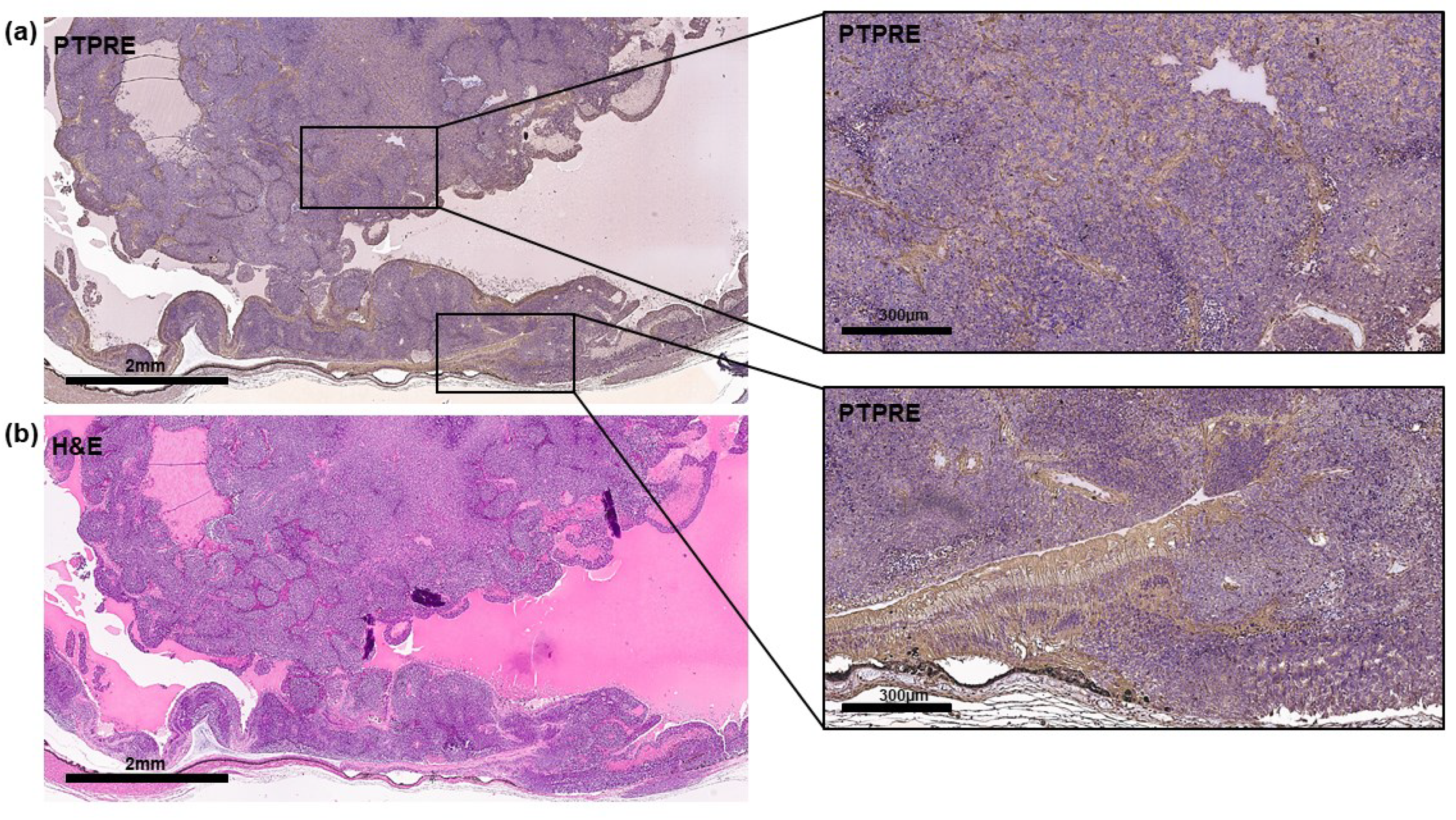
Figure 3.
Genomic bisulfite sequencing of the PTPRE promoter associated CpG sites in etoposide resistant (Etop) and chemosensitive retinoblastoma cell lines. Each circle represents a single CpG site. The average methylation of all CpG dinucleotides analyzed is indicated based on sequencing of at least ten clones. Nearly all CpGs in the PTPRE promoter of the analyzed RB cell lines were demethylated.
Figure 3.
Genomic bisulfite sequencing of the PTPRE promoter associated CpG sites in etoposide resistant (Etop) and chemosensitive retinoblastoma cell lines. Each circle represents a single CpG site. The average methylation of all CpG dinucleotides analyzed is indicated based on sequencing of at least ten clones. Nearly all CpGs in the PTPRE promoter of the analyzed RB cell lines were demethylated.

Figure 4.
MiR631 expression in RB patient tumors (a) and etoposide resistant (Etop) Y79 and WERI cell lines (b) as revealed by Real-time PCR and effects of miR361 overexpression (c) on PTPRE protein expression (c) proliferation (d) and apoptosis levels (e) of Y79_Etop and WERI_Etop cells. Values are means of at least three independent experiments ± SEM. ns p> 0.05; *p< 0.05; **p< 0.01; and ****p< 0.0001 statistical differences compared to the control group calculated by Student’s t-test.
Figure 4.
MiR631 expression in RB patient tumors (a) and etoposide resistant (Etop) Y79 and WERI cell lines (b) as revealed by Real-time PCR and effects of miR361 overexpression (c) on PTPRE protein expression (c) proliferation (d) and apoptosis levels (e) of Y79_Etop and WERI_Etop cells. Values are means of at least three independent experiments ± SEM. ns p> 0.05; *p< 0.05; **p< 0.01; and ****p< 0.0001 statistical differences compared to the control group calculated by Student’s t-test.
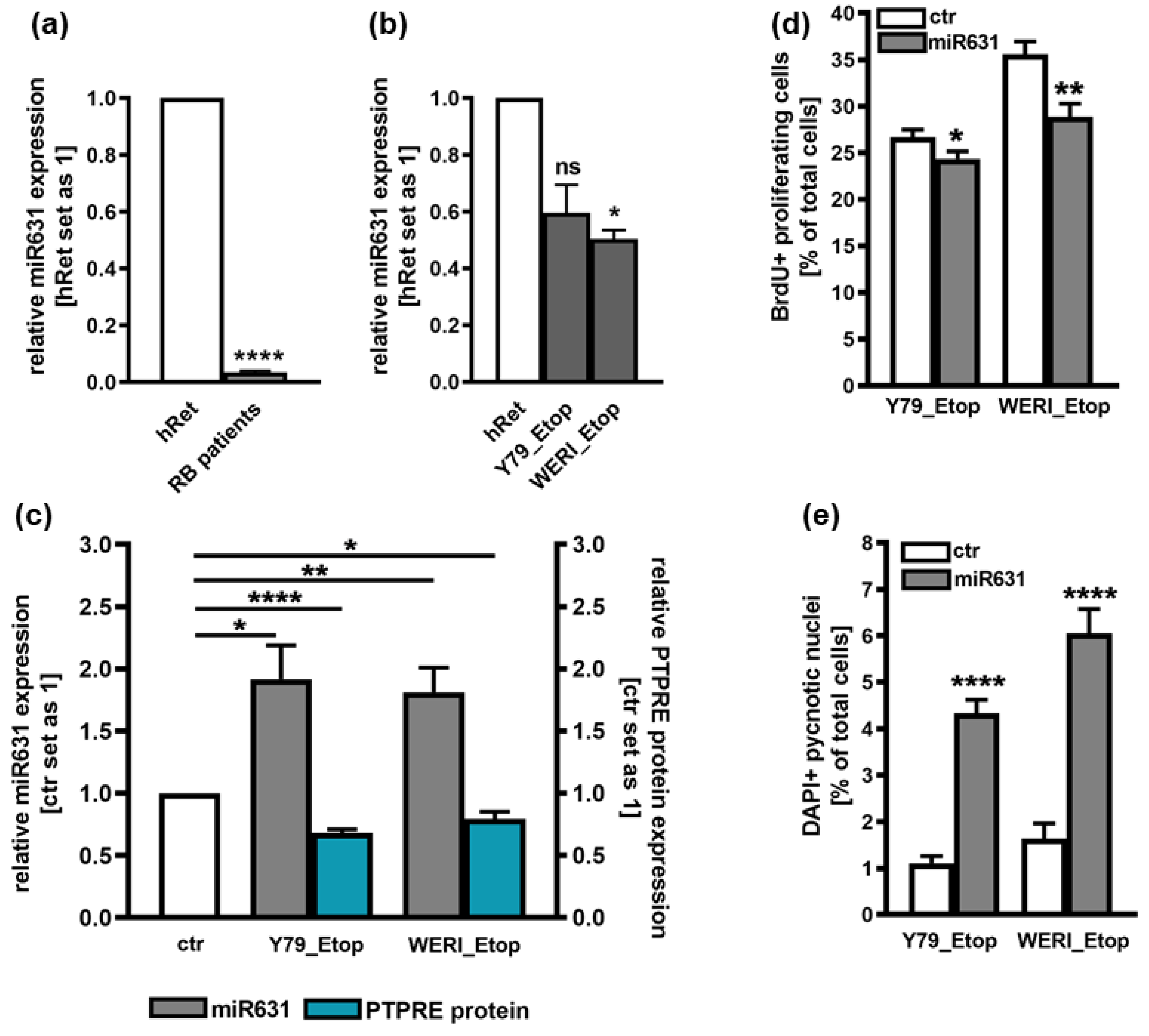
Figure 5.
Effects of FGFR inhibition on cell viability and apoptosis of the etoposide resistant RB cell lines Y79_Etop and WERI_Etop as revealed by WST-1 assays (a) and trypane blue cell counting (b). FGFR inhibition decreases cell viability and increases apoptosis of Y79_Etop and WERI_Etop cells compared to control cells (ctr). Values are means of three independent experiments ± SEM. ns p>0.05; *p< 0.05 statistical differences compared to the control (ctr) group calculated by Student’s t-test.
Figure 5.
Effects of FGFR inhibition on cell viability and apoptosis of the etoposide resistant RB cell lines Y79_Etop and WERI_Etop as revealed by WST-1 assays (a) and trypane blue cell counting (b). FGFR inhibition decreases cell viability and increases apoptosis of Y79_Etop and WERI_Etop cells compared to control cells (ctr). Values are means of three independent experiments ± SEM. ns p>0.05; *p< 0.05 statistical differences compared to the control (ctr) group calculated by Student’s t-test.
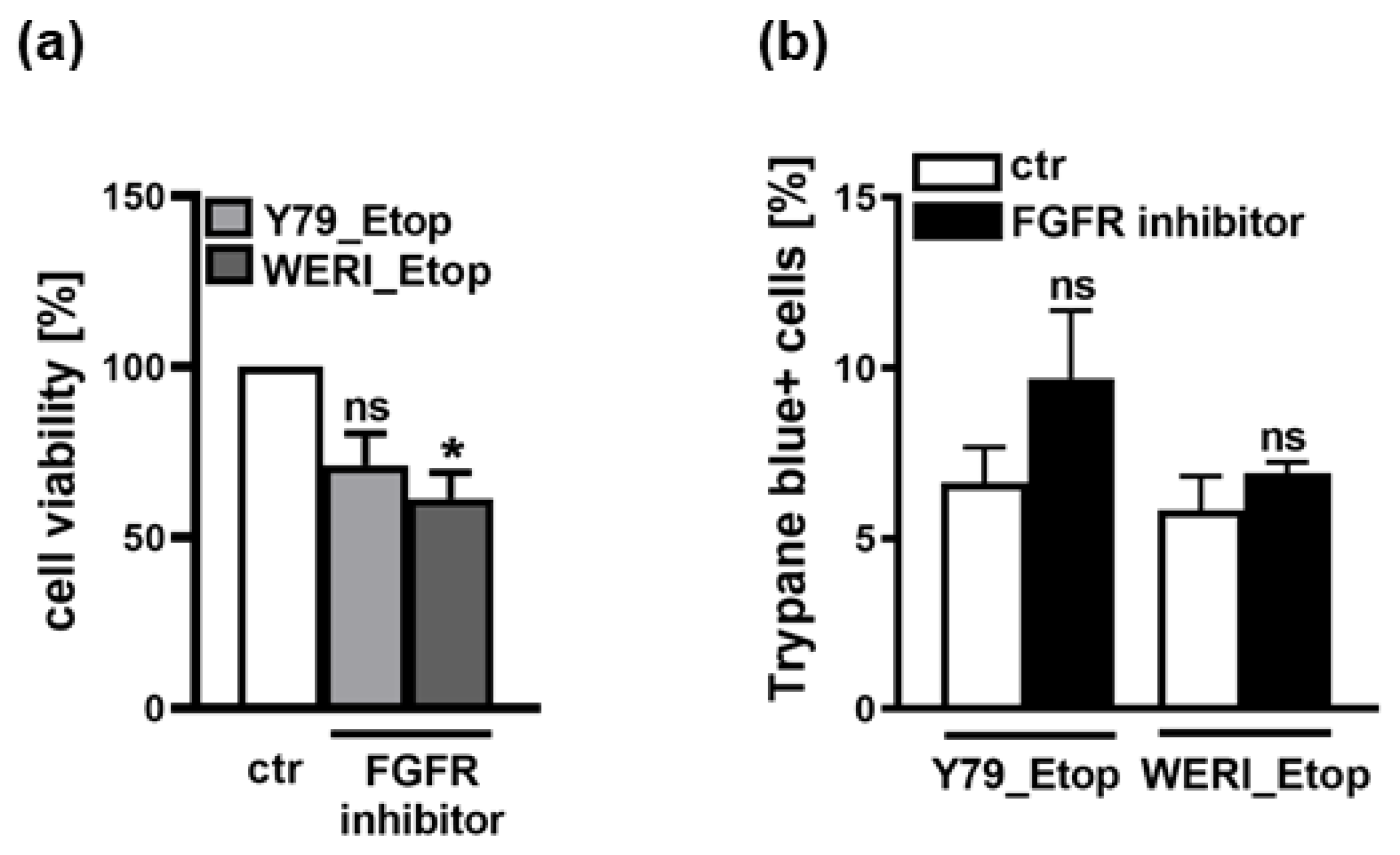
Figure 6.
Verification of PTPRE knockdown efficiency. Efficient stable, lentiviral PTPRE knockdown (KD) in etoposide resistant (Etop) Y79 and WERI cells was verified by quantitative Real-time PCR (a) and Western blot analyses (b). Indicated intensity ratios relative to ß-actin, used as a loading control, were calculated using Micro Manager 1.4 software. Values are means of three independent experiments ± SEM. ****p< 0.0001 statistical differences compared to the control (ctr) group calculated by Student’s t-test.
Figure 6.
Verification of PTPRE knockdown efficiency. Efficient stable, lentiviral PTPRE knockdown (KD) in etoposide resistant (Etop) Y79 and WERI cells was verified by quantitative Real-time PCR (a) and Western blot analyses (b). Indicated intensity ratios relative to ß-actin, used as a loading control, were calculated using Micro Manager 1.4 software. Values are means of three independent experiments ± SEM. ****p< 0.0001 statistical differences compared to the control (ctr) group calculated by Student’s t-test.
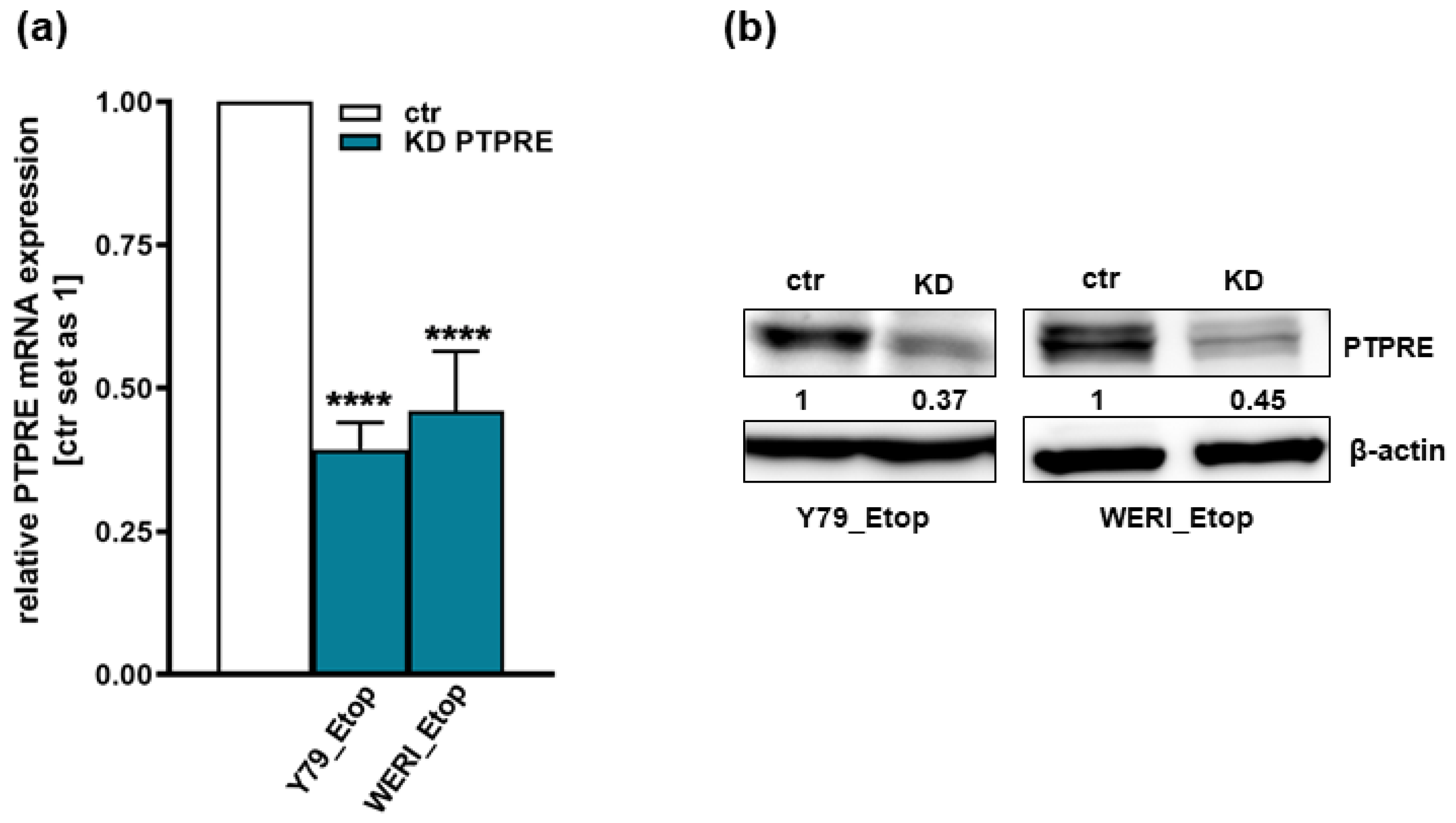
Figure 7.
Effects of PTPRE knockdown (KD) on cell viability, proliferation and growth of the etoposide resistant RB cell lines Y79_Etop and WERI_Etop as revealed by WST-1 assays (a), cell counts from BrdU stains (b) and growth curve analyses (c, d). Stable PTPRE knockdown decreases cell viability, proliferation levels and growth of Y79_Etop and WERI_Etop cells compared to control cells (ctr). Values are means of three independent experiments ± SEM. ns *p> 0.05; **p< 0.01; ***p< 0.001 and ****p< 0.0001 statistical differences compared to the control (ctr) group calculated by Student’s t-test.
Figure 7.
Effects of PTPRE knockdown (KD) on cell viability, proliferation and growth of the etoposide resistant RB cell lines Y79_Etop and WERI_Etop as revealed by WST-1 assays (a), cell counts from BrdU stains (b) and growth curve analyses (c, d). Stable PTPRE knockdown decreases cell viability, proliferation levels and growth of Y79_Etop and WERI_Etop cells compared to control cells (ctr). Values are means of three independent experiments ± SEM. ns *p> 0.05; **p< 0.01; ***p< 0.001 and ****p< 0.0001 statistical differences compared to the control (ctr) group calculated by Student’s t-test.
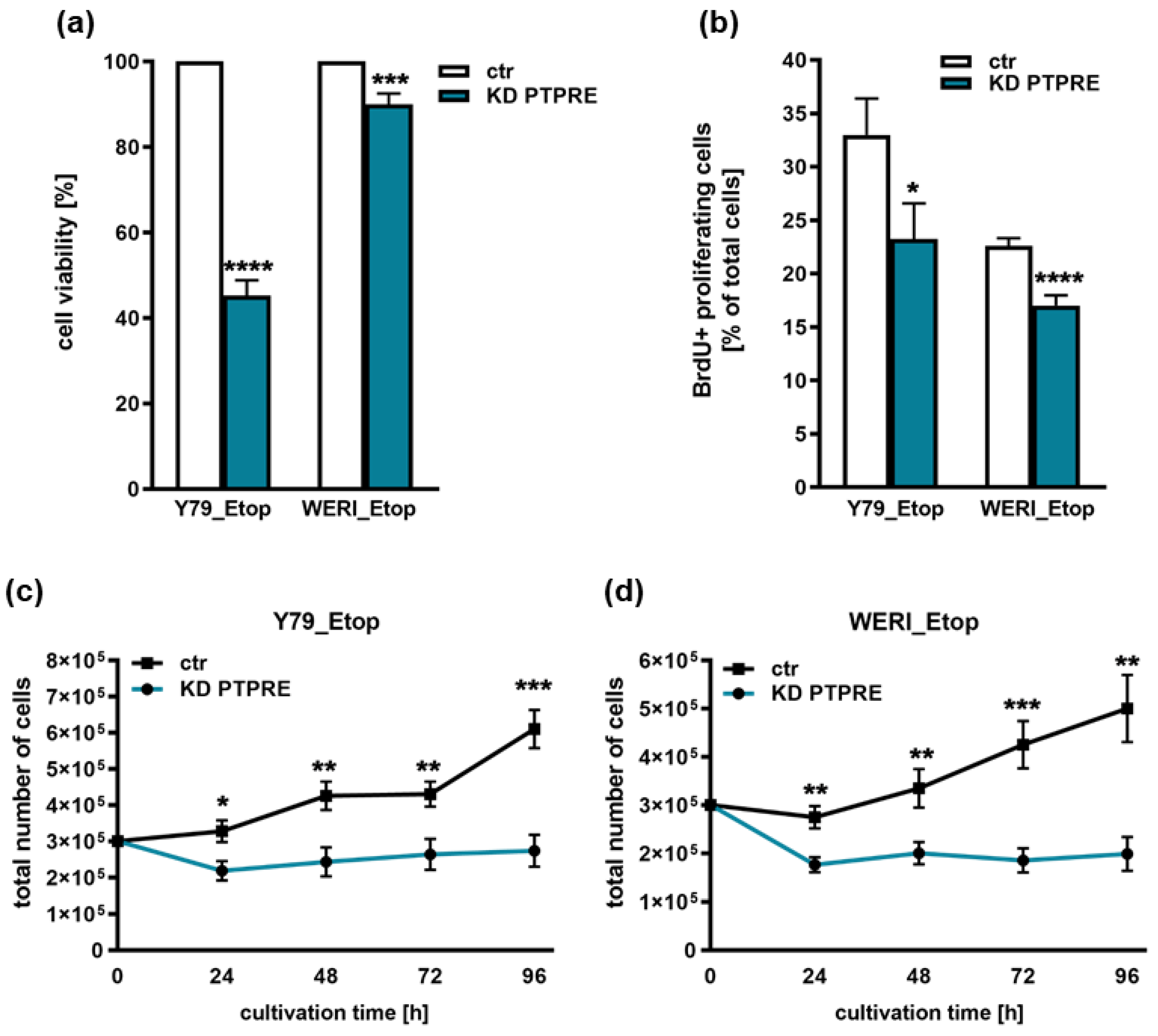
Figure 8.
Effects of PTPRE knockdown (KD) on apoptosis levels of etoposide resistant RB cell lines. PTPRE knockdown increases apoptosis levels of etoposide resistant Y79 and WERI cells compared to control cells (ctr) as revealed by DAPI cell counts after treatment with the broad spectrum caspase inhibitor Boc-D-fmk (a), caspase3/7 assays (b) and immunocytochemical stains against cleaved caspase 3 (red fluorescence with blue DAPI counterstaining; c) and quantification of cleaved caspase 3 positive cells (d). Values are means of three independent experiments ± SEM. ns > 0.05; *p< 0.05; ***p< 0.001 and ****p< 0.0001 statistical differences compared to the control (ctr) group calculated by Student’s t-test or oneway ANOVA with Newman–Keuls post-test.
Figure 8.
Effects of PTPRE knockdown (KD) on apoptosis levels of etoposide resistant RB cell lines. PTPRE knockdown increases apoptosis levels of etoposide resistant Y79 and WERI cells compared to control cells (ctr) as revealed by DAPI cell counts after treatment with the broad spectrum caspase inhibitor Boc-D-fmk (a), caspase3/7 assays (b) and immunocytochemical stains against cleaved caspase 3 (red fluorescence with blue DAPI counterstaining; c) and quantification of cleaved caspase 3 positive cells (d). Values are means of three independent experiments ± SEM. ns > 0.05; *p< 0.05; ***p< 0.001 and ****p< 0.0001 statistical differences compared to the control (ctr) group calculated by Student’s t-test or oneway ANOVA with Newman–Keuls post-test.
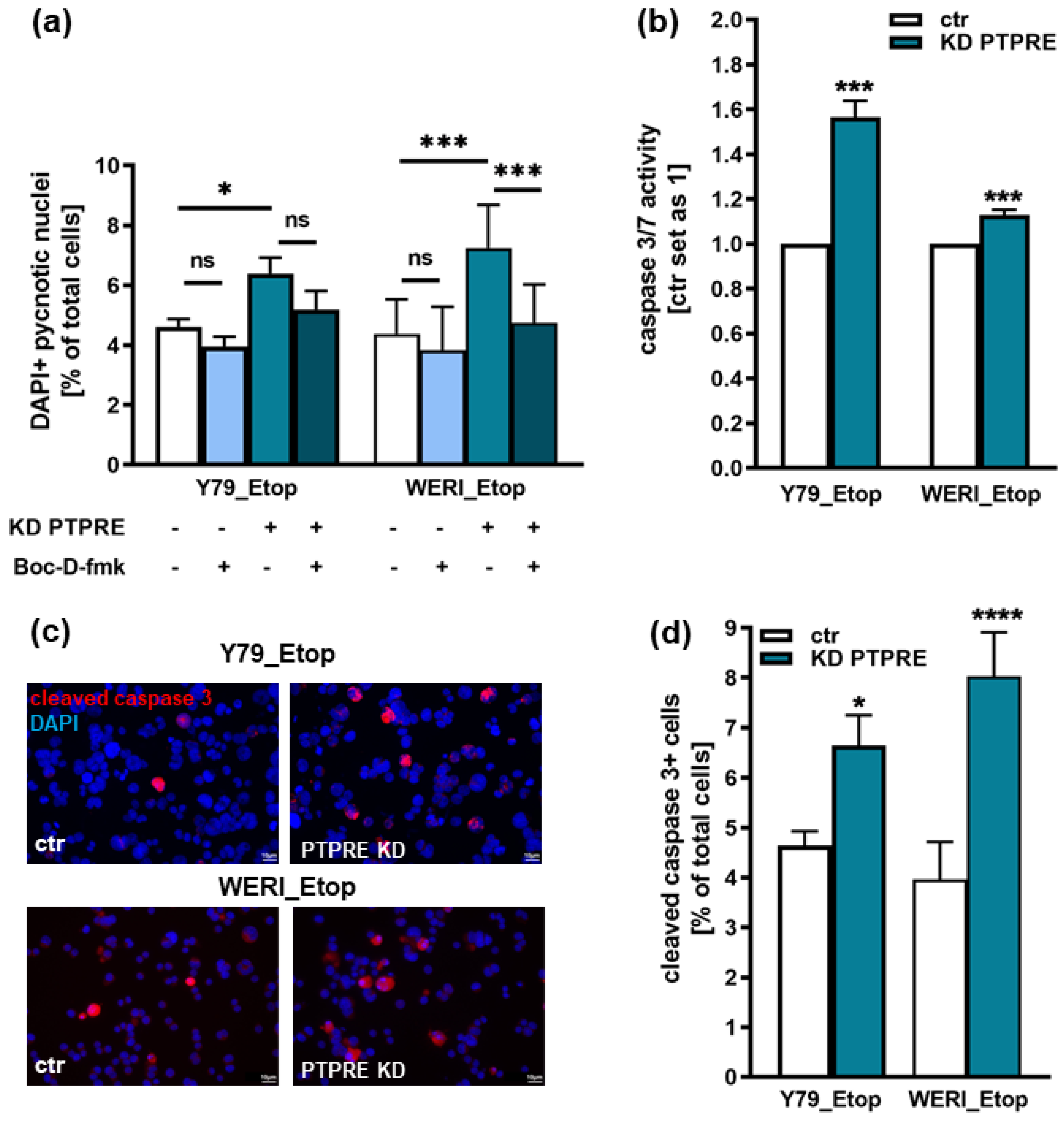
Figure 9.
Effects of PTPRE knockdown (KD) on anchorage independent growth of etoposide resistant Y79 and WERI RB cells. Both etoposide resistant (Etop) RB cell lines showed significantly reduced colony formation capacity (a) and colony sizes (b+c) after PTPRE knockdown as revealed by soft agarose assays. Values are means of three independent experiments ± SEM. ns > 0.05; **p< 0.01; ***p< 0.001 and ****p< 0.0001 statistical differences compared to the control (ctr) group calculated by Student’s t-test.
Figure 9.
Effects of PTPRE knockdown (KD) on anchorage independent growth of etoposide resistant Y79 and WERI RB cells. Both etoposide resistant (Etop) RB cell lines showed significantly reduced colony formation capacity (a) and colony sizes (b+c) after PTPRE knockdown as revealed by soft agarose assays. Values are means of three independent experiments ± SEM. ns > 0.05; **p< 0.01; ***p< 0.001 and ****p< 0.0001 statistical differences compared to the control (ctr) group calculated by Student’s t-test.
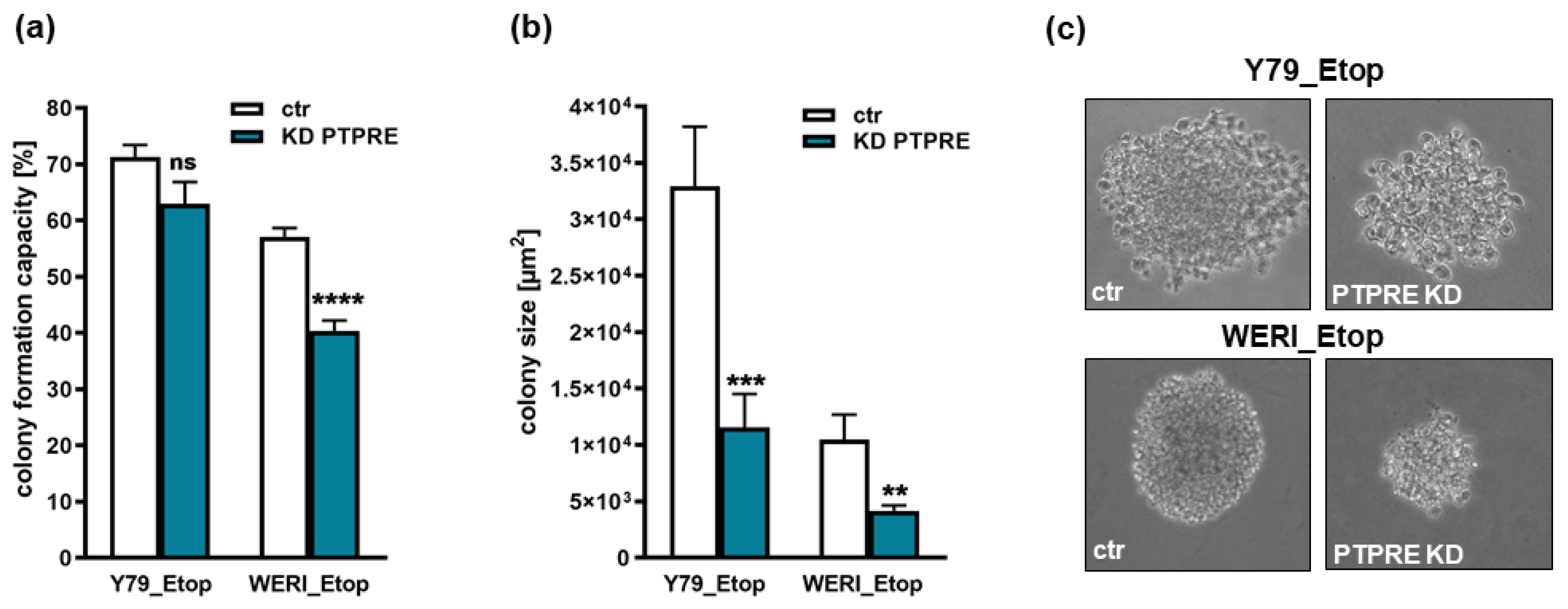
Figure 10.
Effect of lentiviral PTPRE knockdown (KD) on tumor formation of etoposide resistant (Etop) RB cell lines in vivo as revealed by chick chorioallantoic membrane (CAM) assays. Photographs of CAM tumors in situ and ruler measurements (in cm) of excised CAM tumors 7 days after inoculating the Y79_Etop (a) and WERI_Etop (a) RB cells onto the CAM. Quantification of weight (b) and size (c) of CAM tumors developing from PTPRE depleted Y79_Etop and WERI_Etop cell lines in comparison to the control cells (ctr). Values are means of at least three independent experiments ± SEM. ns p> 0.05; *p< 0.05; **p< 0.01 statistical differences compared to the control group calculated by Student’s t-test.
Figure 10.
Effect of lentiviral PTPRE knockdown (KD) on tumor formation of etoposide resistant (Etop) RB cell lines in vivo as revealed by chick chorioallantoic membrane (CAM) assays. Photographs of CAM tumors in situ and ruler measurements (in cm) of excised CAM tumors 7 days after inoculating the Y79_Etop (a) and WERI_Etop (a) RB cells onto the CAM. Quantification of weight (b) and size (c) of CAM tumors developing from PTPRE depleted Y79_Etop and WERI_Etop cell lines in comparison to the control cells (ctr). Values are means of at least three independent experiments ± SEM. ns p> 0.05; *p< 0.05; **p< 0.01 statistical differences compared to the control group calculated by Student’s t-test.
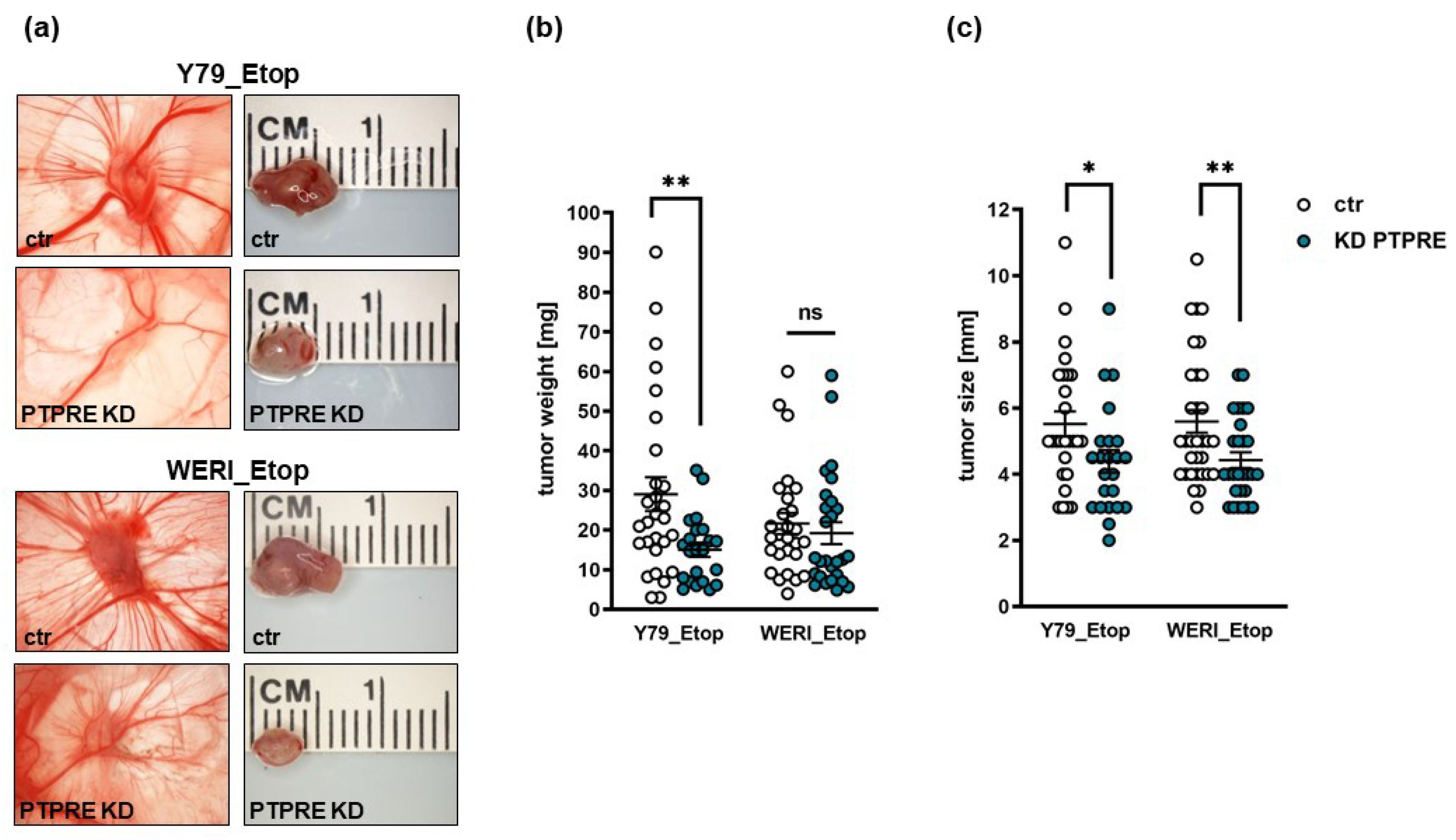
Figure 11.
Effect of PTPRE knockdown (KD) on etoposide re-sensitization of etoposide resistant RB cell lines. Compared to control cells, PTPRE depletion in etoposide (Etop) resistant Y79 and WERI cells induces significant changes in cell viability after etoposide treatment. Etoposide resistant RB cell lines were treated with 3 µmol/ml (Y79_Etop) or 5 µmol/ml (WERI_Etop) etoposide for 7 days prior to WST-1 assays. Values are means of at least three independent experiments ± SEM. **p< 0.01; ***p< 0.001 statistical differences compared to the control group calculated by Student’s t-test.
Figure 11.
Effect of PTPRE knockdown (KD) on etoposide re-sensitization of etoposide resistant RB cell lines. Compared to control cells, PTPRE depletion in etoposide (Etop) resistant Y79 and WERI cells induces significant changes in cell viability after etoposide treatment. Etoposide resistant RB cell lines were treated with 3 µmol/ml (Y79_Etop) or 5 µmol/ml (WERI_Etop) etoposide for 7 days prior to WST-1 assays. Values are means of at least three independent experiments ± SEM. **p< 0.01; ***p< 0.001 statistical differences compared to the control group calculated by Student’s t-test.
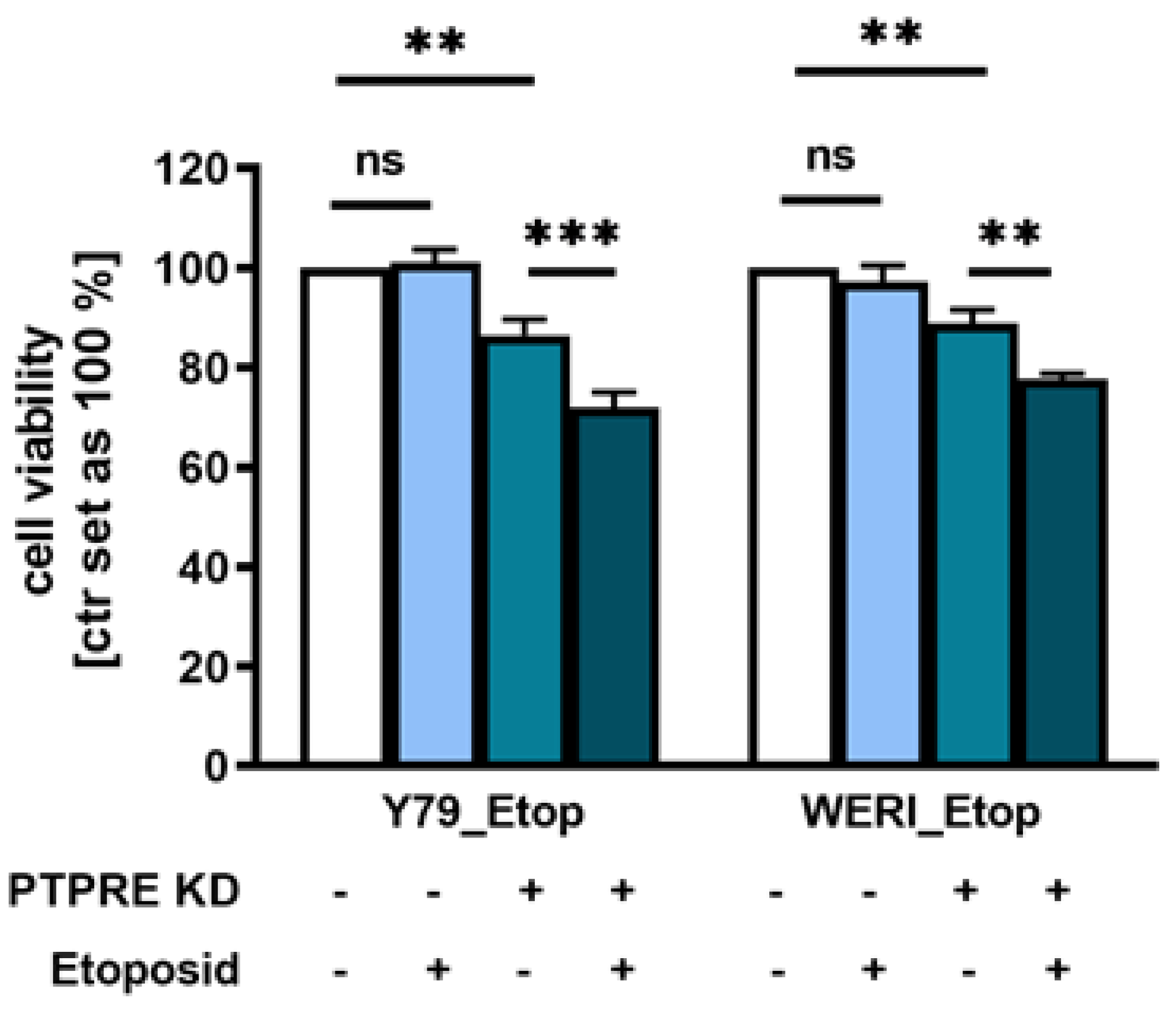
Figure 12.
SRC, ERK1/2, AKT and SGK3 as well as the respective phospho-proteins (pSRC, pERK1/2, pAKT and pSGK3) expression levels after PTPRE knockdown (KD) in the etoposide resistant (Etop) RB cell lines Y79 and WERI as revealed by Western blot analysis. (a) Representative Western blots showing the expression levels of the respective proteins upon PTPRE knockdown in the two RB cell lines. (b-e) Quantification of the phospho-protein/protein ratio of the investigated proteins after PTPRE knockdown in etoposide resistant Y79 and WERI RB cells. Values are means of three independent experiments ± SEM. ns p> 0.05; *p< 0.05; **p< 0.01 and ***p< 0.001; statistical differences compared to the control group calculated by paired Student’s t-test.
Figure 12.
SRC, ERK1/2, AKT and SGK3 as well as the respective phospho-proteins (pSRC, pERK1/2, pAKT and pSGK3) expression levels after PTPRE knockdown (KD) in the etoposide resistant (Etop) RB cell lines Y79 and WERI as revealed by Western blot analysis. (a) Representative Western blots showing the expression levels of the respective proteins upon PTPRE knockdown in the two RB cell lines. (b-e) Quantification of the phospho-protein/protein ratio of the investigated proteins after PTPRE knockdown in etoposide resistant Y79 and WERI RB cells. Values are means of three independent experiments ± SEM. ns p> 0.05; *p< 0.05; **p< 0.01 and ***p< 0.001; statistical differences compared to the control group calculated by paired Student’s t-test.
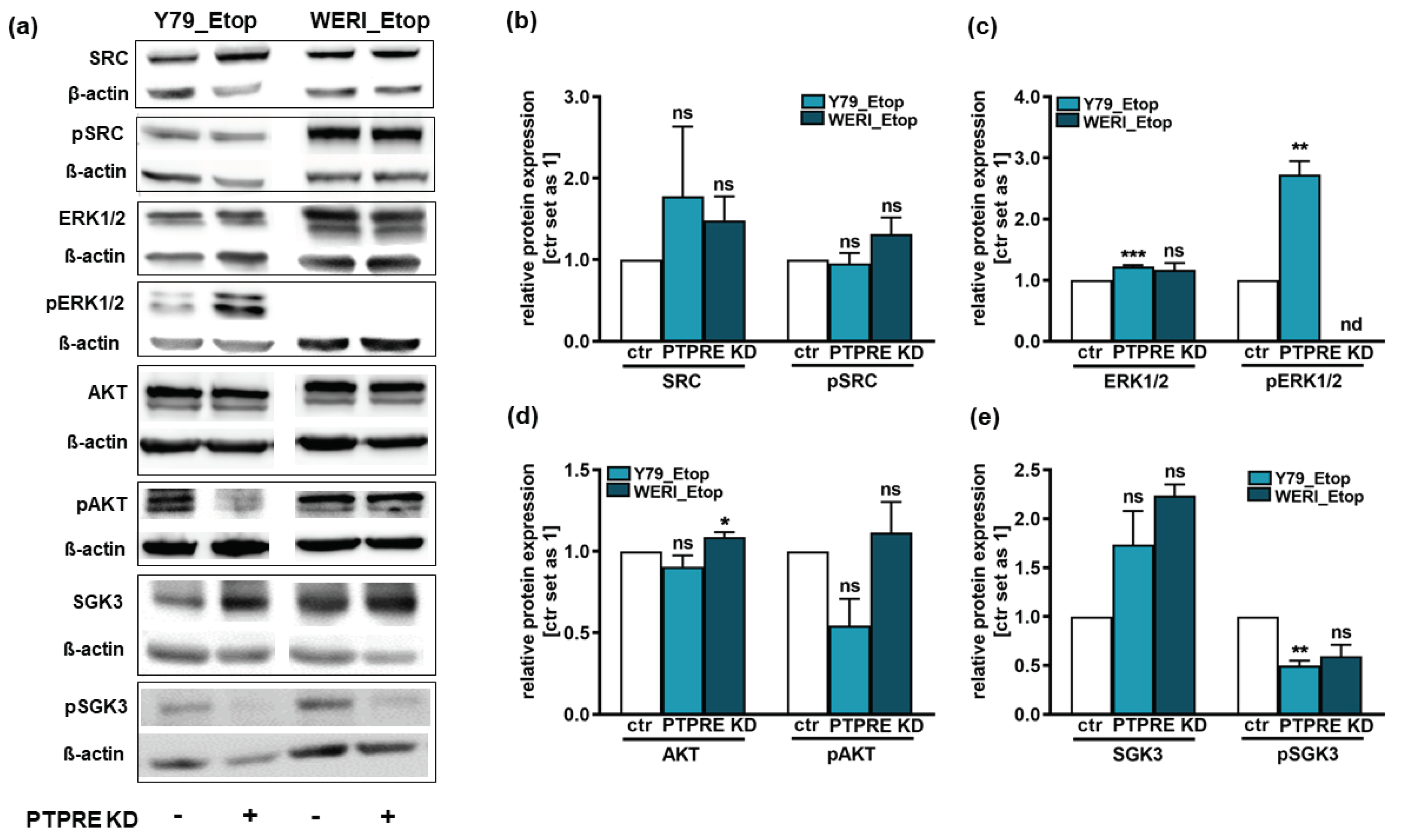

Table 1.
Primary antibodies for Western blot analyses.
| Antibodies | Company | Dilution |
|---|---|---|
| PTPRE (Ma5-25072) | Thermofisher Scientific, Darmstadt, Germany | 1:1000 |
| SRC (#2105) | Cell signaling technology, Danvers, USA | 1:1000 |
| pSRC (#2123) | Cell signaling technology, Danvers, USA | 1:1000 |
| ERK (#9102) | Cell signaling technology, Danvers, USA | 1:1000 |
| pERK (#4370) | Cell signaling technology, Danvers, USA | 1:1000 |
| AKT (#4685) | Cell signaling technology, Danvers, USA | 1:1000 |
| pAKT (#9271) | Cell signaling technology, Danvers, USA | 1:500 |
| SGK3 (Sc-166847) | Santa Cruz Biotechnology, Dallas, USA | 1:500 |
| pSGK3 (#5642) | Cell signaling technology, Danvers, USA | 1:500 |
| FGFb (ab215373) | Abcam, Berlin, Germany | 1:1000 |
| ß-actin (#4967) | Cell signaling technology, Danvers, USA | 1:1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



