
Submitted:
19 March 2024
Posted:
21 March 2024
You are already at the latest version
Abstract
Equine hepacivirus (EqHV, Flaviviridae, Hepacivirus) is a small, enveloped, RNA virus generally causing sub-clinical hepatitis with occasional fatalities. EqHV is reported in equids worldwide, but for Italy data are limited. To address this, a survey study was set up to estimate prevalence at national level and among different production categories (equestrian, competition, work and meat and reproduction) and national macro-regions (North, Central, South and Islands). Data obtained testing 1801 horse serum samples by Real-Time RT PCR were compared within the categories and regions. The NS3 fragment of the PCR-positive samples was sequenced by Sanger protocol for phylogenetic and mutational analysis. The tertiary structure of the NS3 protein was also assessed. The estimated national prevalence was 4.27% [1.97-6.59, 95%CI] and no statistical differences were detected among production categories and macro-regions. The phylogenesis confirmed the distribution in Italy of the three known EqHV subtypes, suggesting also a possible fourth sub-type that however requires further confirmation. Mutational profiles that could also affect the NS3 binding affinity to the viral RNA were detected. The present paper demonstrates that EqHV should be included in diagnostic protocols when investigating causes of hepatitis and in quality control protocols for blood derived products, due to its parental transmission.
Keywords:
equine hepacivirus
; horses
; biomolecular prevalence
; Italy
; phylogenesis
1. Introduction
The Flaviviridae family encompasses over 80 species of small, enveloped, single-stranded RNA viruses classified into four genera: Orthoflavivirus, Pestivirus, Pegivirus and Hepacivirus [1,2]. These viruses are usually host-specific and mainly infect birds and mammals. Horses are susceptible to several Flaviviruses, some of which are relevant zoonotic agents [1]. In the last decade, a recently discovered virus, the Equine Hepacivirus (hereon EqHV, Family: Flaviviridae; Genus: Hepacivirus), initially described as Non-Primate Hepacivirus in 2012 [3], then classified as Hepacivirus A in 2016 [4], and recently renamed Hepacivirus equi [2], gained greater attention due to its high genetic homology with the human Hepatitis C Virus (HCV) [3] and to its global diffusion. As the virus is worldwide present, several studies focused on its transmission routes, which remain elusive and subject of scientific debate: to date only the parenteral transmission [5-8] and sporadic cases of vertical transmission [9,10], were successfully demonstrated, while more data are required to evaluate the potential role of insects [11] and the role of the proximity of individuals in horizontal transmission [9,12-14]. Sexual transmission has not been investigated yet. Noteworthy is that the virus was also found in commercial equine products (serum and plasma) [3-6,15-18], meaning that it could evade sterilizing and control procedures, increasing the risk of its spread by veterinary therapeutic treatments [19,20].The onset of the EqHV infection is relatively rapid: viral RNA can be detected by PCR, one week post-infection, in the absence of clinical signs [5,21], with a high chance of spreading unnoticed within a holding during the first stages of the infection [22]. The outcome of EqHV infection is either a sub-clinical hepatitis, which usually self-clears in a few weeks [23], or a persistent infection which could last up to a year [9,24-27]. Infected individuals usually show mild signs to none, but there are reports of poor performance, jaundice, fatigue, or discomfort in viremic subjects [14,19,24,27,28]. Also, correlations between EqHV infection and increase of specific liver enzymes were assessed [3,5,7,8,21,23,24,27,28]; however, these values are also reported to frequently remain within the reference intervals or mildly increase in viremic animals [8, 21, 29, 30]. Correlation patterns between EqHV infection and a selection of horse characteristics (age, breed, sex, production category) were also investigated. Young horses (<8 years) are often more susceptible [9,31-36] compared to older horses (>10 years) [14, 37]; In thoroughbreds a higher percentage of PCR positivity was described when compared to other breeds [15,21,31,32,33,34,38]; females seem to be more prone to infection [35,36,37] compared to males [30] and competition horses show similar higher trends of susceptibility [31,39-41]. These data which however require further verification, hint how there could be implicit management practices exposing certain individuals to a higher risk of infection.Through the years, several studies have assessed EqHV biomolecular and serological prevalence in Africa [34,36], in North America [3, 20], in South America [30, 35, 39], in Asia [14,26,31,32,37,40-44], and in Australia [45]. Overall, the worldwide biomolecular EqHV prevalence range from <1% to 18.2% and the serological prevalence from 23.9% to 83.7% (taking into account studies which include at least n≥ 100 subjects, sampled and examined within the same time span). In Europe, EqHV prevalence was thoroughly investigated: the first report was on 2012, in UK [29] which highlighted a biomolecular prevalence of 2.1% (3/143); in Germany higher biomolecular prevalence were reported, ranging from 2.4% to 18.2% [15, 21, 33, 46, 47]; in Austria, prevalence range from 0.38% to 4.15% [11], while in France from 5.6% to 6.2% [25, 38]. In Italy, the only study estimating EqHV prevalence was published in 2017 [48], reporting a biomolecular prevalence of 4.7% in horses (91/1932), with the sampling performed only in two restricted Italian geographical areas: the North-East and the South-East, leaving out most of the peninsula and the islands.
The aims of the present study were: estimate EqHV national biomolecular prevalence in horses; verify if any statistical differences exists among the prevalence estimated for production categories (competition, equestrian, work/meat and reproduction) and macro-regions of Italy (North, Center, South and Islands); perform a phylogenetic analysis on the sequences obtained from the positive samples; assess the mutation level of the analyzed sequences and also investigate their effect on the NS3 protein tertiary structure.
2. Materials and Methods
2.1. Study Design and Sampling
The sampling scheme was designed to detect in the Italian horse population, an unknown prevalence (50%), with 95% Confidence Level (CL) and 5% Standard Error (SE). Mules or donkeys were not included in the study due to difficulties in collecting samples representative of the respective populations and funding limitations. The required number of samples for the parameters defined is 384 [49]. As the aims of the study included also an evaluation based on geographical and production categories, with four strata for each, the sample number was multiplied by four, resulting in 1536 samples. For convenience of stratification, 2,000 samples was set for the study. To stratify the sampling for production categories and geographical origin, thus obtaining results comparable with those of other studies, data on all the equine premises in Italy were extrapolated from the Veterinary Information System Database (https://www.vetinfo.it/, Accessed 2017). At that time, the categories were 12, classified by ‘purpose’ and by the absence/presence of mares or stallion on the premises. To simplify this classification, the 12 categories were therefore grouped in four macrocategories described as: equestrian (EQU - not competitive horses and pet horses), Competition (COM - competitive horses used in sport events and races), Work and Meat (W/M - horses used to carry loads/work and for meat consumption) and Reproduction (REP - brood mares/stallions use for breeding purposes). The macroregions defined by Italian National Institute of Statistics (ISTAT) are: North (Liguria, Lombardy, Piedmont, Aosta Valley, Emilia-Romagna, Friuli Venezia Giulia, Trentino South Tyrol, Veneto), Center (Latium, Marche, Tuscany, Umbria), and South (Abruzzo, Basilicata, Calabria, Campania, Molise, Apulia, Sicily, Sardinia). For this study, the two islands, Sicily and Sardinia, were considered separately. Samples were stratified within the macroregions proportionally to the consistency of premises for each production category. Only one sample from each premise was chosen to avoid possible bias due to repetition and holding proximity. Serum samples were randomly collected from those submitted for equine infectious anemia (EIA) surveillance activities, during the 2019-2022 period by the animal health laboratory network, with which the sampling strategy was previously shared. All samples were sent to the National Reference Center for Equine Diseases (NRL-ED) and stored at -80°C until analyzed. Upon arrival, data for each sample, as reported in the sampling form, were registered for production category and geographical position, the latter using QGIS (QGIS.org (2020-2022). QGIS Geographic Information System. Open Source Geospatial Foundation Project. http://qgis.org”). When more samples than necessary were collected, the selection criteria was set as distance among sampled premises ≥ five km.
2.2. Extraction and RNA Amplification
RNA extraction was performed using MagPurix® EVO with the extraction kit Viral/Pathogen Nucleic Acids Extraction Kit B (Zinexts Life Science corp.), and QiaSymphony with the extraction kit DSP Virus/Pathogen Mini Kit (Qiagen) according to manufacturer instructions. Products of extraction and after their analysis were stored at -80°C. A Real-time RT-PCR was set up targeting the 5’UTR fragment of the viral genome that is highly conserved: the eligible primers and probe sequences were sought through a bibliography consultation and chosen from a previously described molecular method which presented both reliability and sensitivity [3]. The selected primers are displayed in Table S1. To perform the analysis, the Luna Mastermix (Luna® Universal Probe One-Step RT-qPCR Kit, New England Biolabs inc.) was used and the Real-Time PCR condition on QuantStudio™ 7 Flex Real-Time PCR System (Applied Biosystems®) were set as follows: incubation at 55°C for 10 minutes, then 95°C for 1 minute, followed by 45 cycles at 95°C for 10 seconds and 60°C for 1 minute. Cycle threshold (Ct) limit was set at 40 to discriminate between PCR positive and negative samples: samples below Ct 40 were considered positive, while samples that scored Ct≥ 40 were either classified as negative, or repeated when results were equivocal/borderline.
2.3. Statistical Analysis
Prevalence were calculated as number of positive sample on the total analyzed at national, macro-regional and production category level. To detect potential statistical differences among the prevelence of macro-region and production categories, a Chi squared test was performed with the online tool Graphpad (https://www.graphpad.com/quickcalcs/contingency1/ Accessed on 10/08/2023).
2.4. Phylogenetic Analysis
For the phylogenetic analysis, a conventional RT-PCR assay was set up targeting the NS3 conserved region of the virus genome that corresponds to the protease/helicase domain of the viral polyprotein. The NS3 possess a binding affinity to viral RNA and may be involved in the viral replication activity. This region was chosen after screening the published bibliography on EqHV phylogeny studies: primers from [47] were used, considering the length of the amplicon produced (around 607bp) and the possibility to compare the sequenced samples with a broader online deposited dataset.
A Veriti™ 96-Well Fast Thermal Cycler (Applied Biosystems™) was used to perform the PCR and the SuperScript™ III One-Step RT-PCR System with Platinum™ Taq DNA Polymerase kit (Invitrogen, Thermo Fisher Scientific) was employed with a primer concentration of 30pmol/µl (0.28µM, 0.33µl). PCR products were visualized and verified for their length in bp in 1.5% agarose gel capillary electrophoresis using QIAxcel Advanced (QIAGEN) following manufacturer's instructions. The samples were then recovered and purified with ExoSap kit (Applied Biosystems,Thermo Fisher Scientific) according to manufacturer's instructions and visualized again on capillary electrophoresis to assure the correct purification and integrity of the amplicons. Sequencing was performed using Sanger Sequencing 3500 Series Genetic Analyzers (Applied Biosystems) following manufacturer's instructions. Each pair of chromatogram trace files was assembled in consensus sequences with the Tracy command line pipeline (version 0.7.3, https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6635-8), which uses a progressive multiple sequence alignment algorithm (https://academic.oup.com/bioinformatics/article/25/9/1118/204548?login=false) obtaining a graph-based alignment built on pairwise overlap alignments. Sequences were edited by BioEdit Software (version 7.2.5, Tom Hall, Raleigh, NC, USA) and aligned by ClustalW Multiple Alignment internal tool. The evolutionary tree for NS3 fragment of EqHV was inferred by using the Maximum Likelihood method based on the Tamura-Nei model using MEGA11 software [50]. Initial tree(s) for the heuristic search were obtained by applying the Neighbor-Joining method to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach). In addition, the results obtained were confirmed by applying Maximum Likelihood model produced by IQ-TREE 2 software [51] based on TPM2+I+G4 evolutionary model chosen for the trees by ModelFinder [52]. Phylogenetic tree topology consistency was checked using the Bootstrap method producing 1000 replicates [50]. Beside the sequence of isolates from the present study, sequences of the EqHV NS3 gene were downloaded from Genbank: some were taken as reference sequences (sub-type 1: JQ434002, JQ434005, JQ434008; sub-type 2, JQ434004, JQ434007; and sub-type 3: JQ434001, JQ434003, JQ434006)[3]; others and more recent sequences were used to corroborate the discrimination of these three sub-types (MK644936 [41] subt1), MZ274312 [44] subt2), MN734124 [37] subt3), mt955622 [8] subt1); several sequences were employed to enrich the analysis with european data (MH027992, MH027996, MH028000, MH028002, MH028005 [47], KF177391 [28], JX948116 [29], KC411810, KC411812 [46]; other asian and american sequences were utilized to enrich the analysis with global data (LC495903, LC440467 [26], LC030422, LC030428, LC030430, LC030431, LC030432 [31], KX056116, KX056117 [32], MF152651, MF152652 (Direct Submission), NC024889 [42], KJ472766 [5], KP325403 [6]; Also, some other recent Italian sequences were kindly provided by [22] (ON653391, ON653392, ON653393) and added to the analysis. Sequence KC411778 of Rodent Hepacivirus was used as outgroup [46].
2.5. Mutational Analysis
Sequences obtained within the setting of this study, and coding for a portion of NS3 protein of EqHV (from aa 355 to aa 505) were analyzed by using SeqScape-v2.6 software (Thermo-Fisher Scientific) and were then aligned by Bioedit 7.0 software [53]. Sequences having a mixture of wild-type and mutant residues at single positions were considered to have the mutantion(s) at that position. The mutation frequency for each codon of EqHV NS3 was defined as amino acid difference respect to EqHV NS3 reference sequence of each specific sub-type (Genbank accession number: JQ434005, JQ434004 and JQ434001 for sub-type 1, 2 and 3, respectively).
I-Tasser software [54] was used to predict the three-dimensional wt structures of Equine hepacivirus NS3 protein. The best model was aligned with the PDB model (1CU1) of the Human hepacivirus (HCV) by TM-align [55] resulting in a RMSD of only 0.26 Å, supporting the robustness of the predicted structures. The structure of EqHV NS3 protein was colored by UCSF Chimera [56].
3. Results
3.1. Study design and sampling
From 2019 to 2022, 1801 horse serum samples were collected and screened by RT-Real Time PCR for the detection of EqHV RNA. The total number of samples collected exceeded the minimum number required of 1536 samples but did not reach the expected numbers of 2000 sera. In some cases and in particular or the Islands (Sicily and Sardinia), the distance proxy (five km distance as set on QGIS) used for the selection of the samples to be tested could not be applied because: a) the geographical distribution of the stables was non homogeneous and the majority of regional holdings were located only in a province, or in a small territory; b) the overall number of regional samples per category was low and the few samples received were registered regardless of the distance; In all of the above mentioned cases, the required number of samples overruled the geographical distance, even if farms closer than one km for the same production category were not included in the present study.
3.2. Real-Time PCR results
National prevalence estimated was 4.28% [1.97-6.59, 95%Confidence Interval (CI)] with 77 samples detected positive. In Table 1, total samples analyzed, total positive samples, prevalence with 95%CI and standard error (SE) within production categories and macro-regions are respectively presented. Geographical distribution of the analyzed samples are shown in Supplementary information from Figure S1 to Figure S4, while that of positive samples is shown in Figure 1.
3.3. Statistical Analysis
3.4. Phylogenetic Analysis
Among the 77 positive samples, NS3 sequence was obtained for 35 of them (length: 497 bp). Phylogenetic analysis was performed using the Maximum Likelihood method, including the reference sequences of sub-types 1, 2 and 3 available in literature [3], to assign a sub-type to each of the sequences. The phylogenetic tree with the highest log likelihood (-6324.5024) is shown in Figure 2. The percentage of trees in which the associated taxa clustered together is shown next to the branches.
The majority of the sequences were classified as sub-type 1 (30/35, 85.7%), respectively followed by sub-type 3 (4/35, 11.4%), and sub-type 2 (1/35, 2.9%). Notably, two potential transmission clusters were observed (all supported by 99% of bootstrap): one composed of five samples and the second of two samples. Both clusters included viral strains infecting animals from the island of Sardinia. A group of sequences seem to cluster in a fourth sub-type that was never reported.
3.5. Mutational Analysis
Among the 77 positive samples, NS3 sequence was obtained for 35 of them (length: 497 bp). The mutational analysis of the EqHV NS3 Serine protease/helicase (aa 348-505), revealed the presence of at least one specific mutation differentiating the viral isolates of our study from the reference sequence of each sub-type in almost all EqHV-infected subjects (94.3%). In particular, complex mutational patterns, characterized by the co-presence of >2 NS3 mutations, were identified in EqHV strains detected in 11 horses (31.4%). In Table 4 and Table 5, prevalence of detected mutations are displayed, stratified according to production categories and macro-regions respectively. Notably, the mutation occurring with the highest frequency among the analyzed viral sequences was T490V (77.1%). In particular, this mutation was identified in EqHV sequences derived from almost all Italian Regions, indicating that it is the dominant viral species across Italy. Moreover, L358I was found in 20% of the overall analyzed EqHV strains, being detected in several Italian Regions (Latium, Lombardy, Sardinia and Sicily), thus highlighting its relevant distribution across our country. Similarly, V377I and V447I were identified in viral strains from two different Regions with an overall prevalence of 8.6% and 5.7%, respectively. Lastly, additional mutations with a peculiar geographic-dependent distribution were also present. In particular, in the present defined sample, S488T and N405K were found to circulate exclusively in EqHV infected horses in Sardinia with a prevalence of 14.3% and 8.6%, respectively. Lastly, other four mutations (S356A, S357A, T358L, D396E) were detected altogether in a single subject. Most of the identified mutations reside within alfa-helixes of the subdomains II and III of the NS3 helicase functional region, as shown in Figure 3 and Table 6.
4. Discussion
Here we present the first study on EqHV prevalence at national level with the aim to provide further data regarding the distribution of this virus in Italy, both geographically and among horse production categories and the results of the phylogenetic and the NS3 tertiary structure analysis.
The study presents some limitations: although it was designed as a cross sectional study, COVID-19 pandemic in 2020 brought activities almost to a complete halt. This obliged us to further extend the sampling time for over a year and a half to reach the required sample size. Thus, the prevalence data presented cannot be considered as punctual, but referred to a time span. Also, the study was designed to proportionally sample the horse population, reflecting its distribution on the Italian territory, to estimate an accurate EqHV biomolecular prevalence in Italian horses production categories between 2019-2022: however, the collection of samples from some provinces and regions was harder during and after the COVID-19 pandemic; nevertheless, the sampling level achieved was considered reliable since the SE resulted <5%, as set in the study design. Slight SE variances were instead observed in the macro-regions comparison, but all values were close to the expected one. Another limitation of the present study was that data on age, breed, and sex of the analyzed horses were not collected and therefore no retrospective comparisons, nor statistical analysis could be inferred on the influence that these risk factors could have played in the study scenario. Investigating these variables would require a greater number of samples that was not achievable with the funding resources available for the project. For the same reason, prevalence in donkeys and mule was not investigated, and priority was given to horses considering that they are the species most present and with the highest economic value.
All considered, this study however presents the first datum of EqHV biomolecular prevalence at national level in Italian horses: the described prevalence of 4.27% is coherent with what previously reported in Italy (91/1932 sera, 4.7%) [48] and within the range of biomolecular prevalence reported both in Europe (<1% - 18.2%) [11, 15, 21, 25, 29, 33, 38, 46, 47, 57, 58] and worldwide (3.2% - 46.2%) [3, 14, 20, 26, 30-32, 34-37, 39-45]. It should be pointed out that higher biomolecular prevalence (40-46.2%) are described in literature [22, 26, 33, 41] when the sample is collected within the same holdings; on the contrary, lower prevalence (3.4-5.6%) are described when the sampling was set to assess the prevalence in a geographic area [11, 25, 40, 48]. Intra-premises prevalence appears higher worldwide: in fact, once introduced in a stable, the virus seems to spread rapidly, with a still unclear transmission route, and because of its asymptomatic development, it is feasible that one or more carriers can infect other horses without showing any evident clinical signs. The sampling was designed to also investigate four horse production categories: equestrian, competition, work/meat, and reproduction which reflect the different categories within which holdings are officially classified by the Italian animal health authorities. This categorization was established to study whether different management practices could play a role in the spread of the virus. As a matter of fact, the literature reports that competition horses and breeding horses are often more susceptible to infection than other categories [31, 35, 36, 37, 39, 40, 41]: in this study the majority of PCR positive samples were grouped in the competition and reproduction ones, although statistical analysis did not highlight any significant difference among the production categories. Further studies are necessary to confirm these results with a higher number of samples, and if any differences would be detected, they could be ascribable to the different management practices (veterinary treatments, animal movement, direct contact with other horses of unknown health status) which expose them to a potentially higher risk scenario, compared to pet or privately owned horses stabled alone; also, it could be due to the intrinsic characteristics of the breeds which are often used for competitive sports and that are valuable for breeding, since it has been highlighted that Thoroughbreds, seem more susceptible to EqHV infections [15, 21, 31, 32, 33, 34, 38]. Nonetheless, the data collected in this study can be useful to highlight and confirm that EqHV infection poses a potential threat for animal health, being spread across all categories, and could have a tangible impact on the sport industry, that has the highest economic turnover: in fact poor performances, lethargy, apathy and fatigue are widely reported [14, 19, 24, 27, 28, 33], with occasional cases of euthanasia, due to poor overall health conditions and severe hepatic damage [19, 21, 24]. Data on EqHV biomolecular prevalence highlight also another issue related to therapies using blood, plasma and other blood derivatives, commercially available, as well as blood transfusions between horses. EqHV was already detected in some commercial blood products [15, 16, 17, 18], so it is crucial to assess both quality control analysis to certify the commercial products as EqHV free (together with the exclusion of other etiological agents that can be relevant for equine health), and include this virus in veterinary diagnostic protocols to control its spread in the equine population. At the present time, there is no therapy available for EqHV, thus biosecurity measures to reduce the spread of the disease on a holding and between holdings are: isolate the suspect case(s), suspend any production of blood products (if on a holding with this purpose), and periodically control the animals to monitor for the presence of the virus. Infection may last for a period of six months [5, 6, 8] after which the subject recovers, and no natural cases of resurgence of infection are reported so far, although re-infection is reported [7, 8]. On the contrary, if the virus can still be detected after six months, the subject is considered chronically infected [5, 6, 24, 26, 27, 28, 59] and should be managed with caution. This study also provides comprehensive data on the phylogenetic characteristics of the isolates from the whole Italian territory, integrating the already available data that were limited to some regions only. For this purpose, we analyzed a portion of the NS3 fragment, which is the serine protease/helicase domain of the viral polyprotein. Although EqHV is a RNA virus, and therefore prone to mutation, the strains of the present study mostly belong to the three known EqHV sub-types (Figure 2). As expected from the literature [30, 37, 44] the majority of the sequences (30) clustered within sub-type 1, while four were identified as sub-type 3 and only one sequence as sub-type 2. Some interesting clusters in sub-type 1 (identified in Figure 2 with grey dotted ellipses) are of particular interest: top to bottom, the first cluster is represented by three very recent sequences from an Italian EqHV outbreak (kindly provided by the authors of [22]), relevant because the authors could follow up the infection of few horses for several months, allowing the tracking of viral mutation within the same subjects: this is the case of ON653391 and ON653393, detected from the same horse sampled six months apart, clearly showing how EqHV is prone to mutations (for more in depth considerations see [22]). The second and the third clusters, instead, include sequences from the present study and are located mid-way and at the bottom of sub-type 1, respectively: the first one includes five sequences from Sardinia (OQ067932, OQ414236, OQ107572, OQ067931, OQ437965) and the second, two sequences, still from Sardinia (OQ312099 and OQ320792): both clusters show a 99 bootstrap value. A retrospective tracing of the subjects allowed to determine that a) the premises of origin are distant from each other (>12km to <250km), apart from two samples which do not belong to the same cluster, nor to the same production category, but which stables of origin are located 2.1 Km apart (Accession Number ID: OQ067932; OQ320792); b) being geographically distributed, they could hardly share the same veterinarian, except from the said two subjects; c) none of the seven horses had a history of movement to or from the other stables, according to the data available on the Veterinary Information System Online Database; d) data on veterinary treatments (such as vaccines, plasma transfusion, etc.) could not be retrieved. The reason why these sequences cluster so close together, then, cannot be surely assessed, but it must be considered that each stable held a discreet number of horses (>1 to <21) at the time of sampling, therefore it cannot be excluded that movement of an EqHV positive horse, different from those sampled, could be the source of infection for each respective cluster. Movements from one stable to another is common practice in the horse industry, either for trade, management, breeding or sport activities: since the virus appears widespread and the sequences are phylogenetically close, regardless of their origin, presumably movements played a role in the spreading of the three sub-types worldwide in the last decade, and probably before. When it comes to horse pre-movement sanitary controls, several diseases are screened, but EqHV is not currently present in routine laboratory diagnosis, at least in Italy, and probably also in most countries of the world. Stricter diagnostic protocols when moving animals should be considered, especially when susceptible breeds or valuable individuals are involved. Further on the phylogenetic analysis, another interesting aspect of the tree should be discussed: included in sub-type 3 there is a branch (the second branch bottom-up, identified by a 92 bootstrap value) which clusters alone and originates from a different node when compared to the other branches of the tree: four sequences from the present study are included in this group (OQ420478, OQ420479, OP947531, OQ024216) along with two sequences retrieved from Genbank (LC030432 [31] and MN734124 [37]). Other recent phylogenetic analyses [22, 26, 37, 44, 60] and the original paper from [31], show a similar outcome: in these papers, in fact, the branch, identified either by LC030432 [31], MN734124 [37], or both, separates from the node which includes the sequences used as reference for sub-type 3 (JQ434001,JQ434003,JQ434006 [3]). This suggests that these sequences could substantially differ from those included in sub-type 3 and from the other sub-types: it could be hypothesized that this branch was never part of the EqHV sub-type 3, but it represented a totally different sub-type which was until now under-represented and that was therefore considered part of sub-type 3. The most accredited criteria to propose new sub-types for EqHV is that described in literature for HCV [4, 61, 62], which were already used to introduce the presence of sub-type 2 [30, 38] and 3 [41] for EqHV. These criteria enlist the need of specific parameters to assess the detection of a new sub-type, such as “complete or nearly complete coding region sequence differing from other sequences by at least 15% of nucleotide positions” and “sequence information from at least two other isolates in core/E1 […] and NS5B” [62] which could not be fulfilled in the present study. However, a more numerous set of sequences, representative of this branch and that could be used as reference are those of the NS3 protein submitted by [31] (LC030420, LC030425, LC030426, LC030427, LC030432), the complete polyprotein sequence by [37] (MN734124) and the new partial NS3 sequences from the present study (OQ420478, OQ420479, OP947531, OQ024216). We are strongly looking forward to further studies and broader sampling from horses worldwide to verify this hypothesis that could update the classification of EqHV.
Regarding the mutational analysis, most of the identified mutations reside within the alfa-helixes of the II and III subdomains of the NS3 helicase functional regions. In particular, these mutations are localized in portions that are critical for the RNA binding and the following unwinding activity of NS3. On this basis, it could be speculated that the reported mutations may play a role in these processes, but further in-depth in silico studies are necessary to determine how their accumulation in these regions impact on NS3 proper folding and its binding affinity to viral RNA, to elucidate potential implications on the viral replication activity.
5. Conclusions
Equine hepacivirus is a recently discovered virus of equids. It affects the liver of infected horses and causes sub-clinical hepatitis, seldom leading to death. Its presence is demonstrated worldwide and since the virus has also been detected in blood products, it represents a threat for animal health in veterinary practice. The present study reports the first data on national prevalence in Italy, demonstrating its circulation at national level. Phylogenetic analysis shows that the italian strains connect with isolates present worldwide, thus the extensive circulation of the virus in confirmed. Moreover, the possible presence of a fourth sub-type is discussed, but further studies are needed to verify this hypotesis. All considered, EqHV remains a relevant virus in equine infectious disease landscape and should be addressed with care, highlighting all the more its importance in diagnostic protocols for hepatitis in horses and in blood products safety control tests.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Equine Hepatic Viruses Consortium (alphabetical order). Figure S1-Collection sites of the serum samples belonging to the Equestrian (EQ) category. Figure S2- Collection sites of the serum samples belonging to the Competition (COMP) category. Figure S3- Collection sites of the serum samples belonging to the Work/Meat (W/M) category. Figure S4-Collection sites of the serum samples belonging to the Reproduction (REP) category: Table S1: Primers and probe used for the RT-Real time PCR and the Endpoint PCR.
Author Contributions
Conceptualization, R.N. and M.T.S.; methodology, R.N., M.T.S. and M.G.S.; software, G.P., R.S, M.C.B., D.L.R. and L.C; validation, R.N., A.C, G.M.; formal analysis, R.N., V.S., R.S, M.C.B., D.L.R. and L.C; investigation, G.P., R.C., R.S, M.C.B., D.L.R.; sampling, the Equine Hepatic Viruses Consortium; data curation, G.P., R.N.; writing—original draft preparation, G.P., R.N.; writing—review and editing, G.P., R.N., V.S., R.S, M.C.B., M.T.S.; project administration, R.N., M.T.S.; funding acquisition, M.T.S. All authors have read and agreed to the published version of the manuscript.
and designed research supervised study and funding acquisition; M.G.S. designed sampling; the Equine Hepatic Viruses Consortium collected samples for research; G.P, R.C. A.C, G.M. performed PCR analysis and sequencing; R.N, G.P., V.S., R.S, M.C.B., D.L.R. and L.C analyzed data; G.P., R.N.,M.T.S, V.S., R.S, M.C.B, D.L.R. wrote and revised the paper.
Funding
This project was funded by the Italian Ministry of Health, project code IZSLT1018.
Institutional Review Board Statement
Not applicable
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, [R.N.], upon reasonable request.
Acknowledgments
The authors would like to thank Professor Gabriella Elia and Dr. Georgia Diakoudi from Department of Veterinary Medicine, University of Bari, Italy for kindly providing the sequences to be included in the phylogenetic tree.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3, . [CrossRef]
- Postler, T.S.; Beer, M.; Blitvich, B.J.; Bukh, J.; de Lamballerie, X.; Drexler, J.F.; Imrie, A.; Kapoor, A.; Karganova, G.G.; Lemey, P.; et al. Renaming of the genus Flavivirus to Orthoflavivirus and extension of binomial species names within the family Flaviviridae. Arch. Virol. 2023, 168, 1–7, . [CrossRef]
- Burbelo, P.D.; Dubovi, E.J.; Simmonds, P.; Medina, J.L.; Henriquez, J.A.; Mishra, N.; Wagner, J.; Tokarz, R.; Cullen, J.M.; Iadarola, M.J.; et al. Serology-Enabled Discovery of Genetically Diverse Hepaciviruses in a New Host. J. Virol. 2012, 86, 6171–6178, . [CrossRef]
- Smith, D.B.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, A.S.; Pletnev, A.; Rico-Hesse, R.; Stapleton, J.T.; et al. Proposed update to the taxonomy of the genera Hepacivirus and Pegivirus within the Flaviviridae family. J. Gen. Virol. 2016, 97, 2894–2907, . [CrossRef]
- Ramsay, J.D.; Evanoff, R.; Wilkinson, T.E., Jr.; Divers, T.J.; Knowles, D.P.; Mealey, R.H. Experimental transmission of equine hepacivirus in horses as a model for hepatitis C virus. Hepatology 2015, 61, 1533–1546, . [CrossRef]
- Scheel, T.K.H.; Kapoor, A.; Nishiuchi, E.; Brock, K.V.; Yu, Y.; Andrus, L.; Gu, M.; Renshaw, R.W.; Dubovi, E.J.; McDonough, S.P.; et al. Characterization of nonprimate hepacivirus and construction of a functional molecular clone. Proc. Natl. Acad. Sci. 2015, 112, 2192–2197, . [CrossRef]
- Pfaender, S.; Walter, S.; Grabski, E.; Todt, D.; Bruening, J.; Romero-Brey, I.; Gather, T.; Brown, R.J.P.; Hahn, K.; Puff, C.; et al. Immune protection against reinfection with nonprimate hepacivirus. Proc. Natl. Acad. Sci. 2017, 114, E2430–E2439, . [CrossRef]
- Tomlinson, J.E.; Wolfisberg, R.; Fahnøe, U.; Patel, R.S.; Trivedi, S.; Kumar, A.; Sharma, H.; Nielsen, L.; McDonough, S.P.; Bukh, J.; et al. Pathogenesis, MicroRNA-122 Gene-Regulation, and Protective Immune Responses After Acute Equine Hepacivirus Infection. J. Hepatol. 2021, 74, 1148–1163, . [CrossRef]
- Gather, T.; Walter, S.; Todt, D.; Pfaender, S.; Brown, R.J.P.; Postel, A.; Becher, P.; Moritz, A.; Hansmann, F.; Baumgaertner, W.; et al. Vertical transmission of hepatitis C virus-like non-primate hepacivirus in horses. J. Gen. Virol. 2016, 97, 2540–2551, . [CrossRef]
- Pronost, S.; Fortier, C.; Marcillaud-Pitel, C.; Tapprest, J.; Foursin, M.; Saunier, B.; Pitel, P.-H.; Paillot, R.; Hue, E.S. Further Evidence for in Utero Transmission of Equine Hepacivirus to Foals. Viruses 2019, 11, 1124, . [CrossRef]
- Badenhorst, M.; de Heus, P.; Auer, A.; Rümenapf, T.; Tegtmeyer, B.; Kolodziejek, J.; Nowotny, N.; Steinmann, E.; Cavalleri, J.-M. No Evidence of Mosquito Involvement in the Transmission of Equine Hepacivirus (Flaviviridae) in an Epidemiological Survey of Austrian Horses. Viruses 2019, 11, 1014, . [CrossRef]
- S. Pronost, et al. Hépacivirus, pégivirus, TDAV: Une nouvelle triade de virus hépatiques chez le ceval?. Pratique vétérinaire équine 197, 24-31 (2018).
- Altan, E.; Li, Y.; Jr, G.S.-S.; Sawaswong, V.; Barnum, S.; Pusterla, N.; Deng, X.; Delwart, E. Viruses in Horses with Neurologic and Respiratory Diseases. Viruses 2019, 11, 942, . [CrossRef]
- Yoon, J.; Park, T.; Kim, A.; Song, H.; Park, B.; Ahn, H.; Go, H.; Kim, D.; Lee, J.; Park, S.; et al. First report of equine parvovirus-hepatitis and equine hepacivirus coinfection in horses in Korea. Transbound. Emerg. Dis. 2021, 69, 2735–2746, . [CrossRef]
- Postel, A.; Cavalleri, J.-M.V.; Pfaender, S.; Walter, S.; Steinmann, E.; Fischer, N.; Feige, K.; Haas, L.; Becher, P. Frequent presence of hepaci and pegiviruses in commercial equine serum pools. Veter- Microbiol. 2015, 182, 8–14, . [CrossRef]
- Paim, W.; Weber, M.; Cibulski, S.; da Silva, M.; Puhl, D.; Budaszewski, R.; Varela, A.; Mayer, F.; Canal, C. Characterization of the viral genomes present in commercial batches of horse serum obtained by high-throughput sequencing. Biologicals 2019, 61, 1–7, . [CrossRef]
- Lu, G.; Huang, J.; Yang, Q.; Xu, H.; Wu, P.; Fu, C.; Li, S. Identification and genetic characterization of hepacivirus and pegivirus in commercial equine serum products in China. PLOS ONE 2017, 12, e0189208–e0189208, . [CrossRef]
- Meister, T.L.; Tegtmeyer, B.; Postel, A.; Cavalleri, J.-M.; Todt, D.; Stang, A.; Steinmann, E. Equine Parvovirus-Hepatitis Frequently Detectable in Commercial Equine Serum Pools. Viruses 2019, 11, 461, . [CrossRef]
- Kopper, J.J.; Schott, H.C.; Divers, T.J.; Mullaney, T.; Huang, L.; Noland, E.; Smedley, R. Theiler's disease associated with administration of tetanus antitoxin contaminated with nonprimate (equine) hepacivirus and equine parvovirus-hepatitis virus. Equine Veter- Educ. 2018, 32, E5–E9, . [CrossRef]
- Tomlinson, J.E.; Kapoor, A.; Kumar, A.; Tennant, B.C.; Laverack, M.A.; Beard, L.; Delph, K.; Davis, E.; Ii, H.S.; Lascola, K.; et al. Viral testing of 18 consecutive cases of equine serum hepatitis: A prospective study (2014-2018). J. Veter- Intern. Med. 2018, 33, 251–257, . [CrossRef]
- Pfaender, S.; Cavalleri, J.M.; Walter, S.; Doerrbecker, J.; Campana, B.; Brown, R.J.; Burbelo, P.D.; Postel, A.; Hahn, K.; Anggakusuma; et al. Clinical course of infection and viral tissue tropism of hepatitis C virus–like nonprimate hepaciviruses in horses. J. Hepatol. 2014, 61, 447–459, . [CrossRef]
- Cardone, R.; Buonavoglia, A.; Lanave, G.; Vasinioti, V.I.; Mininni, V.; Lorusso, E.; Decaro, N.; Martella, V.; Elia, G.; Diakoudi, G. Description of an Equine Hepacivirus Cluster in a Horse Stable in Italy. Transbound. Emerg. Dis. 2023, 2023, 1–7, . [CrossRef]
- Cavalleri, J.V.; Korbacska-Kutasi, O.; Leblond, A.; Paillot, R.; Pusterla, N.; Steinmann, E.; Tomlinson, J. European College of Equine Internal Medicine consensus statement on equine flaviviridae infections in Europe. J. Veter- Intern. Med. 2022, 36, 1858–1871, . [CrossRef]
- Tegtmeyer, B.; Echelmeyer, J.; Pfankuche, V.M.; Puff, C.; Todt, D.; Fischer, N.; Durham, A.; Feige, K.; Baumgärtner, W.; Steinmann, E.; et al. Chronic equine hepacivirus infection in an adult gelding with severe hepatopathy. Veter- Med. Sci. 2019, 5, 372–378, . [CrossRef]
- Pronost, S.; Hue, E.; Fortier, C.; Foursin, M.; Fortier, G.; Desbrosse, F.; Rey, F.; Pitel, P.-H.; Saunier, B. Identification of equine hepacivirus infections in France: Facts and Physiopathological insights. J. Equine Veter- Sci. 2016, 39, S22, . [CrossRef]
- Date, T.; Sugiyama, M.; Lkhagvasuren, D.; Wakita, T.; Oyunsuren, T.; Mizokami, M. Prevalence of equine hepacivirus infection in Mongolia. Virus Res. 2020, 282, 197940, . [CrossRef]
- Elia, G.; Lanave, G.; Lorusso, E.; Parisi, A.; Trotta, A.; Buono, R.; Martella, V.; Decaro, N.; Buonavoglia, C. Equine hepacivirus persistent infection in a horse with chronic wasting. Transbound. Emerg. Dis. 2017, 64, 1354–1358, . [CrossRef]
- Reuter, G.; Maza, N.; Pankovics, P.; Boros, . Non-primate hepacivirus infection with apparent hepatitis in a horse — Short communication. Acta Veter- Hung. 2014, 62, 422–427, . [CrossRef]
- Lyons, S.; Kapoor, A.; Sharp, C.; Schneider, B.S.; Wolfe, N.D.; Culshaw, G.; Corcoran, B.; McGorum, B.C.; Simmonds, P. Nonprimate Hepaciviruses in Domestic Horses, United Kingdom. Emerg. Infect. Dis. 2012, 18, 1976–1982, . [CrossRef]
- Figueiredo, A.S.; Lampe, E.; Espírito-Santo, M.P.D.; Mello, F.C.D.A.; de Almeida, F.Q.; de Lemos, E.R.S.; Godoi, T.L.O.S.; Dimache, L.A.G.; dos Santos, D.R.L.; Villar, L.M. Identification of two phylogenetic lineages of equine hepacivirus and high prevalence in Brazil. Veter- J. 2015, 206, 414–416, . [CrossRef]
- Matsuu, A.; Hobo, S.; Ando, K.; Sanekata, T.; Sato, F.; Endo, Y.; Amaya, T.; Osaki, T.; Horie, M.; Masatani, T.; et al. Genetic and serological surveillance for non-primate hepacivirus in horses in Japan. Veter- Microbiol. 2015, 179, 219–227, . [CrossRef]
- Kim, H.-S.; Moon, H.-W.; Sung, H.W.; Kwon, H.M. First identification and phylogenetic analysis of equine hepacivirus in Korea. Infect. Genet. Evol. 2017, 49, 268–272, . [CrossRef]
- Reichert, C.; Campe, A.; Walter, S.; Pfaender, S.; Welsch, K.; Ruddat, I.; Sieme, H.; Feige, K.; Steinmann, E.; Cavalleri, J.M. Frequent occurrence of nonprimate hepacivirus infections in Thoroughbred breeding horses – A cross-sectional study for the occurrence of infections and potential risk factors. Veter- Microbiol. 2017, 203, 315–322, . [CrossRef]
- Badenhorst, M.; Tegtmeyer, B.; Todt, D.; Guthrie, A.; Feige, K.; Campe, A.; Steinmann, E.; Cavalleri, J.M. First detection and frequent occurrence of Equine Hepacivirus in horses on the African continent. Veter- Microbiol. 2018, 223, 51–58, . [CrossRef]
- Figueiredo, A.S.; Lampe, E.; de Albuquerque, P.P.L.F.; Chalhoub, F.L.L.; de Filippis, A.M.B.; Villar, L.M.; Cruz, O.G.; Pinto, M.A.; de Oliveira, J.M. Epidemiological investigation and analysis of the NS5B gene and protein variability of non-primate hepacivirus in several horse cohorts in Rio de Janeiro state, Brazil. Infect. Genet. Evol. 2018, 59, 38–47, . [CrossRef]
- Abbadi, I.; Lkhider, M.; Kitab, B.; Jabboua, K.; Zaidane, I.; Haddaji, A.; Nacer, S.; Matsuu, A.; Pineau, P.; Tsukiyama-Kohara, K.; et al. Non-primate hepacivirus transmission and prevalence: Novel findings of virus circulation in horses and dogs in Morocco. Infect. Genet. Evol. 2021, 93, 104975, . [CrossRef]
- Wu, et al., First identification and genomic characterization of equine hepacivirus sub-type 3 strain in China. Virus Genes 56, 777–780 (2020).
- Pronost, S.; Hue, E.; Fortier, C.; Foursin, M.; Fortier, G.; Desbrosse, F.; Rey, F.A.; Pitel, P.-H.; Richard, E.; Saunier, B. Prevalence of Equine Hepacivirus Infections in France and Evidence for Two Viral Subtypes Circulating Worldwide. Transbound. Emerg. Dis. 2016, 64, 1884–1897, . [CrossRef]
- B. S. Gemaque, et al., Hepacivirus infection in domestic horses, Brazil, 2011-2013. Emerg. Infect. Dis. 20, 2011–2013 (2014).
- G. Lu, et al., First description of hepacivirus and pegivirus infection in domestic Horses in China: A study in guangdong province, heilongjiang province and Hong Kong district. PLoS One 11, 1–12 (2016).
- Lu, G.; Ou, J.; Sun, Y.; Wu, L.; Xu, H.; Zhang, G.; Li, S. Natural recombination of equine hepacivirus subtype 1 within the NS5A and NS5B genes. Virology 2019, 533, 93–98, . [CrossRef]
- Tanaka, T.; Kasai, H.; Yamashita, A.; Okuyama-Dobashi, K.; Yasumoto, J.; Maekawa, S.; Enomoto, N.; Okamoto, T.; Matsuura, Y.; Morimatsu, M.; et al. Hallmarks of Hepatitis C Virus in Equine Hepacivirus. J. Virol. 2014, 88, 13352–13366, . [CrossRef]
- Hayashi, S.; Tanaka, T.; Moriishi, K.; Hirayama, K.; Yamada, A.; Hotta, K. Seroepidemiology of non-primate hepacivirus (NPHV) in Japanese native horses. J. Veter- Med Sci. 2018, 80, 186–189, . [CrossRef]
- Chen, Y.; Cai, S.; Zhang, Y.; Lai, Z.; Zhong, L.; Sun, X.; Li, S.; Lu, G. First identification and genomic characterization of equine hepacivirus subtype 2 in China. Arch. Virol. 2021, 166, 3221–3224, . [CrossRef]
- C. Fortier, et al., Hepatitis viruses: prevalence of equine parvovirus-hepatitis virus and equine hepacivirus in France and Australia. Equine Vet. J. 53, 68–68 (2021).
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Lukashev, A.N.; Gmyl, A.; Coutard, B.; Adam, A.; Ritz, D.; Leijten, L.M.; van Riel, D.; et al. Evidence for Novel Hepaciviruses in Rodents. PLOS Pathog. 2013, 9, e1003438, . [CrossRef]
- Schlottau, K.; Fereidouni, S.; Beer, M.; Hoffmann, B. Molecular identification and characterization of nonprimate hepaciviruses in equines. Arch. Virol. 2018, 164, 391–400, . [CrossRef]
- Elia, G.; Lanave, G.; Lorusso, E.; Parisi, A.; Cavaliere, N.; Patruno, G.; Terregino, C.; Decaro, N.; Martella, V.; Buonavoglia, C. Identification and genetic characterization of equine hepaciviruses in Italy. Veter- Microbiol. 2017, 207, 239–247, . [CrossRef]
- Thrusfield, M., Christley, R., Brown, H., Diggle, P. J., French, N., Howe, K., Kelly, L., O'Connor, A., Sargeant, J., & Wood, H. (2017). Veterinary Epidemiology: Fourth Edition. (4th ed.) Wiley-Blackwell. [CrossRef]
- K. Tamura, G. Stecher, S. Kumar, MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol.38, 3022–3027 (2021).
- B. Q. Minh, et al., IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol.37, 1530–1534 (2020).
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589, . [CrossRef]
- T.A. Hall, BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95-98 (1999).
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 2015, 12, 7–8, . [CrossRef]
- Zhang, Y.; Skolnick, J. TM-Align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309, . [CrossRef]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; E Ferrin, T. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339–339, . [CrossRef]
- Lyons, S.; Kapoor, A.; Schneider, B.S.; Wolfe, N.D.; Culshaw, G.; Corcoran, B.; Durham, A.E.; Burden, F.; McGorum, B.C.; Simmonds, P. Viraemic frequencies and seroprevalence of non-primate hepacivirus and equine pegiviruses in horses and other mammalian species. J. Gen. Virol. 2014, 95, 1701–1711, . [CrossRef]
- Badenhorst, M.; de Heus, P.; Auer, A.; Tegtmeyer, B.; Stang, A.; Dimmel, K.; Tichy, A.; Kubacki, J.; Bachofen, C.; Steinmann, E.; et al. Active equine parvovirus-hepatitis infection is most frequently detected in Austrian horses of advanced age. Equine Veter- J. 2021, 54, 379–389, . [CrossRef]
- Gather, T.; Walter, S.; Pfaender, S.; Todt, D.; Feige, K.; Steinmann, E.; Cavalleri, J.M.V. Acute and chronic infections with nonprimate hepacivirus in young horses. Veter- Res. 2016, 47, 1–5, . [CrossRef]
- Pacchiarotti, G.; Nardini, R.; Scicluna, M.T. Equine Hepacivirus: A Systematic Review and a Meta-Analysis of Serological and Biomolecular Prevalence and a Phylogenetic Update. Animals 2022, 12, 2486, . [CrossRef]
- Simmonds, P.; Bukh, J.; Combet, C.; Deléage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspé, G.; Kuiken, C.; Maertens, G.; et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. J. Hepatol. 2005, 42, 962–973, . [CrossRef]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. J. Hepatol. 2013, 59, 318–327, . [CrossRef]
Figure 1.
EqHV PCR positive samples detected in this study are shown on the map. Blue dots represent Equestrian (EQU) positive samples, blue diamonds Competition (COM) positive samples, blue triangles Work/Meat (W/M) positive samples and blue squares Reproduction (REP) positive samples. Only regional borders are reported.
Figure 1.
EqHV PCR positive samples detected in this study are shown on the map. Blue dots represent Equestrian (EQU) positive samples, blue diamonds Competition (COM) positive samples, blue triangles Work/Meat (W/M) positive samples and blue squares Reproduction (REP) positive samples. Only regional borders are reported.
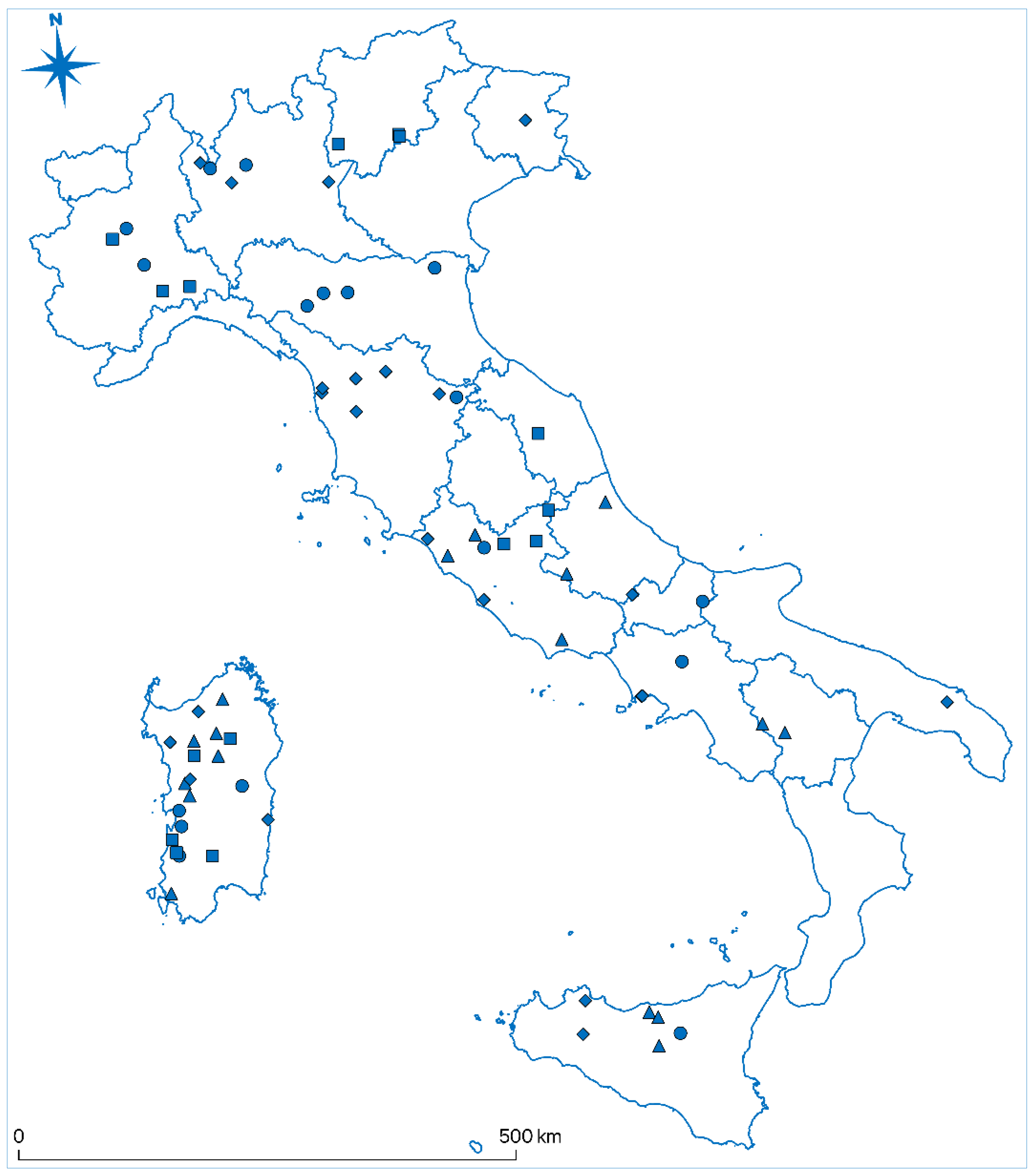
Figure 2.
Phylogenetic analysis of NS3 partial gene of Equine Hepacivirus. The figure reports the phylogenetic tree based on 497-nt fragment of NS3 gene of 35 EqHV strains detected in the present study or retrieved from GenBank database including the reference sequences of sub-types 1 (O and ●), 2 (O and ●) and 3 (O and ●). Molecular evolutionary analyses were performed using Maximum Likelihood method based on the Tamura-Nei model with bootstrap test (n =1000). Bootstrap values >50% are shown. In grey dotted ellipses the potential transmission clusters were observed (all supported by 99% of bootstrap). Sequence KC411778 [46] of Rodent Hepacivirus was used as outgroup. Scale bar indicates nt substitutions per site.
Figure 2.
Phylogenetic analysis of NS3 partial gene of Equine Hepacivirus. The figure reports the phylogenetic tree based on 497-nt fragment of NS3 gene of 35 EqHV strains detected in the present study or retrieved from GenBank database including the reference sequences of sub-types 1 (O and ●), 2 (O and ●) and 3 (O and ●). Molecular evolutionary analyses were performed using Maximum Likelihood method based on the Tamura-Nei model with bootstrap test (n =1000). Bootstrap values >50% are shown. In grey dotted ellipses the potential transmission clusters were observed (all supported by 99% of bootstrap). Sequence KC411778 [46] of Rodent Hepacivirus was used as outgroup. Scale bar indicates nt substitutions per site.
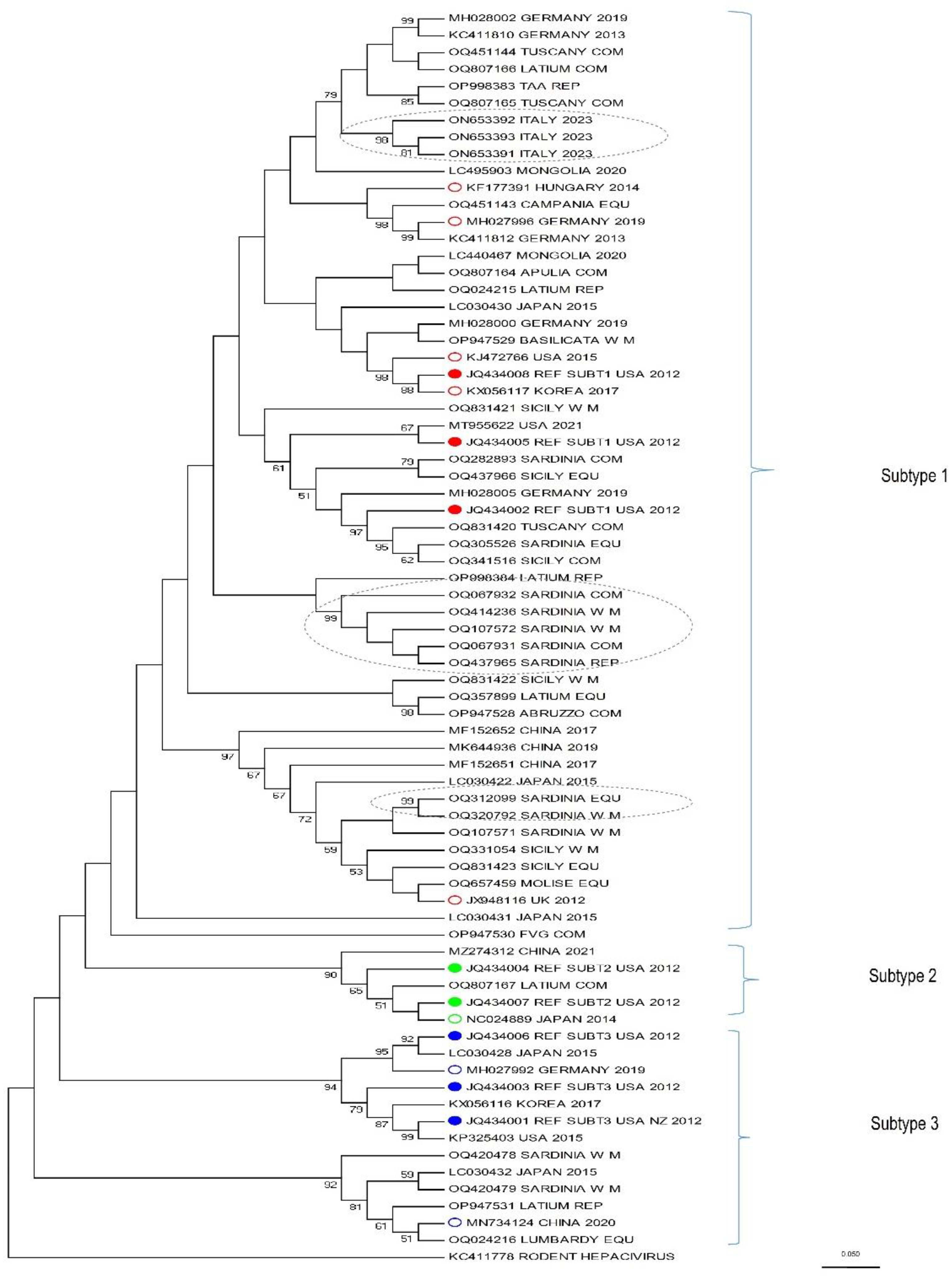
Figure 3.
The three-dimensional structure of Equine hepacivirus NS3 protein constructed by I-Tasser software using the PDB model of the human hepacivirus (HCV 1CU1) as reference. The functional domains are color-coded in the Equine hepacivirus NS3 protein as follows: Serine protease domain (1-180, green), Helicase Subdomain I (181-326 cyan), Helicase Subdomain II (327-481 dodger blue), Helicase Subdomain III (482-631 navy blue) Residues S356, S357, L358, V377, D396, N405, V447, S488, T490, showing their side chains as spheres are reported in grey.
Figure 3.
The three-dimensional structure of Equine hepacivirus NS3 protein constructed by I-Tasser software using the PDB model of the human hepacivirus (HCV 1CU1) as reference. The functional domains are color-coded in the Equine hepacivirus NS3 protein as follows: Serine protease domain (1-180, green), Helicase Subdomain I (181-326 cyan), Helicase Subdomain II (327-481 dodger blue), Helicase Subdomain III (482-631 navy blue) Residues S356, S357, L358, V377, D396, N405, V447, S488, T490, showing their side chains as spheres are reported in grey.
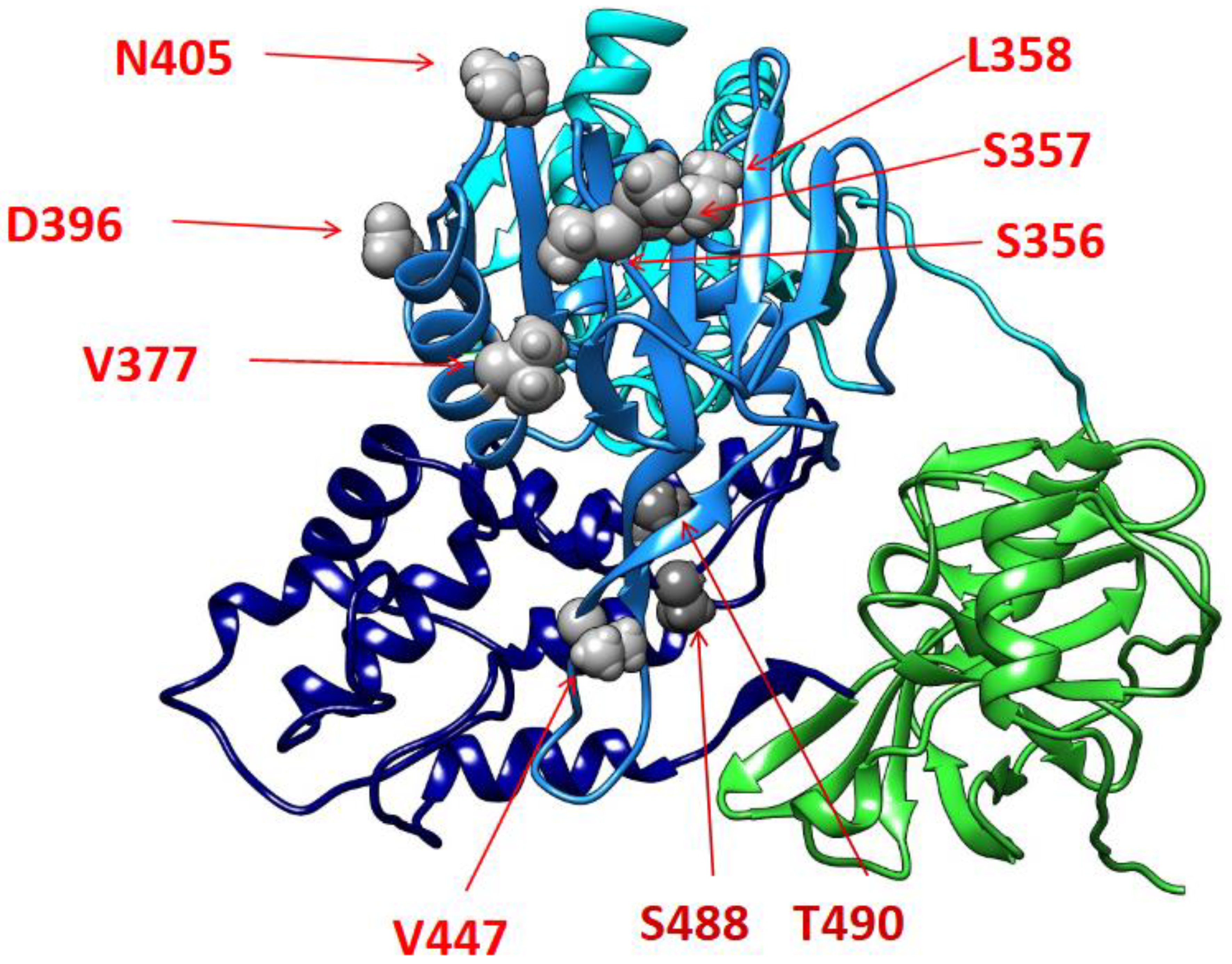

Table 1.
Total analyzed samples, total positive samples, prevalence with 95% CI and SE values within categories (upper table) and macro-region (lower table).
Table 1.
Total analyzed samples, total positive samples, prevalence with 95% CI and SE values within categories (upper table) and macro-region (lower table).
| Analysed samples | Positive samples | P (%) | Lower IC (%) | Upper IC (%) | SE (%) | |
|---|---|---|---|---|---|---|
| Production categories | ||||||
| Equestrian | 497 | 17 | 3.42 | 0 | 7.82 | ± 4.40 |
| Competition | 464 | 23 | 4.96 | 0.41 | 9.51 | ± 4.55 |
| Work/Meat | 442 | 17 | 3.85 | 0 | 8.51 | ± 4.66 |
| Reproduction | 398 | 20 | 5.03 | 0.11 | 9.94 | ± 4.91 |
| Italy macroregions | ||||||
| North | 393 | 18 | 4.58 | 0 | 9.52 | ± 4.94 |
| Center | 315 | 17 | 5.40 | 0 | 10.92 | ± 5.52 |
| South | 435 | 13 | 2.99 | 0 | 7.96 | ± 4.70 |
| Sardinia | 376 | 20 | 5.32 | 0.26 | 10.37 | ± 5.05 |
| Sicily | 282 | 9 | 3.19 | 0 | 9.03 | ± 5.84 |
Table 2.
Chi Square p-values of the pair-comparison among production categories.
| Chi Square P | Reproduction | Work/Meat | Competition |
|---|---|---|---|
| Equestrian | 0.23 | 0.72 | 0.23 |
| Competition | 0.96 | 0.41 | |
| Work/Meat | 0.40 |
Table 3.
Chi Square p-values of the pair-comparison among macro-regions.
| Chi Square P | Sicily | Sardinia | South | Center |
|---|---|---|---|---|
| North | 0.43 | 0.740 | 0.272 | 0.73 |
| Center | 0.23 | 1.00 | 0.130 | |
| South | 1.00 | 0.11 | ||
| Sardinia | 0.25 |
Table 4.
Prevalence of NS3 Mutations in the overall study population (T=35) and stratified according to the different production category. T= total sequences for each column; N= number of sequence showing the mutation indicated in the row heading (%)= percentage of sequence with the given mutation on the total.
Table 4.
Prevalence of NS3 Mutations in the overall study population (T=35) and stratified according to the different production category. T= total sequences for each column; N= number of sequence showing the mutation indicated in the row heading (%)= percentage of sequence with the given mutation on the total.
| NS3 mutation | Overall, N (%) | Competition, N (%) T=12 | Equestrian, N (%) T=8 | Reproduction, N (%), T=5 | Work meat, N (%) T=10 |
|---|---|---|---|---|---|
| T490V | 27 (77.1) | 9 (75) | 6 (75) | 4 (80) | 8 (80) |
| L358I | 7 (20.0) | 1 (8.3) | 2 (25) | 1 (20) | 3 (30) |
| S488T | 5 (14.3) | 2 (16.7) | 0 (0) | 1 (20) | 2 (20) |
| V377I | 3 (8.6) | 0 (0) | 0 (0) | 0 (0) | 3 (30) |
| N405K | 2 (5.7) | 1 (8.3) | 0 (0) | 1 (20) | 0 (0) |
| V447I | 2 (5.7) | 1 (8.3) | 1 (12.5) | 0 (0) | 0 (0) |
| D396E | 1 (2.9) | 1 (8.3) | 0 (0) | 0 (0) | 0 (0) |
| S356A | 1 (2.9) | 1 (8.3) | 0 (0) | 0 (0) | 0 (0) |
| S357A | 1 (2.9) | 1 (8.3) | 0 (0) | 0 (0) | 0 (0) |
| T358L | 1 (2.9) | 1 (8.3) | 0 (0) | 0 (0) | 0 (0) |
Table 5.
Prevalence of NS3 Mutations in the overall study population (N=35) and stratified according to geographical area: (a) Centraly Italy includes 9 EqHV strains isolated from Latium (N=6) and Tuscany (N=3); (b) Southern Italy includes 5 EqHV strains isolated from Apulia, Basilicata, Molise, Campania and Abruzzi (N=1 for each region); (c) Northern Italy includes 3 EqHV strains isolated from Lombardy, Friuli Venezia Giulia and Trentino Alto Adige (N=1 for each region).
Table 5.
Prevalence of NS3 Mutations in the overall study population (N=35) and stratified according to geographical area: (a) Centraly Italy includes 9 EqHV strains isolated from Latium (N=6) and Tuscany (N=3); (b) Southern Italy includes 5 EqHV strains isolated from Apulia, Basilicata, Molise, Campania and Abruzzi (N=1 for each region); (c) Northern Italy includes 3 EqHV strains isolated from Lombardy, Friuli Venezia Giulia and Trentino Alto Adige (N=1 for each region).
| NS3 Mutations | Overall, N (%) N=35 | Sardinia, N (%) (N=12) | Sicily, N (%) (N=6) | Central Italya, N (%) (N=9) | Southern Italyb, N (%) (N=5) |
|---|---|---|---|---|---|
| T490V | 27 (77.1) | 10 (83.3) | 4 (66.7) | 6 (66.7) | 5 (100) |
| L358I | 7 (20.0) | 4 (33.3) | 1 (16.7) | 1 (11.1) | 0 (0) |
| S488T | 5 (14.3) | 5 (41.7) | 0 (0) | 0 (0) | 0 (0) |
| V377I | 3 (8.6) | 2 (16.7) | 1 (16.7) | 0 (0) | 0 (0) |
| N405K | 2 (5.7) | 2 (16.7) | 0 (0) | 0 (0) | 0 (0) |
| V447I | 2 (5.7) | 0 (0) | 1 (16.7) | 1 (11.1) | 0 (0) |
| D396E | 1 (2.9) | 1 (8.3) | 0 (0) | 0 (0) | 0 (0) |
| S356A | 1 (2.9) | 0 (0) | 0 (0) | 1 (11.1) | 0 (0) |
| S357A | 1 (2.9) | 0 (0) | 0 (0) | 1 (11.1) | 0 (0) |
| T358L | 1 (2.9) | 0 (0) | 0 (0) | 1 (11.1) | 0 (0) |
Table 6.
Localization of the identified mutations within the NS3 domains.
| NS3 Mutations | Localization as secondary structure | NS3 Functional Domain |
|---|---|---|
| S356A | Helix 356-358 | Helicase Subdomain II |
| S357A | Helix 356-358 | Helicase Subdomain II |
| T358L | Helix 356-358 | Helicase Subdomain II |
| L358I | Helix 356-358 | Helicase Subdomain II |
| V377I | Helix 371-384 | Helicase Subdomain II |
| D396E | Coil 392-405 | Helicase Subdomain II |
| N405K | Strand 405-410 | Helicase Subdomain II |
| V447I | Helix 447-502 | Helicase Subdomain II |
| S488T | Helix 488-500 | Helicase Subdomain III |
| T490V | Helix 488-500 | Helicase Subdomain III |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



