
Submitted:
21 March 2024
Posted:
26 March 2024
You are already at the latest version
Abstract
Analysis of liquid biopsy samples can reveal a wealth of molecular as well as cellular entities circulating in various body fluids. These entities include circulating tumor cells, circulating cell-free and tumor DNA, RNA, and proteins. Detection and characterization of this genetic and proteomic material can provide crucial information on tumor characteristics, including its heterogeneity, biology, tumor progression, staging, associated gene mutations and the course of therapy. The increased use of liquid biopsy in clinical oncology offers in non-invasive manner easier tumor sampling and continuous monitoring of the disease evolution through repetitive sampling from cancer patients. The continuous monitoring offered by this method is highly relevant to the assessment of a given therapeutic regime and early detection of markers associated with possible drug resistance, which may develop in the course of the therapy.
The use of proteomic approaches, including mass spectrometry-based proteomics for the analysis of biofluids extends for over three decades, yet only in the last few years such methods have found their way to clinical oncology. The non-invasive nature of liquid biopsy and the simple procedures for sample collection made this approach far more attractive than the traditional and well-established tissue biopsy. Such preference becomes more practical in the analysis of solid tumors, particularly in pediatric population. In this review we discuss some works on the emerging role of liquid biopsy analyses in clinical oncology, and how such use allows a better understanding of diagnosis, prognosis, disease evolution and response to therapy of various forms of cancer. We also discuss the role of MS-based methods in the characterization of post-translational modifications of proteins circulating in liquid biopsies.
Keywords:
Liquid biopsy
; clinical oncology
; mass spectrometry-based proteomics body fluids
; circulating biomarkers
; Protein post translational modifications
1. Introduction
Tissue biopsy has been and remains the gold standard for tumors profiling. However, the invasive nature of this method together with other limitations have led to an increased interest in developing less invasive alternative method. Liquid biopsy (LB) has been identified as a complementary and in increasing number of analyses as alternative to tissue biopsy. Currently there is a justified enthusiasm regarding the potential of liquid biopsy as an alternative to tissue biopsy for the molecular profiling of cancers. This enthusiasm however, still needs to be further supported with more robust clinical data., particularly regarding the design of sensitive, specific and reproducible assays for the early detection of various forms of cancer.
The term “liquid biopsy” was first coined over a decade ago [1]. Initially, this method was applied to investigate circulating tumor cells (CTCs) [2,3] and subsequently it was extended to circulating tumor DNA (ctDNA) [4]. It is fair to say that the term liquid biopsy is relatively recent, yet the analytical principle of the method is much older. For almost three decades mass spectrometry-based proteomics has been used to analyze various body fluids in the search for proteins to serve as biomarkers for various disease conditions, including various forms of cancer [5,6,7].
Over the last ten years, molecular profiling of tumors has partially shifted from an invasive surgical sampling to liquid biopsy sampling. The biggest advantage of this shift is the ease of effecting repetitive sampling throughout the course of the disease, which is essential to gather clinical information on the most recent tumor evolution. Such information are crucial for tailoring therapy regimes based on the most recent evolution of the disease. Other reasons for such shift can be partially justified by the following considerations:
- tissue biopsy cannot be performed in certain anatomical sites, which interferes with timely diagnosis/prognosis of many forms of solid tumors, particularly in pediatric patients. Acquiring samples by an invasive method throughout treatment to monitor tumor progress, response to therapy, and relapse pose a major challenge to tumor profiling [8]. Such challenge manifests more clearly in molecular profiling of pediatric solid tumors. For instance, malignant primary brain tumors are the most common cancers in children under the age of 14 years [9]. These forms of cancer are not only difficult to diagnose but also difficult to monitor their response to therapy. Such difficulties are directly linked to tissue biopsy, which is not suitable for repetitive sampling and cannot be performed in certain anatomical locations [10]. Biopsies through surgery have further limitations in terms of time, cost of repeatability and age of patient [11].
- Molecular characterization obtained by tissue biopsy is intrinsically partial and often not informative due to low quantity and/or poor-quality of tumor DNA. On the other hand, tumor cells from primary and metastatic sites, which are shed into various body fluids at sufficient concentration can offer. the chance to analyze samples containing tumor genetic and proteomic materials (see Figure 1). Furthermore, repetitive sampling through liquid biopsies has the potential to track the evolutionary dynamics and heterogeneity of tumors and may indicate very early emergence of resistance to therapy, residual disease and recurrence. It can be said that the evolution of targeted therapy emphasized the need for longitudinal monitoring of molecular changes within tumors to help guiding the therapeutic regimes. In addition, non-invasive, longitudinal assessment of tumors is necessary for confident tracking of potential biomarkers shed into various body fluids of patients [12,13].
- It is becoming evident that liquid biopsies capture part of the tumor shed into a body fluid. Analyses of circulating tumor cells and circulating tumor DNA obtained in a minimally invasive method may furnish molecular information highly relevant to the characterization of the parent tumor of these circulating components. Initial application of liquid biopsies in clinical oncology were limited to the investigation of circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA). Further development of the method extended such investigations to other molecular entities, which can be divided into two groups, the first includes both large and small molecules without cells and/or without a subcellular structure in the body fluid; these include proteins, nucleic acids, lipids, carbohydrates and metabolites.
The second group includes entities with cellular or subcellular structures, including single or clustered circulating tumor cells (CTCs), circulating cancer-related fibroblasts (CAF), immune cells, tumor-educated platelets (TEP) [14], extracellular vehicles (EVs) and circulating mitochondria [15,16]. Early liquid biopsy investigations were limited to blood samples, present day investigations use a wide range of body fluids, including blood, plasma, urine, saliva, seminal plasma, and. cerebrospinal fluid (CSF). Currently, there is a prevailing scientific opinion that data generated by the analysis of these circulating molecular entities in liquid biopsy samples is a major step towards personalized therapy, which is one of the main objectives of precision medicine. [17].
As mentioned above, the diversity of the entities circulating in liquid biopsy samples demands the application of different analytical approaches to obtain meaningful and reliable information regarding the correlation between the detected molecules/cells and the investigated disease(s). These approaches can be divided into genomic and proteomic analysis. Investigating circulating tumor cells (CTCs), cell free DNA (cfDNA), circulating tumor DNA (ctDNA) are examined by genomic technologies, while proteins, peptides and various metabolites are examined by proteomic analysis. It is commonly agreed that liquid biopsy samples are rich with highly informative molecules both small and large. That said, the utility of the generated data in clinical oncology is highly dependent on the initial concentration of a given moiety, the method of enrichment, the efficacy of extraction of the circulating entities and the specificity and sensitivity of the method of detection. On the genomic side, there are a number of well tested technologies, which can be applied to detect extremely low levels of circulating tumor DNA fragments and corelate such levels to tumor mutations [18]. These technologies include, Digital droplet polymerase chain reaction [19], beads, emulsification, amplification and magnetics PCR (BEAMing PCR) [20] and next-generation sequencing (NGS) also known as massively parallel sequencing, which is rapidly being incorporated into oncology practice [21]. Discussing these technologies in more details is outside the scope of this review, however, analysis of (LB) samples and data generated by some of these technologies will be discussed.
Currently, the discovery and validation of cancer biomarkers is drawing intense research activities within clinical oncology. Discovery of biomarkers for the early detection, correct classification of risk groups and treatment efficacies are among the priorities of such activities. Genomics and transcriptomics have identified various cancer-driving genes associated with the initiation and progress of various forms of cancer. Datasets generated by these approaches are central to the area of biomarkers discovery, however such datasets are not necessarily sufficient to have a comprehensive insight into the investigated disease biology. Such comprehensive picture is likely to be achieved through the combination of those datasets to those generated by proteomic analysis.
Traditionally, clinical diagnostics have strongly relied on antibody-based detection strategies, however, these methods are known to have a number of limitations, which encouraged the search for alternative methods. For over 20 years, mass spectrometry (MS) based-proteomics have been used to detect and characterize biological macromolecules both in bodily fluids and in tissues. [22,23]. In the last ten years MS-based methods to investigate liquid biopsy samples has gained a fresh momentum not only for biomarkers discovery but also to understand disease biology. Recent advances in MS instrumentation and the relatively recent combination of the well-established liquid chromatography/MS (LC/MS) with ion mobility (IM) together with more refined protein labelling procedures rendered this analytical platform a key player in the analysis of liquid biopsy samples within clinical oncology research. In this review we attempt to discuss the emerging role of liquid biopsies and how such role providing genetic and proteomic information relevant to biomarkers discovery and to our understanding of the biology of some diseases.
2. Discussion
2.1. Is Liquid Biopsy an Alternative or Complementary to Tissue Biopsy?
Tissue biopsy remains the gold standard for diagnosis and prognosis of various forms of cancer. This surgical method is also used to cure the majority of localized cancers without the need for systematic therapy [24]. That said, this method of sampling has a number of limitations, including invasiveness, which limits repetitive sampling necessary for realistic monitoring of the evolution of a given tumor. Tissue biopsy has the limitation of reflecting a single point in time of a single site of the tumor, which renders the method inadequate for a comprehensive characterization of the tumor under examination [25]. This sampling method which is limited in space and time not only fails to capture the heterogeneity of the tumor but also does not reflect the tumor microenvironment, which is known to influence tumor progress and its response to therapy. In the last ten years there has been a clear shift from tissue biopsy to liquid biopsy. The reasons for such shift have been pointed out earlier in this text.
A number of recent investigations have underlined two issues, which may interfere with the full adoption of liquid biopsy as an alternative to tissue biopsy. The first issue is the diagnostic sensitivity of liquid biopsy, existing evidence shows that patients with early-stage cancers can harbor less than one mutant template molecule per milliliter of plasma [4,26], which is beyond the limit of detection of existing technologies that assess multiple mutations simultaneously [27,28]. The second issue is the identification of the tissue of origin. Because the same gene mutations drive multiple tumor types, liquid biopsies based on genomic analysis alone generally cannot identify the anatomical location of the primary tumor. Although the application of liquid biopsies in cancer research has made a significant progress, today’s diagnosis of many forms of cancer are still mainly based on the analyses of the primary tumor and follow-up is usually performed by imaging techniques which cannot provide detailed information on tumor evolution. Such information necessitates longitudinal monitoring of the molecular changes within tumors in order to precisely guide anti-cancer treatment. Hence, there is a compelling need for the indirect assessment of tumors via less-invasive tumor markers which (LB) sampling can offer [12,13].
2.2. Liquid Biopsy in Pediatric Tumors
Tumors of the central nervous system (CNS) are the most frequent solid tumor types in children, and the major cause for cancer-related mortality in this age group. The biology of these forms of cancer is known to be highly heterogeneous and comprise various subpopulations [29,30] Currently, there is sufficient evidence showing that the combined cerebrospinal fluid (CSF) cytology and radiological neuroimaging is not sensitive enough to follow the development and metastasis of these cancers. Diagnosis, prognosis and patient’s stratification of these malignancies are likely to benefit most from the increasing use of liquid biopsies in clinical oncology. Liquid biopsies are of particular interest for pediatric oncology as they are noninvasive, enabling serial sampling, which allow for more reliable monitoring of a given therapy, avoid procedural sedation and contribute to the identification of patients, which are likely to benefit from targeted therapy [31]. In recent years there has been an increasing use of liquid biopsy as a sampling method., which is having a significant impact on pediatric oncology. Till very recently, investigating pediatric solid tumors relied heavily on the use of the highly invasive tissue biopsy method both for diagnosis and for cure. Apart from excessive invasiveness, surgical biopsy cannot be performed for some anatomical locations, furthermore, the resected tumor samples frequently do not reflect the genetic heterogeneity of the tumor. In recent years liquid biopsies have rapidly evolved to provide greater details about tumor characteristics such as tumor progression, tumor staging, heterogeneity and gene mutations. The incorporation of molecular profiling into the treatment of pediatric CNS tumors contributed to a more personalized approach to therapy. More recently, the World Health Organization (WHO) has begun incorporating molecular profiling into the diagnosis of specific tumor types [32]. The wide spectrum of the circulating analytes in liquid biopsies render them an attractive source of both genetic and proteomic data relevant not only for the profiling of a given tumor but also for the understanding of its biology. That said, liquid biopsy still faces a number of difficulties, which have to be resolved before this powerful method of sampling realizes its full potential. One of these difficulties is how to obtain meaningful and useful clinical information through the detection and characterization of circulating molecular/cellular entities. Circulating. tumor cells (CTCs) can be taken as a representative example of such challenge. These cells are initially released from primary tumors in the tissue, travel through the circulatory system and are implicated in the development of metastasis (or secondary) tumors at distant sites in the body [33]. We know that CTCs circulate in body fluids at extremely low levels, which renders their extraction and enrichment fairly challenging. For example, a patient sample may contain 10 of these circulating cells in a background of 106 white blood cells and 109 red blood cells [34,35]. Moreover, CTCs are known to form cellular aggregates with cells like fibroblasts and platelets. These aggregates have been reported to spread to more distant sites in the body relative to their isolated CTC counterparts. [36,37].
Another limitation of liquid biopsy sampling is the difficulty to identify the tissue(site) of origin of the circulating molecules/cells. Establishing the site of origin is relevant to the process of molecular profiling, which can be enriched by using molecular information derived from different anatomical sites, both at baseline and recurrence. Furthermore, reliable assignment of the tissue of origin can also contribute to the ongoing attempts to understand certain mechanism(s) of multidrug resistance (MDR). these attempts are seeking to address the following question: Is drug resistance to a given therapy is exclusively linked to the cancerous cells or is it a combined effect of these cells as well as their microenvironment.? Currently, data generated using liquid biopsies do not allow reliable correlation between the detected entities and the site of origin. Another issue to be considered in pediatric liquid biopsy analysis is the low mutational burden compared to their adult counterparts. Recent landmark sequencing studies have demonstrated that genetic mutations in most childhood cancers are substantially lower than those in adult cancers [38,39]. Furthermore, fusion genes are more common than in adult cancers, and certain specific mutations found in pediatric cancers are rare in adult counterparts. On the other hand, epigenetic dysregulations such as DNA methylation and histone post-translational modifications (PTMs) tend to have a greater impact on the biology of diseases in childhood. These observations suggest that Liquid biopsy analysis in childhood cancers is likely to be more fruitful by monitoring circulating entities associated with both genetic as well as epigenetic modifications
2.3. Proteomic Analysis of Circulating Proteins in Pediatric Liquid Biopsies
The complexity of the proteome within liquid biopsies is one of the reasons why proteomic analyses of these samples are still lagging behind their genomic counterparts. Currently, many predictive biomarkers of various forms of pediatric cancers are based on the analyses of circulating genomic material obtained by liquid biopsies. MS-based proteomics has long been applied in the search for protein biomarkers in various body fluids, where most analyses focused on serum/plasma and urine. The main challenge in this type of analyses is the absence of specific, sensitive and reproducible assays suitable for the identification and quantification of proteins in complex biological matrices such as blood and plasma. Taking plasma as a representative example of such complexity we encounter a wide dynamic range of more than ten orders of magnitude in protein abundances, with only 22 proteins constituting 99% of the protein content [40]. These proteins concentration range from serum albumin (50 mg/mL), immunoglobulins and coagulation factors down to small protein hormones and cytokines (pg./mL) [41]. This wide dynamic range has made it difficult to study the plasma proteome due to the masking of often low abundant potential disease biomarkers by the few very highly abundant proteins. It is becoming more evident that to obtain meaningful and reliable proteomic data, the assay has to be specific, highly sensitive, capable to detect and quantify the major number of proteins present in the investigated sample and above all reproducible. Another and equally challenging issue in these analyses is the quantification of proteins with certain (PTMs). For example, a single glycosylation site can be occupied by multiple, heterogeneous glycated structures, which inevitably results in different glycoforms of the same protein This effect causes glycopeptide signal distribution and inevitable signal reduction of the individual structures [42]. Another commonly encountered PTMs, which can impact on the reliability of the analyses is the presence of disulfide bonds, a modification, which can be responsible for poor digestion, leading to poor sequence coverage. Furthermore, the MS/MS spectra generated in the presence of disulfide-linked peptides are not easy to interpret [43,44]. More details on some PTMs and their use as potential disease biomarkers are discussed below.
Traditionally, antibody-based detection methods have been used to identify proteins in biofluids. These methods however, have a limited capability in terms of simultaneous identification of high number of proteins. The emerging role of MS-based proteomics not only overcome such limitation but also facilitate high throughput analyses of clinical samples.
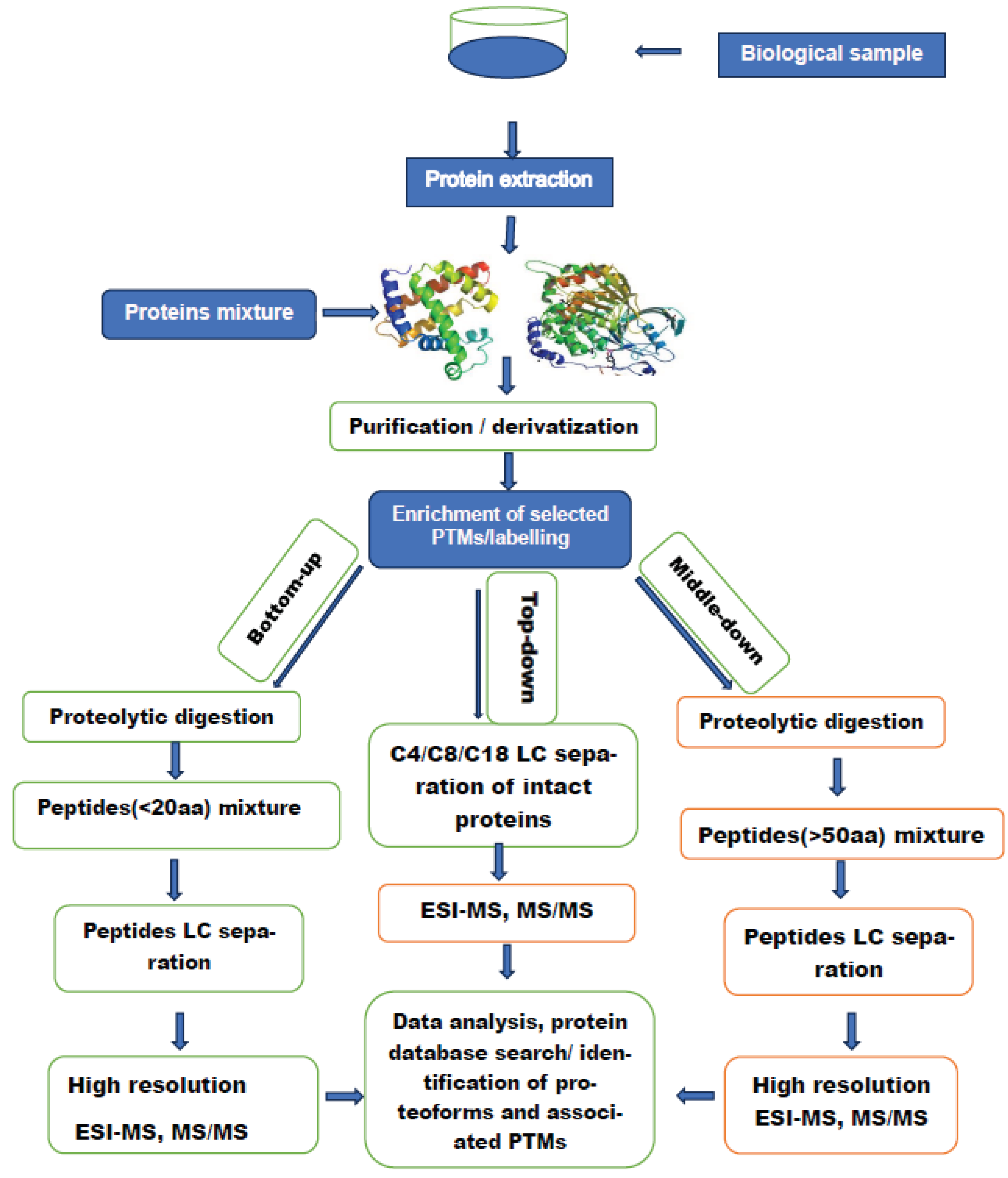
Almost 30 years ago the term “shot gun” was coined by Yates’s group [45]. This approach uses liquid chromatography (LC) coupled to mass spectrometry (MS, MS/MS) to investigate complex protein mixtures. The (LC) phase separates a mixture of peptides derived from enzymatic digestion of a protein mixture. Separated peptides are injected into an electrospray ion source to generate multiply charged ions. This mixture of charged peptides is then separated according to their mass to charge (m/z) ratios and focused into a collision cell to be fragmented and subsequently detected and interpreted. Modern mass spectrometers accommodate various modes of ion activation/fragmentation, including collision gas to perform collision induced dissociation (CID) both at low and high collision energy, electron-based fragmentation, which can be performed through electron transfer (ET) or electron capture (EC) and UV photodissociation (UVP). The experimental arrangement in Figure 2 has three versions, “Top-down”, “middle-down” and” bottom-up”. [46,47]. Understandably, Figure 2. does not include modifications or variations, which can be introduced by individual research groups. For example, Proteolytic digestion of intact proteins can be replaced with 1-D or 2-D gel separation, followed by spot excision and in-gel pyrolytic digestion [48]. The figure describes the three methods in a general manner, therefor additional steps and/or modifications to handle specific analysis are to be expected. For instance, in a typical bottom-up experiment, proteins are digested with trypsin. However, the use of this enzyme to digest certain class of proteins (e.g., histone proteins) can result in very short peptides, which are poorly retained on reversed phase (LC) columns. Furthermore, these short peptides do not acquire sufficient charge states in ESI ion source, leading to extremely poor MS/MS analysis [49]. To attenuate both limitations, derivatization of free amine groups on the N termini and lysines before a trypsin digestion is frequently practiced to mimic an Arg-C digestion, which permits the use of trypsin [50,51]. Another class of proteins, which call for specific modifications is membrane proteins. These proteins are not water soluble which render them unamenable to LC-MS/MS. Analysis of this class of proteins require the use of particular detergents compatible with mass spectrometry [52].
Although the three methods indicated in Figure 2 use LC-MS/MS, bottom-up has emerged as the most diffused method for high throughput in large scale proteomic analysis. That said, the use of trypsin for digestion can in certain situations result in short peptides and the failure to detect and localize labile PTMs. One of the solutions to avoid such limitation is the use of “top-down” method, which analyses intact proteins and thus gives a higher possibility to detect labile PTMs.
To demonstrate the utility of this technique, one group used LC-MS/MS to characterize the CSF proteome of patients with brain tumors to identify potential biomarkers predictive of metastatic spread [53]. Cerebrospinal fluid (CSF) samples derived from 27 children with brain tumors and 13 controls were processed using core-shell hydrogel nanoparticles, a method designed to simultaneously perform size exclusion and affinity chromatography in solution [54]. The main objective of the study was to Identify protein biomarkers predictive of metastatic spread. In the same study a number of the detected proteins were further confirmed using western blotting (WB), reverse-phase protein array (RPPA) and enzyme-linked immunosorbent assay (ELISA). This well-designed study reported the following conclusions: LC-MS/MS measurements revealed the presence of over five hundred non-abundant proteins, almost hundred and fifty of which were missing from the CSF database. The same study reported that six of these proteins could be used to differentiate metastatic cases from controls. Considering the number of samples used in this investigation, results obtained represent a useful example as proof of concept. These positive results have been obtained under difficult analysis conditions. comprehensive analysis of CSF is challenged by low protein levels (0.3-0.7 mg/ml), protein concentration range extends over 12 orders of magnitude, which masks low concentration brain-specific proteins [55]. To attenuate the complexity of the proteins profile in CSF samples the authors used immunoaffinity depletion of high-abundant CSF proteins as well as fractionation and separation prior to MS analysis. To translate the promising results of this study into potential biomarkers for metastatic spread of the investigated disease further analyses involving much higher number of samples is necessary. Such further analyses accompanied with reliable validation at the clinical level may lead to promising biomarker(s).
An earlier study [56] used surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF-MS) [57] to compare protein expression profiles in cerebrospinal fluid from pediatric brain tumors and from pediatric controls. The main steps in SELDI analysis are: Sample deposition into the chromatographic surface, after a given time to allow protein capturing the unbound material is washed out and the captured proteins are covered with adequate laser absorbing matrix and introduced into the ion source of the instrument. In the final step, mass spectra are generated using laser desorption ionization and TOF analyzer. In their study the authors used a total of 32 lumbar CSF samples from newly diagnosed and untreated pediatric brain tumor patients (males and females; average age, 7.2 years) and 70 pediatric control patients (males and females; average age, 8.5 years). Prior to performing mass spectrometry analysis, CSF samples were examined by Cytologic analysis, which showed malignant cells in 10 medulloblastoma cases. CSF from patients with other tumors and from control patients did not show any cytologic abnormalities. Further details regarding sample preparation are given by the authors and we do not see the point in repeating them here. The authors concluded that SELDI-TOF mass spectrometry can be successfully used to find differentially expressed proteins in CSF of pediatric brain tumor and control patients. One of these proteins, apolipoprotein A-II was found highly overexpressed in CSF of pediatric brain tumor patients. This overexpression was tentatively attributed to a disrupted blood–brain barrier in tumor patients. These conclusions merit further considerations. SELDI-TOF is known to be highly suitable for high throughput qualitative screening of proteins in complex biological samples. This technique however, has two major limitations. One of these limitations is linked to the capture surfaces (Chips). These are chemically and/or biochemically modified surfaces designed to capture a given range of proteins within the deposited sample. By definition such range represents a small part of the proteome present in the CSF sample. Historically, SELDI-MS has been associated with low resolution TOF analyzers, simply because the technique was designed to perform high throughput screening of targeted protein profiles in complex biological samples.
2.3.1. Post-Translational Modifications as Disease Biomarkers
The human genome project established the number of human genes to be between 25000 and 30000 [58]. This relatively low number was rather surprising, yet a bigger surprise was revealed by subsequent investigations, which predicted that such low number of genes can express millions of proteins [59,60]. Such high number of gene products is mainly attributed to alternative splicing and post translational modifications (PTMs). Currently there are over 400 PTMs listed in Uniport database [61]. Years of research have established that Protein activities are not only controlled by the rates of protein biosynthesis and protein degradation but also by other processes, including PTMs, which modulate molecular interactions, redirect cellular protein localization, and promote or inhibit interactions with other proteins [62]. The central role of these modifications in diverse cellular functions explains their frequent implication in various human diseases, including various forms of cancer.
Mass spectrometry is considered the technique of choice for the detection and localization of PTMs. This observation is supported by current literature, which indicates that most data available on protein PTMs have been obtained by MS-based proteomic methods [63]. In recent years the role of this technique has been enhanced by a number of developments, including, new generation of high resolution/high mass accuracy instruments, tangible improvements in MS-based proteomic analyses, more efficient enrichment and labelling strategies, and the application of bioinformatic algorithms with more rigorous scoring strategies. PTMs. analyses are commonly conducted using liquid chromatography (LC) coupled to tandem mass spectrometry (MS/MS). More recently there has been more frequent use of ion mobility MS coupled to LC-MS/MS [64]. The coupling of ion mobility is considered an additional dimension in the analyses, which enhances the separation capability and the structural information provided by LC-MS/MS. Improvement in the performance of MS-based proteomics has been also strengthened by the introduction of electron-based, and photon–based ion activation methods [65,66,67,68,69] The use of these methods enhanced the capabilities of MS/MS analysis to detect and localize labile PTMs.
One of the main problems facing MS-based proteomic analysis of body fluids is the masking of low-copy proteins by few abundant proteins. This effect is strongly manifested in blood and in plasma proteomic analyses. Regardless of the identity of the biofluid, these dominant proteins compete for the PTMs, which renders the detection of modified low- copy proteins more challenging. To underline this observation, the following example can be considered representative. MS-based proteomics was used to profile circulating nucleosomes in plasma taken from patients with colorectal cancer (CRC). [70]. Intact circulating nucleosomes were captured using immunoprecipitation, captured nucleosomes were subsequently analyzed by LC- MS/MS. The authors reported 13 PTMs characteristic of (CRC), notably the methylation of Lys 9, Lys 27 and acetylation of Lys27 of histone H3. Although the authors used a relatively low number of samples (9 patients and 9 control), the reported results underline the importance of protein enrichment in samples dominated by few abundant proteins as is the case with plasma. The identification of Lys27 acetylation would have been fairly problematic in the presence of albumin peptides within the investigated digest. This observation is supported by an earlier study [71], where LC-MS/MS was used to investigate cancer patient serum samples following immunoaffinity Enrichment. In their study the authors reported that the abundant serum human albumin was found to be acetylated at 59 different sites. What these data say that without the removal of high abundant proteins, protein digest examined by LC-MS/MS will contain a peptide mixture, including those derived from an abundant protein with 59 acetylation sites. This scenario means that the detection and quantification of an acetylated peptide derived from a low- copy protein is going to be highly challenging.
2.3.2. Investigation of Histone Post Translational Modifications
Traditionally histone PTMs have been investigated using antibody-based approaches such as western blots and chromatin immunoprecipitation. Over the years these methods have furnished a wealth of information on the role and functions of various histone PTMs. These methods however, have a number of critical limitations, which have been pointed out in early works [72,73]. Most known antibodies are highly suitable for the detection of known PTMs but not the novel modifications. Many antibodies are not entirely site-specific and can cross react with similar modifications on different amino acids [74].
In this part of the review, we discuss histone (PTMs), their detection and analysis using MS-based proteomics. The core histones are predominantly globular structures except for their N-terminal ‘‘tails,’’ which are unstructured. The main characteristic of these proteins, and particularly of their tails, is the large number and type of modifications, which these proteins can experience. There are at least eight distinct types of modifications found on histones [75]. The choice to discuss histone PTMs has a number of motives: i) the diversity, high frequency and complexity of these modifications render them highly challenging to most detection techniques, including the powerful gene array technology (Chip on CHIP) [76]. This method can detect the modification status over a range of 2–3 nucleosomes or even on a single nucleosome, but it cannot determine the modification status of different histones within the same nucleosome. This limitation means that this powerful method cannot determine if both copies of a histone are identically modified within a single nucleosome or whether there is a distinct pattern on each. Such limitation can be addressed using MS-based analysis. ii) specific histone (PTMs) has been described as general hallmarks of cancer and can represent potential diagnostic and prognostic biomarkers for some forms of cancer [77,78,79]. The widespread presence of histone aberrations in tumors supports the observation that the enzymes responsible for the addition and removal of histone PTMs are among the most frequently mutated class of genes in cancer [80].
It is now fairly accepted that most cancer cells harbor both genetic as well as epigenetic alterations, with a high number of modifications associated with histone proteins. The core protein components of chromatin in eukaryotes consists of five different Lys- and Arg-rich proteins (H1, H2A, H2B, H3, and H4) [81]. These histone proteins are assembled in octamers named nucleosomes, which are wrapped by DNA. Histone N-terminal tails are exposed outside the nucleosome, and they are heavily modified by dynamic post-translational modifications (PTMs). The heavy presence of Lys- in these tails is one of the reasons for the extensive modifications of these tails. Database on PTMs shows that Lys- undergoes the highest number of PTMs (15 PTM types) [82,83]. Although, histones are known to be highly packed structures, their highly basic-terminal tails are responsible for most of the modifications experienced by these proteins. The high number of different modifications experienced by the tails of H3 and H4 are of particular interest to the question of regulation of gene expression [84,85]. Early studies have focused on histone tail modifications, however, studies in the last decade have furnished new insights into the modifications within the globular structure of histones [86]. These studies have shown that the histone octamer lateral surface is in direct contact with DNA, making it less accessible than the histone tails. The same literature observes that nucleosomes are not static, and their DNA spontaneously unwraps from and rewraps onto the lateral surface, rendering chromatin accessible to modifiers [87]. This observation is supported by many PTMs identified within the histone globular domains map to either the lateral surfaces [88] or to interfaces between the histone proteins that comprise the octamers. Before discussing some examples on the use of MS-based proteomics in the search for PTMs as disease biomarkers, the following observations regarding the methods of analysis are of interest. I) bottom-up and middle-down methods exploit a number of advantages, which peptides have over intact proteins. Peptides are more easily separated by reversed-phase liquid chromatography (RPLC), have a higher ionization cross section within the electrospray ion source and have a much simpler fragmentation pattern, which is not irrelevant to a reliable identification of the investigated protein. The sum of these advantages translates into a robust platform that facilitates high-throughput analysis, which in some cases allow the identification and quantification of thousands of proteins from complex protein lysates [89]. The third workflow, “Top-down” method is less amenable to high throughput analysis, yet it is more efficient in the detection of labile PTMs, which can be missed in methods analyzing peptides rather than intact proteins. ii) bottom-up method relies heavily on protease digestion effected by trypsin. This method of digestion has the advantage of producing basic arginine or lysine at the C-terminal, which is ideal for traditional collision induced dissociation (CID). On the other hand, the high digestion efficiency of this enzyme has the back draw of producing short peptides (<6 amino acids). These short peptides cannot be retained efficiently by reversed-phase LC columns, and second, they do not retain sufficient charge states In ESI to provide meaningful fragments in the MS/MS analysis [23]. Such back draws of the method manifests more heavily in the analysis of proteins particularly rich in Lysine and arginine residues as in the case of histone proteins. The use of bottom*up method to investigate these proteins require various derivatization procedures prior to trypsin digestion [90,91]. The other alternative to performing derivatization option is to use top-down or middle-down methods [92,93].
Over the last ten years there have been intense research activities to understand the role of histone PTMs in the initiation and progression of various diseases. These studies have established that histone post-translational modifications (PTMs) are essential for epigenetic regulation and maintenance of major DNA processes such as replication, transcription, and chromatin remodeling [75,94] Lawrence et al. 2016. Some of these investigations emphasized the central role of proteomic analysis in understanding the link between these modifications and various diseases.
Correlation between some of these PTMs and cancerous cells were reported over twenty years ago [95]. The authors used immunodetection, high-performance capillary electrophoresis and LC-MS, MS/MS to investigate histone H4, isolated from fresh primary tumors and from their normal counterparts. These analyses were compared to results acquired for H4 from cell lines, and found to be very similar. The main conclusions of this study were: first, in normal tissues, 60% of total histone H4 was found monoacetylated compared to 35% in primary tumors. Second, cancer cells had a loss of monoacetylated and trimethylated forms of histone H4, losses which, appeared early and accumulated during the tumorigenic process. The losses occurred predominantly at the acetylated Lys16 and trimethylated Lys20 residues. and were associated with the hypermethylation of DNA repetitive sequences, a well-known characteristic of cancer cells. The same data suggest that the global loss of monoacetylation and trimethylation of histone H4 is a common hallmark of human tumor cells. In a more recent study [96] the authors investigated plasma samples from 106 healthy controls and from the same number of patients diagnosed with CRC within 24 months following participation in The Trøndelag Health Study (The Trøndelag Health Study (HUNT) is a population-based cohort of ∼229,000 individuals recruited in four waves beginning in 1984 in Trøndelag County, Norway) [97]. The authors reported that the detection of known methylated ctDNA markers of CRC is possible up to two years prior to the clinical diagnosis. In other words, some patients could be diagnosed earlier, if ctDNA detection was part of the CRC screening program.
General Remarks
- 4.
- Both research scientists and treating oncologists are in agreement that liquid biopsies (LBs) could be a valid alternative to surgical biopsies as a method for molecular profiling of various forms of cancer. This agreement is based on a number of advantages offered by this emerging method. Which are missing in tissue biopsy. First, noninvasive repetitive sampling throughout the course of the disease, such characteristic is essential to have continuous update on the evolution of the tumor, which gives a robust indication on response to therapy and early warning of emerging drugs resistance. Analysis of genetic and proteomic material circulating in liquid biopsy samples furnish detailed information on the heterogeneity of tumor, which are necessary for correct patient stratification and the choice of therapy. It is interesting to note that Intratumor heterogeneity revealed by multiregion sequencing implies that analysis based on tissue biopsy samples can lead to underestimation of the tumor genomics landscape, which may present major challenges to personalized-medicine and biomarkers development [98] Other advantages of liquid biopsies have been mentioned earlier in the text.
- 5.
- Existing literature suggests that pediatric neuro-oncology is one area, which is likely to benefit most from the increasing use of (LBs) in the molecular analysis of tumors. Due to the high risk associated with tissue biopsy, diagnosis and understanding of pediatric brain tumors have lagged behind other forms of cancer. The shift from surgical biopsies to (LBs) allowed access to certain anatomical zones, which could not be accessed by surgery., and second, serial sampling and major information on the heterogeneity of the disease allow more accurate patient stratification and almost real time monitoring of the evolution of the tumor. Both elements are essential on the path leading to more successful personalized therapy. The application of (LBs) for molecular diagnosis and treatment response in pediatric brain tumors has been demonstrated in four recent studies [99,100,101,102].
- 6.
- We asked in this text whether (LB) is an alternative to tissue biopsy.? Currently, it is immature to talk about complete displacement of tissue biopsy in favor of liquid biopsy. This observation can be supported by the following considerations. Tissue biopsy is not only a sampling method but also a method of cure for the majority of localized cancers without the need for systematic therapy [103]. At the present time, most genetic and proteomic assays designed to characterize circulating entities in (LBs) are not sensitive enough to allow a complete shift from tissue biopsy. In other words, such transition is dependent on the extent of progress in oncology research with all its components to put in place standardized assays of high sensitivity, high specificity, reproducible and easy to transfer and implement in clinical settings The authors believe that the complexity and the vast diversity of tumors will ensure that both methods of sampling will continue to serve oncology research for years to come.
- 7.
- Various works cited in this text suggests that many (LBs) with a focus on early detection [104] of various forms of cancer lack the sensitivity for a reliable detection of early-stage cancers This observation applies to both genetic and proteomic cancer biomarkers. So far information in hand suggest that tumor derived genetic biomarkers are not always shed into the blood stream in early stages, and even when they are shed into the bloodstream, they exist at very low concentrations [105,106]. Similar observations can be made regarding cancer protein biomarkers. For example, prostate specific antigen (PSA) and carcinoma antigen-125 (CA-125) are often not elevated in cancer patients, even in those with advanced stage of the disease [4]. Furthermore, both markers lack specificity as they can be elevated in cancer-free patients [107].
- 8.
- Many forms of cancer can be completely cured if they are detected in their very early stages, Successful cure can be also enhanced if these cancers can be predicted through reliable identification of predictive biomarkers. Most of these potential biomarkers are found in liquid biopsies of both healthy controls and patients. Currently, many predictive biomarkers for various forms of cancer are based on the investigation of genomic material obtained by both liquid and tissue biopsies. Biomarkers based on the proteomic analyses of the same media are still lagging behind their genomic counterparts. The gap between the two approaches has different reasons, including the complexity of the proteome compared to the genome and the extremely wide dynamic range of the investigated proteins. Despite notable progress in proteomic research, the area of validated predictive biomarkers transferable to clinical oncology remains poorly served, there is a prevailing opinion that to enhance the role of the proteomic analyses of body fluids in the search for predictive biomarkers , assays more sensitive , more specific reproducible and easy to transfer to clinical practice are necessary, More rigorous selection and validation of potential disease biomarkers and their efficient transfer to clinical practice. A successful transfer requires rigorous. standardization of the working practices regarding all aspects of (LBS) analyses
- 9.
- Molecular diagnostics based on tumor’s genomic analysis, including those based on (LBs) reveals a complex and highly challenging task for treating oncologists and researchers alike. The evolving list of diagnostic, prognostic and predictive biomarkers in some way interferes with a reliable tracking of the link between actionable genomic alterations and targeted therapies in clinical trials. Most recent literature reports dozens to hundreds of genes, which have to be considered in the process of choosing a given therapy. One of the initiatives to help treating oncologists in decision making has been recently reported [108] The Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy (IPCT) at MD Anderson Cancer Center has developed a knowledge base, available at https://personalizedcancertherapy.org or https://pct.mdanderson.org (PCT). This knowledge base provides information on the function of common genomic alterations and their therapeutic implications.
Conclusions
Current literature on cancers emphasizes the emerging role of liquid biopsy as a non-invasive sampling method for the investigation of various forms of cancer and other diseases. Analysis of molecular and cellular entities shed into various body fluids provide genetic, epigenetic and proteomic data crucial for the understanding of tumor characteristics, including its heterogeneity, its biology, tumor progression, staging, associated gene mutations and the evolution of the disease and its reaction to therapy. The increased use of (LBs) in cancer research is relatively recent, yet the benefits of this method of sampling is already evident in the diagnosis, prognosis and stratification of various forms of cancer, including those in pediatric age. Furthermore, datasets generated by the above analyses represent an important step towards personalized medicine. However, before achieving this ambitious objective, oncology research has to overcome a number of limitations, which have been pointed out within this text, some of these limitations are worth emphasizing i) currently, analyses of (LB) samples cannot identify the tissue of origin of the detected molecules/cells. The difficulty of such identification is linked to the hypothesis that the same gene mutations drive multiple tumor types. liquid biopsies based on genomic analysis alone generally cannot identify the anatomical location of the primary tumor. Establishing the tissue of origin is relevant to the process of molecular profiling, which can be enriched by using molecular information derived from different anatomical sites
ii) Many liquid biopsy strategies being developed for the early detection of cancer lack the sensitivity and specificity required to detect early-stage cancer biomarkers. Existing evidence shows that patients with early-stage cancers can harbor less than one mutant template molecule per milliliter of plasma, which is beyond the limit of detection of existing technologies that monitor multiple mutations simultaneously. The same literature indicates that there is a limited number of assays with sufficient sensitivity and specificity to allow the detection of biomarkers associated with the early stages of cancers, this observation is applicable to both genetic and proteomic biomarkers.
iii) Current information provided by the detection and characterization of circulating molecular/cellular entities in (LB) samples are not sufficient to allow reliable determination of the origin and dynamics of their shedding into the blood stream. Such determination would contribute to a better understanding of the molecular profiles based on the characterization of these entities, furthermore, it is important to establish whether these cells/molecules actually reflect the biology and the physiology of the disease. Such information would be central in guiding oncologists and physicians to a reliable detection and monitoring of the disease.
In conclusion and based on current literature on cancers it is too early to consider liquid biopsy as an alternative to tissue biopsy, instead the two sampling methods will continue to contribute to clinical oncology in tandem for years to come.
Abbreviations
| LB | Liquid biopsy. |
| CTCs | Circulating tumor cells. |
| ctDNA | Circulating tumor DNA. |
| CAF | Cancer-related fibroblasts |
| TEP | Tumor-educated platelets |
| EVs | Extracellular vesicles |
| CSF | Cerebrospinal fluid |
| PCR | polymerase chain reaction |
| DdPCR | Digital droplet polymerase chain reaction |
| BEAMing PCR | Beads, emulsification, amplification and magnetics PCR |
| NGS | Next-generation sequencing |
| LC/MS | Liquid chromatography/mass spectrometry |
| MS/MS | Tandem mass spectrometry |
| IM | Ion mobility |
| WHO | World Health Organization |
| CNS | Central nervous system |
| PTMs | post-translational modifications |
| CID | Collision induced dissociation |
| ET | Electron transfer |
| EC | Electron capture |
| UVPD | Ultra Violet photodissociation |
| WB | Western blotting |
| RPPA | Reverse-phase protein array |
| ELISA | Enzyme-linked immunosorbent assay |
| ESI | Electrospray ionization |
| SELDI | Surface-enhanced laser desorption/ionization |
| TOF-MS | Time-of-flight mass spectrometry |
| CRC | Colorectal cancer |
| RPLC | Reversed-phase liquid chromatography |
| PSA | Prostate specific antigen (PSA) |
| CA-125 | Carcinoma antigen-125 (CA-125) |
References
- Pantel, K.; Alix-Panabières, C. Circulating tumor cells in cancer patients: Challenges and perspectives. Trends in Molecular Medicine 2010, 16, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres,C; Pantel, K. Challenges in circulating tumour cell research. Nat Rev Cancer 2014, 14, 623–631. [CrossRef] [PubMed]
- Pantel, K.; Speicher, M.R. The biology of circulating tumor cells. Oncogene 2016, 35, 1216–1224. [Google Scholar] [CrossRef]
- Bettegowda, C.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci Transl Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Adkins, J.N.; Varnum, S.M.; Auberry, K.J.; Moore, R.J.; Angell, N.H.; Smith, R.D. : Springer, D.L.; Pounds, J.G. Toward a human blood serum proteome: Analysis by multidimensional separation coupled with mass spectrometry. Mol. Cell. Proteomics 2002, 12, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Russell, T.; Wood, G.; Desiderio, D.M. Analysis of the human lumbar cerebrospinal fluid proteome. Electrophoresis 2002, 23, 1185–1196. [Google Scholar] [CrossRef]
- Cazares, L.H.; Adam, B.L.; Ward, M.D.; Nasim, S.; Schellhammer, P.F.; Semmes, O.J.; et al. Normal, benign, preneoplastic, and malignant prostate cells have distinct protein expression profiles resolved by surface enhanced laser desorption/ionization mass spectrometry. Clin Cancer Res 2002, 8, 2541–2552. [Google Scholar]
- Perakis, S.; Speicher, M.R. Emerging concepts in liquid biopsies. BMC Med. 2017, 15, 112. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.K.; Enderle, D.; Noerholm, M.; Breakefield, X.O.; Skog, J.K. Exosome-Based Liquid Biopsies in Cancer: Opportunities and Challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Bounajem, M.T.; Karsy, M.; Jensen, R.L. Liquid biopsies for the diagnosis and surveillance of primary pediatric central nervous system tumors: A review for practicing neurosurgeons. Neurosurg. Focus 2020, 48, E8. [Google Scholar] [CrossRef]
- Azad, T.D.; Jin, M.C.; Bernhardt, L.J.; Bettegowda, C. Liquid biopsy for pediatric diffuse midline glioma: A review of circulating tumor DNA and cerebrospinal fluid tumor DNA. Neurosurg. Focus 2020, 48, E9. [Google Scholar] [CrossRef]
- In ‘t Veld, S.G.J.G.; Wurdinger, T. Tumor-Educated Platelets. Blood 2019, 133, 2359–2364. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A; Lim, A.R.; Ghajar, C.M. Circulating and Disseminated Tumor Cells: Harbingers or Initiators of Metastasis? Mol. Oncol. 2017, 11, 40–61. [CrossRef] [PubMed]
- Alix-Panabières, C. EPISPOT assay: Detection of viable DTCs/CTCs in solid tumor patients. Recent Results Cancer Res. 2012, 195, 69–76. [Google Scholar]
- Di Meo, A.; Bartlett, J.; Cheng, Y.; Pasic, M.D.; Yousef, G. M. Liquid biopsy: A step forward towards precision medicine in urologic malignancies 16:80. Molecular Cancer 2017, 16, 80. [Google Scholar] [CrossRef]
- Conteduca, V.; Wetterskog, D.; Sharabiani, M. T. et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Ann Oncol. 2017, 28, 1508–1516. [Google Scholar] [CrossRef]
- Hindson, C. M.; et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef]
- Denis, J. A.; Guillerm, E.; Coulet, F.; Larsen, A. K.; Lacorte, J. M. The role of BEAMing and digital PCR for multiplexed analysis in molecular oncology in the era of next-generation sequencing. Mol. Diagn. Ther. 2017, 21, 587–600. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos; N, Velculescu, V.E.; Zhou, S.; Diaz Jr, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Li, L.; Sun, C.; Sun, Y.; Dong, Z.; Wu, R.; et al. Tissue expansion. Nature Communications 2022, 13, 7242. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics 2017. CA Cancer J Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneityand branched evolution revealed by multiregion sequencing. N Engl J Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Cohen, J.D.; Javed, A.A.; Thoburn ,C.; Wong ,F.; Tie ,J.; Gibbs ,P.; Schmidt ,C.M.; Yip-Schneider, M.T.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci USA 2017, 114, 10202–10207. [CrossRef]
- Cree, I.A.; Uttley, L,; Woods, H.B.; Kikuchi, H.; Reiman, A.; Harnan, S.; Whiteman, B.L.; Philips, S.T. et al. The evidence base for circulating tumour DNA blood-based biomarkers for the early detection of cancer: A systematic mapping review. BMC Cancer 2017, 1, 697.
- Bardelli, A.; Pantel, K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell. 2017, 31, 172–179. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Ostrom Q., T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan J., S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2013-2017. Neuro-Oncol. 2020, 22, iv1–iv96. [Google Scholar] [CrossRef]
- Tang, K.; ; Gardner, S; Snuderl, M. The Role of Liquid Biopsies in Pediatric Brain Tumors. J Neuropathol Exp Neurol. 2020, 79, 934–940. [CrossRef]
- Louis, D. N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [CrossRef]
- Parkinson, D.R; Petty, B.G.; Compton, C.; Cristofanilli, M.; Deisseroth, A.; Hayes, D.F.; Kapke, G.; Kumar, P.; Lee, J.S. Considerations in the development of circulating tumor cell technology for clinical use. J Trans Med. 2012, 10, 1–20. [Google Scholar] [CrossRef]
- Mansilla, C.; Soria, E.; Ramirez, N. The identification and isolation of CTCs: A biological Rubik’s cube. Crit. Rev. Oncol. Hematol. 2018, 126, 129–134. [Google Scholar] [CrossRef]
- Morgan, T.M. Liquid biopsy: Where did it come from, what is it, and where is it going? Investig Clin Urol. 2019, 60, 139–141. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011, 20, 576–590. [Google Scholar] [CrossRef]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015, 7, 308re308. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 pediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Fruci, D.; Cho, W.C.S.; Nobili, V.; Locatelli, F.; Alisi, A. Drug Transporters and Multiple Drug Resistance in Pediatric Solid Tumors. Curr. Drug Metab. 2016, 17, 308–316. [Google Scholar] [CrossRef]
- Tirumalai, R.S.; Chan, K.C.; Prieto, D.R.A.; Issaq, H.J.; Conrads, T.P.; Veenstra, T.D. Characterization of the low molecular weight human serum proteome. Mol Cell Proteomics 2003, 2, 1096–1103. [Google Scholar] [CrossRef]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol Syst Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Larsen, M. R.; Højrup, P.; Roepstorff, P. Characterization of Gel-separated Glycoproteins Using Two-step Proteolytic Digestion Combined with Sequential Microcolumns and Mass Spectrometry. Mol. Cell. Proteomics 2005, 4, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Mysling, S.; Salbo, R.; Ploug, M.; Jørgensen, T. J. D. Electrochemical reduction of disulfide-containing proteins for hydrogen/deuterium exchange monitored by mass spectrometry. Anal. Chem. 2014, 86, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Bobst, C.E.; Kaltashov, I.A. Enhancing the quality of H/D Exchange measurements with mass spectrometry detection in disulfide-rich proteins using electron capture dissociation. Anal.Chem. 2014, 86, 5225–5231. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; McCormack, A.L.; Schieltz, D.; Carmack, E.; Link, A. Direct analysis of protein mixtures by tandem mass spectrometry. J. Protein Chem. 1997, 16, 495–497. [Google Scholar] [CrossRef]
- Sidoli, S.; Lin, S.; Karch, K.R.; Garcia, B.A. Bottom-Up and Middle-Down Proteomics Have Comparable Accuracies in Defining Histone Post-Translational Modification Relative Abundance and Stoichiometry. Anal. Chem. 2015, 87, 3129–3133. [Google Scholar] [CrossRef]
- Donnelly, D.P.; Rawlins, C.M.; DeHart, C.J.; Fornelli, L.; Schachner, L.; Lin, Z.; Lippens, J.L.; Aluri, K.C.; Sarin, R.; Chen, B.; Lantz, C. et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nature Methods, 2019; 16, 587–594. [Google Scholar]
- Emmalyn, J.; Dupree, M.J.; Hannah, Y.; Marius, M.; Brindusa, A.P. et al. Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of This Field. Proteomes. 2020, 8, 14. [Google Scholar]
- Lu, C.; Coradin, M.; Porter, E.G.; Garcia, B. : Accelerating the Field of Epigenetic Histone Modification Through Mass Spectrometry–Based. Mol Cell Proteomics 2021, 20, 100006. [Google Scholar] [CrossRef]
- Tian, S.; Zheng, S.; Han, Y.; Guo, Z.; Zhai, G.; Bai, X.; He, X.; Fan, E.; Zhang, Y.; Zhang, K. Maleic anhydride labeling-based approach for quantitative proteomics and successive derivatization of peptides. Anal. Chem. 2017, 89, 8259–8265. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B. A.; Mollah, S.; Ueberheide, B. M.; Busby, S. A.; Muratore, T. L.; Shabanowitz, J.; Hunt, D. F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007, 2, 933. [Google Scholar] [CrossRef]
- Sadaf, A.; Byrne, K.H.; Chae, P.S. Chapter Four - Amphipathic Agents for Membrane Protein Study. Methods in Enzymology. 2015, 557, 57–94. [Google Scholar]
- Spreafico, F.; Bongarzone, I.; Pizzamiglio, S.; Magni, R.; et al. Proteomic analysis of cerebrospinal fluid from children with central nervous system tumors identifies candidate proteins relating to tumor metastatic spread. Oncotarget. 2017, 8, 46177–46190. [Google Scholar] [CrossRef]
- Galogahi, F.M.; Zhu, Y.; Hongjie An, H.; Nguyen, N.T. Core-shell microparticles: Generation approaches and applications. Journal of Science: Advanced Materials and Devices 2020, 5, 417–435. [Google Scholar] [CrossRef]
- Schutzer, S.E.; Liu, T.; Natelson, B.H.; Angel, T.E.; Schepmoes, A.A.; Purvine, S.O.; Hixson, K.K.; Lipton, M.S.; Camp, D.G. II; Coyle, P.K.; Smith, R.D.; Bergquist, J. Establishing the Proteome of Normal Human Cerebrospinal Fluid. PLoS ONE. 2010, 5, e10980. [Google Scholar] [CrossRef]
- de Bont, J. M.; den Boer, M. L.; Reddingius, R.E.; et al. Identification of Apolipoprotein A-II in Cerebrospinal Fluid of Pediatric Brain Tumor Patients by Protein Expression Profiling Clinical Chemistry 2006, 52, 1501–1509.
- Hutchens, T.W.; Yip, T.T. New desorption strategies for the mass spectrometric analysis of macromolecules. Rapid Commun Mass Spectrom. 1993, 7, 576–580. [Google Scholar] [CrossRef]
- Consortium, I. H. G. S. Finishing the euchromatic sequence of the human genome Nature 2004, 431, 931–945. 431.
- Gaudet, P.; Michel, P.-A.; Zahn-Zabal, M.; Britan, A.; Cusin, I.; et al. The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res. 2017, 45, D177–D182. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- UniProt, C. Uniport: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Mnatsakanyan, R.; Shema ,G.; Basik ,M.; Batist ,G.; Borchers ,C.H.: Sickmann ,A.; Zahedi, R.P. Detecting post-translational modification signatures as potential biomarkers in clinical mass spectrometry. Expert Rev Proteomics 2018, 15, 515–535. [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; et al. Phospho-Site Plus: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Smith, D.P.; Giles, K.; Bateman, R.H.; Radford, S.E.; Ashcroft, A.E. Monitoring Copopulated Conformational States During Protein Folding Events Using Electrospray Ionization-Ion Mobility Spectrometry-Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2007, 18, 2180–2190. [Google Scholar] [CrossRef]
- Zubarev, R. A.; Horn, D. M.; Fridriksson, E. K.; Kelleher, N. L.; Kruger, N. A.; Lewis, M. A.; Carpenter, B. K.; McLafferty, F. W. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 2000, 72, 563. [Google Scholar] [CrossRef]
- Syka, J.E.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 9528–9533. [Google Scholar] [CrossRef]
- Woodin, R. L.; Bomse, D. S.; Beauchamp, J. L. Multiphoton dissociation of molecules with low power continuous wave infrared laser radiation. J. Am. Chem. Soc. 1978, 100, 3248–3250. [Google Scholar] [CrossRef]
- Bowers, W. D.; Delbert, S. S.; Hunter, R. L.; Mc Iver, R. T. Fragmentation of oligopeptide ions using ultraviolet-laser radiation and fourier-transform mass-spectrometry. J Am Chem. Soc. 1984, 106, 7288–7289. [Google Scholar] [CrossRef]
- Hunt, D. F.; Shabanowitz, J.; Yates, J. R. Peptide sequence- analysis by laser photodissociation fourier-transform mass spectrometry. J.Chem.Soc.Chem.Comm. 1987, 548–550. [Google Scholar] [CrossRef]
- Van den Ackerveken, P.; Lobbens, A.; Turatsinze, J.-V.; Solis-Mezarino, V.; Völker-Albert, M.; Imhof, A.; Herzog, M. A novel proteomics approach to epigenetic profiling of circulating nucleosomes. Nature, Scientific Reports. 2021, 11, 7256. [Google Scholar] [CrossRef]
- Gu, H.; Ren, J.M.; Jia, X.; Levy, T.; Rikova, K.; Yang, V.; Lee, K.A.; Stokes, M.P.; Silva, J.C. Quantitative Profiling of Post-translational Modifications by Immunoaffinity Enrichment and LC-MS/MS in Cancer Serum without Immunodepletion. Molecular & Cellular Proteomics 2016, 15, 692–702. [Google Scholar]
- Britton, L.M.P.; Gonzales-Cope, M.; Zee, B.M.; Garcia, B.A. Breaking the histone code with quantitative mass spectrometry. Expert Review of Proteomics. 2011; 8, 631–643. [Google Scholar]
- Torrente ,M. P.; Zee, B.M.; Young, N.L.; Baliban,R.C.; LeRoy,G.; Christodoulos, A.F.; Hake,S.B. ; Garcia, B.A. Proteomic interrogation of human chromatin. PLoS ONE. 2011, 6, e24747. [Google Scholar]
- Rothbart, S.B.; Dickson, B.M.; Raab, J.R.; Grzybowski, A.T.; Krajewski, K.; Guo, A.H; Strahl, B. D. An Interactive Database for the Assessment of Histone Antibody Specificity. Molecolar Cell. 2015, 59, 502–511. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell. 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Casci, T. Chip on chips. Nat Rev Genet , 2001, 2, 88. [Google Scholar] [CrossRef]
- Fraga, M.F.; Esteban Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Gen. 2005, 37, 4. [Google Scholar] [CrossRef]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef]
- Bauden, M.; Pamart, D.; Ansari, D.; Herzog, M.; Eccleston, M.; Micallef, J.; et al. Circulating nucleosomes as epigenetic biomarkers in pancreatic cancer. Clin Epigenet. 2015, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013, 153, 38. [Google Scholar] [CrossRef]
- Lai, W. K. M.; Pugh, B. F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol. 2017, 18, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the Swissport database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef]
- Huang, K.-Y.; Lee, T.-Y.; Kao, H.-J. et al. dbPTM in exploring disease association and cross-talk of post-translational modifications. Nucleic Acids Res. 2019, 47, D298–D308. [Google Scholar] [CrossRef]
- Oliver, S. S.; Denu, J. M. Dynamic interplay between histone H3 modifications and protein interpreters: Emerging evidence for a "histone language". Chem Bio Chem 2011, 12, 299–307. [Google Scholar] [CrossRef]
- Leroy, G.; Dimaggio, P. A.; Chan, E. Y.; Zee, B. M.; Blanco, M. A.; Bryant, B.; Flaniken, I. Z.; Liu, S.; Kang, Y.; Trojer, P.; Garcia, B. A. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics & Chromatin 2013, 6, 20. [Google Scholar]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications cause and consequence of genome function Nature reviews Genetics Reviews. 2022; 23, 563. [Google Scholar]
- Li, G.; Levitus, M.; Bustamante, C.; Widom, J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005, 12, 46–53. [Google Scholar] [CrossRef]
- Cosgrove, M. S.; Boeke, J. D.; Wolberger, C. Regulated nucleosome mobility and the histone code. Nat. Struct. Mol. Biol. 2004, 11, 1037–1043. [Google Scholar] [CrossRef]
- Cristobal, A.; Marino, F.; Post, H.; Van Den Toorn, H.W.P.; Mohammed, S.; Heck, A.J.R.; et al. Streptomyces erythraeus trypsin for proteomics applications. J. Proteome Res. 2009, 8, 1810–1817. [Google Scholar]
- Han, Y.; Lu, C.; Zhang, K.; Tian, S.; Fan, E.; Chen, L.; He, X.; Zhang, Y. Quantitative characterization of histone post-translational modifications using a stable isotope dimethyl labeling strategy. Anal. Methods 2015, 7, 3779–3785. [Google Scholar] [CrossRef]
- Tian, S.; Zheng, S.; Han, Y.; Guo, Z.; Zhai, G.; Bai, X.; He, X.; Fan, E.; Zhang, Y.; Zhang, K. Maleic anhydride labeling-based approach for quantitative proteomics and successive derivatization of peptides. Anal. Chem. 2017, 89, 8259–8265. [Google Scholar] [CrossRef]
- Zheng, Y.; Fornelli, L.; Compton, P. D.; Sharma, S.; Canterbury, J.; Mullen, C.; Zabrouskov, V.; Fellers, R. T.; Thomas, P. M.; Licht, J. D. Unabridged analysis of human histone H3 by differential top-down mass spectrometry reveals hypermethylated proteoforms from MMSET/NSD2 overexpression. Mol. Cell. Proteomics 2016, 15, 776–790. [Google Scholar] [CrossRef]
- Sidoli, S.; Garcia, B. A. Middle-down proteomics: A still unexploited resource for chromatin biology. Expert Rev. Proteomics 2017, 14, 617–626. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballesta, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Brenne, S. S.; Madsen, P. H.; Pedersen, I.S; Hveem, K.; Skorpen, F.; Krarup, H. B.; Giskeødegård, G.F. Colorectal cancer detected by liquid biopsy 2 years prior to clinical diagnosis in the HUNT study. Journal of Cancer 2023, 129, 861–868. [Google Scholar] [CrossRef]
- Brumpton, B.M.; Graham, S.; Ida Surakka, I.; Skogholt, A.H.; Løset, M.; . Fritsche, L.G.; et al. The HUNT study: A population-based cohort for genetic research. Cell Genom. 2022, 2, 100193. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N Engl J Med 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Liu, A. P. Y.; Smith, K. S.; Kumar, R.; Paul, L.; Bihannic, L.; Lin, T.; et al. Serial assessment of measurable residual disease in medulloblastoma liquid biopsies. Cancer Cell 2021, 39, 1519–1530.e4. [Google Scholar] [CrossRef] [PubMed]
- Cantor, E.; Wierzbicki, K.; Tarapore, R. S.; Ravi, K.; Thomas, C.; Cartaxo, R.; et al. Serial H3K27M cell-free tumor DNA (cf-tDNA) tracking predicts ONC201 treatment response and progression in diffuse midline glioma. Neuro-Oncol. 2022, 24, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Miller, A. M.; Szalontay, L.; Bouvier, N.; Hill, K.; Ahmad, H.; Rafailov, J.; et al. Next-generation sequencing of cerebrospinal fluid for clinical molecular diagnostics in pediatric, adolescent and young adult brain tumor patients. Neuro-Oncol 2022, 24, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Pages, M.; Uro-Coste, E.; Colin, C.; Meyronet, D.; Gauchotte, G.; Maurage, C.-A.; et al. The implementation of DNA methylation profiling into a multistep diagnostic process in pediatric neuropathology: A 2-year real-world experience by the French neuropathology network. Cancers. 2021, 13, 1377. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D; Cohn, A; Tummala, M; Lapham, R; Cosgrove, D.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol. 2021, 32, 1167–1177. [CrossRef]
- Wu, T.M.; Liu, J.B.; Liu, Y.; Shi, Y.; Li, W.; Wang, G.R.; et al. Power and promise of next-generation sequencing in liquid biopsies and cancer control. Cancer Control. 2020, 27, 1–10.
- Campos-Carrillo, A.; Weitzel, J.N.; Sahoo, P.; Rockne, R.; Mokhnatkin, J.V.; Murtaza, M.; et al. Circulating tumor DNA as an early cancer detection tool. Pharmacol Ther. 2020, 207, 107458. [Google Scholar] [CrossRef] [PubMed]
- Adhyam, M. and Gupta, A.K. A Review on the Clinical Utility of PSA in Cancer Prostate. Indian J Surg Oncol. 2012, 3, 120–129. [CrossRef] [PubMed]
- Dumbrava, E.I.; Meric-Bernstam, F. Personalized cancer therapy leveraging a knowledge base for clinical decision-making. Cold Spring Harb Mol Case Stud. 2018, 4, a001578. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Liquid biopsies from cancer patient, various analytes within a typical sample, techniques of analysis and the likely information provided by such analyses.
Figure 1.
Liquid biopsies from cancer patient, various analytes within a typical sample, techniques of analysis and the likely information provided by such analyses.
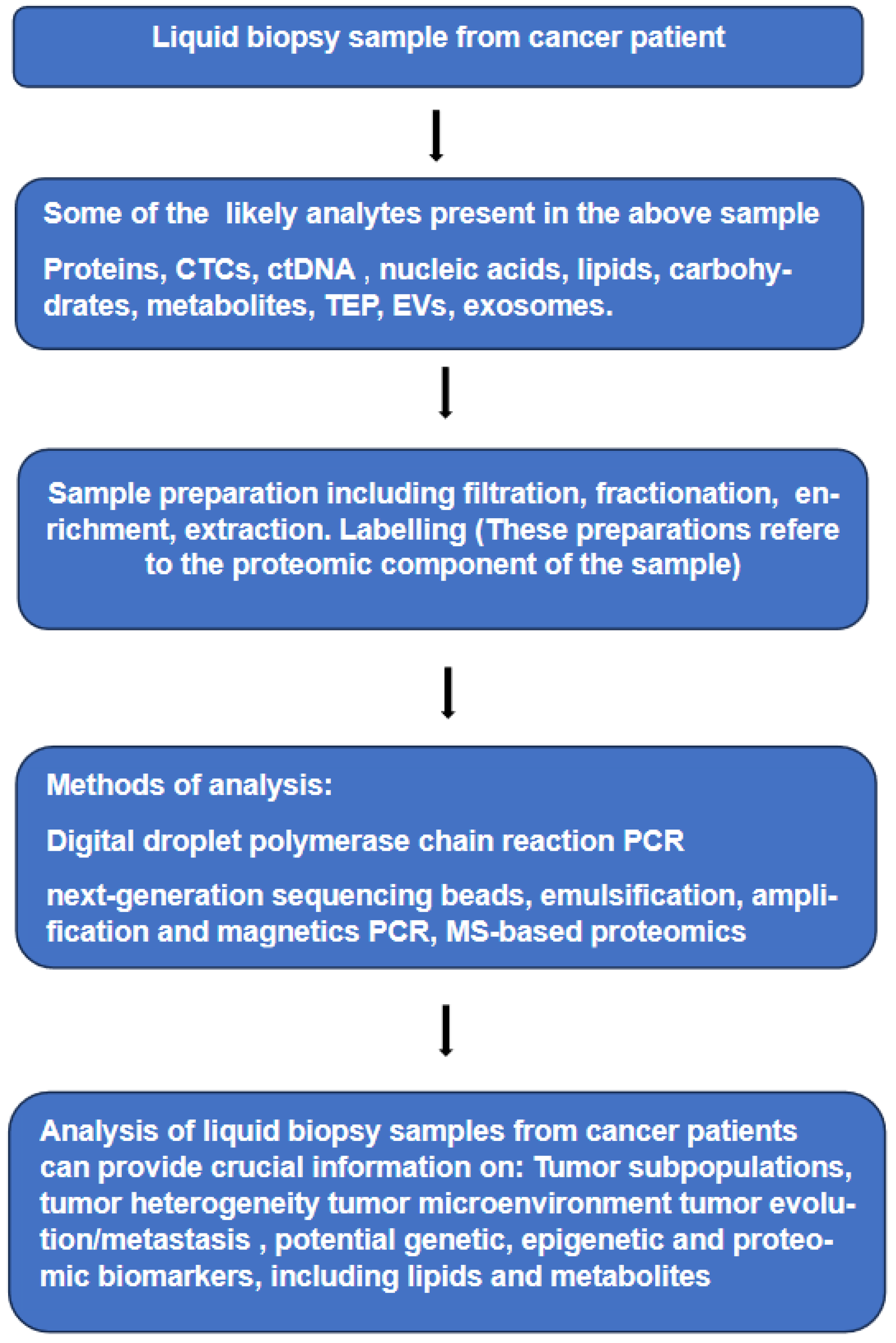
Figure 2.
MS-based proteomic analysis of protein PTMs, schematic representation of the steps used in three methods, commonly used in such analyses.
Figure 2.
MS-based proteomic analysis of protein PTMs, schematic representation of the steps used in three methods, commonly used in such analyses.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



