
Submitted:
20 March 2024
Posted:
21 March 2024
You are already at the latest version
Abstract
Proteomic analysis of purified rabies virus (RABV) revealed 47 entrapped host proteins within viral particles. Out of these, 11 proteins were highly disordered. Our study was particularly focused on five of the RABV-entrapped mouse proteins with the highest levels of disorder, Neuromodulin, Chmp4b, DnaJB6, Vps37B, and Wasl. We extensively utilized bioinformatics tools, such as FuzDrop, D2P2, UniProt, RIDAO, STRING, AlphaFold, and ELM for comprehensive analysis of the intrinsic disorder propensity of these proteins. Our analysis suggested that these disordered host proteins might play a significant role in facilitating the rabies virus pathogenicity, immune system evasion, and the development of the antiviral drug resistance. Our study highlighted the complex interaction of the virus with its host, with the focus on how the intrinsic disorder can play a crucial role in virus pathogenic processes and suggested that these intrinsically disordered proteins (IDPs) and disorder-related host interactions can also be a potential target for the therapeutic strategies.
Keywords:
rabies virus
; intrinsically disordered proteins
; protein-protein interactions
; Neuromodulin
; Chmp4b
; DnaJB6
; Vps37B
; Wasl
1. Introduction
Rabies virus (RABV) also known as Rhabdovirus causes rabies, which is a preventable (through prompt administration of post-exposure prophylaxis (PEP) to victims of bites by rabid animals [1]) but rarely curable disease [2]. Once the symptoms start manifesting the disease is nearly 100% fatal [3]. It was reported that RABV infection causes more than 55,000 deaths worldwide [4].
Rabies virus affects the central nervous system causing acute infection [5]. The transmission of virus usually happens through the bite of a rabid animal [2,3]. The virus has a rod- or bullet-like shape, and its genome is a single-stranded, negative-sense, linear non-segmented enveloped RNA [6]. RABV belongs to the Rhabdoviridae family and genus Lyssavirus, hence, the name rhabdovirus [6,7].
The genome encodes for 5 different proteins named as N (nucleoprotein), P (phosphoprotein), M (matrix protein), G (glycoprotein), and L (polymerase) [6]. The bullet shaped virus enclosed in lipid envelope covered by glycoprotein that facilitates the attachment of the virus to the host cell receptors and thus ensures viral entry. The helical ribonucleocapsid core is composed of viral genome and nucleoprotein [8].
Most often, the exposure to RABV happens due to the bite or the scratches of a rabid animal [2,6]. At the site of injury, the muscle cells of the new host become exposed to the rabid animal saliva, which contains the particles of rabies virus [9,10]. RABV initially replicates in the muscle cells, but its next destination is peripheral nervous system [6,9,10]. The virus binds to the receptors on the nerve endings of peripheral nervous system near the site of infection [11,12]. From here on, RABV moves along the nerves through axonal transport to enter the peripheral nervous system [11]. Then it moves to the main target, the central nervous system [2]. When the RABV is in the central nervous system of the host, it starts to replicate rapidly, spreading to the spinal cord and different parts of the brain causing inflammation of the brain (encephalitis) [2].
The lifecycle of rabies virus as it enters the host cell can be divided into following steps:
- -
- Attachment: At first, G protein of virus attaches itself to the cell surface receptors. [11];
- -
- -
- -
- -
- -
- Translation: Viral mRNA strand is used for the Translation of 5 major proteins (N, P, M, G, and L);
- -
- Assembly: All these viral particles (genome and proteins) assembled into new virions [11];
- -
- Budding: Assembled virions bud off from the cell surface of host cells acquiring its envelope from the host cell membrane [13];
- -
- Release: The mature rabies virus normally releases from the cells through cell lysis and spread through the central nervous system and brain to infect healthy cells [13].
During the assembly of virus progeny, some host proteins become integrated into the mature virion particles, which may help the virus to camouflage as the host cells to escape the immune system [14]. In this article we will focus on the analysis of the intrinsic disorder of such host proteins entrapped in the virus particles. Knowing more about the intrinsic disorder property of these proteins will help us understand about the interaction of viruses with host cells, because intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs) are highly flexible and can change their structure and function in response to different environments [15]. Therefore, protein intrinsic disorder can help viruses to become more adaptable and flexible. We can also learn the strategies of viruses to evade the immune system to help us understand the pathogenesis of rabies virus in greater depth.
In this context, Yan Zhang and colleagues published a paper discussing the host proteins incorporated into RABV particles when they are released from the host cells [16]. The authors purified the viral particles to perform the proteome profiling of RABV. They found out that along with 5 main viral proteins, 49 host proteins are also integrated in viral particles, and 24 of these are directly taking part in viral replication suggesting that the virus hijacks the host cellular machinery and interacts with host proteins for efficient replication [16]. An illustrative example is given by the integration of heat shock protein (HSP70) into a matured RABV virion. Decreasing the expression of HSP70 leads to a substantial reduction in the levels of viral RNAs, proteins, and virions [17]. This suggests that specifically the enveloped viruses utilized the host proteins to carry out their replication [16].
Rabies viruses that belong to the Rhabdoviruses family bud out of host cells using the host endosomal sorting complex required for transport (ESCRT) machinery [18,19]. Hijacking of the host ESCRT machinery plays a vital role in integrating the host proteins in the virus particles [18,19]. Two important proteins in this respect are charged multivesicular body protein 4b (Chmp4b) and Vacuolar protein sorting-associated protein 37B (Vps37b), both play crucial roles in budding process during the virus life cycle [16]. Chmp4b is an essential component of ESCRT III complex, which is responsible for the final stages of budding [16]. Thus, protein is involved in the final detachment of the newly formed virions from the cell membrane of the host cells. On the other hand, the Vps37b is involved with ESCRT I, which is taking part in the initial step of the viral budding process [16]. Therefore, these two proteins can serve as potential therapeutic targets.
The protocol utilized by Zhang and colleagues in this important study is outlined below [16]. The authors used nano-scale liquid chromatography tandem mass spectrometry techniques on purified viral particles to identify 49 virus-associated host proteins [16]. Then, the Western blotting approach was used to validate the presence of these proteins in the matured viral particles [16]. They used RABV, CVS-11 strain to infect mouse Neuro-2A cells [16]. This step was crucial to obtain virus particles for further analysis [16]. In these experiments, cells were infected at 70% confluence, and the virus-containing supernatant was subject to multiple rounds of differential centrifugation [16]. Ultra-centrifugation at 100,000 × g was performed on samples containing bullet-shaped viruses for higher purification [16]. The purified viral particles isolated through centrifugal separation were observed through transmission electron microscopy and cryo-electron microscopy to inspect and verify the purity and structural integrity of viral particles [16].
At the next stage, the deglycosylation of proteins by the PNGase F enzyme was conducted to enhance the sensitivity of peptides that contain glycosylation sites [16,20,21]. The proteins were analyzed through SDS-PAGE to separate them based on their molecular weight [16], followed by Western blotting using a monoclonal antibody against the RABV glycoprotein and a secondary antibody labeled with Alexa Flours 680 for detection [16].
Next, the proteomic analysis was conducted by nano LC-MS/MS on the peptide mixtures obtained through the digestion of deglycosylated protein samples with trypsin [16]. Analysis of proteins through Liquid Chromatography-Tandem Mass Spectrometry allowed for the detailed analysis of peptides, including their structure and sequence [16]. To ensure accuracy and reliability, the data obtained through mass spectrometry were then analyzed using the Andromeda search engine and MaxQuant software against specific databases, such as UniProtKB mouse sequence database and RABV (CVS-11) protein sequences from GenBank [16]. The authors employed intensity-based absolute quantification (iBAQ) for evaluation of the protein abundance and applied FDR (False Discovery Rate) of 0.01 % to ensure the data accuracy [16]. To further increase the confidence, Zhang et al. did not rely solely on one experimental run. Instead, they observed proteins in three different assays [16]. The repetition helped in ruling out false positives and enhanced the credibility of the results. In the identification of high-confidence proteins, each candidate was supposed to fulfill three criteria; first was the consistent identification in three different independently purified virion preparations, second was the abundance threshold exceeding 106, and the third criterion was the presence of at least 2 unique peptides corresponding to each target protein [16].
Then, to investigate the incorporation of host proteins into the virus particles, purified virus particles were treated with protease K to digest proteins, which helps in focusing on proteins that are truly incorporated into the viral particles and in removing the loosely attached proteins [16]. The treated virions were processed to remove any cleaved peptides [16].
To prepare the cell extracts, 2 groups of N2a cell lines were prepared; one treated with CVS11 and the other serves as control. The supernatant of the cell extracts was collected after 72 hours, these liquid parts contain the proteins released from the cells [16]. Both the cell extracts and virus particles the one treated with protease K and untreated were subjected to Western blotting. The goal was to identify the proteins in Virus particles and cell extracts using antibodies [16].
Viral proteins, such as Glycoprotein G, Nucleoprotein N, and matrix protein M were detected using specific mouse polyclonal antibodies, while host cell proteins, such as Hsc70, cofilin, and Chmp4b were probed through additional antibodies [16]. Finally, all these proteins were visualized using the fluorescently labeled secondary antibodies [16]. The overall step was crucial to check whether host proteins were incorporated into the virus particles and do they incorporate in them firmly [16].
Moving on, Zhang et al. performed functional characterization of the 49 incorporated host proteins in the virus particles through the gene ontology database [16]. They were aiming to get a deep understanding of the complex interaction of host cells and RABV, and the functional implications of these proteins in the virion particles like involvement in the viral process like budding [16]. Protein-protein interaction network analysis was carried out, which also strongly suggests that many of these host proteins are involved in viral budding, especially through ESCRT machinery [16]. This implies the possibility that the virus might be exploiting these host proteins, mainly the ones involved in ESCRT machinery, to exit the host cells further assisting the viral pathogenesis [16]. One important aspect was left unexplored by the authors, namely, the intrinsic disorder status of the host proteins entrapped in RABV particles.
Intrinsically disordered proteins (IDPs) are a class of biologically active proteins without unique structures [15,22,23,24,25]. Contrary to traditional ordered proteins IDPs and intrinsically disordered regions (IDRs) lack well-defined three-dimensional structures and exist as highly dynamic conformational ensembles [15,22,23,24,25]. Intrinsic disorder is highly prevalent, and almost 70% of PDB structures have disordered regions [26,27]. IDPs are multifunctional proteins that can have multiple binding partners and are characterized by high sensitivity to subtle changes in local environmental conditions like pH and temperature, being capable of rapid change of their structures in response to the external environment [15,22,23,24,25]. IDPs/IDRs have a large interface area with the dominance of hydrophobic-hydrophobic contact. Unlike ordered proteins, IDPs have a weak hydrophobic core (if any), as their amino acid sequences have low content of hydrophobic and aromatic residues and contain large numbers of charged and polar residues [15,28]. All these properties make intrinsic disorder proteins an integral part of the protein universe with important biological functions that complement functionality of ordered proteins. The flexibility and adaptability of IPDs make them suitable candidates to take part in diverse cellular functions like cell signaling, molecular recognition, and protein-protein interactions [29,30]. At the same time, the adaptable and flexible nature of IDPs also makes them important players in the pathogenesis of various diseases like cancer and neurodegenerative diseases [31,32,33,34,35].
In this study, to analyze the intrinsic disorder status of the host proteins entrapped in RABV, we used the data on the 47 high confidence host proteins reported by Zhang et al. [16]. These entrapped proteins were subjected to multifactorial disorder analysis using a set of commonly used disorder predictors. Then, we conducted more detailed bioinformatics characterization of 5 entrapped proteins with highest levels of predicted disorder.
2. Materials and Methods
2.1. Protein Datasets
UniProt IDs of all mouse proteins analyzed in this study were retrieved from Table 1 of the Zhang et al. research article [16]. These IDs were used to collect amino acid sequences (in FASTA format) of these proteins from UniProt database, which are listed in the Supplementary Table S1. We subjected all these proteins to bioinformatics analysis and selected the most disordered proteins for in-depth research. The selected proteins are Neuromodulin (also known as Growth-associated protein 43 (Gap43), or Calmodulin-binding protein P-57, or Axonal membrane protein GAP-43; UniProt ID: P06837), a charged multivesicular body protein 4b (Chmp4b, UniProt ID: Q9D8B3), Dnaj homolog superfamily B member 6 (Dnajb6, UniProt ID: O54946), a vacuolar protein sorting-associated protein 37B (Vps37b, UniProt ID ; Q8R0J7), and a Neural Wiskott-Aldrich syndrome protein (also known as Actin nucleation-promoting factor WASL; UniProt ID: Q91YD9). The analysis of proteins using various bioinformatics tools discussed below was done by submitting their amino acid sequences in FASTA format to corresponding computational platforms.
2.2. Exploration of the Intrinsic Disorder Predisposition
The susceptibility of our protein data set to intrinsic disorder was evaluated through the RIDAO web platform, which is a convenient bioinformatics tool to generate the disorder profiles of query proteins. RIDAO combines the outputs of six commonly used per residue disorder predictors, such as PONDR® FIT, PONDR® VSL2, PONDR® VL3, PONDR® VLXT, IUPred Short, and IUPred Long to generate an integral disorder profile of an individual query protein or to provide global disorder characterization of a protein dataset [36]. The disorder score was assigned to each residue, with a residue with disorder score equal to or above 0.5 being considered as disordered and a residue with disorder score below 0.5 being predicted as ordered. Residues/regions with disorder scores between 0.15 and 0.5 were considered as ordered but flexible. For each protein, RIDAO also calculated the percent of predicted intrinsically disordered residues (PPIDR), which was used for classification of proteins as ordered (PPIDR < 10%), moderately disordered (10% ≤ PPIDR < 30%), and highly disordered (PPIDR ≥ 30%).
2.3. ELMs: Eukaryotic Linear Motifs
ELM (eukaryotic linear motif) database is a platform used to recognize the SLiMs (short linear motifs) in the proteins [37,38,39,40,41,42,43]. The motifs recognized are special in a way that if the information on the 3D organization of a functional protein is absent, SLiMs still provide a way to evaluate potential functionality of protein, since these functional motifs are linear, which is a unique property because of the intrinsic disorder nature of these motifs [44]. Identification of these motifs helps in the understanding of the functionality of the protein as SLiMs are involved in important interactions and perform regulatory roles [42]. In this study, we found the eukaryotic linear motifs in the aggregation hotspots, droplet-promoting regions, multiple binding modes regions, and molecular recognition feature (MoRF) regions of our selected proteins. The goal was to map the identified ELMs/SLiMs onto these IDR regions. By identifying ELMs, the goal was to deepen our understanding of the functionality of our proteins, and how they interact and play a role within the cellular environment.
2.4. Functional Annotation Derived from Disorder
D2P2 is a special Database of Disordered Protein Prediction designed to facilitate the statistical comparison among different prediction methods to facilitate the analysis of IDPs [45]. Along with disorder predictions, D2P2 also shows localization of MoRF regions, unique disordered binding sites that become ordered at interaction with specific partners, found through the ANCHOR algorithm, PTMs, and also list the SUPERFAMILY domains from evolutionary studies [45].
2.5. FuzDrop Analysis: Identifying LLPS Promoters
We used FuzDrop [46] to predict the likelihood of proteins taking part in spontaneous liquid-liquid phase separation and generates a scoring system based on the sequence of proteins to identify the regions that promote this process. Protein with pLLPS (probability of liquid-liquid phase separation) score of 0.60 or higher are identified as promoters of droplet formation and participants of liquid-liquid phase separation that leads to droplet formation and generates membrane-less organelles that are important for several cellular functions such as stress response and regulation [47,48,49,50].
2.6. Protein-Protein Interaction Network
STRING database strives to incorporate all established and predicted connections among proteins, comprising both physical and functional associations [51,52,53]. Users get to analyze network visualizations, predicted connections, and functional annotations for the analysis of protein. PPI networks of proteins were retrieved by using (the STRING database https://string-db.org assessed on March 10, 2024). For analysis of protein interactions through STRING, we used medium confidence level and 500 interactors in 1st shell to generate PPI network. For the global interactions network, 11 most disordered proteins were subjected to generate a PPI network using the same settings mentioned above. The Functional enrichment data of these proteins can be found in Supplementary Tables S2, S3, and S4.
2.7. CH-CDF Analysis
CH-CDF graph combined the results of two plots Charge-hydropathy (CH) and cumulative distribution function (CDF). CH graph is plotted based on the net charge and hydropathy of proteins, disordered proteins tend to have high net charge and low hydropathy, and they are found to be clustered in the specific area of the plot [54,55]. A linear line is placed to separate these disordered proteins from the ordered [54,55]. CDF plot is based on PONDR scores, plotting PONDR scores to their frequency. PONDR scores tell us about the disorder associated with the protein sequence. For CH plot protein that appear above the linear boundary is considered disordered and the one that appeared below the boundary is considered as ordered [54,55].
For CDF plot the CDF curve for the ordered proteins are plotted below the order-disorder line it is considered as disordered and if it appears above this boundary it is labeled as an ordered protein [54].
CH-CDF plot classified proteins effectively in two categories ordered and disordered, by plotting the average distance of protein from order-disorder boundary (CDF) and the scores obtained through the CH plot [56].
2.8. 3D Structures of Proteins
Alpha Fold, a protein structure database developed by DeepMind exploits an AI system to predict 3D structure of proteins based on the amino acid sequences with high accuracy [57]
3. Results and Discussion
3.1. Global Disorder Analysis of Host Proteins Entrapped in RABV Particles
First, to get an overview of the overall disorder status of host (mouse) proteins entrapped in RABV particles, we analyzed these proteins by a set of commonly used per-residue disorder predictors, such as PONDR® VSL2, PONDR® VL3, PONDR® VLXT, and PONDR® FIT, IUPred Short, and IUPred Long. These predictors were accessed through the Rapid Intrinsic Disorder Analysis Online (RIDAO) platform (available at https://RIDAO.app) [36]. Average Disorder Scores (ADS) and Percentages of Predicted Disordered Residues (PPDR) were computed for each protein, employing the outputs of these per-residue predictors. ADS is a measure of the average disorder for a protein and PPDR is a measure of the proportion of amino acids within a protein that have a predicted disorder score above 0.5.
The results of these analyses are summarized in Supplementary Table S5. These data were used to classify each protein by its disorder status. Of note, since ADS does not share a direct relationship with PPDR, we defined proteins as highly ordered if they had a PPDR of less than 10% or an ADS of less than 0.15. Proteins 10% ≤ PPIDR < 30% or 0.15 ≤ ADS < 0.5 were considered moderately disordered. Proteins with PPDR ≥ 30% and ADS of 0.5 or more were labeled as highly disordered. These categorizations are consistent with the standards set in our previous publications [58,59,60] and are in line with the accepted practice in the field [61]. This approach provides means for a more detailed study of protein structures by clearly identifying varying levels of their structural (dis)organization.
Since the effectiveness and accuracy of PONDR® VSL2 has been proven in the Critical Assessment of protein Intrinsic Disorder [62], we used the outputs of this tool to generate an illustrative representation of global disorder distribution in mouse proteins entrapped in the RABV particles. Results of this analysis are shown in Figure 1A, which indicates that most of the host proteins are predicted as moderately or highly disordered.
In fact, approximately, 27.7% of entrapped host proteins are in the red zone (highly disordered), and additional 27.7 are in the light pink zone (i.e., proteins with PPDRVSL2 ≥ 30% but 0.15 ≤ ADSVSL2 < 0.5). Furthermore, 40.4% of proteins are predicted as moderately disordered: they are located within dark pink area and therefore are characterized by 10% ≤ PPIDRVSL2 < 30% or 0.15 ≤ ADSVSL2 < 0.5. None of these proteins was predicted as highly ordered based on their PPIDRVSL2 and ADSVSL2 data, and only two were placed to the light cyan area, being characterized by PPDRVSL2 < 10% but ADSVSL2 > 0.15. Figure 1A also shows that neuromodulin (UniProt ID: P06837) represents a noticeable exception, being located at the top corner of the red zone and being notably separated from other data points. These observations suggest that neuromodulin has a much higher disorder propensity than the rest of the data set. Detailed characterization of neuromodulin as a highly disordered protein could be of particular interest for further investigation in relation to its unique functional implications in a wide range of biological processes, as well as its disease associations.
To get further insight into structural organization of the entrapped host proteins, we combined the outputs of two binary disorder predictors to their outputs, charge-hydropathy (CH) plot, which classified proteins based on the distribution of charged amino acids and cumulative distribution function analysis. When compared to ordered proteins, disordered proteins often have a lower hydropobicity and higher net charge [63]. The CDF describes the cumulative frequency of disordered proteins along the length of a given protein. If a CDF curve of a given protein is below the order-disorder boundary, this protein is considered to be disordered and ordered if the CDF curve is located above this boundary [54]. The outputs of these binary predictors were used to generate the ∆CH-∆CDF plot presenting us with the global disorder analysis for our sets of proteins [30,56,63,64]. With this technique, we were able to classify proteins based on where they fell on the plot. Quadrant 1 (Q1, bottom right) encompasses proteins that are likely structured. Quadrant 2 (Q2, bottom left) comprises proteins that are either molten globular or hybrid; i.e., proteins that are compact yet lack a distinctive 3D structure or contain noticeable levels of ordered and disordered residues. Quadrant 3 (Q3, top left) includes highly disordered proteins, whereas Quadrant 4 (Q4, top right) captures proteins that are predicted to be disordered according to the CH-plot yet ordered according to the CDF-plot [30,56,63,64]. Therefore, based on their position within the ∆CH-∆CDF phase space, proteins can be classified into ordered with stable structure, molten globule-like (not completely ordered and disordered, with flexible structure), and highly disordered proteins lacking a stable 3D structure.
Figure 1B represents the results of global disorder analysis of the entrapped host proteins in the form of the ∆CH-∆CDF graph. The top left quadrant is designated as Quadrant 3; it is where both binary predictors agree that the protein is unstructured and called as disorder quadrant. Neuromodulin is again acting as an outlier in ∆CH-∆CDF plot, occupying the top most position in Q3. In addition to neuromodulin, this quadrant contains four more highly disordered proteins. Furthermore, 8 entrapped mouse proteins are classified as molten globular or hybrid, whereas all the remaining proteins in this dataset (34 or 72.34%) are placed in the Q1, indicating that they are expected to be mostly ordered. There are no proteins in the upper right quadrant (Q4). Some proteins are located at the boundaries between two quadrants suggesting they may have mixed characteristics attributed to both adjacent quadrants, indicating that these proteins may have flexible structures.
Next, we analyzed the intra-set interactivity of mouse proteins entrapped in RABV viral particles. To this end, we utilized the STRING platform, which generates a protein-protein interaction (PPI) network of predicted associations based on predicted and experimentally-validated information on the interaction partners of a protein of interest [53]. Surprisingly, Figure 2 shows that all 47 proteins analyzed in this study were involved in the formation of a rather dense PPI network, which is characterized by the average node degree of 10.3 and the average local clustering coefficient of 0.651. Protein in this network are involved in 243 PPIs, which significantly exceeds the expected number of interactions (69) for a random set of proteins of the same size and degree distribution drawn from the genome.
Figure 2.
STRING-based analysis of the intra-set interactivity of 47 mouse proteins entrapped in RABV viral particles. In the corresponding network, the nodes correspond to proteins, whereas the edges show predicted or known functional associations. Seven types of evidence are used to build the corresponding network, and are indicated by the differently colored lines: a green line represents neighborhood evidence; a red line – the presence of fusion evidence; a purple line – experimental evidence; a blue line – co-occurrence evidence; a light blue line – database evidence; a yellow line – text mining evidence; and a black line – co-expression evidence [53].
Figure 2.
STRING-based analysis of the intra-set interactivity of 47 mouse proteins entrapped in RABV viral particles. In the corresponding network, the nodes correspond to proteins, whereas the edges show predicted or known functional associations. Seven types of evidence are used to build the corresponding network, and are indicated by the differently colored lines: a green line represents neighborhood evidence; a red line – the presence of fusion evidence; a purple line – experimental evidence; a blue line – co-occurrence evidence; a light blue line – database evidence; a yellow line – text mining evidence; and a black line – co-expression evidence [53].
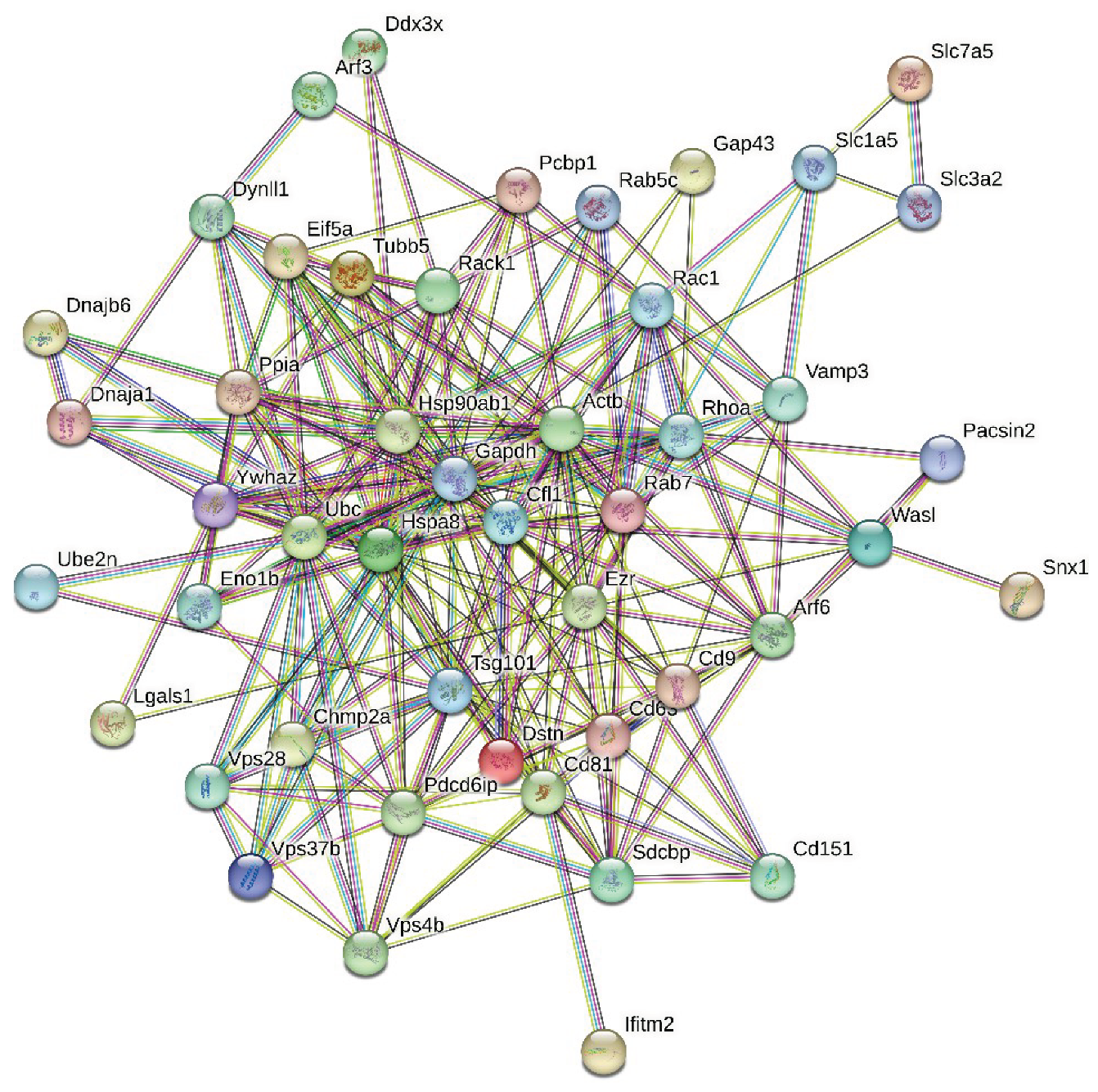
Proteins in this PPI network are involved in the following biological processes (based on the Gene Ontology (GO) classification): Regulation of cellular component organization (GO:0051128; p-value = 4.73e-14); Regulation of transport (GO:0051049; p-value = 1.28e-12); Regulation of localization (GO:0032879; p-value = 4.85e-12); Positive regulation of transport (GO:0051050; p-value = 8.84e-12); and Protein localization (GO:0008104; p-value = 8.84e-12). Among the most significantly enriched molecular functions of these proteins are Protein-containing complex binding (GO:0044877; p-value = 1.53e-07); Protein binding (GO:0005515; p-value = 3.76e-07); G protein activity (GO:0003925; p-value = 6.11e-05); Binding (GO:0005488; p-value = 6.56e-05); and Protein domain specific binding (GO:0019904; p-value = 0.00014). Finally, these proteins are most significantly enriched in the following cellular components: Vesicle (GO:0031982; p-value = 8.04e-13); Melanosome (GO:0042470; p-value = 1.16e-12); Cytoplasmic vesicle (GO:0031410; p-value = 5.80e-12); Cytosol (GO:0005829; p-value = 1.83e-11); and Endosome (GO:0005768; p-value = 2.11e-11).
Next, we looked for the presence of a correlation between the level of intrinsic disorder in a given protein and its interactivity within the intra-set PPI network (i.e., its node degree). Results of this analysis are shown in Figure 3A illustrating that such a correlation is almost absent.
Figure 3A shows that in the analyzed in this study intra-set PPI network, almost half of mouse proteins entrapped in the RABV particles are engaged in more than 12 interactions (i.e., serves as hubs of this network, with hub being defined here as a node with the number of interaction exceeding the average node degree of this network, which is 10.3). However, there is no clear disorder enrichment among hubs. These observations suggest that this intra-set PPI network is almost disorder-neutral. This is a rather interesting and unexpected observation, as typically, there is a strong positive correlation between the protein interactivity and its intrinsic disorder predisposition. In fact, it was indicated in many studies that one of the remarkable functional features of IDPs and IDRs is their extraordinary binding promiscuity [32,65,66,67,68,69]. In fact, IDPs/IDRs are considered as binding “professionals”, which continuously interact with various partners via multiple binding modes [32,65,66,67] and form static, semi-static, dynamic, or fuzzy complexes [68,69], as well as can be engaged in polyvalent interactions [70], where multiple binding sites of one protein are simultaneously bound to multiple receptors on another protein [71]. Often, disorder-based interactions are characterized by a combination of high specificity and low affinity [72], and many IDPs/IDRs can fold (at least partially) as a result of binding to their partners [73,74,75]. The degree of such binding-induced folding can be different in various systems, thereby forming complexes with broad structural and functional heterogeneity [68,69]. Furthermore, some IDPs/IDRs are capable of adopting different structures while forming complexes with different partners, thereby acting as morphing shape-changers [66,75,76,77,78,79,80,81,82,83,84]. Often, significant levels of disorder are retained by IDPs/IDRs in their bound state (at least outside the binding interface) resulting in the formation of so-called fuzzy complexes [85,86,87,88,89,90,91,92]. Therefore, it is not surprising that many IDPs/IDRs serve as hub proteins – nodes in complex PPI networks that have a very large number of connections to other nodes [80,93,94,95,96,97,98].
Next, we analyzed the predisposition of mouse proteins entrapped in the RABV particles to serve as drivers of liquid-liquid phase separation (LLPS) using the FuzDrop platform [46]. Results of this analysis are summarized in Figure 3B showing dependence of the probability of analyzed proteins for spontaneous liquid-liquid phase separation, pLLPS, on their intrinsic disorder status. This analysis revealed that there is a strong positive correlation between PPIDRVSL2 and pLLPS, and that all 7 proteins predicted as droplet drivers (i.e., proteins characterized by pLLPS ≥ 0.60) are also predicted to be highly disordered. It is recognized now that a significant part of cellular processes is determined by the functioning of liquid-droplet-like condensates – membrane-less organelles (MLOs) [99,100,101]. In fact, MLOs are very diverse and commonly found in cytoplasm, nucleus, mitochondria of various eukaryotic cells, in chloroplasts of plant cells, as well as in bacterial cells [102]. Biogenesis of MLOs is driven by the intracellular LLPS processes, which are also known as liquid-liquid demixing phase separation [103,104], and which are strongly dependent on IDPs and IDRs [105,106]. In fact, many of the MLO resident proteins are IDPs or contain IDRs, and the formation of all the MLOs analyzed so far relies on IDPs/IDRs, indicating that intrinsic disorder is important for MLO biogenesis [103].
After subjecting all 47 mouse proteins found in the rabies virus to intrinsic disorder analysis, we selected 11 most disordered proteins for comprehensive analysis, with 5 of these highly disordered proteins being discussed below in detail. The information about the remaining highly disordered proteins can be found in the Supplementary Materials.
3.2. Functional Intrinsic Disorder in the Most Disordered Mouse Proteins Found in the Rabies Virus
3.2.1. Neuromodulin (UniProt ID: P06837)
Neuromodulin is a protein encoded by the gene Gap43. This protein is involved in neuron growth acting as a crucial component of the growth cones present at the tip of elongating axons (https://www.uniprot.org/uniprotkb/P06837/entry)
In mice, neuromodulin is the peripheral membrane protein not entirely embedded in the membrane but associated with it that allows for the dynamic interaction with other membrane proteins. Neuromodulin is transported to the growth cones of neurons. These growth cones are present at the tip of the axons, essential for guiding the direction of neuronal growth during development and regeneration. Several studies have been conducted to elucidate the process by which protein is transported to the growth cones. Zuber et al. suggested that the N-terminal 10 amino acid sequence is sufficient to target the protein to these growth cones [107]. However, later, an experiment conducted with a fusion protein combining neuromodulin and β-galactosidase, which is an enzyme used as a marker in an experiment, revealed observed that only the N-terminal 10 amino acid sequence is not sufficient to transport protein to its target, the protein’s ability to attach to the membrane through palmitoylation at cysteines 3 and 4 is also essential for assembling the protein at the growth cones [108,109]. This also signifies the importance of post-translational modification in the protein.
The mouse neuromodulin is a 227 residue long, highly disordered protein of 23.6 kDa, whose interactions with calmodulin along with neurogranin are crucial for learning and memory formation in the nervous system [110]. This protein, which is also designated as GAP-43 or P-57 neuromodulin, is one of the main presynaptic substrates of protein Kinase C [110,111,112]. Phosphorylation of neuromodulin leads to decreased affinity for calmodulin [110]. Under a low calcium ions conditions, the protein binds to calmodulin through highly unstructured IQ motif (I/L/V) QXXXRXXXX(R/K), which adopts an α-helical confirmation upon binding with calmodulin [110]. Phosphorylation through protein Kinase at serine residues modulates this interaction, influencing the behavior of F actin in the growth cones of neurons [111].
Along with this, this protein consists of ‘Gap junction protein N-terminal region’ (residues 2-31), IQ motif (residues 31-60). Phosphorylation occurs at Ser41, Ser86, Serine96, Thr88, Thr89, Thr89, Thr95, Ser96, Ser103, Thr138, Ser142, Ser144, Ser145, Thr172, Ser192, and Ser 193. Palmitoylation at cysteine residues at positions 3 and 4 (more specifically S-palmitoyl cysteine modification) is important for protein association with the cellular membrane and its location. Loss of these modifications at these sites are mutations associated with PTM and can prevent the protein from properly being lipidated and lead to changes in protein function and location (https://www.uniprot.org/uniprotkb/P06837/entry).
Figure 4 represents the results of the functional disorder analysis of this protein. The per-residue disorder profile generated by RIDAO indicates that neuromodulin is predicted as a highly disordered protein (see Figure 4A). In fact, the PPIDR scores determined using disorder predictors PONDR® FIT, PONDR® VSL2, PONDR® VL3, PONDR® VLXT, IUPred Short, IUPred Long were 100%, 100%, 100%, 90.75%, 96.68%, and 99.56% respectively. The mean disorder profile (MDP) was 100 %, signifying that protein is highly disordered. The residues are predicted to be disordered above the 0.5 threshold, MDP value of 100 % implies that entire neuromodulin is likely to be intrinsically disordered [36].
The D2P2 platform was used to generate a functional disorder profile for Neuromodulin (see Figure 4B). The top section of the image is showing colored bars that represent the disordered regions predicted by each predictor, such as IUPred-L, IUPred-S, PV2, PrDOS, VSL2b, VLXt, Espritz-D, Espritz-X, and Espritz-N [45]. Below these colored bars of predicted disorder is the domain prediction bar exhibiting 3 domains, with one of these domains marked as number 3 being the IQ domain of neuromodulin we discussed above. It ranges from residue 31 to 50 and is known as an IQ calmodulin-binding motif.
The consensus bar of green color is predicted Disorder Agreement between all predictors. According to D2P2 platform, all the predictors agree that disorder regions are found at residue 2-227. This protein is highly unstructured, being the most disordered among all the 47 host proteins analyzed in this study. Moving on with the D2P2 results the yellow zigzagged lines are representing MoRF regions. MoRF regions short for Molecular Recognition Features, which are disordered protein regions that become ordered upon binding to the respective protein partners [113]. Multiple MoRF regions are found at the range 1-9, 32-52, 58-81, 102-109, and 116-227 identified through the ANCHOR algorithm also named as disorder-based binding sites, indicating that neuromodulin has tendency to engage in disorder to order transition-based interactions. Below these MoRF region predictions is the different colored circles with a letter representing the PTMs sites along the length of the protein. Other than this, D2P2 also included the superfamily annotation and Pfam domains indicating the larger family the protein belongs to and the shared structural and functional domain within the family, giving insight into the role of protein and its functional profile.
Figure 4C represents FuzDrop-generated plot showing the sequence distribution of the residue-based droplet-promoting probabilities, pDP. Residues with the pDP values above 0.6 are capable of promoting liquid-liquid phase separation. In neuromodulin, most of the residues have pDP values above the said threshold. Therefore, most of the neuromodulin residues have a high probability of promoting droplet formation. Peaks in the graph indicate the regions that can promote the formation of membrane-less organelles in the cells through liquid-liquid phase separation. Membrane-less organelles are liquid compartments within the cell involved in specific biological functions like in gene regulation, not enclosed by traditional lipid membranes [114]. In neuromoduin, the droplet promoting region, i.e., a region, where it is particularly susceptible to phase separation, is located at the residue 2-127. Furthermore, neuromodulin contains one aggregation hot spot (residues 52-66), which is a region with high probability to promote droplet-formation that is predicted to exhibit muliplicity of binding modes enabling adaptability of interactions to the cellular context. Furthermore, the pLLPS value was predicted for neuromorulin to be 0.9949. Since the proteins with pLLPS ≥ 0.60 are designated as droplet-drivers with a tendency to undergo spontaneous liquid-liquid phase separation, mouse neuromodulin is predicted as a protein with very high droplet-driving potential.
Figure 4D shows a FuzDrop-generated multiplicity of binding modes (MBM) plot, indicating that protein can bind to multiple partners behaving differently in terms of structure and function, either as an ordered or disordered state depending on the type of interactions and its environment. Values of MBM ≥ 0.65 suggest that the residues/regions are context-dependent and are prone to engage in multiple interactions. The bar graph shows positions of MBM regions (residues 9-16 and 40-66), which have the potential to be engaged in multiple binding modes assisting the phase separation.
The interactability of neuromodulin was evaluated using the STRING database.
Figure 4E is revealing that this protein is acting as the central node in the complex PPI network. We used a medium confidence threshold and a maximum limit of 500 interactors to generate this PPI network that contains 145 nodes, each node representing a protein including neuromodulin and 2925 edges (protein-protein interactions). This number of edges in the neuromodulin-centered network significantly exceeds the number of edges expected for a random set of proteins of the same size and degree distribution drawn from the genome (which is 616). The average node degree of this network is observed to be 39, indicating that the average connectivity of each protein in the network is very high, which is further supported by the average local clustering coefficient of 0.659 indicating a high tendency of nodes to cluster together. Finally, the observed p-value of < 1.0e-16 is indicative of the high significance of the generated data, suggesting the PPI network is unlikely to be produced by random chance.
Functional enrichment analysis in terms of the Gene Ontology revealed that proteins in this network are involved in important biological processes, such as Neurogenesis (GO:0022008; p-value = 2.34e-52), Nervous system development (GO:0007399; p-value = 2.34e-52), Generation of neurons (GO:0048699; p-value = 1.74e-48), Neuron differentiation (GO:0030182; p-value = 7.50e-47), and Neuron development (GO:0048666; p-value = 2.92e-43). Most common molecular function of these proteins are Protein binding (GO:0005515; p-value = 1.35e-25), Binding (GO:0005488; p-value = 6.32e-18), Identical protein binding (GO:0042802; p-value = 1.38e-10), Signaling receptor binding (GO:0005102; p-value = 5.78e-10), and Protein-containing complex binding (GO:0044877; p-value = 9.88e-10). Finally, these proteins are cellular components of Axon (GO:0030424; p-value = 2.24e-59), Somatodendritic compartment (GO:0036477; p-value = 3.44e-50), Neuron projection (GO:0043005; p-value = 3.44e-50), Cell body (GO:0044297; p-value = 2.06e-48), and Neuronal cell body (GO:0043025; p-value = 2.87e-46).
Figure 4F illustrates the 3D structure of the protein predicted by AlphaFold, which has the capability of predicting the structure of intrinsic disorder regions. Most of the predicted structure of our protein has low confidence scores and would be in disordered form, when not interacting with the partners. In short, most of the protein would be unstructured in isolation as the average per-residue model confidence score pLDDT is 55.78 for this protein. The only high confidence structural element of this protein is the blue α-helical region (residues 27-52). However, single α-helix cannot exist in isolation and is likely to be induced by binding to specific partner(s) [115]. In line with these considerations, this helical region corresponds to the IQ motif responsible for the calmodulin binding.
Finally, we looked at the localization of ELMs (short functional motifs) within the various regions found in neuromodulin. Results of this analysis are summarized in Table 1.
Data reported in this section indicates that neuromodulin is characterized by a high level of intrinsic disorder with strong functional potential.
3.2.2. Charged Multivesicular Body Protein 4b (Chmp4b, UniProt ID: Q9D8B3)
Charged multivesicular body protein 4b also known as chromatin-modifying protein 4b (Chmp4b) is an essential component of the ESCRT-III (Endosomal Sorting Complex for Transport III) system that plays a significant role in the process of endosomal sorting in the cells [116]. There are five specific ESCRT complexes (ESCRT-0, -I, -II, -III, and the Vps4 complex) characterized by specific functions, such as interaction with ubiquitylated membrane proteins, membrane deformation, and abscission, all related to the topologically unique membrane bending and scission reaction away from the cytoplasm [117]. These ESCRT-driven activities are crucial for the processing of the multivesicular body (MVB) pathway, cytokinesis, and HIV budding [117]. It was indicated that the ESCRT-0, -I, and -II complexes represent stable protein ensembles in the cytoplasm, whereas ESCRT-III complex that includes four core subunits (Vps20/CHMP6, Snf7/CHMP4(A–C), Vps24/CHMP3 and Vps2/CHMP2(A,B)) and three assessor proteins (Did2/CHMP1(A,B), Vps60/CHMP5 and Ist1) is transiently formed on endosomes [117].
The point to be noted is that ESCRT machinery including ESCRT-III is hijacked by HIV viruses in humans, which is critical for the release of HIV from the infected host cells [118]. Defects in ESCRT machinery, which include Chmp4b, play a role in the pathogenesis of neurodegenerative diseases because of their function of clearing out the misfolded proteins from the cells.
Chmp4b is a 224 residue-long protein with the molecular weight of 25KDa, which is encoded by the Chmp4b gene. Post-translational modifications can be found at residues 2 (N-acetylserine), 6 (N6-acetyllysine), 14 (N6-acetyllysine), 14 (Phosphoserine), and 223 (Phosphoserine), and Snf7 domain found between residues 24 and 199 is involved in the multiple functions of the ESCRT machinery. The coiled-coil domain is located between the residues 23 and 183, whereas the N-terminal region (residues 2-153) is involved in the intramolecular interactions with the C-terminal region (residues 154-224). It was indicated that core subunits of the ESCRT-III complex potentially have similar structural organization, where the N-terminal region “consists of two helices (α1, α2) that form a 7 nm hairpin structure important for membrane binding and homo- or hetero-dimerization. In the cytoplasm, the negatively charged C-terminal region (α5 and α6) folds back on the positively charged N-terminal hairpin, which confers an autoinhibitory mechanism that stabilizes the inactive monomers” [117].
The per-residue intrinsic disorder predisposition graph generated based on the outputs of the RIDAO algorithm is shown in Figure 5A. It clearly shows that the mouse Chmp4b is predicted to be a highly disordered protein. In fact, the PPIDR values predicted for this protein by various predictors included in RIDAO are high: PONDR® VLXT: 73.21%; PONDR® VSL2B: 92.41%; PONDR® VL3: 83.48%; PONDR® FIT: 83.48 %; IUPred_Short: 55.36%; and IUPred_Long: 78.12%. The average of all these values is 81.70 %, which indicates that this protein is highly disordered. Most of the disordered residues were observed in the N- and C-terminal tails of the protein (residues 1-81 and 124-224).
Further proof of the highly disordered nature of mouse Chmp4b is given by the outputs of the D2P2 platform (see Figure 5B), which provides a comprehensive functional disorder prediction profile of protein. Predicted disorder agreement is shown in the green colored bars just below the predicted Snf7domain. MoRF regions depicted below the disorder prediction are residue 1-12, 108-118, 141-200, and 214-244. These are the regions that undergo disorder-to-order transition upon binding with their respective partners [119]. Two PTM sites indicated in the D2P2 profile are at Lys107 (Ubiquitination) and at Ser223 (Phosphorylation).
Results of the FuzDrop-based analysis of mouse Chmp4b are summarized in Figure 5C,D. Figure 5C illustrates the droplet-promoting probabilities for each residue. Although the pLLPS value of 0.5154 predicted for this protein is below the 0.60 threshold, Chmp4b is predicted to have two droplet-promoting regions (residues 1-22 and 190-224); i.e., regions with the pDP values above 0.6 threshold, indicating that this protein can serve as a droplet client. The aggregation hot spots (i.e., regions that have a high tendency to aggregate, and therefore can also contribute to the pathogenesis of neurological disorders) are found at residues 54-62, 197-207, and 211-217. The Multiplicity of Binding Modes graph is displayed in Figure 5D revealing the tendency of residues to engage in multiple interactions with various partners.
High MBM predicts that several regions (residues 27-32, 39-82, 183-190, 197-207, and 211-217) can take part in multiple interactions aiding the liquid-liquid phase separation process and be involved in context-dependent interactions (see Figure 5D). These regions consist of residues that behave differently depending on the context of their cellular environment.
Figure 5E depicts the Chmp4b-centered PPI network that includes 100 proteins interconnected through 1,341 edged (edges represent the interactions between proteins). This observed value of edges is much greater than the expected number of edges of 176. The average node degree (which is the average number of connections per protein) predicted for this network is 26.8 and its average local clustering coefficient is 0.806. The PPI enrichment p-value is < 1.0e-16 suggesting that proteins in this Chmp4b-centered PPI network have more interactions among themselves than what would be expected for a random set of proteins of the same size and degree distribution drawn from the genome. GO-based functional enrichment analysis of this network indicated that these proteins are involved in the following biological processes: Ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway (GO:0043162; p-value = 6.73e-43), Multivesicular body sorting pathway (GO:0071985; p-value = 1.62e-33), Vacuolar transport (GO:0007034; p-value = 2.43e-33), Endosome transport via multivesicular body sorting pathway (GO:0032509; p-value = 8.11e-33), and Late endosome to vacuole transport (GO:0045324; p-value = 5.55e-32). Among the most significantly enriched molecular functions of these proteins are Structural constituent of eye lens (GO:0005212; p-value = 4.36e-20), Structural molecule activity (GO:0005198; p-value = 1.15e-18), Structural constituent of cytoskeleton (GO:0005200; p-value = 8.50e-15), GTP binding (GO:0005525; p-value = 3.97e-09), and Ubiquitin-like protein ligase binding (GO:0044389; p-value = 4.28e-07). The most significant enrichment of these proteins is observed in the following cellular components: ESCRT complex (GO:0036452; p-value = 3.59e-45), Endosome (GO:0005768; p-value = 7.27e-29), Endosome membrane (GO:0010008; p-value = 3.37e-28), Late endosome (GO:0005770; p-value = 5.21e-27), and Late endosome membrane (GO:0031902; p-value = 1.09e-26).
Lastly, Figure 5F represents the 3D model generated for mouse Chmp4b by AlphaFold. Surprisingly, although predicted structure mostly represents a set of disjoined α-helices that do not form a core, this model is characterized by a relatively high confidence of above 70% (the structure is mostly consist of structural elements colored in cyan (high confidence, 90 > pLDDT > 70) and blue (very high confidence, pLDDT > 90). As it was already indicated, long α-helical segments cannot exist in isolation. Therefore, it is very likely that structure predicted by AlphaFold corresponds to the bound form of the protein. It is known that the remodeling of membrane in abscission is caused by the polymerization of ESCRT-III components, which are soluble in monomeric autoinhibited state but assemble into membrane-bound filaments with crucial roles in membrane fission, when this autoinhibition is relieved [120]. Therefore, it is likely that formation of ESCRT-III filaments is accompanied by the disorder-to order transition of the core subunits of this complex.
To shed more light on the potential functionality of various regions identified in mouse Chmp4b, we analyzed these proteins using the ELM platform. Results of this analysis are listed in Table 2.
Data reported in Table 2 indicate that the intrinsically disordered regions of Chmp4b involved in promotion of liquid-liquid phase separation, serving as aggregation hot spots, and acting as MoRFs and regions with multiplicity of binding modes are heavily enriched in potentially functional short linear motifs.
3.2.3. DnaJ Homolog Subfamily B Member 6 (DNAJB6; UniProt ID: O54946)
DnaJB6 is a 365-residue long protein with the molecular weight of 99,807 Da, which is involved in the cellular response towards stress, and, being a member of the Hsp40 family chaperone family, act as a co-chaperone of Hsp70 [121]. It has a stimulatory effect on the ATPase activity of the heat shock protein Hsp70. DnaJB6 activity as a co-chaperone indicates its importance in protein folding, repair, and assembly. For example, it plays a role of an endogenous chaperone for huntingtin neuronal protein [121]. Being able to successfully suppress aggregation and toxicity of polyglutamine-containing, aggregation-prone proteins [122,123], DnaJB6 is designated as the antiamyloid chaperone, which is also capable of binding to the amyloid-β peptide fibrils and inhibiting secondary nucleation [124]. Furthermore, this chaperone is related to the biogenesis of the interphase nuclear pore complex (NPCs), binds to phenylalanine-glycine-rich nucleoporins (FG-Nups), and prevents their aggregation in cells and in vitro [125]. Furthermore, it is able to form foci (i.e., likely to phase separate) in close proximity to NPCs [125]. This protein was also shown to play a role in the organization of keratin 8 and 18 (KRT8/KRT18) filaments [126].
The N-terminal half of the protein contains a dnaJ domain (residues 3-69) and contains an Hsp70 interacting region (residues 2-147). Region comprising residues 120-243 has been shown to interact with KRT8 and the C-terminal region (residues 243-365) is expected to be disordered and contains a subregion 273-287 with the compositional bias (enriched in basic and acidic residues). In line with these observations, Figure 6 shows that mouse DnaJB6 protein contains significant levels of functional intrinsic disorder. Based on the RIDAO-based analyses (see Figure 6A), this protein is characterized by the PPIDR values of 50.68% (PONDR® VLXT), 96.44% (PONDR® VSL2B), 89.59% (PONDR® VL3), 76.16% (PONDR® FIT), 43.29% (IUPred_Short), and 52.33% (IUPred_Long). The mean PPIDR value averaged over all these tools is 66.58%. Figure 6A also shows that a highly disordered region was found at the C-terminus region of the protein (residues 253-365).
As per the D2P2 analysis, the consensus IDRs are found at residues 15-98, 106-188, and 197-365 (Figure 6B). Figure 6B also shows that mouse DnaJB6 contains three MoRFs (residues 223-278, 282-298, and 305-365) and includes several PTMs, such as phosphorylation of Ser15, mono-methylation at Arg136, and ubiquitylation at Lys20, Lys 34, Lys 60, Lys 61, and Lys 67.
Figure 6C shows the FuzDrop-generated profile reflecting the LLPS and droplet formation tendency of the protein. Here, the residues with pDP ≥ 0.6 threshold are expected to have the tendency to promote liquid-liquid phase separation. The pLLPS value of 0.9937 for DNAJB6 is extremely high, significantly exceeding a threshold value of 0.6, indicating that this protein is a droplet-driver. This is in a line with the aforementioned capability of DnaJB6 to form foci in the vicinity of NPCs [125]. Figure 6C also shows in DnaJB6, the droplet-promoting regions are predicted at residues 58-94, 119-185, and 233-365. Aggregations hot spots are found at residues 58-69, 83-90, 105-114, 119-131, 156-185, 241-250, 316-323, and 345-353. Figure 6D portrays a multiplicity of binding modes influenced by cellular contexts, such as PTMs and sub-cellular location of protein. The residues with MBM ≥ 0.65 are said to form regions with context-dependent interactions. For DnaJB6, the following regions were predicted to be MBM regions: 14-23, 39-55, 57-69, 83-90, 93-131, 156-203, 206-211,227-237,241-250,316-323, and 345-353.
Figure 6E represents the STRING-generated PPI network of mouse DnaJB6. This network includes 68 interactors and 993 interactions. It is characterized by an average local clustering coefficient of 0.78 and has an average node degree of 29.2. The expected number of edges for the DnaJB6-centerd PPI network is expected to be 209, indicating that the actual network has far more interactions than expected indicating that the members of this network are involved in the significant number of biological processes. A p-value of < 1.0e-16 suggests that the network we are observing in Figure 6E is statistically significant and cannot be generated by a random chance.
Members of the DnaJB6-centerd PPI network are involved in following biological processes: Protein folding (GO:0006457; p-value = 6.61e-43), Chaperone-mediated protein folding (GO:0061077; p-value = 8.46e-27), Protein refolding (GO:0042026; p-value = 1.27e-25), Response to topologically incorrect protein (GO:0035966; p-value = 1.27e-20), and Response to unfolded protein (GO:0006986; p-value = 2.60e-20). Major molecular functions of these proteins are Unfolded protein binding (GO:0051082; p-value = 1.35e-40), Protein folding chaperone (GO:0044183; p-value = 3.88e-38), ATP-dependent protein folding chaperone (GO:0140662; p-value = 2.63e-31), Heat shock protein binding (GO:0031072; p-value = 6.85e-24), and Chaperone binding (GO:0051087; p-value = 1.15e-18). The members of this nework act as cellular components of Cytoplasm (GO:0005737; p-value = 8.10e-14), Chaperone complex (GO:0101031; p-value = 1.59e-13), Cytosol (GO:0005829; p-value = 1.45e-11), Mitochondrial matrix (GO:0005759; p-value = 2.49e-10), and Mitochondrion GO:0005739; p-value = 4.02e-09).
The 3D structural model of the protein predicted by AlphaFold as shown in Figure 6F has an average per-residue model confidence score (pLDDT) of 60.8, indicating overall low confidence. AlphaFold-predicted structure also reveals that the C-terminal region of the protein is highly disordered, whereas the N-terminal region includes two structured domains, a mostly α-helical DnaJ domain (residues 1-104) and a mostly β-structural domain (residues 190-234) containing five antiparallel β-strands (residue 190-199, 202-211, 214-221, 224-230, and 233-234) followed by an α-helix (residues 236-245).
Table 3 lists some of the ELMs predicted in mouse DnaJB6 and shows that in line with its high intrinsic disorder status, this protein has a multitude of potential disorder-based functions.
Note, Table 3 dos not include all ELMs found in mouse DnaJB6, as this protein is predicted to have 57 different ELMs, with many of these being present in multiple copies (there are total of 186 ELM instances in DnaJB6).
3.2.4. Vacuolar Protein Sorting-Associated Protein 37B (Vps37B, UniProt ID: Q8R0J7)
Vps37B alternatively called ESCRT-I complex subunit Vps37B is 285 amino acids along with the molecular mass of 31,056 Da. Vps37B is a component of ESCRT-I complex (Endosomal Sorting Complex required for transport), which is a regulator of the vesicular transport process. As it was already indicated, endosomal sorting complexes required for transport machinery include five complexes with unique but connected functions, ESCRT-0, ESCRT-I, ESCRT-II, ESCRT-III, and the Vps4 complex. Among the many important activities of ancient ESCRT machinery are membrane deformation and scission (budding of the membranes and severing membrane necks from their interface) to form intraluminal vesicles (ILVs) linked to the biogenesis of the multivesicular bodies (MVBs) in endolysosomal sorting, as well as the budding of HIV-1 and other viruses from the plasma membrane of infected cells, and the membrane abscission step in cytokinesis. Furthermore, these complexes are related to the autophagy, cytokinesis, exovesicle release, repair of plasma and intracellular membranes, and enveloped RNA virus budding [127,128,129,130]. ESCRTs are oligomeric complexes that have complementary functions. Major components of the ESCRT-I complex, which is central to all ESCRT pathways and is essential for the MVB sorting of ubiquitylated cargo, are the three core subunits, Tsg101 (Vps23 in Saccharomyces cerevisiae), Vps28, and one of four Vps37 family members (Vps37A, Vps37B, Vps37C, or Vps37D), and a single auxiliary protein (Ubiquitin-associated protein 1 (Ubap1), MVB protein of 12 kDa (Mvb12A or Mvb12B)) [131,132]. The C-terminal half of Vps37, together with the N-terminal half of Vps28 and the C-terminal steadiness box (SB) domain of Vps23, are involved in the assembly of the ESCRT-I complex. The importance of Vps37 for the ESCRT-I structure and functionality is illustrated by the fact that depletion of this protein induces destabilization of the ESCRT-I and promotes strong cellular stress responses [133].
Vps37B contains the aforementioned C-terminal domain (residues 84-173) involved in the assembly of the ESCRT-I complex and a 50-170 region involved in interaction with the ESCRT-III protein IST1 [134]. Furthermore, regions 167-215 and 242-285 are annotated as intrinsically disordered on the corresponding UniProt page (https://www.uniprot.org/uniprotkb/Q8R0J7/entry#family_and_domains). Figure 7 provides support to this idea and shows that C-terminal half of mouse Vps37B is predicted to be highly disordered. Based on the data reported in Figure 7A, mouse Vps37B is characterized by the PPIDR values of 75.09%, 80.35%, 76.84%, 50.18%, 35.09%, and 46.67% as per the outputs of PONDR® VLXT, PONDR® VSL2B, PONDR® VL3, PONDR® FIT, IUPred_Short, and IUP_Long, respectively, and has the MPD (mean predicted disorder)-based PPIDR of 64.56%, classifying this protein as highly disordered.
Figure 7B shows that according to the results of the D2P2 analysis, disordered regions are found at residues 1-9, 13-18, 23-62, 90-102, 113-126, and 149-285 along the length of the protein. Figure 7B represents the disorder consensus bar in blue and green hues. Above this bar are conserved functional domains Modifier of rudimentary (Mod(r)) protein (residues 10-159) and Endosomal sorting complex domain ranging from 104 to 157. Protein is predicted to have 6 MoRFs (residues 133-144, 154-166, 188-202, 218-242, 249-263, and 279-285) and one ubiquitylation site at Lys 45.
Figure 7C,D represent the results of the FuzDrop-based analysis and show that mouse Vps37B is characterized by high probability of spontaneous liquid-liquid phase separation, pLLPS = 0.7062, implying that protein has a high tendency to be involved in droplet formation and can act as a droplet-driver. Figure 7C demonstrates the sequence distribution of residue-based droplet-promoting probabilities and indicates that the Vps37B is expected to contain two droplet promoting regions (DPRs) positioned at residues 157-237 and 244-285. There are also five aggregation hot spots in mouse Vps37B, residues 160-168, 191-213, 218-224, 228-237, and 251-258. Figure 7D represents a multiplicity of binding modes plot and shows that there are 12 regions with context-dependent interactions in this protein, residues 4-14, 16-26, 76-82, 150-155, 160-168, 191-201, 203-213, 218-224, 228-250, 243-248, and 251-258.
The Vps37B-centered PPI network generated by STRING is shown in Figure 7E. This network includes 42 involved in 636 interactions, which is significantly larger than the expected number of interactions 73, indicating that the network structure is not random as its network enrichment p-value is <1.7e-16. With the average node degree of 31.6 and the average local clustering coefficient of 0.903, this PPI network is highly connected. Analysis of this network revealed that among important biological processes ascribed to its members are Ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway (GO:0043162; p-value = 1.09e-52), Vacuolar transport (GO:0007034; p-value = 2.94e-45), Multivesicular body sorting pathway (GO:0071985; p-value = 3.42e-41), Endosome transport via multivesicular body sorting pathway (GO:0032509; p-value = 9.64e-40), and Late endosome to vacuole transport (GO:0045324; p-value = 2.43e-38). Most significantly enriched molecular functions of these proteins are Ubiquitin binding (GO:0043130; p-value = 3.22e-11), Protein binding (GO:0005515; p-value = 2.21e-06), Protein tag (GO:0031386; p-value = 4.31e-05), Protein domain specific binding (GO:0019904; p-value = 4.37e-05), and MIT domain binding (GO:0090541; p-value = 0.00019). Among cellular components significantly enriched in the members of this network are ESCRT complex (GO:0036452; p-value = 9.78e-58), Endosome membrane (GO:0010008; p-value = 8.71e-42), Late endosome membrane (GO:0031902; p-value = 7.17e-40), Late endosome (GO:0005770; p-value = 3.41e-39), and Endosome (GO:0005768; p-value = 1.41e-37).
Figure 7F represents the model of the Vps37B 3D structure generated by AlphaFold. Although this model is characterized by an average per-residue model confidence score (pLDDT) of 74.5, classifying confidence of this model as high, Figure 7F shows that the major structural element is a long stand-alone α-helix (residues 36-100), which physically cannot exist as a sable structure and therefore potentially represents a result of a structure that can be realized in the bound state.
We also looked at the abundance of ELMs in this protein and found that Vps37B has 132 instances of 56 ELMs. Although 25 ELMs (63 instances) were filtered out by ELM server due to the fact that they are located within a globular domain (Modifier of rudimentary (Mod(r)) protein (residues 10-159)), based on the structural model shown in Figure 7F, this region in fact does not form a globular domain (see above), and therefore, all predicted ELMs should be considered here. Figure 8 represents the output of ELM analysis and shows that entire protein is covered by short motifs with various functions, and many ELMs are included in or overlap with disorder-based regions discussed here, MoRFs, DPRs, aggregation hot spots, and MBP regions.
3.2.5. Actin Nucleation-Promoting Factor Wasl (UniProt ID: Q91YD9)
In mammals, the family of Wiskott–Aldrich syndrome protein (WASP) includes five subfamilies, such as WASP (which was the first member of the family discovered as a hematopoietically expressed protein encoded by a gene mutated in the rare X-linked immunodeficiency Wiskott–Aldrich syndrome [135]) and neuronal-WASP (N-WASP; also known as WASL), the three WASP family verprolin homolog isoforms (WAVE1–WAVE3; also known as SCAR1–SCAR3 and WASF1–WASF3), WASP homolog associated with actin, membranes and microtubules (WHAMM), WASP and SCAR homolog (WASH; also known as WASHC1), and junction-mediating regulatory protein (JMY) [136,137]. Members of this family act as regulators of the generation of branched actin filaments that are involved in the multitude of biological processes, such as endocytosis and/or phagocytosis at the plasma membrane, generation of cargo-containing vesicles from organelles including the Golgi, endoplasmic reticulum (ER), and the endo-lysosomal network, as well as formation of lamellipodia and filopodia [137]. WASP family members promote nucleation of seven-subunit actin-related proteins-2/3 (ARP2/3) complex acting as one of the major actin nucleators [138]. Interaction of WASP proteins with APR2/3 complex is determined by the conserved WCA (WH2, connecting and acidic) domain [137].
WASL also known as Neural Wiskott-Aldrich syndrome protein (N-WASP) is a 501- residue long protein with a molecular mass of 54,274 Daltons. Because of its role in actin polymerization, WASL is involved in cytokineis and mitosis and also plays a role in the formation of cell filopodia [139]. WASL interact with WASP activator CDC42 to form and maintain filopodia [140]. Along with cellular functions, WASL is also involved at the nuclear level possibly playing a role in regulating gene transcription [141].
In mouse Wasl, WASP homology 1 (WH1) domain (also known as Ena/VASP Homology domain 1, EVH1) is present at residues 31-138 and the CRIB domain is located at residues 200-213. P21-Rho-binding domain is found in the 199-257 region. WH2 motif also named as the first tandem Wiskott Aldrich syndrome homology region 2 is present in the region from 398 to 424 residues and the second WH2 motif is found at 424-449 position. Furthermore, Wasl contains a long proline-rich region (residues 271-391), and two regions with compositional bias, a region enriched in polar residues (residues 4420459), and acidic region (residues 482-501). PTMs are found at positions 2 (N-acetylserine), 239 (phosphoserine), 253 (Phosphoserine; by FAK1 and TNK2), 304 (Omega-N-methylarginine), and 481 (Phosphoserine) (https://www.uniprot.org/uniprotkb/Q91YD9/entry).
Peculiarities of functional intrinsic disorder of mouse Wasl protein are shown in Figure 9. According to the multifactorial disorder analysis by RIDAO, Wasl is predicted to contain high level of intrinsic disorder, with the C-terminal half of the protein being mostly disordered (see Figure 9A). The overall disorder content of mouse Wasl is exceeding 50%: 60.68% (PONDR® VLXT), 70.46% (PONDR® VSL2), 69.06% (PONDR® VL3), 59.28% (PONDR® FIT), 62.08% (IUPred Short), 72.46% (IUPred Long), and 64.47% (MDP).
Figure 9B represents a functional disorder profile generated by the D2P2 platform. This analysis revealed that disordered regions, where 75% of predictors agree, are found at residues 1-7, 13-15, 135-136, 138-160, 182-199, 260-432, 434-434, 440-467, and 470-501. The ordered N-terminal region corresponds to the PH domain-like (residues 27-138) and WHI domain (residues 28-135). Another region with somehow decreased disorder content corresponds to the P21-Rho-binding domain (PBD, residues 199-258), which is a part of the WASP C-terminal domain (residues 204-300). Finally, two WH2 motifs (residues 398-424 and 426-448), being the parts of a second WASP C-terminal domain (residues 378-489) are also expected to be more ordered than their flanking regions. Note that these two regions correspond to the characteristic dips with mean disorder scores of 0.68 ± 0.11 and 047 ± 0.25 clearly observed in PONDR® VLXT profile (see Figure 9A). Furthermore, Wasl is predicted to contain 8 MoRF regions (residues 167-178, 204-214, 219-271, 327-33, 345-352, 387-449, 458-482, and 491-501), indicating that intrinsic disorder can play an important role in functionality of this protein. Finally, the presence of multiple different PTMs (all located within IDRs) should be emphasized (see colored circles at the bottom of Figure 9B).
Figure 9C represents a graph depicting the probability of residues to promote liquid-liquid phase separation. Droplet-promoting regions (DPRs) are found at residues 127-165, 194-222, 258-402, and 444-501. Figure 9C also shows that the aggregation hot spots are located at residues 4-9, 132-139, 155-161, 194-206, 444-454, 459-466, 470-483, and 487-492. The pLLPS of 0.9796 for this protein is second highest among other proteins considered here, suggesting that the mouse Wasl protein has a very high tendency to promote the formation of membrane-less organelles and potentially acts as a droplet-driver. Figure 9D shows that the multiplicity of binding modes plot, illustrating that Wasl contain 13 regions with MBM value exceeding the 0.65 threshold, residues 4-23, 119-127, 132-139, 155-161, 163-170, 194-206, 220-225, 256-261, 404-471, 428-454, 459-466, 470-483, and 487-493.
STRING analysis revealed that the mouse Wasl forms a dense PPI network that includes 232 nodes linked by 5,283 edges (see Figure 9E). The number of PPIs in this network is much larger than the expected number of edges (917), indicating that this is a statistically significant PPI network with the PPI enrichment p-value of < 1.0e-16. The average node degree is 45.5 and an average local clustering coefficient is 0.595. The top 5 most enriched Biological Processes attributed to the proteins in this network are Actin filament-based process (GO:0030029; p-value = 6.73e-119), Actin cytoskeleton organization (GO:0030036; p-value = 1.35e-117), Cytoskeleton organization (GO:0007010; p-value = 2.58e-96), Regulation of actin cytoskeleton organization (GO:0032956; p-value = 1.33e-91), and Regulation of actin filament-based process (GO:0032970; p-value = 2.26e-90). The most enriched Molecular Functions of the members of this Wasl-centered PPI network are Actin binding (GO:0003779; p-value = 1.01e-71), Cytoskeletal protein binding (GO:0008092; p-value = 1.68e-71), Protein binding (GO:0005515; p-value = 7.97e-48), Actin filament binding (GO:0051015; p-value = 3.00e-40), and Protein-containing complex binding (GO:0044877; p-value = 3.18e-34). The top 5 most enriched Cellular Components, where these proteins are found, are Cytoskeleton (GO:0005856; p-value = 4.13e-75), Cell leading edge (GO:0031252; p-value = 2.82e-74), Actin cytoskeleton (GO:0015629; p-value = 3.67e-73), Cell projection (GO:0042995; p-value = 2.16e-63), and Lamellipodium (GO:0030027; p-value = 1.97e-55).
Figure 9E represents the 3D structural model generated for mouse Wasl by AlphaFold and supports the idea of the high disorder content in this protein. In fact, Figure 9E shows that although Wasl is predicted to have several ordered domains and regions, it also contains multiple regions with low and very low per-residue model confidence score (pLDDT), indicating that such regions can be disordered in isolation. Overall structural model of Wasl is characterized by the average pLDDT value of 69.28, indicating that this structure is generally modeled with low confidence (70 > pLDDT > 50).
At the final stage, we analyzed the presence and distribution of ELMs within the sequence of this protein. Not surprisingly, because of its length and high prevalence of disorder, mouse Wasl was predicted to have 231 instances of 65 ELMs. Results of this analysis are summarized in Figure 10 and show that many ELMs are incorporated in or overlap with disorder-based regions discussed here, MoRFs, DPRs, aggregation hot spots, and MBP regions.
3.3. Global PPI Networks Analysis of the Most Disordered Mouse Proteins Found in the Rabies Virus
Next, we looked at the interconnectivity of the members of a group of the 11 most disordered mouse proteins found in RABV particles. Results of this analysis are shown in Figure 11. When this set was analyzed by STRING using medium confidence of 0.4 for the minimum required interaction score, these proteins were not linked in a single network, but formed two disconnected networks, consisting of 6 and 3 proteins, with two proteins, Vesicle-associated membrane protein 3 (Vamp3) and neuromodulin (Gap43), being the loners (see Figure 11A). Although 11 proteins were connected by 8 interactions within this disjoined network (defining the low node degree of 1.45), they still had more interactions among themselves than what would be expected for a random set of proteins of the same size and degree distribution drawn from the genome (1). When the confidence of the minimum required interaction score was decreased to 0.15 (low confidence), all 11 proteins became engaged in interactions and formed a single PPI network with 25 edges and average node degree of 4.55. These proteins had one common molecule function, Protein Binding (GO:0005515; p-value = 0.0470), being significantly engaged in several biological processes, such as Protein localization (GO:0008104; p-value = 0.00017), Cellular localization (GO:0051641; p-value = 0.00017), Viral release from host cell (GO:0019076; p-value = 0.0011), Ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway (GO:0043162; p-value = 0.0013), and Vesicle-mediated transport (GO:0016192; p-value = 0.0014). These proteins were significantly enriched membes of the following cellular components: ESCRT complex (GO:0036452; p-value = 0.00071), Cytoplasmic vesicle (GO:0031410; p-value = 0.0011), Endosome (GO:0005768; p-value = 0.0014), Cytosol (GO:0005829; p-value = 0.0048), and ESCRT I complex (GO:0000813; p-value = 0.0052).
We also checked the set-centered interactivity of these 11 most disordered mouse proteins found in RABV particles. To this end, we used the multiple proteins search option of STRING platform and selected a custom value of 500 maximum first-shell interactions (note that the number of interactors in STRING is limited to 500) and high confidence level (minimum required interaction score of 0.7). Using these settings resulted in the generation of a well-connected PPI network containing 281 proteins involved in 3,918 interactions (see Figure 12). The average node degree of this network is 20.6 and its average local clustering coefficient is 0.618. The top 5 most enriched biological processes these proteins are involved in are Vesicle-mediated transport (GO:0016192; p-value = 3.41e-80), Cellular localization (GO:0051641; p-value = 3.41e-80), Regulation of cellular component organization (GO:0051128; p-value = 3.22e-76), Protein localization (GO:0008104; p-value = 2.85e-71), and Establishment of localization in cell (GO:0051649; p-value = 1.38e-65). The top 5 enriched biological functions of members of this network are Protein binding (GO:0005515; p-value = 1.34e-77), Protein domain specific binding (GO:0019904; p-value = 8.56e-46), Cytoskeletal protein binding (GO:0008092; p-value = 1.06e-39), SNAP binding (GO:0005484; p-value = 2.99e-38), and Binding (GO:0005488; p-value = 5.52e-36). The top 5 most enriched cellular components, where these proteins are found, are Vesicle (GO:0031982; p-value = 1.74e-73), Cytoplasmic vesicle (GO:0031410; p-value = 6.16e-66), Cell junction (GO:0030054; p-value = 6.16e-66), Cell projection (GO:0042995; p-value = 3.74e-64), and Cytoplasm (GO:0005488; p-value = 3.746e-64).
Application of the k-means clustering (which is an unsupervised machine learning algorithm designed to group the unlabeled datasets into different clusters thereby dividing a set of data into a number of groups depending on how similar and different they are to one another) to this PPI network centered at the 11 most disordered mouse proteins found in RABV particles revealed that the set of 381 proteins can be split on 3 clusters.
The biggest cluster includes 263 proteins involved in 2,312 interactions (see red circles in Figure 12). This sub-network includes many proteins from the regulation of actin cytoskeleton pathway (KEGG pathway ID: mmu04810, p-value = 1.98e-39). The average node degree of this network is 17.6 and its average local clustering coefficient is 0.587. The top 5 enriched molecular functions of the members of this network are Protein binding (GO:0005515; p-value = 5.60e-58), Cytoskeletal protein binding (GO:0008092; p-value = 7.30e-44), Actin binding (GO:0003779; p-value = 6.12e-43), Protein domain specific binding (GO:0019904; p-value = 1.31e-40), and Enzyme binding (GO:0019899; p-value = 1.22e-38). The top 5 most enriched biological processes these proteins are involved in are Regulation of cellular component organization (GO:0051128; p-value = 1.80e-65), Actin filament-based process (GO:0030029; p-value = 1.10e-53), Regulation of cellular component biogenesis (GO:0044087; p-value = 1.35e-52), Actin cytoskeleton organization (GO:0030036; p-value = 5.31e-50), and Cytoskeleton organization (GO:0007010; p-value = 5.51e-48). The top 5 most enriched cellular components, where these proteins can be found, are Cell projection (GO:0042995; p-value = 1.61e-62), Plasma membrane bounded cell projection (GO:0120025; p-value = 2.96e-59), Cytosol (GO:0005829; p-value = 1.36e-51), Cell junction (GO:0030054; p-value = 2.26e-50), and Cytoskeleton (GO:0005856; p-value = 6.03e-48).
The second cluster includes 60 proteins involved in 481 interactions (see green circles in Figure 12) and mostly related to the endocytosis pathway (KEGG pathway ID: mmu04144; p-value = 1.50e-44). This sub-network is characterized by the average node degree of 16 and average local clustering coefficient of 0.813. The top 5 most enriched molecular functions of proteins in this cluster are Ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway (GO:0043162; p-value = 5.74e-45), Vacuolar transport (GO:0007034; p-value = 4.66e-41), Multivesicular body sorting pathway (GO:0071985; p-value = 4.84e-37), Viral budding (GO:0046755; p-value = 2.25e-36), and Endosome transport via multivesicular body sorting pathway (GO:0032509; p-value = 5.26e-36). The top 5 most enriched biological processes assigned to these proteins are Ubiquitin binding (GO:0043130; p-value = 6.09e-08), Protein binding (GO:0005515; p-value = 2.69e-06), L-lactate dehydrogenase activity (GO:0004459; p-value = 4.33e-06), Calcium-dependent protein binding (GO:0048306; p-value = 9.28e-06), and Phospholipid binding (GO:0005543; p-value = 5.52e-05). Proteins of this cluster are most enriched in the following 5 cellular components: ESCRT complex (GO:0036452; p-value = 1.23e-49), Endosome membrane (GO:0010008; p-value = 6.18e-43), Late endosome membrane (GO:0031902; p-value = 1.39e-38), Endosome (GO:0005768; p-value = 8.24e-38), and Cytoplasmic vesicle membrane (GO:0030659; p-value = 1.80e-34).
In the third cluster, there are 58 proteins connected by 875 interactions (see blue circles in Figure 12). Most of the proteins in this cluster are related to the SNARE interactions in vesicular transport pathway (KEGG pathway ID: mmu04130l p-value = 6.94e-62). This sub-network is characterized by the average node degree of 30.2 and the average local clustering coefficient of 0.868. The top 5 most enriched molecular functions of the members of this sub-network are Vesicle-mediated transport (GO:0016192; p-value = 4.17e-59), Membrane fusion (GO:0061025; p-value = 6.79e-59), Vesicle fusion (GO:0006906; p-value = 1.63e-53), Establishment of localization in cell (GO:0051649; p-value = 5.79e-46), and Vesicle organization (GO:0016050; p-value = 7.23e-44). The top 5 most enriched biological processes these proteins are involved in are SNARE binding (GO:0000149; p-value = 1.15e-72), SNAP receptor activity (GO:0005484; p-value = 5.11e-70), Protein-macromolecule adaptor activity (GO:0030674; p-value = 6.13e-46), Syntaxin binding (GO:0019905; p-value = 4.45e-43), and Syntaxin-1 binding (GO:0017075; p-value = 7.64e-16). The top 5 most enriched cellular components, where proteins from this cluster can be found, are SNARE complex (GO:0031201; p-value = 2.90e-91), Transport vesicle (GO:0030133; p-value = 4.17e-43), Exocytic vesicle (GO:0070382; p-value = 3.76e-41), Synaptic vesicle (GO:0008021; p-value = 2.50e-40), and Membrane protein complex (GO:0098796; p-value = 4.14e-40).
To get a hint on the prevalence of intrinsic disorder in host interactors of mouse proteins entrapped in the RABV particle, we applied RIDAO platform to proteins in the aforementioned clusters. Results of this analysis are summarized in Figure 13, which clearly shows that all these protein sets are characterized by the presence of significant levels of intrinsic disorder. In fact, in all these clusters, proteins classified as disordered based on their PPIDR values exceeding the 30% threshold constitute vast majority, and 41% to 55% are expected to be highly disordered (based on their position within the red segment of Figure 13A). Furthermore, from 47.5% to 65.5% proteins in these clusters are located outside the quadrant Q1 and therefore contain significant levels of intrinsic disorder (see Figure 13B).
3.4. The Roles of Intrinsically Disordered Host Proteins in Viral Immune Evasion and Pathogenesis Enhancement
Now, we are going to focus on how the Rabies virus exploited the structural chaos associated with the entrapped host proteins (i.e., their high intrinsic disorder status) to its own benefit. The incorporation of host proteins within viral particles helps them evade the immunity, the antiviral resistance, and eventually results in the enhancement of viral pathogenesis [14]. These functions can be associated with the intrinsic disorder present in these entrapped host proteins. The viruses with incorporated host proteins are less recognizable by the host immune system, the antigens of the incorporated host proteins can mask the viral antigens normally recognizable through the immune system [14,142]. Because of the masking of the viral antigens by host proteins, host antibodies cannot efficiently detect the viral particles to successfully eliminate them [14,143]. Furthermore, because of this mimicry, the immune system can get confused and the effort of finding the mimicking viral particles can sometimes trigger autoimmunity, where the host immune system cells start attacking the own healthy cells leading to the tissue damage [143,144,145].
Viruses can also exploit host receptors to enter other cells effectively, which not only enhances its transmission rate but also increases the range of cell types a virus can infect [146], and the addition of the host proteins to the viral particles can enhance this ability of virus [147]. Molecular mimicry also helps viruses to evade antiviral drugs, making the development of antiviral drugs more complicated. Because these drugs are designed to attack unique viral particles without harming healthy host cells [148], the incorporation of host proteins in viral particles can make it difficult for the antiviral drugs to distinguish between the host cells and viral particles, leading to the increased toxicity and side effects and less effective therapeutic targeting.
In the context of our study, when we add the intrinsic disorder of these entrapped proteins to the picture, we can say that the scenario becomes even more complicated. As we have mentioned in the introduction, IDPs/IDRs lack 3D structure and are highly flexible and adaptable. They can bind to a variety of partners [149] and can facilitate the interaction of viral particles with a wide array of host cells, facilitating viral entry, replication, and overall pathogenesis. The flexible nature of IDPs can also assist viruses to evolve and become more adaptable to their environment. Viruses can manipulate the properties of intrinsically disordered host proteins to escape the environmental pressure created by the host immune system, also making therapeutic strategies more complex. We can hypothesize that these IDPs/IDRs are providing numerous additional functional and evolutionary benefits to the virus.
In short, we can target the interaction of host IDPs/IDRs with the virus to disrupt the viral life cycle. Understanding the role of host IDPs/IDRs in the life cycle of viruses can open new lines of research to develop more effective antiviral therapeutic strategies.
4. Conclusions
The bioinformatics analysis performed on the host proteins incorporated within the rabies viruses offers significant findings regarding the role of host intrinsic disorder in the life cycle of rabies viruses.
Out of 47 host proteins that are entrapped in the viral particles, most were predicted as noticeably disordered. In fact 40.4% and 55.4% of these proteins were predicted as moderately or highly disordered, respectively. Based on the results of the PONDR® VSL2-based disorder analysis, 11 proteins were predicted to be mostly disordered, since they were shown to have PPIDR values exceeding 50% and ADS values exceeding 0.5. Detailed computational analysis of the five most disordered host proteins entrapped in the RABV particles, Neuromodulin, Chmp4b, DnaJB6, Vps37B, and Wasl, revealed several important roles that intrinsic disorder can play in functionality of these proteins. It is also very likely that intrinsic disorder of the host proteins entrapped in the viral particles could be playing essential roles in the pathogenicity of viruses, modulating their mechanisms of immune evasion, promoting the development of antiviral drug resistance and, thereby, contributing to the virus adaptability and evolution.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Supplementary Table S1. Amino acid sequences of proteins analyzed in this study; Supplementary Table S2: Functional enrichment data for 11 highly disordered host proteins focusing on gene ontology highlighting biological process; Supplementary Table S3: Functional enrichment data for 11 highly disordered host proteins focusing on their individual gene ontology highlighting Molecular Function; Supplementary Table S4: Functional enrichment data for 11 highly disordered host proteins focusing on their individual gene ontology highlighting Cellular Component; Supplementary Table S5. Multifactorial analysis of intrinsic disorder predisposition of mouse proteins entrapped in RABV particles; Supplementary Table S6: Localization of ELMs (Eukaryotic Linear Motifs) within the Droplet Promoting Regions, Aggregation Hot-spots and MoRFs of mouse Neuromodulin (UniProt ID: P06837); Supplementary Table S7: Distribution of ELMs (Eukaryotic Linear Motifs) in Droplet Promoting Regions, Aggregation Hot-spots, regions, and MoRFs (Molecular recognition features) of the protein Chmp4b (UniProt ID: Q9D8B3); Supplementary Table S8: Distribution of ELMs (Eukaryotic Linear Motifs) in droplet promoting regions, aggregation hot-spots, regions with multiplicity of binding modes and MoRF (Molecular recognition features) of protein DnaJ homolog subfamily B member 6 (UniProt ID: O54946); Supplementary Table S9: Distribution of ELMs (Eukaryotic Linear Motifs) in droplet promoting regions, aggregation hot-spots, regions with multiplicity of binding modes and MoRF (Molecular recognition features) of Vps37B protein (UniProt ID: Q8R0J7); Supplementary Table S10: Distribution of ELMs (short linear functional motifs) within the sequence of the mouse Wasl protein (UniProt ID: Q91YD9).
Author Contributions
Conceptualization, V.N.U.; methodology, V.N.U.; validation, H.N.A. and V.N.U.; formal analysis, H.N.A. and V.N.U.; investigation, H.N.A. and V.N.U.; data curation, H.N.A. and V.N.U.; writing—original draft preparation, H.N.A. and V.N.U.; writing—review and editing, H.N.A. and V.N.U.; visualization, V,N.U.; supervision, V.N.U. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained within the article and supplementary materials.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Hemachudha, T.; Laothamatas, J.; Rupprecht, C.E. Human rabies: a disease of complex neuropathogenetic mechanisms and diagnostic challenges. Lancet Neurol 2002, 1, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, J.E. Rabies. Available online: https://www.merckmanuals.com/professional/neurologic-disorders/brain-infections/rabies (accessed on March 18).
- Pieracci, E.G.; Pearson, C.M.; Wallace, R.M.; Blanton, J.D.; Whitehouse, E.R.; Ma, X.; Stauffer, K.; Chipman, R.B.; Olson, V. Vital Signs: Trends in Human Rabies Deaths and Exposures - United States, 1938-2018. MMWR Morb Mortal Wkly Rep 2019, 68, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Hampson, K.; Coudeville, L.; Lembo, T.; Sambo, M.; Kieffer, A.; Attlan, M.; Barrat, J.; Blanton, J.D.; Briggs, D.J.; Cleaveland, S.; et al. Estimating the global burden of endemic canine rabies. PLoS Negl Trop Dis 2015, 9, e0003709. [Google Scholar] [CrossRef]
- Brunker, K.; Mollentze, N. Rabies Virus. Trends Microbiol 2018, 26, 886–887. [Google Scholar] [CrossRef]
- Rupprecht, C.E. Rhabdoviruses: Rabies Virus. In Medical Microbiology, 4th ed.; Baron, S., Ed.; Galveston (TX), 1996.
- Horwitz, J.A.; Jenni, S.; Harrison, S.C.; Whelan, S.P.J. Structure of a rabies virus polymerase complex from electron cryo-microscopy. Proc Natl Acad Sci U S A 2020, 117, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Riedel, C.; Vasishtan, D.; Prazak, V.; Ghanem, A.; Conzelmann, K.K.; Rumenapf, T. Cryo EM structure of the rabies virus ribonucleoprotein complex. Sci Rep 2019, 9, 9639. [Google Scholar] [CrossRef]
- Warrell, M.J.; Warrell, D.A. Rabies: the clinical features, management and prevention of the classic zoonosis. Clinical Medicine 2015, 15, 78. [Google Scholar] [CrossRef]
- Singh, R.; Singh, K.P.; Cherian, S.; Saminathan, M.; Kapoor, S.; Manjunatha Reddy, G.; Panda, S.; Dhama, K. Rabies–epidemiology, pathogenesis, public health concerns and advances in diagnosis and control: a comprehensive review. Veterinary Quarterly 2017, 37, 212–251. [Google Scholar] [CrossRef]
- Davis, B.M.; Rall, G.F.; Schnell, M.J. Everything You Always Wanted to Know About Rabies Virus (But Were Afraid to Ask). Annu Rev Virol 2015, 2, 451–471. [Google Scholar] [CrossRef]
- Potratz, M.; Zaeck, L.M.; Weigel, C.; Klein, A.; Freuling, C.M.; Muller, T.; Finke, S. Neuroglia infection by rabies virus after anterograde virus spread in peripheral neurons. Acta Neuropathol Commun 2020, 8, 199. [Google Scholar] [CrossRef]
- Wunner, W.H.; Conzelmann, K.K. Rabies virus. In Rabies, 3rd Edition ed.; AC., J., Ed.; Academic Press/Elsevier: Oxford, UK, 2013; pp. 17-60.
- Burnie, J.; Guzzo, C. The Incorporation of Host Proteins into the External HIV-1 Envelope. Viruses 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci 2013, 22, 693–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Feng, Y.; Tu, Z.; Lou, Z.; Tu, C. Proteomic Profiling of Purified Rabies Virus Particles. Virol Sin 2020, 35, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, X.; Vidy, A.; Fouquet, B.; Blondel, D. Hsp70 protein positively regulates rabies virus infection. J Virol 2012, 86, 4743–4751. [Google Scholar] [CrossRef]
- Chen, B.J.; Lamb, R.A. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 2008, 372, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Vera-Velasco, N.M.; Garcia-Murria, M.J.; Sanchez Del Pino, M.M.; Mingarro, I.; Martinez-Gil, L. Proteomic composition of Nipah virus-like particles. J Proteomics 2018, 172, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.L.; Stone, K.L.; Colangelo, C.M.; Gulcicek, E.E.; Palese, P. Cellular proteins in influenza virus particles. PLoS Pathog 2008, 4, e1000085. [Google Scholar] [CrossRef]
- Uversky, V.N. Functional unfoldomics: Roles of intrinsic disorder in protein (multi)functionality. Adv Protein Chem Struct Biol 2024, 138, 179–210. [Google Scholar] [CrossRef]
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. Intrinsically disordered proteins play diverse roles in cell signaling. Cell Commun Signal 2022, 20, 20. [Google Scholar] [CrossRef]
- Kulkarni, P.; Bhattacharya, S.; Achuthan, S.; Behal, A.; Jolly, M.K.; Kotnala, S.; Mohanty, A.; Rangarajan, G.; Salgia, R.; Uversky, V. Intrinsically Disordered Proteins: Critical Components of the Wetware. Chem Rev 2022, 122, 6614–6633. [Google Scholar] [CrossRef]
- Uversky, V.N. Recent Developments in the Field of Intrinsically Disordered Proteins: Intrinsic Disorder-Based Emergence in Cellular Biology in Light of the Physiological and Pathological Liquid-Liquid Phase Transitions. Annu Rev Biophys 2021, 50, 135–156. [Google Scholar] [CrossRef]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol Life Sci 2015, 72, 137–151. [Google Scholar] [CrossRef]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn 2012, 30, 137–149. [Google Scholar] [CrossRef]
- Dyson, H.J. Making Sense of Intrinsically Disordered Proteins. Biophys J 2016, 110, 1013–1016. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xue, B.; Jones, W.T.; Rikkerink, E.; Dunker, A.K.; Uversky, V.N. A functionally required unfoldome from the plant kingdom: intrinsically disordered N-terminal domains of GRAS proteins are involved in molecular recognition during plant development. Plant Mol Biol 2011, 77, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dave, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: intrinsically disordered proteins and human diseases. Chem Rev 2014, 114, 6844–6879. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim Biophys Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Turoverov, K.K.; Kuznetsova, I.M.; Uversky, V.N. The protein kingdom extended: ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog Biophys Mol Biol 2010, 102, 73–84. [Google Scholar] [CrossRef]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr Opin Struct Biol 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Dayhoff, G.W., 2nd; Uversky, V.N. Rapid prediction and analysis of protein intrinsic disorder. Protein Sci 2022, 31, e4496. [Google Scholar] [CrossRef] [PubMed]
- Puntervoll, P.; Linding, R.; Gemund, C.; Chabanis-Davidson, S.; Mattingsdal, M.; Cameron, S.; Martin, D.M.; Ausiello, G.; Brannetti, B.; Costantini, A.; et al. ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res 2003, 31, 3625–3630. [Google Scholar] [CrossRef]
- Gould, C.M.; Diella, F.; Via, A.; Puntervoll, P.; Gemund, C.; Chabanis-Davidson, S.; Michael, S.; Sayadi, A.; Bryne, J.C.; Chica, C.; et al. ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res 2010, 38, D167–180. [Google Scholar] [CrossRef]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol Biosyst 2012, 8, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, H.; Michael, S.; Weatheritt, R.J.; Davey, N.E.; Van Roey, K.; Altenberg, B.; Toedt, G.; Uyar, B.; Seiler, M.; Budd, A.; et al. ELM--the database of eukaryotic linear motifs. Nucleic Acids Res 2012, 40, D242–251. [Google Scholar] [CrossRef]
- Gouw, M.; Samano-Sanchez, H.; Van Roey, K.; Diella, F.; Gibson, T.J.; Dinkel, H. Exploring Short Linear Motifs Using the ELM Database and Tools. Curr Protoc Bioinformatics 2017, 58, 8–22. [Google Scholar] [CrossRef]
- Kumar, M.; Gouw, M.; Michael, S.; Samano-Sanchez, H.; Pancsa, R.; Glavina, J.; Diakogianni, A.; Valverde, J.A.; Bukirova, D.; Calyseva, J.; et al. ELM-the eukaryotic linear motif resource in 2020. Nucleic Acids Res 2020, 48, D296–D306. [Google Scholar] [CrossRef]
- Kumar, M.; Michael, S.; Alvarado-Valverde, J.; Zeke, A.; Lazar, T.; Glavina, J.; Nagy-Kanta, E.; Donagh, J.M.; Kalman, Z.E.; Pascarelli, S.; et al. ELM-the Eukaryotic Linear Motif resource-2024 update. Nucleic Acids Res 2024, 52, D442–D455. [Google Scholar] [CrossRef]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem Rev 2014, 114, 6733–6778. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)P(2): database of disordered protein predictions. Nucleic Acids Res 2013, 41, D508–516. [Google Scholar] [CrossRef]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc Natl Acad Sci U S A 2020, 117, 33254–33262. [Google Scholar] [CrossRef]
- Hatos, A.; Tosatto, S.C.E.; Vendruscolo, M.; Fuxreiter, M. FuzDrop on AlphaFold: visualizing the sequence-dependent propensity of liquid-liquid phase separation and aggregation of proteins. Nucleic Acids Res 2022, 50, W337–W344. [Google Scholar] [CrossRef]
- Antifeeva, I.A.; Fonin, A.V.; Fefilova, A.S.; Stepanenko, O.V.; Povarova, O.I.; Silonov, S.A.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. Liquid-liquid phase separation as an organizing principle of intracellular space: overview of the evolution of the cell compartmentalization concept. Cell Mol Life Sci 2022, 79, 251. [Google Scholar] [CrossRef]
- Nesterov, S.V.; Ilyinsky, N.S.; Uversky, V.N. Liquid-liquid phase separation as a common organizing principle of intracellular space and biomembranes providing dynamic adaptive responses. Biochim Biophys Acta Mol Cell Res 2021, 1868, 119102. [Google Scholar] [CrossRef]
- Gomes, E.; Shorter, J. The molecular language of membraneless organelles. J Biol Chem 2019, 294, 7115–7127. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011, 39, D561–568. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst 2008, 4, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Djulbegovic, M.B.; Taylor, D.J.; Antonietti, M.; Cordova, M.; Dayhoff, G.; Uversky, V.; Galor, A.; Karp, C.L. Intrinsic disorder and the human tear film proteome. Investigative Ophthalmology & Visual Science 2023, 64, 187–187. [Google Scholar]
- Taylor Gonzalez, D.J.; Djulbegovic, M.; Antonietti, M.; Cordova, M.; Dayhoff, G.W., 2nd; Mattes, R.; Galor, A.; Uversky, V.N.; Karp, C.L. Intrinsic Disorder in the Human Tear Proteome. Invest Ophthalmol Vis Sci 2023, 64, 14. [Google Scholar] [CrossRef] [PubMed]
- Djulbegovic, M.; Uversky, V.N. The aqueous humor proteome is intrinsically disordered. Biochemistry and Biophysics Reports 2022, 29. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem 2011, 112, 3256–3267. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Tosatto, S.C. Critical assessment of protein intrinsic disorder prediction. Nature methods 2021, 18, 472–481. [Google Scholar] [CrossRef]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Pac Symp Biocomput 2012, 128–139. [Google Scholar]
- Xue, B.; Oldfield, C.J.; Van, Y.Y.; Dunker, A.K.; Uversky, V.N. Protein intrinsic disorder and induced pluripotent stem cells. Mol Biosyst 2012, 8, 134–150. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J Mol Graph Model 2001, 19, 26–59. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef]
- Uversky, V.N. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem. Soc. Rev. 2011, 40, 1623–1634. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr Pharm Des 2013, 19, 4191–4213. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett 2015. [Google Scholar] [CrossRef] [PubMed]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angewandte Chemie-International Edition 1998, 37, 2755–2794. [Google Scholar] [CrossRef]
- Schulz, G.E. Nucleotide Binding Proteins. In Molecular Mechanism of Biological Recognition, Balaban, M., Ed.; Elsevier/North-Holland Biomedical Press: New York, 1979; pp. 79–94. [Google Scholar]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv Protein Chem 2002, 62, 25–49. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr Opin Struct Biol 2009, 19, 31–38. [Google Scholar] [CrossRef]
- Meador, W.E.; Means, A.R.; Quiocho, F.A. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science 1993, 262, 1718–1721. [Google Scholar] [CrossRef]
- Kriwacki, R.W.; Hengst, L.; Tennant, L.; Reed, S.I.; Wright, P.E. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci U S A 1996, 93, 11504–11509. [Google Scholar] [CrossRef]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput 1998, 473–484. [Google Scholar]
- Uversky, V.N. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell Mol Life Sci 2003, 60, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets: The roles of intrinsic disorder in protein interaction networks. FEBS Journal 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Dajani, R.; Fraser, E.; Roe, S.M.; Yeo, M.; Good, V.M.; Thompson, V.; Dale, T.C.; Pearl, L.H. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. Embo J 2003, 22, 494–501. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol 2002, 12, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.L.; Oldfield, C.J.; Xue, B.; Meng, J.; Huang, F.; Romero, P.; Uversky, V.N.; Dunker, A.K. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci 2013, 22, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 2008, 9 Suppl 1, S1. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Hazy, E.; Tompa, P. Limitations of induced folding in molecular recognition by intrinsically disordered proteins. Chemphyschem 2009, 10, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.; Aivazian, D.; Stern, L. Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry 2004, 43, 2049–2061. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.B.; Zhuravleva, A.V.; Orekhov, V.Y. Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie 2007, 89, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Permyakov, S.E.; Millett, I.S.; Doniach, S.; Permyakov, E.A.; Uversky, V.N. Natively unfolded C-terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins 2003, 53, 855–862. [Google Scholar] [CrossRef]
- Fuxreiter, M. Fuzziness: linking regulation to protein dynamics. Mol Biosyst 2012, 8, 168–177. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Tompa, P. Fuzzy complexes: a more stochastic view of protein function. Adv Exp Med Biol 2012, 725, 1–14. [Google Scholar] [CrossRef]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett 2015. [Google Scholar] [CrossRef]
- Patil, A.; Nakamura, H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett 2006, 580, 2041–2045. [Google Scholar] [CrossRef]
- Ekman, D.; Light, S.; Bjorklund, A.K.; Elofsson, A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol 2006, 7, R45. [Google Scholar] [CrossRef]
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol 2006, 2, e100. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J Proteome Res 2006, 5, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.P.; Dash, D. Intrinsic disorder in yeast transcriptional regulatory network. Proteins 2007, 68, 602–605. [Google Scholar] [CrossRef]
- Singh, G.P.; Ganapathi, M.; Dash, D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins 2007, 66, 761–765. [Google Scholar] [CrossRef]
- Antifeeva, I.A.; Fonin, A.V.; Fefilova, A.S.; Stepanenko, O.V.; Povarova, O.I.; Silonov, S.A.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. Liquid–liquid phase separation as an organizing principle of intracellular space: overview of the evolution of the cell compartmentalization concept. Cellular and Molecular Life Sciences 2022, 79. [Google Scholar] [CrossRef]
- Turoverov, K.K.; Kuznetsova, I.M.; Fonin, A.V.; Darling, A.L.; Zaslavsky, B.Y.; Uversky, V.N. Stochasticity of Biological Soft Matter: Emerging Concepts in Intrinsically Disordered Proteins and Biological Phase Separation. Trends Biochem Sci 2019, 44, 716–728. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- !!! INVALID CITATION !!! [52-67].
- Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett 2015, 589, 15–22. [Google Scholar] [CrossRef]
- Brangwynne, C.P. Phase transitions and size scaling of membrane-less organelles. J Cell Biol 2013, 203, 875–881. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol 2016, 44, 18–30. [Google Scholar] [CrossRef]
- Brangwynne, Clifford P. ; Tompa, P.; Pappu, Rohit V. Polymer physics of intracellular phase transitions. Nat Phys 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Zuber, M.X.; Strittmatter, S.M.; Fishman, M.C. A membrane-targeting signal in the amino terminus of the neuronal protein GAP-43. Nature 1989, 341, 345–348. [Google Scholar] [CrossRef]
- Liu, Y.C.; Chapman, E.R.; Storm, D.R. Targeting of neuromodulin (GAP-43) fusion proteins to growth cones in cultured rat embryonic neurons. Neuron 1991, 6, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fisher, D.A.; Storm, D.R. Intracellular sorting of neuromodulin (GAP-43) mutants modified in the membrane targeting domain. J Neurosci 1994, 14, 5807–5817. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chichili, V.P.; Zhong, L.; Tang, X.; Velazquez-Campoy, A.; Sheu, F.S.; Seetharaman, J.; Gerges, N.Z.; Sivaraman, J. Structural basis for the interaction of unstructured neuron specific substrates neuromodulin and neurogranin with Calmodulin. Sci Rep 2013, 3, 1392. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Mani, S.; Donovan, S.L.; Schwob, J.E.; Meiri, K.F. Growth-associated protein-43 is required for commissural axon guidance in the developing vertebrate nervous system. J Neurosci 2002, 22, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.R.; Au, D.; Alexander, K.A.; Nicolson, T.A.; Storm, D.R. Characterization of the calmodulin binding domain of neuromodulin. Functional significance of serine 41 and phenylalanine 42. J Biol Chem 1991, 266, 207–213. [Google Scholar] [CrossRef]
- Katuwawala, A.; Peng, Z.; Yang, J.; Kurgan, L. Computational Prediction of MoRFs, Short Disorder-to-order Transitioning Protein Binding Regions. Comput Struct Biotechnol J 2019, 17, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Marnik, E.A.; Updike, D.L. Membraneless organelles: P granules in Caenorhabditis elegans. Traffic 2019, 20, 373–379. [Google Scholar] [CrossRef]
- Mohammed, A.S.; Uversky, V.N. Intrinsic Disorder as a Natural Preservative: High Levels of Intrinsic Disorder in Proteins Found in the 2600-Year-Old Human Brain. Biology (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Zhou, Y.; Bennett, T.M.; Shiels, A. A charged multivesicular body protein (CHMP4B) is required for lens growth and differentiation. Differentiation 2019, 109, 16–27. [Google Scholar] [CrossRef]
- Schmidt, O.; Teis, D. The ESCRT machinery. Curr Biol 2012, 22, R116–120. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Freed, E.O.; van Engelenburg, S.B. A Consensus View of ESCRT-Mediated Human Immunodeficiency Virus Type 1 Abscission. Annu Rev Virol 2017, 4, 309–325. [Google Scholar] [CrossRef]
- Sharma, R.; Sharma, A.; Patil, A.; Tsunoda, T. Discovering MoRFs by trisecting intrinsically disordered protein sequence into terminals and middle regions. BMC Bioinformatics 2019, 19, 378. [Google Scholar] [CrossRef] [PubMed]
- Hagen, C.; Dent, K.C.; Zeev-Ben-Mordehai, T.; Grange, M.; Bosse, J.B.; Whittle, C.; Klupp, B.G.; Siebert, C.A.; Vasishtan, D.; Bauerlein, F.J.; et al. Structural Basis of Vesicle Formation at the Inner Nuclear Membrane. Cell 2015, 163, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, Z.; Cao, Y.; Zhang, S.; Li, H.; Huang, Y.; Ding, Y.Q.; Liu, X. The Hsp40 family chaperone protein DnaJB6 enhances Schlafen1 nuclear localization which is critical for promotion of cell-cycle arrest in T-cells. Biochem J 2008, 413, 239–250. [Google Scholar] [CrossRef]
- Sarparanta, J.; Jonson, P.H.; Golzio, C.; Sandell, S.; Luque, H.; Screen, M.; McDonald, K.; Stajich, J.M.; Mahjneh, I.; Vihola, A.; et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet 2012, 44, 450–455. [Google Scholar] [CrossRef]
- Hageman, J.; Rujano, M.A.; van Waarde, M.A.; Kakkar, V.; Dirks, R.P.; Govorukhina, N.; Oosterveld-Hut, H.M.; Lubsen, N.H.; Kampinga, H.H. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell 2010, 37, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Osterlund, N.; Frankel, R.; Carlsson, A.; Thacker, D.; Karlsson, M.; Matus, V.; Graslund, A.; Emanuelsson, C.; Linse, S. The C-terminal domain of the antiamyloid chaperone DNAJB6 binds to amyloid-beta peptide fibrils and inhibits secondary nucleation. J Biol Chem 2023, 299, 105317. [Google Scholar] [CrossRef]
- Kuiper, E.F.E.; Gallardo, P.; Bergsma, T.; Mari, M.; Kolbe Musskopf, M.; Kuipers, J.; Giepmans, B.N.G.; Steen, A.; Kampinga, H.H.; Veenhoff, L.M.; et al. The chaperone DNAJB6 surveils FG-nucleoporins and is required for interphase nuclear pore complex biogenesis. Nat Cell Biol 2022, 24, 1584–1594. [Google Scholar] [CrossRef]
- Izawa, I.; Nishizawa, M.; Ohtakara, K.; Ohtsuka, K.; Inada, H.; Inagaki, M. Identification of Mrj, a DnaJ/Hsp40 family protein, as a keratin 8/18 filament regulatory protein. J Biol Chem 2000, 275, 34521–34527. [Google Scholar] [CrossRef]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat Rev Mol Cell Biol 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Szymanska, E.; Budick-Harmelin, N.; Miaczynska, M. Endosomal "sort" of signaling control: The role of ESCRT machinery in regulation of receptor-mediated signaling pathways. Semin Cell Dev Biol 2018, 74, 11–20. [Google Scholar] [CrossRef]
- Olmos, Y.; Carlton, J.G. The ESCRT machinery: new roles at new holes. Curr Opin Cell Biol 2016, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H. ESCRTs are everywhere. EMBO J 2015, 34, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Wunderley, L.; Brownhill, K.; Stefani, F.; Tabernero, L.; Woodman, P. The molecular basis for selective assembly of the UBAP1-containing endosome-specific ESCRT-I complex. J Cell Sci 2014, 127, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Stefani, F.; Zhang, L.; Taylor, S.; Donovan, J.; Rollinson, S.; Doyotte, A.; Brownhill, K.; Bennion, J.; Pickering-Brown, S.; Woodman, P. UBAP1 is a component of an endosome-specific ESCRT-I complex that is essential for MVB sorting. Curr Biol 2011, 21, 1245–1250. [Google Scholar] [CrossRef]
- Kolmus, K.; Erdenebat, P.; Szymanska, E.; Stewig, B.; Goryca, K.; Derezinska-Wolek, E.; Szumera-Cieckiewicz, A.; Brewinska-Olchowik, M.; Piwocka, K.; Prochorec-Sobieszek, M.; et al. Concurrent depletion of Vps37 proteins evokes ESCRT-I destabilization and profound cellular stress responses. J Cell Sci 2021, 134. [Google Scholar] [CrossRef]
- Bajorek, M.; Morita, E.; Skalicky, J.J.; Morham, S.G.; Babst, M.; Sundquist, W.I. Biochemical analyses of human IST1 and its function in cytokinesis. Mol Biol Cell 2009, 20, 1360–1373. [Google Scholar] [CrossRef]
- Derry, J.M.; Ochs, H.D.; Francke, U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell 1994, 78, 635–644. [Google Scholar] [CrossRef]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol 2010, 11, 237–251. [Google Scholar] [CrossRef]
- Alekhina, O.; Burstein, E.; Billadeau, D.D. Cellular functions of WASP family proteins at a glance. J Cell Sci 2017, 130, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Goley, E.D.; Welch, M.D. The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol 2006, 7, 713–726. [Google Scholar] [CrossRef]
- Burianek, L.E.; Soderling, S.H. Under lock and key: spatiotemporal regulation of WASP family proteins coordinates separate dynamic cellular processes. Semin Cell Dev Biol 2013, 24, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Stanganello, E.; Hagemann, A.I.; Mattes, B.; Sinner, C.; Meyen, D.; Weber, S.; Schug, A.; Raz, E.; Scholpp, S. Filopodia-based Wnt transport during vertebrate tissue patterning. Nat Commun 2015, 6, 5846. [Google Scholar] [CrossRef] [PubMed]
- Verboon, J.M.; Sugumar, B.; Parkhurst, S.M. Wiskott-Aldrich syndrome proteins in the nucleus: aWASH with possibilities. Nucleus 2015, 6, 349–359. [Google Scholar] [CrossRef]
- Damian, R.T. Molecular mimicry: antigen sharing by parasite and host and its consequences. The American Naturalist 1964, 98, 129–149. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Human Immunodeficiency Virus Proteins Mimic Human T Cell Receptors Inducing Cross-Reactive Antibodies. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Sorci, G.; Cornet, S.; Faivre, B. Immune evasion, immunopathology and the regulation of the immune system. Pathogens 2013, 2, 71–91. [Google Scholar] [CrossRef]
- Rojas, M.; Restrepo-Jimenez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramirez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J Autoimmun 2018, 95, 100–123. [Google Scholar] [CrossRef]
- Maginnis, M.S. Virus-Receptor Interactions: The Key to Cellular Invasion. J Mol Biol 2018, 430, 2590–2611. [Google Scholar] [CrossRef]
- Simmons, R.A.; Willberg, C.B.; Paul, K. Immune evasion by viruses. eLS 2013. [Google Scholar]
- Bule, M.; Khan, F.; Niaz, K. Antivirals: past, present and future. Recent advances in animal virology 2019, 425–446. [Google Scholar]
- Morris, O.M.; Torpey, J.H.; Isaacson, R.L. Intrinsically disordered proteins: modes of binding with emphasis on disordered domains. Open Biol 2021, 11, 210222. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Multifactorial intrinsic disorder analysis of mouse proteins entrapped in RABV particles. A. PONDR® VSL2 Score vs. VSL2 PONDR® (%) analysis. PONDR® VSL2 (%) is a percent of predicted disordered residues (PPDR), i.e., residues with disorder scores above 0.5. PONDR® VSL2 score is the average disorder score (ADS) for a protein. Color blocks indicate regions in which proteins are mostly ordered (blue and light blue), moderately disordered (pink and light pink), or mostly disordered (red). If the two parameters agree, the corresponding part of the background is dark (blue or pink), whereas light blue and light pink reflect areas in which the predictors disagree with each other. The boundaries of the colored regions represent arbitrary and accepted cutoffs for ADS (y-axis) and the percentage of predicted disordered residues (PPDR; x-axis). B. Charge-Hydropathy and Cumulative Distribution Function (CH-CDF) analysis of entrapped host proteins. The CH-CDF plot is a two-dimensional representation that integrates both the CH plot, which correlates a protein's net charge and hydrophobicity with its structural order, and the CDF, which accumulates disorder predictions from the N-terminus to the C-terminus of a protein, offering insight into the distribution of disorder residues. The Y-axis (ΔCH) represents the protein's distance from the CH boundary, indicating the balance between charge and hydrophobicity, while the X-axis (ΔCDF) represents the deviation of a protein's disorder frequency from the CDF boundary. Proteins are then stratified into four quadrants: Quadrant 1 (bottom right) indicates proteins likely to be structured; Quadrant 2 (bottom left) includes proteins that may be in a molten globule state or lack a unique 3D structure; Quadrant 3 (top left) consists of proteins predicted to be highly disordered; Quadrant 4 (top right) captures proteins that present a mixed prediction of being disordered according to CH but ordered according to CDF.
Figure 1.
Multifactorial intrinsic disorder analysis of mouse proteins entrapped in RABV particles. A. PONDR® VSL2 Score vs. VSL2 PONDR® (%) analysis. PONDR® VSL2 (%) is a percent of predicted disordered residues (PPDR), i.e., residues with disorder scores above 0.5. PONDR® VSL2 score is the average disorder score (ADS) for a protein. Color blocks indicate regions in which proteins are mostly ordered (blue and light blue), moderately disordered (pink and light pink), or mostly disordered (red). If the two parameters agree, the corresponding part of the background is dark (blue or pink), whereas light blue and light pink reflect areas in which the predictors disagree with each other. The boundaries of the colored regions represent arbitrary and accepted cutoffs for ADS (y-axis) and the percentage of predicted disordered residues (PPDR; x-axis). B. Charge-Hydropathy and Cumulative Distribution Function (CH-CDF) analysis of entrapped host proteins. The CH-CDF plot is a two-dimensional representation that integrates both the CH plot, which correlates a protein's net charge and hydrophobicity with its structural order, and the CDF, which accumulates disorder predictions from the N-terminus to the C-terminus of a protein, offering insight into the distribution of disorder residues. The Y-axis (ΔCH) represents the protein's distance from the CH boundary, indicating the balance between charge and hydrophobicity, while the X-axis (ΔCDF) represents the deviation of a protein's disorder frequency from the CDF boundary. Proteins are then stratified into four quadrants: Quadrant 1 (bottom right) indicates proteins likely to be structured; Quadrant 2 (bottom left) includes proteins that may be in a molten globule state or lack a unique 3D structure; Quadrant 3 (top left) consists of proteins predicted to be highly disordered; Quadrant 4 (top right) captures proteins that present a mixed prediction of being disordered according to CH but ordered according to CDF.
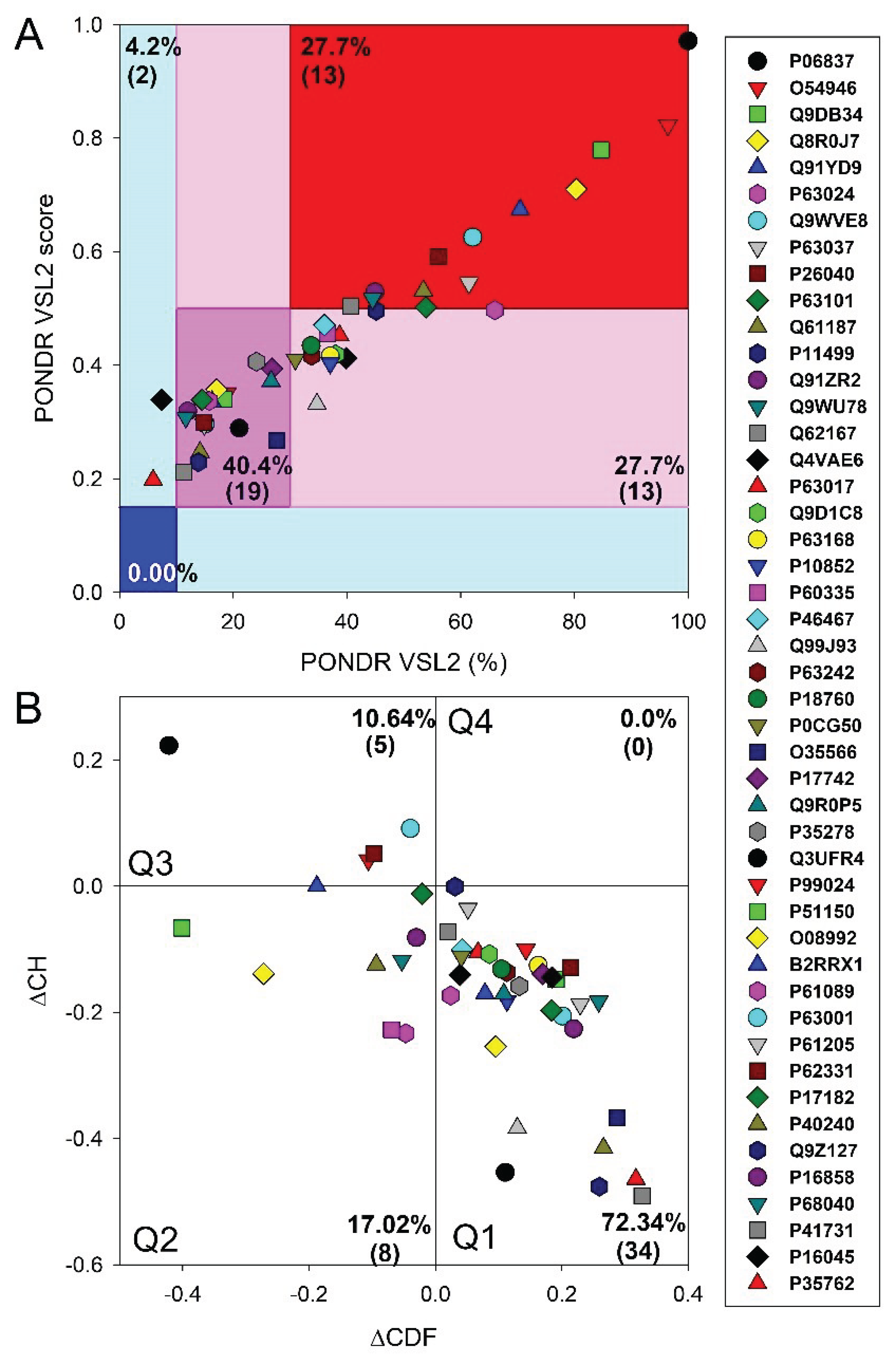
Figure 3.
Correlation between the intrinsic disorder levels in the host proteins entrapped in RABV particles and their interactivity within the intra-set PPI (A) and predisposition for to be involved in liquid-liquid phase separation, LLPS (B). Solid lines in both plots show linear fits of the reported data, whereas short-long-dashed lines represents boundaries between different disorder categories, as well as between hubs and non-hubs (A) and LLPS promoters and other proteins (B). .
Figure 3.
Correlation between the intrinsic disorder levels in the host proteins entrapped in RABV particles and their interactivity within the intra-set PPI (A) and predisposition for to be involved in liquid-liquid phase separation, LLPS (B). Solid lines in both plots show linear fits of the reported data, whereas short-long-dashed lines represents boundaries between different disorder categories, as well as between hubs and non-hubs (A) and LLPS promoters and other proteins (B). .
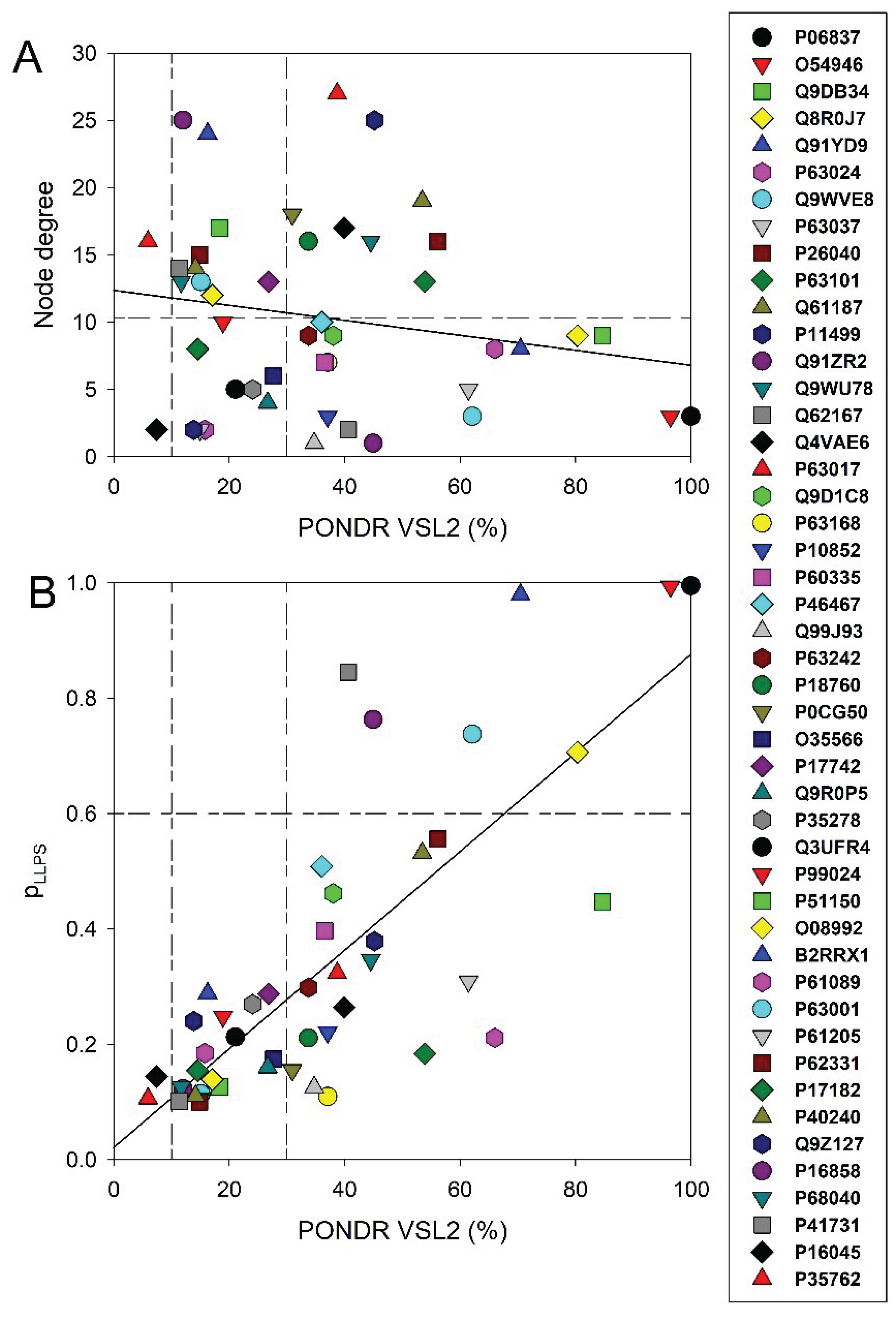
Figure 4.
Functional disorder analysis of mouse neuromodulin (UniProt ID: P06837). A. Per-residue disorder profile generated by RIDAO showing that a major portion of this protein has predicted value of disorder above the established threshold (0.5). B. Functional disorder profile generated for neuromodulin by the D2P2 database showing rhe outputs of several disorder predictors such as VLXT, VSL2b, PrDOS, IUPred and Espritz. The colored bar highlighted by blue and green shade represents the disorder prediction, colored circles below the bar shows the predicting PTMs. C. The FuzDrop-generated plot showing the sequence distribution of the residue-based droplet-promoting probabilities, pDP. D. The FuzDrop-generated plot of the multiplicity of binding modes showing positions of regions that can sample multiple binding modes in the cellular context (sub-cellular localisation, partners, posttranslational modifications)-dependent manner (residues 9-16 and 40-66). E. Protein-protein interaction network generated by STRING. This PPI nework was generated using the minimum required interaction score of 0.4 (medium confidence) and adjusting the value of a maximum number of interactors to 500. Network nodes represent individual proteins and edges represent protein-protein interaction for shared function, with types of Interactions; the blue line represents curated databases, black line for co-expression, and green line for gene neighborhood. F. 3D structural model is predicted through AlphaFold. The structure is colored according to the per-residue model confidence score ranging from orange to blue. Fragments of structure from very low (pLDDT < 50) value to very high confidence (pLDDT > 90), respectively.
Figure 4.
Functional disorder analysis of mouse neuromodulin (UniProt ID: P06837). A. Per-residue disorder profile generated by RIDAO showing that a major portion of this protein has predicted value of disorder above the established threshold (0.5). B. Functional disorder profile generated for neuromodulin by the D2P2 database showing rhe outputs of several disorder predictors such as VLXT, VSL2b, PrDOS, IUPred and Espritz. The colored bar highlighted by blue and green shade represents the disorder prediction, colored circles below the bar shows the predicting PTMs. C. The FuzDrop-generated plot showing the sequence distribution of the residue-based droplet-promoting probabilities, pDP. D. The FuzDrop-generated plot of the multiplicity of binding modes showing positions of regions that can sample multiple binding modes in the cellular context (sub-cellular localisation, partners, posttranslational modifications)-dependent manner (residues 9-16 and 40-66). E. Protein-protein interaction network generated by STRING. This PPI nework was generated using the minimum required interaction score of 0.4 (medium confidence) and adjusting the value of a maximum number of interactors to 500. Network nodes represent individual proteins and edges represent protein-protein interaction for shared function, with types of Interactions; the blue line represents curated databases, black line for co-expression, and green line for gene neighborhood. F. 3D structural model is predicted through AlphaFold. The structure is colored according to the per-residue model confidence score ranging from orange to blue. Fragments of structure from very low (pLDDT < 50) value to very high confidence (pLDDT > 90), respectively.
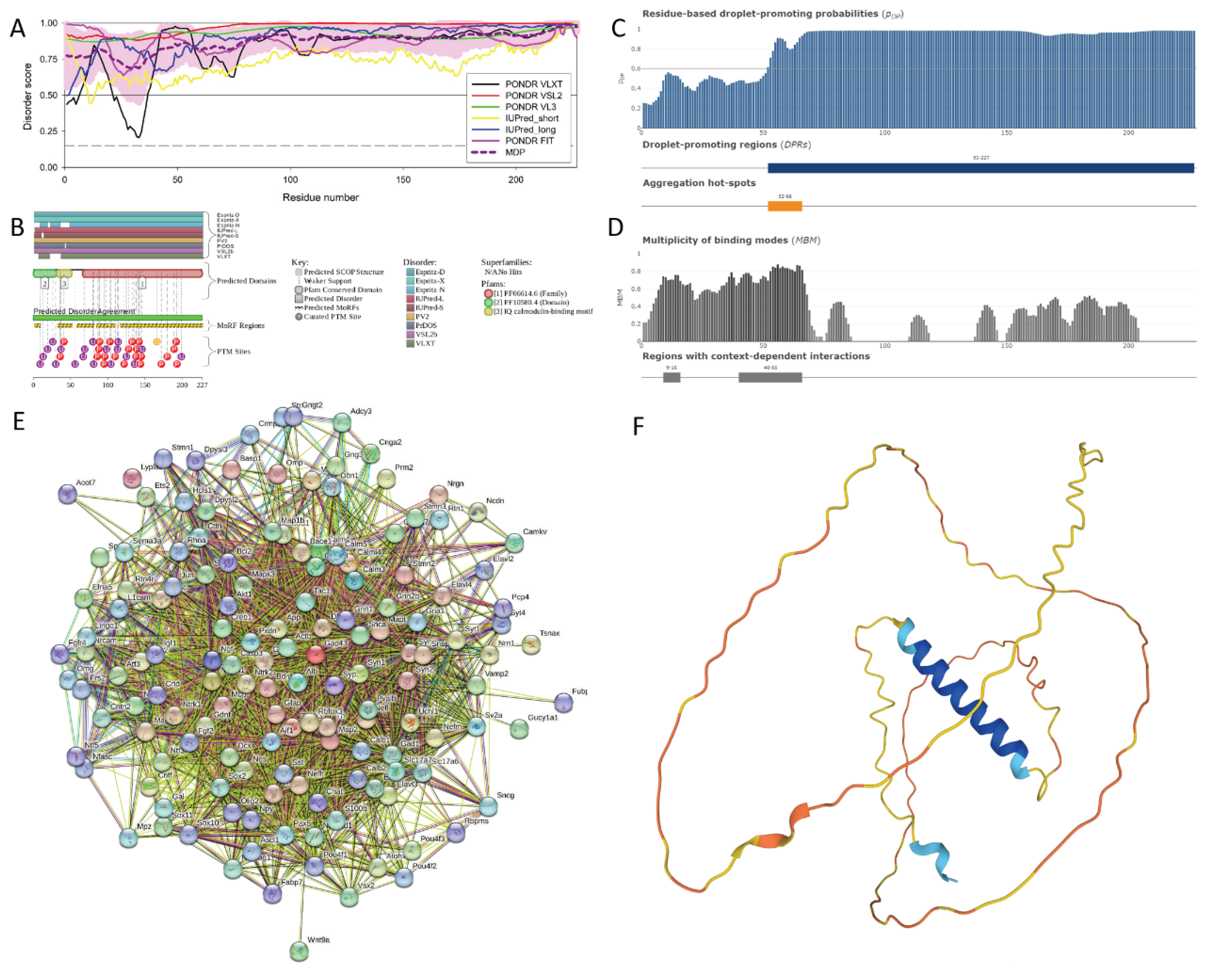
Figure 5.
Functional Disorder Analysis of mouse Chmp4b (UniProt ID: Q9D8B3). A. Per-residue diosrder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. Per-residue LLPS potential as estimated by FuzDrop, demonstrating the tendency of each residue to promote droplet formation. D. Multiplicity of Binding Modes plot generated by FuzDrop. E. The PPI network generated utilizing STRING by adjusting the value of the maximum number of interactors at 500. F. 3D structural model generated by AlphaFold. The structure is colored according to the per-residue model confidence score ranging from orange (very low confidence pLDDT < 50) to blue (very high confidence pLDDT > 90), respectively.
Figure 5.
Functional Disorder Analysis of mouse Chmp4b (UniProt ID: Q9D8B3). A. Per-residue diosrder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. Per-residue LLPS potential as estimated by FuzDrop, demonstrating the tendency of each residue to promote droplet formation. D. Multiplicity of Binding Modes plot generated by FuzDrop. E. The PPI network generated utilizing STRING by adjusting the value of the maximum number of interactors at 500. F. 3D structural model generated by AlphaFold. The structure is colored according to the per-residue model confidence score ranging from orange (very low confidence pLDDT < 50) to blue (very high confidence pLDDT > 90), respectively.
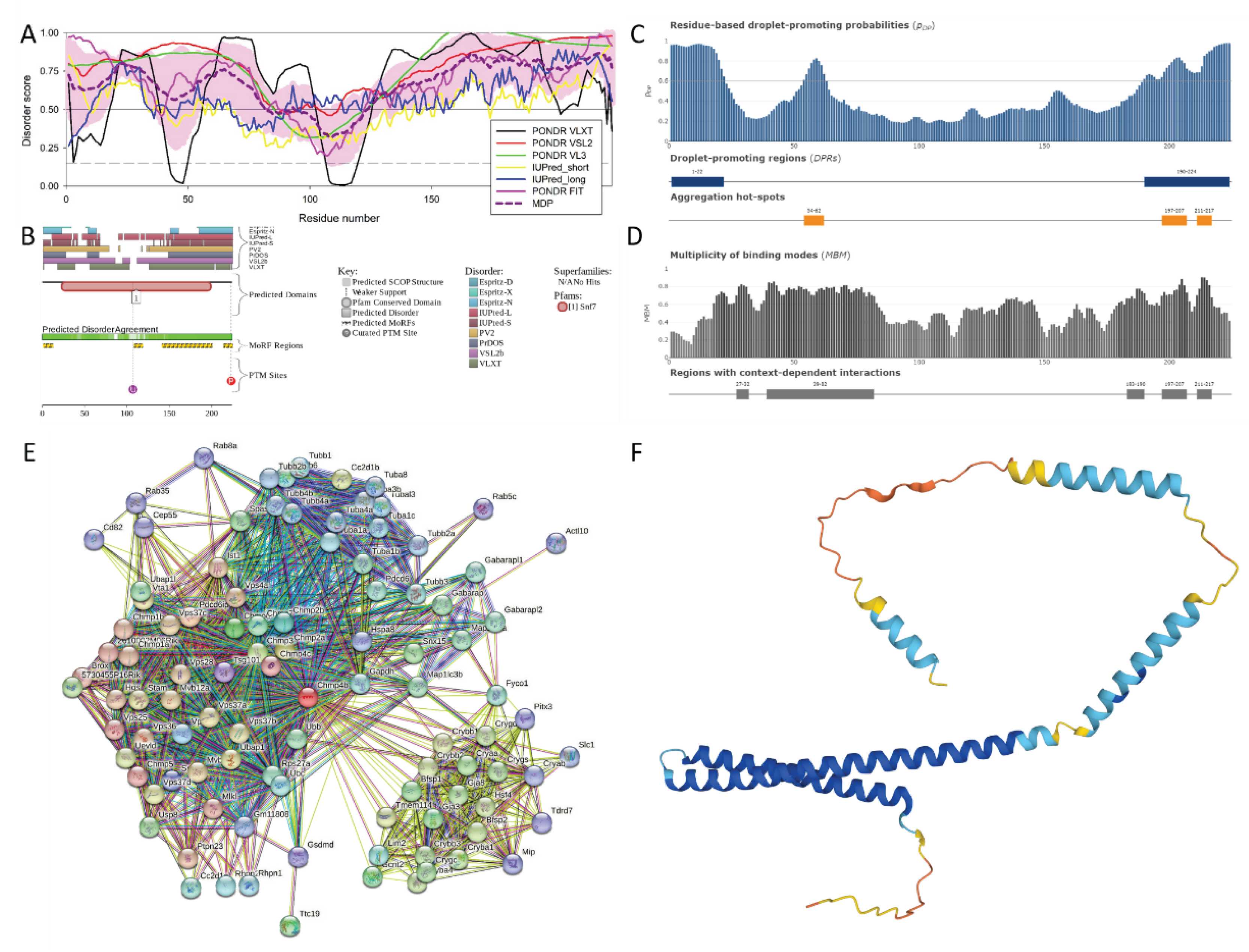
Figure 6.
Functional Disorder Analysis of protein DnaJ homolog subfamily B member 6 (UniProt ID: O54946). A. RIDAO-generated per-residue disorder profile. B. Disorder-based functionality evaluated by D2P2. C. Per-residue LLPS potential as estimated by FuzDrop, demonstrating the tendency of each residue to promote droplet formation. D. Multiplicity of Binding Modes plot generated by FuzDrop. E. The PPI network generated utilizing STRING by adjusting the value of the maximum number of interactors at 500. F. 3D structural model generated by AlphaFold. The structure is colored according to the per-residue model confidence score (pLDDT), with fragments of structure with very low (pLDDT < 50), low (70 > pLDDT > 50, high (90 > pLDDT > 70), and very high confidence (pLDDT > 90) confidence being shown by orange, yelow, cyan, and blue colore, respectively.
Figure 6.
Functional Disorder Analysis of protein DnaJ homolog subfamily B member 6 (UniProt ID: O54946). A. RIDAO-generated per-residue disorder profile. B. Disorder-based functionality evaluated by D2P2. C. Per-residue LLPS potential as estimated by FuzDrop, demonstrating the tendency of each residue to promote droplet formation. D. Multiplicity of Binding Modes plot generated by FuzDrop. E. The PPI network generated utilizing STRING by adjusting the value of the maximum number of interactors at 500. F. 3D structural model generated by AlphaFold. The structure is colored according to the per-residue model confidence score (pLDDT), with fragments of structure with very low (pLDDT < 50), low (70 > pLDDT > 50, high (90 > pLDDT > 70), and very high confidence (pLDDT > 90) confidence being shown by orange, yelow, cyan, and blue colore, respectively.
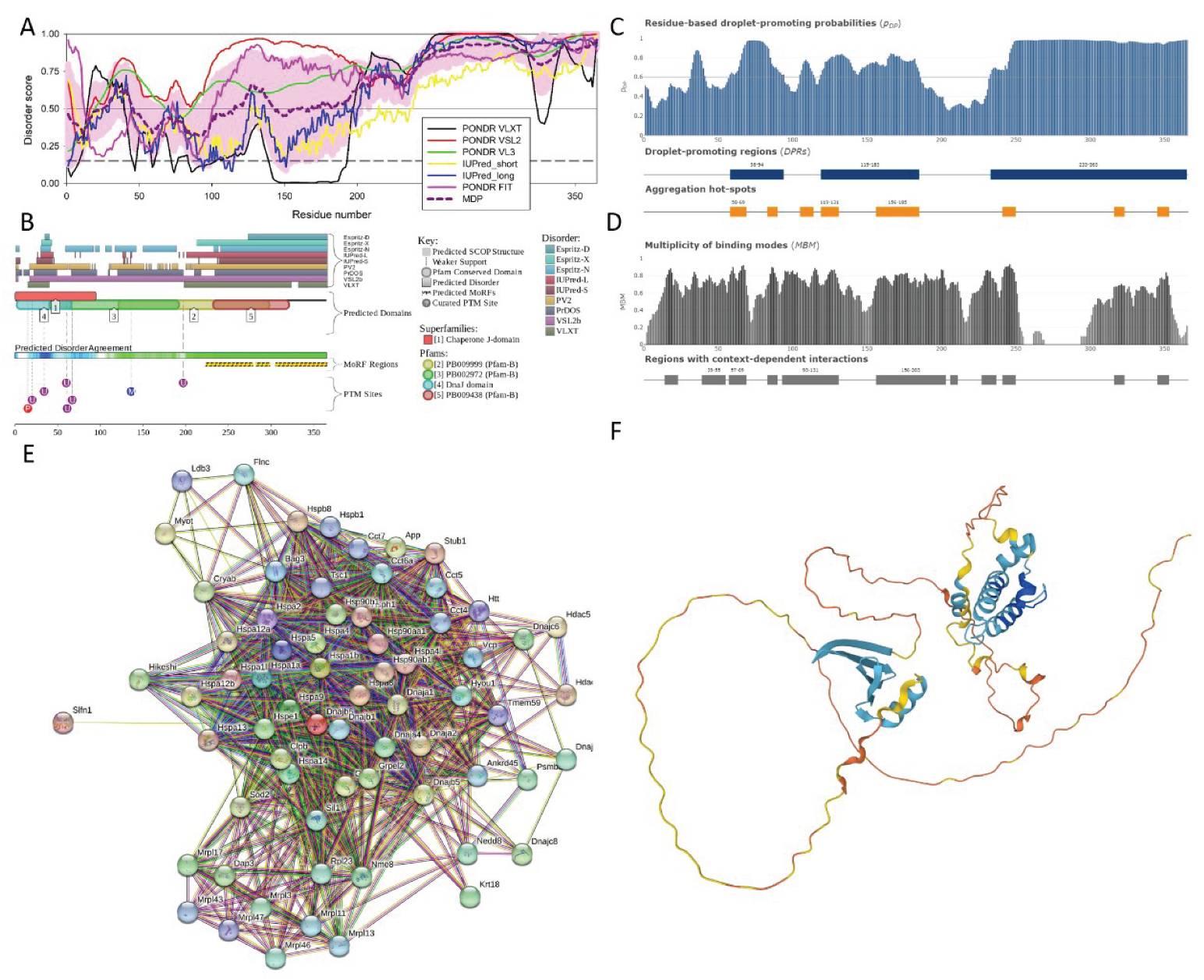
Figure 7.
Functional Disorder Analysis of protein Vps37b (UniProt ID: Q8R0J7). A. Per-residue disorder profile generated by the RIDAO platform. B. Functional disorder profile generated by the D2P2 database. C. Per-residues droplet formation propensity generated by FuzDrop. D. Multiplicity of Binding Modes plot generatwd by FuzDrop. E. Protein-Protein interaction network generatwd for this protein utilizing STRING database. F. 3D structural model predicted by AlphaFold. The structure is colored according to the per-residue model confidence score ranging from orange (pLDDT < 50) to blue (pLDDT > 90).
Figure 7.
Functional Disorder Analysis of protein Vps37b (UniProt ID: Q8R0J7). A. Per-residue disorder profile generated by the RIDAO platform. B. Functional disorder profile generated by the D2P2 database. C. Per-residues droplet formation propensity generated by FuzDrop. D. Multiplicity of Binding Modes plot generatwd by FuzDrop. E. Protein-Protein interaction network generatwd for this protein utilizing STRING database. F. 3D structural model predicted by AlphaFold. The structure is colored according to the per-residue model confidence score ranging from orange (pLDDT < 50) to blue (pLDDT > 90).
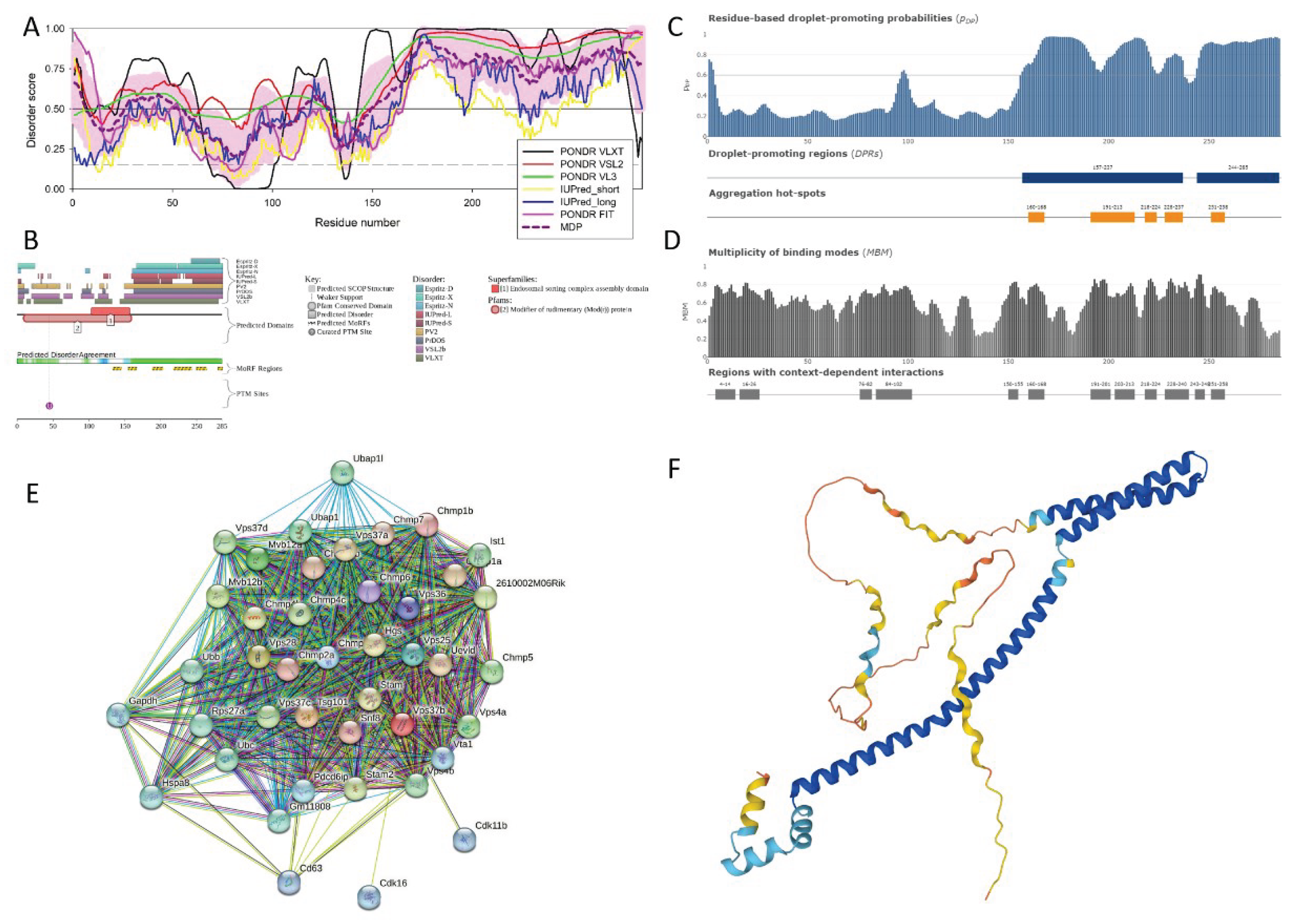
Figure 8.
Distribution of ELMs (short linear functional motifs) within the sequence of the mouse Vps37B protein. Refer to the additional information provided in Supplementary Table S9.
Figure 8.
Distribution of ELMs (short linear functional motifs) within the sequence of the mouse Vps37B protein. Refer to the additional information provided in Supplementary Table S9.
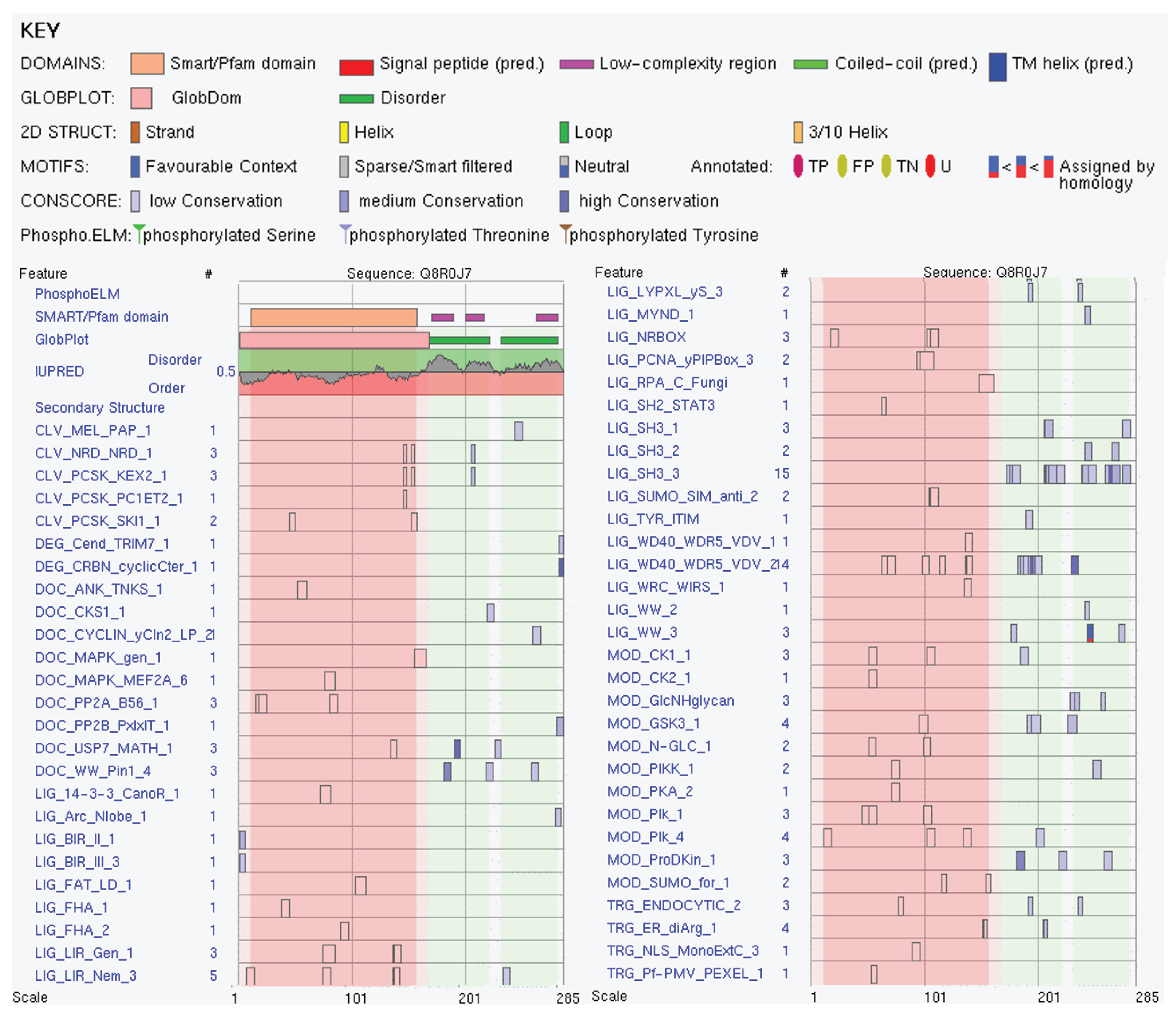
Figure 9.
Functional Disorder Analysis of mouse protein Wasl (UniProt ID: Q91YD9). A. Multiparametric intrinsic disorder profile generated by RIDAO. B. D2P2-generated functional disorder profile. C. Residue-based LLPS propensity. D. Multiplicity of Binding Modes plot. E. Wasl-centered PPI network generatwd utilizing STRING Database by adjusting the value of the maximum number of interactors to 500. F. 3D structural model as predicted by AlphaFold. The structure is colored according to the per-residue model confidence score ranging from orange (very low confidence, pLDDT < 50) to blue (very high confidence, pLDDT > 90).
Figure 9.
Functional Disorder Analysis of mouse protein Wasl (UniProt ID: Q91YD9). A. Multiparametric intrinsic disorder profile generated by RIDAO. B. D2P2-generated functional disorder profile. C. Residue-based LLPS propensity. D. Multiplicity of Binding Modes plot. E. Wasl-centered PPI network generatwd utilizing STRING Database by adjusting the value of the maximum number of interactors to 500. F. 3D structural model as predicted by AlphaFold. The structure is colored according to the per-residue model confidence score ranging from orange (very low confidence, pLDDT < 50) to blue (very high confidence, pLDDT > 90).
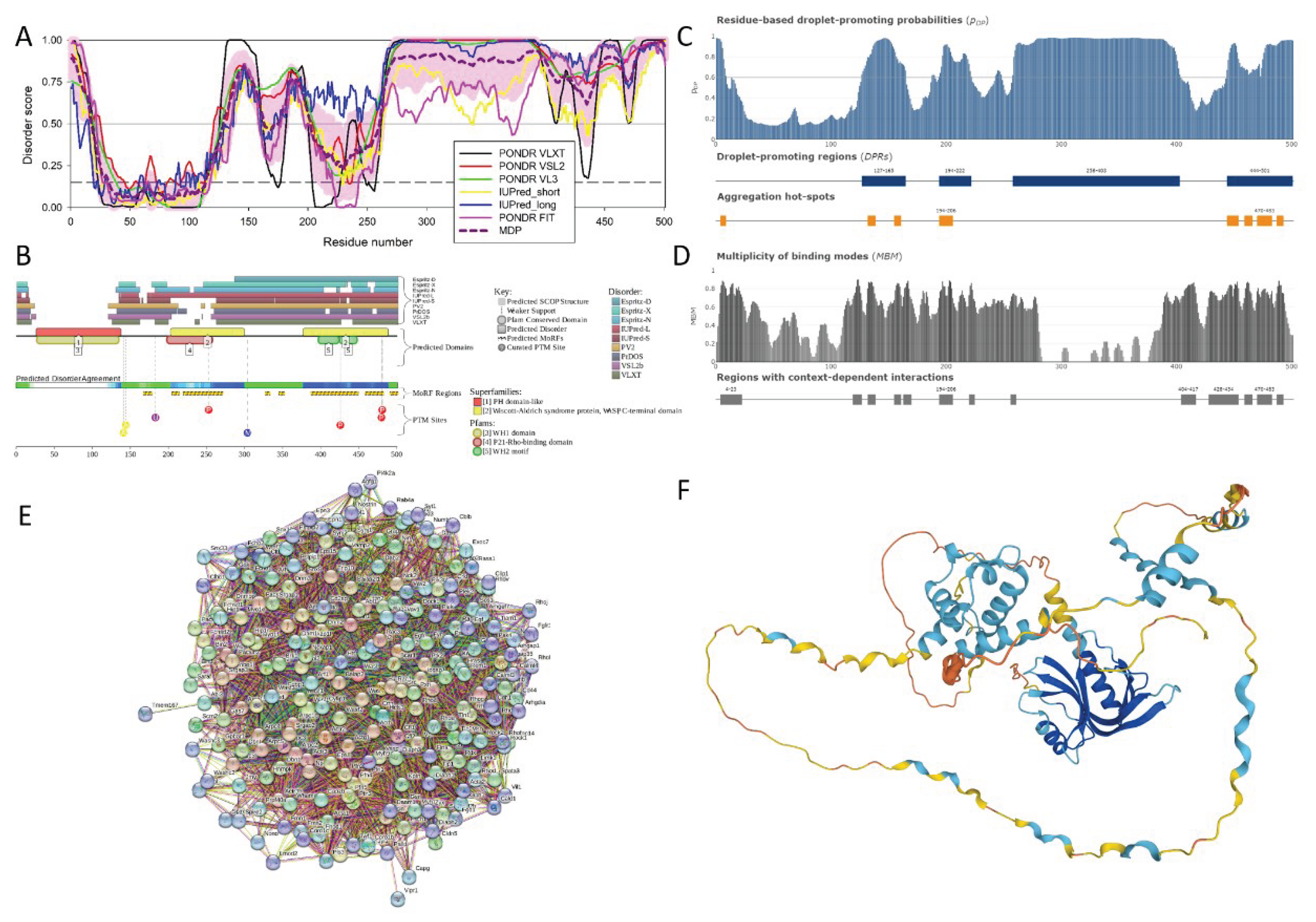
Figure 10.
Distribution of ELMs (short linear functional motifs) within the sequence of the mouse Wasl proten (UniProt ID: Q91YD9). For additional information see Supplementary Table S10.
Figure 10.
Distribution of ELMs (short linear functional motifs) within the sequence of the mouse Wasl proten (UniProt ID: Q91YD9). For additional information see Supplementary Table S10.
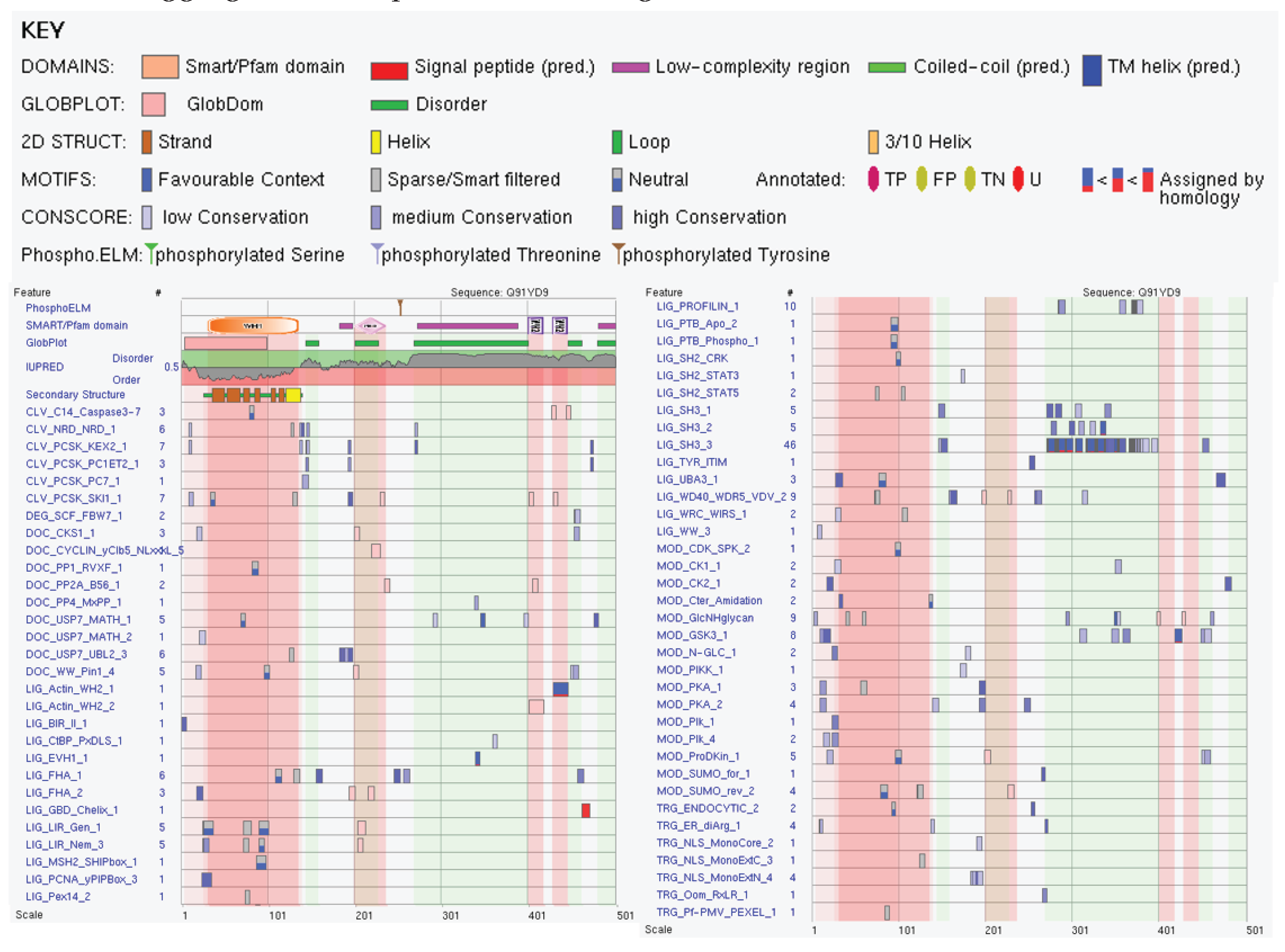
Figure 11.
Intra-set interactivity of 11 most disordered mouse proteins entrapped in the RABV particles. Networks are constructed by STRING using medium confidence of 0.4 (A) and low confidence of 0.15 (B). .
Figure 11.
Intra-set interactivity of 11 most disordered mouse proteins entrapped in the RABV particles. Networks are constructed by STRING using medium confidence of 0.4 (A) and low confidence of 0.15 (B). .
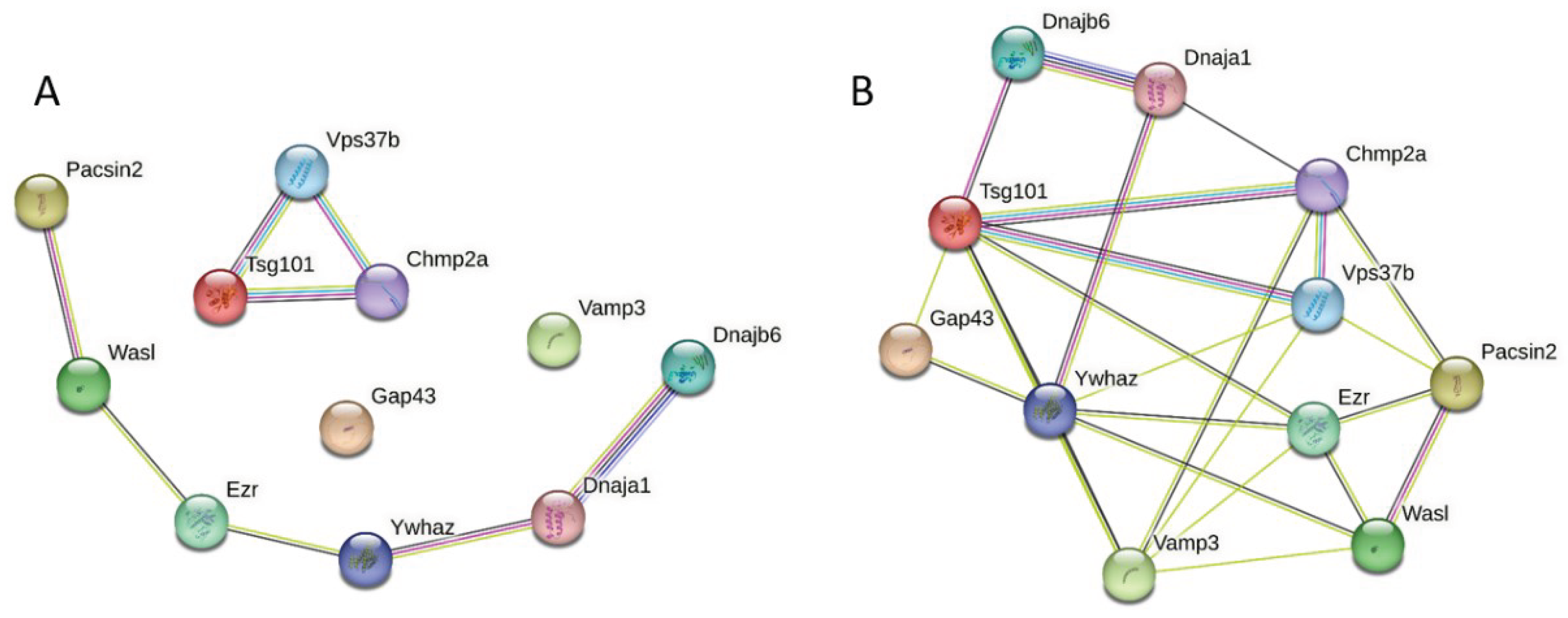
Figure 12.
Global interactivity of the 11 most disordered mouse proteins found in the RBV particles. Using the k-means clustering (the alrotithm, which is included in STRING, automatically assigns data points to one of the K clusters depending on their distance from the center of the clusters) this PPI network can be divided on three clusters. .
Figure 12.
Global interactivity of the 11 most disordered mouse proteins found in the RBV particles. Using the k-means clustering (the alrotithm, which is included in STRING, automatically assigns data points to one of the K clusters depending on their distance from the center of the clusters) this PPI network can be divided on three clusters. .

Figure 13.
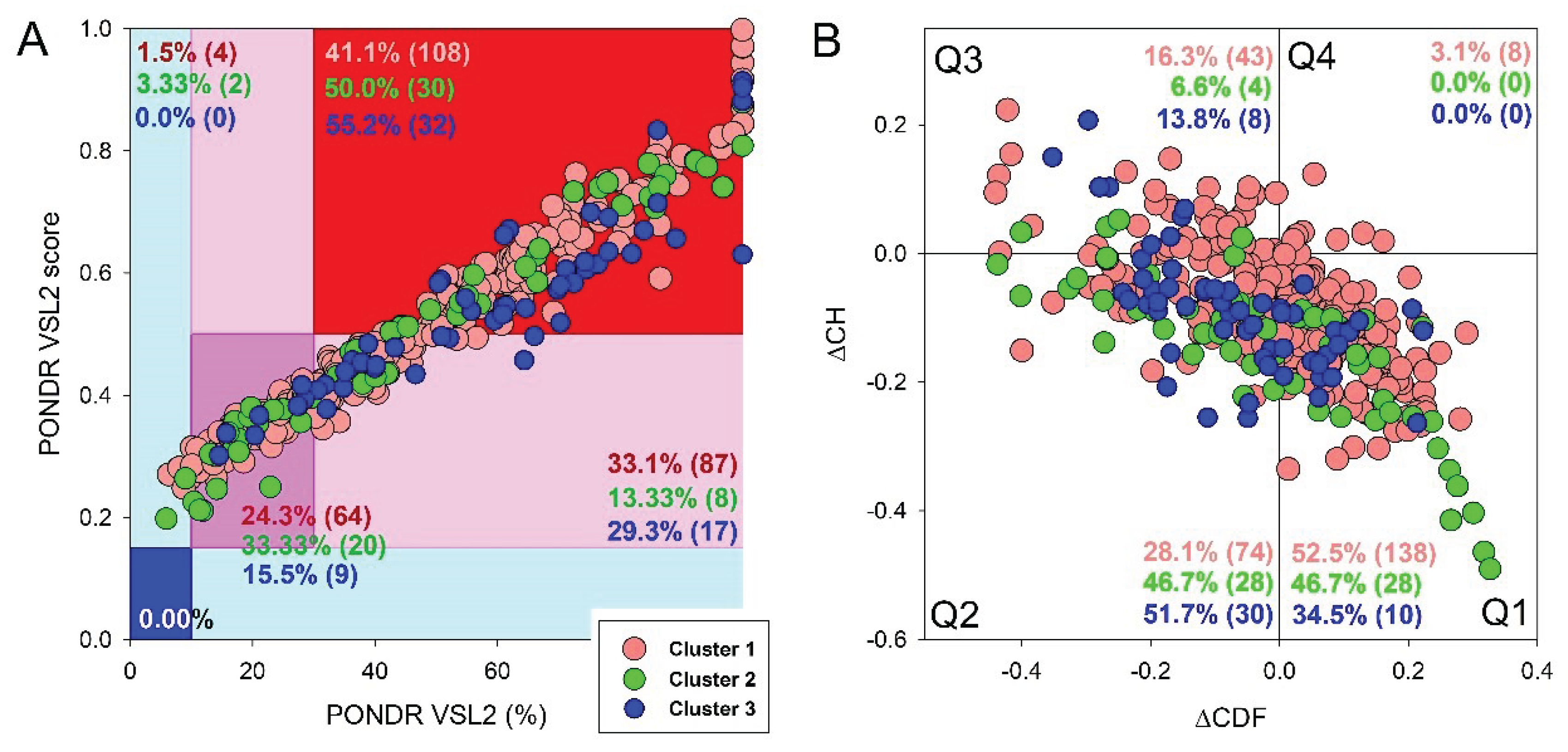

Table 1.
Localization of ELMs (Eukaryotic Linear Motifs) within the Droplet Promoting Regions, Aggregation Hot-spots and MoRFs of mouse Neuromodulin (UniProt ID: P06837). For additional information, see Supplementary Table S6.
Table 1.
Localization of ELMs (Eukaryotic Linear Motifs) within the Droplet Promoting Regions, Aggregation Hot-spots and MoRFs of mouse Neuromodulin (UniProt ID: P06837). For additional information, see Supplementary Table S6.
| Region Type | Region Range | ELM ID | Position |
| Droplet Promoting region | 52-277 | LIG_PDZ_Class_3 | 222-227 |
| LIG_WD40_WDR5_VDV_2 | 219-222 | ||
| 218-222 | |||
| 215-222 | |||
| 155-161 | |||
| 154-161 | |||
| 133-137 | |||
| 132-137 | |||
| 131-137 | |||
| 96-99 | |||
| 95-99 | |||
| 64-66 | |||
| 58-64 | |||
| MOD_GlcNHglycan | 209-212 | ||
| 132-135 | |||
| 127-130 | |||
| 85-88 | |||
| 84-88 | |||
| MOD_SUMO_rev_2 | 203-207 | ||
| 200-207 | |||
| 198-207 | |||
| 196-201 | |||
| 193-201 | |||
| 192-201 | |||
| 191-201 | |||
| 154-159 | |||
| 149-159 | |||
| 122-126 | |||
| 118-126 | |||
| CLV_C14_Caspase3-7 | 197-201 | ||
| DOC_USP7_MATH_1 | 207-211 | ||
| 190-194 | |||
| 119-123 | |||
| MOD_CK2_1 | 190-196 | ||
| 142-148 | |||
| MOD_GSK3_1 | 186-193 | ||
| 135-142 | |||
| MOD_PIKK_1 | 190-196 | ||
| LIG_TRAF6_MATH_1 | 184-192 | ||
| DOC_WW_Pin1_4 | 169-174 | ||
| 139-144 | |||
| 93-98 | |||
| MOD_ProDKin_1 | 169-175 | ||
| 139-145 | |||
| 93-99 | |||
| DOC_USP7_UBL2_3 | 153-157 | ||
| MOD_SUMO_for_1 | 152-155 | ||
| 97-100 | |||
| 25-28 | |||
| MOD_CK1_1 | 142-148 | ||
| 128-134 | |||
| 86-92 | |||
| LIG_BIR_III_2 | 118-122 | ||
| MOD_Plk_2-3 | 107-113 | ||
| MOD_CDK_SPK_2 | 93-98 | ||
| MoRF | 102-109 | MOD_Plk_2-3 | 107-113 |
| MoRF | 58-81 | LIG_WD40_WDR5_VDV_2 | 58-64 |
| 63-66 | |||
| Aggregation Hotspot | 52-66 | LIG_WD40_WDR5_VDV_2 | 58-64 |
| 63-66 | |||
| MoRF | 1-9 | LIG_UBA3_1 | 1-9 |
| LIG_FHA_1 | 6-12 | ||
| MOD_PKA_2 | 5-11 | ||
| CLV_NRD_NRD_1 | 6-8 | ||
| CLV_PCSK_KEX2_1 | 6-8 | ||
| TRG_ER_diArg_1 | 5-7 | ||
| DEG_Nend_Nbox_1 | 1-3 |
Table 2.
Distribution of ELMs (Eukaryotic Linear Motifs) in Droplet Promoting Regions, Aggregation Hot-spots, regions with multiplicity of binding modes, and MoRFs (Molecular recognition features) of the protein Chmp4b (UniProt ID: Q9D8B3). Table summarizes the ELMs mapped onto these regions suggesting potential functional role of these motifs. For additional information, see the Supplementary Table S7.
Table 2.
Distribution of ELMs (Eukaryotic Linear Motifs) in Droplet Promoting Regions, Aggregation Hot-spots, regions with multiplicity of binding modes, and MoRFs (Molecular recognition features) of the protein Chmp4b (UniProt ID: Q9D8B3). Table summarizes the ELMs mapped onto these regions suggesting potential functional role of these motifs. For additional information, see the Supplementary Table S7.
| Region Type | Region range | ELM ID | Position |
| MoRF | 1-12 | LIG_BIR_II_1 | 1-5 |
| LIG_LIR_Nem_3 | 2-7 | ||
| LIG_Pex14_2 | 4-8 | ||
| Droplet-promoting region | 1-22 | LIG_BIR_II_1 | 1-5 |
| LIG_LIR_Nem_3 | 2-7 | ||
| LIG_Pex14_2 | 4-8 | ||
| DOC_WW_Pin1_4 | 18-23 | ||
| MOD_ProDKin_1 | 18-24 | ||
| LIG_FHA_2 | 19-25 | ||
| Region with multiplicity of binding modes | 27-32 | MOD_PKA_2 | 29-35 |
| Region with multiplicity of binding modes | 39-82 | MOD_SUMO_rev_2 | 41-47 |
| TRG_NLS_Bipartite_1 | 55-75 | ||
| DOC_USP7_UBL2_3 | 56-60 | ||
| CLV_PCSK_PC1ET2_1 | 62-64 | ||
| TRG_NLS_MonoExtN_4 | 70-75 | ||
| TRG_NLS_MonoCore_2 | 69-74 | ||
| 70-75 | |||
| TRG_NLS_MonoExtC_3 | 69-74 | ||
| 70-75 | |||
| Aggregation Hotspot | 54-62 | DOC_USP7_UBL2_3 | 56-60 |
| CLV_PCSK_PC1ET2_1 | 62-64 | ||
| MoRF | 108-118 | LIG_SH2_STAP1 | 111-115 |
| LIG_WD40_WDR5_VDV_2 | 111-115 | ||
| CLV_PCSK_SKI1_1 | 114-118 | ||
| MoRF | 141-200 | MOD_GSK3_1 | 143-150 |
| 181-188 | |||
| DOC_PP1_RVXF_1 | 149-156 | ||
| LIG_Pex14_2 | 155-159 | ||
| LIG_WD40_WDR5_VDV_2 | 161-168 | ||
| 162-168 | |||
| 163-168 | |||
| 177-183 | |||
| CLV_PCSK_SKI1_1 | 178-182 | ||
| TRG_Pf-PMV_PEXEL_1 | 178-182 | ||
| LIG_SUMO_SIM_par_1 | 179-184 | ||
| MOD_CK2_1 | 181-187 | ||
| MOD_GlcNHglycan | 182-186 | ||
| 183-186 | |||
| LIG_FHA_1 | 186-192 | ||
| LIG_SH3_3 | 186-192 | ||
| 189-195 | |||
| 194-200 | |||
| 197-203 | |||
| DOC_USP7_MATH_1 | 198-202 | ||
| Region with multiplicity of binding modes | 183-190 | LIG_WD40_WDR5_VDV_2 | 177-183 |
| TRG_Pf-PMV_PEXEL_1 | 178-182 | ||
| LIG_SUMO_SIM_par_1 | 179-184 | ||
| MOD_CK2_1 | 181-187 | ||
| MOD_GlcNHglycan | 182-186 | ||
| 183-186 | |||
| LIG_FHA_1 | 186-192 | ||
| LIG_SH3_3 | 186-192 | ||
| 189-195 | |||
| Region with multiplicity of binding modes | 197-207 | DOC_USP7_MATH_1 | 198-202 |
| LIG_SH3_2 | 200-205 | ||
| CLV_PCSK_SKI1_1 | 202-206 | ||
| DOC_USP7_UBL2_3 | 202-206 | ||
| Aggregation Hotspot | 197-207 | DOC_USP7_MATH_1 | 198-202 |
| LIG_SH3_2 | 200-205 | ||
| CLV_PCSK_SKI1_1 | 202-206 | ||
| DOC_USP7_UBL2_3 | 202-206 | ||
| Droplet-promoting region | 190-224 | LIG_FHA_1 | 186-192 |
| LIG_SH3_3 | 186-192 | ||
| 189-195 | |||
| 194-200 | |||
| 197-203 | |||
| DOC_USP7_MATH_1 | 198-202 | ||
| LIG_SH3_2 | 200-205 | ||
| CLV_PCSK_SKI1_1 | 202-206 | ||
| DOC_USP7_UBL2_3 | 202-206 | ||
| LIG_SH3_4 | 202-209 | ||
| TRG_NESrev_CRM1_2 | 208-217 | ||
| 209-217 | |||
| 210-217 | |||
| 211-217 | |||
| 212-217 | |||
| Aggregation Hotspot | 211-217 | TRG_NESrev_CRM1_2 | 208-217 |
| 209-217 | |||
| 210-217 | |||
| 211-217 | |||
| 212-217 | |||
| MoRF | 214-224 | TRG_NESrev_CRM1_2 | 208-217 |
| 209-217 | |||
| 210-217 | |||
| 211-217 | |||
| 212-217 | |||
| MOD_SUMO_rev_2 | 212-217 |
Table 3.
Distribution of ELMs (Eukaryotic Linear Motifs) in droplet promoting regions, aggregation hot-spots, regions with multiplicity of binding modes and MoRF (Molecular recognition features) of protein DnaJ homolog subfamily B member 6 (UniProt ID: O54946). Table is summarizing the ELMs mapped onto these regions suggesting potential functional role of these motifs. For additional information, see the Supplementary Table S8.
Table 3.
Distribution of ELMs (Eukaryotic Linear Motifs) in droplet promoting regions, aggregation hot-spots, regions with multiplicity of binding modes and MoRF (Molecular recognition features) of protein DnaJ homolog subfamily B member 6 (UniProt ID: O54946). Table is summarizing the ELMs mapped onto these regions suggesting potential functional role of these motifs. For additional information, see the Supplementary Table S8.
| Region Type | Region Range | ELM ID | Position |
| Region with multiplicity of binding modes | 14-23 | DOC_WW_Pin1_4 | 12-17 |
| CLV_NRD_NRD_1 | 23-25 | ||
| Region with multiplicity of binding modes | 39-55 | CLV_NRD_NRD_1 | 43-45 |
| CLV_PCSK_SKI1_1 | 44-48 | ||
| Droplet Promoting Region | 58-94 | LIG_LIR_Nem_3 | 63-68 |
| DOC_WW_Pin1_4 | 83-88 | ||
| DOC_PP4_FxxP_1 | 84-87 | ||
| Region with multiplicity of binding modes | 57-69 | TRG_Pf-PMV_PEXEL_1 | 62-66 |
| LIG_LIR_Nem_3 | 63-68 | ||
| Aggregation hotspot | 58-69 | TRG_Pf-PMV_PEXEL_1 | 62-66 |
| LIG_LIR_Nem_3 | 63-68 | ||
| Droplet Promoting Region | 58-94 | TRG_Pf-PMV_PEXEL_1 | 62-66 |
| DOC_PP4_FxxP_1 | 84-87 | ||
| 94-97 | |||
| Region with multiplicity of binding modes | 83-90 | DOC_WW_Pin1_4 | 83-88 |
| MOD_ProDKin_1 | 83-89 | ||
| DOC_PP4_FxxP_1 | 84-87 | ||
| Aggregation hotspot | 83-90 | DOC_WW_Pin1_4 | 83-88 |
| MOD_ProDKin_1 | 83-89 | ||
| DOC_PP4_FxxP_1 | 84-87 | ||
| Region with multiplicity of binding modes | 93-131 | DOC_PP4_FxxP_1 | 84-87 |
| 94-97 | |||
| CLV_PCSK_SKI1_1 | 102-106 | ||
| Aggregation hotspot | 105-114 | LIG_BRCT_BRCA1_1 | 111-115 |
| LIG_AP2alpha_2 | 109-111 | ||
| Aggregation hotspot | 119-131 | DOC_PP4_FxxP_1 | 116-119 |
| LIG_AP2alpha_1 | 116-120 | ||
| 120-124 | |||
| LIG_AP2alpha_2 | 118-120 | ||
| CLV_NRD_NRD_1 | 127-129 | ||
| CLV_PCSK_KEX2_1 | 127-129 | ||
| Droplet Promoting Region | 119-185 | DOC_PP4_FxxP_1 | 116-119 |
| LIG_AP2alpha_1 | 116-120 | ||
| 120-124 | |||
| CLV_NRD_NRD_1 | 127-129 | ||
| CLV_PCSK_KEX2_1 | 127-129 | ||
| LIG_Arc_Nlobe_1 | 148-152 | ||
| 155-120 | |||
| OC_USP7_MATH_1 | 164-168 | ||
| LIG_BRCT_BRCA1_1 | 177-181 | ||
| Aggregation hotspot | 156-185 | LIG_Arc_Nlobe_1 | 155-159 |
| DOC_WW_Pin1_4 | 160-165 | ||
| OC_USP7_MATH_1 | 164-168 | ||
| LIG_BRCT_BRCA1_1 | 177-181 | ||
| Region with multiplicity of binding modes | 156-203 | OC_USP7_MATH_1 | 164-168 |
| LIG_BRCT_BRCA1_1 | 177-181 | ||
| CLV_PCSK_SKI1_1 | 202-206 | ||
| DOC_USP7_UBL2_3 | 203-207 | ||
| Region with multiplicity of binding modes | 206-211 | CLV_PCSK_SKI1_1 | 202-206 |
| DOC_USP7_UBL2_3 | 203-207 | ||
| CLV_PCSK_KEX2_1 | 207-209 | ||
| MoRF | 223-278 | CLV_NRD_NRD_1 | 245-247 |
| CLV_PCSK_SKI1_1 | 226-230 | ||
| Region with multiplicity of binding modes | 227-237 | CLV_PCSK_SKI1_1 | 226-230 |
| Droplet Promoting Region | 233-365 | CLV_NRD_NRD_1 | 245-247 |
| DEG_ODPH_VHL_1 | 253-264 | ||
| DOC_USP7_MATH_1 | 291-295 | ||
| 293-297 | |||
| 334-338 | |||
| DEG_SCF_FBW7_1 | 271-278 | ||
| 273-278 | |||
| 275-282 | |||
| 277-282 | |||
| 287-294 | |||
| DOC_USP7_UBL2_3 | 310-314 | ||
| 341-345 | |||
| 348-352 | |||
| 352-356 | |||
| 358-362 | |||
| Region with multiplicity of binding modes | 241-250 | CLV_NRD_NRD_1 | 245-247 |
| DOC_CKS1_1 | 248-253 | ||
| Aggregation hotspot | 241-250 | CLV_NRD_NRD_1 | 245-247 |
| DOC_CKS1_1 | 248-253 | ||
| MoRF | 282-298 | DEG_SCF_FBW7_1 | 275-282 |
| 277-282 | |||
| 287-294 | |||
| DOC_WW_Pin1_4 | 279-284 | ||
| 287-292 | |||
| DOC_USP7_MATH_1 | 291-295 | ||
| 293-297 | |||
| LIG_WD40_WDR5_VDV_2 | 290-295 | ||
| Region with multiplicity of binding modes | 316-323 | MOD_CK2_1 | 312-318 |
| DOC_ANK_TNKS_1 | 323-330 | ||
| Aggregation hotspot | 316-323 | MOD_CK2_1 | 312-318 |
| DOC_ANK_TNKS_1 | 323-330 | ||
| Aggregation hotspot | 345-353 | OC_USP7_UBL2_3 | 341-345 |
| 348-352 | |||
| 352-356 | |||
| DOC_USP7_UBL2_3 | 341-345 | ||
| 348-352 | |||
| 352-356 | |||
| TRG_NLS_Bipartite_1 | 345-361 | ||
| 346-261 | |||
| 347-361 | |||
| CLV_NRD_NRD_1 | 345-347 | ||
| CLV_PCSK_KEX2_1 | 345-347 | ||
| Region with multiplicity of binding modes | 345-353 | OC_USP7_UBL2_3 | 341-345 |
| 348-352 | |||
| 352-356 | |||
| DOC_USP7_UBL2_3 | 341-345 | ||
| 348-352 | |||
| 352-356 | |||
| TRG_NLS_Bipartite_1 | 345-361 | ||
| 346-261 | |||
| 347-361 | |||
| CLV_NRD_NRD_1 | 345-347 | ||
| CLV_PCSK_KEX2_1 | 345-347 | ||
| MoRF | 305-365 | DOC_USP7_UBL2_3 | 310-314 |
| 341-345 | |||
| 348-352 | |||
| 352-356 | |||
| 358-362 | |||
| OC_USP7_UBL2_3 | 341-345 | ||
| 348-352 | |||
| 352-356 | |||
| 368-362 | |||
| TRG_NLS_Bipartite_1 | 345-361 | ||
| 346-261 | |||
| 347-361 | |||
| DOC_USP7_MATH_1 | 334-338 | ||
| CLV_NRD_NRD_1 | 345-347 | ||
| CLV_PCSK_KEX2_1 | 345-347 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



