
Submitted:
22 March 2024
Posted:
25 March 2024
You are already at the latest version
Abstract
We hypothesise that anti-inflammatory macrolides can alleviate both the NLRP3 inflammasome activation and accelerated senescence in alveolar macrophages in COPD and/or response to cigarette smoke. Lung tissues from COPD patients and mice chronically exposed to cigarette smoke, cultures of human primary alveolar macrophages and macrophages derived from blood (MDM) or THP-1 monocytes were analysed for markers of macrophage NLRP3 inflammasome activation and accelerated senescence using multifluorescence quantitative confocal microscopy, Western blot, flow cytometry and histochemistry. Cultured macrophages were stimulated with 10% cigarette smoke extract (CSE), with or without presence of non-antibiotic macrolides 2'-desoxy-9-(S)-erythromycylamine and azithromycin-based 2'-desoxy molecule. Cigarette smoke and CSE induced in macrophages significant (p
Keywords:
inflammasome
; NLRP3
; cellular senescence
; antiinflammatory macrolides
; alveolar macrophages
; COPD
1. Introduction
Chronic obstructive pulmonary disease (COPD) is a major global health issue and the third leading cause of death worldwide [1]. The pathologic hallmarks of this disease include sustained chronic inflammation of the small airways leading to lung parenchyma tissue loss (emphysema) and limitation of airflow (shortness of breath) with increased mucus production and bronchiolar obstruction. Immune cells involved in airway inflammation in COPD include alveolar macrophages, neutrophils, T-lymphocytes of the TC1, TH1 and TH17 phenotypes and innate lymphoid cells; in some patients the TH2 and type 2 innate lymphoid cells may become predominant, associated with higher infiltration with eosinophils instead of neutrophils (reviewed in [2]). Alveolar macrophages are the first line responders to airborne irritants and microbes through phagocytosis and coordinators of tissue repair through efferocytosis of apoptotic cells. Constituting 80-90% of the total immune cell population in the lung, these cells are truly ‘orchestrators’ of pro-inflammatory changes leading to COPD [3]. In COPD and response to cigarette smoke, a primary cause of the disease, both phagocytosis and efferocytosis are inhibited in macrophages [4,5,6]. This aberrancy can be associated with various abnormalities, e.g., in LC3-associated phagocytosis [7], mitochondrial functions [8], sphingolipid signalling [9,10] and zinc homeostasis [11].
Accelerated cellular senescence is a characteristic feature of ageing-associated diseases including COPD, Alzheimer’s disease, arthritis and atherosclerosis. Senescent cells adopt pro-inflammatory phenotypes that maintain low-level inflammation, promoting gradual deterioration of organ structures and functions in a process that is described as “inflammageing”. Multiple markers utilized for assessment of cell senescence can be classified according to the targeted organelles and physiologic functions: lysosomal compartment (increased lysosomal mass and activity, accumulation of lipofuscin, SA-β-Gal), cell cycle arrest (p53), morphologic changes (lamin B1 loss and disrupted nuclear membrane), metabolic adaptations (mitochondrial dysfunctions, increase ROS, upregulated pro-survival pathways and chromatin reorganization [12]. As immune cells play central role in inflammageing, the term “immunosenescence” was also applied [13]. In normal ageing and ageing-associated diseases, cells in both the innate and the adaptive compartments of the immune system undergo senescence, and T-lymphocytes are known to be particularly susceptible to this process. Thus, an increase in senescence T-cells may contribute to mechanisms of rheumatic diseases [14,15], COPD [16,17], bronchiolitis obliterans [18] and other chronic inflammatory diseases. Macrophages are the key cell type in inflammageing; their dysfunction in senescence contribute into failure of resolving micro-inflammation and consequently perpetuation of the vicious cycle of inflammation in ageing-associated diseases [19,20]. Although deficient phagocytosis/efferocytosis and pro-inflammatory changes of alveolar macrophages in COPD have been documented as discussed above, whether and how these cells adopt senescence phenotypes have been not formally addressed.
The inflammasome is hypothesized as a master regulator of low-grade inflammation in inflammageing [21]. Inflammasomes are multiprotein complexes, platforms for cleavage (maturation) of IL-1β and other cytokines of the IL-1 family. They have been reported to be involved in mechanisms of chronic inflammatory diseases of the lung, in particular COPD and response to cigarette smoke [22,23,24,25]. Of many inflammasomes discovered, the NLRP3 inflammasome is the most studied for its response to a broad array of stimuli derived from both pathogens and the host cell, in particular from lysosomal dysfunctions. Its activation in COPD and in response to cigarette smoke, remains however an area of debate; current data both supporting and arguing for the NLRP3 inflammasome activation (reviewed in [26]).
With cellular senescence therapy emerging as a novel avenue to COPD management, multiple cellular processes and organelles have been proposed as potential therapeutic targets, including mTOR/autophagy axis, SIRT1 and other sirtuins pathways, mitochondria and antioxidants [27]. We have demonstrated that anti-inflammatory agents such as prednisolone in combination with theophylline, curcumin or resveratrol were able to inhibit pro-inflammatory cytokine production, at the same time restoring decreased SIRT1 expression and steroid sensitivity in CD28-null senescent T- and NKT-like cells from COPD patients [17]. We have also demonstrated that non-antibiotic derivatives of azithromycin and erythromycin macrolides retained their anti-inflammatory effects on cigarette smoke extract (CSE)-stimulated macrophages [28]. Alongside multiple inflammatory markers and markers of cell death induced by cigarette and/or CSE, we noted a significant increase of cleaved caspase-1 and cleaved IL-1β, together with IL-1β release [7,28], suggestive of an inflammasome activation. In this new study, we hypothesize that COPD and/or cigarette smoke-associated inflammation in alveolar macrophages is associated with both accelerated cellular senescence and activation of the NLRP3 inflammasome, and that anti-inflammatory macrolides can target both features. Multiple lines of data were corroborated to demonstrate an increase of the NLRP3 inflammasome pathway and cellular senescence in COPD/cigarette smoke-associated inflammation, and a short-term cell model of CSE-induced inflammasome activation and macrophage senescence was employed to test effects of anti-inflammatory macrolides.
2. Materials and Methods
2.1. Antibodies
Primary antibodies included IL-1β (rabbit polyclonal from Santa Cruz, H53, aa117-269, 1:50 for cell immunofluorescence, IF-C), cleaved IL-1β (rabbit monoclonal, Cell Signalling, Asp116, 1:1000 for Western blot, and goat polyclonal, Santa Cruz, h117, 1:40 for IF-C and m118, 1:40, for paraffin tissue immunofluorescence, IF-P), NLRP3 (rabbit polyclonal, Santa Cruz, H66, 1:40 for IF-C and IF-P), lamin B1 (rabbit polyclonal, Invitrogen, PA5-86096, 1:200 for IF-P and IF-C, 1:20000 for Western blots), p53 (goat polyclonal, Santa Cruz, C19, 1:100 for IF-P, 1:50 for IF-C, and 1:1000 for Western blots), CD9 PE (mouse monoclonal, Immunotech, 1:20 for flow-cytometry and IF-C), CD81 (FITC, mouse monoclonal, Beckton Dickinson, 1:20 for flow-cytometry and IF-C), F4/80 (rat monoclonal, Sapphire, 1:30 for IF-P). Specificity of the NLRP3 and cleaved IL-1β antibodies was shown in previous publications, including peptide blocking experiments (Supplementary Data Text S1 and Figure S1). Other antibodies used for IF-C and IF-P including lamin B1, and p53 were tested on Western blots to show relevant molecular weight bands [25].
As secondary antibodies for immunofluorescence, donkey IgG (F(ab’) fragments directed against rabbit or goat or mouse or rabbit IgG, conjugated with Alexa Fluo (AF) 647, AF594, or AF488 were used (Jackson ImmunoResearch, 1:200). To eliminate cross-species reactions in multi-fluorescence quantitative confocal microscopy (MQCM), all secondary antibodies used in this study were pre-absorbed with cross species IgG by the manufacturer. Secondary antibodies for Western blots were LCR-926-32211:IRDye 800CW Goat anti-Rabbit IgG and LCR-926-68020:IRDye 680LT Goat anti-Mouse IgG (Millennium Science, Australia).
2.2. Human Tissue and Cells
2.2.1. Bronchoscopy Sampling and Subject Selection
Bronchoalveolar lavage (BAL) was obtained via flexible bronchoscopy as we have previously described [4]. Informed consent was obtained and the study protocol was approved by The Royal Adelaide Hospital Research Ethics Committee. Subjects for flow-cytometry analysis of BAL-derived macrophages were 22 age-matched healthy adult volunteers with no history of asthma or allergy and normal lung function and (b) 10 subjects with COPD. Demographic characteristics of COPD vs. non-COPD controls individuals undergoing BAL for this study showed no significant difference between the two groups on age and gender (Supplementary Data Table S1). For short-term cultures of BAL-derived macrophages and stimulation with cigarette smoke extract (CSE), samples were obtained from 7 healthy non-smokers.
2.2.2. BAL Processing for Primary Alveolar Macrophages
The method was described previously [4]. Briefly, 50 mL aliquot of sterile normal saline was administered into the airways using a syringe, followed by aspiration using low suction at room temperature. This process was repeated twice more with additional 50 mL aliquots of saline using the same method. The first aspirated BAL specimen from each patient was excluded to prevent contamination and processed for microbiological testing. The second and third aliquots were collected, stored on ice, and processed within 1 hour of collection. The bronchoalveolar lavage fluid was then centrifuged at 2500rpm for 5 mins. Supernatants were collected and stored at -80°C. Cells were resuspended in 2 mls of RPMI 1640 medium supplemented with 2 mM L-glutamine, 10% FBS, penicillin/gentamicin and incubated for 1 hr to allow macrophages to adhere. Supernatants containing lymphocytes and other non-adherent cell types were separated for parallel studies.
2.2.3. THP-1 Macrophages
THP-1 macrophage-like cells were differentiated from THP-1 monocyte cell line (ATCC, Manassas, VA, USA) as previously described [9,10,11]. Briefly, monocytes cultured at 37°C/5% CO2 in RPMI 1640 medium containing 10% FBS, penicillin/streptomycin, 2 mM l-glutamine, and 0.05 mM ß-mercaptoethanol (Sigma-Aldrich, MO, USA) were seeded at 5 × 105 cells/mL with 50 nM phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) and cultured for 72 h to differentiated into a macrophage-like phenotype.
2.2.4. Monocyte-Derived Macrophages (MDM)
MDM were prepared using blood from 5 consenting healthy non-smoking donors using a protocol described previously [30,31]. Briefly, healthy adult controls were recruited from our volunteer database, comprising non-smokers with no history of respiratory or allergic ailments. Written informed consent was obtained from all healthy subjects following an invitation to participate in the study. The study protocol received approval from the Royal Adelaide Hospital Research Committee (Approval #: #020811d), and all research procedures adhered to relevant guidelines and regulations. For monocyte isolation, whole blood was collected using Lithium-Heparin tubes (Greiner Bio One, Austria). Blood was diluted with plain RPMI 1640 medium at a ratio of 1:2 and layered over LymphoprepTM (STEMCELL Technologies, BC, Canada). The samples were centrifuged at 800×g for 25 minutes with acceleration but no brake. Peripheral blood mononuclear cells (PBMC) were isolated following the manufacturer's instructions. To generate macrophages from monocytes, PBMCs were seeded into plates at a density of 1.4 × 106/mL in plain RPMI 1640 medium and incubated at 37°C with 5% CO2 for 90 minutes to allow monocyte attachment. Unattached cells were removed by aspiration, and the attached cells were washed 3 times with PBS to eliminate any remaining unattached cells. The cells were then cultured in RPMI 1640 medium supplemented with 2 mM L-glutamine, 10% FBS, penicillin/gentamicin, and 20 ng/mL macrophage colony-stimulating factor (M-CSF, Life Sciences) for 12 days, with complete media changes at 4 and 8 days.
2.2.5. Human lung Tissue
Human lung tissue sections were cut from paraffin blocks archived from a previous study [25,29] approved by Royal Adelaide Hospital Ethics Committee; whereby samples of non-tumour tissue were obtained with informed consent from 10 COPD, all smokers/ex-smokers, 4 non-COPD smokers/ex-smokers, and 4 non-smoker/non-COPD patients undergoing lobectomy for management of lung cancer. States of COPD and cancers, and separation of non-tumour tissue from tumour tissue were previously described in [29]. Demographic characteristics of lobectomy patients (Supplementary Data Table S1) revealed no significant differences in ages and gender distribution among the three studied groups.
2.3. Mouse Tissue and Alveolar Macrophages
Paraffin blocks of BALBc mouse lung tissues were banked from a previous study approved by the Institute of Medical and Veterinary Science Animal Ethics Committee [32]. Briefly, animals were chronically exposed to cigarette smoke for 6 weeks (n=6) or sham-smoke control (n=6). Sections cut from archived blocks of lungs of cigarette smoke-exposed and sham-exposed animals were mounted on the same slides in arrays of 4 so that all the 12 animals could be analysed in the same batch. Mouse alveolar macrophages were recognized in paraffin sections by their location in the air sacs of alveoli and morphology (large cells having nearly round or oval shape); the cell type was confirmed by F4/80-positive immunofluorescence and high resolution light microscopy which showed a morphology distinctive from type 2 alveolar cells which also have large size but are typically seen at alveolar junctions and showing lamellar bodies in the cytoplasm [33].
2.4. Cell Models of Cigarette Smoke Exposure and Macrolide Treatment
Cigarette smoke extract (CSE) was prepared following previously described protocol [7; 9-11]. Briefly, smoke from 4 1R5 F research-reference filtered cigarettes (The Tobacco Research Institute, University of Kentucky, Lexington, KY) was bubbled for 5 min per cigarette through 40 mL RPMI 1640 medium containing 10% foetal bovine serum (FBS), 1% penicillin/streptomycin and with 2 mM L-glutamine (all Thermo Fisher Scientific, MA, USA) using a vacuum pump. Following adjustment to pH7.4 the obtained suspension considered as 100% cigarette smoke extract was then aliquoted and stored at −80 °C. Our methods for stimulation of MDM with CSE were described previously [30]. Differentiation of macrophages from a THP-1 cell line (ATCC, Manassas, VA, USA) and stimulation with CSE were also carried out as previously described [9,10,11]. Treatment of CSE-stimulated macrophages with macrolide antibiotics was adapted from a previously described protocol [28]. Briefly, macrophages were differentiated in 8-well chamberslides; wells treated for 24 hours with one of the following: 10% CSE, 10% CSE and 2'-desoxy-9-(S)-erythromycylamine (GS-459755, Gilead Sciences, Seattle, WA, USA) or azithromycin-based 2'-desoxy molecule (GS-560660, Gilead Sciences), or azithromycin (Max Pharma, Surrey Hills, VIC, Australia). In control wells, cells were incubated with vehicle only.
2.5. Immunofluorescence Staining and Multi-Fluorescence Quantitative Confocal Microscopy (MQCM)
Immunofluorescence of cells and tissue sections was carried out as previously described [10,25]. Briefly, before staining, dewaxed paraffin tissue sections were antigen-retrieved with heat in citrate buffer pH6; cells fixed with 2.5% phosphate saline-buffered (PBS) formalin were permeabilized with 0.1% triton X-100. Cells and tissues were incubated at 4oC overnight with cocktails of primary antibodies, then at ambient temperature for 40 minutes with cocktails of secondary antibodies. Blocking with serum-free protein block (DakoCytomation, Dako, Glostrup, Denmark) and multiple washes in 0.05% tween-20/PBS were applied for reduction of background nonspecific fluorescence. For minimal risk of cross-species binding, only secondary antibodies pre-absorbed with cross-species IgG were used.
MQCM imaging and analysis were carried out as detail described [25,34] using a FV3000 confocal system (Olympus, Tokyo, Japan) and ImageJ morphometric software (NIH, MA, USA). Briefly, 5 optical fields were randomly selected in DAPI channel from each chamberslide well or each tissue section for simultaneous scanning in all remaining fluorescence channels. Using ImageJ menus Image/Adjust/Threshold and Analyze/Analyze Particles, uniformly setting at minimal particle size of >10 square pixels (> 3.13 µm2), mean fluorescence intensity (MFI) values were measured from 8-bit monochromatic images of the channels of interest. Average MFI values were corrected for nonspecific fluorescence background including increased autofluorescence in CSE-treated cells, which was measured in the same way from conjugate-only negative control wells in every experiment. For percentage of positive cells, total numbers of cells were counted by DAPI [25,34].
2.6. Colocalization Analysis
Colocalization of NLRP3 and cleaved IL-1β immunofluorescence was analyzed by ImageJ software using two different methods, Pearson’s correlation analysis (Pearson’s R-value and Costes’ P-value by Coloc2 plugin), and Manders’ split coefficients analysis (M1, the fraction of NLRP3 overlapping with cleaved IL-1β immunofluorescence, and M2, the fraction of cleaved IL-1β overlapping with NLRP3 immunofluorescence, both measured in JACoP pluggin).
2.7. Flow-Cytometry
Flow-cytometric analysis of cell surface expression of CD9 and CD81 on BAL-derived alveolar macrophages from COPD and age matched controls was performed using a previously described protocol [35,36,37]. Briefly, 200-μl aliquots of BAL were stained with phycoerythrin (PE)–conjugated monoclonal antibodies to CD9 and fluorescein isothiocyanate (FITC)–conjugated monoclonal antibodies to CD81, cells washed with 0.5% bovine serum albumin in Isoton II (Coulter Immunotech, Hialeah, FL, USA; events were acquired immediately with a FACScalibur flow cytometer (BD Biosciences) and analyzed with CellQuest software (BD Biosciences). The gating strategy for macrophage analysis based on forward and side scatters and CD-14 co-staining was previously described in details [37]. At least 10,000 events were collected.
2.8. Western Blot Analysis
Western blot analysis of macrophage cell lysates was carried out as previously described [7]. Briefly, cells were lysed using M-PER mammalian cell protein lysis reagent containing PMSF protease inhibitor (Sigma Aldrich); protein concentration quantified using BCA protein assay (CA, USA). Protein samples of 10 μg were electrophoresed on 4–12% gradient Bis-Tris gels before being transferred to nitrocellulose membrane. Membranes were blocked in 5% skim milk (Fonterra, NZ) or 5% bovine serum albumin before incubating with primary antibodies overnight. Membranes were incubated with corresponding secondary antibodies and washed three times with TBST and then probed with Bio-Rad software (CA, USA). Band detection was performed using Odyssey ® XF Imaging System with Image Studio Lite (LI-COR Biosciences, USA) for densitometry analysis.
2.9. Histochemistry
Dehydrated paraffin tissue sections were stained with Sudan Black B for lipofuscin in a routine protocol by the Histology Service at the Adelaide Health and Medical Sciences campus of the University of Adelaide.
2.10. Statistical Analysis
Statistical analysis was carried out using the Graphpad Prizm software, non-parametric Mann-Whitney U-test was applied for difference between groups, paired two-tailed Wilcoxon test for induction effects of CSE on macrophages obtained from different donors. Changes were considered significant when p<0.05.
3. Results
3.1. Cigarette Smoke Induced Macrophage NLRP3 Inflammasome Activation
In our previous studies in mouse and cell models, among multiple markers of inflammation, CS and CSE were shown to induce in macrophages a significant increase of cleaved IL-1β and cleaved caspase-1 in cell lysates by Western blot, and release of IL-1β into culture medium by ELISA [7,28], prompting a further investigation on an inflammasome activation. We first reanalysed paraffin lung tissue stored from a previously published mouse model of chronic exposure to cigarette smoke and revealed particulate cytoplasmic immunofluorescence of cleaved IL-1β colocalized with NLRP3 in cells scattered among alveolar tissue. These cells were recognized as alveolar macrophages by localization in alveolar air space and large cytoplasm, which was confirmed by positive F4/80 staining (Figure 1a,b). MQCM analysis demonstrated significant increase of NLRP3/cleaved IL-1β in alveolar tissue of CS-exposed in comparison to sham-exposed control animals, though only cleaved IL-1β but not NLRP3 was seen as particulate (Figure 1c,d). Therefore, human primary alveolar macrophages obtained via bronchioalveolar lavage (BAL) from healthy non-smoker donors were studied in an ex vivo model of short-term exposure to CSE (Figure 1e–i), which was shown to induce significantly upregulated, colocalized immunonofluorescence of NLRP3 and cleaved IL-1β, both of which in particulate forms. Of note, increase of IL-1β by an alternative antibody reactive with total IL-1β was also demonstrated but in homogeneous form (Figure 1e,h). Significant colocalization between NLRP3 and cleaved IL-1β in CSE-treated BAL-derived alveolar macrophages is evidenced by a quantitative analysis revealing Costes P-value equal to 1 and Pearson’s R value ranging from 0.73 to 0.87; the Manders’ split coefficients M1 and M2 revealing high fraction of NLRP3 immunofluorescence overlapping with cleaved IL-1β, and vice versa (Supplementary Figure S2).
To gain more material for further analysis, macrophages were differentiated from the THP-1 monocyte cell line and used as a surrogate model of alveolar macrophages in the same protocol of CSE-stimulation. In previous studies into this model, among multiple markers of inflammation and cell death, a significant increase of cleaved IL-1β and cleaved caspase-1 in cell lysates was shown by Western blot, and release of IL-1β into culture medium by ELISA [7; 28]. In this study, similarly to primary alveolar macrophages, particulate patterns of CSE-induced cleaved IL-1β/NLRP3 immunofluorescence were shown (Figure 2a), quantitative analysis confirming their significant colocalization (Supplementary Data Figure S2b). In difference from primary alveolar macrophages, increase of the total IL-1β in CSE-treated THP-1 macrophages was not confirmed, confocal analysis revealing even a reduction of the total IL-1β in those cells that contained bright particulate cleaved IL-1β (Figure 2b). To confirm that CSE-induced cleavage of IL-1β was associated with the NLRP3 inflammasome activation, upregulation of cleaved IL-1β was shown inhibited after co-treatment of cells with either ZyVad (pan caspase inhibitor) or glyburide (an antagonist of NLRP3) (Figure 2c–e). Thus, taken together with our previously obtained data, multiple lines of new data from analysis of human BAL-derived macrophages, mouse lung sections, and a human cell line strongly support a notion that, at least in the conditions of our experiments, the NLRP3 inflammasome activation could be induced in alveolar macrophages by chronic in vivo exposure to cigarette smoke, and in short term exposure of macrophage cell cultures to CSE.
3.2. Accelerated Cellular Senescence of Alveolar Macrophages in COPD Patients and Mice Chronically Exposed to Cigarette Smoke
In paraffin lung sections of both COPD patients and smokers, a distinctive accumulation of lipofuscin was demonstrated in cytoplasm of alveolar macrophages compared to non-COPD-non-smoker control (Figure 3a). Similar phenomenon was reproduced in the mouse model of chronic exposure to cigarette smoke (Figure 3b). Furthermore, mouse lung tissue sections were analysed by confocal microscopy for lamin B1 and p53 expression in alveolar macrophages (Figure 3c,d). While alveolar macrophages in control animals showed a marginalised nuclear pattern of lamin B1 expected for its association with the nuclear envelope; a dispersed pattern was seen in cells from cigarette smoke-exposed animals; this reduction was confirmed by MQCM analysis (Figure 3d). Immunofluorescence of p53 was mostly localized to cytoplasm. A trend of p53 increase in cigarette smoke-exposed animals which was not significant was also recorded.
We furthermore employed multi-colour flow-cytometry to measure the surface expression of CD9 and CD81 molecules in alveolar macrophages obtained from age-matched COPD patients vs non-COPD non-smoker controls (Supplementary Data Table S1). The results showed that the COPD status was associated with a significantly downregulated surface expression of these tetraspanins (Figure 3e).
3.3. Short-Term Macrophage Cell Culture Model for Double Targeting NLRP3 Inflammasome Activation and Accelerated Cell Senescence
To this end, we set an aim to test whether markers of both accelerated cellular senescence and the NLRP3 inflammasome activation can be induced in a short-term cell culture model of CSE-stimulated macrophages, and if so, whether they can be targeted with anti-inflammatory macrolides. Raising a hypothesis that decreased surface CD9 and CD81 could be markers of accelerated macrophage senescence, the THP-1 macrophage model was first tested, detecting however only low levels of CD9/CD81 immunofluorescence in the basal state (Supplementary Data Figure S3). Primary macrophages derived from peripheral blood monocytes (MDMs) were then employed which revealed bright immunofluorescence of both CD9 and CD81 near the cell surface of control but not CSE-treated cells (Figure 4a and Supplementary Data Movie S1). The CSE-induced macrophage senescence phenotype was confirmed by significantly upregulated expression of p53 shown by Western blotting analysis, and lamin B1 decrease from marginalized nuclear localization shown by confocal microscopy (Figure 4b,d and Supplementary Data Movie S1); this change was associated with increased cleaved IL-1β demonstrated by Western blot analysis (Figure 4c) which was shown colocalized with NLRP3 by confocal microscopy (Supplementary Data Figure S4).
We then embarked to carry out experiments using MDMs to test a hypothesis, that both the NLRP3 inflammasome activation and accelerated cellular senescence induced by short-term CSE stimulation in macrophages could be alleviated by treatment with anti-inflammatory macrolides azithromycin or nonantibiotic GS-459755 and GS-560660. Results of these experiments showed, that treatment with CSE alone induced significant increase of particulate cleaved IL-1β and NLRP3, decrease of the cell surface CD9/CD81, and lamin B1 near the nuclear envelope. While CSE-induced cleaved IL-1β was significantly reversed by all three macrolides, trends of alleviation were observed for other markers which were significant for lamin B1 by G4 and CD9 by G5. These results are summarized in the Figure 5.
4. Discussion and Conclusion
COPD is now believed as an ageing-associated disease, whereby the affected organ, in this case the lung, is ageing faster than the patient’s biological age. While most of studies in this area have been focusing on the epithelial compartment [38,39,40,41,42,43], senescence of immune cells is understudied, studies so far focusing on T-cells [16,17,44,45]. To our knowledge this study is the first to gather multiple evidence of accelerated cellular senescence in alveolar macrophages in COPD and in response to cigarette smoke. Lipofuscin, non-degradable products from oxidatively damaged proteins that accumulated in lysosomes, is a well-accepted marker of cellular ageing for various cell types [12,46]. Increased lipofuscin deposits in alveolar macrophages were indeed already reported in an early study on smoking patients diagnosed with sarcoidosis [47]. Lamin B1 is a protein required for chromatin attachment to the nuclear envelope for normal genome function; its downregulation in ageing is documented, being therefore a negative marker for accelerated cellular senescence [12,48]. Downregulated lamin B1 has been reported in lung samples from COPD and CSE-stimulated human bronchial epithelial cells [41]. The p53 protein, a transcription factor participating in DNA repair and maintaining genome integrity, is generally upregulated in response to cell stress and apoptosis and is another commonly used marker of senescence [12,49]. Elevated expression of p53 was reported in lung samples of patients having COPD/emphysema [50,51], as well as in CSE-stimulated in vitro cells e.g., fibroblasts [52] or endothelial cells [53]. Thus, changes of these markers in the current study are in concordant with previous data, and altogether they consolidate the notion of accelerated cellular senescence in alveolar macrophage macrophages in COPD and/or response to cigarette smoke exposure.
Roles of the tetraspanins CD9 and CD81 in cellular senescence present a controversial question in literature. Although some recent studies into vascular pathology suggest that upregulation of CD9 could be a marker of ageing [54,55], mouse models of CD9/CD81 double knockout were shown to develop a COPD-like syndrome and multiple organ ageing [56,57]. Tetraspanins have complex, multifaceted roles in cell biology. As a key organizer of plasma cell membrane, CD9 may deliver negative regulatory effects on COPD-related inflammation; mechanistically CD9 complexing with TLR4/CD14 has been implicated in downregulating the latter recruitment into the lipid raft domains to activate the pro-inflammatory NFkB signaling [58]. Considering this previously available data [56,57,58], the loss of plasma membrane localization of CD9/CD81 in alveolar macrophages of COPD patients and in CSE-stimulated macrophages ex vivo in this study suggest that their decreased surface expression could possibly be employed as a marker of cellular senescence, at least in the macrophage cell type. Further studies employing clinical cell/tissue samples as well as experimentally induced ageing are required before CD9/CD81 could be used as diagnostic markers.
Whether the NLRP3 inflammasome pathway is activated in COPD and/or response to cigarette smoke exposure remains a debatable issue (see review [26]). While multiple evidence from human [22; 59] and animal studies [59,60,61] lends support for a “Yes” answer, other studies present opposite results [62,63,64,65,66], As a potent protective mechanism but potentially detrimental at excessive activation, the NLRP3 inflammasome pathway is tightly controlled by multiple regulatory pathways acting on post-translational modifications of proteins as well as transcription and translation levels [67,68,69]. In this relation cigarette smoke could inhibit the inflammasome by upregulated catabolism of NLRP3 [64]. Furthermore cigarette smoke-induced depletion of alveolar macrophages [70] could also obscure detection of the inflammasome activation in this cell type. In this line, our previous data showed upregulated cell death evidenced by significant increase of cleaved caspase-3 and PARP in both in alveolar macrophages of cigarette smoke-exposed mice and in CSE-stimulated macrophage cell culture [7]. Consequently, depending on particular clinical or experimental settings, as well as methodologic variations among studies in the field, markers of both upregulation and downregulation of the NLRP3 inflammasome pathway can be observed in COPD and/or response to cigarette smoke exposure. Thus, in our experiments, using the same antibody to IL-1β detected the induction effect of CSE on this parameter only in primary alveolar macrophages, but not THP-1-differentiated macrophages, even though cleavage of IL-1β was demonstrated in both cases by two alternative antibodies, in two different methods. A limitation of this study was that we could not ascertain whether the NLRP3 inflammasome pathway is upregulated in alveolar macrophages of COPD patients. An upregulated expression of the inflammasome-related genes could not also be demonstrated. Nevertheless, we present here multiple lines of evidence to support an activation of the alveolar macrophage NLRP3 inflammasome in response to cigarette smoke exposure, either in an in vivo model of chronic exposure to cigarette smoke, and in cell culture models of short-term treatment with CSE. Importantly, one of these short-term models was employed for further study of whether the cellular senescence is accelerated in parallel with the inflammasome activation.
The key result of this study was that both azithromycin and the two novel anti-inflammatory non-antibiotic macrolides could significantly reverse NLRP3 inflammasome activation and accelerated cellular senescence in CSE-exposed macrophages. A rationale for the use of anti-inflammatory agents to double target the inflammasome and accelerated cellular senescence is the implicated role of the NLRP3 inflammasome as a driver of low-grade inflammation that accelerates cellular senescence [21,71]. We hypothesized that anti-inflammatory macrolides such as azithromycin, at low dose, could be candidates in this direction. Indeed, efficacy of azithromycin in preventing acute exacerbations in COPD has already been demonstrated in a number of clinical trials e.g., [72,73,74]. Mechanistically, azithromycin may intervene in the inflammasome pathway by induction of instability of mRNA for the NLRP3 synthesis [75], or inhibition of the upstream STAT1 and NFkB signalling [76]. As di-cationic molecules, macrolides such as azithromycin and erythromycin are highly accumulated in lysosomes where they may regulate the membrane stability of this organelle [77], which in turn may regulate both the NLRP3 inflammasome activation and cell senescence. While data of this study supports that azithromycin could reverse the NLRP3 inflammasome activation and alleviate markers of accelerated cellular senescence in CSE-exposed macrophages, its clinical use in this regard is limited by a potential for the emergence of antibiotic resistant bacteria following its long-term use [78]. This limitation has stimulated studies of non-antibiotic macrolides by many groups [79,80], including ours [28]. In the present study we present further evidence for the efficacy of these non-antibiotic macrolides, alongside azithromycin, in reversal of both the NLRP3 inflammasome activation and signs of accelerated cellular senescence in CSE-stimulated macrophages. It should be noted that our study was limited to only a short-term in vitro CSE exposure; and whether this approach has positive effects on alveolar macrophages isolated from COPD patients or has long-term efficacy in an in vivo setting remains objectives for future investigations.
In conclusion, our findings support that 1) alveolar macrophages undergo both activation of the NLRP3 inflammasome and accelerated cellular senescence in COPD and/or response to cigarette smoke exposure, and 2) in short-term exposure of macrophages to CSE, application of azithromycin or non-antibiotic macrolides can alleviate both the inflammasome activation and accelerated cellular senescence.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
We thank Gilead Sciences, Inc. for the gift of nonantibiotic macrolides GS-459755 and GS-560660. Excellent technical assistance from Ms Agatha Labrinidis and the staff of Adelaide Microscopy at the University of Adelaide, and Dr James Manavis and the staff of the Histopathology Service at the Adelaide Health and Medical Sciences is acknowledged.
References
- Lindberg, A.; Lindberg, L.; Sawalha, S.; Nilsson, U.; Stridsman,C. ; Lundbäck, B.; Backman, H. Large underreporting of COPD as cause of death-results from a population-based cohort study. Respir. Med. 2021, 186. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Alveolar macrophages as orchestrators of COPD. COPD. 2004, 1, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Scicchitano, R.; Reynolds, P.N.; Holmes, M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol. Cell Biol. 2003, 81, 289–96. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Ahern, J.; Jersmann, H.; Holmes, M.; Reynolds, P.N. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease Am. J. Respir. Cell Mol. Biol. 2007, 37, 748–55. [Google Scholar] [CrossRef]
- Taylor, A.E.; Finney-Hayward, T.K.; Quint, J.K.; Thomas, C.M.; Tudhope, S.J.; Wedzicha, J.A.; Barnes, P.J.; Donnelly, L.E. Defective macrophage phagocytosis of bacteria in COPD. Eur. Respir. J. 2010, 35, 1039–47. [Google Scholar] [CrossRef] [PubMed]
- Asare, P.F.; Tran, H.B.; Hurtado, P.R.; Perkins, G.B; Nguyen, P.; Jersmann, H. Roscioli, E.; Hodge, S. Inhibition of LC3-associated phagocytosis in COPD and in response to cigarette smoke. Ther. Adv. Respir. Dis. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Belchamber, K.B.R.; Singh, R.; Batista, C.M.; Whyte, M.K.; Dockrell, D.H.; Iain Kilty, I.; Robinson, M.J.; Wedzicha, J.A.; Barnes, P.J.; Donnelly, L.E. ; COPD-MAP consortium. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur. Respir. J, 1: 54, 1802. [Google Scholar]
- Barnawi, J.; Tran, H.; Jersmann, H.; Pitson, S.; Roscioli, E.; Hodge, G.; Meech, R.; Haberberger, R.; Hodge, S. Potential Link between the Sphingosine-1-Phosphate (S1P) System and Defective Alveolar Macrophage Phagocytic Function in Chronic Obstructive Pulmonary Disease (COPD). PLoS One, 0122. [Google Scholar] [CrossRef]
- Tran, H.B.; Barnawi, J.; Ween, M.; Hamon, R.; Roscioli, E.; Hodge, G.; Reynolds, P.N.; Pitson, S.M.; Davies, L.T.; Haberberger, R.; Hodge, S. Cigarette smoke inhibits efferocytosis via deregulation of sphingosine kinase signaling: reversal with exogenous S1P and the S1P analogue FTY720. J. Leukoc. Biol. 2016, 100, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Hamon, R.; Homan, C.C.; Tran, H.B.; Mukaro, V.R.; Lester, S.E.; Roscioli, E.; Bosco, M.D.; Murgia, C.M.; Ackland, M.L.; Jersmann, H.P.; Lang, C.; Zalewski, P.D.; Hodge, S.J. Zinc and zinc transporters in macrophages and their roles in efferocytosis in COPD. PLoS One. 2014, 9, e110056. [Google Scholar] [CrossRef]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. [CrossRef]
- Prelog, M.; Schwarzenbrunner, N.; Sailer-Höck, M.; Kern, H.; Klein-Franke, A.; Ausserlechner, M.J.; Koppelstaetter, C.; Brunner, A.; Duftner, C.; Dejaco, C.; Strasak, A.M.; Müller, T.; Zimmerhackl, L.B.; Brunner, J. Premature aging of the immune system in children with juvenile idiopathic arthritis. Arthritis Rheum. 2008, 58, 2153–62. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Shao, L.; Colmegna, I.; Goronzy, J.J.; Weyand, C.M. Telomerase insufficiency in rheumatoid arthritis. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 4360–5. [Google Scholar] [CrossRef] [PubMed]
- Hodge, G.; Jersmann, H.; Tran, H.B.; Holmes, M,; Reynolds, P. N.; Hodge, S. Lymphocyte senescence in COPD is associated with loss of glucocorticoid receptor expression by pro-inflammatory/cytotoxic lymphocytes. Respir. Res. 2015, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Hodge, G.; Tran, H.B.; Reynolds, P.N.; Jersmann, H.; Hodge, S. Lymphocyte senescence in COPD is associated with decreased sirtuin 1 expression in steroid resistant pro-inflammatory lymphocytes. Ther. Adv. Respir. Dis. 1753. [Google Scholar] [CrossRef] [PubMed]
- Hodge, G.; Hodge, S.; Liu, H.; Nguyen, P.; Holmes-Liew, C.L.; Holmes, M. BOS Is Associated With Decreased SIRT1 in Peripheral Blood Proinflammatory T, NK, and NKT-like Lymphocytes. Transplantation. 2019, 103, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- De Maeyer, R.P. H; Chambers. E.S. The impact of ageing on monocytes and macrophages. Immunol. Lett. [CrossRef]
- Campbell, R.A. , Docherty, M-H.; Ferenbach, D.A.; Mylonas, K.J. The Role of Ageing and Parenchymal Senescence on Macrophage Function and Fibrosis. Front. Immunol. 7007. [Google Scholar] [CrossRef]
- Latz, E.; Duewell, P. NLRP3 inflammasome activation in inflammaging. Semin. Immunol. [CrossRef]
- Faner, R.; Sobradillo, P.; Noguera, A. , Gomez, C.; Cruz, T.; López-Giraldo, A.; Ballester, E.; Soler, N.; Arostegui, J.I; Pelegrín, P.; Rodriguez-Roisin, R.; Yagüe, J.; Cosio, B.G.; Juan, M.; Agustí, A. The inflammasome pathway in stable COPD and acute exacerbations. ERJ Open Res. 2016, 2, 00002–2016. [Google Scholar] [CrossRef]
- Colarusso, C.; Terlizzi, M.; Molino, A.; Pinto, A.; Sorrentino, R. Role of the inflammasome in chronic obstructive pulmonary disease (COPD). Oncotarget. 2017, 8, 81813–81824. [Google Scholar] [CrossRef] [PubMed]
- Colarusso, C.; Terlizzi, M.; Molino, A.; Imitazione, P.; Somma, P.; Rega, R.; Saccomanno, A.; Aquino, R.P.; Pinto, A.; Sorrentino, R. AIM2 Inflammasome Activation Leads to IL-1α and TGF-β Release From Exacerbated Chronic Obstructive Pulmonary Disease-Derived Peripheral Blood Mononuclear Cells. Front. Pharmacol, 2: 10. [CrossRef]
- Tran, H.B.; Hamon, R.; Jersmann, H.; Ween, M.P.; Asare, P.; Haberberger, R.; Pant, H.; Hodge, S.J. AIM2 nuclear exit and inflammasome activation in chronic obstructive pulmonary disease and response to cigarette smoke. J. Inflamm. (Lond). 2021, 18, 19. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, J.W.; Kim, R.Y.; Robertson, A.A.B.; Hirota, J.A.; Wood, L.G.; Knight, D.A.; Cooper, M.A.; O'Neill, L.A.J.; Horvat, J.C.; Hansbro, P.M. Inflammasomes in the lung. Mol. Immunol. [CrossRef]
- Barnes, P.J. Targeting cellular senescence as a new approach to chronic obstructive pulmonary disease therapy. Curr. Opin. Pharmacol. [CrossRef]
- Hodge, S.; Tran, H.B.; Hamon, R.; Roscioli, E.; Hodge, G.; Jersmann, H.; Ween, M.; Reynolds, P.N.; Yeung, A. Treiberg, J.; Wilbert, S. Nonantibiotic macrolides restore airway macrophage phagocytic function with potential anti-inflammatory effects in chronic lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L678–L687. [Google Scholar] [CrossRef] [PubMed]
- Cordts, F.; Pitson, S.; Tabeling, C.; Gibbins, I.; Moffat, D.F.; Jersmann, H.; Hodge, S.; Haberberger, R.V. Expression profile of the sphingosine kinase signalling system in the lung of patients with chronic obstructive pulmonary disease. Life Sci. 2011, 89, 806–11. [Google Scholar] [CrossRef]
- Ween, M.P.; White, J.B.; Tran, H.B.; Mukaro, V.; Jones, C.; Macowan, M.; Hodge, G.; Trim, P.J.; Snel, M.F.; Hodge, S., J. The role of oxidised self-lipids and alveolar macrophage CD1b expression in COPD. Sci. Rep. 2021, 11, 4106. [Google Scholar] [CrossRef]
- Hodge, S.; Hodge, G.; Jersmann, H.; Matthews, G.; Ahern, J.; Holmes, M.; Reynolds, P.N. Azithromycin improves macrophage phagocytic function and expression of mannose receptor in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med.
- Tran, H.B.; Jersmann, H.; Truong, T.T.; Hamon, R.; Roscioli, E.; Ween, M.; Pitman, M.R.; Pitson, S.M.; Hodge, G.; Reynolds, P.N.; Hodge, S. Disrupted epithelial/macrophage crosstalk via Spinster homologue 2-mediated S1P signaling may drive defective macrophage phagocytic function in COPD. PLoS One. 2017, 12, e0179577. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.B.; Lewis, M.D.; Tan, L.W.; Lester, S.E.; Baker, L.M.; Ng, J.; Hamilton-Bruce, M.A. , Hill, C.L.; Koblar, S.A.; Rischmueller, M.; Ruffin, R.E.; Wormald, P.J.; Zalewski, P.D.; Lang, C.J. Immunolocalization of NLRP3 Inflammasome in Normal Murine Airway Epithelium and Changes following Induction of Ovalbumin-Induced Airway Inflammation J Allergy (Cairo). 2012;2012:819176. [CrossRef]
- Tran, H.B.; Jakobczak, R.; Abdo, A.; Asare, P.; Reynolds, P.; Beltrame, J.; Hodge, S.; Zalewski, P. Immunolocalization of zinc transporters and metallothioneins reveals links to microvascular morphology and functions. Histochem. Cell Biol. 2022, 158, 485–496. [Google Scholar] [CrossRef]
- Hodge, S.J.; Hodge, G.L.; Holmes, M.; Reynolds, P.N. Flow cytometric characterization of cell populations in bronchoalveolar lavage and bronchial brushings from patients with chronic obstructive pulmonary disease. Cytometry B Clin. Cytom.
- Hodge, S.; Hodge, G.; Ahern, J.; Jersmann, H.; Holmes, M.; Reynolds, P.N. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol.
- Hodge, S.; Matthews, G.; Mukaro. V.; Ahern, J.; Shivam, A.; Hodge, G.; Holmes, M.; Jersmann, H.; Reynolds, P.N. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am. J. Respir. Cell. Mol. Biol. 2011, 44, 673–81. [Google Scholar] [CrossRef]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar cell senescence in patients with pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–93. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; Kojima, J.; Shimizu, K.; Numata, T.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Harada, T.; Nishimura, S.L.; Kaneko, Y.; Nakayama, K.; Kuwano, K. . PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015, 11, 547–59. [Google Scholar] [CrossRef]
- 40. Jang, J-H.; Chand, H.S.; Bruse, S.; Doyle-Eisele, M.; Royer, C.; McDonald, J.; Qualls, C.; Klingelhutz, A.J.; Lin, Y.; Mallampalli, R.; Tesfaigzi, Y.; Nyunoya, T. Connective Tissue Growth Factor Promotes Pulmonary Epithelial Cell Senescence and Is Associated with COPD Severity. COPD. [CrossRef]
- Saito, N.; Araya, J.; Ito, S.; Tsubouchi, K.; Minagawa, S. ; Hara. H.; Ito, A.; Nakano, T.; Hosaka, Y.; Ichikawa, A.; Kadota, T.; Yoshida, M.; Fujita, Y.; Utsumi, H.; Kurita, Y.; Kobayashi, K.; Hashimoto, M.; Wakui, H.; Numata, T.; Kaneko, Y.; Asano, H.; Odaka, M.; Ohtsuka, T.; Morikawa, T.; Nakayama, K.; Kuwano, K. Involvement of Lamin B1 Reduction in Accelerated Cellular Senescence during Chronic Obstructive Pulmonary Disease Pathogenesis J. Immunol. 2019, 202, 1428–1440. [Google Scholar] [CrossRef]
- Sarker, R.S.J.; Conlon, T.M.; Morrone, C.; Srivastava, B.; Konyalilar, N.; Verleden, S.E.; Bayram, H.; Fehrenbach, H.; Yildirim, A.Ö. CARM1 regulates senescence during airway epithelial cell injury in COPD pathogenesis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L602–L614. [Google Scholar] [CrossRef]
- de Vries, M.; Nwozor, K.O.; Muizer, K.; Wisman, M.; Timens, W.; van den Berge, M.; Faiz, A.; Hackett, T-L. ; Heijink, I.H.; Brandsma, C.A. The relation between age and airway epithelial barrier function. Respir. Res. 2022, 23, 43. [Google Scholar] [CrossRef]
- Hodge, G.; Jersmann, H.; Tran, H.B.; Asare, P.F.; Jayapal, M.; Reynolds, P.N.; Holmes, M.; Hodge, S. COPD is associated with increased pro-inflammatory CD28null CD8 T and NKT-like cells in the small airways. Clin. Exp. Immunol. 2022, 207, 351–359. [Google Scholar] [CrossRef]
- Fernandes, J.R.; Pinto, T.N.C.; Arruda, L.B.; da Silva, C.C.B.M.; de Carvalho, C.R.F.; Pinto, R.M.C.; da Silva Duarte, A.J.; Benard, G. Age-associated phenotypic imbalance in TCD4 and TCD8 cell subsets: comparison between healthy aged, smokers, COPD patients and young adults. Immun. Ageing. 2022, 19, 9. [Google Scholar] [CrossRef]
- Jung, T.; Bader, N.; Grune, T. Lipofuscin: formation, distribution, and metabolic consequences. Ann N Y Acad Sci. 2007;1119:97-111. [CrossRef]
- Leuenberger, P.; Vonmoos, S.; Vejdovsky, R. Morphologic changes of alveolar macrophages in smoking sarcoidosis patients. Eur. J. Respir. Dis. Suppl.
- Lukášová, E.; Kovařík, A.; Kozubek, S. . Consequences of lamin B1 and lamin B receptor downregulation in senescence cells. Cells. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules. 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Siganaki, M.; Koutsopoulos, A.V.; Neofytou, E.; Vlachaki, E.; Psarrou, M.; Soulitzis, N.; Pentilas, N.; Schiza, S.; Siafakas, N.M.; Tzortzaki, E.G. Deregulation of apoptosis mediators' p53 and bcl2 in lung tissue of COPD patients. Respir. Res. 2010, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.C.; Vachon-Beaudoin, G.; Parent, J.; Chakir, J.; Milot, J. Increased p53 level, Bax/Bcl-x(L) ratio, and TRAIL receptor expression in human emphysema. Am. J. Respir. Crit. Care Med. 2008, 178, 240–7. [Google Scholar] [CrossRef] [PubMed]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette smoke induces cellular senescence. Am. J. Respir. Cell. Mol. Biol. 2006, 35, 681–8. [Google Scholar] [CrossRef] [PubMed]
- Damico, R.; Simms, T.; Kim, B.S.; Tekeste, Z.; Amankwan, H.; Damarla, M.; Hassoun, P.M. p53 mediates cigarette smoke-induced apoptosis of pulmonary endothelial cells: inhibitory effects of macrophage migration inhibitor factor. Am. J. Respir. Cell. Mol. Biol. 2011, 44, 323–32. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Kim, E.C.; Son, Y.; Lee, D.W.; Park, Y.S.; Choi, J.H.; Cho, K.H.; Kwon, K.S.; Kim, J.R. CD9 induces cellular senescence and aggravates atherosclerotic plaque formation. Cell Death Differ. 2020, 27, 2681–2696. [Google Scholar] [CrossRef]
- Kim, J.R.; Choi, J.H. CD9 expression in vascular aging and atherosclerosis. Histol. Histopathol. 2020, 35, 1449–1454. [Google Scholar] [CrossRef]
- Takeda, Y.; He, P.; Tachibana, I.; Zhou, B.; Miyado, K.; Kaneko, H.; Suzuki, M.; Minami, S.; Iwasaki, T.; Goya, S.; Kijima, T.; Kumagai, T.; Yoshida, M.; Osaki, T.; Komori, T.; Mekada, E.; Kawase, I. Double deficiency of tetraspanins CD9 and CD81 alters cell motility and protease production of macrophages and causes chronic obstructive pulmonary disease-like phenotype in mice. J. Biol. Chem. 2008, 283, 26089–97. [Google Scholar] [CrossRef]
- Jin, Y.; Takeda, Y.; Kondo, Y.; Tripathi, L.P.; Kang, S.; Takeshita, H.; Kuhara, H.; Maeda, Y.; Higashiguchi, M.; Miyake, K.; Morimura, O.; Koba, T.; Hayama, Y.; Koyama, S.; Nakanishi, K.; Iwasaki, T.; Tetsumoto, S.; Tsujino, K.; Kuroyama, M.; Iwahori, K.; Hirata, H.; Takimoto, T.; Suzuki, M.; Nagatomo, I.; Sugimoto, K.; Fujii, Y.; Kida, H.; Mizuguchi, K.; Ito, M.; Kijima, T.; Rakugi, H.; Mekada, E.; Tachibana, I.; Kumanogoh, A. Double deletion of tetraspanins CD9 and CD81 in mice leads to a syndrome resembling accelerated aging. Sci. Rep. 2018, 8, 5145. [Google Scholar] [CrossRef]
- Suzuki, M.; Tachibana, I.; Takeda, Y.; He, P.; Minami, S.; Iwasaki, T.; Kida, H.; Goya, S.; Kijima, T.; Yoshida, M.; Kumagai, T.; Osaki, T. , Kawase, I. Tetraspanin CD9 negatively regulates lipopolysaccharide-induced macrophage activation and lung inflammation. J. Immunol. 2009, 182, 6485–93. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, N.S.; Bracke, K.R.; Dupont, L.L.; Van Pottelberge, G.R.; Provoost, S.; Vanden Berghe, T.; Vandenabeele, P.; Lambrecht, B.N.; Joos, G.F.; Brusselle, G.G. Role of IL-1α and the Nlrp3/caspase-1/IL-1β axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur. Respir. J. 2011, 38, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Eltom, S.; Belvisi, M.G.; Stevenson, C.S.; Maher, S.A.; Dubuis, E.; Fitzgerald, K.A.; Birrell, M.A. Role of the inflammasome-caspase1/11-IL-1/18 axis in cigarette smoke driven airway inflammation: an insight into the pathogenesis of COPD. PLoS One. 2014, 9, e112829. [Google Scholar] [CrossRef] [PubMed]
- Huot-Marchand, S; Nascimento, M.; Culerier, E.; Bourenane, M.; Savigny, F.; Panek, C.; Serdjebi, C.; Le Bert, M.; Quesniaux, V., F., J.; Ryffel, B.; Broz, P.; Riteau, N.; Gombault, A.; Couillin, I. Cigarette smoke-induced gasdermin D activation in bronchoalveolar macrophages and bronchial epithelial cells dependently on NLRP3 Front. Immunol. 2022;13:918507. [CrossRef]
- Di Stefano, A.; Caramori, G.; Barczyk, A.; Vicari, C.; Brun, P.; Zanini, A.; Cappello, F.; Garofano, E.; Padovani, A.; Contoli, M.; Casolari, M.; Durham, A.L.; Chung, K.F.; Barnes, P.J.; Papi, A.; Adcock, I.; Balbi, B. Innate immunity but not NLRP3 inflammasome activation correlates with severity of stable COPD. Thorax. 2014, 69, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Wang, X.; Ge, S.; Chen, W.; Wang, W. Han, X. Long-term cigarette smoking suppresses NLRP3 inflammasome activation in oral mucosal epithelium and attenuates host defense against Candida albicans in a rat model. Biomed. Pharmacother, 1085. [Google Scholar] [CrossRef]
- Han, S-H. ; Jerome, J.A.; Gregory, A., D.; Mallampalli R., K. Cigarette smoke destabilizes NLRP3 protein by promoting its ubiquitination Respir Res. 2017, 18, 2. [CrossRef]
- Buscetta, M.; Di Vincenzo, D.; Miele, M.; Badami, E.; Pace, E.; Cipollina, C. Cigarette smoke inhibits the NLRP3 inflammasome and leads to caspase-1 activation via the TLR4-TRIF-caspase-8 axis in human macrophages FASEB J. 2020, 34, 1819–1832. 34. [CrossRef]
- Buscetta, M.; Cristaldi, M.; Cimino, M.; La Mensa, A.; Dino, P.; Bucchieri, F.; Rappa, F.; Amato, S.; Aronica, T., S.; Pace, E.; Bertani, A.; Cipollina, C. Cigarette smoke promotes inflammasome-independent activation of caspase-1 and -4 leading to gasdermin D cleavage in human macrophages FASEB J. 2022, 36, e22525. 36. [CrossRef]
- Nanda, S.K.; Vollmer, S.; Perez-Oliva, A.B. Posttranslational regulation of inflammasomes, its potential as biomarkers and in the Identification of novel drugs targets. Front. Cell. Dev. Biol. 8875. [Google Scholar] [CrossRef]
- Lopez-Castejon, G. Control of the inflammasome by the ubiquitin system. FEBS J. 2020, 287, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Zangiabadi, S.; Abdul-Sater, A.A. Regulation of the NLRP3 Inflammasome by Posttranslational Modifications. J. Immunol. 2022, 208, 286–292. [Google Scholar] [CrossRef]
- Fleischmann, M; Andrew G Jarnicki, A. G.; Brown, A.S.; Yang, C.; Anderson, G.P.; Garbi, N.; Hartland, E.L.; van Driel, I.R.; Ng, G.Z. Cigarette smoke depletes alveolar macrophages and delays clearance of Legionella pneumophila. Am J Physiol Lung Cell Mol Physiol 2023, 324, L373–L384. [CrossRef]
- Ferrucci, L.; Fabbri, E. . Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Albert, R.K.; Connett, J.; Bailey, W.C.; Casaburi, R.; Cooper, J.A. Jr; Criner, G.J.; Curtis, J.L.; Dransfield, M.T.; Han, M.K.; Lazarus, S.C.; Make, B.; Marchetti, N.; Martinez, F.J.; Madinger, N.E.; McEvoy, C.; Niewoehner, D.E.; Porsasz, J.; Price, C.S.; Reilly, J.; Scanlon, P.D.; Sciurba, F.C.; Scharf, S.M.; Washko, G.R.; Woodruff, P.G.; Anthonisen, N.R.; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N. Engl. J. Med. 2011, 365, 689–98. [Google Scholar] [CrossRef]
- Hodge, S.; Reynolds, P.N. Low-dose azithromycin improves phagocytosis of bacteria by both alveolar and monocyte-derived macrophages in chronic obstructive pulmonary disease subjects. Respirology. 2012, 17, 802–7. [Google Scholar] [CrossRef] [PubMed]
- Vermeersch, K.; Gabrovska, M.; Aumann, J.; Demedts, I. K; Corhay, J-L.; Marchand, E.; Slabbynck, H.; Haenebalcke, C.; Haerens, M.; Hanon, S.; Jordens, P.; Peché, R.; Fremault, A.; Lauwerier, T.; Delporte, A.; Vandenberk, B.; Willems, R.; Everaerts, S.; Belmans, A.; Bogaerts, K.; Verleden, G.M.; Troosters, T.; Ninane, V.; Brusselle, G.G.; Janssens, W. Azithromycin during acute Chronic Obstructive Pulmonary Disease exacerbations requiring hospitalization (BACE). A multicenter, randomized, double-blind, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2019, 200, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Lendermon, E.A.; Coon, T.A.; Bednash, J.S.; Weathington, N.M.; McDyer, J.F.; Mallampalli, R.K. Azithromycin decreases NALP3 mRNA stability in monocytes to limit inflammasome-dependent inflammation. Respir Res. 2017, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Haydar, D.; Cory, T.J.; Birket, S.E.; Murphy, B.S.; Pennypacker, K.R.; Sinai, A.P.; Feola, D.J. Azithromycin polarizes macrophages to an M2 phenotype via inhibition of the STAT1 and NF-κB signaling pathways. J. Immunol. 2019, 203, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Persson, H.L.; Vainikka, L.K.; Sege, M.; Wennerström, U.; Dam-Larsen, S.; Persson, J. Leaky lysosomes in lung transplant macrophages: azithromycin prevents oxidative damage. Respir. Res. 2012, 13, 83. [Google Scholar] [CrossRef]
- Taylor, S.L.; Leong, L.E.X; Mobegi, F.M.; Choo, J.M.; Wesselingh, S.; Yang, I.A.; Upham, J.W.; Reynolds, P.N.; Hodge, S.; James, A.L.; Jenkins, C.; Peters, M.J.; Baraket, M.; Marks, G.B.; Gibson, P.G.; Rogers, G.B.; Simpson, J.L. Long-term azithromycin reduces Haemophilus influenzae and increases antibiotic resistance in severe asthma. Am. J. Respir. Crit. Care Med. 2019, 200, 309–317. [Google Scholar] [CrossRef]
- Kricker, J.A.; Page, C.P.; Gardarsson, F.R.; Baldursson, O.; Gudjonsson, T.; Parnham, M.J. Nonantimicrobial Actions of Macrolides: Overview and Perspectives for Future Development. Pharmacol. Rev. 2021, 73, 233–262. [Google Scholar] [CrossRef]
- Mencarelli, A.; Distrutti, E.; Renga, B.; Cipriani, S.; Palladino, G.; Booth, C.; Tudor, G. ; Guse, J-H.; Hahn, U.; Burnet, M.; Fiorucci, S. Development of non-antibiotic macrolide that corrects inflammation-driven immune dysfunction in models of inflammatory bowel diseases and arthritis. Eur. J. Pharmacol. [CrossRef]
Figure 1.
Cigarette smoke (CS) induced upregulated colocalized NLRP3 and cleaved IL-1β in alveolar macrophages in vivo and in vitro. (a): Representative confocal images of alveolar macrophages in a CS-exposed mouse, macrophages recognized by their large cytoplasm and localization in air space of alveoli, confirmed by F4/80 staining (green). (b): Representative confocal images of cleaved IL-1β (green) and NLRP3 (red) in lung tissue sections of CS-exposed (CS) and sham-exposed control (CTR) animals. Boxed areas in the left images are shown to the right at higher magnification and in monochromatic channels to reveal alveolar macrophages (arrowheads). Blue is DAPI, scale bars are in micrometres. (c, d): MQCM analysis of NLRP3 and cleaved IL-1β in mouse alveolar tissue. Each dot represents averaged MFI value measured from alveolar tissue captured in 5 optical fields of one mouse lung. **, p<0.01, n=6 animals per group. (e, f): Representative confocal images of BAL-derived alveolar macrophages, stimulated with cigarette smoke extract (CSE), vs. vehicle control (CTR). Immunofluorescence of IL-1β (e) and NLRP3 (f) is shown in red, cleaved IL-1β in green, yellow in (f) is merged colour of NLRP3 and cleaved IL-1β colocalization. Insets are boxed areas shown at higher magnification. Results of MQCM analysis of NLRP3, IL-1β and cleaved IL-1β are shown in (g-i); each dot represents average value measured from 5 optical fields of samples from one donor. *, p<0.05, n=6 donors.
Figure 1.
Cigarette smoke (CS) induced upregulated colocalized NLRP3 and cleaved IL-1β in alveolar macrophages in vivo and in vitro. (a): Representative confocal images of alveolar macrophages in a CS-exposed mouse, macrophages recognized by their large cytoplasm and localization in air space of alveoli, confirmed by F4/80 staining (green). (b): Representative confocal images of cleaved IL-1β (green) and NLRP3 (red) in lung tissue sections of CS-exposed (CS) and sham-exposed control (CTR) animals. Boxed areas in the left images are shown to the right at higher magnification and in monochromatic channels to reveal alveolar macrophages (arrowheads). Blue is DAPI, scale bars are in micrometres. (c, d): MQCM analysis of NLRP3 and cleaved IL-1β in mouse alveolar tissue. Each dot represents averaged MFI value measured from alveolar tissue captured in 5 optical fields of one mouse lung. **, p<0.01, n=6 animals per group. (e, f): Representative confocal images of BAL-derived alveolar macrophages, stimulated with cigarette smoke extract (CSE), vs. vehicle control (CTR). Immunofluorescence of IL-1β (e) and NLRP3 (f) is shown in red, cleaved IL-1β in green, yellow in (f) is merged colour of NLRP3 and cleaved IL-1β colocalization. Insets are boxed areas shown at higher magnification. Results of MQCM analysis of NLRP3, IL-1β and cleaved IL-1β are shown in (g-i); each dot represents average value measured from 5 optical fields of samples from one donor. *, p<0.05, n=6 donors.
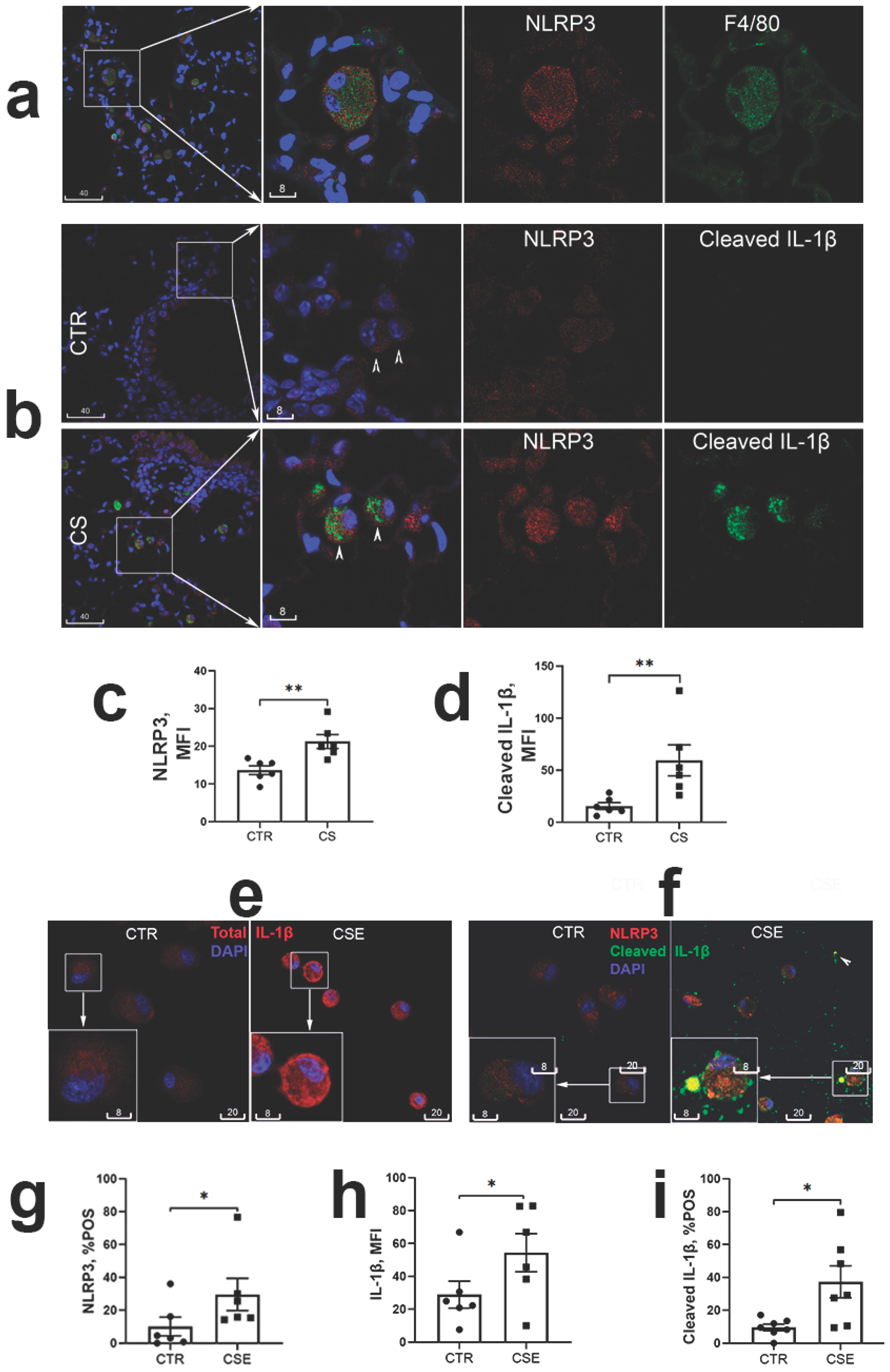
Figure 2.
The NLRP3 inflammasome activation in CSE-stimulated THP-1 macrophages. (a): Representative confocal images of NLRP3 (red, AF594) and cleaved IL-1β (green, AF488, yellow indicating colocalization with NLRP3) in THP-1 macrophages, control (CTR) vs CSE-stimulated. Insets are magnification of boxed areas, shown to the right in monochromatic channels. (b): Co-staining with two different antibodies to IL-1β (aa117-269, red) and cleaved IL-1β (Ala117 neoepitope, green) revealed reduction of total IL-1β in cells having increased cleaved IL-1β. (c-e): Blocking of CSE-induced NLRP3 and cleaved IL-1β immunofluorescence in THP-1 macrophages with Zyvad (ZYV) or Glyburide (GLY). Representative confocal images in (c) depict cleaved NLRP3 (red) and cleaved IL-1b (green, yellow being merged colour of colocalization with NLRP3) in THP-1 macrophages treated with CSE, with or without presence of ZYV/GLY, vs. vehicle control (CTR). (d), Significant reversal of CSE-induced cleaved IL-1β by Zyvad or Glyburide shown by MQCM analysis; *, p<0.05; **, p<0.01; n=3 experiments.
Figure 2.
The NLRP3 inflammasome activation in CSE-stimulated THP-1 macrophages. (a): Representative confocal images of NLRP3 (red, AF594) and cleaved IL-1β (green, AF488, yellow indicating colocalization with NLRP3) in THP-1 macrophages, control (CTR) vs CSE-stimulated. Insets are magnification of boxed areas, shown to the right in monochromatic channels. (b): Co-staining with two different antibodies to IL-1β (aa117-269, red) and cleaved IL-1β (Ala117 neoepitope, green) revealed reduction of total IL-1β in cells having increased cleaved IL-1β. (c-e): Blocking of CSE-induced NLRP3 and cleaved IL-1β immunofluorescence in THP-1 macrophages with Zyvad (ZYV) or Glyburide (GLY). Representative confocal images in (c) depict cleaved NLRP3 (red) and cleaved IL-1b (green, yellow being merged colour of colocalization with NLRP3) in THP-1 macrophages treated with CSE, with or without presence of ZYV/GLY, vs. vehicle control (CTR). (d), Significant reversal of CSE-induced cleaved IL-1β by Zyvad or Glyburide shown by MQCM analysis; *, p<0.05; **, p<0.01; n=3 experiments.
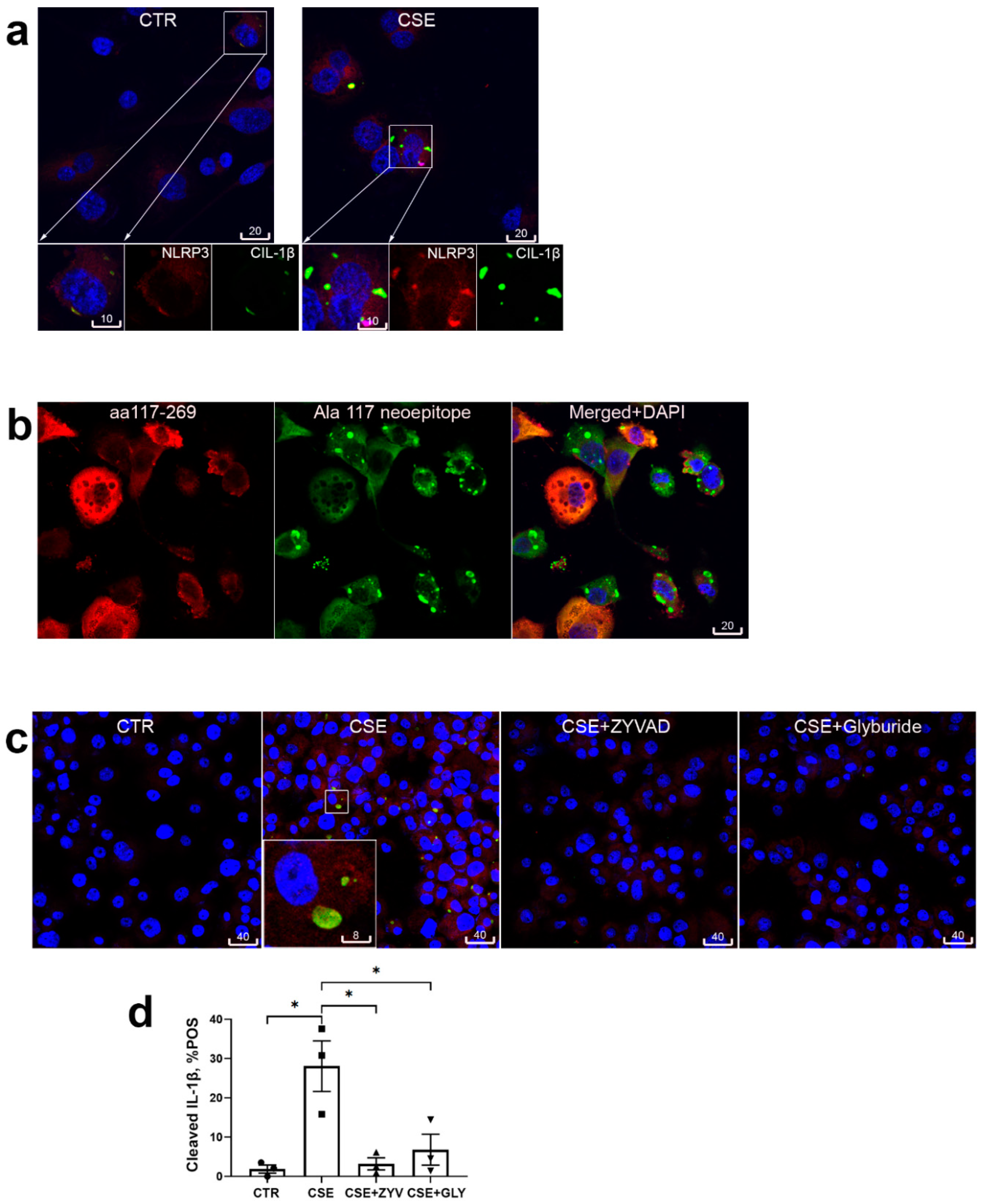
Figure 3.
Markers of accelerated senescence of alveolar macrophages in human COPD and in cigarette smoke-exposed mice. (a): In paraffin lung sections alveolar macrophages (arrows) were densely stained with Sudan Black B for lipufuscin in COPD patients and cigarette smokers (SMK) but not non-smoker non-COPD controls (CTR). *, p<0.05; **, p<0.01. (b): Lipofuscin was densely detected in alveolar macrophages (arrows) of mice chronically exposed to cigarette smoke (CS), but not controls (CTR). **, p<0.01, n=6 per group. (c): Representative confocal images of lamin B1 (red) and p53 (white) in lungs of cigarette smoke-exposed (CS) and sham-exposed (CTR) mice; alveolar macrophages in boxed areas are shown at higher magnification and in monochromatic channels in the insets. (d): MQCM analysis of lamin B1 in nuclear envelope, **, p<0.01, n=6 per group. (e): Flow-cytometric analysis of surface CD81 and CD9 expression in BAL-derived alveolar macrophages of 22 healthy control donors (CTR) and 10 individuals having COPD, *, p<0.05.
Figure 3.
Markers of accelerated senescence of alveolar macrophages in human COPD and in cigarette smoke-exposed mice. (a): In paraffin lung sections alveolar macrophages (arrows) were densely stained with Sudan Black B for lipufuscin in COPD patients and cigarette smokers (SMK) but not non-smoker non-COPD controls (CTR). *, p<0.05; **, p<0.01. (b): Lipofuscin was densely detected in alveolar macrophages (arrows) of mice chronically exposed to cigarette smoke (CS), but not controls (CTR). **, p<0.01, n=6 per group. (c): Representative confocal images of lamin B1 (red) and p53 (white) in lungs of cigarette smoke-exposed (CS) and sham-exposed (CTR) mice; alveolar macrophages in boxed areas are shown at higher magnification and in monochromatic channels in the insets. (d): MQCM analysis of lamin B1 in nuclear envelope, **, p<0.01, n=6 per group. (e): Flow-cytometric analysis of surface CD81 and CD9 expression in BAL-derived alveolar macrophages of 22 healthy control donors (CTR) and 10 individuals having COPD, *, p<0.05.
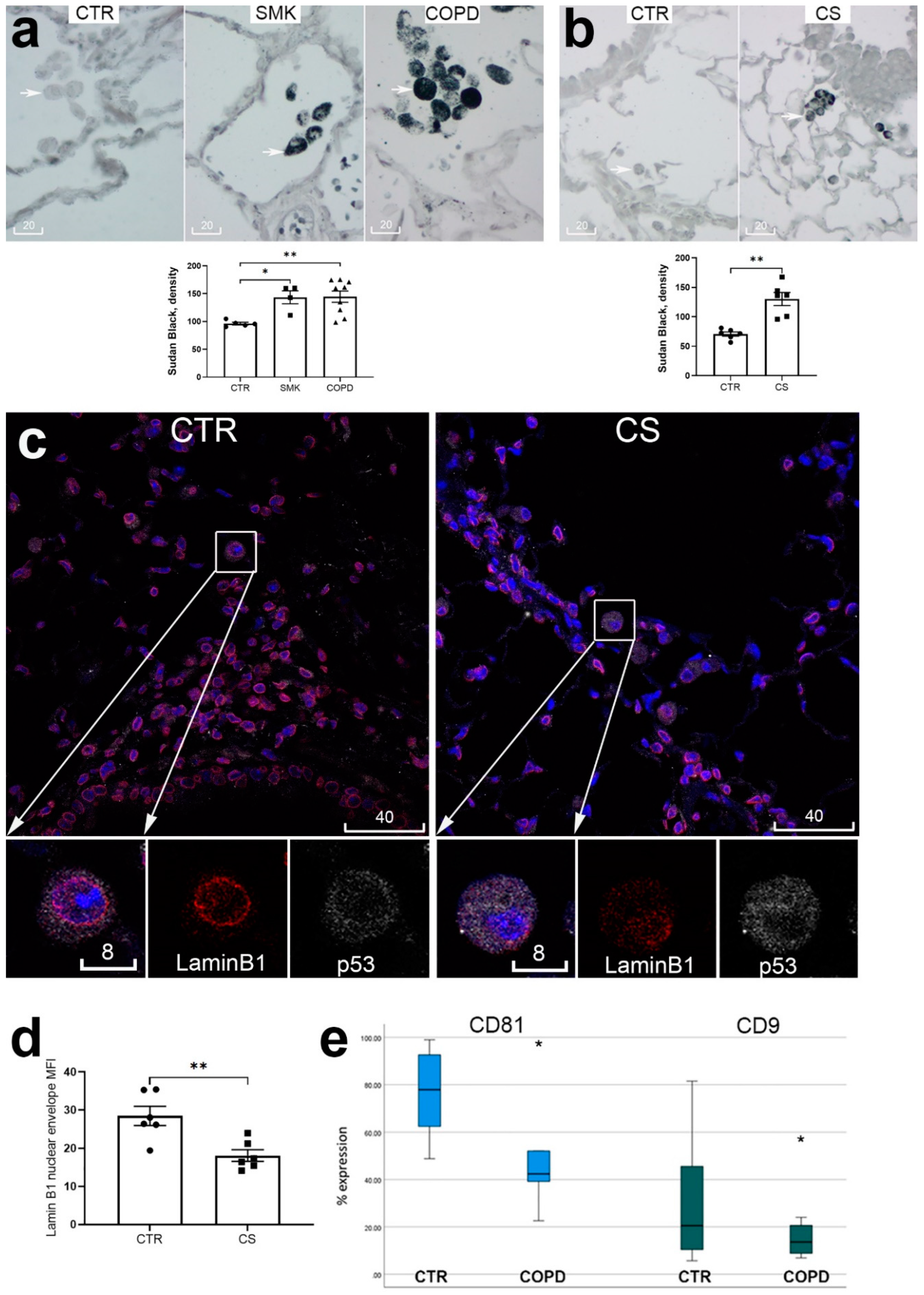
Figure 4.
Human monocyte-derived macrophages (MDM) highly expressed cell surface CD9/81 (a), markers of cell senescence lamin B1 and p53 (b, d), and cleaved IL-1β (c). CD9/81 displayed strong immunolocalization near the cell surface (a, green), whereas lamin B1 near the nuclear envelope (d, red, magenta is colocalization with DAPI). Cigarette smoke extract-exposed MDM showed significant increase of p53 detected by Western blot analysis (b, *, p<0.05, n=6 repeats), decrease of nuclear envelope lamin B1 detected by MQCM (d, red), and increase of cleaved IL-1β detected by Western blot (c, **, p<0.01, n=6 repeats). In representative confocal images in (a) and (d), blue is DAPI, scale bars are in micrometers. NEG, negative staining control.
Figure 4.
Human monocyte-derived macrophages (MDM) highly expressed cell surface CD9/81 (a), markers of cell senescence lamin B1 and p53 (b, d), and cleaved IL-1β (c). CD9/81 displayed strong immunolocalization near the cell surface (a, green), whereas lamin B1 near the nuclear envelope (d, red, magenta is colocalization with DAPI). Cigarette smoke extract-exposed MDM showed significant increase of p53 detected by Western blot analysis (b, *, p<0.05, n=6 repeats), decrease of nuclear envelope lamin B1 detected by MQCM (d, red), and increase of cleaved IL-1β detected by Western blot (c, **, p<0.01, n=6 repeats). In representative confocal images in (a) and (d), blue is DAPI, scale bars are in micrometers. NEG, negative staining control.
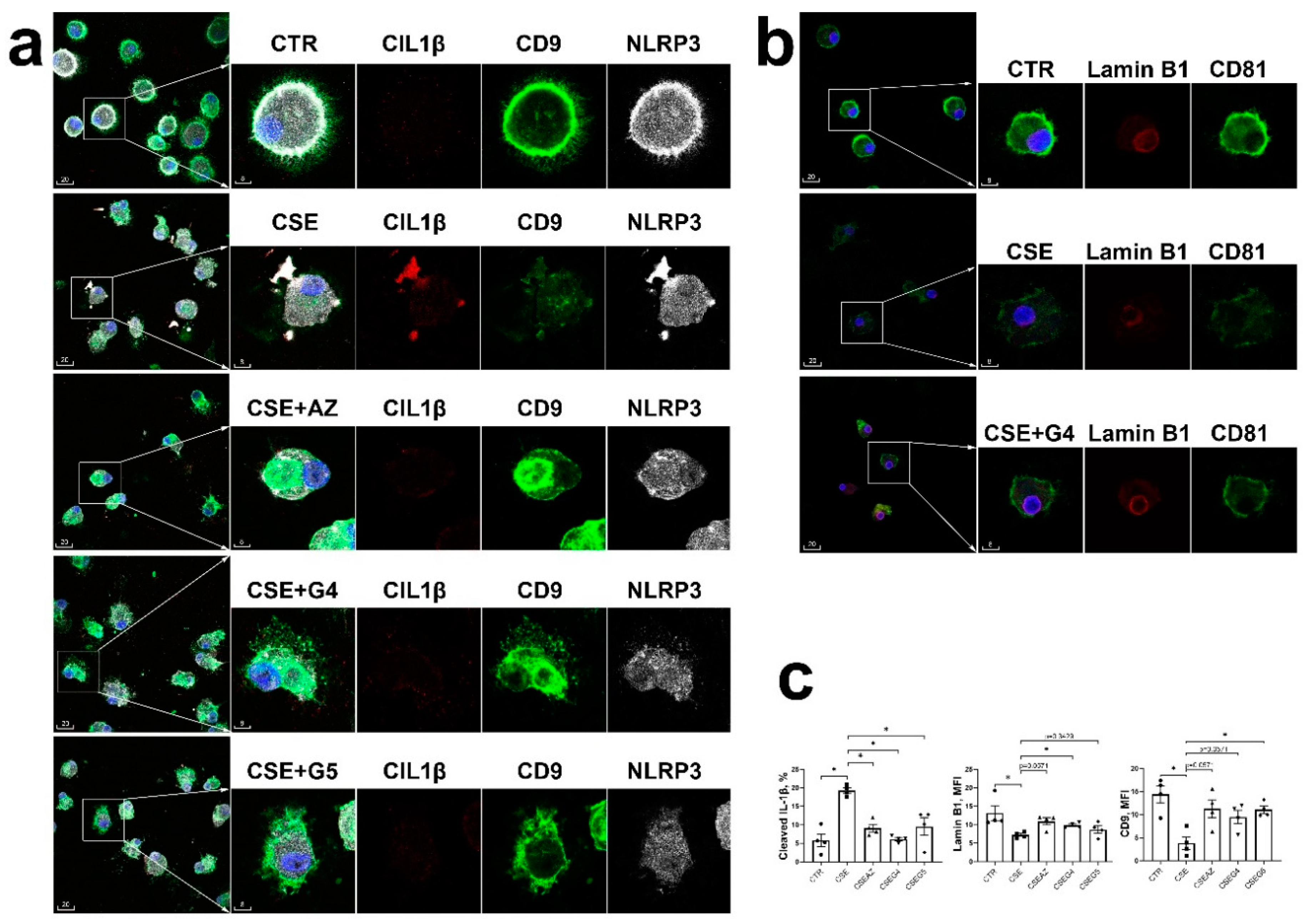
Figure 5.
Cigarette smoke extract-induced upregulated NLRP3 and cleaved IL-1β and markers of acceleration cellular senescence in monocyte-derived macrophages were reversed by anti-inflammatory macrolides. (a): Representative confocal images of cleaved IL-1β (red), NLRP3 (white) and CD9 (green) in control macrophages (CTR), and macrophages exposed to cigarette smoke extract (CSE), with or without presence of azithromycin (AZ), GS-459755 (G4), or GS-560660 (G5). (b): Representative confocal images of lamin B1 (red) and CD81 (green) in control macrophages (CTR), and macrophages exposed to cigarette smoke extract (CSE), with or without presence of G4. In (a) and (b), the boxed areas are shown at higher magnification and in monochromatic channels in the insets. Blue is DAPI. Scale bars are in micrometers. (c): MQCM analysis of parameters measured in experiments shown in (a) and (b). *, p<0.05. n=4 experiments using samples obtained from different donors.
Figure 5.
Cigarette smoke extract-induced upregulated NLRP3 and cleaved IL-1β and markers of acceleration cellular senescence in monocyte-derived macrophages were reversed by anti-inflammatory macrolides. (a): Representative confocal images of cleaved IL-1β (red), NLRP3 (white) and CD9 (green) in control macrophages (CTR), and macrophages exposed to cigarette smoke extract (CSE), with or without presence of azithromycin (AZ), GS-459755 (G4), or GS-560660 (G5). (b): Representative confocal images of lamin B1 (red) and CD81 (green) in control macrophages (CTR), and macrophages exposed to cigarette smoke extract (CSE), with or without presence of G4. In (a) and (b), the boxed areas are shown at higher magnification and in monochromatic channels in the insets. Blue is DAPI. Scale bars are in micrometers. (c): MQCM analysis of parameters measured in experiments shown in (a) and (b). *, p<0.05. n=4 experiments using samples obtained from different donors.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



